

Abstract
Triple-negative breast cancers contain a spectrum of epithelial and mesenchymal phenotypes. SUM-229PE cells represent a model for this heterogeneity, maintaining both epithelial and mesenchymal subpopulations that are genomically similar but distinct in gene expression profiles. We identified differential regions of open chromatin in epithelial and mesenchymal cells that were strongly correlated with regions of H3K27ac. Motif analysis of these regions identified consensus sequences for transcription factors that regulate cell identity. Treatment with the MEK inhibitor trametinib induced enhancer remodeling that is associated with transcriptional regulation of genes in epithelial and mesenchymal cells. Motif analysis of enhancer peaks downregulated in response to chronic treatment with trametinib identified AP-1 motif enrichment in both epithelial and mesenchymal subpopulations. ChIP-seq of JUNB identified subpopulation-specific localization, which was significantly enriched at regions of open chromatin. These results indicate that cell identity controls localization of transcription factors and chromatin modifying enzymes to enhancers for differential control of gene expression. We identified increased H3K27ac at an enhancer region proximal to CXCR7, a G-protein coupled receptor that increased fifteen-fold in expression in the epithelial subpopulation during chronic treatment. RNAi knockdown of CXCR7 inhibited proliferation in trametinib resistant cells. Thus, adaptive resistance to chronic trametinib treatment contributes to proliferation in the presence of the drug. Acquired amplification of KRAS following trametinib dose escalation further contributed to POS cell proliferation. Adaptive followed by acquired gene expression changes contributed to proliferation in trametinib resistant cells, suggesting inhibition of early transcriptional reprogramming could prevent resistance and the bypass of targeted therapy.
Introduction
Intratumor heterogeneity arising from differences in gene regulation and genetic mutation represents a significant challenge to therapeutic durability and prevention of clinical drug resistance. Deep sequencing of tumors has revealed that mutations are not present uniformly throughout the tumor (1). This heterogeneity increases the likelihood that drug tolerant populations can develop and persist through treatment, leading to relapse. Indeed, whole exome sequencing of circulating tumor DNA isolated from breast cancer patients during treatment revealed the enrichment of low frequency mutations that could contribute to therapeutic resistance (2). Single nuclei sequencing of breast cancer tumor cells showed point mutations evolved slowly giving significant clonal diversity, with many of the mutations occurring at low frequency (3). Furthermore, a spectrum of interconvertible phenotypic and functional states has been linked to clinical drug resistance (4,5). This finding is consistent with analysis of circulating tumor cells revealing that the cells are not always committed to a single cell state, but display a mixture of phenotypes that are differentially sensitive to targeted therapy (6). Recent work has demonstrated that chromatin remodeling that alters gene expression plays an essential role in cell state switching, which allows cells to access drug tolerant persister phenotypes (5,7). Cell state switching has been shown to mediate resistance through the transcriptional activation of genes that promote survival or proliferation (8,9). Thus, genetic heterogeneity and cell state plasticity play essential roles in mediating drug resistance and selection of drug tolerant subpopulations.
Triple negative breast cancer (TNBC) is a heterogenous disease clinically characterized by the absence of the estrogen, progesterone and HER2 receptors. Unlike other breast cancer subtypes, there are no FDA-approved targeted therapies currently available for TNBC patients. Instead, these patients are treated with a combination of surgery, radiation therapy, and chemotherapy (10). While TNBC patients have the best pathological complete response rates of any breast cancer subtype, TNBC patients with residual disease have the lowest survival (11). Deep sequencing of TNBC tumors revealed a broad distribution in the number of clonal subpopulations within individual tumors, suggesting that genetic heterogeneity contributes to outcome (12). These results indicate that heterogeneity in TNBC breast cancer poses a significant obstacle to improve patient outcomes. Therefore, identifying durable targeted therapies for TNBC patients is essential.
Despite their initial promise, kinase inhibitors have often been ineffective as monotherapies due to adaptive transcriptional activation of genes that occurs in response to treatment leading to bypass of the targeted kinase inhibition (13,14). The Cancer Genome Atlas Project evaluated 510 breast cancer tumors revealing the overexpression of genes in the MAPK signaling pathway in TNBC patients, including EGFR, KRAS, and BRAF (15). This result led to ongoing clinical trials examining the safety and efficacy of the MEK inhibitors selumetinib and trametinib in breast cancer patients. A window-of-opportunity trial studying TNBC patients demonstrated that these tumors rapidly respond by increasing expression of receptor tyrosine kinases (RTKs) and reactivating MAP kinase signaling (16). Additional work examining the adaptive response to AKT inhibition revealed a similar upregulation of RTKs in response to AKT inhibition in several preclinical models (17). Combining a BET bromodomain inhibitor with targeted therapy abrogated the adaptive transcriptional activation by inhibiting the formation of a functional pTEFb complex (16,18).
Our goal herein was to understand how the chromatin landscape influences the response to targeted therapy in the epithelial and mesenchymal subpopulations of the TNBC cell line SUM-229PE. These cells were isolated from the pleural effusion of a breast cancer patient following treatment with chemotherapy, and express cytokeratins consistent with their origin from luminal breast epithelial cells. A comprehensive histological study of breast cancer cell lines identified two distinct subpopulations that are maintained in the SUM-229PE line that are distinguished by unique morphologies and differential expression of epithelial markers (19). An siRNA screen in these SUM-229PE subpopulations demonstrated that a regulatory subunit of the SWI/SNF chromatin remodeling complex, Smarcd3/Baf60c, is necessary to maintain the mesenchymal phenotype (20). RNAi silencing of the SWI/SNF chromatin-remodeling factor Smarcd3/Baf60c gave a robust conversion of SUM-229PE mesenchymal cells to the epithelial EpCAM+ phenotype, suggesting the mesenchymal and epithelial variation is regulated by the different chromatin states of the two subpopulations (20). Using studies of chromatin accessibility, histone acetylation and RNA expression, we identified differences in the chromatin landscape between the epithelial and mesenchymal subpopulations, and how the landscape changes in response to therapy. These findings provide insight into how resistance emerges during treatment with targeted therapy.
Materials and Methods
Cell Culture
Low-passage SUM-229PE cells (BioIVT) and isolated subpopulations were cultured in Ham’s F-12 Media (Gibco cat. # 11765-054) with 5% fetal bovine serum (VWR cat. # 97068-085), 5 μg/mL insulin (Gibco cat. # 12585-014), 1 μg/mL hydrocortisone (Sigma cat. # H0396), 1% penicillin-streptomycin solution (Gibco cat. # 15140-014) and 5 mM HEPES (Corning cat. # 25-060-CI). SUM-229 POS R1 and POS R2 were continuously cultured in 10 nM trametinib for six weeks, changing the media every three days. These cells were then trypsinized and dose escalated to 30 nM trametinib. POS R1 and POS R2 cells were continuously cultured in Ham’s F-12 media as formulated above, with 30 nM trametinib following dose escalation. SUM-229PE cells originally obtained from Asterand Biosciences in 2011, authenticated by RNA-Seq, and tested for mycoplasma by DNA staining in 2017. Cells were cultured for a maximum of one month before treatment.
FAIRE Sequencing
2.5 million SUM-229 cells were treated with or without 30 nM trametinib for 24 hours. Cells were crosslinked and nuclei were prepared using the Active Motif Chromatin Preparation kit (Active Motif cat. # 53046) according to manufacturer’s instructions except that 0.25 million formaldehyde crosslinked Drosophila S2 cells were spiked into the SUM-229 cells just prior to nuclei preparation. Nuclei were resuspended in 90 μL Lysis Buffer A (21) and 10 μL of MegaShear (Triangle Biotechnology, Inc.) was added to each tube prior to sonication. Sonication was performed in a Covaris E110 instrument for 4 minutes per sample using conditions described in (22). Following sonication, insoluble debris was removed by centrifugation and samples were digested with 200 μg RNase (Qiagen) at 37˚C for 30 minutes. Ten μL of sample was removed for input and digested with 40 mg Proteinase K (Worthington) for 1 hour at 55˚C, followed by 2 hours at 80˚C to reverse the crosslinks, then purified using a silica matrix column (Zymo Research, ChIP DNA Clean and Concentrate kit, cat . # D5201). FAIRE was performed on the remaining 90 μL of sonicated chromatin by purification on a silica matrix column as described in (23). DNA was quantitated using fluorometry (Qubit dsDNA High Sensitivity Assay Kit, Invitrogen). Average peak fragment size of input and FAIRE DNA was confirmed using gel electrophoresis. Approximately 50 ng of fragmented DNA was adapter ligated and purified using the KAPA stranded HyperPrep Kit (Roche cat. # 07962347001) and Illumina TruSeq Indexed Adapters according to the manufacturer’s instructions. Dual size selection was performed after 18 cycles of PCR amplification of the cDNA library according to manufacturer recommendations. A multiplexed pool of libraries was sequenced using the 75-cycle NextSeq 500/550 High Output v2 sequencing kit (Illumina cat. # FC-404-2005) on the Illumina NextSeq500 to yield approximately 5.0 × 107 75 bp single-ended reads per sample. Following removal of adaptor sequences (cutadapt v1.12), reads were filtered for quality using fastq_quality_filter in FASTX-Toolkit (v0.0.12) with options -Q 33, -p 90, and q 20. Reads were then aligned to hg19 using STAR (v2.5.2b). Tracks of FAIRE-seq signal were created by visualizing RPM normalized bigWigs on UCSC genome browser.
Chromatin Immunoprecipitation Sequencing
ChIP-Seq for H3K27ac was performed using the ChIP-grade antibody for histone H3K27ac (Active Motif cat. # 39133). ChIP-Seq for JUNB was performed using the ChIP-grade antibody for JUNB (Cell Signaling cat. #3753S). Fixation and immunoprecipitation were performed as previously described (16,24). See supplementary methods for a detailed protocol.
Identification of Differential Regions of Signal Enrichment in FAIRE and ChIP-Seq
Regions of signal enrichment (ROE) were identified for each experimental condition (MACS2 v2.1.2). The Score per million (SPM) was calculated for each region of enrichment by dividing the MACS2 derived -log10(p-value) by the total -log10(p-value) then by 1,000,000 for each sample. A union set of all ROE was created where any overlapping ROE was resolved by keeping the ROE with the highest SPM value. Raw read counts for ROE were tabulated using the featureCounts function in the Rsubread R package (v. 1.34.7) (25). Differential signal between experimental conditions were identified using DESeq2 (v. 1.24.0) at a padj < 0.05 (26). See supplementary methods for a detailed protocol of FAIRE-Seq, JUNB ChIP-Seq, and H3K27ac ChIP-Seq analysis.
Comparison of Differential Genes in SUM-229PE Subpopulation to Differential Genes in Patient Tumors
Raw RSEM values derived from RNA-seq performed on patient tumors classified as basal-like (patients 1-5) or claudin-low (patient 6) were downloaded from GEO (GSE107502). RSEM values were derived using methods described in previous work (16). Differentially expressed genes were identified in basal-like tumors using DESeq2, where the independent variables were pre- vs. post- treatment and patient number. Differentially expressed genes were also identified in the claudin-low tumor using DESeq2. For the POS vs. basal-like comparison a shared set of test genes was first defined, followed by the isolation of genes with significantly different expression (padj < 0.05), and finally identifying the gene overlap of both experiments. Statistical significance was determined using a Fisher’s exact test. The same approach was applied to NEG v. claudin-low, as well as the POS v. claudin-low, and NEG v. basal-like.
Integration of JUNB ChIP-seq and FAIRE-seq data
Sites of differential JUNB binding present in POS or NEG cells were identified using DESeq2 on MACS2 (default parameters) defined JUNB peaks. The average normalized (RPM) FAIRE signal in both POS and NEG cells was compiled for regions of increased JUNB binding in POS cells (as well as NEG cells) using python library pyBigWig (v0.3.17). Statistical significance for the comparison of FAIRE signal at POS or NEG differential JUNB binding sites was calculated using Mann-Whitney U test.
RNA Sequencing
RNA-Seq libraries were prepared as previously described (16). See supplementary methods for a detailed protocol.
Fluorescence-Activated Cell Sorting
SUM-229PE cells were stained and sorted by fluorescence-activated cell sorting (FACS) as previously described (20). See supplementary methods for a detailed protocol.
Crystal Violet Colony Formation Assay
Cells were fixed and stained as previously described (18). See supplementary methods for a detailed protocol.
RNA Isolation From Fixed Isolated and Parental Cells
Approximately 10 million SUM-229PE cells were treated for 24 hours with 30 nM trametinib or DMSO control. Additionally, 5 million POS cells and 5 million were treated with 30 nM trametinib or DMSO control. Cells were fixed using the MARIS method as previously described (27). See supplementary methods for a detailed protocol.
Xenograft Growth Assay
Female NOD scid gamma mice were given a mammary fat pad injection of 2 × 106 POS, POS R1, NEG, or NEG R cells suspended in 50% Matrigel. See supplementary methods for a detailed protocol.
Beta-Galactosidase Staining
Cells were stained using Senescence β-Galactosidase Staining Kit (Cell Signaling Technology cat. # 9860) according to the manufacturer’s instructions. See supplementary methods for a detailed protocol.
siRNA Transfection
Cells were plated in a 96-well plate and transfected with 25 nM of siRNA targeting KRAS or CXCR7. Individual siRNA from siGENOME smartpools targeting CXCR7 (GE Dharmacon cat. # MQ-013212-03-0002) and KRAS (GE Dharmacon cat. # MQ-005069-00-0002) were resuspended in siRNA buffer (GE Dharmacon cat. # B-002000-UB-100). Cells were transfected using RNAiMAX (Invitrogen cat. # 13778-075) with 25 nM siRNA. siGENOME non-targeting pool #2 was used as control siRNA (GE Dharmacon cat. # D-001206-14-05). Cells were incubated for 24 hours following transfection, then media was aspirated. Cells were treated with 30 nM trametinib for 72 hours, then stained with Hoescht 33342 at 2.5 μg/mL for 30 minutes at 37°C. Plates were imaged using a Thermo Cellomics ArrayScan VTI, capturing 25 frames per well. Cell number was quantified and siRNA target was compared siRNA control.
RNAscope detection of KRAS RNA
KRAS RNA was detected using the RNAscope® 2.5 LS Probe for Hs-KRAS-O1 (ACD Biotechne cat. # 522968) in a Bond RX autostainer (Leica Biosystems) following the manufacturer’s directions. TSA-Cy5 and DAPI were used to visualize RNAScope signal and nuclei, respectively. Slides containing RNAscope fluorescently labeled cells were scanned either in the Aperio ScanScope FL or the Aperio Versa Digital Pathology Scanner using a 20X objective (Leica Biosystems). Images were archived in TPL’s eSlide Manger database (Leica Biosystems). Images were manually annotated for regions of interest using Tissue Studio software (Definiens Inc.Tissue Studio version 2.7 with Tissue Studio Library version 4.4.2). See supplementary methods for a detailed protocol.
Single Cell RNA Sequencing
Single cell RNA Sequencing of approximately 5000 POS cells and 5000 POS R1 cells was performed according to the manufacturer specification (10X Genomics cat. # 1000092, 1000074, 120262). See supplementary methods for a detailed protocol.
Results
SUM-229PE Subpopulations are Genotypically Similar but Epigenetically Distinct
Extending the studies previously performed on TNBC cell lines (19), immunostaining of SUM-229PE cells (Fig. 1A) revealed two subpopulations. One was marked by an elongated phenotype, prominent filamentous actin fibers (green) and perinuclear vimentin (red), while the other displayed a rounded phenotype and stained positive for the epithelial marker EpCAM (blue). Flow cytometry of SUM-229PE cells using epithelial markers EpCAM and CD49f (integrin α6) confirmed two subpopulations: EpCAM, CD49f positive (POS), and EpCAM, CD49f negative (NEG) (Fig. 1B). Following six weeks of isolated culture, less than 1% of the NEG subpopulation stained positive for EpCAM and CD49f. We analyzed whole-exome sequencing of the isolated subpopulations and called variants using a germline mutation caller. Whole-exome analysis yielded similar variant calls as those made with a somatic mutation caller (16). We found that variant read abundance was highly correlated between POS and NEG cells, demonstrating that these cells are genetically similar (Fig. 1C, Supp. Table 1 and 2). Karyotypes of the POS and NEG cells demonstrated similar ploidy and many of the same chromosomal aberrations, although NEG cells contained a higher frequency of dicentric chromosomes (Supp. Table 3). Because of the genomic similarity of the two subpopulations, as well as the conversion of NEG to POS upon loss of Smarcd3, we hypothesized that the distinct phenotypes resulted primarily from differences in the chromatin landscape (20).
Figure 1. TNBC Subpopulations Are Genotypically Similar But Epigenetically Distinct.
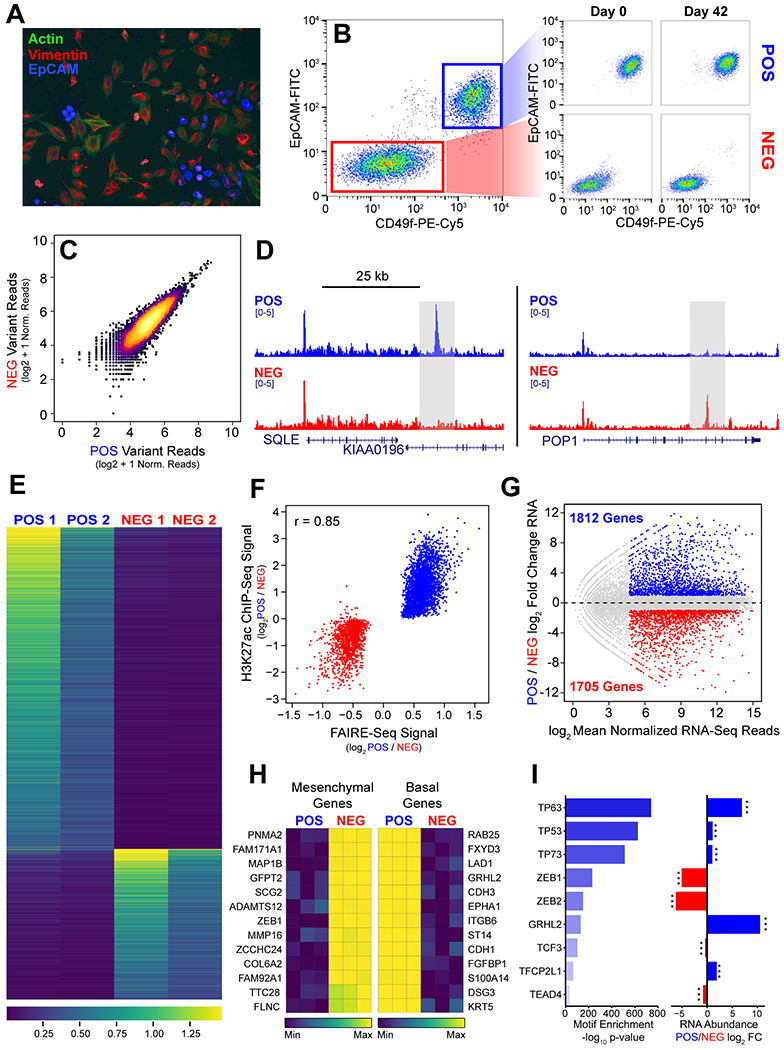
(A) Immunofluorescence of SUM-229PE cells stained for F-actin (green) EpCAM (blue) and vimentin (red). (B) Flow cytometry of SUM-229PE cells stained for EpCAM and CD49f (ITGA6). EpCAM negative subpopulation (NEG) highlighted in red, and positive subpopulation (POS) highlighted in blue. (C) Comparison of the abundance of whole exome sequencing variants in POS and NEG cells relative to hg19. (D) Representative tracks depicting regions of significant differential FAIRE signal at enhancers in the POS and NEG cells (gray box). (E) Heatmap of average normalized FAIRE signal at differential peaks in biological replicates of both POS and NEG cells ranked by -log10 p-value. Blue to yellow represents low to high signal. (F) Average normalized H3K27ac signal at differential FAIRE peaks identified in POS (blue) and NEG (red) cells. Pearson correlation coefficient = 0.85. (G) Differential gene expression in POS cells relative to NEG cells. Significant genes with greater than two-fold difference in expression, normalized RNA reads > 25, and a p-value < 0.05 are highlighted (POS blue, NEG red). (H) Row-mean normalized expression of genes that were the most differentially-expressed in mesenchymal (left) and basal (right) breast cancer gene sets (from Charafe et al.). Maximum expression indicated in yellow. (I) Consensus motifs identified in FAIRE regions enriched in POS cells. Differential RNA expression of genes encoding transcription factors identified by motif analysis (right). *** indicates adjusted p-value < 0.001
We identified regions of enhanced chromatin accessibility by Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE-seq) (21). We identified all regions of FAIRE signal enrichment in the POS and NEG subpopulations by MACS2 and then used DESeq2 to identify subpopulation-specific regions. We identified 3,130 regions (1.6% of 194,421 total) enriched in the POS subpopulation and 1,455 regions (0.7% of 192,421 total) enriched in the NEG subpopulation (Fig. 1D–E).
We then performed ChIP-seq for histone H3 lysine 27 acetylation (H3K27ac) to identify variation in potential enhancers. As before, regions of H3K27ac enrichment were called by MACS2 and regions with differential H3K27ac signal were identified by DESeq2. 17,073 peaks (30% of 57,075 total) were selectively enriched in POS cells (Fig. S1A, blue) and 15,430 peaks (27% of 57,075 total) were selectively enriched in the NEG cells (Fig. S1A, red). We then compared differences in chromatin accessibility with variation in the acetylation signal. Overall, FAIRE signal strongly correlated with H3K27ac signal (Pearson correlation r = 0.85) (Fig. 1F). Previous studies have shown that H3K27ac induces chromatin relaxation, and our data demonstrates that these regions of accessible chromatin are often highly acetylated (28,29).
We also evaluated transcriptional differences between POS and NEG subpopulations by RNA-seq. To assess whether prolonged culture of isolated POS and NEG cells alters the transcriptome, we performed RNA-seq on the isolated cells and recently sorted subpopulations. We found that gene expression in recently sorted subpopulations and POS (r=0.91) and NEG (r=0.95) cells cultured in isolation were highly correlated (Fig. S1B). We then identified genes by DESeq2 that were significantly differentially expressed between the POS and NEG cells. To define the most significant expressed genes, we filtered the DESeq2 results for genes with a minimum of 25 reads and at least a two-fold difference in expression. We identified 1,812 genes enriched in POS cells, and 1,705 genes enriched in NEG cells (Fig. 1G). GO analysis of genes preferentially expressed in POS cells are associated with the maintenance of cell-cell adhesion (Fig. S1C–S1E) and genes expressed in NEG cells are associated with binding of extracellular matrix (Fig. S1F–S1I).
We identified genes that are associated with mesenchymal and basal-like breast cancer cells by comparing gene expression between the basal-like cell line, HCC-1806, and the mesenchymal breast cancer cell line, SUM-159 (data not shown). We observed that POS-specific genes were significantly enriched among HCC-1806 specific genes (hypergeometric test p = 5.74 × 10−126). A similar result was observed when we compared the NEG-specific with SUM-159-specific genes (hypergeometric test p = 5.50 × 10−45) (Fig. S1J). Importantly, the intersections of the converse associations were not significant. We next compared gene expression in POS and NEG cells to gene sets used to classify mesenchymal and epithelial breast cancers (30). NEG cells express genes found in mesenchymal breast cancer (Fig. 1H, left, Fig. S1K), including the transcription factor ZEB1 and matrix protein COL6A2. In contrast, POS cells specifically express genes associated with basal breast cancers (Fig. 1H, right, Fig. S1L), including the transcription factor GRHL2 as well as epithelial markers CDH1 and KRT5.
Our SUM-229PE results are consistent with previous studies that demonstrated subpopulations with distinct gene expression profiles are observed in several breast cancer cell lines (31). We performed a similar analysis on the basal-like breast cancer cell line SUM-149. Flow cytometry analysis (Fig. S1M) and RNA-Seq revealed distinct profiles in these isolated subpopulations (data not shown).
We associated differential chromatin accessibility with potential transcriptional regulators in the POS subpopulation. Enriched motifs were identified by comparing accessible chromatin in POS cells to accessible chromatin common to POS and NEG cells. We identified enrichment of the consensus sequences of epithelial transcription factors p63 and GRHL2 (Fig. 1I, left). Expression of the associated genes TP63 and GRHL2 was upregulated in POS cells relative to NEG cells (Fig 1I, right). Interestingly, analysis also identified the motif for ZEB1, a transcription factor expressed in mesenchymal cells to repress epithelial genes. Although chromatin accessibility was detected at these regions, ZEB1 expression was enriched only in NEG cells (Fig.1I, right). Taken together, these data demonstrate that SUM-229PE heterogeneity is marked by chromatin and transcriptional variation reflecting epithelial and mesenchymal differentiation states.
Differential Remodeling of the Enhancer Landscape is Associated with the Transcriptional Response to Trametinib
To confirm that POS and NEG cells respond to MEK inhibition, we first treated these cells with a clinically relevant dose of 30 nM trametinib for 24 hours and evaluated change in the cell cycle relative to control cells (21). We observed a G1 arrest in POS cells (Fig. 2A, blue), increasing the frequency of cells in G1 from 30% to 87% (Fig. S2A, blue). The impact of trametinib was less prominent in the NEG cells (Fig. 2A, red), increasing the frequency of cells in G1 from 41% to 67% (Fig. S2A, red). We then assessed changes in the enhancer landscape following trametinib treatment. Previous studies have demonstrated that breast cancer cells respond to MEK inhibition by altering the enhancer and transcriptional landscape (14). To assess how acute trametinib treatment impacted the enhancer landscape of the SUM-229PE subpopulations, we performed H3K27ac ChIP-seq following 24 hours of trametinib treatment. We identified regions of H3K27ac signal enrichment using MACS2, and then identified subpopulation-specific regions of acetylation signal by DESeq2. In POS cells, we identified 9,788 H3K27ac peaks that increase in response to trametinib, and 10,136 peaks that decrease (Fig. 2B). In NEG cells, we identified 2,395 H3K27ac peaks that increase and 3,797 peaks that decrease (Fig. 2C). H3K27ac dynamics demonstrated a greater number of gained and lost regions of H3K27ac signal and a wider dynamic range in the POS cells compared to the NEG cells.
Figure 2. Differential Remodeling of the Enhancer Landscape is Associated with the Transcriptional Response to Trametinib.
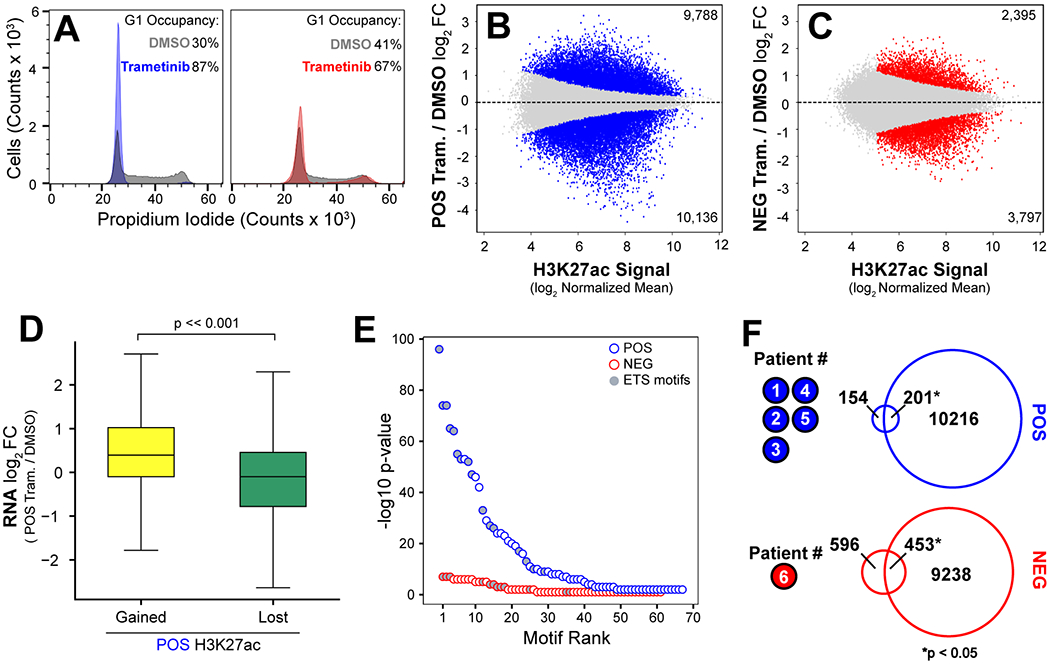
(A) Cell cycle analysis of POS cells (left) treated with DMSO (grey) or 30 nM trametinib (blue) for 24 hours, and NEG cells (right) treated with DMSO (grey) or 30 nM trametinib (red) for 24 hours. (B-C) Regions of differential H3K27ac enrichment in POS cells (blue) and NEG cells (red) after 24 hours of treatment with 30 nM trametinib or DMSO control. Highlighted points indicate regions with an adjusted p-value < 0.05 and log2 fold change less than or greater than 0. Gray points indicate regions without significant change following treatment. (D) Genes proximal to either gained or lost regions of H3K27ac following treatment have significantly different levels of expression (p-value << 0.001 Mann-Whitney U). (E) Significance of motif enrichment at regions of H3K27ac loss following trametinib treatment in POS (blue) and NEG (red) subpopulations. Gray fill indicates ETS family motifs. (F) Overlap of genes differentially expressed in POS cells following 24 hours of trametinib treatment (top, blue) and genes differentially expressed in five TNBC patients with basal-like breast cancer treated with trametinib for 7 days. Intersection of differentially expressed genes in NEG cells following 24 hours of treatment (bottom, red) with genes differentially expressed in a TNBC patient with mesenchymal breast cancer treated for 7 days. Significance determined by Fisher’s Exact Test.
We next compared the transcriptional response to trametinib in SUM-229PE subpopulations to POS and NEG isolated cells. The transcriptomes of FACS-sorted subpopulations were strongly correlated with those that were in culture for 6 weeks (POS, r =0.90 and NEG, r=0.92) (Fig. S2B). We then determined the transcriptional response to acute trametinib treatment in POS and NEG cells relative to untreated controls. We identified 882 uniquely upregulated and 1,022 uniquely downregulated genes in POS cells (Fig. S2C). GO analysis of downregulated genes identified in POS cells are involved in cell cycle regulation and cell division (Fig. S2D), consistent with cell cycle analysis (Fig. 2A, Fig. S2A). Similar to the changes in enhancer landscape, we observed a more dynamic response in the POS cells relative to the NEG cells. We identified 508 uniquely upregulated and 235 uniquely downregulated genes in NEG cells (Fig. S2E). GO analysis of genes upregulated in NEG cells identified the genes previously implicated in trametinib resistance (Fig. S2F) (32).
To assess the impact of trametinib treatment on enhancers, we evaluated the relationship between gene expression and H3K27ac. We observed a significant increase in the expression of genes associated with a gain of H3K27 signal, and significant decrease in the expression of genes associated with loss of H3K27ac signal (Fig. 2D). We hypothesized that sites of decreased H3K27ac found in trametinib-responsive POS cells would indicate reduced activity of transcription factors downstream of MAPK. Therefore, we analyzed H3K27ac peaks downregulated in response to trametinib treatment (compared with regions without change in acetylation). Several MAPK-regulated ETS motifs were highly enriched in H3K27ac peaks downregulated in POS cells (Fig. 2E, blue). However, we did not observe similar enrichment of ETS motifs in NEG cells, suggesting that MEK inhibition primarily effects these transcription factors in POS cells.
We next compared the transcriptional response observed in the POS and NEG subpopulations after trametinib treatment to the transcriptional response in tumors from patients with basal-like and claudin-low breast cancer who participated in a 7-day trametinib window-of-opportunity trial (16). We explored the transcriptional responses to trametinib in our preclinical model with those of patients by comparing differentially expressed genes in the POS cells to basal-like tumors and those in the NEG cells to a claudin-low patient tumor. A significant fraction of genes (n = 201) were shared between the POS transcriptional response and the basal breast cancers (Fig. 2F, blue). A significant fraction of genes (n = 453) in the NEG transcriptional response was shared with those of Patient #6, a claudin-low breast cancer (Fig. 2F, red). Notably, no significant overlap occurred in the converse comparison. These data indicate that the subpopulations of the SUM-229PE cell line are reflective of the variability observed in the drug response of TNBC patients.
Enhancer Remodeling and Transcriptional Response to MEK inhibitor Persist in Chronic Treatment
Recent studies have shown that the frequency of epithelial and mesenchymal subpopulations can change in response to treatment with kinase inhibitors (6). Risom et al. demonstrated that drug treatment may induce cell-state switching in basal-like breast cancer to overcome tumor cell growth arrest (5). This suggests that chronic treatment with targeted therapy applies selective pressure for cells that readily adapt to the presence of the drug. To study how differential chromatin remodeling of epithelial and mesenchymal cells may influence the clinical response to chronic treatment, we interrogated the response of POS and NEG cells during treatment with 10 nM trametinib for five weeks. Cell cycle analysis demonstrated that the S and G2 peaks were almost entirely lost in POS cells (Fig. 3A). In contrast, NEG cells (Fig. 3B) continued to slowly proliferate during the course of chronic treatment. We further quantified proliferation of POS and NEG cells by crystal violet staining during chronic treatment (Fig. 3C). Increased staining at Day 21 and 49 of treatment in NEG cells indicates that these cells continue to proliferate in the presence of the drug, while small colonies begin to emerge in the POS cells by Day 21, suggesting the onset of a resistant phenotype.
Figure 3. Unique Enhancer Remodeling and Transcriptional Response Persists Following Chronic Trametinib Treatment.
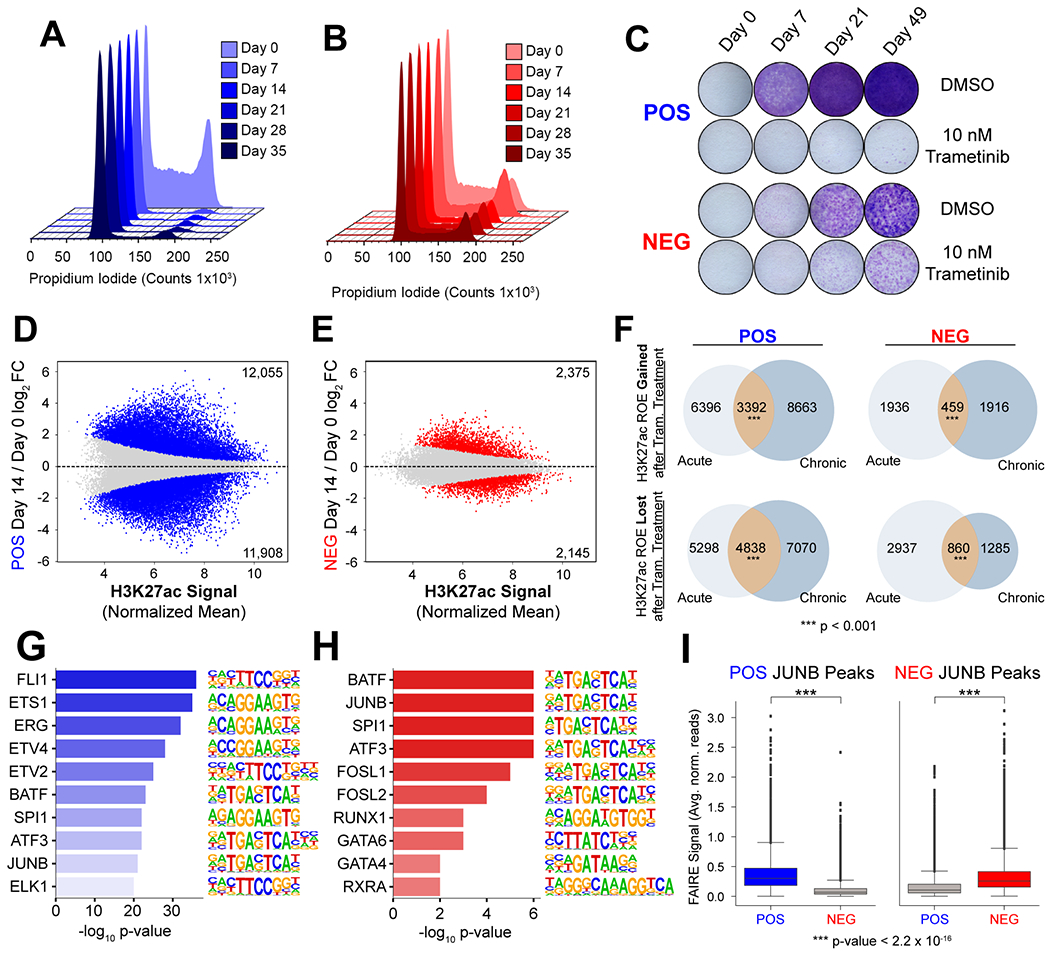
(A) Cell cycle analysis of POS cells during treatment with trametinib from Day 0 to Day 35 (light to dark blue). (B) Cell cycle analysis of NEG cells during treatment with trametinib from Day 0 to Day 35 (light to dark red). (C) Crystal violet staining of POS and NEG cells following treatment with DMSO or 10 nM trametinib for the for the time indicated. (D) Differential H3K27 acetylation in POS cells at Day 14 of trametinib treatment relative to Day 0. Statistically significant regions are highlighted in blue. (E) Differential H3K27 acetylation in NEG cells at Day 14 of trametinib treatment relative to Day 0. Statistically significant regions are highlighted in red. (F) Comparison of regions with differential H3K27 acetylation in response to 24 hours or 14 days of treatment. Significance determined by permutation test. (G) Motif analysis of H3K27ac peaks significantly downregulated at D14 in POS cells in response to trametinib treatment. (H) Motif analysis of H3K27ac peaks significantly downregulated at D14 in NEG cells in response to trametinib treatment. (I) Quantification of FAIRE signal in POS and NEG cells at POS cell specific JUNB peaks (left panel) and NEG cell specific JUNB peaks (right panel).
We evaluated changes in the enhancer landscape by performing H3K27ac ChIP-seq in POS and NEG cells following two weeks of 10 nM trametinib treatment. Similar to that observed following acute treatment, POS cells exhibited a more dynamic response than NEG cells, with 23,963 differential regions of H3K27ac signal (12,055 increased, 11908 decreased) compared to 4,520 (2,375 increased, 2,145 decreased) in the NEG cells (Fig. 3D, E). To determine the degree to which the dynamic enhancer landscape established during the acute phase of treatment is maintained over prolonged treatment, we compared both increased and decreased H3K27ac peaks observed after 24 hours and 14 days of treatment. We found a significant fraction of the remodeled enhancer landscape persists over the course of treatment in both subpopulations at both gained and reduced regions of H3K27ac enrichment (Fig. 3F).
To identify potential transcription factors involved in response to prolonged trametinib treatment, we performed motif analysis of H3K27ac peaks decreased in POS and NEG cells using HOMER. We observed an enrichment of ETS motifs at these regions of H3K27ac signal loss relative to regions unchanged after treatment in POS cells, which were absent from the most enriched motifs in the NEG cells (Fig. 3G–H). Identification of ETS motifs demonstrates a similarity between acute and extended treatment. Motif analysis of H3K27ac peaks increased in POS cells reveals enrichment of p63 and GHRL2, as well as mediators of the integrated stress response, ATF4 and DDIT3 (Fig. S3A). In contrast, we observe enrichment of SRF and SMAD3 in NEG cells (Fig. S3B).
A recent study has demonstrated that chromatin states influence the localization of transcription factors (33). We observed JUNB motif enrichment at dynamic sites of H3K27ac in both POS and NEG cells following prolonged treatment. To determine if subpopulation specific JUNB localization corresponded to differential chromatin accessibility prior to treatment, we performed JUNB ChIP-seq. We then identified JUNB sites that were specific to POS or NEG cells and associated these regions with FAIRE signal. We found that JUNB sites unique to POS cells had significantly increased FAIRE signal relative to the signal at the same regions in NEG cells (Fig 3I, left). We observed a similar trend between JUNB binding and chromatin accessibility at JUNB binding sites unique to NEG cells (Fig 3I, right). These results indicate that subpopulation-specific chromatin accessibility may direct the localization of AP-1 family members.
RNA-seq of NEG and POS cells was used to determine the persistence of the transcriptional response during the course of chronic trametinib treatment. In POS cells, we identified 4,904 genes (37% of expressed genes) that change at least two-fold in response to chronic trametinib treatment (Fig. S3C). Hierarchical clustering of dynamic genes in POS cells revealed two expression patterns: 1735 genes that continuously decrease in expression during chronic treatment (DOWN), and 2644 genes that continuously increase in response to chronic treatment (UP). In the NEG subpopulation, we identified 1441 genes (11% of expressed genes) that change at least two-fold during the course of chronic treatment (Fig. S3D). Hierarchical clustering of differentially expressed genes identified three distinct patterns of gene expression: 416 genes that initially increase in response to trametinib and then return to baseline (UP-DOWN), 441 genes that continuously decrease throughout treatment (DOWN), and 529 genes that sustain increased expression throughout treatment (UP). When comparing the transcriptional response of the POS and NEG subpopulations, we observe a stronger response in POS cells (37% of expressed genes) compared to the NEG (11% of expressed genes). This is correlated with the dynamic changes in the enhancer landscape observed in Fig. 3D–E. While GO analysis of genes downregulated in POS cells during treatment is consistent with growth arrest, we also observed the enrichment of genes that regulate PI3K signaling (Fig. S3E–F). Interestingly, GO analysis of dynamic genes in NEG cells reveals that PDGF signaling regulation occurs transiently (Fig. S3G), but downregulation of MAPK phosphatases persists during chronic treatment (Fig. S3H). We identified continuous increase in genes regulating TGFβ and the cytoskeleton (Fig. S3D, Fig S3I–J), concordant with the enrichment of SMAD3 and SRF observed in motif analysis (Fig S3B). We observed significant overlap in dynamic genes identified in POS and NEG cells, indicating that MEK inhibition culminates in both common and unique responses (Fig. S3K).
Drug Resistant POS Cells Emerge Following Chronic Trametinib Treatment
After approximately three weeks of treatment with 10 nM trametinib, we began to observe small colonies of POS cells, suggesting that these cells had developed resistance (Fig. 3C). To further study the factors that contribute to resistance, we treated POS and NEG cells with 10 nM trametinib for six weeks, then dose-escalated to 30 nM trametinib, and independently isolated drug-resistant cell lines from these cultures. We compared the responses of these drug resistant cell lines (POS R1, POS R2, and NEG R) to naïve POS and NEG cells that had never been exposed to trametinib (POS N and NEG N). Notably, when dose escalated to 30 nM trametinib, NEG R cells growth arrested, but the two independently isolated POS cell lines, POS R1 and POS R2 continued to proliferate (data not shown). Whereas growth arrested NEG cells stained positive for senescence-associated beta-galactosidase activity after dose escalation, (Fig. 4A, NEG R), both the naïve NEG (NEG N) and naïve POS cells (POS N) were beta-galactosidase negative (Fig. 4A). As expected, drug resistant POS cells, were also beta-galactosidase negative (Fig. 4A, POS R). Cell cycle analysis revealed treatment with 30 nM trametinib induced G1 arrest in POS N cells (Fig. 4B, blue) but both POS R cells continued to proliferate (Fig. S4A), with a similar frequency of cells in S phase in POS R1 (51%) and POS R2 (38%) compared to the POS N control (55%) (Fig. S4B).
Figure 4. Drug Resistant POS Cells Emerge Following Chronic Trametinib Treatment.
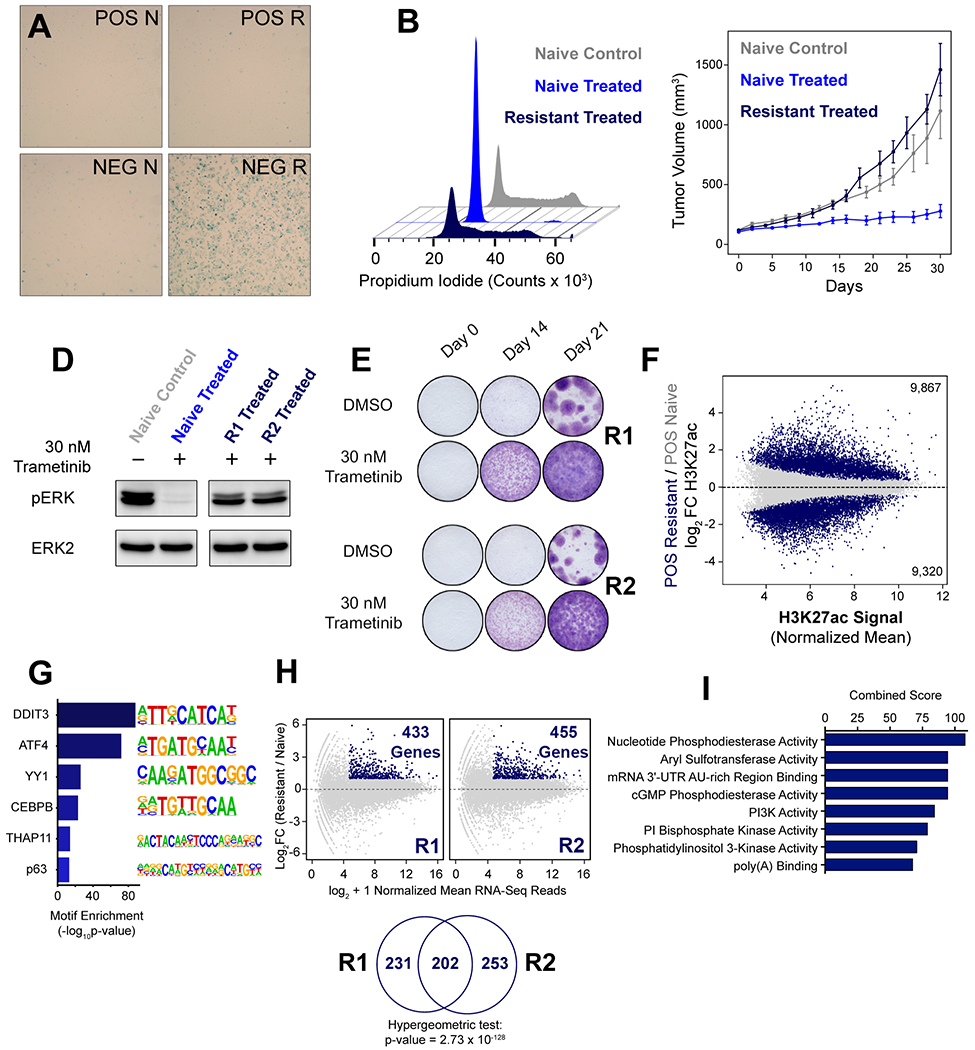
(A) POS and NEG naïve cells (POS N, NEG N, left) were stained for beta-galactosidase and compared to POS and NEG cells that had been conditioned for six weeks in 10 nM trametinib and then dose-escalated to 30 nM trametinib (POS R, NEG R, right). (B) Cell cycle analysis of POS naïve cells treated with DMSO (grey) or 30 nM trametinib (blue) for 24 hours compared to POS R1 cells cultured in 30 nM trametinib (dark blue). (C) 2 million POS naïve cells were injected into the mammary fat pad of nod/scid mice (n=6) and treated with control chow (grey) or trametinib chow (0.2 mpk) (blue). Nod/scid mice were pre-treated with trametinib chow (0.2 mpk) and then injected with 2 million POS resistant cells (dark blue) each (n=6). (D) Immunoblot of phospho-ERK (T202/Y204) in naïve and resistant cells following 24 h treatment as indicated. (E) Crystal violet staining of POS R1 and POS R2 treated continuously with 30 nM trametinib or DMSO for the time indicated. (F) Differential H3K27 acetylation in trametinib resistant cells relative to trametinib naïve cells. Statistically significant regions are shown in dark blue. (G) Motif analysis of H3K27ac peaks significantly upregulated in POS R cells relative to unchanged H3K27ac peaks. (H) MA Plot of genes upregulated in trametinib resistant cells relative to trametinib naïve cells. Significant genes with a minimum mean expression of 25 reads and greater than two-fold difference in expression with a p-value < 0.05 are highlighted in dark blue. (I) GO analysis of the molecular function of the intersection of genes upregulated in POS R1 and R2 cells identified in Fig. 4H.
We further studied the trametinib response of POS and NEG cells in vivo by xenograft injection into the mammary fat pad of NOD scid gamma mice (NSG). NEG naïve and NEG resistant cells failed to establish tumors, and therefore these arms of the study were terminated after ninety days (Fig. S4C, grey and maroon). POS naïve xenografts grew significantly slower on trametinib (Fig. 4C, royal blue) compared to control (Fig. 4C, grey), but POS resistant xenografts (Fig. 4C, dark blue) grew at a rate similar to the POS naive xenografts treated with control (Fig. 4C, grey). These results indicate that POS R xenografts are resistant to trametinib in vivo and continue to grow in the presence of the drug.
We compared activation of ERK1/2 (phosphorylation at T202/Y204) in the POS N cells to POS R1 and POS R2 cells (Fig. 4D). We observed that ERK phosphorylation was lost in POS N cells treated with 30 nM trametinib for 24 hours, but ERK phosphorylation was preserved in the POS R1 and POS R2 cells despite trametinib. We measured proliferation of POS R1 and POS R2 by crystal violet staining of cells following 0, 14, or 21 days of treatment with 30 nM trametinib (Fig. 4E). We observed visible staining at Day 14 of treatment, which intensified at Day 21, indicating these cells continue to proliferate in the presence of the drug.
To assess differences between the POS R enhancer landscape after dose escalation and the POS N enhancer landscape, we identified condition-specific regions of H3K27ac signal by DESeq2. We identified 9,867 increased H3K27ac peaks (18% of the total 53,549) and 9,320 decreased H3K27ac peaks (17% of the total 53,549) in POS R cells compared to POS N control (Fig. 4F). We then determined motifs that were enriched at increased H3K27ac peaks in POS R cells. We identified strong enrichment of DDIT3 and ATF4 motifs (Fig. 4G). We performed DESeq2 analysis of gene expression in POS R1 relative to POS N cells, which revealed 433 genes upregulated in POS R1 cells. We also analyzed POSR2 cells, which identified 455 genes upregulated relative to POS N cells (Fig. 4H, top). We observed a highly significant overlap of genes upregulated in POS R1 and POS R2 cells, suggesting that these cell lines converged on similar resistance mechanisms (Fig. 4H, bottom). GO analysis of the 202 genes upregulated in both POS R1 and POS R2 cells revealed the enrichment of PI3K signaling (Fig. 4I). Hierarchical clustering of these genes identified a class of genes that continuously increased in expression during chronic trametinib treatment (Fig. S4D), indicating that epigenetic remodeling associated with these genes increases transcription, contributing to resistance in POS cells.
G-protein Coupled Receptor CXCR7 Mediates Proliferation in POS Resistant Cells
Several studies have demonstrated that resistance to targeted kinase inhibitors can be acquired by either adaptation or selection of resistant cells (23–27). One of the most significantly upregulated genes in POS R1 and POS R2 cells relative to naïve cells was the G-protein coupled receptor Atypical Chemokine Receptor 3, (CXCR7) and its cognate ligand, Adrenomedullin (ADM) (Fig. 5A). We confirmed the expression of CXCR7 in both POS R1 and POS R2 cells by immunoblot (Fig. 5B). Studies have shown that CXCR7 is a Gi-coupled receptor that can activate MAPK signaling via beta-arrestin to stimulate survival and proliferation of breast cancer cells (28–30). Therefore, we quantified cell proliferation following siRNA knockdown of CXCR7 in POS N and POS R1 cells to determine the effect of CXCR7 on proliferation of trametinib resistant and trametinib naïve cells. Interestingly, CXCR7 knockdown in the POS N cells had no significant effect on proliferation (Fig. 5C). In contrast, CXCR7 knockdown in the POS R1 cells significantly decreased proliferation (Fig. 5D), indicating that CXCR7 plays an essential role in the recovery of proliferation in the presence of trametinib. We verified CXCR7 knockdown by immunoblot of CXCR7 following treatment with an siRNA targeting CXCR7 or an siRNA control (Fig. 5E). We re-examined CXCR7 expression in POS cells during chronic trametinib treatment, which revealed that CXCR7 expression increased approximately five-fold at Day 1 and increased to a maximum expression level at Day 14 (Fig. 5F). Proximal to the gene encoding CXCR7, we identified an enhancer region containing accessible chromatin (Fig. 5G, orange) with JUNB binding (Fig. 5G, green) that correlated with increased H3K27ac during chronic treatment (Fig. 5G, purple).
Figure 5. G-protein Coupled Receptor CXCR7 Mediates Proliferation in POS Resistant Cells.
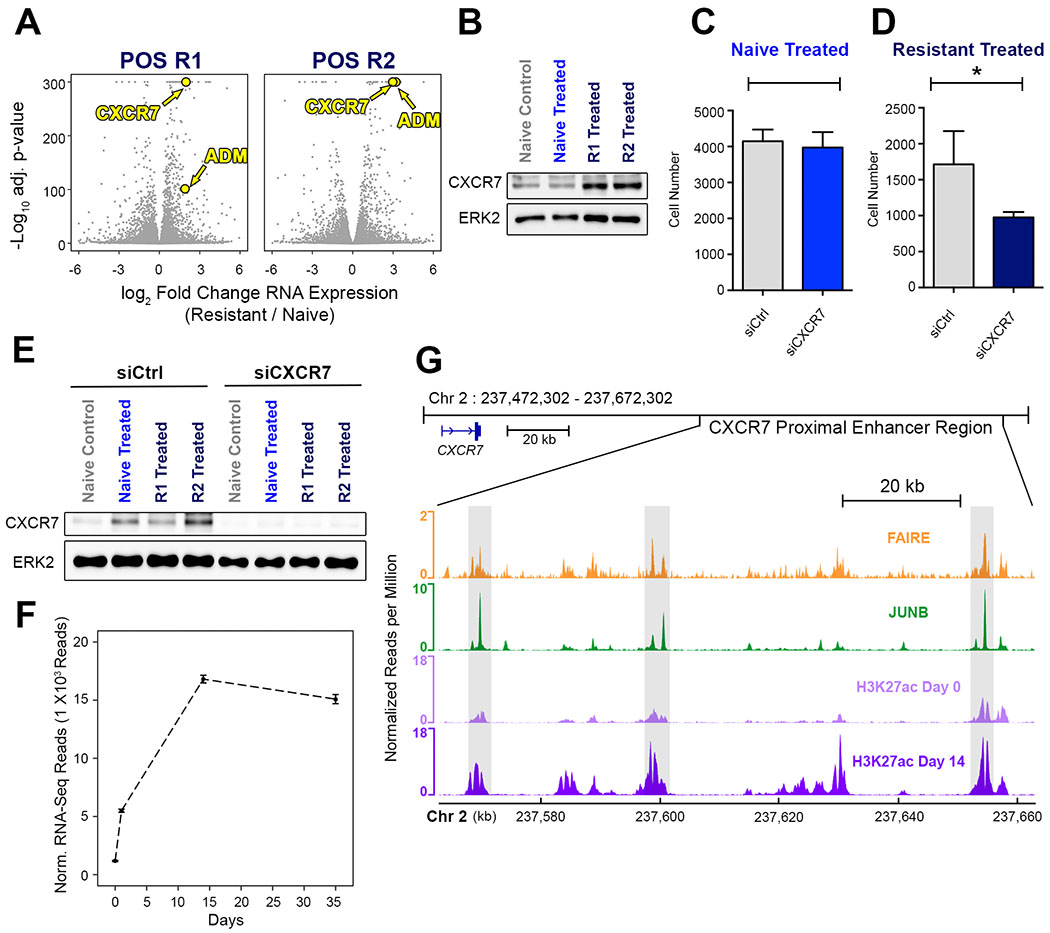
(A) Volcano plot of gene expression in POS R1 and POS R2 cells relative to POS N. Relative CXCR7 and Adrenomedullin (ADM) expression in POS R1 and POS R2 cells highlighted in yellow. (B) Immunoblot for CXCR7 in naïve cells treated for 24 hours with DMSO or 30 nM trametinib compared to POS R1 and POS R2 cells cultured in 30 nM trametinib. (C) POS N cell number was measured by high content imaging after 72 hours of treatment with 30 nM trametinib in combination with control siRNA (grey) or siRNA targeting CXCR7 (blue). (D) POS R1 cell number was measured by high content imaging after 72 hours of treatment with 30 nM trametinib in combination with control siRNA (grey) or siRNA targeting CXCR7 (dark blue). (E) Immunoblot for CXCR7 following treatment of trametinib naïve and trametinib resistant cells with control siRNA or siRNA targeting CXCR7. (F) CXCR7 expression in POS naïve cells during chronic treatment with 10 nM trametinib measured by RNA-Seq. (G) FAIRE-seq (orange) and JUNB ChIP-seq (green) shows JUNB binding at regions of relaxed chromatin (boxed regions). H3K27ac signal (purple) at the enhancer region proximal to CXCR7 increases during chronic treatment.
Wild-type KRAS is Amplified in Trametinib Resistant POS Cells Following Dose Escalation
KRAS was significantly upregulated in POS R1 and POS R2 cells compared to control (Fig. 6A). An immunoblot for KRAS in POS naïve and resistant cells confirmed the upregulation of KRAS in POS R1 and POS R2 cells (Fig. 6B). However, when we re-examined KRAS expression during chronic treatment, we did not observe a substantial increase in KRAS expression (Fig. S5A). We demonstrated that KRAS expression in POS N and POS R1 contributes to proliferation by silencing KRAS (Fig. 6C, Fig. 6D). KRAS depletion was confirmed by immunoblot following treatment with KRAS siRNA (Fig. S5B). We performed KRAS RNA-FISH in POS N (Fig. 6E) and POS R1 (Fig. 6F) to determine if cells with high KRAS expression pre-exist in the POS naïve cells. We observed a broad distribution of KRAS expression in the POS R1 population, with 13% of the cells classified with low expression, 23% classified with medium expression, and 61% classified with high expression (Fig. S5C, dark blue). In comparison, the untreated POS cells had more uniformly low expression, with greater than 99% of cells classified with no expression (Fig. S5C, grey). Furthermore, when we treated cells continuously with 10 nM trametinib, we did not observe a significant increase in KRAS expression in drug-resistant colonies (Fig. 6G and 6H), suggesting that KRAS expression increases following dose escalation in the drug resistant cells. We performed single-cell RNA sequencing on the POS N and POS R1 cells to further quantify single-cell gene expression of KRAS in these two populations, which demonstrated that POS naïve cells have uniformly low KRAS expression (Fig. 6I and S5D, grey), whereas POS resistant cells have a broad distribution of KRAS expression (Fig. 6I and S5D, navy). Previous studies have shown that gene amplification of the wild-type locus of KRAS can mediate resistance to MAPK inhibition (31,32). RT-PCR using gDNA isolated from POS naïve, POS R1 and POS R2 cells revealed a six-fold amplification of wild-type KRAS in the drug resistant cells relative to control (Fig. 6J). These results indicate that POS cells develop treatment resistance through multiple mechanisms, including remodeling H3K27 acetylation to increase expression of genes involved in survival and proliferation signaling, as well as the amplification of wild-type KRAS to reactivate MAPK activity.
Figure 6. Wild-type KRAS is Amplified in Trametinib Resistant POS Cells Following Dose Escalation.
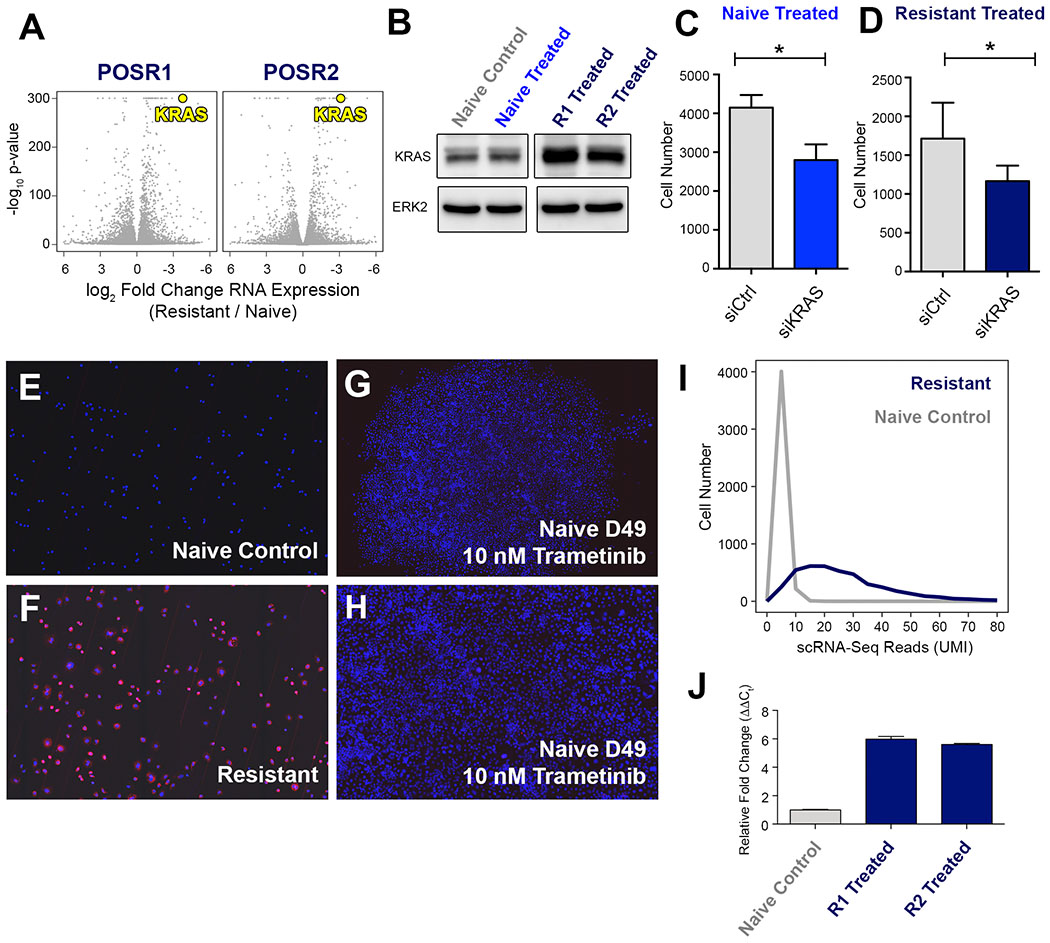
(A) Volcano plot of genes expression in POS R1 and POS R2 cells relative to POS N. Relative KRAS expression in POS R1 and POS R2 cells highlighted in yellow. (B) Immunoblot for KRAS in naïve cells treated for 24 hours with DMSO or 30 nM trametinib compared to POS R1 and POS R2 cells cultured in 30 nM trametinib. (C) POS naive cell number was measured by high content imaging after 72 hours of treatment with 30 nM trametinib in combination with control siRNA (grey) or siRNA targeting KRAS (blue). (D) POS R1 cell number was measured by high content imaging after 72 hours of treatment with 30 nM trametinib in combination with control siRNA (grey) or siRNA targeting KRAS (dark blue). (E) POS cells stained with KRAS RNA-FISH probes at Day 0 of chronic trametinib treatment, viewed at 100X magnification. (F) POS R1 cells stained with KRAS RNA-FISH probes, viewed at 100X magnification. (G) POS cells at Day 49 of 10 nM trametinib treatment, stained with KRAS RNA-FISH probes, viewed at 20X magnification. (H) 100X magnification of colony shown in Fig. 6G. (I) KRAS expression in POS resistant cells (dark blue) and naive cells (grey) measured by scRNA-seq, binned by 10 read intervals of KRAS expression. (J) Copy number PCR targeting KRAS locus in naïve (grey), POS R1, and POS R2 cells (dark blue).
Discussion
Triple negative breast cancer is a heterogeneous disease with no currently available targeted therapy. Immunostaining of TNBC mouse models and patient samples reveals significant tumor heterogeneity (34,35). A recent study of circulating breast cancer tumor cells demonstrated that epithelial and mesenchymal cells have differential sensitivity to combined PI3K and MEK inhibition (6). Furthermore, chronic drug treatment has been shown to select for drug resistant cells. These studies indicate that tumor heterogeneity presents a significant obstacle to the development of durable therapies for the treatment of TNBC. Our goal was to define the differential responses to trametinib in epithelial and mesenchymal subpopulations using a preclinical model of TNBC heterogeneity.
Immunostaining and flow cytometry analysis of breast cancer cell lines revealed remarkable heterogeneity in surface marker expression (19). The two subpopulations of TNBC cell line SUM-229PE are characterized by differential expression of two epithelial surface markers, EpCAM and CD49f. Culture of POS and NEG cells following FACS isolation demonstrated that these phenotypes are stable and model the gene expression of the parental subpopulations. Despite the genomic similarities we observed in POS and NEG cells by whole exome sequencing, FAIRE-Seq identified differential regions of chromatin accessibility that were enriched in binding motifs for transcription factors known to regulate cell identity (36,37). Analysis of differentially expressed genes revealed that POS and NEG cells faithfully represent the basal-like and mesenchymal TNBC phenotypes (30).
We previously demonstrated that siRNA knockdown of Smarcd3/Baf60c, a regulatory subunit of the SWI/SNF chromatin remodeling complex, is sufficient to induce a phenotypic switch from NEG to POS SUM229-PE cells (20). Smarcd3/Baf60c knockdown in NEG cells inhibited expression of two mesenchymal-specific transcription factors, SNAIL and SLUG. Furthermore, we showed ectopic expression of Smarcd3/Baf60c in primary human epithelial cells induced a mesenchymal phenotype (20). Cumulatively, the findings demonstrated that differing chromatin states between POS and NEG subpopulations of SUM-229PE have a strong influence in controlling their respective phenotype.
We used SUM-229PE POS and NEG cells to characterize the regulatory elements associated with the response to acute trametinib treatment. Motif analysis of H3K27ac peaks decreased in POS cells following acute trametinib treatment identified ETS-family transcription factor motifs. ETS-family transcription factors are phosphorylated by MAP kinases to regulate gene transcription downstream of the Ras-Raf-MEK-ERK pathway (38,39). Analysis of decreased H3K27ac peaks in the NEG cells did not identify similar enrichment of ETS-family motifs. The findings are consistent with the sensitivity to MEK inhibition in POS cells, which exhibited downregulation of genes that regulate cell cycle progression. In contrast, NEG cells upregulate genes in response to trametinib that activate bypass pathways to reactivate MAPK signaling.
We observed increased enhancer remodeling in response to acute trametinib treatment in POS cells relative to NEG cells, which persists during chronic trametinib treatment. MAPK-regulated ETS motifs identified in H3K27ac peaks decreased in POS cells remain downregulated during chronic treatment. Additionally, chronic trametinib treatment revealed new motifs previously unobserved in the acute response. Interestingly, the SRF consensus motif is enriched at sites of increased acetylation in NEG cells. Previous studies have shown that SRF activation stimulates both proliferation and cell contractility, concordant with increased expression of genes in these pathways in NEG cells during chronic treatment (40).
While we identified the enrichment of the JUNB motif only in NEG cells in response to acute trametinib treatment, we observed enrichment of the JUNB motif in both POS and NEG cells during chronic treatment. Previous studies have shown that AP-1 binding is strongly correlated with chromatin accessibility (33). JUNB localized to differential sites of chromatin accessibility in POS and NEG cells, which indicated that MAPK inhibition can have unique outcomes in genomically similar subpopulations, dictated by differential localization of common transcription factors.
Drug resistant colonies emerged from a population of slowly proliferating persister POS cells, which survived treatment and adapted to a clinically-relevant trametinib dose (41). In contrast, NEG cells became senescent and failed to develop resistance in vitro or in vivo. Two drug resistant cell lines were independently isolated from POS cells, which were used to identify factors driving survival and proliferation. Motif analysis of H3K27ac peaks increased in POS resistant cells revealed further enrichment of sequences previously identified in response to chronic trametinib treatment. We also observed further enrichment of GO terms associated with PI3K signaling in POS resistant cells, indicating PI3K may cooperate with amplified KRAS to enhance proliferation in the presence of trametinib.
CXCR7 and adrenomedullin were upregulated in POS resistant cells, and RNAi knockdown of CXCR7 inhibited proliferation. Increased transcription correlated with increased H3K27ac at the CXCR7 TSS and a proximal enhancer region. These data suggest that enhancer remodeling at the CXCR7 locus in response to trametinib enhances CXCR7 gene expression, which stimulated proliferation in the presence of trametinib treatment. A clinical study of TNBC patients revealed that 16% of patients had CXCR7 overexpression, and these patients had significantly worse overall survival (42). Immunostaining for CXCR7 in 10 normal, 10 metastatic, and 38 invasive ductal carcinoma samples showed a significant correlation between CXCR7 expression and aggressive breast cancer (42). These results demonstrate that the activation of CXCR7 observed in POS cells highlights a potentially clinically relevant mechanism of tumor aggression and drug resistance in TNBC patients that is regulated by enhancer remodeling.
We identified amplification of KRAS in POS trametinib resistant cells, and KRAS knockdown suppressed growth. We found that KRAS amplification occurred after dose escalation. Clinical studies reveal that KRAS overexpression is feature observed in both TNBC patients and mouse models (15,37). Preclinical studies of the role of wild-type and oncogenic KRAS demonstrate that overexpression of wild-type KRAS reduces proliferation in the context of mutant KRAS, but is sufficient to create drug resistance (43,44). These results demonstrate that KRAS amplification acquired during chronic trametinib treatment is sufficient to stimulate proliferation in the continued presence of the drug.
In this work, we have described different chromatin states in the SUM-229PE POS and NEG cells that contribute to the maintenance of epithelial and mesenchymal phenotypes. These distinct chromatin states dictate selective recruitment of transcription factors and epigenetic modifying enzymes to different genes and their enhancers, resulting in a differential response to targeted drug therapy. Enhancer remodeling in POS cells recruits transcriptional activators to the CXCR7 gene, which promotes proliferation and survival in trametinib resistant cells. We observed the amplification of KRAS after the emergence of drug resistant colonies, indicating adaptive resistance contributes to survival in this context. The progression from adaptive to acquired resistance suggests that inhibiting enhancer remodeling may prevent resistance and the bypass of targeted therapy. We and others have shown that targeting the pTEFb complex and BET proteins with small molecule inhibitors are able to not only inhibit the onset of the initial chromatin reprogramming but reverse the epigenetic changes making the targeted inhibitor therapy durable (16). Inhibiting enhancer remodeling may prevent further proliferation and the acquisition of oncogenic mutations. These results suggest that combining kinase and chromatin remodeling inhibitors may offer a durable treatment strategy to reduce adaptive resistance, and should be tested in clinical trials.
Supplementary Material
Implications Statement.
We defined the differential responses to trametinib in subpopulations of a clinically-relevant in vitro model of TNBC, and identified both adaptive and acquired elements that contribute to the emergence of drug resistance mediated by increased expression of CXCR7 and amplification of KRAS.
Acknowledgements
The authors would like to acknowledge the UNC flow cytometry core facility for technical assistance. We thank Joel Parker for assistance with computational analyses. We also thank UNC TPL facility for technical assistance with RNA-FISH. We acknowledge the LCCC High Throughput Sequencing Facility for technical assistance with scRNA-Seq. This work was made possible by NCI Breast SPORE P50CA058223 to G. Johnson, as well as NIH grant # U01CA238475 and U24DK116204 to G. Johnson. This work was also made possible by NIH grant # R33CA206939 to S. Pattenden and I. Davis. J. Foster was supported by NIH grant # 5T32 GM067553 to the Curriculum in Bioinformatics and Computational Biology at UNC Chapel Hill.
Footnotes
Disclosure of Author Conflict of Interest: Ian Davis is a Scientific Advisory board member and has equity ownership in Triangle Biotechnology, Inc., to which the following technologies used or evaluated in this paper have been licensed: US Pat. No. 9,427,410 and 9,982,290. Samantha Pattenden has equity ownership in Triangle Biotechnology, Inc., to which the following technologies used or evaluated in this paper have been licensed: US Pat. No. 9,427,410 and 9,982,290. Samantha Pattenden is inventor of US Pat. No. 9,982,290.
References
- 1.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N Engl J Med. 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murtaza M, Dawson S- J, Tsui DWY, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. Nature Publishing Group; 2013;497:108–12. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grosselin K, Durand A, Marsolier J, Poitou A, Marangoni E, Nemati F, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nature Genet. Nature Publishing Group; 2019;51:1060–6. [DOI] [PubMed] [Google Scholar]
- 5.Risom T, Langer EM, Chapman MP, Rantala J, Fields AJ, Boniface C, et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat Commun. 2018;9:3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science. 2013;339:580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM, et al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Molecular Cell. 2014;56:219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nature Genet. 2014;46:364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wahba HA, El-Hadaad HA. Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med. 2015;12:106–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, et al. Response to Neoadjuvant Therapy and Long-Term Survival in Patients With Triple-Negative Breast Cancer. Journal of Clinical Oncology. 2008;26:1275–81. [DOI] [PubMed] [Google Scholar]
- 12.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller SM, Goulet DR, Johnson GL. Targeting the Breast Cancer Kinome. J Cell Physiol. 2016;232:53–60. [DOI] [PubMed] [Google Scholar]
- 14.Lim Z- F, Ma PC. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J Hematol Oncol. BioMed Central; 2019;12:134–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, et al. Enhancer Remodeling During Adaptive Bypass to MEK Inhibition Is Attenuated by Pharmacological Targeting of the P-TEFb Complex. Cancer Discovery. American Association for Cancer Research; 2017;CD–16–0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stuhlmiller TJ, Miller SM, Zawistowski JS, Nakamura K, Beltran AS, Duncan JS, et al. Inhibition of Lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep. 2015;11:390–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keller PJ, Lin AF, Arendt LM, Klebba I, Jones AD, Rudnick JA, et al. Mapping the cellular and molecular heterogeneity of normal and malignant breast tissues and cultured cell lines. Breast Cancer Res. BioMed Central Ltd; 2010;12:R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jordan NV, Prat A, Abell AN, Zawistowski JS, Sciaky N, Karginova OA, et al. SWI/SNF Chromatin-Remodeling Factor Smarcd3/Baf60c Controls Epithelial-Mesenchymal Transition by Inducing Wnt5a Signaling. Mol Cell Biol. 2013;33:3011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simon JM, Giresi PG, Davis IJ, Lieb JD. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat Protoc. Nature Publishing Group; 2012;7:256–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiarella AM, Quimby AL, Mehrab-Mohseni M, Velasco B, Kasoji SK, Davis IJ, et al. Cavitation Enhancement Increases the Efficiency and Consistency of Chromatin Fragmentation from Fixed Cells for Downstream Quantitative Applications. Biochemistry. American Chemical Society; 2018;57:2756–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pattenden SG, Simon JM, Wali A, Jayakody CN, Troutman J, McFadden AW, et al. High-throughput small molecule screen identifies inhibitors of aberrant chromatin accessibility. Proc Natl Acad Sci USA. 2016;113:3018–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell. 2013;153:320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- 26.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. BioMed Central; 2014;15:31–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hrvatin S, Deng F, O’Donnell CW, Gifford DK, Melton DA. MARIS: method for analyzing RNA following intracellular sorting. PLoS ONE. 2014;9:e89459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raisner R, Kharbanda S, Jin L, Jeng E, Chan E, Merchant M, et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018;24:1722–9. [DOI] [PubMed] [Google Scholar]
- 29.Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. John Wiley & Sons, Ltd; 2002;3:224–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adélaïde J, Cervera N, et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene. 2006;25:2273–84. [DOI] [PubMed] [Google Scholar]
- 31.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arvey A, Agius P, Noble WS, Leslie C. Sequence and chromatin determinants of cell-type-specific transcription factor binding. Genome Research. 2012;22:1723–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green JE, Shibata MA, Yoshidome K, Liu ML, Jorcyk C, Anver MR, et al. The C3(1)/SV40 T-antigen transgenic mouse model of mammary cancer: ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene. 2000;19:1020–7. [DOI] [PubMed] [Google Scholar]
- 35.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. Nature Publishing Group; 2016;13:674–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carroll DK, Carroll JS, Leong C-O, Cheng F, Brown M, Mills AA, et al. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol. 2006;8:551–61. [DOI] [PubMed] [Google Scholar]
- 37.Cieply B, Riley P, Pifer PM, Widmeyer J, Addison JB, Ivanov AV, et al. Suppression of the epithelial-mesenchymal transition by Grainyhead-like-2. Cancer Research. 2012;72:2440–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plotnik JP, Budka JA, Ferris MW, Hollenhorst PC. ETS1 is a genome-wide effector of RAS/ERK signaling in epithelial cells. Nucleic Acids Res. 2014;42:11928–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Selvaraj N, Kedage V, Hollenhorst PC. Comparison of MAPK specificity across the ETS transcription factor family identifies a high-affinity ERK interaction required for ERG function in prostate cells. Cell Commun Signal. 2015;13:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gualdrini F, Esnault C, Horswell S, Stewart A, Matthews N, Treisman R. SRF Co-factors Control the Balance between Cell Proliferation and Contractility. Molecular Cell. 2016;64:1048–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. The Lancet Oncology. 2012;13:773–81. [DOI] [PubMed] [Google Scholar]
- 42.Wani NA, Nasser MW, Ahirwar DK, Zhao H, Miao Z, Shilo K, et al. C-X-C motif chemokine 12/C-X-C chemokine receptor type 7 signaling regulates breast cancer growth and metastasis by modulating the tumor microenvironment. Breast Cancer Res. 2014;16:1259–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bahcall M, Awad MM, Sholl LM, Wilson FH, Xu M, Wang S, et al. Amplification of Wild-type KRAS Imparts Resistance to Crizotinib in MET Exon 14 Mutant Non–Small Cell Lung Cancer. Clin Cancer Res. American Association for Cancer Research; 2018;24:5963–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ambrogio C, Köhler J, Zhou Z- W, Wang H, Paranal R, Li J, et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell. 2018;172:857–868.e15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.