

Abstract
The α-ketoglutarate (αKG)-dependent oxygenases catalyze a diverse range of chemical reactions using a common high-spin FeIV=O intermediate and, in most reactions, abstracts a hydrogen atom from the substrate. Previously, the FeIV=O intermediate in the αKG-dependent halogenase SyrB2 was characterized by nuclear resonance vibrational spectroscopy (NRVS) and density functional theory (DFT) calculations, which demonstrated that it has a trigonal-pyramidal geometry with the scissile C-H bond of the substrate calculated to be perpendicular to the Fe-O bond. Here, we have used NRVS and DFT calculations to show that the FeIV=O complex in taurine dioxygenase (TauD), the αKG-dependent hydroxylase in which this intermediate was first characterized, also has a trigonal bipyramidal geometry, but with an aspartate residue replacing the equatorial halide of the SyrB2 intermediate. Computational analysis of hydrogen atom abstraction by square pyramidal, trigonal bipyramidal, and six-coordinate FeIV=O complexes in two different substrate orientations (one more along [σ channel] and another more perpendicular [π channel] to the Fe-O bond) reveals similar activation barriers. Thus, both substrate approaches to all three geometries are competent in hydrogen atom abstraction. The equivalence in reactivity between the two substrate orientations arises from compensation of the promotion energy (electronic excitation within the d manifold) required to access the π channel by the significantly larger oxyl character present in the pπ orbital oriented towards the substrate, which leads to an earlier transition state along the C-H coordinate.
Graphical Abstract
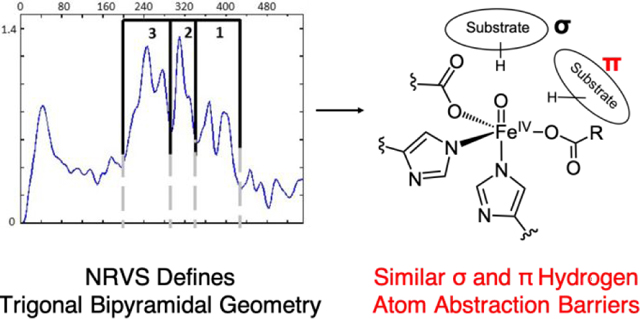
1. Introduction
Mononuclear nonheme iron enzymes catalyze a diverse range of chemical reactions that play key biological roles in O2 homeostasis, antibiotic, natural product and neurotransmitter biosynthesis, bioremediation and DNA repair.1–7 These enzymes employ a high-spin (HS) FeII center to activate dioxygen and form reactive intermediates that initiate substrate oxidation.8,9 In general, they share a common 2-His/1-carboxylate facial triad motif that leaves three coordination positions available for oxygen and/or cosubstrate binding, while the halogenases have a halide ligand replacing the carboxylate.10,11 A large subset of these enzymes utilize α-ketoglutarate (αKG) as a cosubstrate to provide two of the four electrons necessary for dioxygen reduction to generate reactive HS FeIV=O intermediates that can abstract hydrogen from strong C-H bonds (bond dissociation energy ~100 kcal mol−1).12,13 Determining the electronic and geometric structure of these high-valent intermediates is crucial to understanding the factors that govern their subsequent reactivity.
The αKG-dependent enzymes catalyze hydroxylation/halogenation, ring expansion/closure as well as desaturation using a common FeIV=O intermediate that initiates substrate attack.14,15 The first enzymatic FeIV=O intermediate to have been characterized was generated in the O2 reaction of taurine dioxygenase (TauD intermediate J [TauD-J]), which is an αKG-dependent enzyme that catalyzes the degradation of sulfonic acids to increase the bioavailability of sulfur.16,17 Since the observation of TauD-J, ferryl intermediates have been generated in other αKG-dependent and mononuclear enzymes,18–25 including in the halogenase SyrB2, which catalyzes the biosynthesis of the plant pathogen syringomycin.3,26 Unlike TauD, which has a 2-His/Asp facial triad primary coordination sphere, SyrB2 has a halide (either Cl− or Br−) in place of the carboxylate residue.27,28 Using nuclear resonance vibrational spectroscopy (NRVS), we previously characterized the electronic and geometric structure of the SyrB2 FeIV=O as a HS trigonal bipyramidal intermediate with the oxo along the C3 axis.29
From studies on model complexes, we demonstrated that HS FeIV=O intermediates are capable of hydrogen atom abstraction reactivity through either a σ channel, where the substrate C-H bond is along the Fe-O bond, or a π channel, where the substrate is perpendicular to the Fe-O bond.30 For the SyrB2 FeIV=O intermediate, magnetic circular dichroism (MCD) data revealed a dπ* → dσ* excitation, which reflects a very activated dπ* frontier molecular orbital (FMO) oriented perpendicular to the Fe-O bond and available for hydrogen atom abstraction along the π channel.31 By correlating the SyrB2 MCD data with a HS FeIV=O model complex, we calculated enhanced hydrogen atom abstraction reactivity for SyrB2 with the substrate perpendicular to the Fe-O bond (an orientation critical to its halogenation reactivity32). Whereas in SyrB2 reactivity occurs through the π channel, more generally, the orientation of substrate is governed by both its binding pocket in different enzymes28,33–35 as well as the orientation of the FeIV-oxo bond after O2 activation and O-O cleavage.29,36 From Figure 1, the αKG and taurine bound TauD FeII active site has the substrate docked over the active site pocket with an angle of 140° (C-Fe-Hisaxial).28,37 On the other hand, the recent crystal structure of the halogenase BesD shows the substrate docked at an angle of 88°, which is perpendicular to the vacant O2 binding position on the iron center.35 Thus, due to such geometric differences in substrate binding pockets, the factors governing differential hydrogen atom abstraction reactivity between the σ and π pathways is important to understand.
Figure 1.
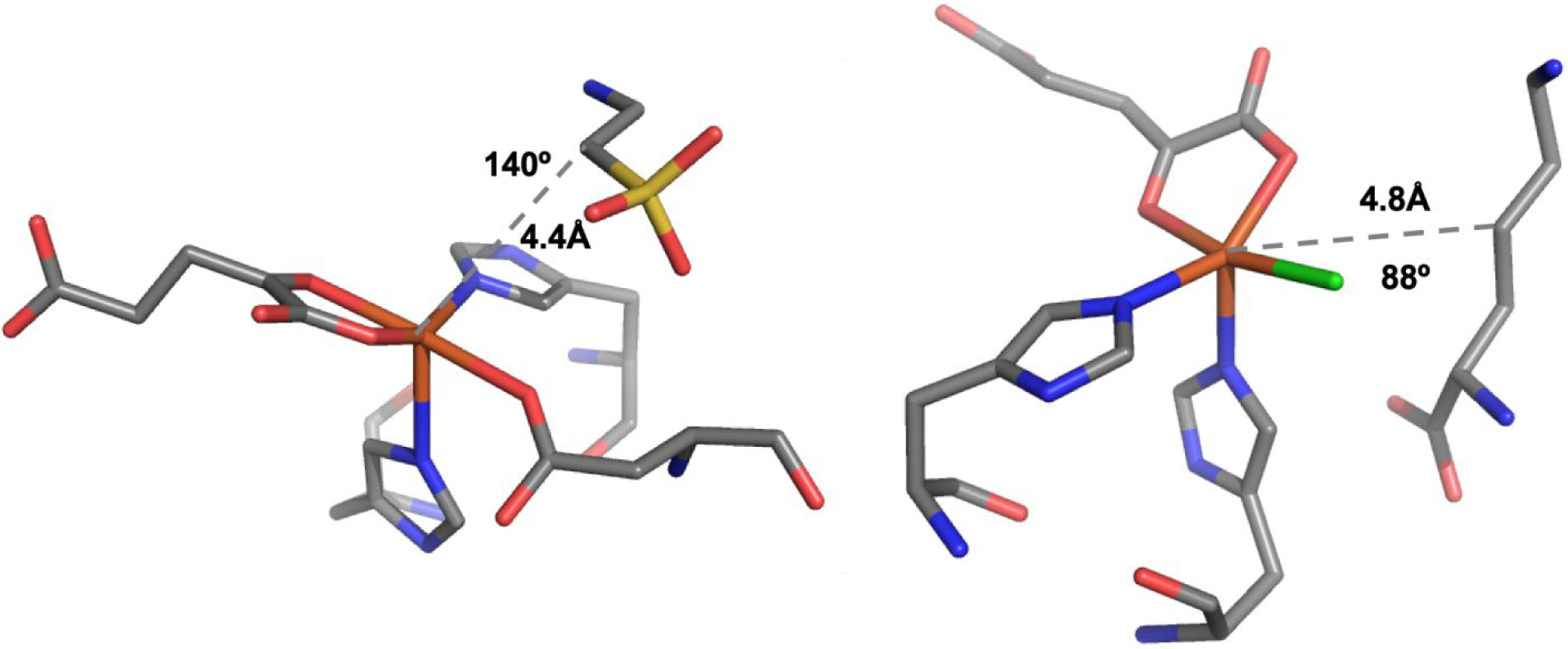
(Left) Substrate orientation in the αKG and taurine bound TauD compared to (Right) αKG and lysine bound BesD.
Because all αKG-dependent enzymes are known or believed to form reactive FeIV=O species and generally have 2-His/1-carboxylate facial triad ligation, the effect of replacing the halide in SyrB2 with a carboxylate is necessary to evaluate. Thus, we used NRVS to determine and analyze the electronic and geometric structure of the FeIV=O intermediate in TauD and compared it to the ferryl-oxo intermediate in SyrB2. After generating a set of FeIV=O active site models of TauD-J, we evaluated a subset of these models, including those with 6-coordinate distorted octahedral and 5-coordinate trigonal bipyramidal and square pyramidal geometries, for their relative reaction barriers in the key hydrogen atom abstraction step. The reactivity of these structures was evaluated with two substrate orientations in the TauD active site pocket to understand the σ and π contributions to reactivity. This study defines the geometric structure of TauD-J and evaluates the importance of geometry, spin state and σ vs π substrate orientation on hydrogen atom abstraction reactivity.
2. Experimental and Computational Details
2.1. Experimental Details
Preparation of 57Fe TauD-J NRVS Sample.
The sample of the ferryl intermediate of TauD was prepared according to literature.38 The procedures and apparatus used in preparation of freeze quench samples have been described.39 An anoxic solution containing 7 mM TauD, 9.5 mM aKG, 9.5 mM 1,1,2,2-[2H4]-taurine (d4-taurine, from C/D/N isotopes), 6.3 mM 57Fe(II) [prepared by dissolution of Fe(0) in 1 M H2SO4, as previously described]40 and 0.025 mM chlorite dismutase (prepared as previously described)41 in 90 mM Tris-HCl, 10% (v/v) glycerol buffer (pH 7.6) was constituted in an anoxic chamber. This solution was mixed at 5 °C with 0.25 equivalent volume of an anoxic solution of 75 mM sodium chlorite in 50 mM Tris-HCl, 10% (v/v) glycerol buffer (pH 7.6), resulting in concentrations of 5 mM TauD•57Fe(II)•aKG•d4-taurine, 0.020 mM Cld and 15 mM chlorite immediately after mixing. The reaction was allowed to proceed for 0.30 s before it was terminated by expulsion into a 50 mL conical tube containing liquified ethane. The ethane was evaporated under modest vacuum while the sample was cooled in liquid nitrogen. After this drying procedure, the powder was stored under liquid nitrogen. Prior to data collection, it was cryogenically packed into an NRVS cell. From Mössbauer spectroscopic data (Figure S1), the NRVS samples studied contained 66% FeIV, 23% FeII (i.e., [FeII/αKG/taurine/TauD]) and ~10% of FeII (likely from adventitious oxidation of unbound FeII).
NRVS Data Collection and Processing.
57Fe NRVS data on the FeIV-containing intermediate J and on the TauD•FeII•αKG•taurine reactant complex were collected at BL09XU at SPring-8. The NRVS energy scale was calibrated using a biferric model complex (Figure S2).42 Samples were maintained at ~60 K on a copper sample mount in a liquid helium cryostat. Individual scans were collected between 0 – 550 cm−1 and were summed using the PHOENIX software package43 until acceptable signal-to-noise ratio in the inelastic peaks was achieved (at least 10:1; Figure S2). PHOENIX was also used to subtract the elastic peak from the raw data and convert the observed counts spectrum into a partial vibrational density of states (PVDOS) spectrum. For the final PVDOS spectrum of intermediate J (presented below), the FeII component (23%) was subtracted, and the resulting spectrum was renormalized to make the area under the spectrum equal to 3.44 Data for TauD-J were collected on two different samples that showed similar spectra; these two data sets were averaged together to improve the signal-to-noise ratio of the final spectrum
2.2. Cluster Models and DFT Calculations
The initial structural model of the active site in the TauD•FeII•αKG•taurine complex (Figure 2) was generated from a crystal structure (PDB code: 1OS7). The ferryl models were generated by computationally evaluating the O2 activation pathway as described in Ref. 29. All structures along the O2 activation trajectory were optimized using the BP86 functional (which has been shown to accurately reproduce NRVS data29,45), in combination with Grimme’s empirical correction to dispersion (denoted D3),46 the def2-SVP basis set47 and the COSMO48,49 solvation model (with a dielectric constant εr=4 mimicking the protein environment), as implemented in Turbomole 6.6.50 The calculations were expedited by expanding the Coulomb integrals in an auxiliary basis set, the resolution-of-identity (RI-J) approximation.51,52 The same level of approximation (i.e., RI-BP86-D3/def2-SVP/COSMO, with εr=4) was used for the vibrational analysis and the simulation of NRVS spectra of the FeIV=O active-site models (simulations of NRVS spectra performed using GenNRVS53). Note that this methodology reproduces the NRVS spectra of structurally characterized FeIV=O model complexes previously published (Figure S3).45
Figure 2.
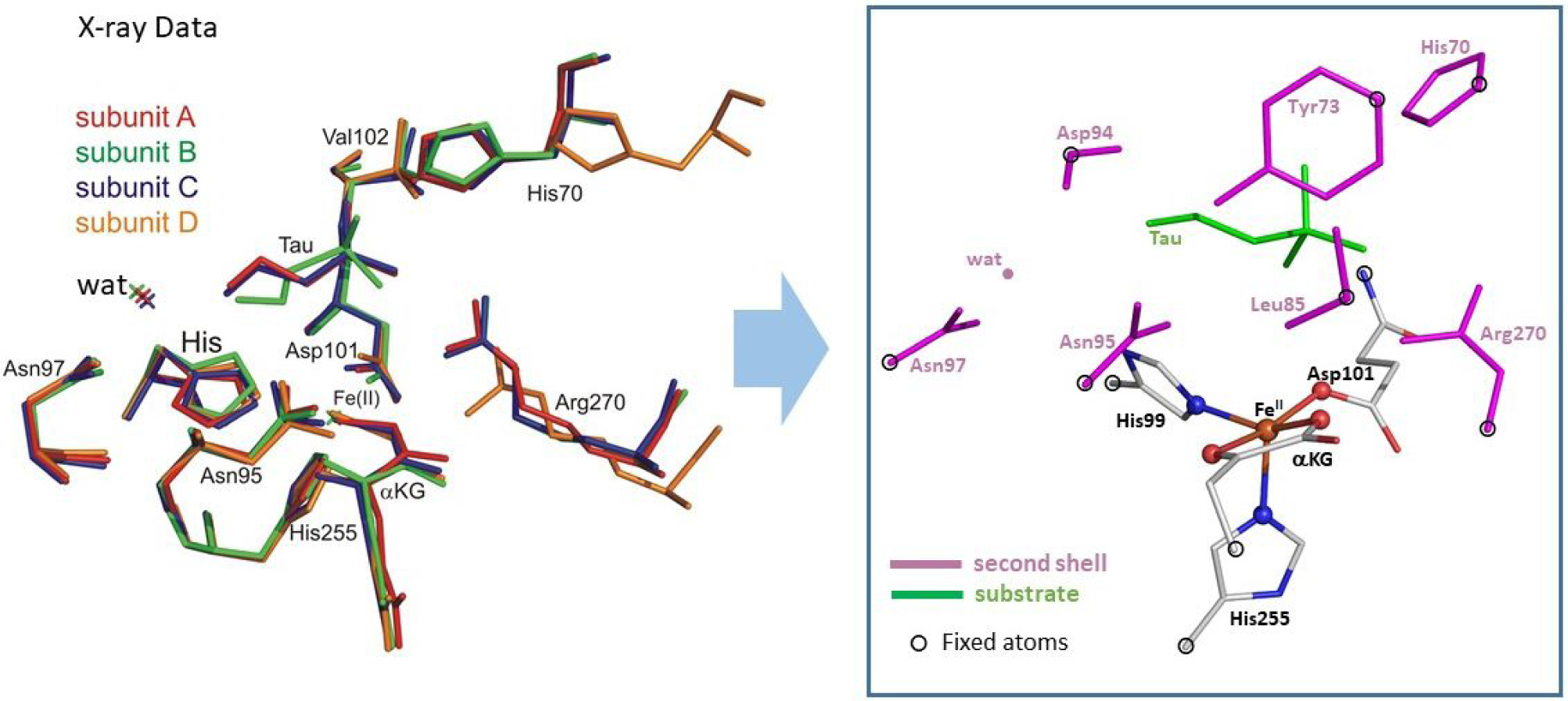
Left: The overlay of four αKG-bound FeII subunits in the crystal structure of TauD (the taurine substrate is absent in subunit D), as taken from PDB 1OS7.28 Right: The cluster model was constructed from subunit C. Hydrogens have been omitted for clarity.
For the hydrogen atom abstraction reaction coordinates, geometry optimizations were performed using the B3LYP-D3/def2-SVP/CPCM(εr=4) method (employing the B3LYP54 functional, which has been widely used to calculate FeIV=O reactivity [geometric parameters and corresponding NRVS spectra of BP86 vs B3LYP optimized structures are similar as demonstrated in Figure S4],29,30,55 and PCM56 solvation model), as implemented in the Gaussian 09 software package.57 For the optimized structures, the Gibbs free energy changes, ΔG, were calculated using the equation:
where the ΔEel term is the energy difference between two structures obtained from single-point B3LYP-D3/def2-TZVP/CPCM(εr=4) calculations, while the Δ[EZPVE +pV − RTlnQ] term corresponds to thermal energy corrections obtained from frequency calculations carried out at the same level of theory as the optimizations. Molecular orbital compositions were calculated using QMForge,58 and molecular orbital contours were generated using LUMO59 and/or IQmol.60
3. Results and Analysis
3.1. Nuclear Resonance Vibrational Spectrum (NRVS) of TauD-J.
The NRVS partial vibrational density-of-states (PVDOS) spectrum of TauD-J presented in Figure 3A was generated by subtracting the ferrous component from the raw data and renormalizing as described in Figure S2. This spectrum exhibits five distinct features in the 200–440 cm−1 region. We have considered this spectrum in three energy regions: region 1 (440–340 cm−1), region 2 (340–285 cm−1) and region 3 (285–200 cm−1), in analogy to the analysis of NRVS PVDOS data reported in Ref. 29 for the FeIV=O intermediate in the halogenase SyrB2 (cf. Figure S5). Region 1 contains two well-resolved peaks at 367 and 395 cm−1, which are less intense (by a factor of ~2) as compared to regions 2 and 3. Region 2 contains the most intense peak at 309 cm−1, and region 3 has two features - at 246 and 270 cm−1. The energy and intensity distribution of the spectrum in Figure 3A are very similar to those of the NRVS spectrum of the (SyrB2)FeIV=O species reported in Ref.29 and compared in Figure S5.
Figure 3.
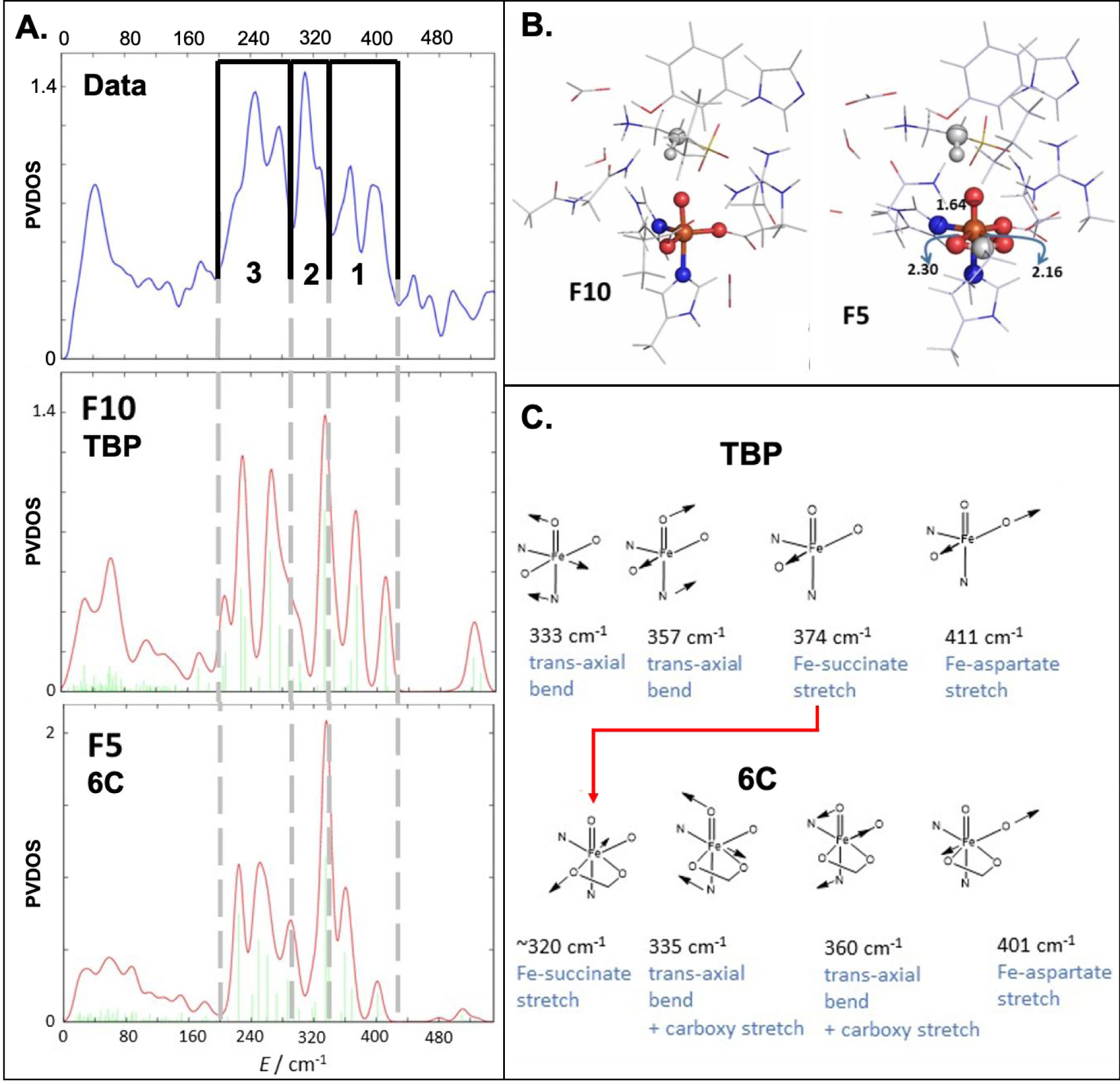
(A) (Top) Experimental NRVS PVDOS spectrum of TauD-J. The spectrum is divided in three regions 3, 2 and 1 as indicated; Simulated NRVS spectra for (Middle) TBP (F10) and (Bottom) 6C (F5) geometries. (B) Structures of the TBP F10 and 6C F5 complexes highlighting the change in denticity of the succinate ligand; (C) The vibrational mode assignments in regions 1 and 2 for the TBP and 6C geometries. The red arrow shows the decrease in energy of the Fe-succinate stretch associated with the monodentate carboxylate in TBP going bidentate in 6C.
3.2. DFT Simulations to Model NRVS Vibrational Data.
To generate structural candidates for the TauD-J, the reaction of the TauD•FeII•αKG•taurine complex (Figure 2) with O2 was analyzed by density functional theory (DFT) calculations. The O2 reaction coordinate (Figure S6), which is analogous to that described in Refs29 and61, leads to the six-coordinate (6C) FeIV=O product (F1; Figure S7). However, the calculated NRVS spectrum of F1 is not consistent with the experimental spectrum (cf. Figure S7). Specifically, the most intense transition is calculated to shift out of region 2 to higher energy (note the energy of the most intense feature in Figure S7 at 350 cm−1), so that its maximum intensity is in region 1. The result is that the calculated spectrum has too much intensity in region 1 relative to the experimental data. From this 6C FeIV=O model, other 6C candidates were generated through rotation of the C-C bond of the bound succinate and/or alteration of the H-bonding interactions between the first-shell (oxo and carboxylate) groups and the second-shell residues (F2–F5 in Figure S8–S11). However, none of these 6C models could reproduce the experimental NRVS pattern (in having the dominant feature in region 2), thus eliminating the 6C complexes as viable candidates for the structure of the TauD-J complex.
By decreasing the denticity of the bound succinate in F1 from bidentate to monodentate, we generated a series of five-coordinate (5C) trigonal bipyramidal (TBP) and square pyramidal (SP) structures (F6–F12 in Figure S12–S18). In comparison to the spectra calculated from the 6C structures, some of the NRVS spectra of these five-coordinate (5C) FeIV=O structural models agree much better with the experimental vibrational data (Figure S12–S18). Amongst these models, F7 (SP, Figure S13) and F10 (TBP, Figures 3 and S16) are two structural candidates that reproduce the experimental data in Figure 3A particularly well. Between these two candidates, the F10 TBP structure is the best candidate, as it reproduces the experimental integrated intensities in regions 1 and 2 (Figure 3A, middle) more accurately than the square pyramidal structure F7 (Figure S13). From Figure 3B, this F10 model contains an FeIV center in a 5C TBP coordination environment with a relatively wide angle between the equatorial monodentate succinate and Asp101 carboxylate groups (OSucc-Fe-OAsp = 140°). Compared with the experimental PVDOS data, its simulated NRVS spectrum is characterized by five spectral features, with similar relative intensities in each region compared to experiment, that fit well in the three experimental energy regions, with the most intense peak in region 2 and two distinct peaks located in each of the other two regions (cf. Figure 3A, middle). Note that minor differences in energy positions are due to interactions with second sphere residues as explained below. The relative intensity contributions in the three regions compared to the total intensity integrated over the range of 0–540 cm−1 are also well reproduced by this structural model (regions 1:2:3 are 17, 16 and 33% [calcd] vs. 17, 16 and 28% [expt]).
Comparing the experimental NRVS spectrum with the calculated F10 5C-TBP spectrum (cf. Figure 3A) allows the assignment of the five major experimental peaks as follows: (i) the 395 and 367 cm−1 peaks in the high energy region 1 correspond to the Fe-aspartate and Fe—succinate stretching modes, respectively; (ii) the 309 cm−1 peak in region 2 is due to a trans-axial bending mode (as shown in Figure 3C, there are 2 trans-axial bends; the one at higher energy is calculated to be weak and to overlap with the succinate stretch); (iii) the 270 and 246 cm−1 peaks in the low energy region 3 are attributed to Fe—His stretching modes. In comparing the experimental and calculated TBP spectra (Figure 3A, top and middle), it is important to note the ~20 cm−1 difference in energy of the trans-axial bend in region 2, where the TBP calculated spectrum is higher in energy. As shown in Figure 3C (top left), this mode displaces the iron atom between the succinate and aspartate ligands towards the second-sphere arginine residue (as shown in Figure 3B, left). Comparison of all the 5C calculated spectra (Figure S12–S18) reveals that the energy of this mode is sensitive to the distance between the arginine and the oxo unit. Thus, when the guanidinium group is closer to the oxo unit, the trans-axial bend is lower in energy, implying that the 20 cm−1 discrepancy between experiment and calculation is not due to a first-sphere coordination effect. Energies of other features also show some deviation, <15 cm−1, from experiment, which are due to second sphere effects not included in the modeling and small differences in metal-ligand bond lengths due to functional choice. Thus, the five peak pattern, i.e. 2 Fe-His modes in region 3, 1 transaxial bend in region 3 and 2 terminal carboxylates in region 1, is reproduced by the F10 model along with the same intensity ratios. This peak pattern, similar energies in the different regions and the intensity ratios were the criteria for our structural assignment.
Figure 3B and 3C also show that a change of the succinate ligand from a monodentate to a bidentate coordination mode (F10 TBP to F5 6C) is associated with a decrease of ~50 cm−1 in the energy of the Fe—succinate stretching mode (red arrow in Figure 3C). This lower energy stretching mode overlaps with the intense trans-axial bend in region 2 and produces a feature that does not correlate with the observed NRVS peak pattern. The difference in the Fe-O(succinate) stretch in the 5C versus 6C structures reflects the fact that, in going from monodentate to bidentate, the Fe-O bond length increases by ~0.3 Å. Thus, the higher energy region 1 is sensitive to Fe-carboxylate stretches and is a diagnostic probe of the 5C monodentate vs. 6C bidentate environment in FeIV centers. As two vibrations are observed in the NRVS data in Figure 3 region 1, this correlation demonstrates that both the facial triad carboxylate and the succinate carboxylate coordinate in monodentate fashions to the FeIV=O.
While NRVS spectroscopy strongly supports a 5C TBP geometry of the ferryl active site in TauD, recent crystal structures of TauD and a second αKG-dependent dioxygenase, VioC, with a vanadium-oxo complex bound62,63 and Mössbauer correlations63,64 have suggested a distorted 6C environment, with an asymmetrically disposed bidentate carboxylate providing the distant sixth ligand. The experimental Mössbauer data for intermediate J are δ = 0.30 mm s−1 and ΔEQ = −0.90 mm s−1.16 In line with previously reported computations of Mössbauer parameters for various intermediate J models, the 5C models presented in this study have calculated isomer shifts ~0.05 mm•s−1 less than those calculated for the 6C models and ~0.1 mm•s−1 less than the experimental values (the ΔEQ is less sensitive to 5C vs. 6C environment - see Table S1).64 Furthermore, the 5C SP structures have calculated isomer shifts that are even less than those of the 5C TBP structures (Table S1). Thus, none of these structures (neither 6C nor 5C) has a calculated isomer shift that matches the experiment value. We believe that this minor discrepancy is explained by the fact that the TauD-J intermediate models are rich in carboxylate ligands, whereas the calibration set from Figure S19 and Ref. 65 was dominated by model complexes with nitrogen ligation.
3.3. Calculated Hydrogen Atom Abstraction Reactivity
TauD-J, formed by reaction of O2 with the TauD•FeII•αKG•taurine complex, abstracts a hydrogen atom from the C1 position of taurine with a rate constant of 13 s−1 (correlating to a ΔG≠ = 14.8 kcal/mol at 278 K).66 Previous studies demonstrated that, in general, the FeIV=O unit can abstract hydrogen through a σ channel, in which the substrate electron goes to the dz2 orbital, or a π channel, in which the substrate electron goes to a dπ* orbital (Figure 4B).30 Having generated a series of possible structures for the FeIV=O intermediate in TauD to correlate to its NRVS data, which assigned TauD-J as having a TBP structure, we evaluated a subset of these structures (Figure 4C) for their relative hydrogen atom abstraction reactivities. In order to elucidate the geometric and electronic factors that contribute to reactivity, we evaluated the hydrogen abstraction reaction coordinates of these three structures (6C, TBP and SP) with two different substrate orientations (Figure 4D). These two substrate orientations were prepared along the O2 reaction coordinate of the TauD•FeII•αKG•taurine crystal structure,28 where a C-C bond rotation changes the FeIV=O/taurine C-H angle from 140° (more σ) to 120° (more π). Using these six different active site structures, the effects of coordination number, spin state (i.e., at the TS the Fe-O has FeIII-oxyl character with the FeII either S = 5/2 or S = 3/2)30 and σ/π-like substrate positioning were evaluated to assess the factors that influence hydrogen atom abstraction reaction barriers.
Figure 4.
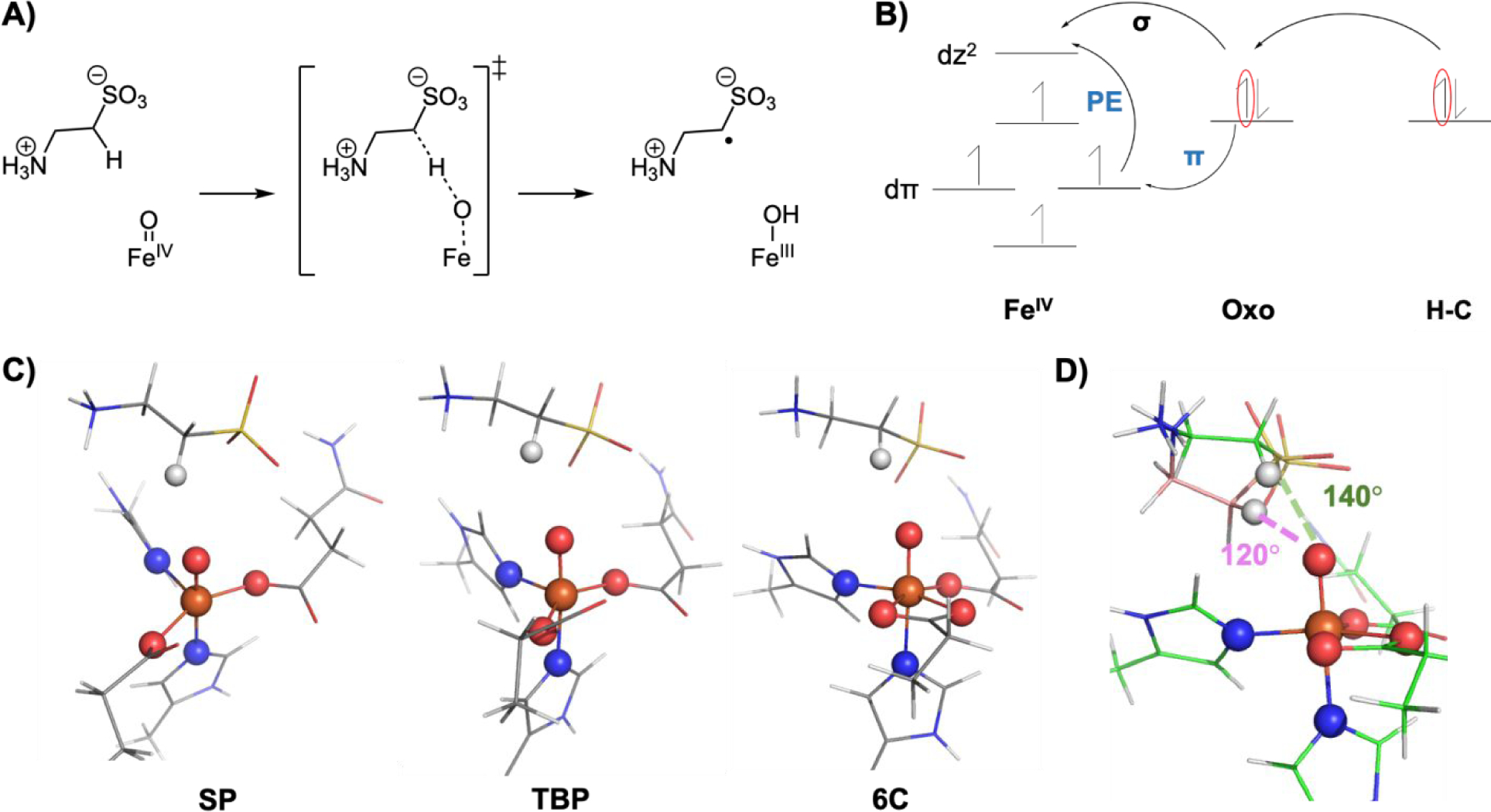
A) Hydrogen atom abstraction reaction coordinate depicting the FeIV=O reactant complex, the transition state and the FeIII-OH product complex. B) Electron flow during hydrogen atom abstraction reactivity where the FeIV=O first polarizes to a FeIII-oxyl and transfers an electron to either dσ* or dπ* orbital depending on substrate orientation. There is an additional promotion energy (PE) for the π pathway. C) SP, TBP and 6C geometries evaluated for HAA reactivity. D) The 140° and 120° orientations evaluated in this study.
3.3.1. Computational Results
Table 1 summarizes the thermodynamics, reaction barriers and tunneling contributions for the HS (SFe = 5/2) hydrogen atom abstraction reaction coordinates. Table 2 summarizes the structural distortions and their associated energies to reach the transition state in each reaction coordinate in the absence of interactions between the Fe-O and the C-H of the substrate. The SFe = 3/2 pathways (Table S2) are 5 – 10 kcal mol−1 thermodynamically less favorable to form the FeIII-OH product and have higher reaction barriers than the SFe = 5/2 coordinates and are predicted not to contribute to reactivity. For all combination of intermediate geometry and substrate positioning, the abstraction of the substrate hydrogen atom is calculated to be approximately thermoneutral in the SFe = 5/2 manifold (Table 1), demonstrating that driving force is not a primary determinant of reactivity. The geometric difference between the 5C and 6C complexes is the denticity of the succinate ligand. The similarity in thermodynamics observed for the hydrogen abstraction reaction in these complexes indicates that the differential effect of succinate stabilization in the FeIV reactant and the FeIII product is the same. Thus, the resulting O-H bond in the ferric product is ~88 kcal mol−1 for all active site structures.
Table 1.
Summary of calculated HAA reaction coordinates comparing different geometries (SP, TBP and 6C) and substrate angles (140° and 120°) on the high-spin S = 5/2 iron spin surface.
| Geometry | Substrate Angle | ΔG0 (kcal mol−1) | ΔG# (kcal mol−1) | Tunneling Contribution (kcal mol−1) | ΔG#tunneling (kcal mol−1) |
|---|---|---|---|---|---|
| SP | 140° | −0.1 | 18.6 | −2.6 | 16.0 |
| 120° | 0.9 | 18.7 | −1.9 | 16.8 | |
| TBP | 140° | −2.0 | 18.9 | −3.1 | 15.8 |
| 120° | −1.1 | 18.0 | −1.9 | 16.2 | |
| 6C | 140° | −0.3 | 16.4 | −2.2 | 14.2 |
| 120° | 1.3 | 16.5 | −0.7 | 15.8 |
Table 2:
Summary of C-H and Fe-O elongation and distortion energies going from the reactant complex to the transition state for HS HAA reaction coordinates.
| Sp140 | SP120 | TBP140 | TBP120 | 6C140 | 6C120 | |
|---|---|---|---|---|---|---|
| ΔC-H (Å) | 0.23 | 0.18 | 0.23 | 0.19 | 0.2 | 0.15 |
| C-H Distortion Energy (kcal/mol)* | 12.6 | 9.0 | 12.8 | 9.8 | 9.9 | 6.0 |
| ΔFe-O (Å) | 0.16 | 0.20 | 0.16 | 0.18 | 0.17 | 0.23 |
| Fe-O Distortion Energy (kcal/mol)* | 8.4 | 10.9 | 8.0 | 10.7 | 8.5 | 14.4 |
Distortion energy calculations were performed with small model structures fixed at reactant and transition state geometries with the substrate and Fe=O fragments separated.
Table 1 also shows that the ΔG‡ values are all within 2 kcal mol−1, with the hydrogen atom abstraction barriers being slightly lower for the 6C geometries than for the 5C complexes and the two different substrate orientations being similar for each geometry. Additionally, the tunneling contributions to the barriers (−RT1n(τ); τ - tunneling factor) were calculated using the Eckart model.67,68 The 140° complexes have larger tunneling contributions than the 120° and the 5C complexes tunnel more than the 6C complexes. These correlate well with the extent of C-H distortion needed to reach the TS geometry (Table 2); transition states with more elongated C-H bonds are expected to have larger tunneling contributions. Finally, all six reaction coordinates have large calculated kinetic isotope effects (KIEs), consistent with the KIE of 50 observed from experiment (Table S3).66 Thus, from the last column in Table 1, which includes the tunneling contributions to hydrogen atom abstraction reactivity, all three geometries and both substrate orientations (more σ for 140° vs more π for 120°) have comparable reactivities.
3.3.2. Equivalence of σ and π barriers
A particularly interesting result in Table 1 is that, for a given structure, both σ and π channels have comparable barriers, even though the π channel involves an additional promotion energy, as indicated (PE) in Figure 4B, to generate a HS FeIII-oxyl intermediate. Starting from the FeIV=O reactant, the Fe-O and C-H bonds elongate (Figure 4A), the Fe-O bond polarizes and all six reaction coordinates in Table 1 have FeIII-oxyl electronic structures at their transition states.30 Assessing the lowest occupied molecular orbital (LUMO) at the transition state in the 140° and 120° complexes (Figure 5 and S20), the 120° complexes have their C-H vectors more perpendicular to their Fe-O vectors compared to the 140° complexes. The angle between the Fe-O and C-H-O vectors in the 140° complexes is 35°, which corresponds to 60% along oxo pz (σ channel) and 40% along px (π channel), while the angle for the 120° complexes is 75°, which corresponds to 20% along oxo pz and 80% along px. Comparing the six reaction coordinate transition states in Table 2, the 120° oriented complexes are all earlier in their C-H coordinates and later in their Fe-O coordinates relative to the 140° complexes. These differences in C-H and Fe-O elongations correlate with the relative distortion energies observed for these structures (Table 2). Assessing the σ and π channel contributions within each structural type, the C-H and Fe-O distortion energies compensate, and thus the two substrate orientations have similar barriers.
Figure 5:
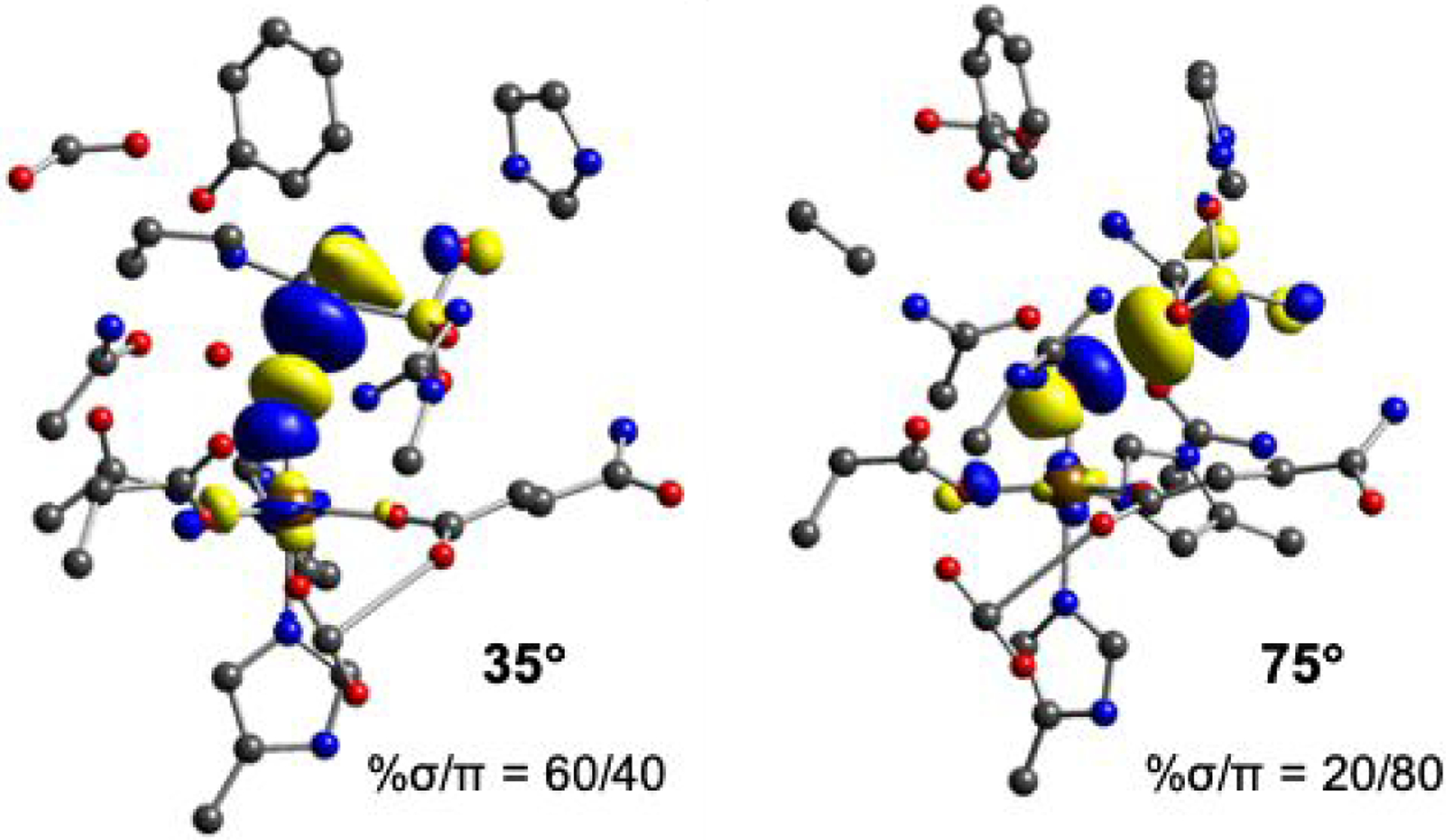
LUMO contours at the transition state for the (left) 140° SP complex and (right) 120° SP complexes. The σ and π contributions to each structure were calculated using the angle between the Fe-O and O-H-C vectors, where the Fe-O vector is along the oxygen pz and the perpendicular vector is along the oxygen px orbital. The corresponding angles are 35° for the 140° complexes, which corresponds to 60% σ and 40% π, and 75° for the 120° complexes, which corresponds to 20% σ and 80% π.
As noted in Figure 4B, the π channel has an additional promotion energy (PE) associated with a dπ* → dσ* excitation required to abstract an α-electron from the perpendicular substrate C-H to produce the high-spin (S=5/2) FeIII-OH product. The change in this promotion energy was evaluated in going from the reactant to the transition state Fe-O bond length (Figure 6). The high-spin excited state with a dπ* hole was generated by elongation of the Fe-O bond to the transition state bond length. In going from the reactant (1.62 Å) to the transition state (1.82 Å) Fe-O bond length, the promotion energy decreases from 33 to 7 kcal mol−1 (separate ground and excited state energy changes given in Figure S21). This reduction in energy enables the promotion of the dπ* electron into dσ* and enables hydrogen atom abstraction reactivity through the π channel. Additionally, from the inset in Figure 6, the π channel has more oxo pπ character (80%, reflecting a more polarized FeIII-oxyl reactant state) than the pσ character involved in the σ channel (only 30% in the reactant), which enables an earlier transition state along the C-H coordinate and results in a lower distortion energy (Table 2). Thus, the very similar σ and π channel reaction barriers is due to a more reactive π channel, which compensates the C-H and Fe-O distortion energies needed to enable π channel reactivity. Finally, we note that there is a small ligand field effect in going from a 6C to 5C geometry, reflected in the ~2 kcal mol−1 lower barriers (before tunneling corrections) for 6C in Table 1 that arises because the required promotion energy is less in the 6C than in the 5C geometry (Figure S22).
Figure 6:
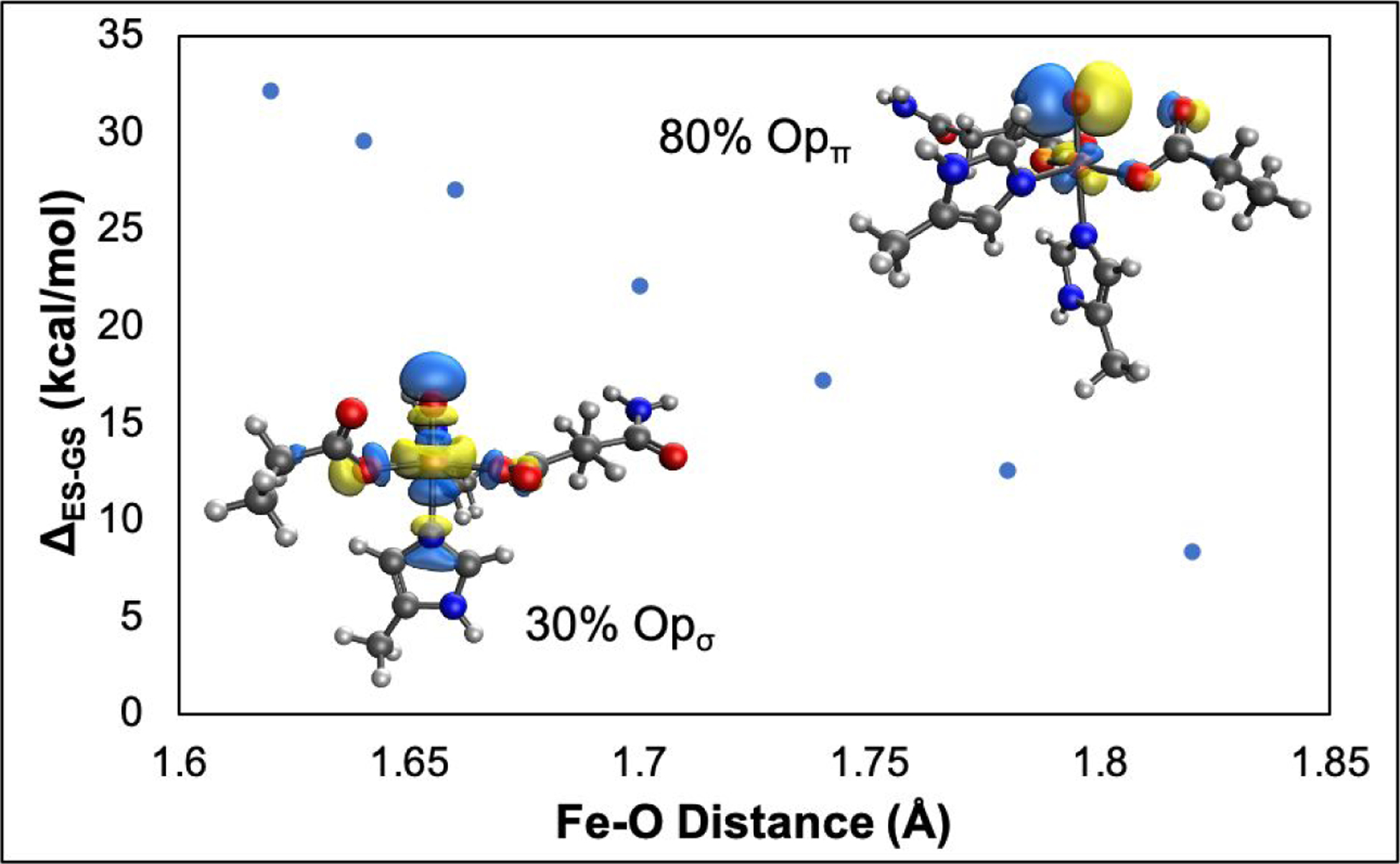
Evolution of the energy difference between the ground state (da* hole) and the excited state (dπ* hole) with elongation of the Fe-O bond. (Inset, bottom left) The contour of the ground state LUMO that has 30% ‘O’ character in dσ*. (Inset, top right) The contour of the excited state LUMO, which has 80% ‘O’ character in dπ*. Both contours are at an Fe-O bond length of 1.62Å.
4. Discussion
In this study, we have defined the structure of the FeIV=O intermediate in TauD using a NRVS/DFT methodology and assessed the structural dependence of hydrogen atom abstraction reactivity of six ferryl geometries and substrate orientations. The NRVS data collected on TauD-J are remarkably similar to those of the FeIV=O in SyrB2 (Figure S5). From evaluation of 12 structural candidates, we conclude that TauD-J has a 5C trigonal bipyramidal geometry, similar to what we observed in SyrB2. In the TauD-J and SyrB2 FeIV=O NRVS data, the intensity distribution in both experimental spectra are comparable, and the trans-axial bend within the 285–340 cm−1 region produces the most intense peak, as it has the largest iron displacement. The main difference in the spectra are in the 200–285 cm−1 and 340–440 cm–1 regions, where SyrB2 has only one peak in the high-energy region compared to the two peaks observed for TauD-J (Figure S5). From the analyses of all the structural candidates and their corresponding NRVS spectra, we identified a peak in the 340–440 cm−1 region that is characteristic of a monodentate carboxylate stretch. This stretching mode is very sensitive to denticity and is a direct probe of the carboxylate coordination mode in FeIV=O intermediates, as a 6-coordinate site with a bidentate carboxylate shifts this mode to the lower energy 285–340 cm−1 region. Because SyrB2 has a halide ligand instead of the facial triad aspartate ligand, a single feature, ascribed to the monodentate succinate, is present in the high energy 340–440 cm−1 region, with the Fe-halide vibration shifted to the low energy region, due to its greater mass. On the other hand, TauD-J has the two vibrations in this high-energy region, due to the additional presence of the monodentate facial triad aspartate, which defines its 5C geometry. Thus, despite the differences in the first coordination spheres of the ferryl complexes in TauD and SyrB2, this study highlights the structural similarity between the intermediates in the halogenases and hydroxylases, and, by extension, other αKG-dependent enzymes. Additionally, due to its ability to identify first sphere metal-ligand vibrations in high-valent iron intermediates, this study also demonstrates that NRVS is a powerful active site structural probe, in general, for mononuclear and binuclear nonheme iron enzymes.
Having generated SP, TBP and 6C active site structures to analyze the NRVS data for TauD-J, we further evaluated the effect of geometry, substrate orientation and iron spin state on the hydrogen atom abstraction reaction coordinates for facial triad FeIV=O intermediates. First, all the SFe = 5/2 pathways were thermodynamically more favorable and had lower reaction barriers than the SFe = 3/2 pathways. For SP, TBP and 6C geometries, the thermodynamic driving force and the reaction barriers for hydrogen abstraction were comparable, demonstrating that these differences in geometric structure are not primary determinants of hydrogen atom abstraction reactivity. Furthermore, by generating two different calculated substrate orientations in the TauD active site, we could evaluate the relative σ and π contributions to reactivity. Our analysis showed that the σ and π reaction coordinates within each geometry have similar reaction barriers. This finding is an important one, as the π channel has, relative to the σ channel, an additional promotion energy that is associated with a dπ* to dσ* excitation (Figure 4B) required to form a HS FeIII-oxyl intermediate and the HS FeIII-OH product. From analysis of the frontier molecular orbitals involved in hydrogen atom abstraction reactivity, we find that the dπ orbital within the π channel has more oxo character (reflected in the oxo pπ orbital coefficients in Figure 6, inset) compared to dσ orbital involved in the σ channel. This increased oxo character in the π channel results in better overlap and earlier transition states along the C-H coordinate (see inset in Figure 6). Additionally, elongating the Fe-O bond of the FeIV=O reactant to that in the transition state results in a decreased energy gap between dσ* and dπ*, which diminishes the promotion energy. Thus, both substrate orientations have similar computational hydrogen atom abstraction reaction barriers; due to its larger oxo character, the π channel polarizes more quickly than the σ channel, which results in an earlier transition state along the C-H coordinate, leading to a lower distortion energy that compensates the promotion energy required for accessing the π channel (Figure 7). In the αKG-dependent enzymes, since there is flexibility with respect to enzyme-cofactor-substrate positioning, our insights have demonstrated that hydrogen atom abstraction reactivity along both the σ and π channels are comparable and lead to an FeIII-OH/substrate radical. The σ channel would lead to the ferric hydroxide oriented toward the substrate radical, while the π channel has the OH oriented away from the carbon radical. As we demonstrated for SyrB2,32 the π channel positions the halide towards the carbon radical, which leads to halogenation of the native substrate, not the thermodynamically favored hydroxylation. Thus, our study has demonstrated that the flexibility of σ and π hydrogen atom abstraction reactivity by FeIV=O intermediates enables non-hydroxylation outcomes, which is crucial for the αKG-dependent enzymes. The versatile reactivity of the αKG-dependent enzymes is due to the relative positioning of the FeIII-OH/substrate radical and it is likely that use of the π channel for hydrogen atom abstraction will be found to enable other reaction outcomes in the broad repertoire of this class of nonheme iron enzymes.
Figure 7:
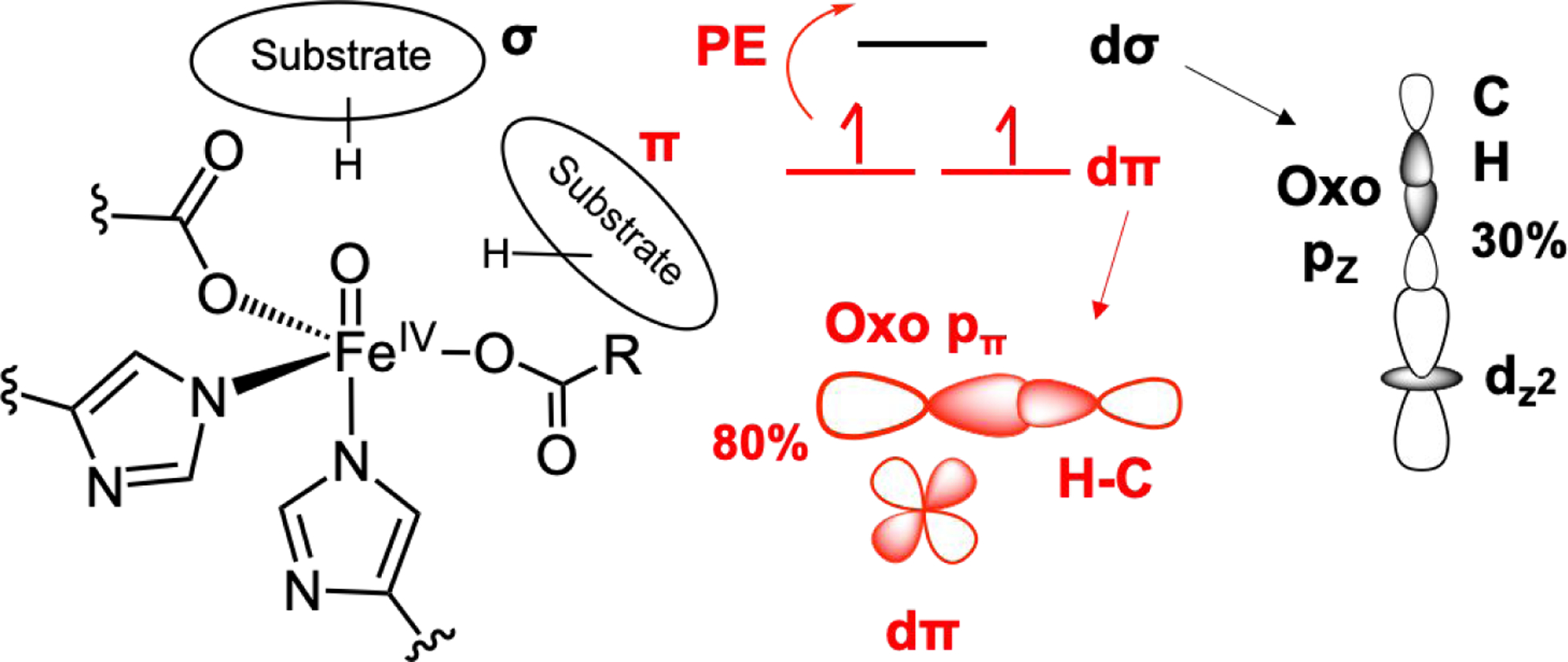
(Left) Substrate orientation relative to FeIV=O along the σ and π channels. (Right) Reactivity along π channel requires an additional promotion energy (PE), which is compensated by an earlier TS along the C-H reaction coordinate due to a more polarized oxo (80% in π channel vs 30% in σ channel).
Supplementary Material
Acknowledgments
This research was supported by the US NIH Grants GM 40392 (to E.I.S.), GM 127079 (to C.K.) and GM 55365 (to L.M.K.D. and J.M.B.). This project was also supported by MSMT CR (LTAUSA19148 to M.S.). The synchrotron experiments were performed at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI; proposal no. 2011B1267).
Footnotes
Supporting Information. The supplementary section contains the Mössbauer spectrum of the NRVS sample, details on the processing of the NRVS data, the simulations for all the NRVS structural models and tables and figures relevant to the reaction coordinate calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Lando D; Peet DJ; Gorman JJ; Whelan DA; Whitelaw ML; Bruick RK FIH-1 Is an Asparaginyl Hydroxylase Enzyme That Regulates the Transcriptional Activity of Hypoxia-Inducible Factor. Genes Dev. 2002, 16 (12), 1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Gerken T; Girard CA; Tung Y-CL; Webby CJ; Saudek V; Hewitson KS; Yeo GSH; McDonough MA; Cunliffe S; McNeill LA; Galvanovskis J; Rorsman P; Robins P; Prieur X; Coll AP; Ma M; Jovanovic Z; Farooqi IS; Sedgwick B; Barroso I; Lindahl T; Ponting CP; Ashcroft FM; O’Rahilly S; Schofield CJ The Obesity-Associated FTO Gene Encodes a 2-Oxoglutarate-Dependent Nucleic Acid Demethylase. Science 2007, 318 (5855), 1469–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vaillancourt FH; Yin J; Walsh CT SyrB2 in Syringomycin E Biosynthesis Is a Nonheme FeII α-Ketoglutarate- and O2-Dependent Halogenase. Proc. Natl. Acad. Sci 2005, 102 (29), 10111–10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Baldwin JE; Abraham E The Biosynthesis of Penicillins and Cephalosporins. Nat. Prod. Rep 1988, 5 (2), 129. [DOI] [PubMed] [Google Scholar]
- (5).Vaillancourt FH; Haro M-A; Drouin NM; Karim Z; Maaroufi H; Eltis LD Characterization of Extradiol Dioxygenases from a Polychlorinated Biphenyl-Degrading Strain That Possess Higher Specificities for Chlorinated Metabolites. J. Bacteriol 2003, 185 (4), 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Keenan BG; Wood TK Orthric Rieske Dioxygenases for Degrading Mixtures of 2,4-Dinitrotoluene/Naphthalene and 2-Amino-4,6-Dinitrotoluene/4-Amino-2,6-Dinitrotoluene. Appl. Microbiol. Biotechnol 2006, 73 (4), 827–838. [DOI] [PubMed] [Google Scholar]
- (7).Mishina Y; He C Oxidative Dealkylation DNA Repair Mediated by the Mononuclear Non-Heme Iron AlkB Proteins. J. Inorg. Biochem 2006, 100 (4), 670–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Solomon EI; Goudarzi S; Sutherlin KD O2 Activation by Non-Heme Iron Enzymes. Biochemistry 2016, 55 (46), 6363–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Solomon EI; Iyer SR Geometric and Electronic Structural Contributions to Fe/O2 Reactivity. Bull. Jpn. Soc. Coord. Chem 2019, 73, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kal S; Que L Dioxygen Activation by Nonheme Iron Enzymes with the 2-His-1-Carboxylate Facial Triad That Generate High-Valent Oxoiron Oxidants. JB/C J. Biol. Inorg. Chem 2017, 22 (2–3), 339–365. [DOI] [PubMed] [Google Scholar]
- (11).Solomon EI; Light KM; Liu LV; Srnec M; Wong SD Geometric and Electronic Structure Non-Heme Iron Enzymes. Acc. Chem. Res 2013, 46, 2725–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Krebs C; Galonić Fujimori D; Walsh CT; Bollinger JM Jr. Non-Heme Fe(IV)-Oxo Intermediates. Acc. Chem. Res 2007, 40, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bollinger JM Jr.; Chang W; Matthews ML; Martinie RJ; Boal AK; Krebs C CHAPTER 3 Mechanisms of 2-Oxoglutarate-Dependent Oxygenases: The Hydroxylation Paradigm and Beyond. 2-Oxoglutarate-Dependent Oxygenases; Schofield C, Hausinger R, Eds.; The Royal Society of Chemistry: Cambridge, 2015, pp 95–122. [Google Scholar]
- (14).Hausinger RP FeII/α-Ketoglutarate-Dependent Hydroxylases and Related Enzymes. Crit. Rev. Biochem. Mol. Biol 2004, 39, 21–68. [DOI] [PubMed] [Google Scholar]
- (15).Hausinger RP CHAPTER 1 Biochemical Diversity of 2-Oxoglutarate-Dependent Oxygenases. 2-Oxoglutarate-Dependent Oxygenases; Schofield C, Hausinger R, Eds.; The Royal Society of Chemistry: Cambridge, 2015, pp 1–58. [Google Scholar]
- (16).Price JC; Barr EW; Tirupati B; Bollinger JM; Krebs C The First Direct Characterization of a High-Valent Iron Intermediate in the Reaction of an α-Ketoglutarate-Dependent Dioxygenase: A High-Spin Fe(IV) Complex in Taurine/α-Ketoglutarate Dioxygenase (TauD) from Escherichia Coli. Biochemistry 2003, 42 (24), 7497–7508. [DOI] [PubMed] [Google Scholar]
- (17).Eichhorn E; van der Ploeg JR; Kertesz MA; Leisinger T Characterization of α-Ketoglutarate-Dependent Taurine Dioxygenase from Escherichia Coli. J. Biol. Chem 1997, 272 (37), 23031–23036. [DOI] [PubMed] [Google Scholar]
- (18).Hoffart LM; Barr EW; Guyer RB; Bollinger JM Jr.; Krebs C Direct Spectroscopic Detection of a C-H-Cleaving High-Spin Fe(IV) Complex in a Prolyl-4-Hydroxylase. Proc. Natl. Acad. Sci 2006, 103 (40), 14738–14743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Pan J; Wenger ES; Matthews ML; Pollock CJ; Bhardwaj M; Kim AJ; Allen BD; Grossman RB; Krebs C; Bollinger JM Evidence for Modulation of Oxygen Rebound Rate in Control of Outcome by Iron(II)- and 2-Oxoglutarate-Dependent Oxygenases. J. Am. Chem. Soc 2019, 141 (38), 15153–15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Dunham NP; Chang W; Mitchell AJ; Martinie RJ; Zhang B; Bergman JA; Rajakovich LJ; Wang B; Silakov A; Krebs C; Boal AK; Bollinger JM Two Distinct Mechanisms for C-C Desaturation by Iron(II)- and 2-(Oxo)Glutarate-Dependent Oxygenases: Importance of α-Heteroatom Assistance. J. Am. Chem. Soc 2018, 140 (23), 7116–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Peck SC; Wang C; Dassama LMK; Zhang B; Guo Y; Rajakovich LJ; Bollinger JM; Krebs C; van der Donk WA O-H Activation by an Unexpected Ferryl Intermediate during Catalysis by 2-Hydroxyethylphosphonate Dioxygenase. J. Am. Chem. Soc 2017, 139 (5), 2045–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Galonić Fujimori D; Barr EW; Matthews ML; Koch GM; Yonce JR; Walsh CT; Bollinger JM; Krebs C; Riggs-Gelasco PJ Spectroscopic Evidence for a High-Spin Br-Fe(IV)-Oxo Intermediate in the α-Ketoglutarate-Dependent Halogenase CytC3 from Streptomyces. J. Am. Chem. Soc 2007, 129 (44), 13408–13409. [DOI] [PubMed] [Google Scholar]
- (23).Tamanaha E; Zhang B; Guo Y; Chang W; Barr EW; Xing G; Clair J St.; Ye S; Neese F; Bollinger JM; Krebs C Spectroscopic Evidence for the Two C-H-Cleaving Intermediates of Aspergillus Nidulans Isopenicillin N Synthase. J. Am. Chem. Soc 2016, 138 (28), 8862–8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Eser BE; Barr EW; Frantom PA; Saleh L; Bollinger JM Jr.; Krebs C; Fitzpatrick PF Direct Spectroscopic Evidence for a High-Spin Fe (IV) Intermediate in Tyrosine Hydroxylase. J. Am Chem. Soc 2007, 129, 11334–11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Panay AJ; Lee M; Krebs C; Bollinger JM; Fitzpatrick PF Evidence for a High-Spin Fe(IV) Species in the Catalytic Cycle of a Bacterial Phenylalanine Hydroxylase. Biochemistry 2011, 50 (11), 1928–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Matthews ML; Krest CM; Barr EW; Vaillancourt FH; Walsh CT; Green MT; Krebs C; Bollinger JM Jr. Substrate-Triggered Formation and Remarkable Stability of the C-H Bond-Cleaving Chloroferryl Intermediate in the Aliphatic Halogenase, SyrB2. Biochemistry 2009, 48, 4331–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Blasiak LC; Vaillancourt FH; Walsh CT; Drennan CL Crystal Structure of the Non-Haem Iron Halogenase SyrB2 in Syringomycin Biosynthesis. Nature 2006, 440 (7082), 368–371. [DOI] [PubMed] [Google Scholar]
- (28).O’Brien JR; Schuller DJ; Yang VS; Dillard BD; Lanzilotta WN Substrate-Induced Conformational Changes in Escherichia Coli Taurine/α-Ketoglutarate Dioxygenase and Insight into the Oligomeric Structure. Biochemistry 2003, 42 (19), 5547–5554. [DOI] [PubMed] [Google Scholar]
- (29).Wong SD; Srnec M; Matthews ML; Liu LV; Kwak Y; Park K; Bell III CB; Alp EE; Zhao J; Yoda Y; Kitao S; Seto M; Krebs C; Bollinger JM; Solomon EI Elucidation of the Fe(IV)=O Intermediate in the Catalytic Cycle of the Halogenase SyrB2. Nature 2013, 499, 320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Srnec M; Wong SD; England J; Que L Jr.; Solomon EI π -Frontier Molecular Orbitals in S=2 Ferryl Species and Elucidation of Their Contributions to Reactivity. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 14326–14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Srnec M; Wong SD; Matthews ML; Krebs C; Bollinger JM; Solomon EI Electronic Structure of the Ferryl Intermediate in the α-Ketoglutarate Dependent Non-Heme Iron Halogenase SyrB2: Contributions to H Atom Abstraction Reactivity. J. Am. Chem. Soc 2016, 138 (15), 5110–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Srnec M; Solomon EI Frontier Molecular Orbital Contributions to Chlorination versus Hydroxylation Selectivity in the Non-Heme Iron Halogenase SyrB2. J. Am. Chem. Soc 2017, 139 (6), 2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Harlos K; Schofield CJ; Zhang Z; Ren J; Stammers DK; Baldwin JE Structural Origins of the Selectivity of the Trifunctional Oxygenase Clavaminic Acid Synthase. Nat. Struct. Biol 2000, 7 (2), 127–133. [DOI] [PubMed] [Google Scholar]
- (34).Elkins JM; Hewitson KS; McNeill LA; Seibel JF; Schlemminger I; Pugh CW; Ratcliffe PJ; Schofield CJ Structure of Factor-Inhibiting Hypoxia-Inducible Factor (HIF) Reveals Mechanism of Oxidative Modification of HIF-1a. J. Biol. Chem 2003, 278, 1802–1806. [DOI] [PubMed] [Google Scholar]
- (35).Neugebauer ME; Sumida KH; Pelton JG; McMurry JL; Marchand JA; Chang MCY A Family of Radical Halogenases for the Engineering of Amino-Acid-Based Products. Nat. Chem. Biol 2019, 15 (10), 1009–1016. [DOI] [PubMed] [Google Scholar]
- (36).Neidig ML; Decker A; Choroba OW; Huang F; Kavana M; Moran GR; Spencer JB; Solomon EI Spectroscopic and Electronic Structure Studies of Aromatic Electrophilic Attack and Hydrogen-Atom Abstraction by Non-Heme Iron Enzymes. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 12966–12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Martinie RJ; Pollock CJ; Matthews ML; Bollinger JM; Krebs C; Silakov A Vanadyl as a Stable Structural Mimic of Reactive Ferryl Intermediates in Mononuclear Nonheme-Iron Enzymes. Inorg. Chem 2017, 56 (21), 13382–13389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Dassama LMK; Yosca TH; Conner DA; Lee MH; Blanc B; Streit BR; Green MT; DuBois JL; Krebs C; Bollinger JM Jr. O2-Evolving Chlorite Dismutase as a Tool for Studying O2-Utilizing Enzymes. Biochemistry 2012, 51 (8), 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Moënne-Loccoz P; Krebs C; Herlihy K; Edmondson DE; Theil EC; Huynh BH; Loehr TM The Ferroxidase Reaction of Ferritin Reveals a Diferric μ−1,2 Bridging Peroxide Intermediate in Common with Other O2-Activating Non-Heme Diiron Proteins. Biochemistry 1999, 38 (17), 5290–5295. [DOI] [PubMed] [Google Scholar]
- (40).Bollinger JM; Tong WH; Ravi N; Huynh BH; Edmonson DE; Stubbe J Mechanism of Assembly of the Tyrosyl Radical-Diiron(III) Cofactor of E. Coli Ribonucleotide Reductase. 2. Kinetics of The Excess Fe2+ Reaction by Optical, EPR, and Mössbauer Spectroscopies. J. Am. Chem. Soc 1994, 116 (18), 8015–8023. [Google Scholar]
- (41).Streit BR; DuBois JL Chemical and Steady-State Kinetic Analyses of a Heterologously Expressed Heme Dependent Chlorite Dismutase. Biochemistry 2008, 47 (19), 5271–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Park K; Tsugawa T; Furutachi H; Kwak Y; Liu LV; Wong SD; Yoda Y; Kobayashi Y; Saito M; Kurokuzu M; Seto M; Suzuki M; Solomon EI Nuclear Resonance Vibrational Spectroscopy and DFT Study of Peroxo-Bridged Biferric Complexes: Structural Insight into Peroxo Intermediates of Binuclear Non-Heme Iron Enzymes. Angew. Chem. Int. Ed. Engl 2013, 52, 1294–1298. [DOI] [PubMed] [Google Scholar]
- (43).Sturhahn W CONUSS and PHOENIX: Evaluation of Nuclear Resonant Scattering Data. Hyperfine Interact 2000, 125 (1–4), 149–172. [Google Scholar]
- (44).Sage JT; Paxson C; Wyllie GRA; Sturhahn W; Durbin SM; Champion PM; Alp EE; Scheidt WR Nuclear Resonance Vibrational Spectroscopy of a Protein Active-Site Mimic. J. Phys. Condens. Matter 2001, 13 (34), 7707–7722. [Google Scholar]
- (45).Bell III CB; Wong SD; Xiao Y; Klinker EJ; Tenderholt AL; Smith MC; Rohde J-U; Que L; Cramer SP; Solomon EI A Combined NRVS and DFT Study of FeIV=O Model Complexes: A Diagnostic Method for the Elucidation of Non-Heme Iron Enzyme Intermediates. Angew. Chemie Int. Ed 2008, 47 (47), 9071–9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Grimme S; Antony J; Ehrlich S; Krieg H A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys 2010, 132 (15), 154104. [DOI] [PubMed] [Google Scholar]
- (47).Weigend F; Ahlrichs R Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys 2005, 7 (18), 3297. [DOI] [PubMed] [Google Scholar]
- (48).Klamt A; Schüürmann G COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc., Perkin Trans 2 1993, No. 5, 799–805. [Google Scholar]
- (49).Schäfer A; Klamt A; Sattel D; Lohrenz JCW; Eckert F COSMO Implementation in TURBOMOLE: Extension of an Efficient Quantum Chemical Code towards Liquid Systems. Phys. Chem. Chem. Phys 2000, 2 (10), 2187–2193. [Google Scholar]
- (50).Ahlrichs R; Bär M; Häser M; Horn H; Kölmel C Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett 1989, 162 (3), 165–169. [Google Scholar]
- (51).Eichkorn K; Treutler O; Öhm H; Häser M; Ahlrichs R Auxiliary Basis Sets to Approximate Coulomb Potentials. Chem. Phys. Lett 1995, 240 (4), 283–290. [Google Scholar]
- (52).Eichkorn K; Weigend F; Treutler O; Ahlrichs R Auxiliary Basis Sets for Main Row Atoms and Transition Metals and Their Use to Approximate Coulomb Potentials. Theor. Chem. Accounts Theory, Comput. Model. (Theoretica Chim. Acta) 1997, 97 (1–4), 119–124. [Google Scholar]
- (53).Tenderholt A gennrvs, Python script, Version 28, 2009. Available at http://www.stanford.edu/group/solomon/gennrvs/gennrvs.py.txt.
- (54).Becke AD Density‐functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98 (7), 5648–5652. [Google Scholar]
- (55).Ye S; Neese F Nonheme Oxo-Iron(IV) Intermediates Form an Oxyl Radical upon Approaching the C-H Bond Activation Transition State. Proc. Natl. Acad. Sci 2011, 108 (4), 1228–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Cossi M; Rega N; Scalmani G; Barone V Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem 2003, 24 (6), 669–681. [DOI] [PubMed] [Google Scholar]
- (57).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09 Revision E.01; Gaussian Inc.: Wallingford CT, 2009. [Google Scholar]
- (58).Tenderholt AL QMForge, Version 2.4, https://qmforge.net.
- (59).Kieber-Emmons MT LUMO, Version 1.0.3, 2014. [Google Scholar]
- (60).Gilbert A IQmol, Version 2.10.0, 2016. [Google Scholar]
- (61).Diebold AR; Brown-Marshall CD; Neidig ML; Brownlee JM; Moran GR; Solomon EI Activation of α-Keto Acid-Dependent Dioxygenases : Application of an {FeNO}7/{FeO2}8 Methodology for Characterizing the Initial Steps of O2 Activation. J. Am. Chem. Soc 2011, 133, 18148–18160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Davis KM; Altmyer M; Martinie RJ; Schaperdoth I; Krebs C; Bollinger JM; Boal AK Structure of a Ferryl Mimic in the Archetypal Iron(II)- and 2-(Oxo)-Glutarate-Dependent Dioxygenase, TauD. Biochemistry 2019, 58 (41), 4218–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mitchell AJ; Dunham NP; Martinie RJ; Bergman JA; Pollock CJ; Hu K; Allen BD; Chang W-C; Silakov A; Bollinger JM Jr.; Krebs C; Boal AK Visualizing the Reaction Cycle in an Iron(II)- and 2-(Oxo)-Glutarate-Dependent Hydroxylase. J. Am. Chem. Soc 2017, 139 (39), 13830–13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Sinnecker S; Svensen N; Barr EW; Ye S; Bollinger JM; Neese F; Krebs C Spectroscopic and Computational Evaluation of the Structure of the High-Spin Fe(IV)-Oxo Intermediates in Taurine: α-Ketoglutarate Dioxygenase from Escherichia Coli and Its His99Ala Ligand Variant. J. Am. Chem. Soc 2007, 129 (19), 6168–6179. [DOI] [PubMed] [Google Scholar]
- (65).Sinnecker S; Slep LD; Bill E; Neese F Performance of Nonrelativistic and Quasi-Relativistic Hybrid DFT for the Prediction of Electric and Magnetic Hyperfine Parameters in 57Fe Mössbauer Spectra. Inorg. Chem 2005, 44 (7), 2245–2254. [DOI] [PubMed] [Google Scholar]
- (66).Price JC; Barr EW; Hoffart LM; Krebs C; Bollinger JM Jr. Kinetic Dissection of the Catalytic Mechanism of Taurine:α-Ketoglutarate Dioxygenase (TauD) from Escherichia Coli. Biochemistry 2005, 44 (22), 8138–8147. [DOI] [PubMed] [Google Scholar]
- (67).Maldonado-Domínguez M; Bím D; Fučík R; Čurík R; Srnec M Reactive Mode Composition Factor Analysis of Transition States: The Case of Coupled Electron-Proton Transfers. Phys. Chem. Chem. Phys 2019, 21 (45), 24912–24918. [DOI] [PubMed] [Google Scholar]
- (68).Eckart C The Penetration of a Potential Barrier by Electrons. Phys. Rev 1930, 35 (11), 1303–1309. 10.1103/PhysRev.35.1303. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.