

Summary
Axons in the mature central nervous system (CNS) fail to regenerate after axotomy, partly due to the inhibitory environment constituted by reactive glial cells producing astrocytic scars, chondroitin sulfate proteoglycans and myelin debris. We investigated this inhibitory milieu, showing it is reversible and depends on glial metabolic status. We show that glia can be reprogrammed to promote morphological and functional regeneration after CNS injury in Drosophila via increased glycolysis. This enhancement is mediated by the glia derived metabolites: L-lactate and L-2-hydroxyglutarate (L-2HG). Genetically/pharmacologically increasing or reducing their bioactivity promoted or impeded CNS axon regeneration. L-lactate and L-2HG from glia acted on neuronal metabotropic GABAB receptors to boost cAMP signaling. Local application of L-lactate to injured spinal cord promoted corticospinal tract axon regeneration, leading to behavioral recovery in adult mice. Our findings revealed a metabolic switch to circumvent the inhibition of glia while amplifying their beneficial effects for treating CNS injuries.
Keywords: axon regeneration, glia, metabolism, lactate, 2-hydroxyglutarate, GABA B receptor, cAMP, functional recovery, spinal cord injury (SCI), central nervous system (CNS), inhibitory environment
Graphical Abstract
eTOC Blurb
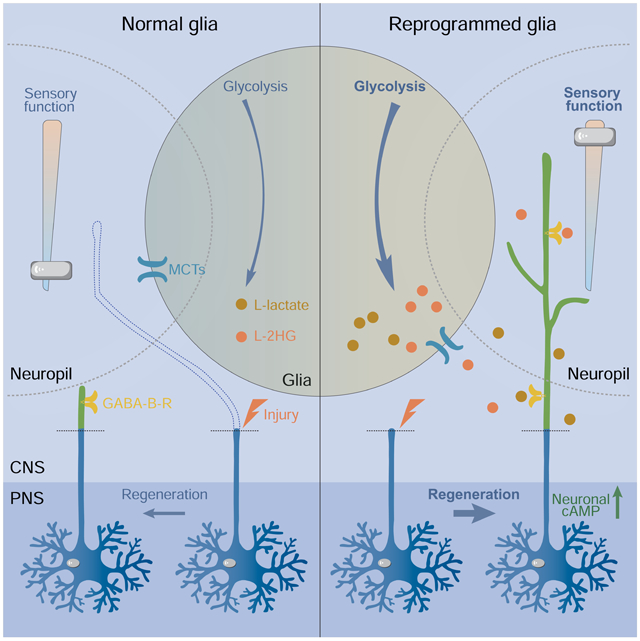
Li et al. reveal that the glial metabolic status is related to the failure of axon regeneration in the CNS. They show that the inhibitory environment can be essentially reversed by elevated aerobic glycolysis in glia, leading to axon regeneration in the CNS, which may be evolutionarily conserved.
Introduction
Regarded as the holy grail in regenerative medicine, achieving axon regeneration and functional recovery in the CNS after injury or in neurodegenerative diseases remains a daunting task. Besides the diminished intrinsic growth capacity of mature neurons, the CNS environment has been demonstrated as inhibitory to axon regrowth compared with the permissive peripheral nervous system (PNS) (David and Aguayo, 1981; Smith et al., 1986). To understand the underlying mechanisms, previous studies have conventionally focused on the inhibitory milieu associated with the lesion site. It is principally constituted by the scar-forming reactive glial cells and the upregulated repulsive chondroitin sulfate proteoglycans (CSPGs), leading to stalled axon regrowth (Liuzzi and Lasek, 1987; Silver and Miller, 2004). The lesion site naturally becomes the first blockade which regenerating axons encounter, and has been the focus of therapeutic interventions (Anderson et al., 2018; Lang et al., 2015). However, contrary to the proposed inhibitory role of the glial scar, experimental evidence has demonstrated that preventing astroglial scar formation following CNS injury does not result in increased regrowth (Anderson et al., 2016; Faulkner et al., 2004; Wanner et al., 2013), suggesting beneficial effects of the astroglial scar on tissue integrity and axon regrowth (Adams and Gallo, 2018; Rolls et al., 2009). In the peripheral nerve grafts experiments, axons regenerated through the grafts and contributed to functional restoration in injured spinal cords (Alilain et al., 2011; Tom et al., 2009). Intriguingly however, even without traumatic injury in the spinal cord, axons elongated long distance in the peripheral nerve grafts but stalled when they reentered the CNS (David and Aguayo, 1981), suggesting that the CNS environment is not permissive to axon regrowth. This is supported by observations from two-photon laser induced single sensory axon injury experiment in the spinal cord. With small damage and little scar, sensory axons also exhibited limited regeneration unless their intrinsic growth capacity was increased by preconditioning, even though they could originally regenerate in the PNS (Ylera et al., 2009). The interesting phenomenon that sensory axons can regenerate in the PNS, but not in the CNS, is conserved in Drosophila (Song et al., 2019; Song et al., 2012; Song et al., 2015). Other than peripheral nerve grafts, transplanted fetal rat spinal cord tissue could help axon regrowth after adult spinal cord injury (Reier et al., 1986), which is consistent with the fact that regeneration occurs in some mammals such as opossums when the CNS is still immature (Ferretti et al., 2003), indicating that the CNS environment is capable of supporting axon regeneration in a specific period. This may be associated with the status of glial cells because reintroducing an immature CNS environment by implantation of immature astrocytes promoted axon regeneration (Smith et al., 1986).
Thus, in this study we asked if, by approaching a specific status of glial cells, the inhibitory CNS environment can be reversed to support axon regeneration. Using a Drosophila CNS injury paradigm, we assessed CNS axon regeneration after manipulating various functional pathways in glial cells. We found that activation of PI3K, EGFR or cell-cycle regulation pathways in glia was able to promote axon regeneration. Moreover, combined activation of PI3K and EGFR pathways in glial cells drastically converted the inhibitory environment and unprecedented elongation of axon regrowth in the CNS was observed. Excitingly, we discovered that the “specific status” is associated with glial glucose metabolism and metabolic rewiring in glial cells is sufficient to promote axon regeneration in the CNS. Importantly, elevated L-lactate and L-2HG, by a metabolic switch to aerobic glycolysis in glia, act on neuronal GABAB receptors in an unexpected inverse activation manner and increase cAMP signaling for neuronal regrowth. We further demonstrated that L-lactate stimulates CNS axon regeneration after spinal cord injury (SCI) in adult mice. While the glia-neuron lactate shuttle hypothesis has been studied widely and glial glycolysis has been shown to be important for neuronal survival in Drosophila (Volkenhoff et al., 2015), our study established the first link between glial glycolysis and neuroregeneration via molecular signaling, and shed light on the role of glial glycolysis in determining the CNS inhibitory environment.
Results
Identification of glial pathways enhancing axon regeneration in the Drosophila CNS
To assess axon regeneration in the CNS, we employed an established injury model (Song et al., 2019; Song et al., 2012; Song et al., 2015) by severing axons of class IV dendritic arborization (C4da) neurons in the ventral nerve cord (VNC) of Drosophila larvae using a two-photon laser (Figure 1A and 1B). After lesion, C4da neuron axons retracted out of the neuropil region at the degeneration stage (Figure S1A) and glial-scar-like structures were formed at the injury sites (Song et al., 2012) (Figure S1B). Given the robust regenerative capability of C4da axons in the periphery, the injured axons regrew back to the neuropil border within 24 hours after injury (h AI). However, they showed limited regrowth within the neuropil region (Figure S1C), indicating that the fly CNS environment inhibits axon regeneration. Axon regeneration is quantified by normalized regeneration length and regeneration percentage (Figure S2, see STAR Methods).
Figure 1. Reprogramming glial cells promotes axon regeneration in the Drosophila CNS.

(A) Images show axon projection of C4da neurons in the VNC of WT Drosophila larvae (ppk-CD4tdGFP; repo-Gal4, UAS-mRFP). Axons are labeled with GFP, glial cells are labeled with RFP, and the neuropil is demarcated by the orange dashed lines.
(B) Schematics of the Drosophila CNS injury model (WT). Left panel shows projection of C4da neuron axons in the larval VNC and the injury sites for one segment (red concentric circles); middle panel shows axons degenerating out of the neuropil (orange region) at 8 h AI; right panel shows injured axons regenerating to the boundary of the neuropil at 24 h AI.
(C) Quantification of normalized regenerated axon length for the screened pathways when manipulated in glial cells, n = 24, 22, 19, 18, 26, 16, 14, 14, 14, 12, 10, 12, 18, 62 lesioned segments from 12, 11, 10, 9, 13, 8, 7, 7, 7, 6, 5, 6, 9, 31 larvae respectively for each genotype, WT (ppk-CD4tdGFP; repo-Gal4, UAS-mRFP) as control, one-way ANOVA with Dunnett’s test. See also Table S1.
(D) Axon regrowth of C4da neurons induced by expressing Pi3K92ECA and EgfrCA individually or together in glia under the control of repo-Gal4, alrm-gal4, TIFR-Gal4 or GMR54H02-Gal4 at 24 h AI on segment A3 in the VNC. Schematics above the images show glia cohorts labeled by different promoters; diagrams below the images show regenerated axons (red) in the neuropil (demarcated by the orange dashed lines); the red dotted circles show injury sites.
(E) Quantification of normalized regenerated axon length for genotypes shown in (D), n = 62, 24, 22, 32, 34, 24, 20 lesioned segments from 31, 12, 11, 16, 17, 12, 10 larvae respectively for each genotype, one-way ANOVA with Dunnett’s test.
(F) Quantification of regeneration percentage for genotypes shown in (D), Fisher’s exact test.
*P < 0.05, **P < 0.01 and ***P < 0.001. Scale bars, 20 μm. Data are expressed as mean ± s.e.m. CA, constitutively active; DN, dominant negative. See also Figure S1.
To determine whether a state of glia to support axon regeneration can be achieved, we performed a glial overexpression screen (with repo-Gal4) of 13 candidate genes representing various cellular functional pathways, by assessing axon regeneration in the CNS. We found that activation of PI3K, EGFR or cell-cycle regulation pathways in glia were able to significantly increase axon regeneration length compared to wild-type (WT) (Figure 1C and Table S1). Strikingly, we found that activation of PI3K and EGFR simultaneously in glia enabled axons to regrow extensively in the neuropil region with regeneration percentage > 90% at 24 h AI, in contrast to < 10% in WT, and ~ 5-fold increase of regeneration length (Figure 1D-1F and S1C-S1E). Compared with Pten deficiency in neurons, which only showed enhanced regeneration at 72 but not 24 h AI in our previous study (Song et al., 2012), activation of both PI3K and EGFR pathways in glia appears to render faster and more robust CNS axon regeneration. We consider activating glial PI3K and EGFR pathways as reprogramming glia to a state which essentially changes the CNS environment for axon regeneration. Intriguingly, even though reprogrammed glial cells by activating PI3K and EGFR pathways (RGCPE, specifically with repo-Gal4>Pi3K92ECA, EgfrCA (constitutively active)) promotes C4da neuron axons regeneration in the CNS, it did not change the limited regeneration capacity of Class III dendritic arborization (C3da) neurons in the PNS (Song et al., 2019; Song et al., 2012; Song et al., 2015) (Figure S1F-S1H), suggesting that RGCPE specifically impacts on the CNS environment to promote axon regeneration.
Given the important roles of astrocytes in the neuropil of Drosophila larvae (Doherty et al., 2009), we asked whether reprogrammed astrocytes underlay the enhanced axon regeneration induced by RGCPE. Although astrocyte specific activation of PI3K and EGFR pathways (with alrm-Gal4>Pi3K92ECA, EgfrCA) partially recapitulated the axon regeneration phenotype induced by RGCPE, their activation in other types of glia (with TIFR-Gal4: ensheathing, cortex, wrapping and surface glia; GMR54H02-Gal4: cortex and surface glia) showed similar beneficial effects (Figure 1D-1F). Flies with these GAL4 drivers alone showed similar axon regeneration with the WT (Figure S2E and S2F). Therefore, no specific types of glia are required for RGCPE to promote axon regeneration and reprogrammed glia do not require direct contact with the regenerating axons, indicating RGCPE exerts influence on axons by releasing factors into the CNS environment.
Reprograming glia promotes functional recovery after CNS injury
To investigate whether the regenerated axons induced by reprogrammed glia contributed to functional recovery, we developed a behavioral paradigm (Figure 2A and 2B) based on the findings that C4da neurons control the larval escaping behavior triggered by thermal stimuli (Tracey et al., 2003). We scored the stereotypical larval circling behavior in response to an obnoxious thermal stimulus (Figure 2B and STAR Methods). If the larva produced two or more circles, it was scored as 1, otherwise as 0. The trial was repeated 4 times for each larva, with 4 as the highest score and 0 as the lowest. Larvae with a score of 2 or more would mean that they showed at least two successful responses and were regarded as functionally recovered. Without injury, there was no difference between WT and larvae with RGCPE in their response to thermal stimuli (Figure S3A).
Figure 2. Reprogramming glial cells promotes functional recovery after injury in the Drosophila CNS.

(A and B) Schematics of the behavioral recovery paradigm. Lesion of the axons of C4da neurons corresponding to the A7 and A8 segments (A) leads to impaired circling behavior in response to a heat probe applied specifically to the A7 and A8 segments in the thermonociception behavior test (B).
(C) Injuring C4da neuron axon bundles at A7-A8 or A6-A7 in the VNC leads to impaired thermonociceptive response specifically at segment A7-A8 or A6-A7, without affecting neighboring segments, n = 36 larvae and unpaired two-tailed Student's t-test for injury on A7 and A8, n = 17 larvae and one-way ANOVA with Dunnett’s test for injury on A6 and A7.
(D) Behavioral scores of each larvae in which glial cells express Pi3K92ECA and EgfrCA by different promoters, compared to WT, n = 36, 30, 25, 20, 21 larvae for each genotype, each circle represents one larva, two-way ANOVA with Holm-Sidak's test.
(E) Functional recovery percentage of larvae with genotypes shown in (D), Fisher’s exact test.
*P < 0.05 and ***P < 0.001. Data are expressed as mean ± s.e.m. See also Figure S3.
In the VNC, each C4da neuron axon segment is connected with the corresponding body segment from which the axon bundles originate. Injuring axon segments abdominal (A) 7 and A8 in the VNC caused loss of thermonociceptive response on body segments A7 and A8 without affecting other segments, and injuring on axon segments A7 and A6 only affected body segments A7 and A6, while A8 and other segments functioned normally (Figure 2C). This suggests that the projections of C4da neuron axons in the CNS appear to show an anteroposterior somatosensory pattern with a one-to-one relationship to the larval body segments. In the functional recovery test for WT and larvae with RGCPE, we severed axon segments A7 and A8 in the VNC, which impaired thermonociception in response to the heat probe applied locally to body segments A7 and A8 at 8 h AI (Figure 2A and 2D, Movie S1 and S3). At 24 h AI, the larvae with RGCPE displayed 53% recovery compared to 6% in WT (Figure 2D, 2E and S3C, Movie S2 and S4). Increased number of larvae recovered at 48 h AI in both groups, but the RGCPE group showed a robust lead (Figure S3B). Activation of PI3K and EGFR pathways in subgroups of glia (by alrm-Gal4>Pi3K92ECA, EgfrCA, TIFR-Gal4>Pi3K92ECA, EgfrCA or GMR54H02-Gal4>Pi3K92ECA, EgfrCA) is also sufficient to increase the recovery percentage compared with WT (Figure 2D, 2E and S3C). Our results indicate that the axon regeneration induced by RGCPE leads to functional restoration after CNS injury.
Glycolysis in reprogrammed glia is required for axon regeneration
To investigate the downstream signaling in RGCPE responsible for the induced axon regeneration, we tested Myc, which was reported as an essential output of EGFR-PI3K activation in Drosophila (Read et al., 2009). We used a loss-of-function (LOF) allele, Myc4, which is recessive lethal (Pierce et al., 2004). Axon regeneration induced by RGCPE was substantially reduced in Myc4 heterozygotes (Figure 3A-3C). Moreover, glia-specific knockdown of Myc by RNAis decreased the regeneration phenotypes by RGCPE (Figure 3A-3C), suggesting that Myc mediates the pro-regeneration effects of RGCPE. This was corroborated by the gain-of-function analysis that reprogramming glia by expression of Myc (with repo-Gal4>Myc) recapitulated the phenotypes of axon regeneration and functional recovery by RGCPE (Figure 3E-3G and 4A-4D). As c-Myc is known to transcriptionally regulate genes in the glycolysis pathway (Yeung et al., 2008), we found that key glycolysis pathway genes were significantly upregulated in the CNS with RGCPE, including Ldh (lactate dehydrogenase) (Figure 3D). Furthermore, we found that Ldh functions downstream of RGCPE because heterozygotes of two Ldh mutants (Li et al., 2017) – Ldh16/+ and Ldh17/+, and glia-specific knockdown of Ldh reduced axon regeneration induced by RGCPE (Figure 3A-3C). Consistently, glia-specific knockdown of two additional glycolysis pathway genes, Hex-A and Pfk, attenuated the axon regeneration phenotype induced by RGCPE (Figure 3A-3C). Moreover, after reprogramming glial cells by Myc, glia-specific knockdown of Ldh, Hex-A or pfk confirmed that they are downstream of Myc and required for axon regeneration induced by reprogrammed glial cells by Myc (RGCM, specifically with repo-Gal4>Myc) (Figure 3E-3G). These findings demonstrate that upregulated glycolysis in RGCPE via Myc is crucial for controlling axon regeneration.
Figure 3. Glycolysis in glia is essential for the axon regeneration induced by reprogramming glial cells.

(A) Axon regrowth of C4da neurons in flies with glial mCherry expression as control (UAS-mCherry; repo-Gal4, UAS-Pi3K92ECA, UAS-EgfrCA; ppk-CD4tdGFP), Myc mutation, glial Myc RNAi, Ldh mutation, glial Ldh RNAi, glial Hex-A RNAi and glial pfk RNAi in the repo-Gal4>Pi3K92ECA, EgfrCA background at 24 h AI on segment A3 in the VNC. Diagrams below the images show regenerated axons (red) in the neuropil (demarcated by the orange dashed lines), and the red dotted circles show injury sites.
(B) Quantification of normalized regenerated axon length for genotypes shown in (A), with two RNAis for each gene knockdown, and two Ldh mutant allels, n = 28, 22, 20, 24, 22, 26, 18, 17, 22, 22, 16, 16 lesioned segments from 14, 11, 10, 12, 11, 13, 9, 9, 11, 11, 8, 8 larvae respectively for each genotype, one-way ANOVA with Dunnett’s test.
(C) Quantification of regeneration percentage for genotypes in (B), Fisher’s exact test.
(D) Mean FPKM (RNA-seq) fold change of genes in the glycolysis pathway, glial overexpression of Pi3K92ECA and EgfrCA versus control (repo-Gal4, UAS-mRFP). (E) Axon regrowth of C4da neurons in flies with glial mCherry expression as control (UAS-mCherry; repo-Gal4, UAS-Myc; ppk-CD4tdGFP), glial Ldh RNAi, Hex-A RNAi or pfk RNAi in the repo-Gal4>Myc background at 24 h AI on segment A3 in the VNC.
(F) Quantification of normalized regenerated axon length for genotypes shown in (E), with two RNAis for each gene knockdown, n = 40, 20, 28, 22, 21, 20, 20 from 20, 10, 14, 11, 11, 10, 10 larvae respectively lesioned segments for each genotype, one-way ANOVA with Dunnett’s test.
(G) Quantification of regeneration percentage for genotypes in (F), Fisher’s exact test.
*P < 0.05, **P < 0.01 and ***P < 0.001. Scale bars, 20 μm. Data are expressed as mean ± s.e.m. CA, constitutively active. See also Figure S4.
Figure 4. Elevated glycolysis in glia is sufficient to promote axon regeneration in the CNS via glial L-lactate efflux.

(A) Axon regrowth of C4da neurons in flies with glial Myc expression, glial Ldh expression, Pdha mutation, glial Pdha RNAi, glial muc RNAi and glial Mpc1 RNAi at 24 h AI on segment A3 in the VNC. Diagrams below the images show regenerated axons (red) in the neuropil (demarcated by the orange dashed lines), and the red dotted circles show injury sites.
(B) Quantification of normalized regenerated axon length for the genotypes of two separated groups. Left: WT, Myc expression in glia or C4da neurons, Ldh expression in glia or C4da neurons; right (WT as control): Pdha heterozygous and homozygous mutants, glial Pdha RNAis, glial muc RNAis and glial Mpc1 RNAis, with two RNAis for each gene knockdown, n = 62, 20, 31, 24, 28, 24, 28, 22, 28, 27, 20, 28, 32 lesioned segments from 31, 10, 16, 12, 14, 12, 14, 11, 14, 14, 10, 14, 16 larvae respectively for each genotype, one-way ANOVA with Holm-Sidak's test.
(C) Quantification of regeneration percentage for genotypes in (B), Fisher’s exact test.
(D) Functional recovery percentage of larvae (WT as control) in which glial cells express Myc or Ldh, n = 36, 17, 26 larvae for each genotype, Fisher’s exact test.
(E) Schematic shows blocking genes which functionally connect glycolysis and the TCA cycle will lead to increase of the end-product of glycolysis, L-lactate.
(F) Lactate level revealed by Laconic driven by repo-Gal4. Quantifications of fluorescence intensity ratio of mTFP/Venus from glial cells in the VNC, glial IFP expression as control (UAS-IFP; repo-Gal4, UAS-Laconic), n = 8, 7, 9 VNCs for each genotype, one-way ANOVA with Dunnett’s test.
(G) Image and diagram showing C4da neuron axon regrowth of flies with glial sln RNAi in the background of repo-Gal4>Pi3K92ECA, EgfrCA at 24 h AI on segment A3 in the VNC.
(H) Quantification of normalized regenerated axon length for the two glial sln RNAis in the background of repo-Gal4>Pi3K92ECA, EgfrCA, glial mCherry expression as control (UAS-mCherry; repo-Gal4, UAS-Pi3K92ECA, UAS-EgfrCA; ppk-CD4tdGFP), n = 28, 26, 18 from 14, 13, 9 larvae respectively lesioned segments for each genotype, one-way ANOVA with Dunnett’s test.
(I) Quantification of regeneration percentage for genotypes in (H), Fisher’s exact test.
(J) Metabolites level in the hemolymph collected from larvae of repo-Gal4>Pi3K92ECA, EgfrCA (n = 8) normalized to those in WT (n = 10), unpaired two-tailed Student's t-test for each metabolite.
(K) Axon regrowth of C4da neurons at 24 h AI on segment A3 in the VNC of WT flies injected with PBS, L-lactate (final cone., ~100 mM sodium L-lactate + 15 mM ethyl L-lactate) right after injury.
(L) Quantification of normalized regenerated axon length for WT flies injected with PBS, sodium L-lactate only (final con., ~150 mM) and L-lactate (final conc., ~100 mM sodium L-lactate + 15 mM ethyl L-lactate) right after injury, n = 22, 18, 22 lesioned segments from 11, 9, 11 larvae respectively for each group, one-way ANOVA with Dunnett’s test.
(M) Quantification of regeneration percentage for treatment groups in (L), Fisher’s exact test.
*P < 0.05, **P < 0.01, ***P < 0.001 and ns, not significant. Scale bars, 20 μm. Data are expressed as mean ± s.e.m. CA, constitutively active. See also Figure S4.
Upregulating glycolysis in glia is sufficient to promote axon regeneration in the CNS
While Myc intrinsically regulates neuronal regeneration (Belin et al., 2015), overexpressing Myc in C4da neurons only slightly increased axon regeneration compared with its overexpression in glia (Figure 4A-4C). As glycolysis functions downstream of Myc in glia for axon regeneration, we wondered if upregulating glycolysis in glia is sufficient to promote axon regeneration in the CNS. Firstly, we tested axon regeneration by targeting the pyruvate dehydrogenase complex (PDC), because inhibition of PDC contributes to high glycolysis rate (McFate et al., 2008). We found that glia-specific knockdown of Pdha (pyruvate dehydrogenase E1 α subunit) or muc/DLAT (dihydrolipoamide S-acetyltransferase, E2 component of PDC) elevated axon regeneration (Figure 4A-4C and 4E). Interestingly, heterozygous mutants of PdhaG0334, but not homozygotes, enhanced axon regeneration (Figure 4A-4C). To understand this seemingly discrepancy, we expressed Myc in glia by the Q-system (repo-QF>Myc) and RNAis for Pdha in neurons by the GAF4/UAS-system (ppk-Gal4>RNAis) simultaneously, and found that neuronal Pdha is required for axon regeneration in the CNS induced by reprogrammed glial cells by Myc (RGCM-Q, specifically with repo-QF>Myc) (Figure 5N and 5O). Furthermore, glia-specific knockdown of Mpc1 (mitochondrial pyruvate carrier, the homolog of human MPC1), a known repressor of the “Warburg effect” (Schell et al., 2014), promoted axon regeneration (Figure 4A-4C), whereas knockdown of CG9399 or CG9396 (the homologs of human MPC2) in glia did not significantly affect axon regeneration in the CNS (Figure S4C and S4D). In addition, sima/HIF-1α (hypoxia-inducible factor 1-α) is a known transcriptional regulator of glycolysis, and in our initial screen, we demonstrated that, although expressing sima in glia did not show significant increase of axon regeneration length, it increased regeneration percentage to ~ 43% (Figure 1C and Table SI). We also confirmed that it does not mediate the axon regeneration phenotypes induced by RGCPE (Figure S4E and S4F). Strikingly, expressing Ldh in glia, but not in C4da neurons, sufficiently promoted axon regeneration and functional recovery (Figure 4A-4D). Therefore, upregulation of glycolysis specifically in glia is sufficient to enhance CNS axon regeneration.
Figure 5. L-2HG mediates the axon regeneration induced by reprogramming glial cells and is sufficient to promote axon regeneration in the CNS.

(A) Axon regrowth of C4da neurons in flies with glial L2HGDH overexpression in the repo-Gal4>Pi3K92ECA, EgfrCA background at 24 h AI on segment A3 in the VNC. Diagrams below the images show regenerated axons (red) in the neuropil (demarcated by the orange dashed lines), and the red dotted circles show injury sites.
(B) Quantification of normalized regenerated axon length for flies with glial mCherry overexpression as control (UAS-mCherry; repo-Gal4, UAS-Pi3K92ECA, UAS-EgfrCA; ppk-CD4tdGFP) and L2HGDH overexpression in the repo-Gal4>Pi3K92ECA, EgfrCA background, n = 28, 24 lesioned segments from 14 and 12 larvae for each genotype, unpaired two-tailed Student's t-test.
(C) Quantification of regeneration percentage for genotypes in (B), Fisher’s exact test.
(D) Axon regrowth of C4da neurons in flies with glial L2HGDH overexpression in the repo-Gal4>Myc background at 24 h AI on segment A3 in the VNC.
(E) Quantification of normalized regenerated axon length for flies with glial mCherry overexpression as control (UAS-mCherry; repo-Gal4, UAS-Myc; ppk-CD4tdGFP) and L2HGDH overexpression in the repo-Gal4>Myc background, n = 40, 28 lesioned segments from 20 and 14 larvae for each genotype, unpaired two-tailed Student's t-test.
(F) Quantification of regeneration percentage for genotypes in (E), Fisher’s exact test.
(G) Axon regrowth of C4da neurons in flies with L2HGDH heterozygous mutants, glial L2HGDH RNAi and glial L2HGDH RNAi + Pdha RNAi at 24 h AI on segment A3 in the VNC.
(H) Quantification of normalized regenerated axon length for flies (WT as control) with L2HGDH heterozygous and transheterozygous mutants, glial L2HGDH RNAis (two RNAis), and glial L2HGDH RNAi + Pdha RNAi, n = 62, 16, 26, 26, 20, 20 lesioned segments from 31, 8, 13, 13, 10, 10 larvae respectively for each genotype, one-way ANOVA with Dunnett’s test.
(I) Quantification of regeneration percentage for genotypes in (H), Fisher’s exact test.
(J) Diagrams show structures of L-lactate, L-2HG (L-2-hydroxyglutarate), L-2HB (L-2-hydroxybutyrate) and L-tartrate, which share the α-hydroxycarboxylic acid group.
(K) Axon regrowth of C4da neurons at 24 h AI on segment A3 in the VNC of WT flies injected with L-2HG (final cone., ~100 mM disodium L-2HG + 5 mM octyl L-2HG), L-2HB (~100 mM sodium L-2HG + 15 mM ethyl 2HB) and L-tart (~100 mM sodium L-tart + 15 mM diethyl L-tart) right after injury.
(L) Quantification of normalized regenerated axon length for WT flies injected with PBS, L-2HG (final conc., ~100 mM disodium L-2HG + 5 mM octyl L-2HG), L-2HB (~100 mM sodium L-2HG + 15 mM ethyl 2HB) and L-tart (~100 mM sodium L-tart + 15 mM diethyl L-tart) right after injury, n = 22, 24, 29, 22 lesioned segments from 11, 12, 15, 11 larvae respectively for each genotype, one-way ANOVA with Dunnett’s test.
(M) Quantification of regeneration percentage for treatment groups in (L), Fisher’s exact test.
(N and O) Neuronal Pdha and L2HGDH are required for axon regeneration induced by glial overexpression of Myc. Normalized regenerated axon length (N) and regeneration percentage (O) of flies with ppk-Gal4 only as control (ppk-Gal4; repo-QF, QUAS-Myc; ppk-CD4tdGFP), neuronal Pdha or L2HGDH RNAis in the repo-QF>Myc background at 24 h AI, two RNAis for each gene, n = 28, 30, 32, 30, 30 lesioned segments from 14, 15, 16, 15, 15 larvae respectively for each genotype, one-way ANOVA with Dunnett’s test for regeneration length (N), Fisher’s exact test for regeneration percentage (O).
(P and Q) Normalized regenerated axon length (P) and regeneration percentage (Q) of flies (WT as control) with glial or neuronal IdhR195H overexpression, n = 62, 17, 28 lesioned segments from 31, 9, 14 larvae respectively for each genotype, one-way ANOVA with Tukey's test for regeneration length (P), Fisher’s exact test for regeneration percentage (Q).
*P < 0.05, **P < 0.01, ***P < 0.001 and ns, not significant. Scale bars, 20 μm. Data are expressed as mean ± s.e.m. CA, constitutively active. See also Figure S5.
L-lactate mediates glial reprogramming and promotes axon regeneration in the CNS
As shown above, LOF of Ldh in glia attenuated the axon regeneration phenotypes induced by RGCPE or RGCM, whereas its glial overexpression promoted axon regeneration. These data suggest that the Ldh-associated metabolites are critical for regulating axon regeneration. Therefore, we used Laconic, a FRET-based lactate sensor (San Martin et al., 2013), to assess lactate level locally in the VNC glia. Both RGCPE and RGCM-Q increased FRET ratio (mTFP/Venus) compared with control which expressed IFP (infrared fluorescence protein) (Yu et al., 2014), indicative of elevated lactate concentrations (Figure 4F). Consistently, Ldh activity in CNS tissues was increased in the RGCPE group, as measured by the L-lactate/pyruvate ratio (Figure S4K). Moreover, we found that the monocarboxylate transporter (MCT) in glia, which is responsible for shuttling molecules containing one carboxylate group (C(=O) OH) such as lactate across the plasma membrane, is required for the axon regeneration induced by RGCPE. We tested fly homologs of MCTs that are expressed in nervous system of human and mice, Sln (Silnoon) and out (outsiders) (Liu et al., 2017). Glial knockdown of Sln attenuated the enhanced axon regeneration induced by RGCPE (Figure 4G-4I), whereas knockdown of out in glia did not significantly reduce axon regeneration length (Figure S4G and S4H). This result suggests that Sln is involved in regulating the efflux of lactate from glia, which is important for the regeneration phenotypes induced by RGCPE. We then measured the levels of extracellular metabolites in the larval hemolymph by mass spectrometry and detected elevated levels of L-lactate, L-2HG and pyruvate in the RGCPE group compared with WT (Figure 4J), while concentrations of their enantiomers D-lactate and D-2HG remained very low (Figure S4I, S4J and S4L). Accumulation of L-2HG was likely due to increased Ldh activity because Ldh also catalyzes the conversion of α-ketoglutarate to L-2HG (Intlekofer et al., 2015; Li et al., 2017).
We have demonstrated that glia-specific knockdown of components of PDC or Mpc1 leads to increased axon regeneration in the CNS, and PDC deficiency or inhibiting MPC has been reported to cause lactate accumulation (Brown et al., 1994; Flores et al., 2017; Taylor et al., 2004). Next, we evaluated whether L-lactate is sufficient to promote CNS axon regeneration. At first, we injected sodium L-lactate into larvae, but failed to promote axon regeneration compared with the phosphate-buffered saline (PBS) control, probably because of insufficient penetration into the CNS due to the blood-brain barrier (BBB). We thus injected a mixture of ethyl-L-lactate and sodium L-lactate to harness the superior cell-membrane permeability of ethyl-L-lactate and the osmotic gradient of sodium L-lactate. Indeed, incubating fly CNS explant in vitro with this mixture increased the glial lactate concentration (Figure S4M). Consistently, injecting the mixture into larvae significantly promoted CNS axon regeneration (Figure 4K-4M). Our results indicate that glia reprogramming leads to L-lactate buildup, which needs to be transported out of glia to promote axon regeneration in the CNS. Moreover, increasing L-lactate in the CNS is sufficient to boost axon regeneration.
L-2HG mediates glial reprogramming and promotes axon regeneration in the CNS
In addition to L-lactate, we noticed that, in the hemolymph from larvae with RGCPE, L-2HG increased ~1.8-fold compared with WT. The physiological and pathological role of L-2HG is largely unknown. L-2-hydroxyglutarate dehydrogenase (L2HGDH) deficiency leads to L-2HG aciduria, which affects the CNS (Barth et al., 1992; Ma et al., 2017), and L-2HG has been associated with normal development in Drosophila (Li et al., 2017). The known functions of L-2HG includes intracellularly regulating methylation on DNA, and inhibiting ATP synthase and mTOR signaling (Fu et al., 2015; Tyrakis et al., 2016). To investigate L-2HG role here, we overexpressed L2HGDH in glia to break down L-2HG and detected significantly reduced axon regeneration induced by RGCPE (Figure 5A-5C) or RGCM (Figure 5D-5F), suggesting the necessity of elevated F-2HG. To test if increasing F-2HG is sufficient to promote axon regeneration in the CNS, we knocked down glial L2HGDH and detected increased axon regeneration (Figure 5G-5I). However, when we used mutants of L2HGDH, a situation similar to Pdha was observed. Only L2HGDH14/+ heterozygotes, but not L2HGDH14/12 trans-heterozygotes which results in more severe L2HGDH FOF (Li et al., 2017), displayed increased axon regeneration (Figure 5G-5I), because neuronal L2HGDH is also required for axon regeneration induced by RGCM-Q (Figure 5N and 5O). Moreover, injecting a mixture of disodium L-2HG and octyl-L-2HG into larvae to increase BBB permeability promoted axon regeneration (Figure 5K-5M). Taken together, L-2HG mediates the axon regeneration induced by reprogrammed glia and increasing L-2HG in the CNS is sufficient to promote axon regeneration. Interestingly, we found that increasing neuronal L-2HG level by knockdown of L2HGDH in neurons negatively regulated axon regeneration, which is consistent with the finding that L-2HG inhibits mTOR signaling (Fu et al., 2015; Hunt et al., 2019). This indicates that L-2HG accumulation in or out of neurons exerts different effects on neuroregeneration.
Compounds consisted of a similar structure with lactate and 2HG promote axon regeneration
Knockdown of both L2HGDH and Pdha in glia, however, did not induce further axon regeneration compared with individual knockdown (Figure 5G-5I), suggesting that L-2HG and L-lactate likely converge on or operate in the same machinery for regulating axon regeneration. Based on the chemical structural similarity between L-lactate and L-2HG, we speculated that other compounds that contain the carboxylic group with a hydroxyl group on the adjacent carbon may also impact CNS axon regeneration (Figure 5J). We thus tested two additional such compounds, L-2-hydroxybutyrate (L-2HB) and L-tartrate, and found that injecting flies with a salt and ester mixture for either led to similar if not better pro-regeneration capability compared to L-lactate and L-2HG (Figure 5K-5M). Moreover, we tested a different 2HG enantiomer, D-2HG, which shares targets with L-2HG (Fu et al., 2015). Idh-R195H is a gain-of-function mutation of Idh (equivalent to human IDH1-R132H) which converts α-ketoglutarate to D-2HG and leads its accumulation (Reitman et al., 2015). We found that overexpressing it in glia significantly increased the CNS axon regeneration, but not in neurons, suggesting that D-2HG regulates axon regeneration non-cell autonomously as L-2HG (Figure 5P and 5Q). Even though L-lactate from glia can fuel neurons, L-tartrate cannot be utilized as an energy source in Drosophila adults (Hassett, 1948) or larvae (Figure S5A), indicating its role as a signaling molecule. These results raise the possibility that L-lactate and L-2HG may function as extracellular signaling molecules when promoting axon regeneration.
L-lactate and L-2HG enhance axon regeneration in the CNS by upregulating neuronal cAMP signaling via GABAB receptor
To understand the neuronal signaling downstream of glial metabolic rewiring, we performed neuronal-specific knockdown of candidate genes responding to L-lactate in flies with RGCM-Q by assessing axon regeneration. A well-known receptor for lactate, HCAR1 (Cai et al., 2008; Liu et al., 2009), does not have homologs in Drosophila. We thus tested MESK2 and CG2082 (the fly homologs of NDRG3, which responds to lactate intracellularly) (Lee et al., 2015), and found they did not affect axon regeneration length induced by RGCM-Q when knocked down in neurons (Figure S6A). Intriguingly, we identified the GABAB receptor, a metabotropic GABA receptor, as responsible in C4da neurons for the axon regeneration induced by RGCM-Q. The axon regeneration induced by RGCM-Q was diminished after GABA-B-R1 knockdown in C4da neurons (Figure 6A-6C). We also tested a LOF allele of GABA-B-R1 – GABA-B-R1MI03255, generated by a MiMIC (Venken et al., 2011) insertion in the first exon, causing reduced transcription (Figure S6B and S6C). GABA-B-R1MI03255 is homozygous lethal, but larvae can survive to third instar. In GABA-B-R1MI03255 homozygotes, the axon regeneration induced by RGCM-Q was significantly reduced (Figure 6A-6C). Attenuated axon regeneration induced by RGCPE was also observed in GABA-B-R1MI03255 heterozygotes (Figure 6D-6F). Moreover, C4da neuron knockdown of GABA-B-R2, which co-assembles with GABA-B-R1 to form the functional GABAB receptor (Jones et al., 1998), also decreased axon regeneration induced by RGCM-Q (Figure 6A-6C). Since GABAB receptor is also present in glia of Drosophila (Muthukumar et al., 2014), we knocked down GABA-B-R1 or GABA-B-R2 in glia, which, however, did not impact axon regeneration induced by RGCPE (Figure S6D and S6E). These results demonstrate that neuronal GABAB receptors are necessary for the axon regeneration phenotype induced by reprogrammed glia. We also tested GABA-B-R3 and Rdl (ligand-gated chloride channel responding to GABA), but knockdown of them in neurons did not affect axon regeneration induced by RGCM-Q (Figure S6A). Utilizing GABA-B-R1MI01930-GFSTF.0, a GFP-tagged GABA-B-R1 allele, we found that the GABA-B-R1 proteins were concentrated in the neuropil and expressed by C4da neurons, and that GABA-B-R1 could be detected in C4da neuron axons before and after injury (Figure 6G and S6F).
Figure 6. Axon regeneration induced by reprogramming glial cells depends on GABAB receptor regulating cAMP in neurons.

(A) Axon regrowth of C4da neurons in flies with ppk-Gal4 only as control (ppk-Gal4; repo-QF, QUAS-Myc; ppk-CD4tdGFP), GABA-B-R1 mutation, C4da neuron GABA-B-R1 RNAi and C4da neuron GABA-B-R2 RNAi in the background of repo-QF>Myc at 24 h AI on segment A3 in the VNC (regenerated axons are in red; the neuropil is demarcated by the orange dashed lines; the red dotted circles show injury sites).
(B) Quantification of normalized regenerated axon length for flies with ppk-Gal4 only as control (ppk-GalA; repo-QF, QUAS-Myc; ppk-CD4tdGFP), GABA-B-R1 mutation, C4da neuron GABA-B-R1 RNAis and C4da neuron GABA-B-R2 RNAis in the background of repo-QF>Myc, two RNAis for each gene knockdown, n = 28, 27, 24, 26, 26, 28 lesioned segments from 14, 14, 12, 13, 13, 14 larvae respectively for each genotype, one-way ANOVA with Dunnett’s test.
(C) Quantification of regeneration percentage for genotypes in (B), Fisher’s exact test.
(D) Axon regrowth of C4da neurons in flies with GABA-B-R1 mutation in the repo-Gal4>Pi3K92ECA, EgfrCA background at 24 h AI on segment A3 in the VNC.
(E) Quantification of normalized regenerated axon length for control (repo-Gal4, UAS-Pi3K92ECA, UAS-EgfrCA; ppk-CD4tdGFP) and GABA-B-R1MI03255/+ heterozygotes in the repo-Gal4> Pi3K92FCA, EgfrCA background, n = 32, 30 lesioned segments from 16 and 15 larvae for each genotype, unpaired two-tailed Student's t-test.
(F) Quantification of regeneration percentage for genotypes in (E), Fisher’s exact test.
(G) Images showing co-localization of GABA-B-R1 and C4da neuron axons before and at 24 h AI. GABA-B-R1 is present in the axon terminal (arrow head) after injury. GABA-B-R1 is labeled by GABA-B-R1MI01930-GFSTF.0, a GFP-tagged GABA-B-R1 allele, and C4da neuron axons are labeled by ppk-CD4tdTomato.
(H) Left, representative images of an axon segment in the VNC revealed by ppk-Gal4 driving cAMPr expression in C4da neurons, with glial Myc expression, glial Myc expression and GABA-B-R1 mutation, and repo-QF only as control (ppk-Gal4, UAS-cAMPr; repo-QF). Right, quantification of fluorescence intensity for cAMPr in the VNC axons of genotypes showed on the left, normalized to control, n = 12, 12, 16 segments from 6, 6, 8 larvae respectively for each genotype, one-way ANOVA with Dunnett’s test.
(I) Quantification of fluorescence intensity changes for cAMPr in the VNC axons at 2 hours after injection of PBS, L-lac (final con., ~100 mM sodium L-lactate + 15 mM ethyl L-lactate) or L-2HG (~100 mM disodium L-2HG + 5 mM octyl L-2HG) into control (ppk-Gal4, UAS-cAMPr) or GABA-B-R1 mutants. Data is presented as ΔF (fluorescence at 2 hours – fluorescence at 0 hour) / F0 (fluorescence at 0 hour), n = 18, 12, 12, 22, 16, 14 segments from 9, 6, 6, 11, 8, 7 larvae respectively for each genotype, two-way ANOVA with Dunnett’s test.
(J) GABA-B receptor activation by L-lactate, L-2HG and SKF97541 (agonist for GABA-B receptor) at a concentration range from 0.1-100 mM (L-lactate, L-2HG, see Figure S6 for SKF97541), which is revealed by 35S-GTPγS assay for G protein coupled receptors with Human recombinant GABBR1a and GABBR2. Activation by SKF97541 is defined as positive. X-axis shows as Log10.
*P < 0.05, **P < 0.01, ***P < 0.001 and ns, not significant. Scale bars, 20 μm. Data are expressed as mean ± s.e.m. CA, constitutively active. See also Figure S6.
By expressing cAMPr, a cAMP sensor (Hackley et al., 2018), in C4da neurons, we further discovered that RGCM-Q increases neuronal cAMP concentration through GABAB receptor (Figure 6H). cAMP is well known to promote axon regeneration in the CNS (Neumann et al., 2002; Pearse et al., 2004; Qiu et al., 2002). Because reprogrammed glial cells promoted axon regeneration by releasing L-lactate and L-2HG, we asked whether they could directly affect neuronal cAMP levels via GABAB receptor. We injected the salt and ester mixture of L-lactate or L-2HG into larvae and measured cAMPr fluorescence change. Both L-lactate and L-2HG increased neuronal cAMP concentration, which was abolished in GABA-B-R1MI03255/+ heterozygotes (Figure 6I and S6G), indicating that both L-lactate and L-2HG can elevate neuronal cAMP through GABAB receptor. However, GABAB receptors are known to activate Gαi/o •type G proteins, which inhibit adenylyl cyclase and are conserved in Drosophila (Gassmann and Bettler, 2012). To determine how L-lactate and L-2HG interact with GABAB receptors, we performed the 35S-GTPγS binding assay on recombinant human GABAB receptors, GABBR1a and GABBR2. Surprisingly, we found that, when using concentrations of L-lactate or L-2HG over 3 or 1 mM, respectively, which is relevant to their physiological concentrations in the hemolymph (Figure S4L), they both inversely activated GABAB receptors compared with the activation induced by its agonist SKF97541 (Figure 6J and S6H). L-2HG showed more potency than L-lactate, which is consistent with the axon regeneration phenotype. This result demonstrates that L-lactate and L-2HG can regulate GABAB receptors by direct binding and explained how they lead to increased neuronal cAMP levels through GABAB receptors.
Intraspinal L-lactate treatment after SCI promotes regrowth of corticospinal tract (CST) axons and recovery of locomotor function in adult mice
To determine whether lactate is effective for stimulating axon regeneration after CNS injury in mammals, we performed dorsal over-hemisection at thoracic 7 (T7) segment in adult mice and locally applied two doses of saline (control) or L-lactate (mixture of sodium and ester) immediately and 6 days after SCI (Figure 7A and 7B). We evaluated regeneration of injured CSTs labeled by tracer biotin dextran amine 5 weeks after SCI. CSTs are important for controlling voluntary movements (Deumens et al., 2005), but particularly refractory to regeneration (Blesch and Tuszynski, 2009; Pearse et al., 2004; Raineteau and Schwab, 2001). In contrast to controls, animals treated with L-lactate exhibited remarkable CST axon regeneration into the lesion area and caudal spinal cord (Figure 7C-7E). We carefully checked the CST axons in caudal spinal cord and confirmed that they meet the previously-defined morphological criteria of regenerating axons (Steward et al., 2003). We quantified the immunostaining for GFAP from parasagittal sections of lesioned spinal cord. Treatment with L-lactate did not alter the size of either the astrocyte scar border or spared but reactive neural tissue (O'Shea et al., 2017) (Figure S7A). Immunostaining for ALDH1L1, another widely-used marker for astrocytes and glial scar (Hackett et al., 2018; Yang et al., 2011), also indicated the similarity of reactive astrocytes tissue between the two groups (Figure S7B).
Figure 7. Local treatments with lactate stimulated regeneration of injured CST axons into the caudal spinal cord and recovery of locomotor function in adult mice.

(A and B) Timeline (A) and schematic (B) of the spinal cord injury experiment in adult mice (lactate treatment, first dose: 155 mM sodium lactate + 5 mM ethyl lactate; second dose: 300 mM sodium lactate + 20 mM ethyl lactate).
(C) Camera Lucida drawings indicate BDA-labeled CST axons from all the parasagittal sections of two representative mice, one from the saline group (control) and the other from the lactate (Lac) group. Lesion center (LC) (red dotted line).
(D) Quantification of CST axon fibers which were traced from all parasagittal sections of the spinal cord 0-4 mm caudal to the lesion, and is presented as the total length of CST axons from each bin box of 0.8-mm spinal cord caudal to the lesion center (n = 3 in saline, 5 in lactate, two-way ANOVA).
(E) Images of parasagittal sections around the LC from the saline and lactate groups with BDA-labeled CST axons (red) and immunostaining for GFAP (green). Dorsal is up in all sections.
(F) Graph indicates the locomotor BMS scores in SCI mice treated with saline or lactate (n = 5 in saline, 7 in lactate, two-way ANOVA).
(G) Graph indicates grid walk errors in 2 groups of mice 5 weeks after SCI (n = 5 in saline, 7 in lactate, unpaired two-tailed Student's t-test).
(H) Graph shows grasping rate of the hindpaws in two groups of mice 5 weeks after SCI (n = 5 in saline, 7 in lactate, unpaired two-tailed Student's t-test).
*P < 0.05, **P < 0.01 and ***P < 0.001. Scale bars, 200 μm. Data are expressed as means ± s.e.m.
We also evaluated functional recovery in SCI mice during a 5-week period of observation. Four hours after SCI, all groups of mice had similar severities of injury, with locomotor BMS scores of approximately 2.5 (Figure 7F). Several weeks after injury, saline-treated controls showed partial recovery, which reached a stable level by 2-5 weeks after SCI. However, the BMS scores in L-lactate treated mice maintained a better recovery trend at 1-5 weeks after SCI (Figure 7F). Evaluation of hindlimb grid walk indicated that L-lactate-treated SCI mice made fewer errors, more correctly placing their hindpaws on the grid 5 weeks after SCI than did the corresponding injury controls (Figure 7G). Moreover, lactate-treated mice performed better in the touch-grasping test by significantly increasing hindlimb grasping rate 5 weeks after SCI (Figure 7H). To conclude, local L-lactate treatment significantly improved regrowth of descending fiber tracts and recovery of locomotor functions in adult mammals with SCI.
We have analyzed the cellular expression of GABAB receptors in adult rodents with and without CNS injury. First, we investigated the single-cell transcriptome data from the mouse CNS (the Allen Mouse Brain Atlas (Lein et al., 2007)). Interestingly, we found that both Gabbrl and Gabbr2 are mainly expressed in neurons (Lopez-Bendito et al., 2002; Zhou et al., 2017), and in particular, Gabbr2 expression is detected in astrocytes and OPCs (oligodendrocyte precursor cells) albeit at a low level (Figure S7C). In oligodendrocytes, endothelial cell, pericytes, VLMCs (vascular and leptomeningeal cells), microglia, VMCs (vascular mesenchymal cells) and PVMs (perivascular macrophages), Gabbr2 expression is not detected (Figure S7C). The observation of low level expression of Gabbr2 in astrocytes and no-detection in microglia, is also confirmed in a bulk RNA-seq dataset of astrocytes and microglia from mouse brain (Figure S7D) (Pan et al., 2020). It is well established that heteromeric assembly of GABBR1 and GABBR2 is important for the G protein function and cell surface trafficking of GABAB receptors (Jones et al., 1998; Kaupmann et al., 1998). Loss of function of either GABBR1 or GABBR2 will lead to the collapse of GABAB receptors (Gassmann and Bettler, 2012), and unbalanced expression of GABBR1 and GABBR2 subunits is suggested to reduce receptor function. This effect would be amplified in GABAB receptor because homo-dimerization of GABBR1 or GABBR2 can occur with similar affinities compared to their hetero-dimerization, and they fail to localize on membrane as receptors (Villemure et al., 2005). Therefore, cell types in which Gabbr2 are not detected, such as oligodendrocytes, endothelial cells, microglia and perivascular macrophages, are likely unable to sense L-lactate through GABAB receptors. Although Gabbr2 is expressed in astrocytes and OPCs, the abundance is limited. Moreover, ratio of Gabbr2 to Gabbr1 in astrocytes is < 0.3 and that in OPCs is < 0.06, whereas the ratio is > 0.8 for neurons (Figure S7C), indicating, at most, weak activity of GABAB receptors in OPCs. To further investigate GABAB receptor in astrocytes, another dataset describing the transcriptome of astrocytes at different locations in the CNS was analyzed, not only confirming that the ratio of Gabbr2 to Gabbr1 in astrocytes from brain is low, but also manifesting that the ratio in astrocytes from spinal cord is even lower (Figure S7E) (Itoh et al., 2018). Surprisingly, we uncovered that SCI would lead to a further downregulation of the ratio of Gabbr2 to Gabbrl in astrocytes near the lesion site (< 0.05) (Figure S7F) and the expression levels of both genes in an additional dataset (Anderson et al., 2016). However, SCI had no effect on the ratio of Gabbr2 to Gabbr1 in CST neurons (Figure S7G), but partially reduced the abundance of Gabbrl and Gabbr2 at 10 days after injury, which returned to normal level at 14 and 21 days after injury (Figure S7J, non-regenerating) (Poplawski et al., 2020). Based on these analyses, we hypothesize that locally treated L-lactate can function through neuronal GABAB receptors to promote axon regeneration, and it is unlikely to impact on other cell types by the same mechanism. Furthermore, we detected increased GABAB receptor expression around the lesion site in the L-lactate treated mice, specifically with 39% increase of Gabbr1 and 35% increase of Gabbr2 in CST axons around the lesion site compared with the control mice (Figure S7H). Together with the result showing increase of regenerated CST axons induced by L-lactate (Figure 7D), these observations suggest a relationship between higher average level of neuronal GABAB receptors and more regenerated axons in L-lactate treated mice, supporting our hypothesis.
In addition to our evidence demonstrating the importance of GABAB receptor to axon regeneration, we found that, in an integrated systems analysis on SCI (Squair et al., 2018), Gabbr1 and Gabbr2 are both in a highly conserved gene coexpression module, which is associated with cell types of neurons and inversely-correlated with injury severity. Specific analysis of Gabbr1 and Gabbr2 abundance with injury severity, a prediction of functional recovery, shows a strong inverse correlation (Spearman’s correlation coefficient for Gabbr1 = −0.832, Gabbr2 = −0.926), indicating that GABAB receptor expression is correlated with the outcome of recovery from SCI (Figure S7I). In another study, expression of Gabbr1 and Gabbr2 were both increased in regenerating CST neurons induced by neural progenitor cell graft after SCI, compared to the lesion only group (Figure S7J) (Poplawski et al., 2020). These results suggest that GABAB receptor is important for axon regeneration after SCI and are consistent with the function of GABAB receptor induced by L-lactate which we have demonstrated in this study.
Discussion
Efforts have been made trying to understand the genetic programs capable of altering the permissive or inhibitory properties of glia, for examples, acidic fibroblast growth factor (aFGF) signaling attenuates astrocytic pathology after SCI (Tsai et al., 2008), and transforming growth factor-α (TGF-α) increases laminin at the lesion sites supporting axon regrowth after SCI (White et al., 2008). We showed that reprogramming glial cells by activation of the PI3K and EGFR pathways promotes axon regrowth and functional recovery in the CNS, indicating that the inhibitory CNS environment can be reversed by transitioning of glial status. Our findings further revealed that the metabolic switch to aerobic glycolysis in reprogrammed glia is critical for axon regeneration and that the elevated metabolites associated with Ldh are sufficient to boost axon regeneration. It is well known that the glial environment of the mature CNS is different from that of the embryonic nervous system in terms of axon regeneration (Yiu and He, 2006). In addition to myelin formation and the termination of the period of experience-driven plasticity, the difference in glial glucose metabolism between the mature and immature CNS might also contribute to their distinctive environments for axon regeneration, as cell proliferation during the embryonic stage relies on aerobic glycolysis (Miyazawa and Aulehla, 2018).
Using our Drosophila CNS injury paradigm, we were able to better resolve the CNS environment. Focusing on the C4da neurons, which are able to regenerate robustly after PNS but not CNS injury, enabled us to study the CNS inhibitory environment aside from the neuronal intrinsic growth capacity. We showed that the inhibitory region to axon regrowth in fly CNS is restricted to the neuropil. By carefully defining the “regeneration percentage” (Figure S2D, see also STAR Methods), we were able to determine whether the trend of axon regrowth was towards the “right place”, which exhibits obvious repulsion to the regenerating axons in WT. However, this barrier appears to be lifted in the CNS with glial metabolic reprograming. Aerobic glycolysis is regulated by various signaling pathways, such as FGFR, HIF and Hedgehog (Liu et al., 2018; Teperino et al., 2012; Yeung et al., 2008), which were tested in our initial screen. Indeed, even though they did not significantly promote axon regeneration length, increased regeneration percentage was observed. Interestingly, PI3K signaling in glia plays an important role in responding to axonal injury (Doherty et al., 2014) and EGFR in reactive astrocytes is upregulated after SCI (White et al., 2008). Therefore, reprogramming glia by co-activation of the PI3K and EGFR pathways characterized here may actually amplify their beneficial glia-responses to axonal injury.
Comparison of axon regeneration induced by the same pro-regeneration manipulations in glia versus neurons, such as Myc and Ldh overexpression, suggests that the effect of the extrinsic environment may be more dominant at least in our injury paradigm. In addition, we reported that glial knockdown of PDC, L2HGDH and Mpc1 all supported axon regeneration in the CNS. Intriguingly, however, their germline deficiencies are all related to neurodegenerative diseases (Brown et al., 1994; Grenell et al., 2019; Ma et al., 2017). Consistently, we found attenuated axon regeneration with neuronal knockdown of Pdha or L2HGDH, or in their homozygous mutants. This suggests that neuronal loss of these genes contributes to neurodegeneration caused by germline deficiency. More importantly, these observations indicate that metabolites exert different effects on neurons extracellularly and intracellularly. L-2HG accumulation in neurons reduces axon regeneration as shown in our experiments and affects neuronal function (Hunt et al., 2019). This is likely due to their inhibition of ATP synthase and mTOR signaling (Fu et al. 2015). However, elevated extracellular L-2HG derived from glia promotes axon regeneration, so does D-2HG. The Yin and Yang functions of these genes and metabolites in glia versus neurons should guide us to develop new regenerative therapies in the future.
Even though what determines the inhibitory feature of the CNS environment for axon regeneration remains complex and unresolved, our data, especially that the metabolic switch is sufficient to promote axon regeneration, raise the hypothesis that the metabolic status of glial cells may underlie the inhibition. We have identified L-lactate and L-2HG as key mediators in the CNS environment, however, no gain-of-function experiments directly associated with metabolites could fully recapitulate the regeneration phenotype induced by RGCPE, and we noticed that axon regeneration induced by glial Myc overexpression is significantly higher than that by glial Ldh overexpression (Figure 4A-4C), suggesting the existence of additional factors from RGCPE and RGCM. Based on the similarity of the chemical structures of L-lactate and L-2HG, we found that compounds containing the carboxylic group with a hydroxyl group on the adjacent carbon, such as 2-hydroxybutyrate and tartrate, were also able to promote axon regeneration, suggesting that other metabolites, such as malate or citrate that contains a similar structure, may also be efficacious. Because the mechanism that we identified associates with receptor binding, we speculate that the overall size and the presence of other chemical groups of these metabolites may be vital for the pro-regeneration capacity, and thus their activities need to be individually evaluated. L-lactate has been reported as an energy source to neurons (Magistretti and Allaman, 2018) and L-2HG can also produce FADH2 when catalyzed by L2HGDH, but we identified a novel signaling role of L-lactate and L-2HG in regulating axon regeneration based on the following observations. First, GABAB receptors responded to the metabolites by upregulating neuronal cAMP levels, and axons failed to regenerate without GABAB receptor after glial reprogramming. Second, D-2HG, enantiomer of L-2HG, promoted axon regeneration only when produced in glia but not neurons. Third, L-tartrate promoted axon regeneration, but it cannot be utilized as an energy source in Drosophila adults (Hassett, 1948) or larvae (Figure S5A). Furthermore, we performed the knockdown of Sln or out in C4da neurons when overexpressing Myc in glia, and found that, while knockdown of out did not impact axon regeneration, knockdown of Sln appeared to reduce the regeneration percentage slightly (Figure S5B and S5C). Given that Sln accounts for an important part of MCT-mediated neuronal influx of metabolites from glia while out to a lesser extent (Liu et al., 2017), this result suggests that energetic supplement of metabolites from glia is necessary but does not play a dominant role in C4da neuron axon regeneration, supporting the critical extracellular signaling role of the metabolites. According to our GTPγS binding assay, L-lactate and L-2HG could inversely activate GABAB receptors. This effect is thought to stabilize the GPCR in its G protein unbound form and thus inhibit its spontaneous activation (Strange, 2002). Effects of L-lactate and L-2HG require relative high concentrations (over 1 mM). It is reported that concentrations of L-lactate range between 2-5 mM within the brain (Magistretti and Allaman, 2018), and we also found that L-lactate and L-2HG concentrations in the WT larvae hemolymph are both ~ 1 mM. It seems likely that the metabolites only show the signaling role when the concentration is above their physiological level. Interestingly, gamma-hydroxybutyrate was reported as a ligand for GABAB receptors, also functioning at millimole concentration (Lingenhoehl et al., 1999). The extent to which L-lactate and L-2HG can inversely activate GABAB receptors in vivo depends on the constitutive activity of GABAB receptors. From our cAMPr experiments, the L-lactate mixture (~100 mM sodium +15 mM ester) and the L-2HG mixture (~100 mM disodium + 5 mM ester) were able to increase cAMP levels via GABAB receptors. The portion of the metabolites mixture that can effectively penetrate into the CNS should be the ester. Thus, it is likely that no more than 15 mM lactate or 5 mM L-2HG is required to increase cAMP level significantly in C4da neurons. Collectively, these results suggest that L-lactate and L-2HG function as signaling molecules during axon regeneration. Therefore, BBB permeable L-lactate and L-2HG analogs represent potential therapeutic targets for treating neural injury including SCI.
To understand how the glial environment affects axon regeneration, we started the investigation by screening various pathways in glia. Using Drosophila larvae we were able to easily alter the whole CNS environment by manipulating gene expression in all types of glia. However, in the mice SCI model, a similar experimental strategy would require considerable more efforts and may run into complications. For example, the screened genes may cause unpredicted effects when extensively expressed in mice glial cells (Lee et al., 2014; Liu and Neufeld, 2004), which can affect the viability of animals, especially given that even Drosophila adults with RGCPE or RGCM cannot survive. The larval injury model thus provides a platform with high-tolerance of genetic manipulations, which is critical for identification of the phenotype. Drosophila larvae are fully functional with homeostasis in their nervous system despite the fact that they are in the developmental stage (Newman et al., 2017; Yuan et al., 2011). As we have demonstrated, they exhibit the CNS inhibitory environment for regenerating axons that closely resembles the condition in the mature CNS. Moreover, Drosophila larvae possess other characteristics making them irreplaceable in our study, such as their translucent body wall which facilitates precise axon injury and regeneration assessment in vivo, short turnaround (2-3 days), and genetic tools available to map the downstream effectors. As we identified specific metabolites capable of altering the CNS inhibitory environment in flies, we tested if L-lactate is effective in the mice SCI model. Local treatment of L-lactate significantly promotes CST axon regeneration and functional recovery, indicating that the inhibitory environment of mice CNS is also likely associated with the metabolism state of glia and that it can be partially reversed by elevating L-lactate concentration. Interestingly, ketogenic diet was reported to improve the functional recovery after SCI in both human and rodents (Streijger et al., 2013; Yarar-Fisher et al., 2018), which is related to the increased expression of MCTs in cells such as astrocytes around the injury site. Given our results, it is likely that elevated MCTs change the metabolic environment in the spinal cord.
Sln was reported to transport lactate and butyrate and is important for early larval development, as its homozygous mutants all die before pupation (Jang et al., 2008). On the other hand, out regulates programmed cell death in germ cells (Coffman et al., 2002; Yamada et al., 2008). As monocarboxylate transporters (MCTs), they are both important for the metabolic homeostasis of the nervous system. Glial knockdown (driven by repo-Gal4) of Sln, but not out, is lethal in Drosophila adults, whereas neuronal knockdown (driven by nsyb-Gal4) of either leads to folded wings (Volkenhoff et al., 2015). Moreover, knockdown of either genes in neurons in adult flies reduces lipid droplets accumulation in photoreceptor glia induced by stress via disrupting lactate transport (Liu et al., 2017). We found that glial Sln in flies is important for axon regeneration induced by reprogrammed glia, while out seems to function to a lesser degree. This could be due to higher abundance of Sln than out in the VNC glia (Allen et al., 2020). MCTs transport not only lactate, but also other monocarboxylates such as butyrate, pyruvate and ketone bodies. Thus our result that glial knockdown of Sln impacts axon regeneration suggests possible involvement of other metabolites, which warrants further investigation.
Severed CNS axons fail to regenerate, resulting in permanent functional deficits in human patients, including SCI. Developing successful regenerative strategies for rewiring CNS axons is extremely important for neuroscience research. Both reduced intrinsic growth capability of neurons and extrinsic inhibitory environment contribute to the regeneration failure in adult mammals (Lu et al., 2014; Sharma et al., 2012). Numerous approaches have been reported to promote axon regeneration of adult CNS (Anderson et al., 2018; Belin et al., 2015; Kurimoto et al., 2010; Lang et al., 2015; Liu et al., 2010; Moore et al., 2009; Norsworthy et al., 2017; Park et al., 2008), but treatments that can be translated into clinical use are very limited . It is thus required to identify novel strategies for stimulating CNS axon regeneration. Our study demonstrated that administration of L-lactate locally to the lesion site of adult mammals promotes significant axon regeneration and functional recovery. L-lactate can influence axon regeneration by targeting GABAB receptor which is widely expressed in neurons. Although we did not specifically look into the potential effects of L-lactate on other cell types by other mechanisms after SCI, it is possible that they can contribute in addition to the route through neuronal GABAB receptors. For example, M2 macrophage is reported to promote CNS repair after injury (Kigerl et al., 2009), and intriguingly, lactate has been shown to induce polarization of macrophages towards the M2 state (Colegio et al., 2014; Zhang et al., 2020).
Limitations of Study
Although we have demonstrated that L-lactate and L-2HG are involved in the axon regeneration induced by reprogrammed glia, our data suggest that additional factors from the reprogrammed glia affect axon regeneration, which warrants further investigation. In the mice SCI experiment, we treated mice with two doses of L-lactate immediately and 6 days after SCI, and found that the treatment was able to promote axon regeneration and functional recovery. While the immediate treatment after injury is effective, it would be interesting to determine whether treating mice months after the injury (in the chronic phase) would result in a similar outcome. We provide evidence showing the relationship between neuronal GABAB receptors and axon regeneration. However, our results do not address to what extent the neuronal GABAB receptors contribute to the observed phenotype. Even though using mice in which neurons have downregulation of GABAB receptors may help address this question, we cannot completely exclude the possibility that L-lactate treatment may also promote axon regeneration through indirect pathways involving other cell types.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yuanquan Song (songy2@email.chop.edu).
Materials Availability
The D. melanogaster line generated in this study (UAS-Laconic) is available from the lead contact upon request.
Data and Code Availability
The accession number for the RNA-seq data reported in this paper is GEO: GSE144655.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila
The full list of fly genotypes used can be found in the Key Resources Table. Randomly selected male and female larvae were used. For sensory axon lesion in Drosophila, fly larvae were raised at 25°C on grape juice agar plates, and at 48 hours after egg laying (h AEL), larvae were collected for axon lesion. For behavioral assay, RNA-seq analysis and other associated experiments, Drosophila usage was detailed in the related sections in METHODS DETAILS. “WT” referred to one strain (ppk-CD4tdGFP; repo-Gal4, UAS-mRFP).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-GFAP | Sigma Aldrich | Cat#G3893; RRID:AB_477010 |
| Rabbit anti-ALDH1L1 | Abcam | Cat#ab87117; RRID:AB_10712968 |
| Mouse anti-GABA B Receptor 1 | Abcam | Cat#ab55051; RRID:AB_941703 |
| Rabbit anti-GABA B Receptor 2 | Novus Biologicals | Cat#NBP2-16569 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sodium L-lactate | Sigma Aldrich | Cat#71718 |
| Ethyl L-lactate | Sigma Aldrich | Cat#69799 |
| Disodium L-2-Hydroxyglutarate | Sigma Aldrich | Cat#90790 |
| octyl-L-2HG | Cayman Chemical | Cat#1391194-64-1 |
| L-2-Hydroxybutyric acid | Sigma Aldrich | Cat#54918 |
| Ethyl 2-Hydroxybutyrate | Fisher Scientific | Cat#H02295G |
| L-tartaric acid | Sigma Aldrich | Cat#251380 |
| Diethyl L-tartrate | Sigma Aldrich | Cat#W237809 |
| Critical Commercial Assays | ||
| Quick-RNA MiniPrep Plus Kit | Zymo Research | Cat#R1058 |
| TSA Plus Cyanine 3 System | PerkinElmer | Cat#NEL744B001KT |
| Deposited Data | ||
| Raw and analyzed RNA-seq data | This paper | GEO: GSE144655 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: ppk-CD4-tdGFP | Han et al., 2011 | N/A |
| D. melanogaster: ppk-CD4-tdTomato | Han et al., 2011 | N/A |
| D. melanogaster: ppk-Gal4 | Grueber et al., 2003 | N/A |
| D. melanogaster: repo-Gal4 | Sepp et al., 2001 | N/A |
| D. melanogaster: UAS-EgfrCA (dEGFRλ) | Read et al., 2009 | N/A |
| D. melanogaster: UAS-Pi3K92ECA (Dp110CAAX) | Read et al., 2009 | N/A |
| D. melanogaster: alrm-Gal4 | Doherty et al., 2009 | N/A |
| D. melanogaster: TIFR-Gal4 | Ziegenfuss et al., 2012 | N/A |
| D. melanogaster: Ldh16 | Li et al., 2017 | N/A |
| D. melanogaster: Ldh17 | Li et al., 2017 | N/A |
| D. melanogaster: L2HGDH12 | Li et al., 2017 | N/A |
| D. melanogaster: L2HGDH14 | Li et al., 2017 | N/A |
| D. melanogaster: UAS-L2HGDH | Li et al., 2017 | N/A |
| D. melanogaster: UAS-cAMPr | Hackley et al., 2018 | N/A |
| D. melanogaster: GMR54H02-Gal4: w1118; P{GMR54H02-GAL4}attP2 | Bloomington Drosophilia Stock Center | BDSC: 45784 |
| D. melanogaster: repo-QF: y1 w*; Pin1/CyO; P{ET-QF2.GU}repo/TM6B, Tb1 | Bloomington Drosophilia Stock Center | BDSC: 66477 |
| D. melanogaster: UAS-btlCA: w*; P{UAS-btl.λ}2 | Bloomington Drosophilia Stock Center | BDSC: 29045 |
| D. melanogaster: UAS-htlCA: y1 w*; P{UAS-htl.λ.M}40-22-2 | Bloomington Drosophilia Stock Center | BDSC: 5367 |
| D. melanogaster: UAS-stg: w*; P{UAS-stg.HA}2 | Bloomington Drosophilia Stock Center | BDSC: 56562 |
| D. melanogaster: UAS-sima: w*; P{UAS-sima.B}2 | Bloomington Drosophilia Stock Center | BDSC: 9582 |
| D. melanogaster: UAS-ci: w*; P{UAS-ci.HA.wt}3 | Bloomington Drosophilia Stock Center | BDSC: 32570 |
| D. melanogaster: UAS-foxo: w1118; P{UASp-foxo.S}3 | Bloomington Drosophilia Stock Center | BDSC: 42221 |
| D. melanogaster: UAS-p53.H159N: y1 w1118; P{UAS-p53.H159N.Ex}2 | Bloomington Drosophilia Stock Center | BDSC: 8420 |
| D. melanogaster: Myc4: Myc4/FM7i, P{ActGFP}JMR3 | Bloomington Drosophilia Stock Center | BDSC: 64769 |
| D. melanogaster: UAS-mCherry: y1 sc* v1 sev21; P{UAS-mCherry.VALIUM10}attP2 | Bloomington Drosophilia Stock Center | BDSC: 35787 |
| D. melanogaster: UAS-Myc: w1118; P{UAS-Myc.Z}132 | Bloomington Drosophilia Stock Center | BDSC: 9674 |
| D. melanogaster: QUAS-Myc: y1 w1118; P{QUAS-Myc.P}7/CyO | Bloomington Drosophilia Stock Center | BDSC: 30009 |
| D. melanogaster: UAS-IdhR195H: w1118; P{UAS-Idh.R195H.FLAG}3 | Bloomington Drosophilia Stock Center | BDSC: 56203 |
| D. melanogaster: GABA-B-R1MI03255: y1 w*; Mi{MIC}GABA-B-R1MI03255/SM6a | Bloomington Drosophilia Stock Center | BDSC: 36226 |
| D. melanogaster: GABA-B-R1MI01930-GFSTF.0: y1 w67c23; Mi{PT-GFSTF.0}GABA-B-R1MI01930-GFSTF.0 | Bloomington Drosophilia Stock Center | BDSC: 60522 |
| D. melanogaster: Ldh RNAi#2: y1 v1; P{TRiP.HMS00039}attP2 | Bloomington Drosophilia Stock Center | BDSC: 33640 |
| D. melanogaster: UAS-sima RNAi#2: y1 sc* v1 sev21; P{TRiP.HMS00832}attP2 | Bloomington Drosophilia Stock Center | BDSC: 33894 |
| D. melanogaster: MESK2 RNAi: y1 v1; P{TRiP.JF03312}attP2 | Bloomington Drosophilia Stock Center | BDSC: 29380 |
| D. melanogaster: L2HGDH RNAi#2: y1 v1; P{TRiP.HMC03444}attP40 | Bloomington Drosophilia Stock Center | BDSC: 51870 |
| D. melanogaster: UAS-GABA-B-R1 RNAi#1: y1 v1; P{TRiP.HMC03388}attP2 | Bloomington Drosophilia Stock Center | BDSC: 51817 |
| D. melanogaster: Myc RNAi#1: w1118; P{GD1419}v2947 | VDRC Stock Center | VDRC: v2947 |
| D. melanogaster: Myc RNAi#2: P{KK103869}VIE-260B | VDRC Stock Center | VDRC: v106066 |
| D. melanogaster: Ldh RNAi#1: w1118; P{GD6887}v31192/TM3 | VDRC Stock Center | VDRC: v31192 |
| D. melanogaster: HexA RNAi#1: w1118; P{GD9964}v21054 | VDRC Stock Center | VDRC: v21054 |
| D. melanogaster: HexA RNAi#2: P{KK100831}VIE-260B | VDRC Stock Center | VDRC: v104680 |
| D. melanogaster: Pfk RNAi#1: w1118; P{GD1508}v3016 | VDRC Stock Center | VDRC: v3016 |
| D. melanogaster: Pfk RNAi#2: P{KK101887}VIE-260B | VDRC Stock Center | VDRC: v105666 |
| D. melanogaster: Pdha RNAi#1: w1118; P{GD12103}v40410 | VDRC Stock Center | VDRC: v40410 |
| D. melanogaster: Pdha RNAi#2: P{KK101856}VIE-260B | VDRC Stock Center | VDRC: v107209 |
| D. melanogaster: muc RNAi#1: P{GD16843}v48941 | VDRC Stock Center | VDRC: v48941 |
| D. melanogaster: muc RNAi#2: P{KK102893}VIE-260B | VDRC Stock Center | VDRC: v110735 |
| D. melanogaster: sima RNAi#1: P{KK102226}VIE-260B | VDRC Stock Center | VDRC: v106187 |
| D. melanogaster: Mpc1 RNAi#1: w1118; P{GD3944}v15858 | VDRC Stock Center | VDRC: v15858 |
| D. melanogaster: Mpc1 RNAi#2: P{KK102734}VIE-260B | VDRC Stock Center | VDRC: v103829 |
| D. melanogaster: CG9399 RNAi: P{KK109212}VIE-260B | VDRC Stock Center | VDRC: v101455 |
| D. melanogaster: CG9396 RNAi: P{KK112662}VIE-260B | VDRC Stock Center | VDRC: v104068 |
| D. melanogaster: L2HGDH RNAi#1: w1118; P{GD5322}v30737 | VDRC Stock Center | VDRC: v30737 |
| D. melanogaster: Sln RNAi#1: w1118; P{GD1940}v4607 | VDRC Stock Center | VDRC: v4607 |
| D. melanogaster: Sln RNAi#2: P{KK104306}VIE-260B | VDRC Stock Center | VDRC: v109464 |
| D. melanogaster: out RNAi#1: w1118; P{GD3448}v51157 | VDRC Stock Center | VDRC: v51157 |
| D. melanogaster: out RNAi#2: P{KK104187}VIE-260B | VDRC Stock Center | VDRC: v108364 |
| D. melanogaster: GABA-B-R3 RNAi: P{KK105961}VIE-260B | VDRC Stock Center | VDRC: v108036 |
| D. melanogaster: CG2082 RNAi: w1118; P{GD8635}v19120 | VDRC Stock Center | VDRC: v19120 |
| D. melanogaster: Rdl RNAi: P{KK104293}VIE-260B | VDRC Stock Center | VDRC: v100429 |
| D. melanogaster: GABA-B-R1 RNAi#2: P{VSH330042}attP40 | VDRC Stock Center | VDRC: v330042 |
| D. melanogaster: GABA-B-R2 RNAi#1: w1118; P{GD699}v1784 | VDRC Stock Center | VDRC: v1784 |
| D. melanogaster: GABA-B-R2 RNAi#2: P{KK100020}VIE-260B | VDRC Stock Center | VDRC: v110268 |
| D. melanogaster: UAS-Grn: M{UAS-grn.ORF.3xHA.GW}ZH-86Fb | FlyORF | FlyORF: F001916 |
| D. melanogaster: UAS-Smox: M{UAS-Smox.ORF.3xHA.GW}ZH-86Fb | FlyORF | FlyORF: F000025 |
| D. melanogaster: UAS-Su(H): M{UAS-Su(H).ORF.3xHA.GW}ZH-86Fb | FlyORF | FlyORF: F001922 |
| D. melanogaster: UAS-Stat92E: M{UAS-Stat92E.ORF.3xHA}ZH-86Fb | FlyORF | FlyORF: F000750 |
| D. melanogaster: UAS-Ldh: M{UAS-Ldh.ORF.3xHA.GW}ZH-86Fb | FlyORF | FlyORF: F002924 |
| D. melanogaster: PdhaG0334: y1 w* P{lacW}PdhaG0334 P{neoFRT}19A/FM7c; P{ey-FLP.N}5 | Kyoto Stock Center | DGRC: 111869 |
| D. melanogaster: UAS-Laconic | This paper | N/A |
| Mouse: C57BL/6J | The Jackson Laboratory | IMSR_JAX:000664 |
| Oligonucleotides | ||
| GABA-B-R1 forward primer: cgcttccctcatcaactcatc | This paper | N/A |
| GABA-B-R1 reverse primer: gtataattcacctatcacgcaaagg | This paper | N/A |
| rp49 forward primer: cagtcggatcgatatgctaagctg | This paper | N/A |
| rp49 reverse primer: taaccgatgttgggcatcagatac | This paper | N/A |
| Recombinant DNA | ||
| plasmid: UAS-Laconic | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
| GraphPad Prism 7 | GraphPad software | N/A |
| Zen | Zeiss | N/A |
| Photoshop | Adobe | N/A |
| Illustrator | Adobe | N/A |
| Microsoft Excel 2016 | Microsoft Corporation | N/A |
| Heatmapper | http://www.heatmapper.ca/ | N/A |
| Metascape | https://metascape.org/gp/index.html#/main/step1 | N/A |
| Superheat | https://rlbarter.github.io/superheat/ | N/A |
| Other | ||
Mice
All studies and procedures involving animal subjects were performed under the approval of the Institutional Animal Care and Use Committee (IACUC) at Temple University. C57BL/6J mice were obtained from the Jackson Laboratory. 10 weeks old female mice were randomly assigned to experimental groups. All mice were housed in an animal facility and maintained in a temperature and light controlled environment with an alternating 12-hour light/dark cycle. The animals had no prior history of drug administration, surgery or behavioral testing. All the behavioral tests were performed by two people who were unaware of animal identifications.
METHOD DETAILS
Sensory axon lesion and imaging in Drosophila
Class IV da neuron (C4da) axon lesion in the VNC were performed in live fly larvae as previously described(Li et al., 2018; Song et al., 2019; Song et al., 2012; Song et al., 2015) with minor modifications. Briefly, fly larvae were raised at 25°C on grape juice agar plates and C4da neurons were labeled with ppk-CD4-tdGFP or ppk-CD4-tdTomato. At 48 hours after egg laying (h AEL), larvae were anesthetized by ether and injured on segments A3 and A6 of axon projections in the VNC by two-photon laser (930 nm, −1,950 mW) mounted on a Zeiss LSM 880 confocal microscope. Larvae were kept at 25°C on grape juice agar plates and imaged at 8, 24 and/or 48 h AI on confocal microscope. We ablated two commissure junction sites for each segment (red centric circles in Figure 1B and red dotted circle in representative images and diagrams). The lesion cores were −3.5 μm in diameter.
Quantification of sensory axon regeneration
Quantification was modified from the methods previously described (Li et al., 2018; Song et al., 2019; Song et al., 2012; Song et al., 2015). For the regenerated axons in the VNC, we used the two-following metrics. Regeneration percentage, which shows the percent of lesioned segments that showed obvious axon regeneration. The criterion for regeneration is theoretically associated with the potential of the regenerated axon to contribute to functional recovery. In practice, we considered the axons as regenerated when they extended their terminals into the region where C4da neuron axon terminals and synapses originally locate (region between the two black dotted lines shown in Figure S2D). For each lesioned segment, it is scored as regenerated as long as one axon bundle has regenerated. Regeneration length, which traces axon regrowth in the neuropil and measures the total length (shown in Figure S2B and S2C) normalized by the distance between segments A4 and A5 (shown in Figure S2A).
Sensory behavioral assay in Drosophila larvae
Fly larvae with C4da neurons labeled by ppk-CD4-tdGFP were raised at 25°C on grape juice agar plates. At 48-72 h AEL, larvae were injured on A7 and A8 axon segments in the VNC (shown in Figure 2A). Larvae were kept at 25°C on grape juice agar plates. A 47°C heat probe was used to stimulate the body segments (typically A7-A8 were the injured experimental segments and A4-A5 were the uninjured control segments), and the circling behavior was monitored. If a larva displayed two or more circles, it was scored as 1, otherwise as 0. Four trials were performed for each larva (4 is the highest score and 0 is the lowest). The behavioral test was performed at 8 h AI to confirm the injury. At 24 and/or 48 h AI, larvae were tested again to determine whether the thermonociception recovered.
Transmission electron microscopy
EM samples were prepared by following the procedures described previously(Han et al., 2014) with slight modifications. At 24 h AI, larvae expressing ppk-CD4-tdGFP; repo-Gal4>mRFP were dissected in cold 0.1 M Sorensen’s phosphate buffer (SPB) and the VNC with brain were fixed in 0.1 M Na-cacodylate buffer (pH 7.4) containing 2% glutaraldehyde for 1 hour on ice. After washing with cacodylate buffer, samples were post-fixed with 2% OsCO4 in SPB for 1.5 h, and rinsed three times in water followed by en-bloc staining in 1% uranyl acetate in water for 1 h at room temperature. Tissues were then rinsed five times with water followed by ethanol dehydration. After dehydration, tissues were rinsed three times in propylene oxide followed by PELCO Eponate-12 resin embedding (Ted Pella. Inc, USA Cat#18012). Serial sections of 85 nm thickness were cut and collected with standard procedures. The sections were imaged without post-embedding staining on a FEI Titan Themis 200 TEM (Thermo Fisher Scientific, USA) housed in the Imaging Suite of CUNY graduate Center-ASRC.
RNA sequencing analysis
Fly larvae were harvested at 96 h AEL and washed with PBS for three times. The CNS of larvae (VNC and brain) were dissected out and underwent RNA purification with Quick-RNA miniprep plus kit (Zymo). Integrity of eluted RNA was analyzed by NanoDrop (Thermo Scientific) and 2100 Agilent Bioanalyzer (Agilent) prior to subsequent experiments. Approximately 1 μg total RNA for each genotype was used. Oligo (dT) magnetic beads were used to select mRNA, then cDNA was synthesized from mRNA fragments by random N6 primers, followed by a second strand cDNA synthesis. After end-repair and adaptor ligation, the double stranded DNA was denatured and circularized on single strand to form the final library. All samples were analyzed by a BGISEQ-500 sequencer (Single End, 50). Quality controlled reads were mapped by Bowtie2(Langmead et al., 2009) and referenced to genome by HISAT(Kim et al., 2015). Individual gene expression levels in the Figure 3D are shown as mean FPKM (fragments per kilobase of transcript sequence per million mapped fragments). Deferentially expressed genes heatmap (Figure S4A) was drawn on Heatmapper.ca (Babicki et al., 2016). GO analysis on the deferentially expressed genes (Figure S4B) was performed on Metascape.org (Zhou et al., 2019). Additional details are described in the figure legends of Figure S4. We used R for published datasets analysis and R package “superheat” for the heatmaps showed in Figure S7.
Chemical compounds injection into fly larvae
Chemical compounds were injected into larvae right after injury (Figure 4K-4M and 5K-5M), or right after the first imaging session (Figure 6I and S6E). Sodium L-lactate (Sigma Aldrich), ethyl L-lactate (Sigma Aldrich), disodium L-2-Hydroxyglutarate (Sigma Aldrich), octyl-L-2HG (Cayman Chemical), L-2-Hydroxybutyric acid (Sigma Aldrich), ethyl 2-Hydroxybutyrate (Fisher Scientific), L-tartaric acid (Sigma Aldrich) and diethyl L-tartrate (Sigma Aldrich) were used. Sodium L-lactate, disodium L-2-Hydroxyglutarate, L-2-Hydroxybutyric acid and L-tartaric acid were dissolved in water and pH was adjust to 7.2 before using. Final concentration of chemical compounds injected into larvae were calculated based on the volume estimated from larvae weight. Injection was performed by glass micropipettes and Hamilton syringes.
Mass spectrometry measurement of metabolites
For hemolymph samples, larvae at 96 h AEL were carefully washed by PBS more than three times. The anterior cuticle was cut open using fine pointed forceps to let out the hemolymph. Extra centrifugation was performed to remove the hemocytes. 8 μL hemolymph were collected for each sample. For the CNS (VNC and brain) samples, they were dissected out and frozen immediately at −78.5°C. 50-100 larval CNS tissues were pooled for each sample. Measurement directly on hemolymph and the CNS would reflect the status of metabolites in specific tissue compared to using whole larvae as samples. However, to collect enough tissues for the measurement, the processing time is prolonged significantly, which could lead to more changes of metabolites from the living state. Thus, speeding up the dissection and freezing the tissue immediately after dissection at low temperature will help decrease the effects from the increased processing time. Enantiomers of 2-HG and lactate were measured as described(Struys et al., 2004). Briefly, samples were derivatized with freshly made DATAN reagent (50 g/L) dissolved in a mixture of methylene chloride and glacial acetic acid (4:1). After incubation for 30 min at 75°C, samples were dried down under nitrogen at room temperature. 100 uL of water were added to the samples, and after centrifugation, samples were injected into the LC-MS system (Agilent triple quad 6410). Separation of D and L isomers of 2-HG and lactate were accomplished by using C18 column (Agilent). [U-13C] lactate was used an internal standard. Organic acids levels were determined by GC-MS as previously described (Weinberg et al., 2000).
Laconic imaging and analysis in flies
Dissected VNCs with glia expressing laconic were placed on slides and analyzed using a confocal microscope. For the microscope setup, a 25x objective was used, with excitation at 458 nm and emission at 485 ± 15 nm (mTFP) and 528 ± 15 nm (Venus). For data analysis, over 20 regions of interest (ROIs) were imaged for each sample. FRET from mTFP to Venus of laconic decreases when lactate concentration increases, so ratio of mTFP/Venus were calculated (San Martin et al., 2013). All images were quantified with Fiji/ImageJ.
Fly larvae survival assay
Larvae raised at 25°C on regular grape juice agar plates were collected at 48 h AEL, and cultured on 2.5% agar plates with various chemical compounds. Fresh-made agar plates with chemicals (solutions were adjusted to pH 7.2 before use) were replaced every 24 hours, and larvae were counted. Agar plates with purified water was used as control.
cAMPr imaging and analysis in flies
Larvae with cAMPr expression in C4da neurons were imaged live. Confocal microscope was used with 488 nm excitation and standard GFP emission settings for cAMPr. Fluorescence intensity at segments A3 and A6 were analyzed. All images were quantified with Fiji/ImageJ.
Semi-quantitative RT-PCR for GABA-B-R1
Semi-quantitative RT-PCR was performed for GABA-B-R1, rp49 (Ribosomal protein 49) in fly larvae. The GABA-B-R1 transcripts were amplified with primers F 5′-cgcttccctcatcaactcatc-3′ and R 5′-gtataattcacctatcacgcaaagg-3′; rp49 was amplified with F 5′-cagtcggatcgatatgctaagctg-3′ and R 5′-taaccgatgttgggcatcagatac-3′.
35S-GTPγS binding assay
The scintillation proximity assay with the buffering system (20 mM HEPES pH 7.4, 100 mM NaCl, 10 μg/ml saponin, 5 mM MgCl2) was used. For reactions (100μl per well), 50 μl L-lactate or L-2HG solution in assay buffer with specific concentrations; 10 μl assay buffer; 20 μl pre-mixed membrane preparation from Chinese hamster ovary K1 cells transfected with human recombinant GABBR1a and GABBR2 and GDP (final conc. 10 μM), membrane was prepared as described previously (Galvez et al., 1999); 10 μl 35S-GTPγS (PerkinElmer) diluted in assay buffer (0.1 nM) mixed with 10 μl PVT-WGA beads (Perkin Elmer) diluted in assay buffer (0.5 mg/10 μl) right before assay started, were added to the wells of an Optiplate (Perkin Elmer). The plates were covered with a Topseal (Perkin Elmer), mixed on an orbital shaker for 2 min, and incubated for 1 hour at room temperature. Then the plates were centrifuged for 10 min at 2000 rpm, incubated at room temperature for another 1 hour and counted for 1 minute per well by a PerkinElmer TopCount reader.
Mice SCI, drug treatments and CST tracing
All studies and procedures involving animal subjects were performed under the approval of the Institutional Animal Care and Use Committee (IACUC) at Temple University. To lesion the spinal cord of C57BL/6J mice (10 weeks old), we exposed the dorsal spinal cord by T6-7 laminectomy. A dorsal over-hemisection (1 mm in depth, and approximately 1.5 mm in dorsoventral diameter) was performed at T7 with a 30-gauge needle and microscissors to completely sever the dorsal spinal cord, including all the CST axons. The lesion depth of 1 mm was ensured by passing a marked 30-gauge needle at least 5 times across the dorsal spinal cord. The lesion area was covered by a piece of gelfoam (2.5x2.5x2.5 mm) after injury and 20 uL of saline or lactate mix of salt and ester was applied to the gelfoam 5 min after SCI. Six days after injury, the gelfoam was replaced gently and the same amount of drug was reapplied to the lesion area. Three weeks after SCI, the mice received BDA (10 kDa) tracer injections into 5 sites of the sensorimotor cortex (anterior-posterior coordinates from Bregma in mm: 1.0, 0.5, 0, −0.5, −1.0, all at 1.0 mm lateral and at a depth of 1.0 mm). Mice were perfused 2 weeks after BDA injection and fixed spinal cords were dissected for histology as below.
Mice histology and axon quantification
The spinal cord extending from 0 to 4 mm rostral to and caudal to the lesion (8 mm long, containing the injury site) was cut parasagittally (30 μm). The spinal cord 5-7 mm rostral to and caudal to injury was transversely sectioned (30 μm). All the parasagittal sections were processed for BDA tracer with the TSA signal amplification systems (PerkinElmer). To visualize the lesion area, all parasagittal sections were also stained for GFAP (Sigma) with an anti-mouse Alexa647-conjugated secondary antibody. Some transverse sections rostral to and caudal to the lesion were used for processing BDA tracer.
To compare axon numbers in the caudal spinal cord between different groups, we determined the length of BDA-labeled CST axons in all parasagittal sections of spinal cord from 0 to 4 mm caudal to the lesion epicenter in each animal. The injury center was determined as the midpoint of histological abnormalities produced by lesion cavitation, reactive astrocytes, and morphological changes of injured axons. The CST axons caudal to the lesion were traced manually in each of the parasagittal sections and their total length inside of several bin boxes at 0.8, 1.6, 2.4, 3.2 and 4 mm caudal to the lesion center was measured using Photoshop and ImageJ. We measured the areas of GFAP+ astrocyte scar border and GFAP+ reactive astrocytes in spared but reactive neural tissue from all the parasagittal sections of lesioned spinal cord in each animal. The astrocyte scar border and the spared but reactive neural tissue were defined as the areas of densely overlapped GFAP+ astrocytic processes around the lesion epicenter and obvious increase of GFAP immunoreactivity surrounding the scar tissue, respectively. Antibodies for GFAP (Sigma, G3893), ALDH1L1 (Abcam, ab87117), GABBR1 (Abcam, ab55051) and GABBR2 (Novus, NBP2-16569) were used.
Mice behavioral evaluations
To determine functional recovery, we evaluated locomotion alterations during 5 weeks of survival by measuring locomotor Basso mouse scale (BMS) scores at four hours and two days after SCI, and weekly thereafter, and grid walk performance at 5 weeks after SCI. We also measured grasping rate of the hindpaws. The BMS scores were evaluated while mouse was walking in an open field and confirmed from digital video records. The grid walk errors were counted from videotapes played at a slow speed (4 separate trials per test) and averaged from different trials. Contact-evoked grasping rate was measured by lowering the hindpaws toward a wire cage lid and determining the percent of times that it was grasped successfully. All the behavioral tests were performed by two people who were unaware of animal identifications.
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical methods were used to predetermine sample sizes, and the statistical analyses were done afterwards without interim data analysis. No data points were excluded. For the experiments done in Drosophila, data collection and analyses were not performed blind to the conditions of the experiments. For in vivo experiments in mice, spinal cord injury, treatment, behavioral assay, tissue processing and imaging were performed in a blind manner. Fisher’s exact test was used to compare percentages. One-way ANOVA followed by multiple comparison test was performed for comparisons among three or more groups of samples. Two-tailed unpaired Student’s t-tests were performed for comparison between two groups of samples. Two-way ANOVA analyses were used to assess significance of multiple data points. The data meet the assumptions of the tests. Data distribution was assumed to be normal, but this was not formally tested. Each experiment has been successfully reproduced.
Supplementary Material
Movie S1. Control larva at 8 h AI, related to Figure 2. Confirm completed injury on A7 and A8 segments at 8 h AI in WT.
Movie S2. Control larva at 24 h AI, related to Figure 2. There is no recovery on A7 and A8 segments at 24 h AI in WT.
Movie S3. Larva with RGCPE at 8 h AI, related to Figure 2. Confirm completed injury on A7 and A8 segments at 8 h AI in larva with RGCPE.
Movie S4. Larva with RGCPE at 24 h AI, related to Figure 2. A7 and A8 segments recovered from injury at 24 h AI in larva with RGCPE.
Highlights.
Activation of the PI3K and EGFR pathways in glia promotes CNS axon regeneration
Enhanced glial glycolysis mediates the induced axon regeneration via metabolites
L-lactate and L-2HG are sufficient to boost CNS axon regeneration
The metabolites act on neuronal GABAB receptors to elevate cAMP as inverse agonists
Acknowledgements
We thank C. Potter, J. Blau and J. Tennessen for fly lines; Bloomington Stock Centre, VDRC and DGRC for fly stocks; Ilana Nissim, Y. Daikhin and O. Horyn for performing the analysis of lactate, 2HG and organic acids in the Metabolomics Core Facility, Children’s Hospital of Philadelphia; S. Zhang, T. Wang, J. Baker and A. Scott-Hansen for imaging consultation and technical support; members of the Song lab for helpful discussions. Y.S. is a recipient of the National Institute of Neurological Disorders and Stroke (NINDS) Pathway to Independence Award. This work was supported by an IDDRC New Program Development Award (CHOP/Penn), an NINDS K99/R00 award (R00NS088211), an NIH grant (1R01NS107392) and a Craig H. Neilsen Foundation research grant to Y.S.; by the Sloan Foundation CSURP program to Y.C. and K.V.; by a T32-MH014654 training grant to P.H.; by research grants to S.L. from NIH (R01NS105961, 1R01NS079432 and 1R01EY024575) and from Shriners Research Foundation (SHC-85100, SHC-86200-PHI-16 and 85112-PHI-18); Y.H. is supported by a PSC-CUNY grant.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams KL, and Gallo V (2018). The diversity and disparity of the glial scar. Nat Neurosci 21, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alilain WJ, Horn KP, Hu H, Dick TE, and Silver J (2011). Functional regeneration of respiratory pathways after spinal cord injury. Nature 475, 196–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen AM, Neville MC, Birtles S, Croset V, Treiber CD, Waddell S, and Goodwin SF (2020). A single-cell transcriptomic atlas of the adult Drosophila ventral nerve cord. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MA, Burda JE, Ren Y, Ao Y, O'Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, and Sofroniew MV (2016). Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MA, O'Shea TM, Burda JE, Ao Y, Barlatey SF, Bernstein AM, Kim JH, James ND, Rogers A, Kato B, et al. (2018). Required growth facilitators propel axon regeneration across complete spinal cord injury. Nature 561, 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babicki S, Arndt D, Marcu A, Fiang Y, Grant JR, Maciejewski A, and Wishart DS (2016). Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 44, W147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth PG, Hoffmann GF, Jaeken J, Lehnert W, Hanefeld F, van Gennip AH, Duran M, Valk J, Schutgens RB, Trefz FK, et al. (1992). F-2-hydroxyglutaric acidemia: a novel inherited neurometabolic disease. Ann Neurol 32, 66–71. [DOI] [PubMed] [Google Scholar]
- Belin S, Nawabi H, Wang C, Tang S, Latremoliere A, Warren P, Schorle H, Uncu C, Woolf CJ, He Z, et al. (2015). Injury-induced decline of intrinsic regenerative ability revealed by quantitative proteomics. Neuron 86, 1000–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesch A, and Tuszynski MH (2009). Spinal cord injury: plasticity, regeneration and the challenge of translational drug development. Trends Neurosci 32, 41–47. [DOI] [PubMed] [Google Scholar]
- Brown GK, Otero LJ, LeGris M, and Brown RM. (1994). Pyruvate dehydrogenase deficiency. J Med Genet 31, 875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai TQ, Ren N, Jin L, Cheng K, Kash S, Chen R, Wright SD, Taggart AK, and Waters MG (2008). Role of GPR81 in lactate-mediated reduction of adipose lipolysis. Biochem Biophys Res Commun 377, 987–991. [DOI] [PubMed] [Google Scholar]
- Coffman CR, Strohm RC, Oakley FD, Yamada Y, Przychodzin D, and Boswell RE. (2002). Identification of X-linked genes required for migration and programmed cell death of Drosophila melanogaster germ cells. Genetics 162, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegio OR, Chu NQ, Szabo AF, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S, and Aguayo AJ (1981). Axonal elongation into peripheral nervous system "bridges" after central nervous system injury in adult rats. Science 214, 931–933. [DOI] [PubMed] [Google Scholar]
- Deumens R, Koopmans GC, and Joosten EA (2005). Regeneration of descending axon tracts after spinal cord injury. Prog Neurobiol 77, 57–89. [DOI] [PubMed] [Google Scholar]
- Doherty J, Logan MA, Tasdemir OE, and Freeman MR (2009). Ensheathing glia function as phagocytes in the adult Drosophila brain. J Neurosci 29, 4768–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J, Sheehan AE, Bradshaw R, Fox AN, Lu TY, and Freeman MR (2014). PI3K signaling and Stat92E converge to modulate glial responsiveness to axonal injury. PLoS Biol 12, e1001985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, and Sofroniew MV (2004). Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci 24, 2143–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti P, Zhang F, and O'Neill P (2003). Changes in spinal cord regenerative ability through phylogenesis and development: lessons to be learnt. Dev Dyn 226, 245–256. [DOI] [PubMed] [Google Scholar]
- Flores A, Schell J, Krall AS, Jelinek D, Miranda M, Grigorian M, Braas D, White AC, Zhou JF, Graham NA, et al. (2017). Factate dehydrogenase activity drives hair follicle stem cell activation. Nat Cell Biol 19, 1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Chin RM, Vergnes L, Hwang H, Deng G, Xing Y, Pai MY, Li S, Ta L, Fazlollahi F, et al. (2015). 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab 22, 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez T, Parmentier ML, Joly C, Malitschek B, Kaupmann K, Kuhn R, Bittiger H, Froestl W, Bettler B, and Pin JP (1999). Mutagenesis and modeling of the GABAB receptor extracellular domain support a venus flytrap mechanism for ligand binding. J Biol Chem 274, 13362–13369. [DOI] [PubMed] [Google Scholar]
- Gassmann M, and Bettler B (2012). Regulation of neuronal GABA(B) receptor functions by subunit composition. Nat Rev Neurosci 13, 380–394. [DOI] [PubMed] [Google Scholar]
- Grenell A, Wang Y, Yam M, Swarup A, Dilan TF, Hauer A, Linton JD, Philp NJ, Gregor E, Zhu S, et al. (2019). Foss of MPC1 reprograms retinal metabolism to impair visual function. Proc Natl Acad Sci U S A 116, 3530–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber WB, Jan FY, and Jan YN (2003). Different levels of the homeodomain protein cut regulate distinct dendrite branching patterns of Drosophila multidendritic neurons. Cell 112, 805–818. [DOI] [PubMed] [Google Scholar]
- Hackett AR, Yahn SF, Lyapichev K, Dajnoki A, Lee DH, Rodriguez M, Cammer N, Pak J, Mehta ST, Bodamer O, et al. (2018). Injury type-dependent differentiation of NG2 glia into heterogeneous astrocytes. Experimental neurology 308, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackley CR, Mazzoni EO, and Blau J (2018). cAMPr: A single-wavelength fluorescent sensor for cyclic AMP. Sci Signal 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Jan LY, and Jan YN (2011). Enhancer-driven membrane markers for analysis of nonautonomous mechanisms reveal neuron-glia interactions in Drosophila. Proc Natl Acad Sci U S A 108, 9673–9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Song Y, Xiao H, Wang D, Franc NC, Jan LY, and Jan YN (2014). Epidermal cells are the primary phagocytes in the fragmentation and clearance of degenerating dendrites in Drosophila. Neuron 81, 544–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassett CC (1948). The utilization of sugars and other substances by Drosophila. Biol Bull 95, 114–123. [PubMed] [Google Scholar]
- Hunt RJ, Granat L, McElroy GS, Ranganathan R, Chandel NS, and Bateman JM (2019). Mitochondrial stress causes neuronal dysfunction via an ATF4-dependent increase in L-2-hydroxyglutarate. J Cell Biol 218, 4007–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Dematteo RG, Venneti S, Finley FW, Lu C, Judkins AR, Rustenburg AS, Grinaway PB, Chodera JD, Cross JR, et al. (2015). Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab 22, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N, Itoh Y, Tassoni A, Ren E, Kaito M, Ohno A, Ao Y, Farkhondeh V, Johnsonbaugh H, Burda J, et al. (2018). Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc Natl Acad Sci U S A 115, E302–E309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Fee G, and Chung J (2008). LKB1 induces apical trafficking of Silnoon, a monocarboxylate transporter, in Drosophila melanogaster. J Cell Biol 183, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, Huang FY, et al. (1998). GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature 396, 674–679. [DOI] [PubMed] [Google Scholar]
- Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R, et al. (1998). GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature 396, 683–687. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, and Popovich PG (2009). Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 29, 13435–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Fangmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto T, Yin Y, Omura K, Gilbert HY, Kim D, Cen LP, Moko F, Kugler S, and Benowitz LI (2010). Long-distance axon regeneration in the mature optic nerve: contributions of oncomodulin, cAMP, and pten gene deletion. J Neurosci 30, 15654–15663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang BT, Cregg JM, DePaul MA, Tran AP, Xu K, Dyck SM, Madalena KM, Brown BP, Weng YL, Li S, et al. (2015). Modulation of the proteoglycan receptor PTPsigma promotes recovery after spinal cord injury. Nature 518, 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fangmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DC, Sohn HA, Park ZY, Oh S, Kang YK, Lee KM, Kang M, Jang YJ, Yang SJ, Hong YK, et al. (2015). A lactate-induced response to hypoxia. Cell 161, 595–609. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Seo BR, Choi EJ, and Koh JY (2014). The role of reciprocal activation of cAbl and Mst1 in the oxidative death of cultured astrocytes. Glia 62, 639–648. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Li D, Li F, Guttipatti P, and Song Y (2018). A Drosophila In Vivo Injury Model for Studying Neuroregeneration in the Peripheral and Central Nervous System. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chawla G, Hurlburt AJ, Sterrett MC, Zaslaver O, Cox J, Karty JA, Rosebrock AP, Caudy AA, and Tennessen JM (2017). Drosophila larvae synthesize the putative oncometabolite L-2-hydroxyglutarate during normal developmental growth. Proc Natl Acad Sci U S A 114, 1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingenhoehl K, Brom R, Heid J, Beck P, Froestl W, Kaupmann K, Bettler B, and Mosbacher J (1999). Gamma-hydroxybutyrate is a weak agonist at recombinant GABA(B) receptors. Neuropharmacology 38, 1667–1673. [DOI] [PubMed] [Google Scholar]
- Liu B, and Neufeld AH (2004). Activation of epidermal growth factor receptor causes astrocytes to form cribriform structures. Glia 46, 153–168. [DOI] [PubMed] [Google Scholar]
- Liu C, Wu J, Zhu J, Kuei C, Yu J, Shelton J, Sutton SW, Li X, Yun SJ, Mirzadegan T, et al. (2009). Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem 284, 2811–2822. [DOI] [PubMed] [Google Scholar]
- Liu J, Chen G, Liu Z, Liu S, Cai Z, You P, Ke Y, Lai L, Huang Y, Gao H, et al. (2018). Aberrant FGFR Tyrosine Kinase Signaling Enhances the Warburg Effect by Reprogramming LDH Isoform Expression and Activity in Prostate Cancer. Cancer Res 78, 4459–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, et al. (2010). PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nature neuroscience 13, 1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, MacKenzie KR, Putluri N, Maletic-Savatic M, and Bellen HJ (2017). The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab 26, 719–737 e716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liuzzi FJ, and Lasek RJ (1987). Astrocytes block axonal regeneration in mammals by activating the physiological stop pathway. Science 237, 642–645. [DOI] [PubMed] [Google Scholar]
- Lopez-Bendito G, Shigemoto R, Kulik A, Paulsen O, Fairen A, and Lujan R (2002). Expression and distribution of metabotropic GABA receptor subtypes GABABR1 and GABABR2 during rat neocortical development. Eur J Neurosci 15, 1766–1778. [DOI] [PubMed] [Google Scholar]
- Lu Y, Belin S, and He Z (2014). Signaling regulations of neuronal regenerative ability. Current opinion in neurobiology 27, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Sun R, Jiang B, Gao J, Deng W, Liu P, He R, Cui J, Ji M, Yi W, et al. (2017). L2hgdh Deficiency Accumulates 1-2-Hydroxyglutarate with Progressive Leukoencephalopathy and Neurodegeneration. Mol Cell Biol 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, and Allaman I (2018). Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci 19, 235–249. [DOI] [PubMed] [Google Scholar]
- McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, et al. (2008). Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem 283, 22700–22708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa H, and Aulehla A (2018). Revisiting the role of metabolism during development. Development 145. [DOI] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, and Goldberg JL (2009). KLF family members regulate intrinsic axon regeneration ability. Science (New York, N.Y 326, 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthukumar AK, Stork T, and Freeman MR (2014). Activity-dependent regulation of astrocyte GAT levels during synaptogenesis. Nat Neurosci 17, 1340–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, and Basbaum AI (2002). Regeneration of sensory axons within the injured spinal cord induced by intragangbonic cAMP elevation. Neuron 34, 885–893. [DOI] [PubMed] [Google Scholar]
- Newman ZL, Hoagland A, Aghi K, Worden K, Levy SL, Son JH, Lee LP, and Isacoff EY (2017). Input-Specific Plasticity and Homeostasis at the Drosophila Larval Neuromuscular Junction. Neuron 93, 1388–1404 e1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norsworthy MW, Bei F, Kawaguchi R, Wang Q, Tran NM, Li Y, Brommer B, Zhang Y, Wang C, Sanes JR, et al. (2017). Sox11 Expression Promotes Regeneration of Some Retinal Ganglion Cell Types but Kills Others. Neuron 94, 1112–1120 e1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea TM, Burda JE, and Sofroniew MV (2017). Cell biology of spinal cord injury and repair. J Clin Invest 127, 3259–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Ma N, Yu B, Zhang W, and Wan J (2020). Transcriptomic profiling of microglia and astrocytes throughout aging. J Neuroinflammation 17, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, et al. (2008). Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science (New York, N.Y 322, 963–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, and Bunge MB (2004). cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med 10, 610–616. [DOI] [PubMed] [Google Scholar]
- Pierce SB, Yost C, Britton JS, Loo LW, Flynn EM, Edgar BA, and Eisenman RN. (2004). dMyc is required for larval growth and endoreplication in Drosophila. Development 131, 2317–2327. [DOI] [PubMed] [Google Scholar]
- Poplawski GHD, Kawaguchi R, Van Niekerk E, Lu P, Mehta N, Canete P, Lie R, Dragatsis I, Meves JM, Zheng B, et al. (2020). Injured adult neurons regress to an embryonic transcriptional growth state. Nature 581, 77–82. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, and Filbin MT (2002). Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 34, 895–903. [DOI] [PubMed] [Google Scholar]
- Raineteau O, and Schwab ME (2001). Plasticity of motor systems after incomplete spinal cord injury. Nat Rev Neurosci 2, 263–273. [DOI] [PubMed] [Google Scholar]
- Read RD, Cavenee WK, Furnari FB, and Thomas JB (2009). A drosophila model for EGFR-Ras and PI3K-dependent human glioma. PLoS Genet 5, e1000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reier PJ, Bregman BS, and Wujek JR (1986). Intraspinal transplantation of embryonic spinal cord tissue in neonatal and adult rats. J Comp Neurol 247, 275–296. [DOI] [PubMed] [Google Scholar]
- Reitman ZJ, Sinenko SA, Spana EP, and Yan H (2015). Genetic dissection of leukemia-associated IDH1 and IDH2 mutants and D-2-hydroxyglutarate in Drosophila. Blood 125, 336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls A, Shechter R, and Schwartz M (2009). The bright side of the glial scar in CNS repair. Nat Rev Neurosci 10, 235–241. [DOI] [PubMed] [Google Scholar]
- San Martin A, Ceballo S, Ruminot I, Lerchundi R, Frommer WB, and Barros LF (2013). A genetically encoded FRET lactate sensor and its use to detect the Warburg effect in single cancer cells. PLoS One 8, e57712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell JC, Olson KA, Jiang L, Hawkins AJ, Van Vranken JG, Xie J, Egnatchik RA, Earl EG, DeBerardinis RJ, and Rutter J (2014). A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol Cell 56, 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepp KJ, Schulte J, and Auld VJ (2001). Peripheral glia direct axon guidance across the CNS/PNS transition zone. Dev Biol 238, 47–63. [DOI] [PubMed] [Google Scholar]
- Sharma K, Selzer ME, and Li S (2012). Scar-mediated inhibition and CSPG receptors in the CNS. Experimental neurology 237, 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, and Miller JH (2004). Regeneration beyond the glial scar. Nat Rev Neurosci 5, 146–156. [DOI] [PubMed] [Google Scholar]
- Smith GM, Miller RH, and Silver J (1986). Changing role of forebrain astrocytes during development, regenerative failure, and induced regeneration upon transplantation. J Comp Neurol 251, 23–43. [DOI] [PubMed] [Google Scholar]
- Song Y, Fi D, Farrelly O, Miles L, Li F, Kim SE, Lo TY, Wang F, Li T, Thompson-Peer KL, et al. (2019). The Mechanosensitive Ion Channel Piezo Inhibits Axon Regeneration. Neuron 102, 373–389 e376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Ori-McKenney KM, Zheng Y, Han C, Jan FY, and Jan YN (2012). Regeneration of Drosophila sensory neuron axons and dendrites is regulated by the Akt pathway involving Pten and microRNA bantam. Genes Dev 26, 1612–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Sretavan D, Salegio EA, Berg J, Huang X, Cheng T, Xiong X, Meltzer S, Han C, Nguyen TT, et al. (2015). Regulation of axon regeneration by the RNA repair and splicing pathway. Nat Neurosci 18, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squair JW, Tigchelaar S, Moon KM, Fiu J, Tetzlaff W, Kwon BK, Krassioukov AV, West CR, Foster LJ, and Skinnider MA (2018). Integrated systems analysis reveals conserved gene networks underlying response to spinal cord injury. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Zheng B, and Tessier-Lavigne M (2003). False resurrections: distinguishing regenerated from spared axons in the injured central nervous system. The Journal of comparative neurology 459, 1–8. [DOI] [PubMed] [Google Scholar]
- Strange PG (2002). Mechanisms of inverse agonism at G-protein-coupled receptors. Trends Pharmacol Sci 23, 89–95. [DOI] [PubMed] [Google Scholar]
- Streijger F, Plunet WT, Lee JH, Liu J, Lam CK, Park S, Hilton BJ, Fransen BL, Matheson KA, Assinck P, et al. (2013). Ketogenic diet improves forelimb motor function after spinal cord injury in rodents. PLoS One 8, e78765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struys EA, Jansen EE, Verhoeven NM, and Jakobs C (2004). Measurement of urinary D- and L-2-hydroxyglutarate enantiomers by stable-isotope-dilution liquid chromatography-tandem mass spectrometry after derivatization with diacetyl-L-tartaric anhydride. Clin Chem 50, 1391–1395. [DOI] [PubMed] [Google Scholar]
- Taylor MR, Hurley JB, Van Epps HA, and Brockerhoff SE (2004). A zebrafish model for pyruvate dehydrogenase deficiency: rescue of neurological dysfunction and embryonic lethality using a ketogenic diet. Proc Natl Acad Sci U S A 101, 4584–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teperino R, Amann S, Bayer M, McGee SL, Loipetzberger A, Connor T, Jaeger C, Kammerer B, Winter L, Wiche G, et al. (2012). Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell 151, 414–426. [DOI] [PubMed] [Google Scholar]
- Tom VJ, Sandrow-Feinberg HR, Miller K, Santi L, Connors T, Lemay MA, and Houle JD (2009). Combining peripheral nerve grafts and chondroitinase promotes functional axonal regeneration in the chronically injured spinal cord. J Neurosci 29, 14881–14890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey WD Jr., Wilson RI, Laurent G, and Benzer S (2003). painless, a Drosophila gene essential for nociception. Cell 113, 261–273. [DOI] [PubMed] [Google Scholar]
- Tsai MC, Shen LF, Kuo HS, Cheng H, and Chak KF (2008). Involvement of acidic fibroblast growth factor in spinal cord injury repair processes revealed by a proteomics approach. Mol Cell Proteomics 7, 1668–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, You J, Chia GS, Sim J, Doedens A, et al. (2016). S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 540, 236–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, Schulze KL, Haelterman NA, Pan H, He Y, Evans-Holm M, Carlson JW, Levis RW, Spradling AC, Hoskins RA, et al. (2011). MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nature Methods 8, 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemure JF, Adam L, Bevan NJ, Gearing K, Chenier S, and Bouvier M (2005). Subcellular distribution of GABA(B) receptor homo- and hetero-dimers. Biochem J 388, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkenhoff A, Weiler A, Letzel M, Stehling M, Klambt C, and Schirmeier S (2015). Glial Glycolysis Is Essential for Neuronal Survival in Drosophila. Cell Metab 22, 437–447. [DOI] [PubMed] [Google Scholar]
- Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, and Sofroniew MV (2013). Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci 33, 12870–12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg JM, Venkatachalam MA, Roeser NF, and Nissim I (2000). Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc Natl Acad Sci U S A 97, 2826–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RE, Yin FQ, and Jakeman LB (2008). TGF-alpha increases astrocyte invasion and promotes axonal growth into the lesion following spinal cord injury in mice. Exp Neurol 214, 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Davis KD, and Coffman CR (2008). Programmed cell death of primordial germ cells in Drosophila is regulated by p53 and the Outsiders monocarboxylate transporter. Development 135, 207–216. [DOI] [PubMed] [Google Scholar]
- Yang Y, Vidensky S, Jin L, Jie C, Lorenzini I, Frankl M, and Rothstein JD (2011). Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia 59, 200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarar-Fisher C, Kulkarni A, Li J, Farley P, Renfro C, Aslam H, Bosarge P, Wilson L, and Barnes S (2018). Evaluation of a ketogenic diet for improvement of neurological recovery in individuals with acute spinal cord injury: a pilot, randomized safety and feasibility trial. Spinal Cord Ser Cases 4, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SJ, Pan J, and Lee MH (2008). Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer. Cell Mol Life Sci 65, 3981–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu G, and He Z (2006). Glial inhibition of CNS axon regeneration. Nat Rev Neurosci 7, 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylera B, Erturk A, Hellal F, Nadrigny F, Hurtado A, Tahirovic S, Oudega M, Kirchhoff F, and Bradke F (2009). Chronically CNS-injured adult sensory neurons gain regenerative competence upon a lesion of their peripheral axon. Curr Biol 19, 930–936. [DOI] [PubMed] [Google Scholar]
- Yu D, Gustafson WC, Han C, Fafaye C, Noirclerc-Savoye M, Ge WP, Thayer DA, Huang H, Kornberg TB, Royant A, et al. (2014). An improved monomeric infrared fluorescent protein for neuronal and tumour brain imaging. Nat Commun 5, 3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, Xiang Y, Yan Z, Han C, Jan LY, and Jan YN (2011). Light-induced structural and functional plasticity in Drosophila larval visual system. Science 333, 1458–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Muri J, Fitzgerald G, Gorski T, Gianni-Barrera R, Masschelein E, D'Hulst G, Gilardoni P, Turiel G, Fan Z, et al. (2020). Endothelial Factate Controls Muscle Regeneration from Ischemia by Inducing M2-like Macrophage Polarization. Cell Metab 31, 1136–1153 e1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YQ, Chen SP, Liu DQ, Manyande A, Zhang W, Yang SB, Xiong BR, Fu QC, Song ZP, Rittner H, et al. (2017). The Role of Spinal GABAB Receptors in Cancer-Induced Bone Pain in Rats. J Pain 18, 933–946. [DOI] [PubMed] [Google Scholar]
- Ziegenfuss JS, Doherty J, and Freeman MR (2012). Distinct molecular pathways mediate glial activation and engulfment of axonal debris after axotomy. Nat Neurosci 15, 979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Control larva at 8 h AI, related to Figure 2. Confirm completed injury on A7 and A8 segments at 8 h AI in WT.
Movie S2. Control larva at 24 h AI, related to Figure 2. There is no recovery on A7 and A8 segments at 24 h AI in WT.
Movie S3. Larva with RGCPE at 8 h AI, related to Figure 2. Confirm completed injury on A7 and A8 segments at 8 h AI in larva with RGCPE.
Movie S4. Larva with RGCPE at 24 h AI, related to Figure 2. A7 and A8 segments recovered from injury at 24 h AI in larva with RGCPE.
Data Availability Statement
The accession number for the RNA-seq data reported in this paper is GEO: GSE144655.