

Abstract
T cell receptors (TCRs) orchestrate cellular immunity by recognizing peptides presented by a range of major histocompatibility complex (MHC) proteins. Naturally occurring TCRs bind the composite peptide/MHC surface, recognizing peptides that are structurally and chemically compatible with the TCR binding site. Here we describe a molecularly evolved TCR variant that binds the human class I MHC protein HLA-A2 independent of the bound peptide, achieved by a drastic perturbation of the TCR binding geometry that places the molecule far away from the peptide binding groove. This unique geometry is unsupportive of normal T cell signaling. A substantial divergence between affinity measurements in solution and in two dimensions between proximal cell membranes leads us to attribute the lack of signaling to steric hindrance that limits binding in the confines of a cell-cell interface. Our results provide an example of how receptor binding geometry can impact T cell function and provide further support for the view that germline-encoded residues in TCR binding loops evolved to drive productive TCR recognition and signaling.
Keywords: T cell receptor, peptide/MHC complex, binding geometry, crystal structure, signaling, 2D affinity
INTRODUCTION
Heterodimeric αβ T cell receptors (TCRs) play a key role in cell-mediated immune responses of adaptive immunity as they are responsible for recognizing antigenic peptides bound and presented by proteins of the major histocompatibility complex (MHC proteins). TCRs recognize the composite peptide/MHC surface primarily using six complementarity determining region (CDR) loops. Despite considerable diversity in these loops, most naturally occurring TCRs show a relatively conserved docking topology, binding the peptide/MHC complex “head on” at the top of the MHC peptide binding groove, with the α chain CDR loops near the N-terminal half of the peptide and β chain CDR loops near the C-terminal half of the peptide1. This is associated with what is often described as the canonical “diagonal” binding or crossing angle, determined by the orientation of the TCR α and β variable domains relative to the peptide2. Although there is a wide variation in this angle as well as other geometrical descriptors used to describe how TCRs engage peptide/MHC complexes, TCRs nonetheless bind with geometries that ensure their CDR loops can sample peptide features and allow T cells to respond accordingly3.
Emerging data indicate that some unusual TCRs that bind peptide/MHC with geometries outside of the traditional diagonal binding mode initiate altered immunological signaling. For example, a recently described TCR that bound with high affinity yet skewed geometry failed to signal4, and TCRs that bind with a reversed diagonal polarity (β chain over the peptide N-terminus, α chain over the C-terminus) have limited function5. A class of γδ TCRs that bind the non-classical MHC protein MR1 have been described that bind away from the antigen binding cleft, on the side of the heavy chain opposite from where the β2-microgloublin (β2m) subunit binds, yet the functionality of these receptors is unclear6. How T cell signaling might change with TCR binding geometry is not yet understood. However, an impact of receptor geometry on the construction of or transmission through a large signaling complex, involving multiple chains of the CD3 components as well as the CD4/CD8 coreceptors, would not be surprising.
Molecular evolution has proven a powerful tool to study the determinants of TCR binding properties. Recently, we used yeast display technology to shift the specificity of the well-characterized TCR A6 away from the nonameric HTLV-1 Tax antigen (sequence LLFGYPVYV) to the anchor modified decameric MART-1 tumor antigen (sequence ELAGIGILTV), both presented by the class I MHC protein HLA-A*0201 (HLA-A2)7. Our subsequent deconstruction of the amino acid changes introduced via the molecular evolution process revealed how the mutations altered TCR compatibility with the two different ligands. The change in TCR specificity was associated with a new, unusual binding geometry; however, the TCR still focused on the peptide and T cell signaling was maintained8.
Interestingly, the same molecular evolution experiment yielded TCR variants that displayed a lack of peptide specificity. For example, the RD2-MART-1-S3–4 variant of the A6 TCR (hereafter termed S3–4) bound both the Tax and MART-1 antigen, as well as the unrelated WT1 tumor antigen (sequence RMFPNAPYL)7. Although TCRs are inherently capable of recognizing large sets of peptides9, 10, in this case the parental TCR A6 showed no recognition of the MART-1 or WT1 peptides. Moreover, the cross-recognition of such a diverse set of peptides, which adopt different paths through the HLA-A2 binding groove and have different lengths, charges, and other physical characteristics, is surprising11–13.
Here, we explored the basis for the lack of specificity with the S3–4 variant of the A6 TCR. We found that the six mutations in the TCR shift the binding mode from the top of the MHC binding groove to the side of the protein, opposite from where the CD8 coreceptor binds. The shift in binding is due to the coordinated introduction of new interactions, removal of steric and electrostatic clashes, and destabilization of the traditional binding geometry. Although the binding of S3–4 to the side of HLA-A2 occurs with a relatively high solution affinity, characteristic of TCRs that bind viral antigens, this altered binding mode was unsupportive of normal T cell signaling. A more evolved variant with an even higher affinity also did not signal. Despite the high solution affinity, measurements of TCR-MHC interactions between cells using two-dimensional (2D) methods revealed a very low 2D affinity, characteristic of a TCR that binds with a solution affinity orders of magnitude weaker. This discrepancy between solution and 2D measurements suggests that S3–4 does not support T cell signaling due to steric limitations that hinder binding to peptide/MHC in a cell-cell interface.
Although S3–4 is an engineered TCR variant, our results provide an example of how an outlier receptor binding geometry can impact T cell function. In addition, our data show that the germline CDR loop mutations are required for the altered binding mode of S3–4. The absence of similar sequences in human TCR variable genes provides new support for the idea that the variable genes of the TCRs have evolved alongside MHC genes to promote productive receptor binding, achieved in part by an evolved biochemical compatibility for top-down TCR-pMHC binding. Beyond these new insights, we suggest that S3–4 may be of use as a molecular chaperone to facilitate studies of poorly stable peptide/MHC complexes or as a probe for identifying properly assembled class I MHC complexes in solution and on cells.
MATERIALS AND METHODS
Protein expression and purification
Expression and refolding of soluble constructs of the S3–4 scTv, A6 c134 TCR, DMF5 TCR, and HLA-A2 were performed as previously described14. Briefly, the S3–4 scTv, the A6 c134 α and β chains, the DMF5 α and β chains, the HLA-A2 heavy chain, and β2m were generated in Escherichia coli as inclusion bodies, which were isolated and denatured in 8 M urea and 6 M guanidinium-HCL. The S3–4 scTv and TCR α/β chains were diluted in 1 L of TCR refolding buffer (50 mM Tris (pH 8), 2 mM EDTA, 2.5 M urea, 9.6 mM cysteamine, 5.5 mM cystamine, 0.2 mM PMSF). Diluted TCR α and β chains were mixed at a 1:1 ratio. HLA-A2 and β2m were diluted in 1 L of MHC refolding buffer (100 mM Tris (pH 8), 2 mM EDTA, 400 mM L-arginine, 6.3 mM cysteamine, 3.7 mM cystamine, 0.2 mM PMSF) at a 1:1 ratio in the presence of 10-fold excess peptide. Refolding was allowed to proceed overnight at 4 °C. After refolding, samples were dialyzed against 10 mM Tris-Cl pH 8.15 for 36–48 hours at room temperature for the TCRs and at 4 °C for the S3–4 and peptide/MHC complexes. Refolded protein was purified by anion exchange followed by size exclusion chromatography. Peptides were purchased commercially from AAPPTEC or Atlantic Peptides at >80% purity and stored at −80 °C in DMSO until use. All experiments with the MART-1 peptide used the anchor modified decamer. Mutagenesis of S3–4 and the DMF5 TCR was performed commercially (Genewiz) and confirmed via sequencing. Peptide/HLA-A2 tetramers were purchased commercially (MBL International).
Surface plasmon resonance
Surface plasmon resonance experiments were performed with a Biacore T200 instrument using Series S CM5 sensor chips as previously described14. For S3–4 and DMF5 binding experiments, S3–4 or DMF5 was immobilized on the sensor chip at 1000–3000 response units via standard amine coupling and peptide/MHC injected as analyte. Steady-state experiments were performed at 25 °C at a flowrate of 10 μL/min, with responses subtracted from an activated then deactivated blank flow cell. To test if S3–4 binds peptide/MHC in a peptide independent manner, A6 c134 was immobilized at a chip density of ~3000 RU. Every sample injection cycle consisted of 20 μM Tax/HLA-A2 to maintain the TCR-peptide/MHC complex on the surface, followed by 500 seconds of buffer, then varying concentrations of S3–4, with responses again corrected for responses from a blank flow cell. SPR data was processed in BiaEvaluation 4.1 and analyzed in OriginPro 9.0 using a 1:1 binding model and global fitting15. The kinetic titration of S3–4MQ binding to Tax/HLA-A2 was performed as previously described16. Briefly, the TCR was coupled to a sensor surface at approximately 500 response units. A series of five injections of increasing concentration of Tax/HLA-A2 were used at a flow rate of 30 μL/min at 25°C. Data were fit with a 1:1 kinetic titration model with drift using BIAevaluation 4.1.
Crystallization, data collection, and structure refinement
For producing the S3–4-Tax/HLA-A2 complex for crystallization, Tax/HLA-A2 was first generated in a 1 L refolding reaction as described above, then inclusion bodies for S3–4 were added for a final S3–4:Tax/HLA-A2 molar ratio of 1:3 in MHC refolding buffer. S3–4 was allowed to refold in the presence of Tax/HLA-A2 overnight at 4 °C. The refolding solution was dialyzed against 10 mM Tris-Cl pH 8.15 for forty-eight hours at 4 °C and the complex purified by anion exchange followed by size exclusion chromatography. The purified complexes were maintained in 10 mM MES, 50 mM NaCl, pH 7.0. Crystals were grown in 10% w/v PEG 20,000, 0.1 M MES (pH 6.5). Crystallization was performed using hanging-drop vapor diffusion using a Mosquito robot. For cryoprotection, crystals were transferred into 20% glycerol with 80% mother liquor for 30 seconds and immediately flash frozen in liquid nitrogen. Diffraction data were collected at the SER-CAT (22-ID) beamline with MAR CCD detector at the Advanced Photon Source, Argonne National Laboratory. Data reduction was performed with HKL200017. The structure was solved by molecular replacement with Phaser in Phenix18, using PDB entry 4FTV19 and the HLA-A2 heavy chain and TCR variable domains as search models. Rigid-body refinement followed by NCS torsion-angle restraints, translation/libration/screw refinement, and multiple steps of restrained refinement were performed using Phenix Refine20. Anisotropic and bulk solvent corrections were considered throughout the refinement process. Evaluation of models and fitting to maps were performed using COOT21. Molprobity was used to evaluate the structure during and after refinement22. Simulated annealing/composite omit maps were calculated with CNS23 as implemented in Discovery Studio 2019.
Cells lines and media
GP2–293, PG13, T2, and Jurkat 76 cell lines were kindly provided by Michael Nishimura (University of Loyola Stritch School of Medicine). The Jurkat cell subline JX-17 was obtained from American Type Cell Culture (ATCC). GP2 cells were maintained in DMEM media supplemented with 2 mM L-glutamine; PG13 cells were cultured in IDMEM; and T2, Jurkat 76, and JX-17 cells were maintained in RPMI-1640 media. All cell culture media was supplemented with 10% FBS, 100 units/mL penicillin, and 100 ug/mL streptomycin. Generation of Jurkat 76 CD8+/CD34− cells has been previously described24. JX-17 with knock-down expression of β2m (JX-17KO) were generated by transfection of the pLKO.1-GFP expressing the sgRNAβ2M then collecting GFP positive cells via FACS25.
Retroviral transduction
Supernatant containing retrovirus was prepared from stable retroviral producer PG13 cells expressing full-length αβ DMF5, S3–4, or HCV1406 from a modified SAMEN retroviral vector as previously described26. Retrovirus was induced from producer PG13 cells by seeding 8 × 106 cells per T-75 flask overnight, then treating cells with IDMEM supplemented with 10% FBS, 10 mM HEPES, and 1 mM sodium butyrate. Twenty-four hours post induction, supernatants containing retrovirus were 0.45 μm filtered and used to transduce Jurkat 76 cells by spinoculation. TCR-transduced cells were first sorted with anti-CD34-MicroBeads then with anti-CD3-MicroBeads using the Midi-MACS magnetic separation system (Myltenyi Biotech). Cell surface protein analysis of TCR-transduced cells was performed by flow cytometry using a Beckman Coulter FC 500 instrument. Cells were stained with either anti-human CD3 (BioLegend), anti-human TCR αβ (BD Pharmigen), iTag MART-1/HLA-A2 tetramer, anti-human HLA-A/B/C (BioLegend), or anti-human CD34 (BioLegend) as recommended by the manufacturer. Flow cytometry data were analyzed with FlowJo.
Cytokine release assays
TCR-transduced Jurkat 76 and JX-17 cells were measured for antigen reactivity by co-culturing with peptide-pulsed T2 cells. In brief, T2 cells were pulsed with increasing concentrations (1 nM to 100 μM) of cognate peptide for 2 hours at 37 °C, 5% CO2 then mixed at a 1:1 ratio of responder and stimulator cells (1 × 105) in 96-well, U-Bottom tissue-culture plates in 200 μL of complete medium. PMA at 25 ng/mL was added to Jurkat 76 cells to increase sensitivity to stimulation. Co-culture supernatants were collected after 20 hours and assayed for IL-2 cytokine release by ELISA (BioLegend). Stimulation of TCR-transduced Jurkat 76 cells with anti-CD3 antibodies was performed with the antibody OKT3 at 1 μg/mL. Measurements were performed in triplicate with values reported as the average ± one standard deviation.
TCR 2D affinity measurements
The 2D affinities of cultured Jurkat 76 cells expressing full-length DMF5WT, DMF5AW or S3–4 were measured using the previously characterized 2D micropipette adhesion frequency assay27, 28. Briefly, the adhesion frequency between the receptor (TCR or S3–4 on Jurkat 76 cells) and the ligand (biotinylated peptide/MHC coated onto human RBCs) aspirated onto opposing micropipettes was observed using an inverted Zeiss microscope. Using an electronically controlled piezoelectric actuator the T cell and pMHC coated RBCs were brought into contact and separated 50 times while the contact area (Ac) and time were kept constant. Following retraction of the T cell, adhesion (binding of TCR-peptide/MHC) was observed as a distention of the RBC membrane, allowing for quantification of the adhesion frequency (Pa) at equilibrium. Surface peptide/MHC (ml) and TCR (mr) densities were determined by flow cytometry along with BD QuantiBRITE PE beads for standardization as previously described29, 30. The relative 2D affinities were calculated using the equation: AcKa = −ln [1-Pa(1)]/mrml, with the reported value determined as the geometric mean of the data. Peptide/MHC used for the 2D measurements incorporated mutations in the α3 domain to abrogate CD8 binding31. Biotinylation of HLA-A2 was performed as follows: 100 μM peptide/MHC complexes expressing AviTag sequence were incubated in approximately 1 mL PBS supplemented with 20 μL of 100 mM ATP, 20 μL of 50 μM BirA enzyme, 5 μL of 1M MgCl2, and 3 μL of 50 mM D-biotin. Samples were incubated at 30 °C for 1 hour with shaking. Additional BirA and biotin were then added and the mixture incubated for an additional hour. Samples were purified from excess biotin by size-exclusion chromatography.
Deep mutational scanning
The design and creation of single codon libraries was performed as described previously8. Briefly, S3–4 was cloned as a single chain (Vβ-linker-Vα) for yeast display, and single codon libraries were created using degenerate NNK primers and overlap extension PCR. All single-codon libraries were pooled and added to digested PCT302 vector and transfected into yeast, creating a library of independent transformants via homologous recombination. The estimated library size was 3.8 × 105, which exceeded the potential diversity of the library by greater than 200-fold. This library was then stained and sorted via FACS with peptide/HLA-A2 tetramer at a concentration of 5 nM. The top 1% for each staining condition were collected. DNA from sorted and unsorted libraries were isolated per manufacturer’s protocol (Zymo Research). Single-chain TCR DNA was then amplified in two rounds. The first round added necessary overhangs for Illumina sequencing; the second added adapters for annealing to MiSeq chip. Enrichment ratios were calculated using modified scripts from Enrich software32. Enrichment ratios were calculated based on sorted/naïve sequence frequencies. For the G102α residue within the CDR3α domain, substitutions to Thr, Asn, Gln, His, Lys, Pro, Tyr, Trp, Ile, Leu, Met, Stop were depleted in the naïve library and ratios could not be calculated. Residues were identified as depleted in the scan. For residues in which sorted mutants were completely depleted, a representative average negative value based on stop codon enrichment was used.
RESULTS
S3–4 binds HLA-A2 independent of which peptide is presented
The S3–4 single-chain TCR variant (scTv) of the A6 TCR was generated in a directed evolution experiment designed to shift the peptide specificity of the well-characterized A6 TCR from the viral Tax antigen to the MART-1 tumor antigen7. The library from which S3–4 evolved incorporated mutations in three CDR loops, utilizing both degenerate and “either/or” mutations in CDR1α, CDR3α, and CDR3β. S3–4 possesses six mutations distributed across these three loops (Fig. S1). The three mutations found in the CDR1α loop in S3–4 are not encoded in any known TCR Vα gene33.
To characterize S3–4, we generated recombinant S3–4 scTv and tested its ability to bind different peptide/HLA-A2 complexes using surface plasmon resonance (SPR). S3–4 bound the Tax peptide (LLFGYPVYV) presented by HLA-A2 with an affinity (KD) of 9 μM, approximately 4-fold weaker than the parental A6 TCR (Fig. 1A). Changing proline 6 to alanine in the Tax peptide resulted in a slightly stronger KD of 5 μM (Fig. 1B). Strong recognition of the Tax P6A variant is notable, as the substitution weakens binding by the parental A6 TCR to nearly undetectable levels34. S3–4 also bound other peptide/HLA-A2 complexes, recognizing HLA-A2 presenting the WT1 (RMFPNAPYL), HCV NS3 (KLVALGINAV), and MART-1 (ELAGIGILTV) peptides with KD values ranging from 5 to 15 μM (Fig. 1C–E). Excluding the Tax and Tax P6A variant, these peptides are divergent in sequence and include both nonamers and decamers that adopt different paths through the HLA-A2 peptide binding groove11, 13, 35. S3–4 thus recognizes HLA-A2 independently of the bound peptide, albeit with a small (≤3-fold) variation in KD.
Figure 1.
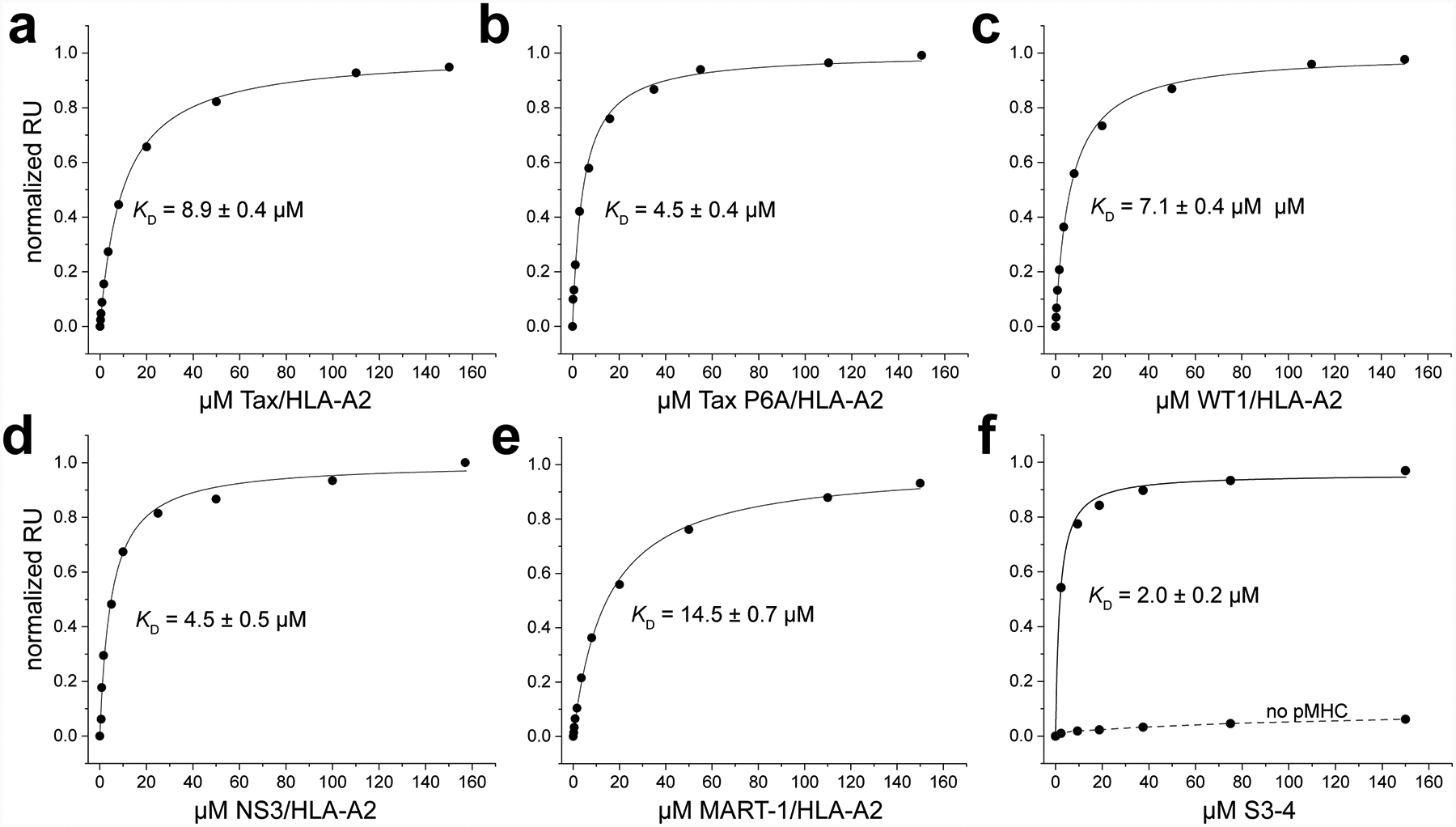
S3–4 binds peptide/HLA-A2 complexes independently of the bound peptide and does not compete for the HLA-A2 peptide binding groove. A-E) Equilibrium titrations with purified protein performed by SPR for different peptide/HLA-A2 complexes binding immobilized S3–4. Binding affinities and associated error are indicated in each inset. The results indicate relatively high affinity binding regardless of peptide, with the small variation resulting from an allosteric effect with different peptides. F) Blocking the peptide and peptide-binding domain of the Tax/HLA-A2 complex using the nM-binding high affinity c134 variant of the A6 TCR does not hinder binding of S3–4. Saturating amounts of Tax/HLA-A2 was injected over an A6 c134 surface to create a traditional TCR-peptide/HLA-A2 complex immobilized on the surface, followed by a normal titration of S3–4, which yielded a KD of 2 μM as indicated. The dashed line indicates responses measured without first injecting Tax/HLA-A2 over the A6 c134 surface, confirming that S3–4 binds the peptide/MHC.
Naturally occurring TCRs bind peptide/MHC complexes “head on” with respect to the peptide binding groove, interacting with both the peptide and binding groove α helices1. To assess whether the mutations in S3–4 altered how the receptor binds, we designed an experiment in which the traditional binding interface was blocked. The c134 variant of A6 binds Tax/HLA-A2 traditionally but with an affinity of 4 nM (approximately 500-fold stronger than wild type A6) and a half-life in excess of one hour36. We coupled A6 c134 to a SPR sensor chip and followed this with an injection of saturating amounts of Tax/HLA-A2. This yielded a peptide/HLA-A2 sensor surface with the peptide and MHC peptide binding groove blocked by A6 c134. We then performed a typical binding experiment by titrating in increasing concentrations of S3–4. Clear binding was observed, yielding a KD of 2 μM (Fig. 1F). Negligible binding was detected in a control experiment in which S3–4 was titrated in alone over the A6 c134 surface. Inability to compete for binding with a conventionally-binding TCR was also observed by yeast surface display, in which S3–4 did not compete with a high affinity TCR specific for the binding WT1/HLA-A237 (Fig. S2, and see below). These experiments demonstrate that the loss of peptide specificity with S3–4 results from a binding orientation that places the TCR away from the MHC peptide binding groove and still permits engagement by normal TCRs.
The small variation in S3–4 affinity toward different peptide/HLA-A2 complexes is consistent with our previous studies demonstrating that different peptides can tune the high frequency dynamics of class I MHC proteins38. These variations in peptide-MHC motions would be expected to translate into subtle allosteric effect on proteins that bind HLA-A2 away from the peptide binding groove38, 39. Similar allosteric effects have been observed with the Ly49C NK receptor, which shows peptide dependent binding to class I MHC despite binding away from the peptide binding groove40.
S3–4 binds HLA-A2 on the side of the protein avoiding the peptide binding groove
To investigate the exact way S3–4 binds HLA-A2, we crystallized and determined the structure of the S3–4 scTv bound to HLA-A2 presenting the Tax peptide to a resolution of 3.1 Å (Table 1). Consistent with the binding data indicating a loss of peptide specificity and an alternative docking site, the structure revealed that S3–4 binds on the “side” of the HLA-A2 protein, rotated approximately 90° from the usual TCR binding site on top of the peptide binding groove (Fig. 2A). The S3–4 binding site is unique to known class I MHC co-receptors: it is on the opposing face (rotated 180°) from where the CD8 coreceptor and the LIR-1 NK receptor bind HLA-A2 and opposite from where the Ly49C NK receptor binds murine class I proteins40 (Fig. 2B). It is rotated 120° away from where a recently described class of γδ TCRs binds the non-classical MHC protein MR-16. This unique site is composed of non-polymorphic residues on the HLA-A2 α3 domain, the β2-microglobulin subunit (β2m), and the polypeptide connecting the end of the α2 helix to the floor of the peptide binding groove (Fig. 2C). The distributed nature of the binding site across both the α3 domain and β2m explains why S3–4 remains specific for class I MHC, as seen in the original yeast display experiments and our control titrations (e.g., Fig. 1F).
Table 1.
X-ray data collection and refinement statistics.
| S3–4-Tax/HLA-A2 | |
|---|---|
| Data Collection | |
| Space group | P 61 |
| Unit cell dimensions | |
| a, b, c (Å) | 236.65, 236.65, 63.15 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 50.00–3.10 (3.15–3.10) |
| Rmerge | 0.276 (1.978) |
| I/σI | 14.7 (1.7) |
| Completeness (%) | 100 (100) |
| Total reflections | 1385353 |
| Unique Reflections | 37072 (1889) |
| Redundancy | 11.0 (10.2) |
| Refinement | |
| Resolution (Å) | 44.72–3.14 |
| Rwork/Rfree | 0.189/0.239 |
| No. atoms / AU | |
| Protein | 9752 |
| B-factors (Å) | 103.0 |
| R.M.S deviations | |
| Bond length (Å) | 0.002 |
| Bond angles (°) | 0.468 |
| Ramachandran favored (%) | 97.15 |
| Ramachandran outliers (%) | 0.08 |
| PDB identifier | 6UZ1 |
Figure 2.
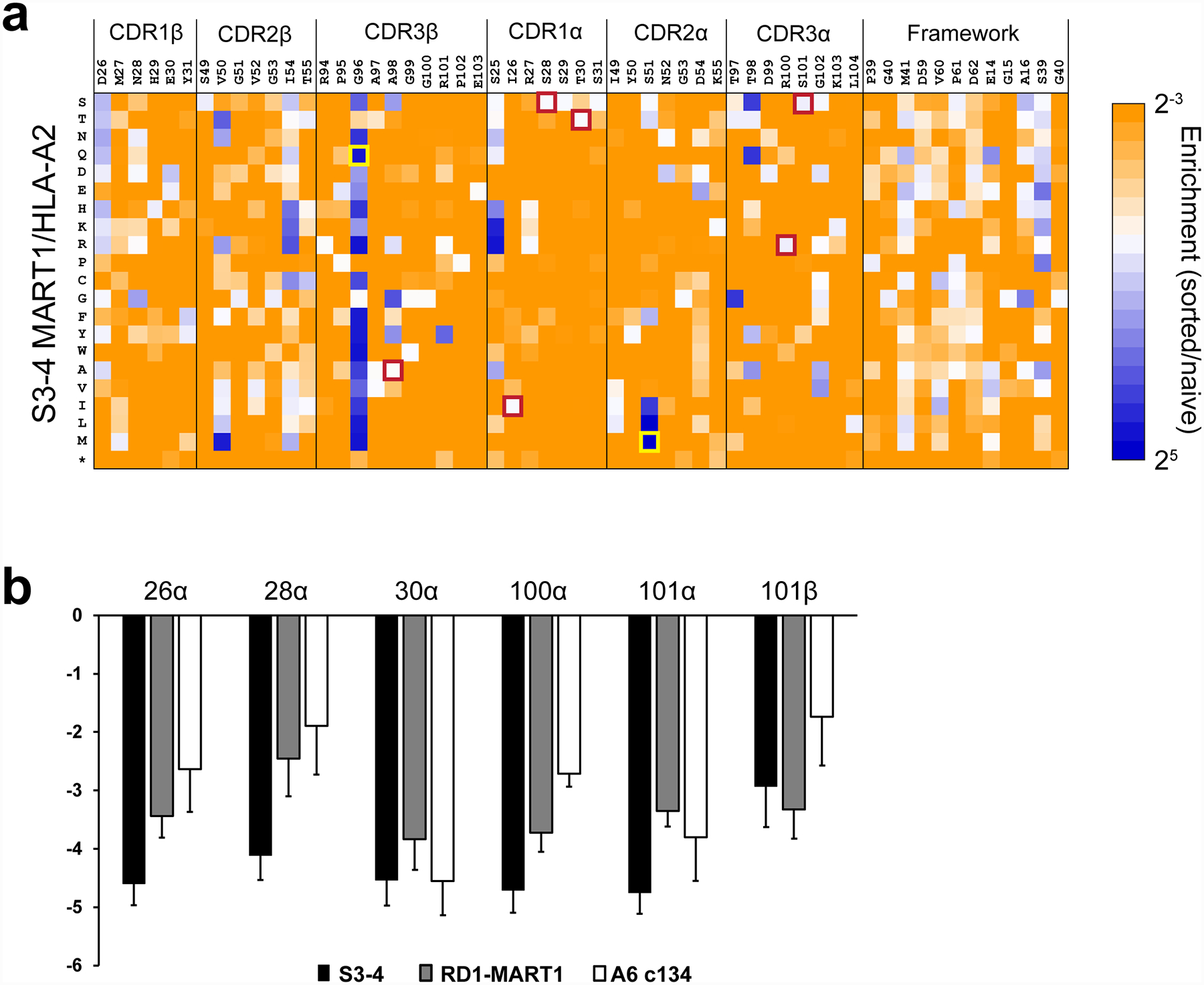
S3–4 binds HLA-A2 on the side, opposite the binding site of the CD8 coreceptor. A) Overview of the complex between S3–4 and Tax/HLA-A2. Electron density for the peptide and S3–4 from a simulated annealing/composite OMIT map is shown, contoured at 1σ. The Vα domain of S3–4 is in orange, the Vβ domain in green, the HLA-A2 heavy chain is grey, and β2m is yellow. This color scheme is maintained throughout the figure. B) As in panel A but also showing the binding sites of the CD8 coreceptor (purple) and the LIR-1 NK receptor (magenta). C) Detailed view of the S3–4 binding site on the side of HLA-A2, composed of residues in the α3 domain, β2m, and the polypeptide connecting the α2 helix to the floor of the peptide binding groove. Contacted amino acids are in red; β2m is colored yellow.
The S3–4-Tax/HLA-A2 structure allowed us to examine how the six mutations altered binding of the TCR. Two major contributions are the introduction of new interactions with the side of HLA-A2 and the elimination of steric or electrostatic clashes that would occur with the new positioning. One new set of interactions arises from the mutation of Ser100 in CDR3α to arginine. The new arginine extends into a cleft formed between the end of the HLA-A2 α2 helix and the α3 domain, interacting with Thr178, Arg181, and Tyr209 of HLA-A2 (Fig. 3A). These interactions are facilitated by the mutation of Trp101α to serine, as the presence of the larger tryptophan would overlap with the side chain of Arg181. The side chain of the new serine at 101α does not interact with HLA-A2, but given its smaller size likely facilitates a conformational change in CDR3α relative to its conformation in the wild type A6 TCR that helps with the insertion of Arg100α into the cleft41. In the CDR1α loop, the mutation of aspartic acid to isoleucine at position 26 allows hydrophobic packing with Pro50 in the HLA-A2 heavy chain, but also removes electrostatic repulsion that would exist between the original aspartic acid and Glu53 of HLA-A2. Consequently, Glu53 of HLA-A2 now hydrogen bonds with the CDR1α loop backbone (Fig. 3B).
Figure 3.
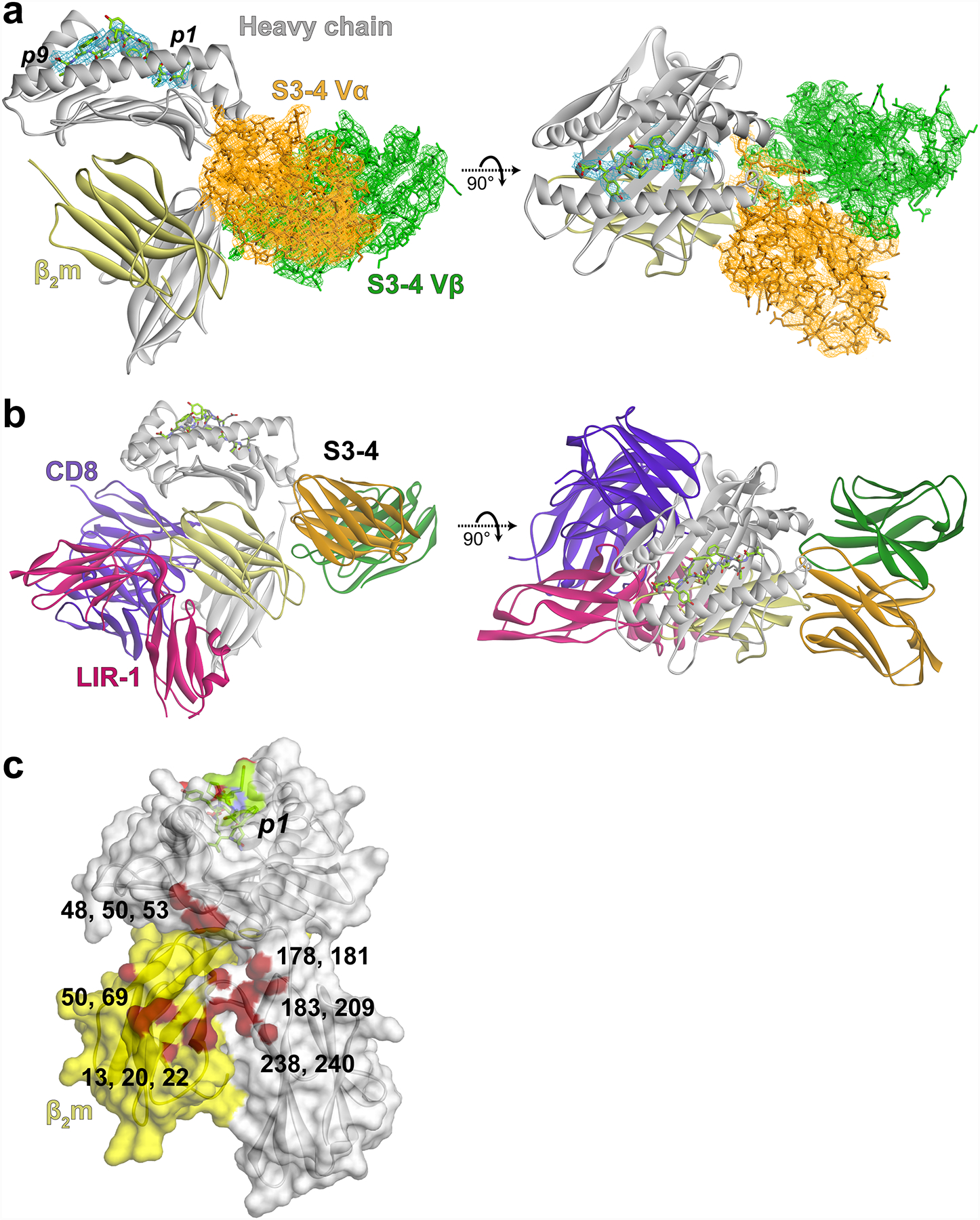
New and altered molecular interactions in the interface between S3–4 and HLA-A2. A) The new arginine at position 100 in CDR3α extends into a cleft on the side of HLA-A2, interacting with Thr178, Arg181, and Tyr209. These interactions are facilitated by replacement of the bulky Trp101α with serine. The S3–4 Vα domain is orange, the Vβ domain green, and the HLA-A2 heavy chain is gray. The wild-type A6 TCR, modeled by superimposing its variable domains onto those of S3–4, is colored light blue. This color scheme is maintained throughout the figure. B) Mutation of Asp26 in CDR1α to isoleucine allows hydrophobic packing with Pro50 of HLA-A2 and removes electrostatic repulsion that would exist between the original aspartic acid and Glu53 of HLA-A2, which interacts with the backbone of Arg27 and Ser28 in CDR1α. C) Mutation of Leu98 in CDR3β to alanine and an associated conformational change allows the loop to avoid steric clashes with Asp183 and Lys186 of HLA-A2. D) In addition to forming new interactions and allowing the TCR to avoid steric and electrostatic clashes, the mutations in S3–4 would destabilize the normal TCR binding mode. Replacement of Gln30 in CDR1α, for example, would remove key hydrogen bonds between the TCR and the peptide backbone. Replacement of Ser100 in CDR3α with arginine would result in clashes with the center of the peptide.
In the TCR β chain, the CDR3β loop is sandwiched between the end of the HLA-A2 α1 helix, the α3 domain, and β2m. The mutation in CDR3β substitutes leucine for an alanine at position 98. The smaller alanine does not directly interact with HLA-A2, but instead allows the loop to avoid steric clashes with the side chain of Asp183 of HLA-A2 (Fig. 3C). The CDR3β loop also undergoes a conformational change relative to the conformation seen in the wild type TCR41, in this case driven by the need to avoid a steric clash with Lys186.
We previously evaluated the binding of the high affinity A6 c134 variant to Tax/HLA-A2 using deep mutational scanning8. Retrospective analysis of this data indicates that each of the six mutations in S3–4 would weaken binding of the A6 TCR to Tax/HLA-A2. Examining these mutations in the context of the A6-Tax/HLA-A2 structure is instructive. Mutating Gln30 in CDR1α to threonine, for example, removes two hydrogen bonds between the TCR and the backbone of the peptide (Fig. 3D). Previous experiments revealed these hydrogen bonds contribute significantly to the binding of the wild-type A6 TCR42, and they are common in TCR structures with HLA-A243. Mutating Ser100 in CDR3α to arginine would not remove interactions but instead result in steric clashes with the center of a class I MHC-presented peptide. Thus, in addition to introducing new interactions and eliminating clashes, the mutations in S3–4 destabilize the traditional TCR binding mode.
As indicated in Fig. S1, the mutations in S3–4 are distributed across CDR1α (3 mutations), CDR3α (2 mutations), and CDR3β (1 mutation). As the mutations in the hypervariable loops appear to play prominent roles in binding (e.g., Ser100α to arginine as shown in Fig. 3A), we were curious as to whether the hypervariable loop mutations alone could facilitate altered TCR binding. Such an outcome would be of interest, as these or similar hypervariable CDR3 loop sequences could arise naturally, contributing to the non-productive development of unusually-binding TCRs. We thus generated a variant of S3–4 that possessed only the CDR3α and CDR3β hypervariable loop mutations (Ser100α→Arg, Trp101α→Ser, and Leu98β→Ala). Although we were able to produce this protein, we were unable to detect binding to different peptide/HLA-A2 complexes by SPR, including the complex with the Tax peptide (Fig. S3A). We also generated a variant which possessed only the mutations in the CDR1α germline loop (Asp26α→Ile, Gly28α→Ser, and Gln30α→Thr). In the background of the high affinity A6 c134 variant, the CDR1α germline variant bound the Tax/HLA-A2 complex 20-fold weaker than A6 c134 and produced no detectable binding to the MART-1/HLA-A2 complex, thus indicating a traditional TCR binding mode (Fig. S3B). Together, these results indicate that the germline and hypervariable loop mutations act cooperatively to destabilize the traditional TCR binding mode and imbue a “sideways” binding mode, and that the hypervariable loop mutations are necessary but not sufficient for altered binding.
Deep mutational scanning of S3–4 confirms the collective action of mutations
To obtain a comprehensive view of how individual residues in S3–4 CDR contribute to non-canonical binding, we performed deep mutational scanning of the S3–4 protein. As we demonstrated previously with several TCRs8, 44, 45, deep mutational scanning permits the analysis of how mutations to each of the 20 amino acids in various positions throughout a TCR impact binding. To compare S3–4 with these other conventional, peptide-specific TCRs, we used yeast surface display of the S3–4 scTv to generate single codon libraries across all CDR loops, and, as controls, 12 framework residues located in exposed regions of the variable domains. The S3–4 libraries were selected by binding to four different peptide/HLA-A2 tetramers, followed by sorting of the top 1% positive populations. Each of these populations was subjected to deep sequencing and the number of sequences containing each single substitution was compared to the unselected libraries to assess if the substitution yielded similar, reduced, or higher expression levels compared to the unselected library (presented as enrichment ratios, log2). Overall, we assessed 1083 single amino acid variants of S3–4 (57 positions × 19 non-wild-type amino acids), with the four different probes: Tax/HLA-A2, MART-1/HLA-A2, WT1/HLA-A2, and Survivin/HLA-A2.
Sequence enrichment data obtained using the MART-1/HLA-A2 complex are shown in Fig. 4A, with data for other peptide/HLA-A2 complexes in Fig. S4A. Notably, the data were nearly identical regardless of which peptide was used. This is clear through visual comparison of each dataset, or by a direct comparison of the enrichment ratios, which correlated well between datasets (Fig. S4B). The near-identity between the data regardless of peptide is consistent with the structural data and the peptide-independence observed in the direct binding experiments.
Figure 4.
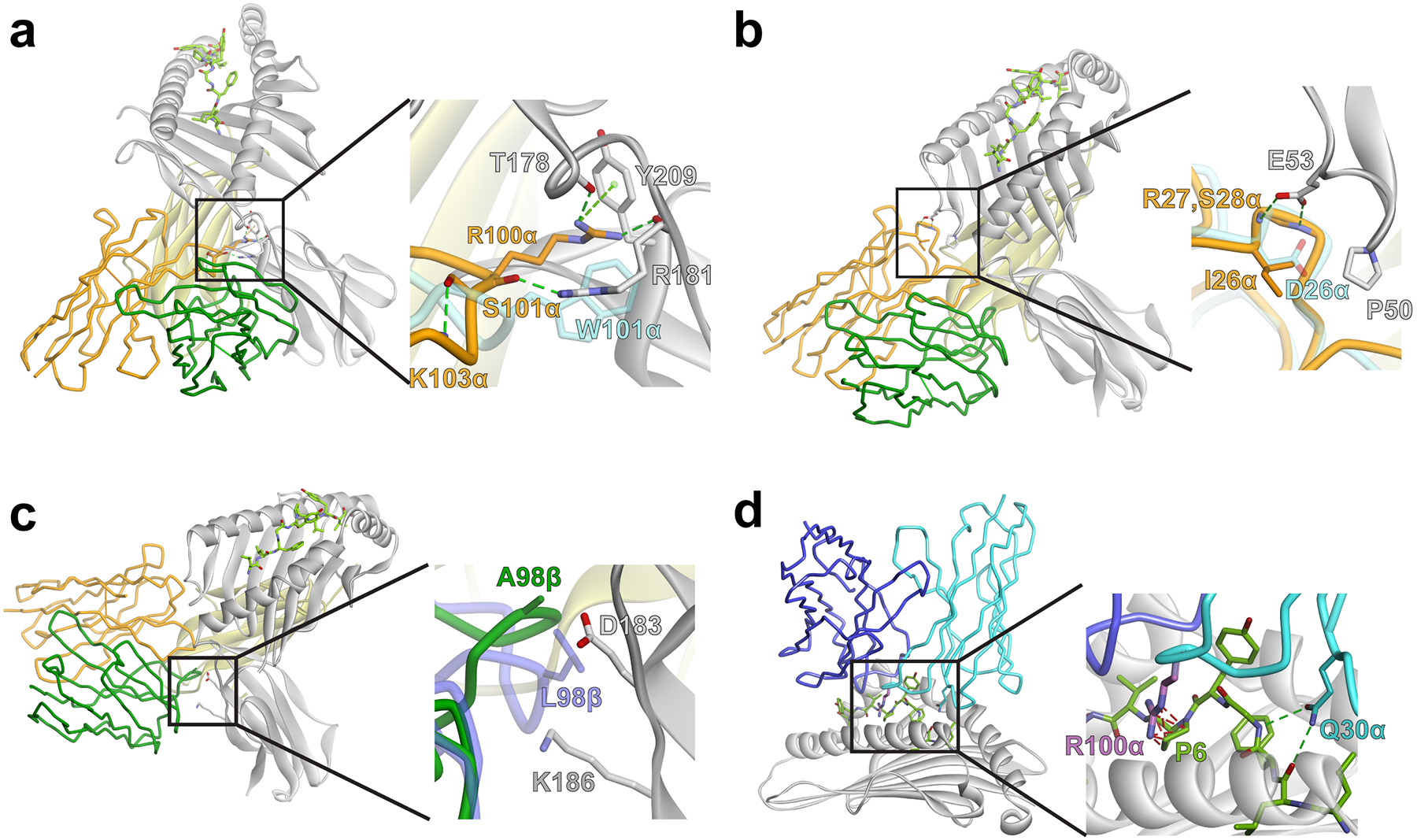
Deep mutational scanning of S3–4 demonstrates the collective importance of the six mutations in conferring peptide-independent binding. A) All single amino acid substitutions in the positions indicated were expressed in a yeast display library of S3–4. Mutants that bound with the top 1% fluorescence signal for MART-1/HLA-A2 at 5 nM were collected by FACS. Sequences were then analyzed using next-generation sequencing. The sequence enrichment compared with the naive library was calculated as a log2 ratio. Enrichment ratios are color-coded from ≤ 2−3 (orange) to ≥ 25 (blue). Stop codons are denoted with an asterisk. For the G102α residue, substitutions to T, N, Q, H, K, P, Y, W, I, L, M, Stop were completely depleted in the naïve library and therefore enrichment values could not be calculated; these residues are identified as negatively enriched (orange) in the scan. The cells boxed in red indicate those amino acids present in S3–4 and, for all positions except 99β, reflect the most frequently observed amino acid. The cells boxed in yellow indicate mutations incorporated to generate a higher affinity version of S3–4. B) Average enrichment values for all non-wild type substitutions were calculated and compared among S3–4, RD1-MART1, and the A6 c134 TCR (RD1-MART1 and A6 c134 enrichment values from previous data8). Error bars represent standard deviation of the average enrichment.
In examining the deep mutational scanning data, we found that S3–4 variants containing substitutions for each of the mutations seen in S3–4 (Fig. S1) were highly depleted. Indeed, for five out of six of the mutations in S3–4 (I26α, S28α, T30α, R100α, and S100α), only the mutated amino acid was well tolerated (Fig. 4A). We calculated the average enrichment values of all 19 substitutions at each of these six positions and compared them to the deep mutational scanning data previously collected for two conventional binding TCRs: the A6 c134 variant described above and the specificity switched RD1-MART1 variant8 (Fig. 4B). By this analysis, while the wild-type residues of A6 c134 and RD1-MART1 were significant for binding their respective ligands, S3–4 showed greater depletion values for substitutions at four of the six positions: Ile26α, Ser28α, Arg100α, and Ser101α. This data confirms that the unique binding geometry and peptide-independence of S3–4 arises from the collective action of the mutations distributed across these CDR loops. The important role of the mutations in CDR1α in S3–4 is particularly highlighted by the number and magnitude of substitutions with negative enrichment values compared to the other CDRs and the framework residues (Fig. 4A and Fig. S4).
Generation of a higher affinity S3–4 variant
Previously, we showed that deep mutational scanning data could be used to generate high affinity TCR variants based on those mutations that yielded the most positive enrichment values44, 45. We tested if this were possible with S3–4, selecting highly enriched mutations to combine with those already present in S3–4. Yeast display titrations indicated that combining Ser51→Met in CDR2α and Gly96→Gln in CDR3β with the six S3–4 mutations indeed led to improved affinity. We produced this high affinity version (termed S3–4MQ) recombinantly and confirmed enhanced binding by SPR, measuring a KD of 42 nM toward Tax/HLA-A2 in a kinetic titration, nearly 200-fold stronger than “wild type” S3–4 (Fig. S5A, B).
T cells expressing full length S3–4 and its high affinity do not trigger in a physiological context
Physiological T cell signaling is dependent on TCR engagement of peptide/MHC complexes, and there is evidence that unusual receptor binding geometries can inhibit or otherwise alter signaling4, 5. We were thus interested in whether the binding geometry of S3–4 would impact TCR signaling. We generated full length versions of the “wild type” S3–4, as well as the high affinity S3–4MQ variant, in retroviral vectors to express the α and β chains46. Jurkat cells were efficiently transduced as evidenced by staining for CD3 and the TCR Cβ domain (Figs. 5A, B). Interestingly, S3–4 and S3–4MQ transduced cells could not be stained by HLA-A2 tetramers loaded with the MART-1 tumor antigen, although control cells expressing the DMF5 TCR that binds MART-1/HLA-A2 with an affinity near that of S3–4 were efficiently stained (Fig. 5C).
Figure 5.
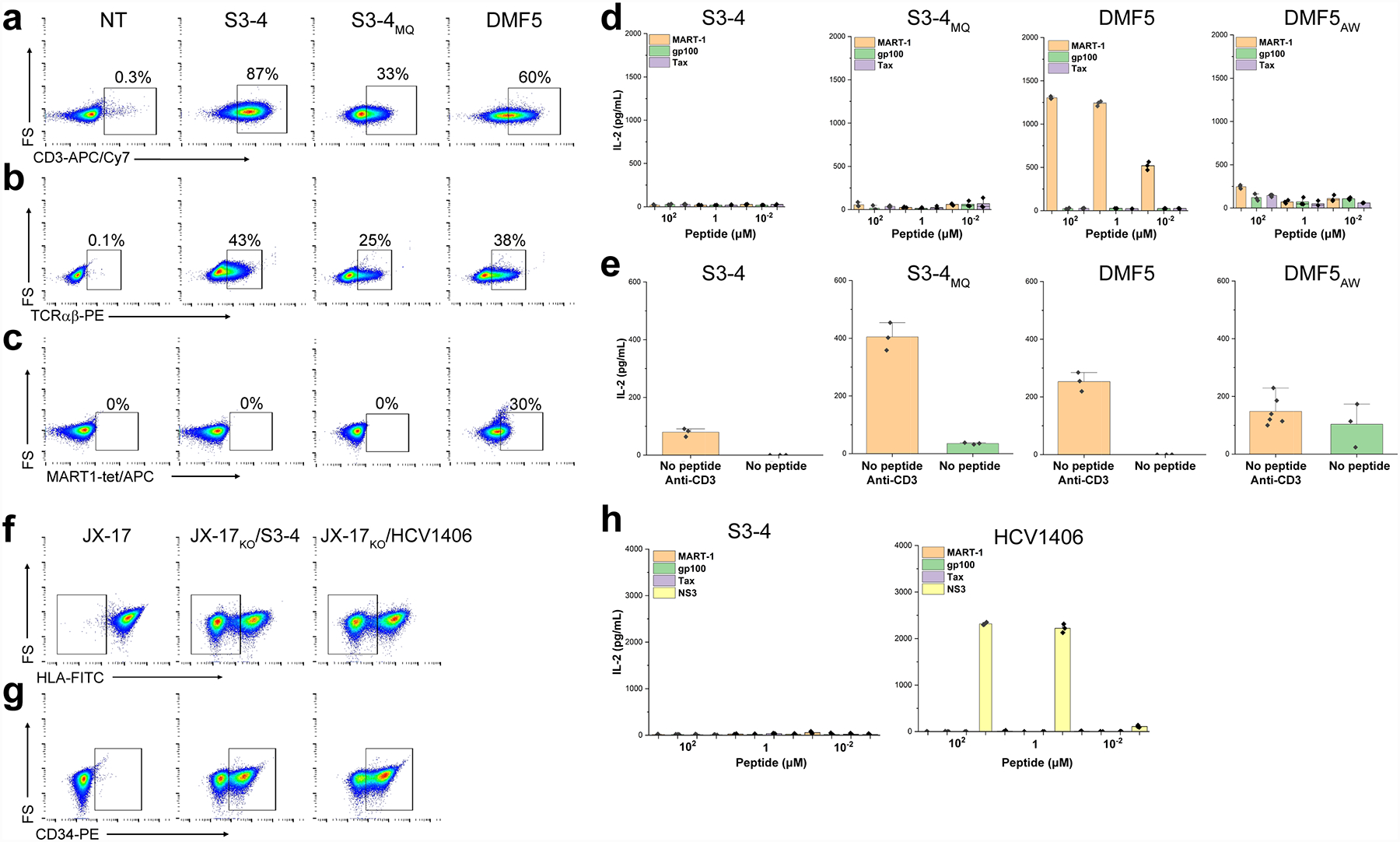
S3–4 does not support normal TCR signaling. A, B) Jurkat cells transduced with “wild type” S3–4 or the high affinity S3–4MQ version are efficiently stained by the anti-CD3 UCHT1 antibody (panel A) and the anti-human TCR αβ antibody IP26 (panel B), indicating TCR surface expression. C) Jurkat cells expressing S3–4 or S3–4MQ are not stained by 4 μM of MART-1/HLA-A2 tetramers unlike cells expressing DMF5. D) Co-culturing HLA-A2+ T2 cells with S3–4 or S3–4MQ expressing Jurkat cells with increasing concentrations of the MART-1, gp100, or Tax peptide does not result in T cell signaling as measured by IL-2 production, although signaling is observed with DMF5-transduced cells co-cultured with the MART-1 peptide. The millimolar affinity DMF5AW variant yields negligible IL-2 signaling. E) Although Jurkat cells expressing S3–4 or S3–4MQ are not triggered by peptide/HLA-A2 complexes, they can be triggered by the anti-CD3 antibody OKT3 when co-cultured with T2 cells in the absence of exogenous peptide (left), as can cells expressing the DMF5 (middle) and DMF5AW TCRs (right). F) Knockout of β2m in JX-17 Jurkat variants leads to heterogenous cell populations in which approximately 50% of the cells cannot be stained with the anti-HLA antibody W6/32. Left panel shows JX-17 cells prior to β2m knockout. Middle and right panels show cells after β2m knockout and transduction with S3–4 and the HCV1406 TCRs, with the boxed region showing cells lacking HLA-A2. G) Confirmation of JX-17 cell TCR transduction by staining for the transduction marker CD34t. H) Co-culturing HLA-A2+ T2 cells and β2m knockout JX-17 cells expressing S3–4 with increasing concentrations of the various peptides does not result in T cell signaling as measured by IL-2 production although the same experiment yields efficient signaling when β2m knockout JX-17 cells expressing the HCV1406 TCR are co-cultured with the HCV NS3 peptide. NT, non-transduced negative control.
The inability to stain with tetramers suggests that the S3–4 binding geometry presents a structural or steric limitation such that the TCR is unable to access its binding site on HLA-A2 when confined to a cell surface. However, there are well-documented instances of functional TCRs which cannot be stained with tetramer47, 48. We thus asked if the Jurkat cells expressing S3–4 or S3–4MQ recognized peptide/HLA-A2 complexes in functional experiments. Co-culturing S3–4 or S3–4MQ positive Jurkat cells with antigen presenting HLA-A2+ T2 cells and increasing concentrations of the MART-1, gp100, or Tax peptide did not lead to cytokine production. Reflecting the tetramer staining experiments, Jurkat cells expressing the DMF5 TCR responded in a dose-dependent fashion to the MART-1 peptide (Fig. 5D). Although S3–4 or S3–4MQ did not signal in response to peptide/HLA-A2, the assembled TCR complex on the Jurkat cell surface was functionally competent, as shown by the production of IL-2 using the anti-CD3 antibody OKT3 (Fig. 5E).
One potential concern is that S3–4 could engage self-MHC on the Jurkat cell surface in cis or trans and lead to aberrant functional results. If this were the case, we would expect to see high level basal cytokine secretion in S3–4 or S3–4MQ transduced Jurkats compared to DMF5 transduced Jurkats, which was not observed, despite the cells being functionally competent as demonstrated by OKT3 stimulation. Nonetheless, we repeated these experiments in the Jurkat variant JX-17 which used CRISPR-based knock-out of β2m expression with sgRNA25. Deploying this system led to a population of cells in which approximately 50% of the cells lacked β2m and thus cell-surface expression of class I MHC (Fig. 5G). The mixed JX-17 population would result in approximately 32% of cells demonstrating the HLA-A2−, S3–4+ phenotype, a reduced level, but still sufficient to detect cytokine production49. The results of these experiments were the same as with the parental Jurkat cells: although both S3–4 and a control TCR were efficiently expressed (Fig. 5G), only cells expressing the control TCR (in this case, the HCV1406 TCR that recognizes the HCV NS3 peptide) responded to peptide in functional experiments measuring IL-2 production (Fig. 5H). We conclude that, despite binding peptide/HLA-A2 with high solution affinities that typify CD8-independent T cell recognition50, in a physiological context neither S3–4 nor the higher affinity S3–4MQ variant is capable of being functionally triggered by peptide/MHC.
Lack of T cell triggering is attributable to steric hindrance from the S3–4 binding site on HLA-A2
Although S3–4 does not support normal T cell signaling, the underlying mechanism is not immediately apparent. To study this, we measured the 2D affinity between S3–4 and HLA-A2. 2D affinity was determined by measuring the interactions between TCR-transduced Jurkat cells and red blood cells displaying peptide/HLA-A2 complexes with a micropipette assay27. S3–4 recognized HLA-A2 presenting the HCV NS3 peptide with a 2D affinity of 2.3 × 10−5 AcKa μM4. (Fig. 6A). For comparison, we also probed the interaction between the DMF5 TCR and HLA-A2 presenting the MART-1 tumor antigen. This interaction proceeded with a much stronger 2D affinity of 2.7 × 10−3 AcKa μM4, approximately two orders of magnitude tighter than that measured for S3–4 binding NS3/HLA-A2. These 2D affinities do not track with their solution affinities: the binding of S3–4 to NS3/HLA-A2 and the binding of DMF5 to MART-1/HLA-A2 both proceed with KD values close to 5 μM (Fig. 1D and ref.14). Thus, when compared to DMF5 binding MART-1/HLA-A2, S3–4 binds NS3/HLA-A2 approximately 100-fold weaker in a 2D setting despite binding the two ligands almost identically in solution (Fig. 6A).
Figure 6.

S3–4 binds very weakly in 2D and fails to signal likely because of steric incompatibility between opposing cell membranes. A) Measurements of 2D affinity between S3–4 and NS3/HLA-A2, the wild type DMF5 TCR and MART-1/HLA-A2, and the millimolar binding DMF5AW variant and MART-1/HLA-A2. B) Schematic showing how, in the absence of extreme membrane or protein re-organization, S3–4 binding to a peptide/MHC complex would lead to overlap between opposing cell membranes. The model of membrane-bound HLA-A2 was generated by conjoining the HLA-A2 ectodomain to a single membrane spanning α helix, based on prior findings that class I MHC proteins extend away from the membrane surface to display peptides to surveilling T cells53.
We next designed a mutant of the DMF5 TCR that binds MART-1/HLA-A2 with similar 2D affinity as S3–4 for peptide/HLA-A2. We combined previously studied mutations at distal sites shown to act independently in the DMF5 TCR51, mutating tyrosine 50 in the α chain to alanine and leucine 98 in the β chain to tryptophan, referred to as DMF5AW. DMF5AW bound to MART-1/HLA-A2 with a 2D affinity of 3.1 × 10−5 AcKa μM4, near the 2D affinity measured for S3–4 binding NS3/HLA-A2 (Fig. 6A). Consistent with S3–4 Jurkat data, DMF5AW did not recognize MART-1/HLA-A2 in a functional experiment measuring IL-2 production (Fig. 5D; Fig. S6). We were unable to detect binding of DMF5AW to MART-1/HLA-A2 in a traditional SPR experiment (Fig. S3C), and MART-1/HLA-A2 tetramers stained DMF5AW weakly compared to wild type (Fig. S6). In contrast, S3–4 bound to NS3/HLA-A2 with a solution affinity in the low micromolar range (Fig. 1D). These results show that, despite binding well in solution, in the confines of a cell-cell interface, S3–4 has hindered access to HLA-A2, translating into a very weak 2D affinity and a corresponding lack of detectable signaling.
Mapping the structure of S3–4 bound to Tax/HLA-A2 into the structure of the recently determined cryoEM structure of the entire TCR/CD3 complex52 allowed us to conceptualize a mode of hindrance between S3–4 and HLA-A2 that is consistent with the data. It has been previously shown that class I MHC proteins extend away from the antigen presenting cell surface53. We generated a simple model reflecting this by joining the structure of the HLA-A2 ectodomain to the structure a single membrane spanning α helix matching the length of the helix in class I MHC proteins. This modeling showed that, barring extreme membrane bending or reorganization of the TCR/CD3 complex, achieving the S3–4 binding orientation would be prohibitive due to interference between cell membranes (Fig. 6B). Owing to the size of the cells relative to the protein, tilting HLA-A2 on the membrane-connecting linker to increase the accessibility of the binding site would not relieve the steric hindrance.
DISCUSSION
Naturally occurring TCRs bind peptide/MHC complexes and interact directly with the peptide. Although an individual TCR can recognize a large number of peptides, often structurally related, they still show specificity for a subset of peptides that are structurally and physically compatible with the TCR binding site9. Here we describe a S3–4, an engineered TCR variant generated via molecular evolution that binds well to the class I MHC protein HLA-A2 regardless of peptide. This curious and unusual loss of specificity is achieved by the molecule binding to the “side” of HLA-A2, opposite the binding site of the CD8 coreceptor. The altered binding mode is a product of the collective action of mutations in germline and hypervariable CDR3 loops. The mutations act by introducing new interactions with the MHC protein, eliminating steric and electrostatic clashes that otherwise would occur, and destabilizing the traditional TCR binding mode.
In some instances, TCRs that bind with geometries that deviate from the traditional binding mode have shown altered signaling properties4, 5. In those cases the mechanism underlying the altered signaling is not clear, but has been hypothesized to involve influences on the structure, stability, and signaling dynamics of a multi-protein T cell signaling complex, which includes the TCR and its bound peptide/MHC as well as the associated CD3 signaling modules, coreceptor, and possibly higher order TCR-peptide/MHC oligomers. Here, we found that S3–4 could not trigger and thus did support normal signaling, nor did a S3–4 variant that bound in solution with nanomolar-level affinity. The simplest explanation for the loss of signaling is suggested by a divergence between solution and 2D affinity measurements. In solution, S3–4 bound with strong affinity, typical of co-receptor independent anti-viral TCRs50. Yet in 2D format when opposing cells must be brought into proximity, the TCR bound very weakly, resembling a TCR that binds with a solution affinity in the non-stimulatory millimolar range. When the S3–4 complex is modeled into the recently determined intact TCR/CD3 complex52, the constraints between opposing cell membranes suggest a source of steric hindrance that would lead to the observed divergence between solution and 2D affinity, and ultimately the lack of signaling. Although S3–4 was generated by molecular evolution, outside of the constraints of normal immune system development and function, it nonetheless provides an instructive example of how an outlier receptor binding geometry can impact T cell function. We might infer that other instances in which T cell function varies with receptor binding geometry also reflect steric consequences, albeit perhaps in more subtle and sophisticated fashions.
The extent to which MHC-restriction is influenced by co-evolution of TCR and MHC genes has been extensively discussed54–59. Recently, we described how TCR/MHC co-evolution has produced sequences and structures in TCR germline loops that are compatible with the topologies and chemistries of MHC binding grooves60. Our observation that the mutations in the S3–4 CDR1α loop are required for altered binding and are not found in known human Vα genes supports this suggestion. Considering that mutations such as those in the S3–4 hypervariable CDR3 loops could theoretically arise in any naturally occurring TCR, TCR-MHC coevolution and the resulting compatibility thus likely bias against the generation of TCRs with unproductive binding geometries such as seen here.
In summary, we have described S3–4, a molecularly evolved TCR variant that binds HLA-A2 independently of the presented peptide, achieved by a drastic perturbation from the typical TCR binding geometry. The variant does not support signaling, even when high affinity mutations are introduced. S3–4 thus reveals a limit to functionally competent T cell receptor binding geometries. The impacts of the mutations reiterate the importance of TCR-MHC co-evolution and the inherent compatibility that emerges. Furthermore, the small but significant differences in binding among different peptide/HLA-A2 complexes demonstrate how the tuning of class I MHC molecular properties by peptides can allosterically impact protein binding. Lastly, given the conserved nature of the binding site across human class I MHC proteins, together with the high affinity of S3–4 (and S3–4MQ), we suggest that S3–4 may be of use as a molecular chaperone for studying weak complexes, or as a probe for identifying properly assembled class I MHC complexes in solution and on cells.
Supplementary Material
Funding Sources
Supported by grants R35GM118166 from NIGMS/NIH to BMB, R01CA178844 from NCI/NIH to DMK, and R01AI147641 from NIAID/NIH to BDE. JAA and AMR were supported by grants TL1TR001107 and TL1TR002531, respectively, from NCATS/NIH. DTH was supported by grant F30CA180723 from NCI/NIH. X-ray diffraction data were collected at SER-CAT, supported by its member institutions and grants S10RR25528 and S10RR028976 from the NIH. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract number W-31-109-Eng-38.
Footnotes
Supporting Information
Figures S1–S6.
Accession Codes
Coordinates and structure factors were deposited in the Protein Data Bank as entry 6UZ1.
REFERENCES
- [1].Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, and McCluskey J (2015) T Cell Antigen Receptor Recognition of Antigen-Presenting Molecules, Annual Review of Immunology 33, 169–200. [DOI] [PubMed] [Google Scholar]
- [2].Rudolph MG, Stanfield RL, and Wilson IA (2006) How TCRs Bind MHCs, Peptides, and Coreceptors, Annu Rev Immunol 24, 419–466. [DOI] [PubMed] [Google Scholar]
- [3].Singh NK, Abualrous ET, Ayres CM, Noe F, Gowthaman R, Pierce BG, and Baker BM (2020) Geometrical characterization of T cell receptor binding modes reveals class-specific binding to maximize access to antigen, Proteins 88, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Adams Jarrett J., Narayanan S, Liu B, Birnbaum Michael E., Kruse Andrew C., Bowerman Natalie A., Chen W, Levin Aron M., Connolly Janet M., Zhu C, Kranz David M., and Garcia KC (2011) T Cell Receptor Signaling Is Limited by Docking Geometry to Peptide-Major Histocompatibility Complex, Immunity 35, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gras S, Chadderton J, Del Campo Claudia M., Farenc C, Wiede F, Josephs Tracy M., Sng Xavier Y. X., Mirams M, Watson Katherine A., Tiganis T, Quinn Kylie M., Rossjohn J, and La Gruta Nicole L. (2016) Reversed T Cell Receptor Docking on a Major Histocompatibility Class I Complex Limits Involvement in the Immune Response, Immunity 45, 749–760. [DOI] [PubMed] [Google Scholar]
- [6].Le Nours J, Gherardin NA, Ramarathinam SH, Awad W, Wiede F, Gully BS, Khandokar Y, Praveena T, Wubben JM, Sandow JJ, Webb AI, von Borstel A, Rice MT, Redmond SJ, Seneviratna R, Sandoval-Romero ML, Li S, Souter MNT, Eckle SBG, Corbett AJ, Reid HH, Liu L, Fairlie DP, Giles EM, Westall GP, Tothill RW, Davey MS, Berry R, Tiganis T, McCluskey J, Pellicci DG, Purcell AW, Uldrich AP, Godfrey DI, and Rossjohn J (2019) A class of γδ T cell receptors recognize the underside of the antigen-presenting molecule MR1, Science 366, 1522–1527. [DOI] [PubMed] [Google Scholar]
- [7].Smith SN, Wang Y, Baylon JL, Singh NK, Baker BM, Tajkhorshid E, and Kranz DM (2014) Changing the peptide specificity of a human T-cell receptor by directed evolution, Nat Commun 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Harris Daniel T., Singh Nishant K., Cai Q, Smith Sheena N., Vander Kooi, Craig W, Procko E, Kranz David M., and Baker Brian M. (2016) An Engineered Switch in T Cell Receptor Specificity Leads to an Unusual but Functional Binding Geometry, Structure 24, 1142–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Singh NK, Riley TP, Baker SCB, Borrman T, Weng Z, and Baker BM (2017) Emerging Concepts in TCR Specificity: Rationalizing and (Maybe) Predicting Outcomes, The Journal of Immunology 199, 2203–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sewell AK (2012) Why must T cells be cross-reactive?, Nat Rev Immunol 12, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sliz P, Michielin O, Cerottini J-C, Luescher I, Romero P, Karplus M, and Wiley DC (2001) Crystal Structures of Two Closely Related but Antigenically Distinct HLA-A2/Melanocyte-Melanoma Tumor-Antigen Peptide Complexes, J Immunol 167, 3276–3284. [DOI] [PubMed] [Google Scholar]
- [12].Khan AR, Baker BM, Ghosh P, Biddison WE, and Wiley DC (2000) The structure and stability of an HLA-A*0201/octameric tax peptide complex with an empty conserved peptide-N-terminal binding site, J Immunol 164, 6398–6405. [DOI] [PubMed] [Google Scholar]
- [13].Borbulevych OY, Do P, and Baker BM (2010) Structures of native and affinity-enhanced WT1 epitopes bound to HLA-A*0201: Implications for WT1-based cancer therapeutics, Molecular Immunology 47, 2519–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Riley TP, Hellman LM, Gee MH, Mendoza JL, Alonso JA, Foley KC, Nishimura MI, Vander Kooi CW, Garcia KC, and Baker BM (2018) T cell receptor cross-reactivity expanded by dramatic peptide–MHC adaptability, Nat Chem Biol 14, 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Blevins SJ, and Baker BM (2017) Using Global Analysis to Extend the Accuracy and Precision of Binding Measurements with T cell Receptors and Their Peptide/MHC Ligands, Frontiers in Molecular Biosciences 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pierce BG, Hellman LM, Hossain M, Singh NK, Vander Kooi CW, Weng Z, and Baker BM (2014) Computational design of the affinity and specificity of a therapeutic T cell receptor, PLoS Comput Biol 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Otwinowski Z, and Minor W (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode, Methods in Enzymology 276, 307–326. [DOI] [PubMed] [Google Scholar]
- [18].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software, Journal of Applied Crystallography 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Borbulevych OY, Baxter TK, Yu Z, Restifo NP, and Baker BM (2005) Increased Immunogenicity of an Anchor-Modified Tumor-Associated Antigen Is Due to the Enhanced Stability of the Peptide/MHC Complex: Implications for Vaccine Design, J Immunol 174, 4812–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine, Acta Crystallographica Section D 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot, Acta Crystallographica Section D 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallographica Section D 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Brunger AT (2007) Version 1.2 of the Crystallography and NMR system, Nature Protocols 2, 2728–2733. [DOI] [PubMed] [Google Scholar]
- [24].Callender GG, Rosen HR, Roszkowski JJ, Lyons GE, Li M, Moore T, Brasic N, McKee MD, and Nishimura MI (2006) Identification of a hepatitis C virus–reactive T cell receptor that does not require CD8 for target cell recognition, Hepatology 43, 973–981. [DOI] [PubMed] [Google Scholar]
- [25].Chi S, Weiss A, and Wang H (2016) A CRISPR-Based Toolbox for Studying T Cell Signal Transduction, Biomed Res Int 2016, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Norell H, Zhang Y, McCracken J, Martins da Palma T, Lesher A, Liu Y, Roszkowski JJ, Temple A, Callender GG, Clay T, Orentas R, Guevara-Patiño J, and Nishimura MI (2010) CD34-based enrichment of genetically engineered human T cells for clinical use results in dramatically enhanced tumor targeting, Cancer Immunology, Immunotherapy 59, 851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, and Zhu C (2010) The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness, Nature 464, 932–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kolawole EM, Andargachew R, Liu B, Jacobs JR, and Evavold BD (2018) 2D Kinetic Analysis of TCR and CD8 Coreceptor for LCMV GP33 Epitopes, Frontiers in Immunology 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Martinez RJ, Andargachew R, Martinez HA, and Evavold BD (2016) Low-affinity CD4+ T cells are major responders in the primary immune response, Nature Communications 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Andargachew R, Martinez RJ, Kolawole EM, and Evavold BD (2018) CD4 T Cell Affinity Diversity Is Equally Maintained during Acute and Chronic Infection, J Immunol 201, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Purbhoo MA, Boulter JM, Price DA, Vuidepot A-L, Hourigan CS, Dunbar PR, Olson K, Dawson SJ, Phillips RE, Jakobsen BK, Bell JI, and Sewell AK (2001) The Human CD8 Coreceptor Effects Cytotoxic T Cell Activation and Antigen Sensitivity Primarily by Mediating Complete Phosphorylation of the T Cell Receptor ζ Chain, Journal of Biological Chemistry 276, 32786–32792. [DOI] [PubMed] [Google Scholar]
- [32].Fowler DM, and Fields S (2014) Deep mutational scanning: a new style of protein science, Nat Meth 11, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Robinson J, Mistry K, McWilliam H, Lopez R, Parham P, and Marsh SG (2011) The IMGT/HLA database, Nucleic acids research 39, D1171–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baker BM, Gagnon SJ, Biddison WE, and Wiley DC (2000) Conversion of a T Cell Antagonist into an Agonist by Repairing a Defect in the TCR/Peptide/MHC Interface. Implications for TCR Signaling, Immunity 13, 475–484. [DOI] [PubMed] [Google Scholar]
- [35].Reiser J-B, Legoux F, Gras S, Trudel E, Chouquet A, Léger A, Le Gorrec M, Machillot P, Bonneville M, Saulquin X, and Housset D (2014) Analysis of Relationships between Peptide/MHC Structural Features and Naive T Cell Frequency in Humans, The Journal of Immunology 193, 5816–5826. [DOI] [PubMed] [Google Scholar]
- [36].Cole DK, Malkit S, Scott DR, Rizkallah Pierre J., Borbulevych OY, Todorov PT, Moysey RK, Jakobsen Bent K., Boulter JM, Baker Brian M., and Li Y (2013) Increased peptide contacts govern high affinity binding of a modified TCR whilst maintaining a native pMHC docking mode, Frontiers in Immunology 4, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, Lever M, Dushek O, Schmitt TM, Greenberg PD, and Kranz DM (2018) Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains, Journal of immunology (Baltimore, Md. : 1950) 200, 1088–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ayres CM, Abualrous ET, Bailey A, Abraham C, Hellman LM, Corcelli SA, Noé F, Elliott T, and Baker BM (2019) Dynamically Driven Allostery in MHC Proteins: Peptide-Dependent Tuning of Class I MHC Global Flexibility, Frontiers in Immunology 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hawse WF, Champion MM, Joyce MV, Hellman LM, Hossain M, Ryan V, Pierce BG, Weng Z, and Baker BM (2012) Cutting edge: Evidence for a dynamically driven T cell signaling mechanism, J Immunol 188, 5819–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dam J, Guan R, Natarajan K, Dimasi N, Chlewicki LK, Kranz DM, Schuck P, Margulies DH, and Mariuzza RA (2003) Variable MHC class I engagement by Ly49 natural killer cell receptors demonstrated by the crystal structure of Ly49C bound to H-2Kb, Nat Immunol 4, 1213–1222. [DOI] [PubMed] [Google Scholar]
- [41].Scott DR, Borbulevych OY, Piepenbrink KH, Corcelli SA, and Baker BM (2011) Disparate Degrees of Hypervariable Loop Flexibility Control T-Cell Receptor Cross-Reactivity, Specificity, and Binding Mechanism, Journal of Molecular Biology 414, 385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Piepenbrink KH, Blevins SJ, Scott DR, and Baker BM (2013) The basis for limited specificity and MHC restriction in a T cell receptor interface, Nat Commun 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Baker BM, Scott DR, Blevins SJ, and Hawse WF (2012) Structural and dynamic control of T-cell receptor specificity, cross-reactivity, and binding mechanism, Immunological Reviews 250, 10–31. [DOI] [PubMed] [Google Scholar]
- [44].Harris DT, Wang N, Riley TP, Anderson SD, Singh NK, Procko E, Baker BM, and Kranz DM (2016) Deep Mutational Scans as a Guide to Engineering High Affinity T Cell Receptor Interactions with Peptide-bound Major Histocompatibility Complex, Journal of Biological Chemistry 291, 24566–24578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sharma P, and Kranz DM (2018) Subtle changes at the variable domain interface of the T-cell receptor can strongly increase affinity, Journal of Biological Chemistry 293, 1820–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Holst J, Vignali KM, Burton AR, and Vignali DAA (2006) Rapid analysis of T-cell selection in vivo using T cell-receptor retrogenic mice, Nat Meth 3, 327-327. [DOI] [PubMed] [Google Scholar]
- [47].Rubio-Godoy V, Dutoit V, Rimoldi D, Lienard D, Lejeune F, Speiser D, Guillaume P, Cerottini J-C, Romero P, and Valmori D (2001) Discrepancy between ELISPOT IFN-γ secretion and binding of A2/peptide multimers to TCR reveals interclonal dissociation of CTL effector function from TCR-peptide/MHC complexes half-life, Proceedings of the National Academy of Sciences 98, 10302–10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lyons GE, Roszkowski JJ, Man S, Yee C, Kast WM, and Nishimura MI (2006) T-cell receptor tetramer binding or the lack there of does not necessitate antigen reactivity in T-cell receptor transduced T cells, Cancer Immunol Immunother 55, 1142–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang Y, Liu Y, Moxley KM, Golden-Mason L, Hughes MG, Liu T, Heemskerk MH, Rosen HR, and Nishimura MI (2010) Transduction of human T cells with a novel T-cell receptor confers anti-HCV reactivity, PLoS Pathog 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Stone JD, and Kranz D (2013) Role of T cell receptor affinity in the efficacy and specificity of adoptive T cell therapies, Frontiers in Immunology 4, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hellman LM, Foley KC, Singh NK, Alonso JA, Riley TP, Devlin JR, Ayres CM, Keller GLJ, Zhang Y, Vander Kooi CW, Nishimura MI, and Baker BM (2019) Improving T Cell Receptor On-Target Specificity via Structure-Guided Design, Molecular Therapy 27, 300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dong D, Zheng L, Lin J, Zhang B, Zhu Y, Li N, Xie S, Wang Y, Gao N, and Huang Z (2019) Structural basis of assembly of the human T cell receptor–CD3 complex, Nature 573, 546–552. [DOI] [PubMed] [Google Scholar]
- [53].Celia H, Wilson-Kubalek E, Milligan RA, and Teyton L (1999) Structure and function of a membrane-bound murine MHC class I molecule, Proceedings of the National Academy of Sciences 96, 5634–5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rangarajan S, and Mariuzza R (2014) T cell receptor bias for MHC: co-evolution or co-receptors?, Cell. Mol. Life Sci, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yin L, Scott-Browne J, Kappler JW, Gapin L, and Marrack P (2012) T cells and their eons-old obsession with MHC, Immunological Reviews 250, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Garcia KC, Gapin L, Adams JJ, Birnbaum ME, Scott-Browne JP, Kappler JW, and Marrack P (2012) A Closer Look at TCR Germline Recognition, Immunity 36, 887–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mazza C, and Malissen B (2007) What guides MHC-restricted TCR recognition?, Semin Immunol 19, 225–235. [DOI] [PubMed] [Google Scholar]
- [58].Collins E, and Riddle D (2008) TCR-MHC docking orientation: natural selection, or thymic selection?, Immunologic Research 41, 267–294. [DOI] [PubMed] [Google Scholar]
- [59].Kranz DM (2009) Two mechanisms that account for major histocompatibility complex restriction of T cells, F1000 Biol Rep 1, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Blevins SJ, Pierce BG, Singh NK, Riley TP, Wang Y, Spear TT, Nishimura MI, Weng Z, and Baker BM (2016) How structural adaptability exists alongside HLA-A2 bias in the human αβ TCR repertoire, Proceedings of the National Academy of Sciences 113, E1276–E1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.