

Abstract
Recent studies have revealed that ARHGEF7 is upregulated in many malignant tumors, but the underlying molecular mechanisms to this response remain to be fully elucidated. In this study, we confirm that ARHGEF7 physically interacts with KLHL2, which was previously identified to be an E3 ubiquitin ligase. KLHL2 is capable of promoting ARHGEF7 degradation via the ubiquitin-proteasome pathway. We identify that the Kelch domain of KLHL2 is necessary for binding with ARHGEF7 and downstream activities. In addition, we find that ARHGEF7 is overexpressed in clear cell renal cell carcinoma (ccRCC) specimens, and that the level of expression negatively correlates with that of KLHL2. Moreover, we utilize knockdown loss-of-function assays to demonstrate that ARHGEF7 in 786-O and A498 cell lines can act as a regulator of cell proliferation, migration and invasion, and that these effects can be reversed by KLHL2 inactivation. Taken together, our data suggest that ARHGEF7 is a putative oncogene that functions via an interaction with KLHL2, and control of ARHGEF7 can be a potential future target to inhibit tumor progression.
Keywords: KLHL2, ARHGEF7, renal cell carcinoma, protein degradation, ubiquitination
Introduction
Renal cell carcinoma (RCC) is the third most common urologic cancer worldwide, and has characteristics of both high morbidity and mortality rates [1,2]. Patients with clear cell renal cell carcinoma (ccRCC) typically display insidious early and few symptoms, making it difficult to detect using conventional techniques. Many patients show a localized progression or distant metastasis when they are initially diagnosed with RCC [3]. Traditional treatment options have had little to no effect for patients with advanced kidney cancer, and targeted agents, such as sorafenib and sunitinib, have only shown slight improvements to survival [4]. Therefore, there is an urgent need to explore additional diagnostic and prognostic markers for the treatment of ccRCC.
The Rho guanine nucleotide exchange factor 7 (ARHGEF7), also known as βPix (Pax-interacting exchange factor beta) or Cool1 (Cloned out of library 1) has multiple functional domains, including the Src homology 3 (SH3) domain, the DBL and plekstrin homology (DH-PH) domain, and the calponin homology (CH) domain [5]. ARHGEF7 has been shown to play an important role in protein polymerization in the trans-Golgi network (TGN) [6]. A previous study has analyzed the single nucleotide polymorphism (SNP) array of colon cancer to shows that ARHGEF7 is upregulated in metastatic tumor compared to primary tumor [7]. Moreover, others have shown that ARHGEF7 plays an important role in cell migration through the regulation of cytoskeleton remodeling and interactions with a variety of proteins, including Abl-interactor (Abi)-1 and p21-activated kinase (PAK2) [8,9]. ARHGEF7 promotes the extension of lamellipodia protrusion by polymerizing actin filaments [10] and varying cell polarization for directed cell migration [11]. Furthermore, ARHGEF7 enhances cell spreading by activating the FAK invigorating RAC1 protein [12]. Zhou et al. described previously that ARHGEF7 plays an important role in the selective signaling of small G proteins and interactions with the PR domain of many proteins, which regulates cytoskeletal rearrangements [13]. ARHGEF7 is highly expressed in many tumors, and promotes the development and metastasis of tumors such as breast cancer and colorectal cancer [14,15].
Ubiquitination is a dynamic and reversible post-transcriptional modification of proteins, resulting in regulation of protein levels [16]. The ubiquitination cascade requires three key components: E1 ubiquitin-activating, E2 ubiquitin-conjugating, and E3 ligase-catalyzing [17]. The E3 ubiquitin ligases recognize specific sites for ubiquitin attachment, which can be reversed by deubiquitinating enzymes (DUB) [18]. Kelch-like 2 (KLHL2), a member of the Kelch-related superfamily, contains a BTB structural domain, a BACK structural domain and a Kelch structural domain [19], and can specifically identify substrate proteins WNK3 and NPCD [20,21]. In general, the Kelch-related superfamily of proteins contains two main domains: a BTB/POZ domain in the N-terminus and a Kelch repeat in the carboxyl terminus [22]. A large number of the BTB-Kelch proteins are believed to be composed of distinct substrate adapters, enabling the identification of a wide range of substrates for ubiquitylation [23]. A previous study has shown that KLHL2 is overexpressed in the smooth muscle cells of arteries in mice, and that AngII can rapidly decrease KLHL2 protein levels by autophagy-induced degradation of KLHL2, resulting in an increase in WNK3 levels and downstream activation [24].
In this study, we demonstrate that ARHGEF7 is a KLHL2-interacting protein, and a substrate for ubiquitination and subsequent proteasomal degradation to promote the neoplastic transformation of ccRCC. Furthermore, our studies reveal that this effect can be abrogated by KLHL2 depletion in ccRCC cells. Therefore, we present a functional insight into the understanding of the KLHL2-ARHGEF7 axis in the development and tumorigenesis of ccRCC.
Materials and methods
Cell culture and transfection
786-O, A498 and 293T cells were obtained from the American Type Culture Collection (ATCC). 786-O cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) 100 U/ml penicillin, and 100 μg/ml streptomycin, and incubated in a 5% CO2 humidified incubator at 37°C. A498 and 293T cells were cultured in DMEM medium supplemented with 10% FBS. All transient transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
Expression constructs
The KLHL2 and ARHGEF7 cDNAs were amplified from 293T cDNA library, and subcloned into pCMV-FLAG or pCMV-Myc expression vectors, respectively. KLHL2 and ARHGEF7 mutants as described were subcloned into pCMV-FLAG expression vectors. All the constructs were confirmed by DNA sequencing.
RNA interference
Non-specific control siRNA and siRNAs for human KLHL2 and ARHGEF7 were purchased from bio-tend. siRNAs transfection of cells was performed. The siRNA oligos sequences were listed in Table 1.
Table 1.
siRNA oligos sequences
| Gene | sequence |
|---|---|
| si-Control | 5’-ACAGACUUCGGAGUACCUGTT-3’ |
| si-KLHL2#1 | 5’-GAACGAAUUAAGAAGUCAATT-3’ |
| si-KLHL2#2 | 5’-GCAAAGAGAGUUAGAAUAATT-3’ |
| si-ARHGEF7#1 | 5’-GAGCUCGAGAGACACAUGGTT-3’ |
| si-ARHGEF7#2 | 5’-GGAUAUUAGUGUCGUGCAATT-3’ |
Antibodies
Commercially available antibodies for Western Blotting were as follows: KLHL2 (NBP-02939; NOVUS BB), ARHGEF7 (A1108, Abclonal), Myc (9E10; Sigma), FLAG (M2; Sigma), HA (MM5-101R; Millipore), and Actin (AC-74; Abclonal).
Immunoprecipitation
To immunoprecipitate the ectopically expressed Flag-tagged proteins, transfected cells were lysed 24 h post-transfection in BC100 buffer. The whole-cell lysates were immunoprecipitated with the monoclonal anti-Flag antibody-conjugated M2 agarose beads (Sigma) at 4°C overnight. After three washes with Flag lysis buffer, followed by two washes with BC100 buffer, the bound proteins were eluted using Flag-Peptide (Sigma)/BC100 for 3 h at 4°C. The eluted material was resolved by SDS-PAGE. To immunoprecipitate the endogenous proteins, cells were lysed with 1× cell lysis buffer (Cell Signaling), and the lysate was centrifuged. The supernatant was precleared with protein A/G beads (Sigma) and incubated with indicated antibodies overnight. Thereafter, protein A/G beads were applied, all at 4°C. After 2 h of incubation, pellets were washed five times with lysis buffer and resuspended in sample buffer and analyzed by SDS-PAGE.
Western blot
Cell lysates or immunoprecipitates were subjected to SDS-PAGE and proteins were transferred to nitrocellulose membranes (GE Healthcare). The membranes were blocked in Tris-buffered saline (TBS, pH 7.4) containing 5% non-fat milk and 0.1% Tween-20, washed twice in TBS containing 0.1% Tween-20, and incubated with primary antibody for 2 h and followed by secondary antibody for 1 h at room temperature. The proteins of interest were visualized using ECL chemiluminescence system (Santa Cruz).
Quantitative RT-PCR
Total RNA was isolated from transiently transfected cells using the TRIzol reagent (Tiangen), and cDNA was reversed-transcribed using the Superscript RT kit (TOYOBO), according to the manufacturer’s instructions. PCR amplification was performed using the SYBR Green PCR master mix Kit (TOYOBO). All quantization was normalized to the level of endogenous control GAPDH. The primer sequences were listed in Table 2.
Table 2.
Sequence of primers
| Gene name | Forward (5’-3’) | Reverse (5’-3’) |
|---|---|---|
| KLHL2 | TAATACCGAAAAACACTGCC | TTCTAACTCTCTTTGCTCGG |
| ARHGEF7 | CGCAAACCTGAACGGAAGCCTT | GTTTTGGCGCTGGTGCAGTAAG |
| GAPDH | GCACCGTCAAGGCTGAGAAC | TGGTGAAGACGCCAGTGGA |
CCK-8 assay
Cell proliferation rate was determined using Cell Counting Kit-8 (CCK-8) according to the manufacturer’s protocol (Vazyme). Briefly, the control or siRNAs transfected cells were seeded onto 96-well plates at a density of 1000 cells per well. During a 7-day culture periods, 10 μl of the CCK-8 solution was added to cell culture once a day, and incubated for 1 h. The resulting color was assayed at 450 nm using a microplate absorbance reader (Bio-Rad). Each assay was carried out in triplicates.
Cell migration and invasion assay
Cell migration was assessed using 24-well transwell unit with polycarbonate membrane (pore size, 8 μm) (Corning) according to the manufacturer’s protocol. The membrane was coated with Matrigel basement membrane matrix (1 μg/μl) (BD Bioscience) for 24 h. Then Cells (0.5-2.5×104) were seeded into the upper chamber in a serum-free medium. The lower chamber was filled with a medium containing 10% FBS. After 24 h of incubation, the cells in the upper chamber were removed, and the cells were fixed in 4% paraformaldehyde, stained with crystal violet for 20 min. After being washed with water three times, digital images were obtained from the membranes, and cell areas were selected using Scan Scope CS system (Aperio Technologies). The migrating cells were quantified in five randomly selected fields in each membrane, and the average value was defined as a migration or invasion index on three independent membranes. For invasion, the membranes utilized were Matrigel-coated invasion chambers (BD Biosciences) that were pre-hydrated in serum-free medium.
Immunohistochemistry
Tissue microarrays (TMAs) were made using above 150 paired tissues in Shanghai Outdo Biotech Company (Shanghai, China), including tumor and adjacent normal tissues. The immunohistochemistry (IHC) was performed by the streptavidin-peroxidase method (Zymed Laboratories Inc., San Francisco, CA, USA). The KLHL2 antibody was purchased from NOVUS (NBP2-17080, USA) and diluted into 1:2000. The ARHGEF7 antibody was purchased from Abclonal (A1108, China) and diluted into 1:1000. IHC score were determined by both the intensity and percentage of tumor cell. An intensity score of 0 to 3 was assigned for the intensity of tumor cells (0, none; 1, weak; 2, intermediate; 3, strong). A proportional score was given by the estimated proportion of positive tumor cells in percentage. To assess the average degree of staining within a tumor, multiple regions were analyzed, and at least 100 tumor cells were assessed. The cytoplasmic expression was assessed by H-score system. The formula for the H-score is: Histoscore = ∑(I × Pi), where I = intensity of staining and Pi = percentage of stained tumor cells, producing a cytoplasmic score ranging from 0 to 200. The scoring was independently assessed by two assessors (AWHC and JHMT) who were not aware of the clinical outcomes.
Statistical analysis
Data were expressed as the means ± SEM. All experiments were carried out with three or more replicates. Statistical analyses were performed by Student’s t-test for most studies, and performed by using GraphPad Prism v7.0 software. A P < 0.05 was considered statistically significant.
Results
ARHGEF7 interacts with KLHL2
We initially searched for ARHGEF7-associated proteins to identify interacting partners in cells. The Proteomic database identified that KLHL2 is co-purified in a complex with ARHGEF7 (https://www.ncbi.nlm.nih.gov/gene/8874). To verify whether KLHL2 is an authentic bona fide ARHGEF7 interactor, myc-KLHL2 and FLAG-ARHGEF7 constructs were co-expressed in 293T cells, followed by co-immunoprecipitation (Co-IP) with an anti-FLAG antibody. As shown in Figure 1A, myc-KLHL2 is immunoprecipitated by FLAG-ARHGEF7, suggesting an exogenous interaction between these two proteins. Complementary results were obtained to show that FLAG-KLHL2 was immunoprecipitated by myc-ARHGEF7 (Figure 1B). FLAG-KLHL2 was also capable of immunoprecipitating endogenous ARHGEF7 (Figure 1C). Endogenous KLHL2 protein was immunoprecipitated by ARHGEF7, suggesting endogenous binding between these two proteins (Figure 1D). In like manner, we performed immunoprecipitation using anti-KLHL2 antibody in cell lysates isolated from 786-O cells, suggesting KLHL2 can interact with ARHGEF7 at the endogenous level (Figure 1E). To investigate whether KLHL2 co-localizes with ARHGEF7 in vivo, 786-O cells were immunostained with KLHL2 and/or ARHGEF7 antibody, and visualized by confocal microscopy. As shown in Figure 1F, KLHL2 and ARHGEF7 were co-localized in the cytoplasm. Taken together, these results indicate that KLHL2 forms a protein complex with ARHGEF7 in cells.
Figure 1.
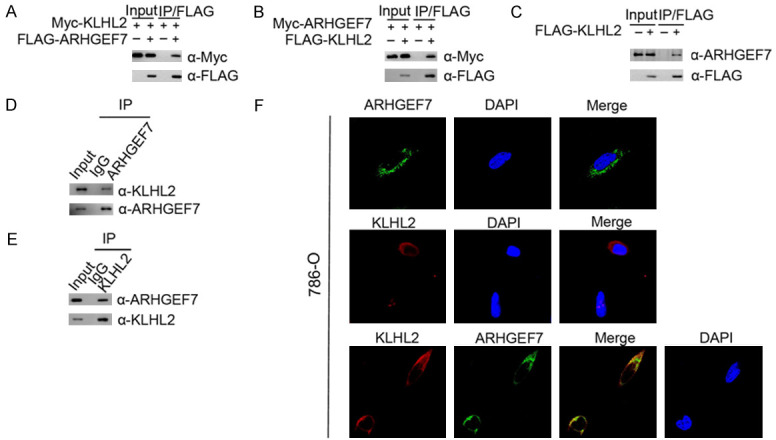
KLHL2 directly interacts with ARHGEF7 in the cytoplasm. A. 293T cells were co-transfected with Myc-KLHL2 and FLAG-ARHGEF7 constructs. After 24 h, cell lysates were prepared for Co-IP with anti-FLAG antibody and Western blotting. B. Similar Co-IP assay was performed between Myc-ARHGEF7 and FLAG-KLHL2. C. Western blot and co-IP samples of anti-FLAG antibody obtained from 293T cells infected with expressing FLAG-KLHL2 or control. The cells were treated with 20 μM MG132 for 8 h before harvesting. D. After treated with 20 μM MG132 for 4 h, 786-O cell lysates were prepared for Co-IP with anti-ARHGEF7 antibody and Western blot analyses with indicated antibodies. E. Similar Co-IP assay was performed with anti-KLHL2. F. Confocal microscopy of 786-O cells stained with ARHGEF7 and KLHL2 antibodies. DAPI (blue channel) represents nuclear staining. Red, green and blue channel images were captured by (LSM710, Zeiss).
KLHL2 degrades ARHGEF7 via the Kelch domain
KLHL2 consists of three structural domains: a CUL3-binding BTB domain, a BACK domain at the C-terminus, and an ubiquitin substrate-binding Kelch domain at the N-terminus [19]. To identify the interacting domain with ARHGEF7, we constructed three deletion mutants of KLHL2 corresponding to the structural domains respectively, which are named the M1, M2, and M3 plasmids (Figure 2A). We performed a Co-IP assay to detect the ability of the overexpressed ARHGEF7, either the wild type (WT) or the three deletion mutants, to still function and bind to ARHGEF7 in 293T cells. As shown in Figure 2B, full-length KLHL2 (KLHL2-WT), M1, and M2 all efficiently interacts with ARHGEF7, but the binding was abrogated completely with M3. Based on these observations, we sought to verify that the N-terminal Kelch region of KLHL2 is required for its interaction with ARHGEF7 in vivo. We initially evaluated whether KLHL2-mediated ubiquitination could promote ARHGEF7 degradation. As expected, ectopic expression of wild-type KLHL2 (WT) or the M1 and M2 mutants effectively reduced ARHGEF7 protein expression, but the M3 mutant had no effect on co-expressed ARHGEF7 (Figure 2C). Furthermore, KLHL2-WT, M1, and M2, but not the M3 mutant, promoted ARHGEF7 degradation in a dose-dependent manner, indicating that the Kelch domains is required for KLHL2-mediated ARHGEF7 degradation (Figure 2D, 2E). This result validates our conjecture that the C-terminal Kelch domain of KLHL2 is essential for its interaction with ARHGEF7 protein.
Figure 2.
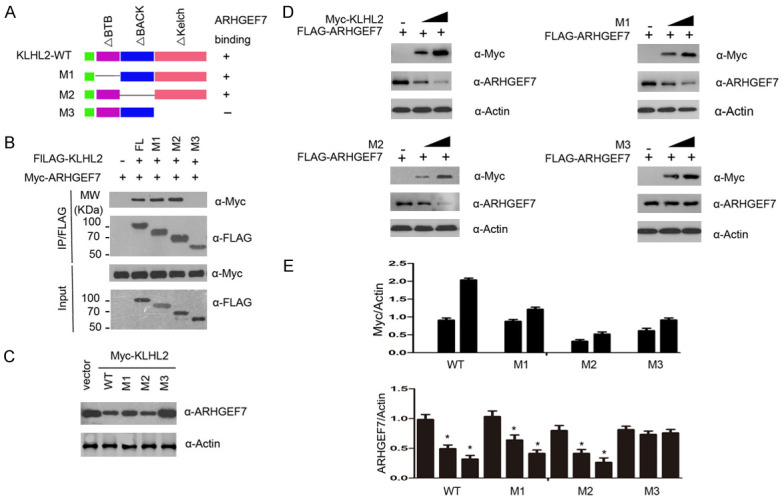
Identification of the region in KLHL2 that required for KLHL2-mediated ARHGEF7 protein degradation. A. Schematic representation of KLHL2 deletion mutants used in the study. B. 293T cells were co-transfected with Myc-ARHGEF7 and the indicated plasmids. Cell lysates were prepared and subjected to immunoprecipitation using the anti-FLAG antibody, followed by WB analyses with the indicated antibodies. C. Ectopically expressed the indicated plasmids promoted endogenous ARHGEF7 protein degradation. D. 293T cells were transfected with FLAG-ARHGEF7 and the indicated plasmids (Myc-KLHL2 WT, M1, M2, and M3) in dose-dependent manners. 24 h after transfection, cells were harvested for WB analyses. E. The quantification of immunoblot is shown in the lower panel. The mean values (S.D.) of three independent experiments are shown.
ARHGEF7 is a substrate for KLHL2-dependent ubiquitination
We next sought to investigate whether KLHL2 regulated the protein levels of ARHGEF7 via the ubiquitin proteasome pathway. Myc-KLHL2 and Flag-ARHGEF7 were co-transfected into 293T cells, followed by treatment with DMSO, MG132, Bortezomib or MLN4924. MG132, Bortezomib and MLN4924 all inhibited the change in expression level of the ARHGEF7 protein (Figure 3A). Next, we depleted endogenous KLHL2 protein levels with two specific siRNAs in 786-O cells, and observed that ARHGEF7 protein was significantly up-regulated (Figure 3B). To exclude the possibility that this was a transcriptional effect, we measured mRNA levels of KLHL2 and ARHGEF7 in 786-O cells. In contrast to the significant decrease in KLHL2 mRNA levels, ARHGEF7 mRNA levels in si-KLHL2 786-O cells stayed relatively similar to that of controls (Figure 3C), suggesting that the effect of KLHL2 on ARHGEF7 was not mediated at the mRNA level. Further results showed that knockdown of KLHL2 could significantly extended the half-life of endogenous ARHGEF7 protein, and stabilize the levels of the ARHGEF7 protein (Figure 3D, 3E). Therefore, we propose that KLHL2 regulates ARHGEF7 expression levels at the protein stage. By knocking down Cul1, Cul2 and Cul3, Cul4, Cul5 or ROC1 in 786-O cells, we found that the protein expression level of ARHGEF7 was significantly up-regulated after Cul3 was knocked down, while the expression level of ARHGEF7 was not changed by the change in expression of other Culling family proteins (Figure 3F). Therefore, we believe that in the Culling family, only Culling 3 directly mediates the ARHGEF7 levels through the ubiquitin-dependent proteasomal activity. To further identify whether ARHGEF7 was degraded through KLHL2-mediated poly-ubiquitination, we note that little or no ARHGEF7 polyubiquitination was observed by knocking down KLHL2 (Figure 3G). We show that the KLHL2-CUL3-ROC1 complex catalyzed ARHGEF7 ubiquitination in vitro (Figure 3H). Following HA-ubiquitin and FLAG-ARHGEF7 co-expression in 293T cells with different doses of KLHL2-WT, M1, M2 or M3, we note that the ARHGEF7 protein was robustly polyubiquitinated by KLHL2-WT, M1, M2, but not the M3 mutant in a dose-dependent manner (Figure 3I). Taken together, these data demonstrate that the KLHL2-CUL3-ROC1 E3 ubiquitin ligase complex regulates ARHGEF7 protein stability through the ubiquitin-dependent proteasomal degradation pathway.
Figure 3.

The KLHL2-CUL3-ROC1 complex targets ARHGEF7 for ubiquitination. A. KLHL2 regulates ARHGEF7 protein level through the ubiquitin-proteasome pathway. 293T cells were transfected with FLAG-ARHGEF7 and increasing amounts of Myc-KLHL2 constructs. Twenty-four hours after transfection, cells were treated with 20 μM MG132, Bortezomib 100 mM, MLN4924, or DMSO for 6 h before cell lysates were prepared for WB analyses. B. Western blot of the indicated proteins in cell lysates from 786-O cells transfected with siRNAs and control. C. 786-O cells were transfected with the control or two independent KLHL2 siRNAs, respectively, 48 h after transfection, cells were harvested for qRT-PCR analysis. GAPDH was used for normalization. The mean values (S.D.) of three independent experiments are shown. D, E. KLHL2 knockdown prolonged the half-life of ARHGEF7 protein. 786-O cells were transfected with control or KLHL2 siRNA, 48 h after transfection, 786-O cells were treated with 30 μM cycloheximide for the indicated periods. Cells were harvested for Western blotting analyses. F. Western blot of indicated proteins in cell lysates from 786-O cells transfected with indicated siRNAs. G. KLHL2 was depleted by siRNAs in 293T cells transiently expressing HA-Ub and FLAG-ARHGEF7. FLAG-ARHGEF7 protein was immunoprecipitated and subjected to WB analyses. H. Western blot of the products of in vitro ubiquitination assay performed by incubating the reconstituted KLHL2-CUL3-ROC1 E3 ligase complex with E1, E2, Ub, and His-ARHGEF7 at 30°C for 2 h. I. The indicated plasmids were transfected into 293T cells. Twenty-four hours after transfection, cells were treated with 20 μM MG132 for 6 h. ARHGEF7 proteins were immunoprecipitated with anti-Flag antibody and analysed by WB.
KLHL2 suppresses renal cancer cell growth, migration and invasion in an ARHGEF7-dependent manner
Accumulating evidence suggests that ARHGEF7 expression is upregulated in various types of human cancers [14,25,26]. Because of this, we were keen to identify whether ARHGEF7 is an important mediator of a KLHL2 depletion-induced malignant transformation in 786-O and A498 cells. As determined by the CCK-8 assay, the growth rate of ARHGEF7-depleted 786-O and A498 cells was slower that of the control cells, while depletion of KLHL2 could markedly promote cell growth and the phenotypes, which can be rescued by ARHGEF7 co-depletion (Figure 4A, 4B). Similarly, cell migration and invasion were performed using a trans-well assay. Knockdown of KLHL2 resulted in a significant increase in the migration and invasion of 786-O and A498 cells, but these effects were reversed by ARHGEF7 inactivation (Figure 4C-F). Therefore, we hypothesize that KLHL2 may play negative roles in cell proliferation, migration, and invasion of 786-O and A498 cells at least in part by downregulating ARHGEF7 expression.
Figure 4.
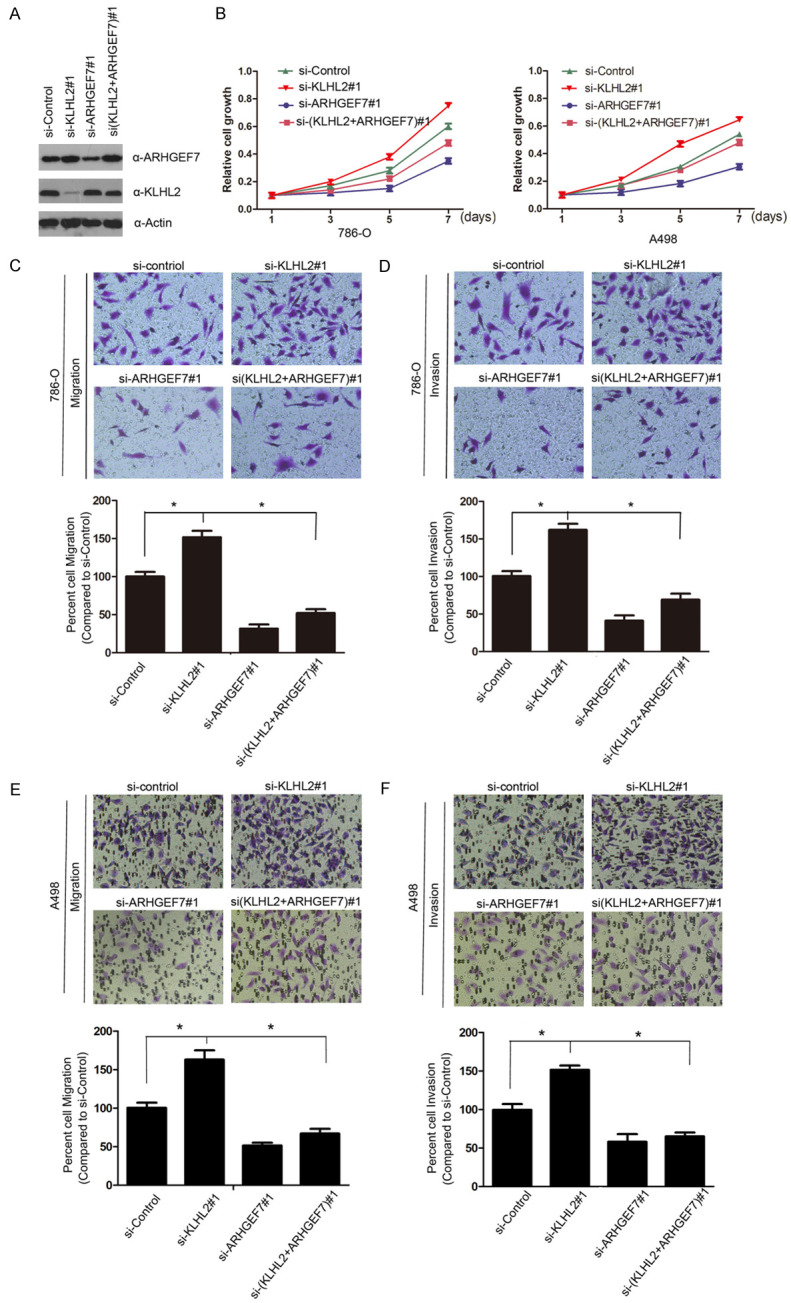
KLHL2 suppresses cell proliferation, migration and invasion partially dependent on ARHGEF7. A, B. Western blot (left panel) and cell proliferation assay (right panel) of 786-O and A498 cells transfected with control or indicated siRNAs. Standard deviation (S.D.) of at least three independent experiments is shown to indicate statistical significance. * represent P < 0.05. C-F. 786-O and A498 cells were transfected with control or indicate siRNAs. Migration and invasion assays were performed using Trans-well chambers without or with Matrigel as described before. Representative images of cell migration and invasion assays were shown on the upper panel, and the quantitative analysis is shown on the lower panel. The mean values (S.D.) of three independent experiments are shown (*, P < 0.05).
KLHL2 protein is negatively correlated with ARHGEF7 in ccRCC patients
We next analyzed the expression of KLHL2 and ARHGEF7 by immunohistochemistry (IHC) in human renal cancer specimens obtained from a cohort of patients (n = 72, normal renal tissue and cancer tissue specimens). IHC staining was evaluated by measuring both the percentage of positive cells and staining intensity (Figure 5A). ARHGEF7 was remarkably overexpressed in renal cancer tissues compared to normal tissue. In contrast, KLHL2 was expressed at a lower level in tumor tissues (Figure 5B). We next evaluated the association of expression levels with clinical pathological characteristics, including tumor stage (stage I+II vs stage III+IV) and differentiation (G1+G2 vs G3+G4). As shown in Figure 5C, 5D, a strong correlation was seen between ARHGEF7 expression and tumor stage (P = 0.0142) or differentiation (P = 0.0007); however, KLHL2 protein was not correlated to tumor stage (P = 0.0932) and differentiation (P = 0.1215). Furthermore, the protein expression levels of KLHL2 and ARHGEF7 were negatively correlated with clinical stage renal cell carcinoma samples from an analysis of expression in KLHL2 and ARHGEF7 levels in 72 paired tissues from ccRCC patients by immunohistochemistry. Clinical and pathologic characteristics of the patients are shown in Table 3. In the 72 paired ccRCC cases, ARHGEF7 protein was greatly overexpressed in 59.7% of tumor tissues, and KLHL2 was found to be under-expressed in 70.89% of tumor tissues (Figure 5E). Moreover, statistical analysis indicated that ARHGEF7 was negatively correlated with KLHL2 expression in this cohort of patient samples (r=-0.3532, P=0.0023) (Figure 5F). These data suggest that ARHGEF7 and KLHL2 protein levels are negatively correlated in human renal cell carcinoma specimens.
Figure 5.
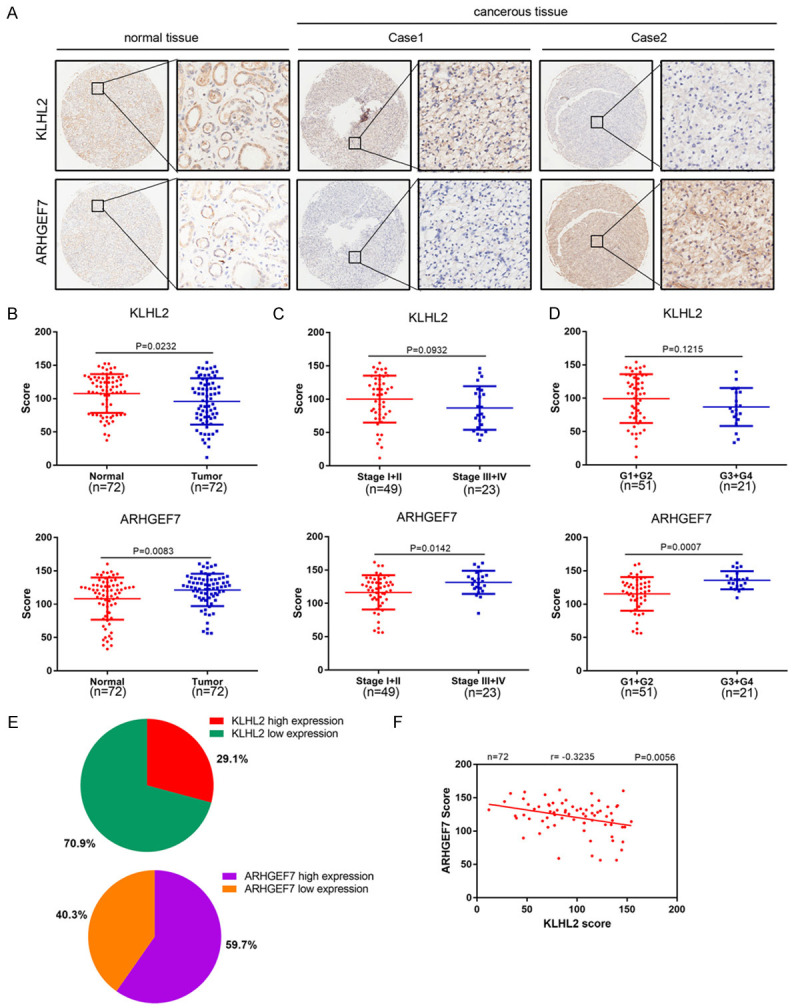
ARHGEF7 and KLHL2 protein are negatively correlated in renal cancer specimens. A. Representative images of IHC analysis of ARHGEF7 and KLHL2 protein expression on tissue microarray (n = 72 paired renal cancer tissue specimens). B. Box plots of ARHGEF7 and KLHL2 protein expression based on their staining index in paired tumor tissue and normal renal tissues. C, D. Box plots of ARHGEF7 and KLHL2 protein expression based on their IS in renal cancer specimens at different clinical stages. E. KLHL2 and ARHGEF7 staining patterns in 72 paired ccRCC cases. F. Correlation analysis of the IS of expression levels of ARHGEF7 and KLHL2 proteins in human renal cancer specimens.
Table 3.
Patient characteristics and clinicopathological factors by KLHL2 and ARHGEF7 expression
| KLHL2 high | KLHL2 low | p value | ARHGEF7 high | ARHGEF7 low | p value | |
|---|---|---|---|---|---|---|
| No. of patients | 21 (29.1%) | 51 (70.9%) | 43 (59.7%) | 29 (40.3%) | ||
| Age | 0.739 | 0.704 | ||||
| ≥60 | 7 (33.3%) | 22 (43.1%) | 19 (44.2%) | 10 (34.5%) | ||
| <60 | 14 (66.7%) | 29 (56.9%) | 24 (55.8%) | 19 (65.5%) | ||
| Gender | 0.130 | 0.492 | ||||
| Male | 16 (76.2%) | 30 (58.8%) | 27 (62.8%) | 19 (65.5%) | ||
| Female | 5 (23.8%) | 21 (41.2%) | 16 (37.2%) | 10 (34.5%) | ||
| Size | 0.434 | 0.430 | ||||
| ≥4 cm | 18 (85.7%) | 41 (80.4%) | 36 (83.7%) | 23 (78.3%) | ||
| <4 cm | 3 (14.3%) | 10 (19.6%) | 7 (16.3%) | 6 (20.7%) | ||
| Primary T stage | 0.223 | 0.026 | ||||
| Low (pT1/pT2) | 17 (81.0%) | 35 (68.6%) | 16 (37.2%) | 25 (86.2%) | ||
| High (pT3/pT4) | 4 (19.0%) | 16 (31.4%) | 27 (62.8%) | 4 (13.8%) | ||
| TNM stage | 0.108 | 0.024 | ||||
| Low (pT1/pT2) | 17 (81.0%) | 32 (62.7%) | 25 (58.1%) | 24 (82.8%) | ||
| High (pT3/pT4) | 4 (19.0%) | 19 (37.3%) | 18(41.9%) | 5 (17.2%) | ||
| Fuhrman grade | 0.015 | 0.000 | ||||
| Low (G1/G2) | 19 (90.5%) | 32 (62.7%) | 24 (55.8%) | 27 (93.1%) | ||
| High (G3/G4) | 2 (9.5%) | 19 (37.3%) | 19 (62.8%) | 2 (6.9%) |
Discussion
ARHGEF7, a guanine nucleotide exchange factor of Rho GTPases, is a novel tumor initiator with increased expression in a variety of tumor types, including lung, breast and colon cancers [14,25,26]. Previous studies have showed that properties of ARHGEF7 promoted the invasion and metastasis of colorectal adenocarcinoma, which is in accordance with the involvement of ARHGEF7 in cell migration and metastasis to other malignancies [25,27]. However, the role of ARHGEF7 in ccRCC remained unknown. In this study, IHC analysis showed that ARHGEF7 was overexpressed in renal cancer tissues, compared with that of paired normal tissues. ARHGEF7 functions as an oncogene in cell migration, mobility, attachment and cell spread in vivo [10,28,29]. Lee et al. has shown that ARHGEF7’s regulation in the separation of bundled actin filaments is restricted [30]. The depletion of bundled actin filaments leads to a change of shifting from ARHGEF7 to FA, which increases actin polymerization and therefore increasing cell mobility [10]. Several reports have utilized mutational analyses to show that ARHGEF7 is important for neurite outgrowth, and that its overexpression causes an increase in neurogenesis [31-33].
In this study, we identify the ubiquitination of ARHGEF7 protein to be regulated by the E3 ligase KLHL2. Wild type KLHL2 recognizes and promotes ARHGEF7 ubiquitination proteasomal degradation. However, truncations of KLHL2 are defective in ARHGEF7 protein degradation and ubiquitination, suggesting the crucial role of certain domains. KLHL2 is a member of the BTB-Kelch protein family, which was recently described to be components of the multi-protein complex known as Culling RING E3 ubiquitin ligases (CRL). CRLs serve to identify and tag proteins for ubiquitination [34-36]. Although KLHL2 is predominantly expressed in brain tissue [25], little is known about its role in ccRCC [37]. Based on analysis of the cancer genome atlas database, KLHL2 mRNA expression was down-regulated in ccRCC compared with that of normal tissue, while the expression of ARHGEF7 was flipped. Immunohistochemistry analysis also revealed that the expression levels of KLHL2 and ARHGEF7 are of inverse correlation in ccRCC. These studies indicate that KLHL2 is poorly expressed in ccRCC.
In summary, our results support the notion that ARHGEF7 functions as an oncogene in ccRCC. Future studies could lead to potential treatments that exploit the targeting of ARHGEF7 in ccRCC.
Acknowledgements
This study was completely supported by the National Natural Science Foundation of China (81570607).
Disclosure of conflict of interest
None.
Abbreviations
- ARHGEF7
Rho guanine nucleotide exchange factor 7
- KLHL2
Kelch-like protein
- ccRCC
clear cell renal cell carcinoma
- UB
ubiquitination enzymes
- Co-IP
co-immunoprecipitation
- IHC
Immunohistochemical
- WB
Western Blotting
References
- 1.Wallis CJD, Klaassen Z, Bhindi B, Ye XY, Chandrasekar T, Farrell AM, Goldberg H, Boorjian SA, Leibovich B, Kulkarni GS, Shah PS, Bjarnason GA, Heng DYC, Satkunasivam R, Finelli A. First-line systemic therapy for metastatic renal cell carcinoma: a systematic review and network meta-analysis. Eur Urol. 2018;74:309–321. doi: 10.1016/j.eururo.2018.03.036. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 3.Srigley JR, Delahunt B, Eble JN, Egevad L, Epstein JI, Grignon D, Hes O, Moch H, Montironi R, Tickoo SK, Zhou M, Argani P ISUP Renal Tumor Panel. The international society of urological pathology (ISUP) vancouver classification of renal neoplasia. Am J Surg Pathol. 2013;37:1469–1489. doi: 10.1097/PAS.0b013e318299f2d1. [DOI] [PubMed] [Google Scholar]
- 4.Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, Larkin J, Ficarra V. Sequential use of sorafenib and sunitinib in advanced renal-cell carcinoma (RCC): an Italian multicentre retrospective analysis of 189 patient cases. Nat Rev Dis Primers. 2017;3:17009. doi: 10.1038/nrdp.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oh WK, Yoo JC, Jo D, Song YH, Kim MG, Park D. Cloning of a SH3 domain-containing proline-rich protein, p85SPR, and its localization in focal adhesion. Biochem Biophys Res Commun. 1997;235:794–798. doi: 10.1006/bbrc.1997.6875. [DOI] [PubMed] [Google Scholar]
- 6.Anitei M, Stange C, Parshina I, Baust T, Schenck A, Raposo G, Kirchhausen T, Hoflack B. Protein complexes containing CYFIP/Sra/PIR121 coordinate Arf1 and Rac1 signalling during clathrin-AP-1-coated carrier biogenesis at the TGN. Nat Cell Biol. 2010;12:330–340. doi: 10.1038/ncb2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munoz-Bellvis L, Fontanillo C, Gonzalez-Gonzalez M, Garcia E, Iglesias M, Esteban C, Gutierrez ML, Abad MM, Bengoechea O, De Las Rivas J, Orfao A, Sayagues JM. Unique genetic profile of sporadic colorectal cancer liver metastasis versus primary tumors as defined by high-density single-nucleotide polymorphism arrays. Mod Pathol. 2012;25:590–601. doi: 10.1038/modpathol.2011.195. [DOI] [PubMed] [Google Scholar]
- 8.Campa F, Machuy N, Klein A, Rudel T. A new interaction between Abi-1 and betaPIX involved in PDGF-activated actin cytoskeleton reorganisation. Cell Res. 2006;16:759–770. doi: 10.1038/sj.cr.7310091. [DOI] [PubMed] [Google Scholar]
- 9.Reddy PN, Radu M, Xu K, Wood J, Harris CE, Chernoff J, Williams DA. p21-activated kinase 2 regulates HSPC cytoskeleton, migration, and homing via CDC42 activation and interaction with beta-Pix. Blood. 2016;127:1967–1975. doi: 10.1182/blood-2016-01-693572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuo JC, Han X, Hsiao CT, Yates JR 3rd, Waterman CM. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for beta-Pix in negative regulation of focal adhesion maturation. Nat Cell Biol. 2011;13:383–393. doi: 10.1038/ncb2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osmani N, Peglion F, Chavrier P, Etienne-Manneville S. Cdc42 localization and cell polarity depend on membrane traffic. J Cell Biol. 2010;191:1261–1269. doi: 10.1083/jcb.201003091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang F, Lemmon CA, Park D, Romer LH. FAK potentiates Rac1 activation and localization to matrix adhesion sites: a role for betaPIX. Mol Biol Cell. 2007;18:253–264. doi: 10.1091/mbc.E06-03-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou K, Wang Y, Gorski JL, Nomura N, Collard J, Bokoch GM. Guanine nucleotide exchange factors regulate specificity of downstream signaling from Rac and Cdc42. J Biol Chem. 1998;273:16782–16786. doi: 10.1074/jbc.273.27.16782. [DOI] [PubMed] [Google Scholar]
- 14.Bae JY, Ahn SJ, Lee JE, Kim JE, Han MR, Han W, Kim SW, Shin HJ, Lee SJ, Park D, Noh DY. BetaPix-a enhances the activity of phospholipase Cgamma1 by binding SH3 domain in breast cancer. J Cell Biochem. 2005;94:1010–1016. doi: 10.1002/jcb.20357. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Davis C, Shah S, Hughes D, Ryan JC, Altomare D, Pena MM. IL-33 promotes growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis. Mol Carcinog. 2017;56:272–287. doi: 10.1002/mc.22491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 17.Kumari N, Jaynes PW, Saei A, Iyengar PV, Richard JLC, Eichhorn PJA. The roles of ubiquitin modifying enzymes in neoplastic disease. Biochim Biophys Acta Rev Cancer. 2017;1868:456–483. doi: 10.1016/j.bbcan.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 18.Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 19.Canning P, Cooper CD, Krojer T, Murray JW, Pike AC, Chaikuad A, Keates T, Thangaratnarajah C, Hojzan V, Ayinampudi V, Marsden BD, Gileadi O, Knapp S, von Delft F, Bullock AN. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J Biol Chem. 2013;288:7803–7814. doi: 10.1074/jbc.M112.437996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasagi Y, Takahashi D, Aida T, Nishida H, Nomura N, Zeniya M, Mori T, Sasaki E, Ando F, Rai T, Uchida S, Sohara E. Impaired degradation of medullary WNK4 in the kidneys of KLHL2 knockout mice. Biochem Biophys Res Commun. 2017;487:368–374. doi: 10.1016/j.bbrc.2017.04.068. [DOI] [PubMed] [Google Scholar]
- 21.Tseng LA, Bixby JL. Interaction of an intracellular pentraxin with a BTB-Kelch protein is associated with ubiquitylation, aggregation and neuronal apoptosis. Mol Cell Neurosci. 2011;47:254–264. doi: 10.1016/j.mcn.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angel P, Allegretto EA, Okino ST, Hattori K, Boyle WJ, Hunter T, Karin M. Oncogene jun encodes a sequence-specific trans-activator similar to AP-1. Nature. 1988;332:166–171. doi: 10.1038/332166a0. [DOI] [PubMed] [Google Scholar]
- 23.Bochtler M, Ditzel L, Groll M, Hartmann C, Huber R. The proteasome. Annu Rev Biophys Biomol Struct. 1999;28:295–317. doi: 10.1146/annurev.biophys.28.1.295. [DOI] [PubMed] [Google Scholar]
- 24.Zeniya M, Morimoto N, Takahashi D, Mori Y, Mori T, Ando F, Araki Y, Yoshizaki Y, Inoue Y, Isobe K, Nomura N, Oi K, Nishida H, Sasaki S, Sohara E, Rai T, Uchida S. Kelch-like protein 2 mediates angiotensin ii-with no lysine 3 signaling in the regulation of vascular tonus. J Am Soc Nephrol. 2015;26:2129–2138. doi: 10.1681/ASN.2014070639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu HW, Chen YQ, Huang CM, Liu CY, Chiou A, Wang YK, Tang MJ, Kuo JC. beta-PIX controls intracellular viscoelasticity to regulate lung cancer cell migration. J Cell Mol Med. 2015;19:934–947. doi: 10.1111/jcmm.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lei X, Deng L, Liu D, Liao S, Dai H, Li J, Rong J, Wang Z, Huang G, Tang C, Xu C, Xiao B, Li T. ARHGEF7 promotes metastasis of colorectal adenocarcinoma by regulating the motility of cancer cells. Int J Oncol. 2018;53:1980–1996. doi: 10.3892/ijo.2018.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Han M, Whetsell W Jr, Wang J, Rich J, Hallahan D, Han Z. Tax-interacting protein 1 coordinates the spatiotemporal activation of Rho GTPases and regulates the infiltrative growth of human glioblastoma. Oncogene. 2014;33:1558–1569. doi: 10.1038/onc.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koh CG, Manser E, Zhao ZS, Ng CP, Lim L. Beta1PIX, the PAK-interacting exchange factor, requires localization via a coiled-coil region to promote microvillus-like structures and membrane ruffles. J Cell Sci. 2001;114:4239–4251. doi: 10.1242/jcs.114.23.4239. [DOI] [PubMed] [Google Scholar]
- 29.Lee SH, Eom M, Lee SJ, Kim S, Park HJ, Park D. BetaPix-enhanced p38 activation by Cdc42/Rac/PAK/MKK3/6-mediated pathway. Implication in the regulation of membrane ruffling. J Biol Chem. 2001;276:25066–25072. doi: 10.1074/jbc.M010892200. [DOI] [PubMed] [Google Scholar]
- 30.Lee CS, Choi CK, Shin EY, Schwartz MA, Kim EG. Myosin II directly binds and inhibits Dbl family guanine nucleotide exchange factors: a possible link to Rho family GTPases. J Cell Biol. 2010;190:663–674. doi: 10.1083/jcb.201003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin EY, Lee CS, Cho TG, Kim YG, Song S, Juhnn YS, Park SC, Manser E, Kim EG. betaPak-interacting exchange factor-mediated Rac1 activation requires smgGDS guanine nucleotide exchange factor in basic fibroblast growth factor-induced neurite outgrowth. J Biol Chem. 2006;281:35954–35964. doi: 10.1074/jbc.M602399200. [DOI] [PubMed] [Google Scholar]
- 32.Shin EY, Woo KN, Lee CS, Koo SH, Kim YG, Kim WJ, Bae CD, Chang SI, Kim EG. Basic fibroblast growth factor stimulates activation of Rac1 through a p85 betaPIX phosphorylation-dependent pathway. J Biol Chem. 2004;279:1994–2004. doi: 10.1074/jbc.M307330200. [DOI] [PubMed] [Google Scholar]
- 33.Shin EY, Shin KS, Lee CS, Woo KN, Quan SH, Soung NK, Kim YG, Cha CI, Kim SR, Park D, Bokoch GM, Kim EG. Phosphorylation of p85 beta PIX, a Rac/Cdc42-specific guanine nucleotide exchange factor, via the Ras/ERK/PAK2 pathway is required for basic fibroblast growth factor-induced neurite outgrowth. J Biol Chem. 2002;277:44417–44430. doi: 10.1074/jbc.M203754200. [DOI] [PubMed] [Google Scholar]
- 34.Furukawa M, He YJ, Borchers C, Xiong Y. Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat Cell Biol. 2003;5:1001–1007. doi: 10.1038/ncb1056. [DOI] [PubMed] [Google Scholar]
- 35.Krek W. BTB proteins as henchmen of Cul3-based ubiquitin ligases. Nat Cell Biol. 2003;5:950–951. doi: 10.1038/ncb1103-950. [DOI] [PubMed] [Google Scholar]
- 36.Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J. 2004;23:1681–1687. doi: 10.1038/sj.emboj.7600186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soltysik-Espanola M, Rogers RA, Jiang S, Kim TA, Gaedigk R, White RA, Avraham H, Avraham S. Characterization of Mayven, a novel actin-binding protein predominantly expressed in brain. Mol Biol Cell. 1999;10:2361–2375. doi: 10.1091/mbc.10.7.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]