

Abstract
HQP8361 (MK8033) is a novel and selective MET kinase inhibitor that has completed a phase I clinical trial. AZD9291 (osimertinib) represents the first-approved third generation EGFR-tyrosine kinase inhibitor (EGFR-TKI) for the treatment of non-small cell lung cancer (NSCLC) with activating EGFR mutations and resistant T790M mutation, but faces the giant challenge of acquired resistance developed in patients in the clinic. The current study focuses on determining the activity and mechanism of action of HQP8361 as a single agent and in combination with AZD9291 against human NSCLC cells, particularly those with acquired resistance to AZD9291. The majority of human NSCLC cell lines tested had very low levels of MET and p-MET and were insensitive to HQP8361. However, AZD9291-resistant (AR) cell lines with high levels of MET and p-MET responded to HQP8361 single agent and particularly to the combination of HQP8361 and AZD9291. The HQP8361 and AZD9291 combination synergistically decreased the survival of these HCC827/AR cell lines with enhanced induction of apoptosis that involved alteration of Bim and Mcl-1 levels via modulating their degradation. Moreover, the combination also very effectively inhibited the growth of HCC827/AR xenografts in nude mice. These preclinical findings support the potential of HQP8361 in the treatment of NSCLCs with MET amplification or highly activated MET and, when combined with AZD9291, in overcoming acquired resistance to EGFR-TKIs due to MET amplification.
Keywords: EGFR, MET, AZD9291, resistance, HQP8361, lung cancer
Introduction
The MET (c-MET) proto-oncogene is located in the long arm of human chromosome 7 and encodes MET (c-MET) protein that functions as a membrane tyrosine kinase receptor. The binding of MET to its ligand hepatocyte growth factor (HGF), secreted by stromal cells, induces dimerization and activation of the receptor, subsequent recruitment of several intracellular effector adaptor proteins such as growth factor receptor-bound protein 2 (GRB2), GAB1, SRC, and PI3K, and eventual activation of downstream signaling pathways [1]. The HGF/MET signaling pathway is highly regulated and plays an important role in cell proliferation, survival, embryogenesis, and cellular migration and invasion [2,3]. The most common variants in the HGF/MET signaling pathway in human non-small cell lung cancer (NSCLC) patients include point mutations, amplification, exon 14 skipping mutations, and fusion [4].
MET gene amplification has been recognized as an important mechanism accounting for the emergence of acquired resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) including the third generation EGFR-TKI, AZD9291 (osimertinib). MET gene amplification is detectable in approximately 5-22% of EGFR-mutant (EGFRm) NSCLC patients with acquired resistance to first generation EGFR-TKIs [5-7], and to AZD9291 and other third generation EGFR-TKIs [1]. Mechanistically, it has been suggested that MET amplification activates EGFR-independent phosphorylation of ErbB3 and downstream activation of the PI3K/AKT pathway, bypassing the suppression of an EGFR inhibitor [8]. Thus, co-inhibition of both EGFR and MET would be required to overcome resistance to EGFR-TKIs caused by MET amplification [1]. In line with this, our previous preclinical study suggested that monotherapy with AZD9291 or other third generation EGFR-TKI will likely be ineffective for the treatment of EGFRm NSCLCs with acquired resistance to first or second generation EGFR-TKIs due to MET gene amplification and protein hyper-activation and that effective treatment will require combination with MET inhibition [9]. In the clinic, patients with MET amplification after AZD9291 resistance tend to have inferior median progression-free survival and median overall survival compared to patients without the appearance of or increase in MET amplification [10]. Clinical pilot studies show that patients with EGFRm NSCLC with newly acquired MET amplification after AZD9291 resistance experienced partial response when treated with the combination of AZD9291 and crizotinib (a MET/ALK dual inhibitor) [10,11].
HQP8361 (also named MK8033) is a novel selective MET kinase inhibitor that has completed a Phase I study in solid tumors and will move forward to a Phase II trial as a monotherapy in MET amplified solid tumors including NSCLC, gastric cancer and hepatocellular carcinoma (https://www.ascentagepharma.com/product-candidates/tki-candidates/HQP-8361/). The current study focused on assessing the single agent activity of HQP8361 in human NSCLC cells and particularly its combinatorial activity with AZD9291 against EGFRm NSCLCs with acquired resistance to AZD9291. Findings from this study may provide strong preclinical support for the design and conduct of HQP8361 clinical trials and for its clinical application for overcoming AZD9291-resistance in NSCLC patients with EGFR mutations.
Materials and methods
Reagents
HQP8361 was provided by Ascentage Pharm (Rockville, MD). AZD9291, crizotinib (PF02341066) and other reagent and antibodies were the same as described previously [9].
Cell lines and cell culture
Human NSCLC cell lines including those resistant cell lines used in this study, were described in our previous studies [9,12,13]. HCC827/AR0.5 and HCC827/AR2 were established through exposing HCC827 cells to 0.5 μM or 2 μM AZD9291 once every 3 days for roughly 3 months. Bim knockout (KO) and ectopic Mcl-1 expression in HCC827/AR cell lines were established using the same procedures described in our previous studies [14,15]. These cell lines were not genetically authenticated. They were cultured in RPMI 1640 containing 5% fetal bovine serum at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Detection of MET gene copy number
Extraction of genomic DNA and the procedure for detection of MET gene copy number were the same as described previously [9].
Cell survival assays
Cell numbers in 96-well plates were measured by the sulforhodamine B (SRB) assay as previously described [16]. Combination index (CI) for drug interaction (e.g., synergy) was calculated using CompuSyn software (ComboSyn, Inc.; Paramus, NJ). The effects of the given drug treatments on colony formation in plates were determined as previously described [17].
Detection of apoptosis
Apoptosis was evaluated with an annexin V/7-AAD apoptosis detection kit (BD Biosciences; San Jose, CA) following the manufacturer’s instructions. Protein cleavages were also detected by Western blot analysis as additional indicators of apoptosis.
Western blot analysis
Preparation of whole-cell protein lysates and Western blot analysis were described previously [18].
Animal xenograft and treatments
Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Emory University and conducted as described in our previous study [9]. In brief, 6 × 106 HCC827/AR cells suspended in serum-free medium were injected subcutaneously into the flank region of five-week-old nude mice, which were purchased from Charles River Labs (Wilmington, MA). When the average tumor size reached 80 mm3 at day 8, the mice were divided into four groups (6 mice/group) based on tumor volumes and body weights for the following treatments: vehicle, AZD9291 (5 mg/kg, once daily; og), HQP8631 (25 mg/kg, once daily; og), and their combination. Tumor volumes were measured using caliper measurements once every two or three days and calculated with the formula V = π (length × width2)/6. At the end of the treatments, mice were sacrificed with CO2. The tumors were then removed, weighed, and frozen in liquid nitrogen. Certain portions of tumor tissues were homogenized in protein lysis buffer for preparation of whole-cell protein lysates for Western blotting to detect the given proteins.
Statistical analysis
The statistical significance of differences (e.g., in tumor sizes or weights) between two tested groups was analyzed with two-sided unpaired Student’s t tests (for equal variances) or with Welch’s corrected t test (unequal variances) by use of Graphpad InStat 8 software. Results were statistically significant at P < 0.05.
Results
HQP8361 is effective in suppressing the growth of AZD9291-resistant EGFRm NSCLC cell lines with hyper-activated MET
To determine the single agent activity of HQP8361 against the growth of human NSCLC cells, we first analyzed its effects on the growth of a panel of human NSCLC cell lines including those with acquired resistance to AZD9291. Among 18 cell lines tested, only the AZD9291-resistant cell line, HCC827/AR, responded to HQP8361 with an IC50 of around 1 μM (Figure 1A). By examining MET and p-MET levels in these cell lines, we found that HCC827/AR cells possessed the highest levels of both MET and p-MET, whereas other cell lines expressed low levels of MET and undetectable levels of p-MET (Figure 1B and 1C). We established two additional AZD9291-resistant cell lines from HCC827 named HCC829/AR0.5 and HCC827/AR2 through repeated treatment of HCC827 cells with fixed high concentrations of AZD9291 at 0.5 μM and 2 μM, respectively (Figure 1D). Like HCC827/AR cells, both HCC829/AR0.5 and HCC827/AR2 exhibited high levels of MET and p-MET (Figure 1E) with amplified MET gene (Figure 1F) and responded better than HCC827 cells to HQP8361 (Figure 1G). These data clearly suggest that HQP8361 has single agent activity only in cell lines with hyper-activated MET or amplified MET gene.
Figure 1.

Human NSCLC cell lines are in general insensitive to HQP8361 (A and G) except those with MET amplification and high levels of MET and p-MET (B-F). (A, D and G) The indicated cell lines seeded in 96-well plates were exposed to varied concentrations of HQP8361 (A and G) or AZD9191 (D) for 3 days. Cell numbers were estimated with sulforhodamine B (SRB) assay. The data are means ± SDs of four replicate determinations. (B, C and E) The indicated cell lines with similar cell densities were harvested for preparation of whole-cell protein lysates and then used for Western blotting to detect the indicated proteins. SE, shorter exposure. (F) MET gene copy numbers in the given cell lines were determined from genomic DNA using qPCR. The data are means ± SEs of two experiments. P, parental.
HQP8361, when combined with AZD9291, synergistically decreases the survival of AZD9291-resistant cell lines with hyper-activated MET and enhances induction of apoptosis
Given that MET amplification is an important mechanism accounting for the emergence of acquired resistance to EGFR-TKIs including AZD9291 and that targeting MET can overcome acquired AZD9291 resistance caused by this mechanism [1,9], we then examined the effects of the HQP8361 and AZD9291 combination on the survival and apoptosis induction of different AZD9291-resistant cell lines. Treatment with each agent alone minimally decreased the survival of HCC827/AR cells, while the combination very effectively decreased survival with CIs < 1 (Figure 2A), indicating synergistic effect. However, the combination was ineffective in both PC-9/AR and PC-9/3M cell lines (Figure 2A). Consistently, the combination enhanced induction apoptosis in HCC827/AR cells, but not in PC-9/AR cells as measured by both annexin V-positive cells (Figure 2B) and PARP cleavage (Figure 2C). Colony formation assay, which allows repeated treatment of cells, also demonstrated that the combination of HQP8361 and AZD9291 was much more effective than either single agent alone in inhibiting the formation and growth of HCC827/AR cells (Figure 2D). Similarly, the HQP8361 and AZD9291 combination synergistically decreased the survival (Figure 2E) and enhanced the induction of apoptosis (Figure 2F and 2G) of both HCC827/AR0.5 and HCC827/AR2 cells. These findings suggest that the combination of HQP8361 and AZD9291 is very effective against AZD9291-resistant cells with MET amplification or MET hyper-activation.
Figure 2.
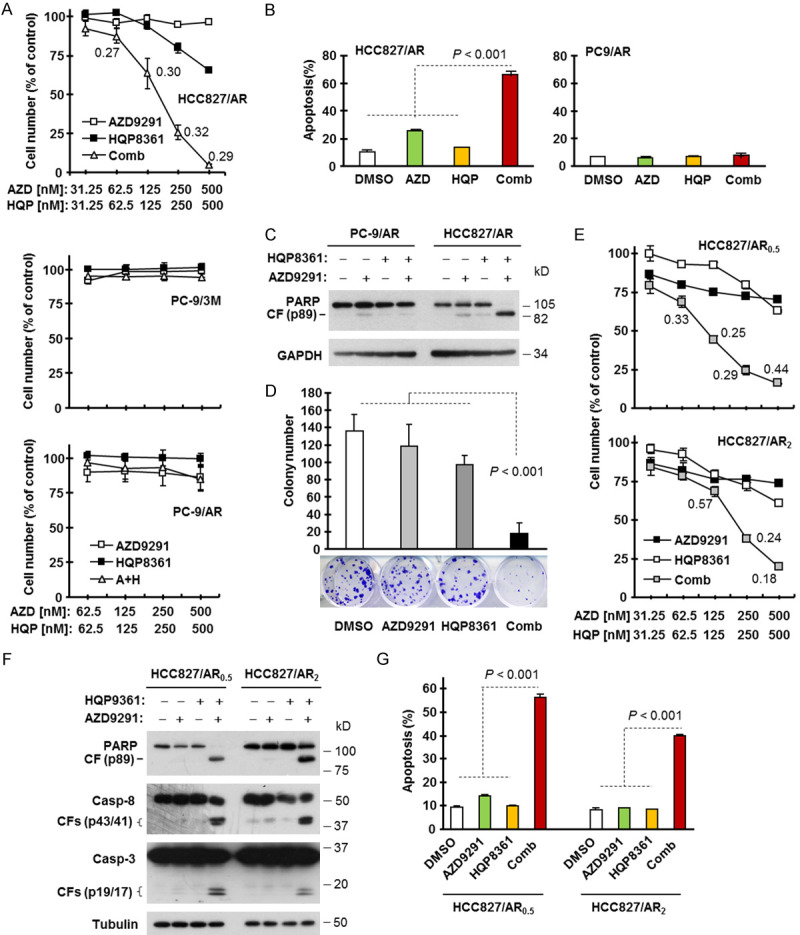
HQP8361 combined with AZD9291 synergistically decreases cell survival (A and E), induces apoptosis (B, C, F and G) and suppresses colony formation and growth (D) in c-Met activated AZD9291-resistant NSCLC cell lines. (A and E) The indicated cell lines seeded in 96-well plates were treated with varied concentrations of HQP8361 alone, AZD9291 alone and their respective combinations. After 3 days, cell numbers were determined with sulforhodamine B (SRB) assay. The data are means ± SDs of four replicate determinations. The numbers inside the graphs are CIs. (B, C, F and G) The indicated cell lines were treated with DMSO, 200 nM AZD9291, 250 nM HQP8361 and AZD9291 plus HQP8361 for 48 h. Apoptosis was evaluated with Western blotting for detection of PARP and caspase cleavage (C, F) and with annexin V/flow cytometry (B, G). Data in (B and G) are means ± SDs of duplicate determinations. (D) HCC827/AR cells seeded in 12-well culture plates were treated with DMSO, 100 nM AZD9291, 100 nM HQP8361 or the combination of AZD9291 and HQP8361; these treatments were repeated every 3 days. After 12 days, the plates were stained with crystal violet dye, counted and then photographed. The data are means ± SDs of triplicate determinations. The statistical difference between two given groups was analyzed with the two-sided unpaired Student’s t test.
The combination of HQP8361 and AZD9291 enhances Bim elevation and Mcl-1 reduction accompanied with augmented suppression of mTOR signaling
To understand the mechanisms by which the combination of HQP8361 and AZD9291 enhances induction of apoptosis, we conducted time-course analyses of Bim and Mcl-1 alterations, two key events that mediate AZD9291-induced apoptosis in EGFRm NSCLC cells [12], in HCC827/AR cells in response to the combination treatment. As presented in Figure 3A, Mcl-1 levels were reduced the most and Bim levels were increased the most in cells exposed to the combination, in comparison with levels in cells exposed to either HQP8361 or AZD9291 alone. These effects occurred early at 8 h and extended to 36 h after treatment in parallel with increased PARP cleavage. Consistently, enhanced Bim elevation and Mcl-1 reduction by the HQP8361 and AZD9291 combination was also observed in HCC827/AR0.5 cells (data not shown). Similar effects on Mcl-1 and Bim were also observed in cells exposed to the combination of crizotinib (another known MET inhibitor) and AZD9291, although the reduction of Mcl-1 was greater in HCC827/AR cells treated with HQP8361 and AZD9291 combination than in those exposed to crizotinib and AZD9291 combination because crizotinib alone apparently decreased Mcl-1 levels (Figure 3B).
Figure 3.
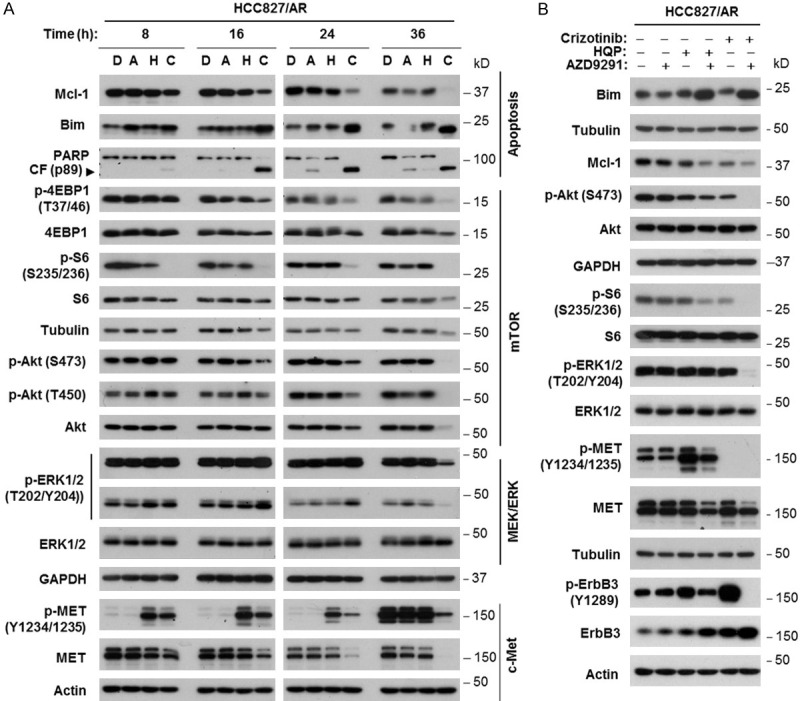
The combination of AZD9291 and HQP8361 enhances modulation of apoptosis-associated protein levels, MET levels and mTOR signaling (A) in HCC827/AR cells, with effects on ERK and MET phosphorylation different from those of the HQP8361 and crizotinib combination (B). HCC827/AR cells were exposed to DMSO (D), 200 nM AZD9291 (A), 250 nM HQP8361 (H), 200 nM crizotinib, AZD9291 plus HQP8361 (C) or AZD9291 plus crizotinib for varied times as indicated (A) or 18 h (B). The cells were then harvested for preparation of whole-cell protein lysates and subsequent Western blot analysis.
Moreover, we assessed the impact of the HQP8361 and AZD9291 combination on signaling pathways downstream of EGFR and MET that may modulate Bim and Mcl-1 levels in this cell line. Interestingly, the combination did not enhance suppression of ERK1/2 phosphorylation until a very late time point (e.g., 36 h; Figure 3A). This differed from the effect of the crizotinib and AZD9291 combination, which substantially enhanced reduction of p-ERK1/2 levels (Figure 3B). Augmented inhibition of S6 phosphorylation (S235/236) was clearly detected early at 8 h, whereas enhanced suppression of Akt (both S473 and T450) and 4EBP1 (T37/46) phosphorylation was observed a little later, at or after 16 h post treatment. We noted that the combination exerted much more substantial suppression of S6 phosphorylation than suppression of Akt and 4EBP1 phosphorylation (Figure 3A). Strong suppression of S6 phosphorylation by the combination was also observed in HCC827/AR0.5 cells (data not shown). Similar effects were seen with the combination of crizotinib and AZD9291 (Figure 3B). Nonetheless, the combination of HQP8361 and AZD9291 enhanced suppression of mTOR signaling in HCC827/AR cells.
We noted that treatment with HQP8361 alone increased the levels of p-MET in both HCC827/AR and HCC827/AR0.5 cells; this effect could be suppressed when combined with AZD9291. Further, the combination of HQP8361 and AZD9291 clearly decreased total MET levels (Figure 3A and data not shown). In contrast, crizotinib alone abolished MET phosphorylation (Figure 3B). However, both HQP8361 and particularly crizotinib increased the levels of both ErbB3 and p-ErbB3 in HCC827/AR cells. When combined with AZD9291, increased ErbB3 phosphorylation was abolished despite further increase in ErbB3 levels (Figure 3B).
The combination of HQP8361 and AZD9291 regulates Bim and Mcl-1 levels through modulation of their protein stability
Considering that AZD9291 combined with crizotinib enhances the elevation of Bim levels by delaying its degradation [9], we next analyzed whether the combination of HQP8361 and AZD9291 affects the degradation of Bim and Mcl-1, resulting in enhanced Bim elevation and Mcl-1 reduction in HCC827/AR cells. By conducting a CHX chase assay, we observed that Bim was degraded much more slowly and Mcl-1 was degraded much faster in cells exposed to the combination of HQP8361 and AZD9291 than in those exposed to DMSO, HQP8361 or AZD9291 alone (Figure 4). These results demonstrate that the combination of HQP8361 and AZD9291 slows Bim degradation while facilitating Mcl-1 degradation in HCC827/AR cells.
Figure 4.
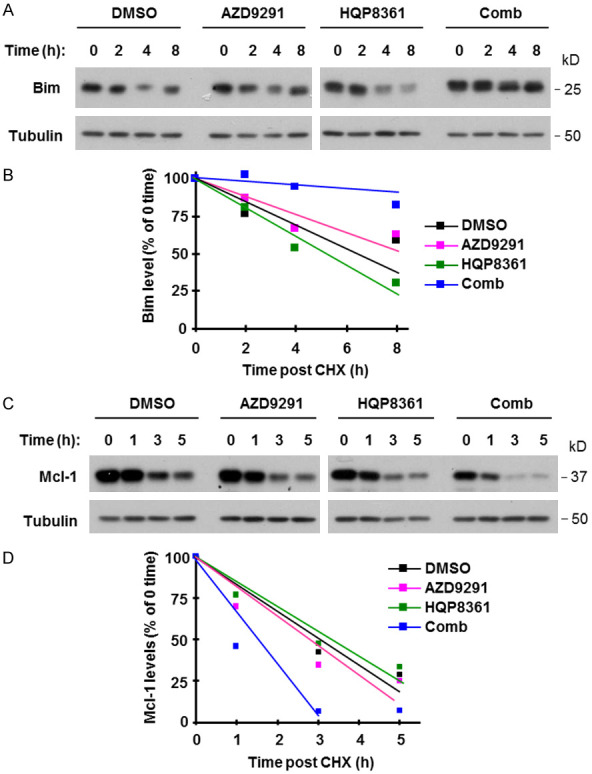
The combination of AZD9291 and HQP8361 stabilizes Bim protein (A and B), but induces Mcl-1 protein degradation (C and D) in HCC827AR cells. HCC827/AR cells were treated with DMSO, 200 nM AZD9291, 250 nM HQP8361 or the combination of AZD9291 and HQP8361 for 8 h and then exposed to 10 ug/ml CHX. At the indicated times after addition of CHX, Western blotting was used to detect the indicated proteins (A and C). NIH image J software was used to quantify band intensities. Bim and Mcl-1 levels are shown as percentage of levels at 0 time post CHX treatments (B and D).
Enhanced Bim elevation and Mcl-1 contribute to augmented induction of apoptosis in HCC827/AR cells
To demonstrate the role of Bim elevation in enhancing apoptosis by the HQP8361 and AZD9291 combination, we generated Bim knockout cell lines from HCC827/AR cells and then examined the impact of Bim loss on the induction of apoptosis by the HQP8361 and AZD9291 combination. As presented in Figure 5A and 5B, the combination of HQP8361 and AZD9291 enhanced the induction of apoptosis as measured by PARP cleavage and annexin V-positive cells in the control HCC827/AR cells as demonstrated above, but these effects were significantly attenuated in the two tested Bim KO cell lines. In HCC827/AR cells expressing ectopic Mcl-1, the combination of HQP8361 and AZD9291 induced much less cleavage of PARP and caspase-3 and significantly fewer annexin V-positive cells than in vector-control cells (Figure 5C and 5D), indicating that enforced Mcl-1 expression in HCC827/AR cells compromises the ability of the HQP8361 and AZD9291 combination to enhance the induction of apoptosis.
Figure 5.
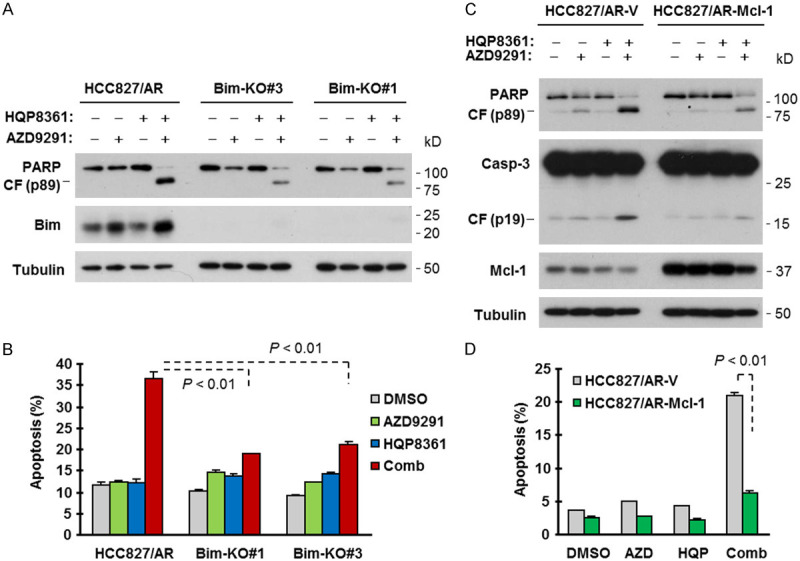
Bim knockout (A and B) or enforced expression of ectopic Mcl-1 (C and D) compromises the activity of HQP8361 and AZD9291 in enhancing the induction of apoptosis in HCC827/AR cells. The indicated cell lines with Bim knockout (A) and Mcl-1 overexpression (C) were exposed to DMSO, 200 nM AZD9291, 250 nM HQP8361 or HQP8361 plus AZD9291 for 48 h. The cells were then harvested for preparation of whole-cell lysates and subsequent detection of the tested proteins by Western blotting (A and C) and for detection of apoptotic cells with annexin V/flow cytometry (B and D). The data are means ± SDs of duplicate determinations. The statistical difference between two given groups was analyzed with the two-sided unpaired Student’s t test.
The combination of HQP8361 and AZD9291 significantly inhibits the growth of HCC827/AR tumors in vivo with modulation of the levels of Bim, Mcl-1 and other protein markers
We lastly determined the effect of the HQP8361 and AZD9291 combination on the growth of HCC827/AR xenografts in nude mice. While treatment with HQP8361 or AZD9291 alone had limited inhibitory effect on the growth of HCC827/AR tumors, the combination of HQP8361 and AZD9291 potently and significantly retarded the growth of HCC827/AR tumors in comparison with either single agent treatment in terms of tumor sizes (Figure 6A and 6B) and weights (Figure 6C). Mice receiving the combination treatment had comparable body weights with those in other groups (Figure 6D), indicating that the combination does not accordingly increase toxicity while enhancing anti-tumor activity.
Figure 6.
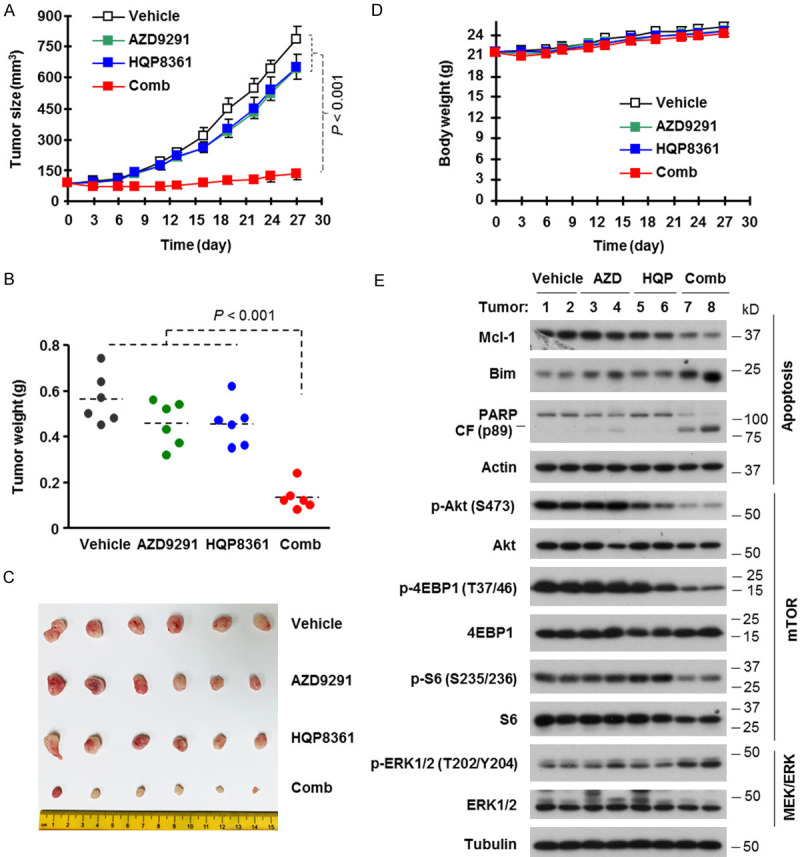
The combination of AZD9291 and HQP8361 effectively inhibits the growth of HCC827/AR xenografts (A-C) without enhancing mouse toxicity (D) and modulates the levels of Bim, Mcl-1 and other proteins in vivo (E). HCC827/AR xenografts were treated (once daily) with vehicle, 5 mg/kg AZD9291 (og), 25 mg/kg HQP8361 (og) or AZD9291 plus HQP8361. Tumor sizes and mouse body weights were measured once every two or three days (A and D). At the end of the treatments, mice were sacrificed and tumors were removed, weighed (B) and photographed (C). The data are means ± SE (n = 6). Whole-tumor protein lysates were prepared from two tumors randomly in each group to detect the indicated proteins by Western blotting (E). CF, cleaved fragment. The statistical difference between two given groups was analyzed with the two-sided unpaired Student’s t test.
We also analyzed alterations of several protein markers in these tumor tissues and found that the combination of AZD9291 and HQP8361 effectively decreased Mcl-1 levels and increased the levels of Bim and cleaved PARP, whereas each single agent had limited or no modulatory effects on the levels of these proteins (Figure 6E). These results indicate that the HQP8361 and AZD9291 combination enhances the modulation of Bim and Mcl-1 levels accompanied with augmented induction of apoptosis in vivo. In agreement with the data generated in cell cultures, the combination of HQP8361 and AZD9291 was more effective than each single agent in decreasing the levels of p-S6, p-4EBP1 and p-Akt, but did not affect p-ERK levels (Figure 6E), indicating that the combination also effectively enhances the suppression of mTOR signaling in vivo.
Discussion
In the current study, we found that among a panel of 18 human NSCLC cell lines, the novel MET selective inhibitor HQP8361 showed single agent activity against the growth of the only cell line, HCC827/AR, with acquired resistance to AZD9291 and other EGFR-TKIs due to MET gene amplification and MET hyper-activation [9]. Other NSCLC lines tested, including 4 cell lines with acquired resistance to AZD9291, had low levels of MET and particularly p-MET and were insensitive to HQP8361. These findings together suggest that HQP8631 may have single agent activity only in cancer cells with high levels of MET (e.g., due to gene amplification) or hyper-activated MET. AZD9291-resistant cell lines such as HCC827/AR with MET amplification or hyper-activated MET protein fall into this category, implying that EGFRm NSCLCs with acquired resistance to AZD9291 due to MET gene amplification and protein hyper-activation may be responsive to HQP8361 monotherapy. This is supported by our data generated from two additional HCC827-derived AZD9291-resistant cells lines (HCC827/AR0.5 and HCC827/AR2) with MET amplification/hyper-activated MET and responsiveness to HQP8361.
MET inhibition has been suggested to be a potential therapeutic strategy for overcoming acquired resistance caused by MET amplification [1,9]. Indeed, HQP8361 synergized with AZD9291 in decreasing cell survival and inducing apoptosis only in HCC827/AR cell lines including HCC827/AR0.5 and HCC827/AR2, but not in other AZD9291-resistant cell lines without activated MET. Our in vivo study also demonstrated very promising activity of the HQP8361 and AZD9291 combination against the growth of HCC827/AR tumors and enhanced induction of apoptosis in these tumors. Hence, HQP8361, when combined with AZD9291, shows great potential for overcoming acquired resistance to AZD9291 and possibly other EGFR-TKIs due to MET gene amplification or protein hyper-activation, likely via enhancement of apoptosis. Therefore, MET detection is critical and may guide the selection of patients with acquired resistance to EGFR-TKIs who may benefit from treatment with the HQP8361 and AZD9291 combination.
We previously reported that AZD9291 combined with another MET inhibitor, such as crizotinib or SGX523, enhances the induction of apoptosis in HCC827/AR cells and effectively suppresses the growth of HCC827/AR tumors [9]. Mechanistically, HQP8361, when combined with AZD9291, shares some common mechanisms with crizotinib or SGX523 in enhancing Bim elevation by delaying Bim degradation and augmenting suppression of Akt, S6 and ErbB3 phosphorylation [9]. However, HQP8361 possesses some features that are distinct from those of crizotinib and/or SGX523. The combination of crizotinib or SGX523 with AZD9291 effectively decreased p-ERK1/2 levels in HCC827/AR cells [9]; but this was not the case for the combination of HQP8361 and AZD9291 as demonstrated both in vitro and in vivo. Treatment with crizotinib alone decreased Mcl-1 levels but when combined with AZD9291 caused little or no further decrease in Mcl-1 levels [9]. In contrast, HQP8361 alone did not apparently decrease Mcl-1 levels, but rapidly and robustly decreased Mcl-1 levels when combined with AZD9291. Another clear difference between HQP8361 and crizotinib or SGX523 is that HQP8361 alone increased p-MET levels, whereas both crizotinib and SGX523 potently abolished MET phosphorylation [9]. However, HQP8361 combined with AZD9291 reduced the levels of p-MET induced by HQP8361 and interestingly decreased MET levels. Albeit with slightly varied mechanisms, these MET inhibitors effectively overcome acquired resistance to AZD9291 or other EGFR-TKIs caused by MET amplification when combined with AZD9291.
Phosphorylation of 4EBP1 and/or S6 (or p70S6K) is known to be the downstream readout of mTOR complex 1 (mTORC1) activity, whereas Akt phosphorylation (at both S437 and T450) is the readout of mTORC2 [19-21]. In this study, treatment with the HQP8361 and AZD9291 combination enhanced suppression of the phosphorylation of 4EBP1, pS6 and Akt both in vitro and in vivo, indicating that the combination effectively inhibits both mTORC1 and mTORC2 signaling pathways in AZD9291-resistant cells and tumors in the background of MET gene amplification and/or MET protein hyper-activation. It has been suggested that MET amplification and protein hyper-activation causes EGFR-TKI resistance mechanistically by activating EGFR-independent phosphorylation of ErbB3 and downstream activation of the PI3K/AKT pathway, providing a bypass pathway in the presence of an EGFR-TKI [8]. Given that the HQP8361 and AZD9291 combination also effectively suppressed ErbB3 phosphorylation, it is reasonable to speculate that the suppression of mTORC1 and mTORC2 signaling by the combination is likely to be secondary to ErbB3 suppression.
We previously established the HCC827/AR cell line through exposure to a gradually increasing concentration of AZD9291, starting with a low concentration (e.g. 10 nM), over a long period of time [9]. In this study, we used a different strategy to establish two additional AZD9291-resistant lines (i.e., HCC827/AR0.5 and HCC827/AR2) through repeated exposure of HCC827 cells directly to fixed high concentrations of AZD9291 at 500 nM, which is close to the clinically achievable steady-state plasma level of AZD9291 in NSCLC patients receiving a 80 mg/day dosage [22], and at 2 μM, respectively, for roughly 3 months. This procedure may better recapitulate the clinical situation where acquired resistance develops during treatment with AZD9291. These cell lines showed similar features with substantially elevated levels of MET and p-MET due to MET amplification and increased sensitivities to HQP8361 or HQP8361 and AZD9291 combination. The establishment of these new cell lines provides additional models for studying AZD9291 resistance.
In summary, the current study provides preclinical evidence in support of the potential efficacy of the novel MET inhibitor, HQP8361, in the treatment of NSCLCs with highly activated MET as a monotherapy and of EGFRm NSCLC with resistance to AZD9291 or other EGFR-TKIs due to MET amplification or protein hyper-activation when combined with AZD9291. Thus, the future validation of HQP8361 for these indications in the clinic is warranted.
Acknowledgements
We are grateful to Dr. Anthea Hammond in our department for editing the manuscript. This study was supported by NIH/NCI R01 CA223220 (to SYS) and UG1 CA233259 (to SSR, TKO and SYS), Emory Winship Cancer Institute lung cancer research pilot funds (to SYS) and Lee Foundation Award to the Winship Lung Cancer Program for supporting the pilot project. DY and YL are visiting medical students participating in the Xiangya-Emory Visiting Medical Student Program. TKO, SSR and SYS are Georgia Research Alliance Distinguished Cancer Scientists.
Disclosure of conflict of interest
SSR is on consulting/advisory boards for AstraZeneca, BMS, Merck, Roche, Tesaro and Amgen. TKO is on consulting/advisory boards for Novartis, Celgene, Lilly, Sandoz, Abbvie, Eisai, Takeda, Bristol-Myers Squibb, MedImmune, Amgen, AstraZeneca and Boehringer Ingelheim. No potential conflicts of interest were disclosed for other authors.
References
- 1.Wang Q, Yang S, Wang K, Sun SY. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J Hematol Oncol. 2019;12:63. doi: 10.1186/s13045-019-0759-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang J, Babic A. Regulation of the MET oncogene: molecular mechanisms. Carcinogenesis. 2016;37:345–355. doi: 10.1093/carcin/bgw015. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Xia M, Jin K, Wang S, Wei H, Fan C, Wu Y, Li X, Li X, Li G, Zeng Z, Xiong W. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer. 2018;17:45. doi: 10.1186/s12943-018-0796-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma PC. MET receptor juxtamembrane exon 14 alternative spliced variant: novel cancer genomic predictive biomarker. Cancer Discov. 2015;5:802–805. doi: 10.1158/2159-8290.CD-15-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tartarone A, Lerose R. Clinical approaches to treat patients with non-small cell lung cancer and epidermal growth factor receptor tyrosine kinase inhibitor acquired resistance. Ther Adv Respir Dis. 2015;9:242–250. doi: 10.1177/1753465815587820. [DOI] [PubMed] [Google Scholar]
- 6.Juchum M, Gunther M, Laufer SA. Fighting cancer drug resistance: opportunities and challenges for mutation-specific EGFR inhibitors. Drug Resist Updat. 2015;20:10–28. doi: 10.1016/j.drup.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Remon J, Moran T, Majem M, Reguart N, Dalmau E, Marquez-Medina D, Lianes P. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: a new era begins. Cancer Treat Rev. 2014;40:93–101. doi: 10.1016/j.ctrv.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 9.Shi P, Oh YT, Zhang G, Yao W, Yue P, Li Y, Kanteti R, Riehm J, Salgia R, Owonikoko TK, Ramalingam SS, Chen M, Sun SY. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016;380:494–504. doi: 10.1016/j.canlet.2016.07.021. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Li L, Han R, Jiao L, Zheng J, He Y. Clinical analysis by next-generation sequencing for NSCLC patients with MET amplification resistant to osimertinib. Lung Cancer. 2018;118:105–110. doi: 10.1016/j.lungcan.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 11.Zhu VW, Schrock AB, Ali SM, Ou SI. Differential response to a combination of full-dose osimertinib and crizotinib in a patient with EGFR-mutant non-small cell lung cancer and emergent MET amplification. Lung Cancer (Auckl) 2019;10:21–26. doi: 10.2147/LCTT.S190403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi P, Oh YT, Deng L, Zhang G, Qian G, Zhang S, Ren H, Wu G, Legendre B Jr, Anderson E, Ramalingam SS, Owonikoko TK, Chen M, Sun SY. Overcoming acquired resistance to AZD9291, a third-generation EGFR inhibitor, through modulation of MEK/ERK-dependent bim and Mcl-1 degradation. Clin Cancer Res. 2017;23:6567–6579. doi: 10.1158/1078-0432.CCR-17-1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zong D, Gu J, Cavalcante GC, Yao W, Zhang G, Wang S, Owonikoko TK, He X, Sun SY. BRD4 levels determine the response of human lung cancer cells to BET degraders that potently induce apoptosis through suppression of Mcl-1. Cancer Res. 2020;80:2380–2393. doi: 10.1158/0008-5472.CAN-19-3674. [DOI] [PubMed] [Google Scholar]
- 14.Qian G, Yao W, Zhang S, Bajpai R, Hall WD, Shanmugam M, Lonial S, Sun SY. Co-inhibition of BET and proteasome enhances ER stress and Bim-dependent apoptosis with augmented cancer therapeutic efficacy. Cancer Lett. 2018;435:44–54. doi: 10.1016/j.canlet.2018.07.033. [DOI] [PubMed] [Google Scholar]
- 15.Ren H, Koo J, Guan B, Yue P, Deng X, Chen M, Khuri FR, Sun SY. The E3 ubiquitin ligases beta-TrCP and FBXW7 cooperatively mediates GSK3-dependent Mcl-1 degradation induced by the Akt inhibitor API-1, resulting in apoptosis. Mol Cancer. 2013;12:146. doi: 10.1186/1476-4598-12-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun SY, Yue P, Dawson MI, Shroot B, Michel S, Lamph WW, Heyman RA, Teng M, Chandraratna RA, Shudo K, Hong WK, Lotan R. Differential effects of synthetic nuclear retinoid receptor-selective retinoids on the growth of human non-small cell lung carcinoma cells. Cancer Res. 1997;57:4931–4939. [PubMed] [Google Scholar]
- 17.Wang X, Hawk N, Yue P, Kauh J, Ramalingam SS, Fu H, Khuri FR, Sun SY. Overcoming mTOR inhibition-induced paradoxical activation of survival signaling pathways enhances mTOR inhibitors’ anticancer efficacy. Cancer Biol Ther. 2008;7:1952–1958. doi: 10.4161/cbt.7.12.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao W, Yue P, Zhang G, Owonikoko TK, Khuri FR, Sun SY. Enhancing therapeutic efficacy of the MEK inhibitor, MEK162, by blocking autophagy or inhibiting PI3K/Akt signaling in human lung cancer cells. Cancer Lett. 2015;364:70–78. doi: 10.1016/j.canlet.2015.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle. 2011;10:2305–2316. doi: 10.4161/cc.10.14.16586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sparks CA, Guertin DA. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene. 2010;29:3733–3744. doi: 10.1038/onc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown K, Comisar C, Witjes H, Maringwa J, de Greef R, Vishwanathan K, Cantarini M, Cox E. Population pharmacokinetics and exposure-response of osimertinib in patients with non-small cell lung cancer. Br J Clin Pharmacol. 2017;83:1216–1226. doi: 10.1111/bcp.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]