

Abstract
Breast cancer remains a complex disease resulting in high mortality in women. A subset of cancer stem cell (CSC)-like cells expressing aldehyde dehydrogenase 1 (ALDH1) and SOX2/OCT4 are implicated in aggressive biology of specific subtypes of breast cancer. Targeting these populations in breast tumors remain challenging. We examined xenografts from three poorly studied triple negative (TN) breast cancer cells (MDA-MB-468, HCC70 and HCC1806) as well as HMLEHRASV12 for stem cell (SC)-specific proteins, proliferation pathways and dual-specific phosphatases (DUSPs) by quantitative real-time PCR (qRT-PCR), immunoblot analysis and immunohistochemistry. We found that pERK1/2 remained suppressed in TN xenografts examined at various stages of growth, while the levels of pp38 MAPK and pAKT was upregulated. We found that DUSP was involved in the suppression of pERK1/2, which was MEK1/2 independent. Our in vitro assays, using HMLEHRASV12 xenografts as a positive control, confirmed increased phosphatase activity that specifically influenced pERK1/2 but not pp38MAPK or pJNK levels. Family members of DUSPs examined, showed increase in DUSP9 expression in TN xenografts. Increased DUSP9 expression in xenografts was consistently associated with upregulation of SC-specific proteins, ALDH1 and SOX2/OCT4. HRAS driven HMLEHRASV12 xenografts as well as mammospheres from TN breast cancer cells showed inverse relationship between pERK1/2 and increased expression of DUSP9 and CSC traits. In addition, treatment in vitro, with MEK1/2 inhibitor, PD 98059, reduced pERK1/2 levels and increased DUSP9 and SC-specific proteins. Depletion of subsets of SOX2/OCT4 by fluorescence-activated cell sorting (FACS), as well as pharmacological and genetic reduction of DUSP9 levels influenced ALDH1 and SOX2/OCT4 expression and reduced mammosphere growth in vitro as well as tumor growth in vivo. Collectively our data support the possibility that DUSP9 contributed to stem cell-like cells that could influence TN breast tumor growth. Conclusion: Our study shows that subsets of TN breast cancers with MEK1/2 independent reduced pERK1/2 levels will respond less to MEK1/2 inhibitors, thereby questioning their therapeutic efficacy. Our study also demonstrates context-dependent DUSP9-mediated reduced pERK1/2 levels could influence stem cell-like traits in TN breast tumors. Therefore, targeting DUSP9 could be an attractive target for improved clinical outcome in a subset of basal-like breast cancers.
Keywords: Triple negative breast cancer, cancer stem cells, ERK1/2, DUSP9
Introduction
Breast cancer, a complex disease, is the most commonly diagnosed cancer among American women [1-3]. One in eight women will be diagnosed with the disease in their lifetime [2]. High mortality rates from breast cancer, which kills 44,000 women in US per year, is second only to lung cancer [2]. Molecular heterogeneity has been identified within breast cancer based on gene expression and genomic or proteomic profiling [4]. Among the various subtypes, basal-like breast cancers, which lack estrogen receptor (ER), progesterone receptor (PR) and HER2/neu protein, have an aggressive clinical behavior and worst prognosis [5]. African American (AA) and younger women frequently develop basal-like breast cancer, 70-80% of which are triple negative for ER, PR and HER2/neu [6]. These triple negative breast cancers (TNBC) are very heterogeneous, have a high content of mammary cancer stem cells (MCSCs) and are refractive to most treatments, thereby causing high mortality rates [7].
MCSCs are a small population of cancer cells with high potency for self-renewal, differentiation, tumor initiation and progression [8-10]. Current thinking is that some CSCs arise from stem cells (SCs), which acquire mutations to become founder cells of origin of breast cancer [11]. Cancer cells can also acquire a stem cell-like phenotype induced by epithelial to mesenchymal transition (EMT), chemotherapy or targeted therapy [12]. Clearly, CSCs are heterogeneous and have a high degree of plasticity that enables their organization in various hierarchical fashions to promote tumor development [13]. Expression of cell surface markers CD44/CD24 and high aldehyde dehydrogenase (ALDH) activity have facilitated detection and isolation of subsets of CSCs from breast tumors [14]. In addition, some aggressive breast tumors express embryonic stem cell (ESC)-associated transcriptional regulators SOX2/OCT4, which are essential for maintaining pluripotency [15]. Although a number of mutations and deregulated pathways have been identified in CSCs, druggable targets remain elusive. Despite limited success, there is a huge effort to target TN cancer cells including MCSCs to reduce tumor burden and bring about longterm stabilization of the disease.
Most existing therapies for TNBC target deregulated PTEN/AKT and MAPK pathways for better clinical outcomes [16,17]. Deregulated PTEN/AKT pathway promote aggressive tumor characteristics in many subtypes of breast cancer [18,19]. However, therapies targeting either PTEN/AKT or Ras/Raf/MEK/ERK1/2 pathway individually or in combination have failed to reduce tumor progression [20,21]. Most of the clinical trials with MEK1/2 inhibitors have proved ineffective in part because their efficacy is limited only to in vitro assays using a large panel of breast cancer cells [22].
Various phosphatases, particularly the dual specific phosphatases (DUSP), could also contribute to reduce the therapeutic efficacy of MEK1/2 inhibitors in cancer treatment. Phosphatases regulate both duration, magnitude and spatio-temporal profiles of MAPK activation, which is crucial in determining the physiological outcomes of cells [23]. DUSP dephosphorylate threonine/serine and or tyrosine residues of the T-X-Y motif within the kinase activation loop to regulate MAPK activity in normal tissues [23]. There are reports of aberrant expression of DUSPs in some subtypes of breast cancer, but their influence in AA breast cancers remain poorly understood. Some DUSPs including DUSP9 implicated in pluripotency could play a role in AA breast cancers, which contain high levels of undifferentiated cancer stem-like cells. DUSP9 been shown to maintain murine ESC pluripotency and self-renewal status by controlling appropriate ERK activity [24]. DUSP9, which was downstream of BMP4/Smad 1/5 signaling axis and steadily attenuated ERK1/2 activity in murine ESCs to reduce spontaneous differentiation [24]. In addition, DUSP9 has also been demonstrated to modulate DNA hypomethylation in female mouse pluripotent stem cells [25]. However, the role of DUSP9 in influencing human ESCs in any cancer has not been investigated.
In this study, we sought to determine relationship between ERK1/2 kinases, phosphatases and stem cell-like traits in xenografts from understudied AA TNBC cells. We used xenografts from HRAS overexpressing HMLEHRASV12 cell line as a positive control for most of our experiments. To our surprise, we found a novel mechanism operating in xenografts from these TN breast cancer cells, where pERK1/2 remained suppressed in a MEK1/2 independent manner. Interestingly, we found dual specific phosphatase 9 (DUSP9)-mediated suppression of pERK1/2 increased stem cell-like traits in TN breast tumors.
Methods
Human cell lines
AA breast cancer cells HCC-1806, HCC-70, MDA-MB-468, MDA-MB-231, and MCF7 were purchased from American Type Culture Collection (ATCC) in 2013. These cells were propagated in RPMI 1640 containing 10% FBS [26]. The ATCC uses Promega PowerPlex 1.2 system and the Applied Biosystems Genotyper 2.0 software for analysis of amplicon. We have not done further testing in our lab. The HMLEHRASV12 cell line, generated by transforming HMLE (human mammary epithelial) cells with HRAST24 oncogene, was collected in 2011 from Robert Weinberg (Whitehead Institute, Cambridge, MA). HMLEHRASV12 cell line was cultured in DMEM-F12 containing 10% FBS.
Xenograft formation
Established breast cancer cell lines HCC1806, HCC70, MDA-MB-468 and HMLEHRASV12 (2×106 cell/100 μL), mixed with Matrigel (1:1) were implanted subcutaneously in the dorsolateral abdominal region of nude mice (Harlan laboratories Inc.) for xenograft growth. To generate transplantable xenografts, primary xenografts were dissociated with 0.2% collagenase (type IV, Sigma-Aldrich) for 1 hour at 37°C, propagated for 48 hours in vitro and further implanted (1×106 cells/injection) subcutaneously in nude mice. Tumor growth was monitored weekly using calipers for length and width measurements, for the duration of the experiment. Tumor volume was calculated as ½ (length × width2) [27]. Tumors excised at various time points were either fixed for paraffin embedding/fixing or immunoblot and qRT PCR analysis. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Charles R. Drew University of Medicine and Science (Los Angeles, CA; permit number: 1-1103-261).
Immunoblot analysis
Xenografts at various time points were homogenized with tissue protein extraction reagent (T-PER) (Thermo Fisher Scientific, Rockford, IL) and protein concentrations were determined using Pierce BCA Protein assay kit (Thermo Fisher Scientific, Cat# 88667) and measured using the Spectra Max spectrophotometer (Model Spectra Max 190) at 545 nm. Cell or tissue lysates (100 µg) were resolved on 10%-15% SDS-PAGE gels and electro-transferred to polyvinylidiene difluoride (PVDF) (BioRad, Hercules, CA, # 1620177). The membranes were incubated with the following primary antibodies at 1:1,000 dilutions: ERK1/2 (Cell Signaling Technology, # 9102), pERK1/2 (Cell Signaling Technology, # 9106), MEK1/2, (Cell Signaling Technology, # 9126), pMEK1/2 (Cell Signaling Technology, # 9121), DUSP9 (Abcam, ab54941), ALDH1 (Abcam, ab9883), OCT4 (Stem Cell Technologies, # 60059), SOX2 (Abcam, ab97959), CD44, (Cell Signaling Technology, # 3570) AKT (Cell Signaling Technology, # 2920), pAKT (Cell Signaling Technology, # 9271), p38MAPK (Cell Signaling Technology, # 9212), pp38 MAPK (Cell Signaling Technology, # 9211), β-actin (Santa Cruz BioTech, sc81178) or GAPDH (Millipore, MAB374). The membranes were further incubated with appropriate rabbit (Cell Signaling Technology, # 7074) or mouse (Cell Signaling Technology, # 7076) horseradish peroxidase-linked F (ab) fragment secondary antibodies (1:1000) for 1 h. Immunoreactive bands were visualized by enhanced chemiluminescence (ECL) detection system (Amersham, Pittsburgh, PA) as described previously [27].
Quantitative real-time PCR
Total RNA was extracted by Trizol reagent and quantified using the Nano Drop. Equal amounts (2 μg) of RNA were reverse transcribed using RNA High Capacity cDNA kit (Applied Biosystems, Foster City, CA). The Power Sybr Green PCR master mix was used with 7500 fast real-time PCR system (Applied Biosystems). Human and mouse PCR primer sequence were obtained from Primer Bank DNA Core facility (http://pga.mph.harvard.edu/primerbank, MGH Harvard, Cambridge, MA) [28]. Sequence of primers used in the study are listed in Table 1.
Table 1.
Species-specific primer sequences used in this study
| Primer | Species | Sequence (5’-3’) |
|---|---|---|
| DUSP1 | Human | F: AGTACCCCACTCTACGATCAGG |
| R: GAAGCGTGATACGCACTGC | ||
| DUSP2 | Human | F: GGGCTCCTGTCTACGACCA |
| R: GCAGGTCTGACGAGTGACTG | ||
| DUSP4 | Human | F: GGCGGCTATGAGAGGTTTTCC |
| R: TGGTCGTGTAGTGGGGTCC | ||
| DUSP5 | Human | F: TGTCGTCCTCACCTCGCTA |
| R: GGGCTCTCTCACTCTCAATCTTC | ||
| DUSP6 | Human | F: GAAATGGCGATCAGCAAGACG |
| R: CGACGACTCGTATAGCTCCTG | ||
| DUSP7 | Human | F: GACGTGCTCGGCAAGTATG |
| R: GGATCTGCTTGTAGGTGAACTC | ||
| DUSP8 | Human | F: TCAGCTCCGTCAACATCTGC |
| R: CGCGTGCTCTGGTCATAGA | ||
| DUSP9 | Human | F: TTCCGCCAATTTGGAGAGCC |
| R: TGCTTGTAGTGAAAGTCACCATT | ||
| DUSP10 | Human | F: ATCGGCTACGTCATCAACGTC |
| R: TCATCCGAGTGTGCTTCATCA | ||
| DUSP16 | Human | F: GCCCATGAGATGATTGGAACTC |
| R: CGGCTATCAATTAGCAGCACTTT | ||
| STYX | Human | F: TGCTATGAAAAGCAAGCTACCTG |
| R: CCCATTTGTAAGCTCCCATCAA | ||
| Dusp9 | Mouse | F: CCTGTGTGAAACCAGCTTCAG |
| R: CAGCTCAAGGTGTCACGGTC | ||
| SOX2 | Human | F: GCCGAGTGGAAACTTTTGTCG |
| R: GGCAGCGTGTACTTATCCTTCT | ||
| OCT4 | Human | F: CAAAGCAGAAACCCTCGTGC |
| R: TCTCACTCGGTTCTCGATACTG | ||
| ALDH1A | Human | F: GCACGCCAGACTTACCTGTC |
| R: CCTCCTCAGTTGCAGGATTAAAG | ||
| Gapdh | Mouse | F: AGGTCGTGTTGAACGGATTTG |
| R: GGGGTCGTTGATGGCAACA | ||
| β-ACTIN | Human | F: CTCCTTTAATGTCACCACGAT |
| R: CATGTACGTTGCTATCCAGGC |
F: Forward; R: Reverse.
Phosphatase assay
Small and large HMLEHRASV12, HCC1806 and HCC70 xenografts were dissociated enzymatically and mechanically in T-PER buffer. The total protein concentration was determined using the Pierce BCA Protein assay kit (Thermo Fisher Scientific). Lysate (200 μg) from small (S) and large (L) xenografts after various treatments were incubated in 37°C water bath for 1 hour following which the samples were subjected to immunoblot analysis. The membranes were probed for ERK1/2, pERK1/2, DUSP9, ALDH1, OCT3/4 and β-actin.
Flow cytometry
Dissociated tumor cells were filtered through 70 μm cell strainer and incubated with FITC or Texas Red-conjugated CD44, SOX2 or OCT4, and subjected to flow cytometric analysis. To examine ALDH1 expressing cells, ALDEFLUOR kit (Stem Cell Technologies) was used to isolate the population with high ALDH1 enzymatic activity as described before [26]. To distinguish between ALDH (+) and ALDH (-) cells, a fraction of cells were incubated under identical conditions in the presence of a 10-fold molar excess of the ALDH inhibitor, diethylaminobenzaldehyde (DEAB). Cells were analyzed in a BD FACS Aria II High Speed Cell Sorter (Becton Dickinson). For depletion of SOX2/OCT4 population, HCC1806 cells incubated with FITC-SOX2 (clone 6G1.2, FITC conjugate, cat# FCMAB112F, Millipore Sigma) and PE-conjugated-OCT4 (clone 3A 2A20, PE, cat# 60093PE, Stem Cell Technology) antibodies for 2 h followed by cell sorting using BD FACS Aria II High Speed Cell Sorter (Becton Dickinson).
Mammosphere formation
Cell lines were trypsinized and suspended in mammosphere media comprised of DMEM supplemented with 10 μg/mL insulin and 25 ng/mL fibroblast growth factor. Single cells from each of the cell lines were plated on ultralow attachment 6-well plates at a density of 50,000 cells/well (Costar TM 3471). The spheres were harvested at 24 hours and 48 hours and subjected to various analysis.
Histochemistry
Immediately after excision from mice, pieces of the xenografts were fixed in 5% formalin overnight after which they were dehydrated in ethanol and embedded in paraffin. Tumor sections (5-6 μm) were deparaffinized, subjected to epitope retrieval, H2O2 treatment, permeabilization with 0.1% triton and probed for various antigens as described previously [28].
Double immunofluorescence
Tumors were fixed in 10% neutral buffered formalin overnight and embedded in paraffin for sectioning. Sections were cut at 5 μm. Tumor sections were deparaffinized, and antigen retrieval was performed with Nuclear Decloaker (Biocare Medical) using a microwave. Paraffin embedded sections of xenografts were analyzed for co-localization of DUSP9 and OCT3/4 as previously described (28). Briefly, sections were blocked with 0.5% normal donkey serum in PBST (PBS+0.3% Triton-X100) for one hour at room temperature. Sections were then incubated anti-DUSP9 antibody (2 hr) followed by FITC conjugated secondary (1 hr). After the washes, the slides were further incubated with anti-OCT3/4/for (2 hr) followed by Texas Red conjugated secondary antibody (1 hr). After the washes (3 times 5 min each) with PBS the slides were further counterstained with DAPI and mounted in prolong anti-fade solution (Molecular Probes) and visualized under florescent microscope.
Patient derived xenografts (PDX)
Human tumor samples were obtained from the Cooperative Human Tissue Network (CHTN; http://chtn.nci.nih.gov) and National Disease Research Institute (NDRI; http://ndriresource.org/). Studies were performed under research protocol approved by the Charles R. Drew University of Medicine and Science Institutional Review Board (permit number: 09-08-2229-06) [28]. Human tumors used in the study are listed in Table 2.
Table 2.
Characteristics of human tumor samples used in the study
| Sample | Age | Lymph node | Pathological Status | Histological grade | Tumor size (cm) |
|---|---|---|---|---|---|
| T1 | 50 | no info | no info | no info | no info |
| T2 | 46 | negative | IIA | invasive mammary carcinoma, ductal | 3.8 |
| T3 | 67 | negative | IIA | invasive ductal carcinoma | 2.8* |
| T4 | 42 | positive | IIIA | invasive mammary carcinoma, ductal | 5.4 |
| T5 | 58 | positive | IIB | mammary carcinoma ductal | 3.5 |
| T6 | 50 | negative | IIA | invasive mammary carcinoma, ductal | 3.1 |
| T7 | 68 | positive | IIB | invasive ductal carcinoma | 4.0 |
| T8 | 44 | positive | IIIB | invasive metaplastic carcinoma | 8.5** |
| T9 | 77 | negative | IIA | invasive mammary carcinoma, ductal | 3.5 |
African American (AA) triple negative (TN) breast tumor patients and sample (T1-T9) characteristics. PDX1 and PDX2 were generated using T3 (*) and T8 (**) samples respectively.
Dusp9 knockdown
Following unique 29 mer human Dusp9 shRNAs constructs in retroviral RFP vectors were purchased from OriGene Technologies (Rockville, MD; Cat# TF313343). The sequences in the shRNA expression cassettes were shRNA1 AACGATGCCTATGACCTGGTCAAGAGGAA (Accession # NM_001395), and shRNA2 (OriGene; TF313343B): GTCACTGTGGCCTACCTCATGCAGAAGCT (Accession # NM_001395.1). Scrambled non-specific sequence cassette 5’ GCACTACCAGAGCTAACTCAGATAGTACT 3’) (Scam shRNA; CAT# TR30012) was used in parallel as a negative control. All shRNA transfections were performed with 1 μg DNA using Turbofectin 8.0 as a transfection reagent (OriGene Technologies) according to the manufacturer’s instructions.
Statistical analysis
Data are presented as mean ± SD, and between-group differences were analyzed using ANOVA. If the overall ANOVA revealed significant differences, then pair-wise comparisons between groups were performed by Newman-Keuls multiple comparison test. All comparisons were two-tailed, and p values <0.05 were considered statistically significant. The experiments were repeated at least three times, and data from representative experiments are shown.
Results
MEK1/2 independent reduced pERK1/2 in xenografts from breast cancer cells and patient tumors
We initially examined for MAPK and AKT pathways in the xenografts from TN breast cancer cells, HCC70, HCC1806 and MDA-MB-468. Cells propagated in vitro were implanted (2×106 cell/injection) subcutaneously (abdominal, dorsolateral) in immuno-deficient mice for engraftment and xenograft growth. Xenografts excised at various stages of growth were homogenized and subjected to immunoblot analysis. Compared to high pERK1/2 levels in pre-implanted HCC70 breast cancer cells, there was reduced pERK1/2 in both small (X1-X3) and large (X4-X6) xenografts that were excised at 1-3w or 4-6w respectively (Figure 1A). However, the total ERK1/2 levels in the cells before implantation and the xenografts remained comparable (Figure 1A). Surprisingly, persistence of reduced pERK1/2 appeared to be independent of upstream pMEK levels, which although varied between xenografts did not appear to influence downstream pERK levels (Figure 1A). In sharp contrast to reduced pERK1/2, the levels of pp38 MAPK as well as pAKT were upregulated in the xenografts at various stages of growth (Figure 1A). A similar trend was observed for TN breast cancer cell lines HCC1806 (Figure 1B) and MDA-MB-468 (data not shown).
Figure 1.
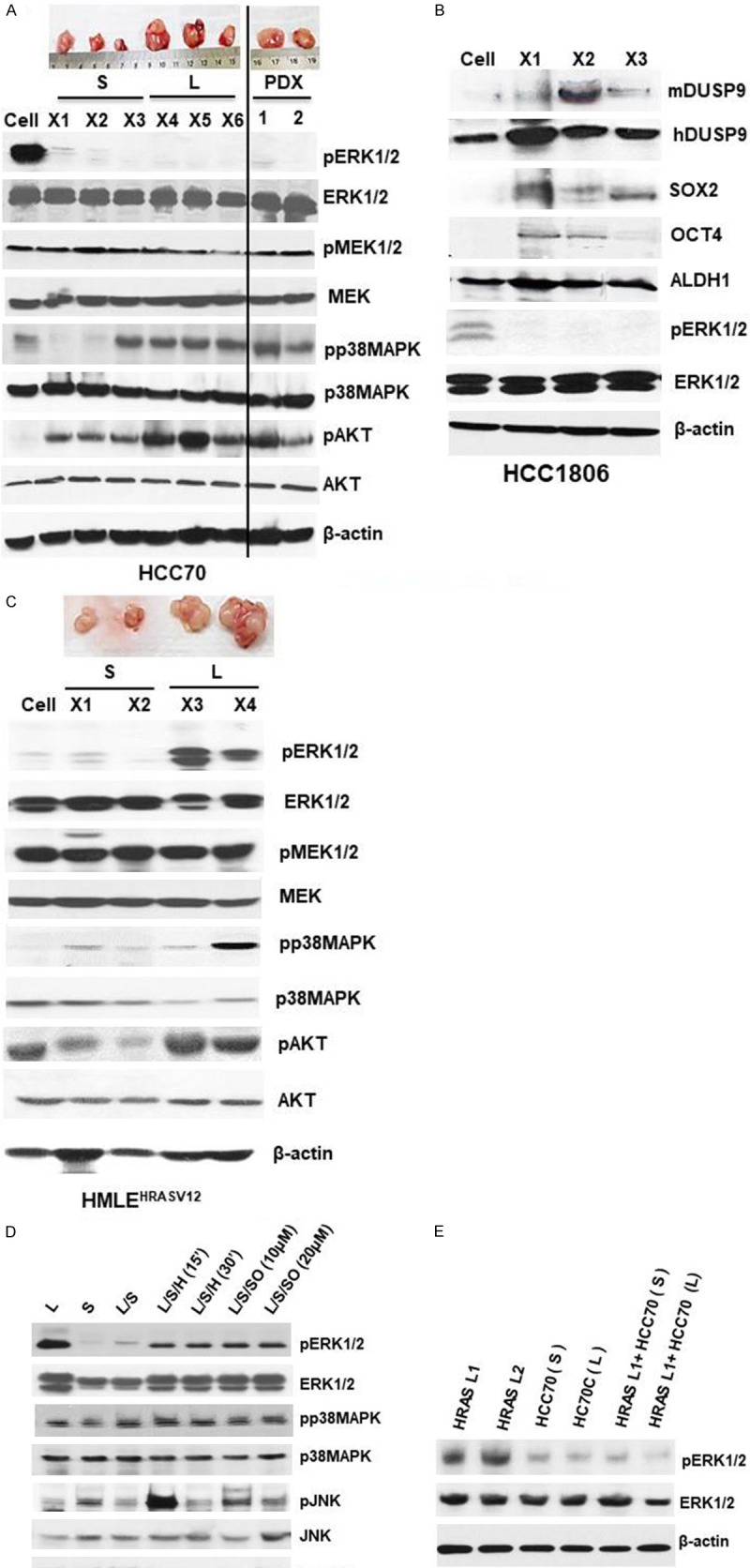
ERK1/2 dephosphorylation in early and late AA TN xenografts and patient-derived xenografts. 100 µg of total lysates obtained from HCC70 cells, or HCC70 xenografts from small (S) (2 weeks) and large (L) (8 weeks) growth and patient-derived xenografts (PDX) (4 months) (A), or HCC1806 xenografts (B) or HMLEHRASV12 (HRAS) (C) were analyzed by western blot to compare various proteins involved in MAP kinase signaling pathways. (D) 100 µg of total cell lysates obtained from large (L), small (S), L/S (equal amounts of L and S lysates incubated at 37°C for 1 hr), L/S/H (L+S mixture heated at 100°C for 15-30 mins), and L/S/I (L+S mixture incubated with 10 or 20 µM sodium orthovanadate and incubated at 37°C for 1 hr) xenografts obtained from HRAS were analyzed by western blot analysis. (E) 100 µg of total cell lysates obtained from HRAS late tumors (HRAS L1 and HRAS L2), and either small (TN/S) or large (TN/L) HCC70 tumors alone or in combination with HRAS L1 followed by incubated at 37°C for 1 hr and analyzed for total ERK1/2 or phosphorylated ERK1/2 by western blot analysis. Experiments were replicated three times and a representative panel has been shown.
To understand the implication of reduced pERK1/2 during xenograft growth, we examined the xenografts from HMLEHRASV12 cell line, which are mammary epithelial cells overexpressing HRAS [29]. We found presence of MEK independent reduced pERK1/2 expression only in small xenograft growth (1-2 weeks, 0.1-0.3 gm), while pERK1/2 was upregulated in large (4-6 weeks, 0.4-0.7 gm) xenografts (Figure 1C). Levels of total ERK1/2 remained comparable in both small (S) and large (L) HMLEHRASV12 xenografts (Figure 1C). Since pERK1/2 levels in TN xenografts, in sharp contrast to HMLEHRASV12, remained reduced throughout the growth, we further examined patient derived xenografts (PDX) from AA TN breast tumors. We obtained PDX by subcutaneous implantation of pieces of fresh TN breast tumors from AA patients in nude mice, and allowed to grow for 2 or 4 months. Immunoblot analysis showed MEK independent presence of reduced pERK1/2 in the PDX from AA TN breast tumors (Figure 1A). Characteristics of human tumors used in the study to generate PDX are marked with (*, **) in Table 2.
Since reduction of pERK1/2 was independent of MEK, we further examined whether phosphatases contributed to the reduced pERK1/2 levels observed in the xenografts. For this purpose, we incubated in vitro, lysates from HMLEHRASV12 small xenografts (SX) that had low pERK1/2 with those from large xenografts (LX) where pERK1/2 was upregulated (Figure 1D). Co-incubation with SX (37°C for 1 h) reduced pERK1/2 levels in LX lysates as analyzed by immunoblot analysis. Heat denaturation of SX lysates prior to incubation with LX lysates attenuated the reduction of pERK1/2, indicating involvement of a phosphatase (Figure 1D). Incubation of lysates in the presence of a general protein tyrosine phosphatase (PTP) inhibitor, sodium orthovanadate (SO) [30], attenuated the decline of pERK1/2, further confirming phosphatase activity. There was no prominent change in the levels of total ERK1/2, JNK, pJNK, p38MAPK or pp38MAPK during incubation of these xenograft lysates (Figure 1D). To determine the contribution of active phosphatases in suppression of pERK1/2 levels, we incubated TN and HMLEHRASV12 LX xenograft lysates. We found reduction of pERK1/2 levels in LX HMLEHRASV12 upon incubation with lysates from AA TN xenografts (2 or 4 weeks) (Figure 1E). Our above data indicate involvement of phosphatases in MEK1/2 independent suppression of pERK1/2 TN and HMLEHRASV12 xenografts.
Increased expression of DUSP9 in the xenografts from breast cancer cell lines and patient derived xenografts
Phosphatases regulate both duration and magnitude of MAPK activation, which is crucial in determining the physiological outcomes of cells [23,31]. To identify the specific phosphatase involved in suppressing pERK1/2 in the xenografts, we initially examined the expression of known dual specific phosphatases (DUSPs) in HMLEHRASV12 and HCC-70 xenografts by qPCR. DUSP9 expression was increased significantly in HMLEHRASV12 and TN xenografts when compared to respective cancer cells (Figure 2A, 2D). We did not detect any changes in the expression of 1, 2, 4, 5, 6, 8, 10 and 16 DUSPs in xenografts when compared to the breast cancer cells. However, we found DUSP9 expression remained consistently high throughout the growth of TN xenografts, which was in sharp contrast to the transient spikes in DUSP9 in HMLEHRASV12 xenografts (Figure 2A, 2B, 2D, and 2E). Small HMLEHRASV12 xenografts expressed higher DUSP9 when compared to the larger xenografts excised at later time points (Figure 2B). Since the host also influence tumor growth, we used host (mouse) specific DUSP9 primers to determine host contribution of the phosphatase. Species-specific primers showed additional increase of DUSP9 expression of host origin in the xenografts (Figure 2C, 2F). To confirm expression of DUSP9 in tumor cells, we implanted green florescent protein (GFP) labeled HCC1806 cells subcutaneously in nude mice for xenograft growth. The xenografts therefore had GFP expressing tumor cells along with host tissues, which did not express any florescence. Paraffin embedded sections of these xenografts when immunolabeled with DUSP9/Texas red conjugated antibodies, was found to express considerable amount of DUSP9 originating both from the host (red) as well as the tumor cells (yellow) (Figure 2G).
Figure 2.

Increased expression of DUSP9 in tumor xenografts and reduced ERK1/2 phosphorylation. (A) Quantitative gene expression analysis using real-time PCR analysis of various dual-specificity phosphatases 1-10 (Dusp1-10) in HRAS cells, and small (S) and large (L) xenografts. Data are expressed as fold change compared to the cells. Quantitative gene expression analysis of Dusp9 in 3 Small (S1-S3) and 3 large (L1-L3) stage xenografts using (B) human and (C) mouse specific primer sets. (D) Quantitative gene expression analysis using real-time PCR analysis of various dual-specificity phosphatases 1-10 (Dusp1-10) in HCC70 cells, and small (S) and large (L) xenografts. Data are expressed as fold change compared to the cells. Quantitative gene expression analysis of Dusp9 in 3 small (S1-S3) and 3 large (L1-L3) xenografts using (E) human and (F) mouse specific primer sets. (G) GFP-labeled HCC1806 cells were subcutaneously implanted in nude mice for xenograft growth. Paraffin-embedded sections from 8-week xenografts were immunolabeled for DUSP9 with Texas Red conjugated secondary antibody. GFP-labeled tumor cells (green) expressing DUSP9 (yellow) as well as host cells expressing DUSP9 in xenografts (red) was analyzed under fluorescent microscope (Olympus). (H) 100 µg of total lysates obtained from large HRAS (HRAS L), equal amounts of HRAS L plus HCC70 L mixed and incubated at 37°C for 1 hr (HRAS L+HCC-70 L) were analyzed by western blot analysis for detection of total and phosphorylated ERK1/2 and DUSP9 protein levels. (I) 100 µg of total lysates obtained from PDX and large HRAS (HRAS L1 and HRAS L2) either alone or mixed together and incubated at 37°C for 1 hr (PDX+HRAS L1 and PDX+HRAS L2), and mixed lysates plus phosphatase inhibitor (PDX+HRAS L1+I) or mixed lysates plus heat (PDX+HRAS L1+H) were analyzed by western blot analysis for detection of total and phosphorylated ERK1/2 and DUSP9 protein levels. Experiments were replicated 3 times and a representative panel has been shown. **P≤0.01; *P≤0.05.
Immunoblot analysis showed inverse relationship between DUSP9 expressions and pERK1/2 levels HMLEHRASV12 xenografts (Figure 2H). We further examined the levels and activity of DUSP9 in PDX from TN breast tumors. PDX had high DUSP9 expression as well as lower pERK1/2 when compared to late HMLEHRASV12 xenografts where lower DUSP9 was associated with higher pERK1/2 levels (Figure 2I). Pre-heating of the PDX lysates attenuated the reduction of high pERK1/2 levels upon co-incubation (Figure 2I). In addition, treatment with phosphatase inhibitor, sodium orthovanadate (SO), in co-incubation experiments with PDX, attenuated the decline of pERK1/2 while the levels of total ERK1/2 did not change (Figure 2I). The above data indicates consistent expression of high DUSP9 expression in xenografts from TN breast cancer cells.
Upregulation of DUSP9 was associated with increase in CSC-like traits in TN xenografts
There are reports that DUSP9 controls appropriate ERK1/2 activity, which appears critical for determining SC fate [24,32]. We further examined DUSP9 and SC-specific proteins (ALDH1, CD44, OCT4 and SOX2) in HCC70 and HMLEHRASV12 xenografts at various stages of growth. While the pre-implanted HCC70 cells expressed DUSP9, its elevated expression in xenografts was associated with increased expression OCT4 and SOX2 (Figure 3A). In contrast, there was no change in ALDH1 expression, which remained consistently high in both pre-implanted cells as well as in the xenografts (Figure 3A). We were unable to detect in HCC70 xenografts, any significant expression of CD44, which is a known marker for a subset of CSCs (Figure 3A). This persistent expression of DUSP9 as well as the increase in SOX2 and OCT4 in TN xenografts, sharply contrasted with those from HMLEHRASV12 xenografts where status of ERK1/2 activation dictated the expression of CSC-specific proteins (Figure 3B). In S HMLEHRASV12 xenografts, increased DUSP9 expression and reduced pERK1/2 was associated with high levels of ALDH1 and OCT4, while L xenografts with high pERK1/2 levels had up regulation of CD44 (Figure 3B). PCR analysis of xenografts from all three TN breast cancer cell lines showed consistent increase in expression of SOX2 in all the xenografts examined (Figure 3C). We further examined DUSP9 and SOX2/OCT4 expression in xenografts from another TN breast cancer cell line HCC1806 and found similar association (Figure 3D).
Figure 3.
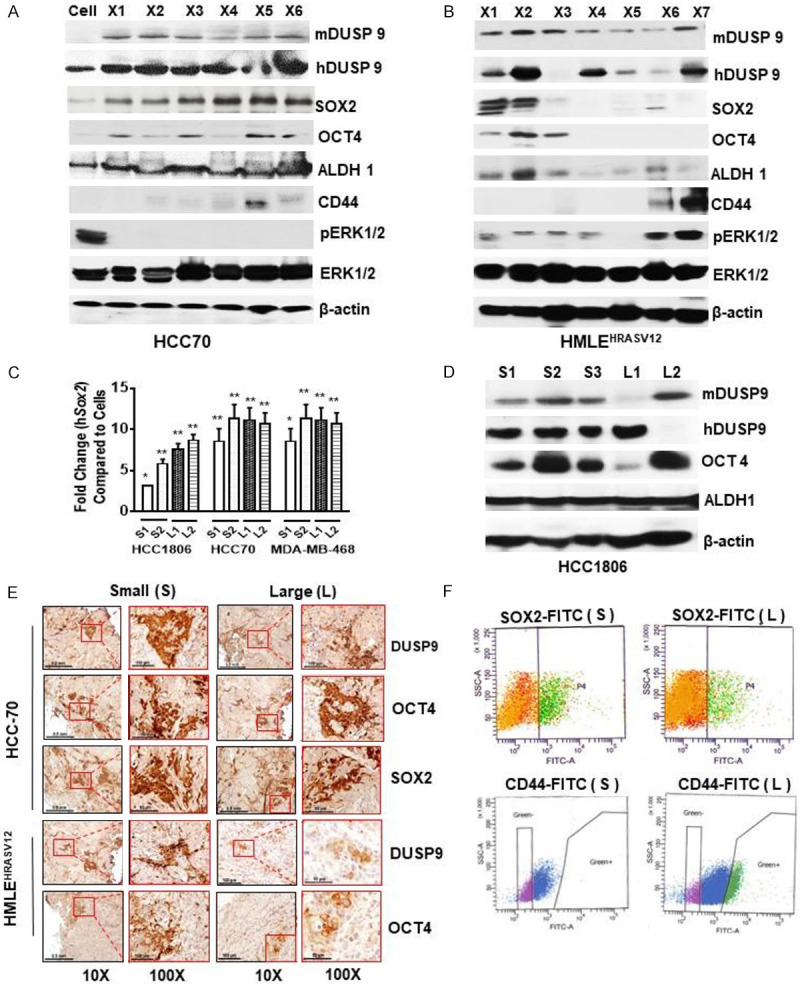
Induction of DUSP9 and ERK1/2 dephosphorylation coincides with cancer stem cell-like characteristics. (A, B) 100 µg of total lysates obtained from xenografts and analyzed by western blot analysis for both human and mouse DUSP9, total and pERK1/2, and various breast cancer stem cell-specific proteins in HCC70 (A) and HMLEHRASV12 (B) xenografts. (C) Quantitative gene expression analysis for SOX2 by real-time quantitative PCR in three TN cell lines. Small, S1, S2 (2 weeks) and large L1, L2 (8 weeks). (D) 100 µg of total lysates obtained from small (S1, S2, S3) and large (L1, L2) xenografts were analyzed by western blot analysis for the detection of DUSP9 (human and mouse), OCT4 and ALDH1 protein expression. Experiments were replicated 3 times and a representative panel has been shown. (E) Immunohistochemical (IHC) analysis of small and large HCC70 and HMLEHRASV12 xenografts using anti-DUSP9, anti-OCT4, or anti-SOX2 antibodies (1:100 dilutions). (F) 1×106 cells obtained from single cell suspension of xenografts was analyzed for SOX2 (HCC70) or CD44 (HRAS) by cytometric analysis.
Immunohistochemistry confirmed significant expression of DUSP9, OCT4 and SOX2 in both S and L TN but only in S HMLEHRASV12 xenografts (Figure 3E). FACS analysis of small/large xenografts showed 6-8% SOX2 expressing cells (Figure 3F, Upper Panel). Furthermore, FACS analysis showed up-regulation (22.2%) of CD44 expressing cells only in L HMLEHRASV12 xenografts (Figure 3F, Lower Panel).
We further dissociated HCC70 xenografts and plated them in low attachment plates for mammosphere growth. Real time qPCR analysis showed mammospheres, when compared to other DUSPs, specifically expressed DUSP9 (Figure 4A). Immunoblot analysis showed that DUSP9 as well as OCT4 and SOX2 protein levels was upregulated while pERK1/2 levels reduced in the mammospheres, when compared to cells grown on high attachment plates (Figure 4B). We further examined cells on plastic and mammospheres for nuclear/cytosolic pERK1/2. Interestingly, pERK1/2 was mostly nuclear in mammospheres, while distribution in cells on plastic was even between cytosolic and nuclear fraction (Figure 4C). In addition, when compared to normal epithelial cells, pERK1/2 was more stable in HRAS transformed cells and mammospheres from these cells as well as breast cancer cells (Figure 4D, 4E).
Figure 4.
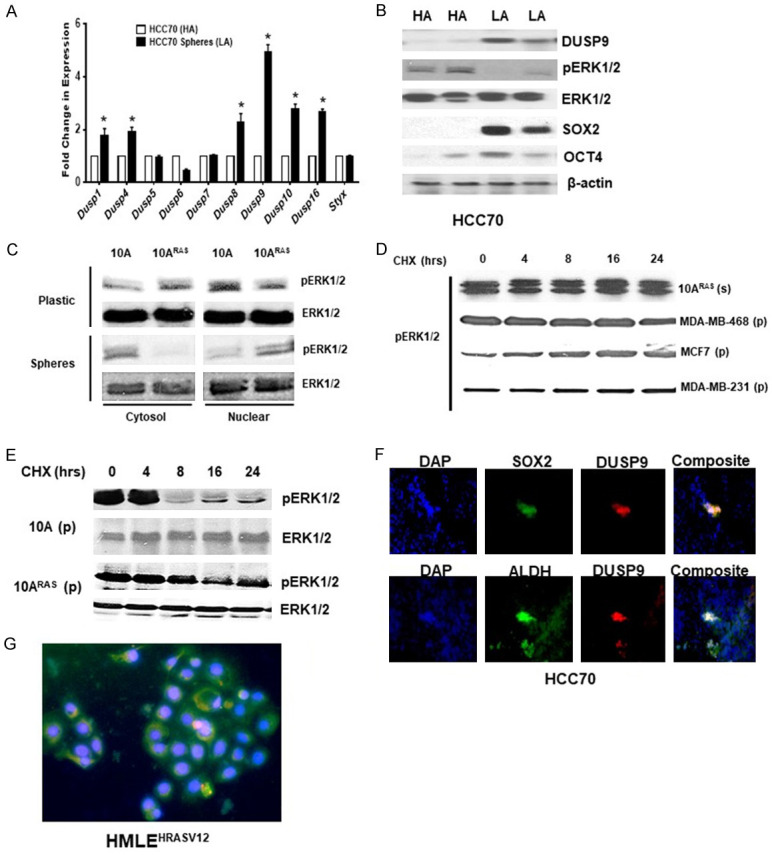
Increased expression of DUSP9 and stem cell-specific proteins in mammospheres. A. Real-time quantitative gene expression analysis of various DUSPs in HCC70 cells grown under high (HA) or low (LA) attachment conditions. B. Western blot analysis of total cell lysates (60 µg) obtained from HCC70 cells grown under high (HA) or low (LA) conditions. Experiment was repeated three times and a representative panel is shown. C. Nuclear and cytosolic fractionation of MCF10AHRASV12 cells on high attachment plates or as mammospheres was subjected to immunoblot analysis using anti-pERK1/2/ERK1/2 antibodies. D. Breast cancer cell lines MCF10AHRASV12 (10ARAS), MDA-MB-468, MCF7 and MDA-MB-231 as mammospheres on high attachment were treated with cycloheximide (CHX) (100 µg/ml) for various time points (0-24 hrs) and subjected to immunoblot analysis using anti-pERK1/2 antibody. E. MCF10AHRASV12 and MCF10A treated with cycloheximide (CHX) (100 µg/ml) for various time pointsre subjected to immunoblot analysis using anti-pERK1/2 and ERK1/2 antibodies. F. Sections of HCC70 xenografts were analyzed by double immunofluorescence using anti-SOX2 (green) or anti-DUSP9 (red). Nuclear staining is shown by DAPI. Co-localization of SOX2 and DUSP9 is shown in yellow. *, P≤0.05; **, P≤0.01 (n=3). G. Double immunofluorescence of paraffin embedded S HMLEHRASV12 xenografts showing co-localization of DUSP9 (green) and SOX2 (red).
We further confirmed by performing double immunofluorescence that DUSP9 co-expressed SOX2/OCT4 expressing cells in HCC70 xenografts (Figure 4F). In addition, double immunofluorescence of S HMLEHRASV12 xenografts showed co-localization of DUSP9 with OCT4 and SOX2 expressing cells (Figure 4G). Our data indicate that increased expression of DUSP9 was associated with CSC-like traits in TN xenografts.
Downregulation of DUSP9 reduced SC-specific proteins and TN breast tumor growth
We examined the effect of reducing DUSP9 in TN breast cancer cells by pharmacological inhibition and ShRNA treatment. HCC1806 cells treated with general phosphatase inhibitor, sodium orthovanadate (SO), showed reduced levels of SOX2/OCT4 and ALDH1 expression, which also coincided with increased levels of pERK1/2 (Figure 5A). We further reduced the levels of DUSP9 by treatment of HCC1806 cells with DUS9 shRNA, where compared to Scrambled (Sc) shRNA, DUSP9 shRNA1 and shRNA2 reduced DUSP9 levels by 76% and 62% respectively (Figure 5B). Furthermore, with reduction of DUSP9, there was simultaneous reduction of OCT4 (64%, 72%) and ALDH1 (64%, 55%) respectively (Figure 5B). DUSP9 shRNA treated cells had reduced ability to form mammospheres when compared to sc-shRNA treated cells (Figure 5C, 5D). In addition, treatment of HCC1806 with MEK1/2 inhibitor, PD 98059 (20 µg/ml) reduced the levels of pERK1/2 while increasing SOX2, OCT4 and ALDH1 expression (Figure 5E). We further depleted SOX2/OCT4 expressing cells from HCC1806 by FACS, before implanting them subcutaneously in nude mice. Tumor volume from control and SOX2/OCT4 depleted cells was measured over the course of 8 weeks as described previously [27] (Figure 5F). Tumor volume from SOX2/OCT4 depleted cells was lower at 2 wks. (65%), 4 wks. (55%), 6 wks. (40%) and 8 wks. (31%) when compared to control cells (Figure 5F, 5G). At 8 weeks tumors from euthanized mice were weighed and those after SOX2/OCT4 depletion were 50% smaller compared to control (Figure 5H). We further treated HCC1806 with DUSP9 shRNA or scrambled (scram) RNA before implanting subcutaneously in immunodeficient mice. Tumor volume from scram and shRNA treated cells was measured over the course of 8 weeks (Figure 5I). Growth of tumors from DUSP9 shRNA treated cells was lower at 2 (64%), 4 (64%), 6 (67%) and 8 weeks (48%), when compared to control (Figure 5I). The mice were euthanized at 8 weeks and tumor weight was examined (Figure 5J, 5K). We found 65% reduction in tumor weight from cells treated with DUSP9 shRNA when compared to control (Figure 5K).
Figure 5.

Effect of pharmacological inhibition or shRNA-mediated blockade of Dusp9 expression on key cancer stem cell characteristics, mammosphere formation and xenograft growth. A. 100 µg of total cell lysates obtained from HCC1806 cells treated with various concentration of sodium orthovanadate (0-1000 µM) for 48 hrs and protein expression of key breast cancer stem cell markers, total and phosphorylated ERK1/2, and DUSP9 were analyzed by western blot analysis. B. HCC-1806 cells was subjected to shRNA-mediated inhibition of Dusp9 expression using standard conditions and protein expression for DUSP9, OCT4/SOX2, and pERK1/2 were analyzed by western blot analysis using 50 µg of total cell lysates. Control cells were treated with scrambled RNA (scRNA) while experimental cells with two different Dusp9 shRNAs (1 and 2). Experiment was repeated two times and a representative data has been shown. C. Scrambled shRNA and Dusp9-shRNA1 (2×104) cells were plated on low attachment plates and photomicrographs of mammospheres are shown. D. Number of mammospheres were counted per field (20 fields) and average numbers are shown. Experiment was repeated two times. E. HCC1806 cell line, in-vitro was treated with various concentrations of PD inhibitor and subjected to immunoblot analysis for pERK1/2/ERK1/2, DUDP9 and MCSC markers (SOX2, OCT4 and ALDH1). GAPDH was used as loading control. F. Control or SOX2- and OCT4-depleted HCC-1806 cells (2×106) were injected into nude mice and tumor volume was analyzed over a period of 8 weeks. *P≤0.05; **P≤0.01. G. Representative tumor from both groups are shown at 8 weeks (n=5). H. Analysis of tumor weights (mg) from both groups are shown after 8 weeks (n=5). *P≤0.05. I. scRNA or Dusp9-shRNA treated HCC1806 cells (1×106) were injected into nude mice and tumor volume was analyzed over a period of 8 weeks. *P≤0.05; **P≤0.01; ***P≤0.001. J. Representative tumor from both groups are shown at 8 weeks (n=6). K. Analysis of tumor weights (mg) from both groups are shown after 8 weeks. (n=6). *P≤0.05.
Oncostatin M increases the expression of DUSP9 and SC-specific proteins
Since DUSP9 expression increased during engraftment of xenografts, we examined whether adipocytes in the host microenvironment influenced the process. For this purpose, HCC70 cells were co-incubated with 3T3-L1 cells for 48 h after which expression of DUSPs as well as SC-specific proteins were examined. We found significant increase in DUSP9 of cancer cell origin, while the expression of other DUSPs remained unchanged upon incubation of cancer cells with 3T3 L1 cells (Figure 6A, 6B). There was also increase in ALDH1 and SOX2 in tumor cells during co-incubation with 3T3-L1 cells (Figure 6C). We also examined the effect of oncostatin M, a secretion from mature adipocytes, on expression of DUSP9 and OCT4/SOX2 in HCC1806 cells. We found that oncostatin M (20 µM) treatment increased the expression of DUSP9 as well as OCT4/SOX2 (Figure 6D, 6E). We further examined AA TN breast tumors for the expression of DUSP9 and pERK1/2 by immunoblot analysis (Figure 6F). We have listed information of patient tumors used in the study in Table 2.
Figure 6.

Oncostatin M (OSM) increases the expression of DUSP9 and embryonic stem cell markers. HCC70 cells were grown either alone or in combination with mouse 3T3-L1 preadipocyte cells under standard adipogenic conditions (for 6-9 days), total RNA was isolated from the cells and gene expression of various Dusp variants human (A) and mouse (B) or (C) Aldh1 and Sox2 were analyzed by quantitative real-time PCR analysis using human specific Aldh1 or Sox2 primer sets. HCC1806 cells were treated with various concentrations (0-20 µM) of oncostatin M (Osm) for 48 hrs and gene expression levels of DUSPs (D) or stem cell-specific proteins (E) were analyzed by quantitative real-time PCR using human specific primer sets. *P≤0.05; **, P≤0.01 (n=3). (F) Western blot analysis for AA TNBC for DUSP9 and ERK1/2. **P≤0.01 (n=3).
Discussion
Given that heterogeneity and plasticity are hallmarks of most cancers, improved technologies including next generation deep sequencing have revealed unexpectedly higher levels of heterogeneity in TNBCs [33]. High levels of genotypic and phenotypic heterogeneity in TNBC has been associated with enhanced vigor of cancer cells, reduced response to chemotherapy and poor prognosis [34,35]. Intriguingly, each of the many subtypes of TNBC has distinct subsets of CSCs, which express ALDH1, CD44, or SOX2/OCT4, which are transcriptional regulators of pluripotency [36-38]. ALDH1/CD44 expressing small subset of CSCs in breast tumors has the potential to promote initiation/growth of xenografts in immunodeficient mouse models [8-10]. In addition, high-grade basal-like breast tumors have a subset of MCSCs that express ES-like gene signatures that determine aggressive tumor behavior [39-41]. Microenvironment that contributes to tumor growth in vivo contrasts sharply to in vitro proliferation driven by growth factor enriched serum. In this study, we found pERK1/2 levels remained reduced in the xenografts from all the three AATN breast cancer cells examined. Our study also shows that in addition to ALDH1, there was also a consistent increase in SOX2/OCT4 expression during xenograft growth. We show for the first time that DUSP9 was up regulated in mammospheres and specifically in ALDH1/SOX2 expressing cells in xenografts/PDX, and it’s down regulation reduced CSC-like traits in vitro and xenograft growth in vivo. In addition, we also found that patient derived xenografts (PDX) from AA TN breast tumors expressed high levels of active DUSP9. Interestingly, both the implanted tumor cells as well as the host known to influence breast tumor growth contributed to DUSP9 expression.
There are reports of DUSP9 identification in a Walktrap-GM module, which is a random-walk-based community detection algorithm that identifies biological modules predisposing to tumor growth [42]. Walktrap was reported with 22-hepatocellular carcinoma and 32 colorectal samples in addition to prognosis in 198 breast cancer patients [42]. DUSP9 play a key role in the MAPK pathway along with cell cycle and proliferation regulators as well as key transcription factor modules [42]. DUSP9 also been shown to maintain murine ESC pluripotency and self-renewal status by controlling appropriate ERK activity [24]. DUSP9, which was downstream of BMP4/Smad1/5 signaling axis and steadily attenuated ERK1/2 activity in murine ESCs to reduce spontaneous differentiation [24]. Suppression of ERK1/2 signaling in mouse embryo in vivo by small molecules inhibitors or in vitro by MEK1/2 inhibitors, promoted ground state pluripotency of mESCs [43,44]. However, role of pERK1/2 in maintenance of pluripotency in human ESCs remains poorly understood [45]. A study shows that human pluripotent stem cells lack the transcription factor circuitry that governs ground state of mESCs and inhibition of ERK1/2 or protein kinase C resets this machinery [46]. However, in sharp contrast to the importance of ERK1/2 suppression in pluripotency maintenance, a study shows PKCδ-mediated transient ERK1/2 activation increased ESC proliferation [47]. Interestingly, a study on non-human primate, common marmoset shows that PI3K/AKT pathway promoted renewal of ESCs [48]. We found that along with DUSP9 and SOX2 there was an increase in pAKT in all the xenografts examined.
Role of DUSPs in human breast tumors remains confusing since both overexpression as well as down-regulation of specific DUSPs have been reported [49,50]. DUSP1 or MKP1 was overexpressed in breast tumors where they increase tamoxifen resistance and their inhibition reduced breast tumor growth [51]. In sharp contrast, there are reports that DUSP4 loss promoted cancer stem cell-like phenotype and increased aggressive properties in basal B subtype of breast cancer [49]. However, loss of DUSP4 increased only a subset of MCSCs that expressed CD44 in claudin low basal B tumors [49]. In addition, EGFR-mediated up regulation of pERK1/2 studies have shown, using both in vitro and in vivo models that increase of CD44 (+) MCSC population was mostly in mesenchymal-like breast cancer cells of Caucasian origin [52]. There are reports that ERK1/2-mediated increase of CD44 in various cell types and cancers contribute to various pathological behaviors [53]. In our study, sustained up regulation of p ERK1/2 and CD44 only in the late stage large HMLEHRASV12 xenografts, could be promoting tumor growth. In TN breast tumor xenografts examined, although ALDH1 and SOX2 cells were high, we could not detect much of pERK1/2 or CD44 suggesting their minimal involvement in growth of these tumors. Nonetheless, active ERK1/2 has been strongly associated with various aspects of breast tumor development including epithelial to mesenchymal transition (EMT) [54]. Most of the ERK1/2-mediated EMT studies performed under in vitro conditions on breast cancer cells were very different from in vivo tumor microenvironment [55]. In addition, ERK1/2-mediated EMT studies conducted in vivo as well as most other in vivo studies examined Caucasian breast cancer cell lines [56].
In sharp contrast to our results, a study conducted in clear renal cell carcinoma used qPCR and immunohistochemistry to show lower expression of DUSP9 was associated with poor prognosis [57]. Similarly, another study also found higher expression of DUSP9, which resulted in microtubule disruption, cell death and tumor suppression in squamous cell carcinoma (SCC) [58]. We do not have an explanation for this except that breast tumors are heterogeneous with diverse cell types, each of which function optimally at varying levels of pERK1/2, which in context dependent manner could induce cell proliferation or differentiation. It appears that AA TN breast tumors could have a unique subset of CSCs that are sensitive to ERK1/2-mediated differentiation and DUSP9-mediated appropriate control of its activity was essential for maintenance of this population and tumor progression. Our in vitro assays show both small and large AA TN xenografts exhibited high active DUSPs suggesting involvement of many DUSPs in controlling pERK1/2 levels in diverse cell types during tumor development.
Our study demonstrates DUSP9-mediated ERK1/2 suppression could be an important event that could influence breast tumor growth. We found low pERK1/2 levels in small xenografts was associated with increase in ESC specific transcriptional regulators, SOX2 and OCT4, which control pluripotency. Our findings are consistent with reports from other studies where although KRAS oncogene promoted pulmonary adenocarcinoma, pERK1/2 remained undetected during tumor initiation or in most tumors [59]. Only a subset of late-stage tumors in the context of a p53 loss showed activation of these kinases, where it up regulated p19Arf, a potent activator of tumor suppressor p53 in mice [60]. In general, each of the subtypes of breast cancer is a dynamic evolving disease where CSC-like cells along with many other recruited cells with stem-like properties, contribute to its development. Although the bulk of breast tumors consist of more differentiated cells, resident CSC-like cells together with adipose derived stem cells (ADSCs) mainly from the microenvironment as well as mesenchymal stem cells (MSCs) recruited from distant sites of the host, determine aggressive behaviors like angiogenesis, migration and invasion [61].
Due to small number of AA TN tumors analyzed and limited information in breast cancer genomic data bases about ethnic differences, we are unable to make a firm conclusion whether DUSP9-mediated ERK1/2 suppression is exclusively restricted to a particular race. However, our study strongly indicate in AA TN breast cancer cells and tumors expressed high levels of ALDH1/SOX2/OCT4 suggesting increase in CSC-like characteristics associated with DUSP9 expression.
Acknowledgements
We acknowledge Dr. Robert Weinberg (Whitehead Institute, Cambridge, MA) for HMLEHRASV12 cells. Fresh and frozen breast tumor specimen and sections were obtained from Cooperative Human Tissue Network (CHTN) and National Disease Research Interchange (NDRI). This work was supported by National Institute of Health grants SC1CA165865 (SP), SC1CA232319 (SP), SC1AG049682 (RS), Urban Health Institute grant S21MD000103 and Accelerating Excellence in Translational Science (AXIS) U54MD007598 to Charles R. Drew University of Medicine and Science.
Disclosure of conflict of interest
None.
Abbreviations
- CSCs
Cancer Stem Cells
- SC
Stem Cell
- ALDH1
Aldehyde dehydrogenase 1
- TN
Triple Negative
- DUSPs
Dual-specific phosphatases
- FACS
Fluorescence-activated cell sorting
- ER
Estrogen Receptor
- PR
Progesterone Receptor
- MCSCs
Mammary Cancer Stem Cells
- ESC
Embryonic Stem Cell
- PDX
Patient Derived Xenografts
References
- 1.Ghoncheh M, Pournamdar Z, Salehiniya H. Incidence and mortality and epidemiology of breast cancer in the world. Asian Pac J Cancer Prev. 2016;17:43–46. doi: 10.7314/apjcp.2016.17.s3.43. [DOI] [PubMed] [Google Scholar]
- 2.DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64:52–62. doi: 10.3322/caac.21203. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Razzaghi H, Troester MA, Gierach GL, Olshan AF, Yankaskas BC, Millikan RC. Association between mammographic density and basal-like and luminal A breast cancer subtypes. Breast Cancer Res. 2013;15:R76. doi: 10.1186/bcr3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. 2009;9(Suppl 2):S73–81. doi: 10.3816/CBC.2009.s.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Killelea BK, Yang VQ, Wang SY, Hayse B, Mougalian S, Horowitz NR, Chagpar AB, Pusztai L, Lannin DR. Racial differences in the use and outcome of neoadjuvant chemotherapy for breast cancer: results from the national cancer data base. J. Clin. Oncol. 2015;33:4267–4276. doi: 10.1200/JCO.2015.63.7801. [DOI] [PubMed] [Google Scholar]
- 8.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charafe-Jauffret E, Ginestier C, Bertucci F, Cabaud O, Wicinski J, Finetti P, Josselin E, Adelaide J, Nguyen TT, Monville F, Jacquemier J, Thomassin-Piana J, Pinna G, Jalaguier A, Lambaudie E, Houvenaeghel G, Xerri L, Harel-Bellan A, Chaffanet M, Viens P, Birnbaum D. ALDH1-positive cancer stem cells predict engraftment of primary breast tumors and are governed by a common stem cell program. Cancer Res. 2013;73:7290–7300. doi: 10.1158/0008-5472.CAN-12-4704. [DOI] [PubMed] [Google Scholar]
- 11.Rios AC, Capaldo BD, Vaillant F, Pal B, van Ineveld R, Dawson CA, Chen Y, Nolan E, Fu NY, Jackling FC, Devi S, Clouston D, Whitehead L, Smyth GK, Mueller SN, Lindeman GJ, Visvader JE. Intraclonal plasticity in mammary tumors revealed through large-scale single-cell resolution 3D imaging. Cancer Cell. 2019;35:618–632. e616. doi: 10.1016/j.ccell.2019.02.010. [DOI] [PubMed] [Google Scholar]
- 12.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rye IH, Trinh A, Saetersdal AB, Nebdal D, Lingjaerde OC, Almendro V, Polyak K, Børresen-Dale AL, Helland Å, Markowetz F, Russnes HG. Intratumor heterogeneity defines treatment-resistant HER2+ breast tumors. Mol Oncol. 2018;12:1838–1855. doi: 10.1002/1878-0261.12375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapucuoğlu N, Bozkurt KK, Başpınar Ş, Koçer M, Eroğlu HE, Akdeniz R, Akçil M. The clinicopathological and prognostic significance of CD24, CD44, CD133, ALDH1 expressions in invasive ductal carcinoma of the breast: CD44/CD24 expression in breast cancer. Pathol Res Pract. 2015;211:740–747. doi: 10.1016/j.prp.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 15.Nishi M, Sakai Y, Akutsu H, Nagashima Y, Quinn G, Masui S, Kimura H, Perrem K, Umezawa A, Yamamoto N, Lee SW, Ryo A. Induction of cells with cancer stem cell properties from nontumorigenic human mammary epithelial cells by defined reprogramming factors. Oncogene. 2014;33:643–652. doi: 10.1038/onc.2012.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borah N, Gunawardana S, Torres H, McDonnell S, Van Slambrouck S. 5,6,7,3’,4’,5’-Hexamethoxyflavone inhibits growth of triple-negative breast cancer cells via suppression of MAPK and Akt signaling pathways and arresting cell cycle. Int J Oncol. 2017;51:1685–1693. doi: 10.3892/ijo.2017.4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato N, Wakabayashi M, Nakatsuji M, Kashiwagura H, Shimoji N, Sakamoto S, Ishida A, Lee J, Lim B, Ueno NT, Ishihara H, Inui T. MEK and PI3K catalytic activity as predictor of the response to molecularly targeted agents in triple-negative breast cancer. Biochem Biophys Res Commun. 2017;489:484–489. doi: 10.1016/j.bbrc.2017.05.177. [DOI] [PubMed] [Google Scholar]
- 18.Guerrero-Zotano A, Mayer IA, Arteaga CL. PI3K/AKT/mTOR: role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev. 2016;35:515–524. doi: 10.1007/s10555-016-9637-x. [DOI] [PubMed] [Google Scholar]
- 19.Cheng TD, Ambrosone CB, Hong CC, Lunetta KL, Liu S, Hu Q, Yao S, Sucheston-Campbell L, Bandera EV, Ruiz-Narváez EA, Haddad S, Troester MA, Haiman CA, Bensen JT, Olshan AF, Palmer JR, Rosenberg L. Genetic variants in the mTOR pathway and breast cancer risk in African American women. Carcinogenesis. 2016;37:49–55. doi: 10.1093/carcin/bgv160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dry JR, Pavey S, Pratilas CA, Harbron C, Runswick S, Hodgson D, Chresta C, McCormack R, Byrne N, Cockerill M, Graham A, Beran G, Cassidy A, Haggerty C, Brown H, Ellison G, Dering J, Taylor BS, Stark M, Bonazzi V, Ravishankar S, Packer L, Xing F, Solit DB, Finn RS, Rosen N, Hayward NK, French T, Smith PD. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244) Cancer Res. 2010;70:2264–2273. doi: 10.1158/0008-5472.CAN-09-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, Santos ED, Chen Z, dos Santos O, Ayral-Kaloustian S, Venkatesan A, Hollander I. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res. 2011;17:3193–3203. doi: 10.1158/1078-0432.CCR-10-1694. [DOI] [PubMed] [Google Scholar]
- 22.Liu H, Scholz C, Zang C, Schefe JH, Habbel P, Regierer AC, Schulz CO, Possinger K, Eucker J. Metformin and the mTOR inhibitor everolimus (RAD001) sensitize breast cancer cells to the cytotoxic effect of chemotherapeutic drugs in vitro. Anticancer Res. 2012;32:1627–1637. [PubMed] [Google Scholar]
- 23.Ludwik KA, Campbell JP, Li M, Li Y, Sandusky ZM, Pasic L, Sowder ME, Brenin DR, Pietenpol JA, O’Doherty GA, Lannigan DA. Development of a RSK inhibitor as a novel therapy for triple-negative breast cancer. Mol Cancer Ther. 2016;15:2598–2608. doi: 10.1158/1535-7163.MCT-16-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maiello MR, D’Alessio A, Bevilacqua S, Gallo M, Normanno N, De Luca A. EGFR and MEK blockade in triple negative breast cancer cells. J Cell Biochem. 2015;116:2778–2785. doi: 10.1002/jcb.25220. [DOI] [PubMed] [Google Scholar]
- 25.Mirzoeva OK, Das D, Heiser LM, Bhattacharya S, Siwak D, Gendelman R, Bayani N, Wang NJ, Neve RM, Guan Y, Hu Z, Knight Z, Feiler HS, Gascard P, Parvin B, Spellman PT, Shokat KM, Wyrobek AJ, Bissell MJ, McCormick F, Kuo WL, Mills GB, Gray JW, Korn WM. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009;69:565–572. doi: 10.1158/0008-5472.CAN-08-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez L, Thames E, Kim J, Chaudhuri G, Singh R, Pervin S. Increased sensitivity of African American triple negative breast cancer cells to nitric oxide-induced mitochondria-mediated apoptosis. BMC Cancer. 2016;16:559. doi: 10.1186/s12885-016-2547-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pervin S, Tran L, Urman R, Braga M, Parveen M, Li SA, Chaudhuri G, Singh R. Oxidative stress specifically downregulates survivin to promote breast tumour formation. Br J Cancer. 2013;108:848–858. doi: 10.1038/bjc.2013.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh R, Parveen M, Basgen JM, Fazel S, Meshesha MF, Thames EC, Moore B, Martinez L, Howard CB, Vergnes L, Reue K, Pervin S. Increased expression of beige/brown adipose markers from host and breast cancer cells influence xenograft formation in mice. Mol Cancer Res. 2016;14:78–92. doi: 10.1158/1541-7786.MCR-15-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tárrega C, Pulido R. A one-step method to identify MAP kinase residues involved in inactivation by tyrosine- and dual-specificity protein phosphatases. Anal Biochem. 2009;394:81–86. doi: 10.1016/j.ab.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Theodosiou A, Ashworth A. MAP kinase phosphatases. Genome Biol. 2002;3:REVIEWS3009. doi: 10.1186/gb-2002-3-7-reviews3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Fei T, Zhang J, Zhu G, Wang L, Lu D, Chi X, Teng Y, Hou N, Yang X, Zhang H, Han JD, Chen YG. BMP4 signaling acts via dual-specificity phosphatase 9 to control ERK activity in mouse embryonic stem cells. Cell Stem Cell. 2012;10:171–182. doi: 10.1016/j.stem.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 33.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su Y, Subedee A, Bloushtain-Qimron N, Savova V, Krzystanek M, Li L, Marusyk A, Tabassum DP, Zak A, Flacker MJ, Li M, Lin JJ, Sukumar S, Suzuki H, Long H, Szallasi Z, Gimelbrant A, Maruyama R, Polyak K. Somatic cell fusions reveal extensive heterogeneity in basal-like breast cancer. Cell Rep. 2015;11:1549–1563. doi: 10.1016/j.celrep.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 35.Koren S, Bentires-Alj M. Breast tumor heterogeneity: source of fitness, hurdle for therapy. Mol Cell. 2015;60:537–546. doi: 10.1016/j.molcel.2015.10.031. [DOI] [PubMed] [Google Scholar]
- 36.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Honeth G, Bendahl PO, Ringnér M, Saal LH, Gruvberger-Saal SK, Lövgren K, Grabau D, Fernö M, Borg A, Hegardt C. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008;10:R53. doi: 10.1186/bcr2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao FQ, Misra Y, Li DB, Wadsworth MP, Krag D, Weaver D, Tessitore J, Li DW, Zhang G, Tian Q, Buss K. Differential expression of Oct3/4 in human breast cancer and normal tissues. Int J Oncol. 2018;52:2069–2078. doi: 10.3892/ijo.2018.4341. [DOI] [PubMed] [Google Scholar]
- 40.Gwak JM, Kim M, Kim HJ, Jang MH, Park SY. Expression of embryonal stem cell transcription factors in breast cancer: Oct4 as an indicator for poor clinical outcome and tamoxifen resistance. Oncotarget. 2017;8:36305–36318. doi: 10.18632/oncotarget.16750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W, Sun L, Yang X, Wang Y, Zhang Y, Shang Y. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J Biol Chem. 2008;283:17969–17978. doi: 10.1074/jbc.M802917200. [DOI] [PubMed] [Google Scholar]
- 42.Petrochilos D, Shojaie A, Gennari J, Abernethy N. Using random walks to identify cancer-associated modules in expression data. Bio Data Min. 2013;6:17. doi: 10.1186/1756-0381-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nichols J, Silva J, Roode M, Smith A. Suppression of Erk signalling promotes ground state pluripotency in the mouse embryo. Development. 2009;136:3215–3222. doi: 10.1242/dev.038893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burdon T, Stracey C, Chambers I, Nichols J, Smith A. Suppression of SHP-2 and ERK signalling promotes self-renewal of mouse embryonic stem cells. Dev Biol. 1999;210:30–43. doi: 10.1006/dbio.1999.9265. [DOI] [PubMed] [Google Scholar]
- 45.Liu X, Chen M, Li L, Gong L, Zhou H, Gao D. Extracellular Signal-regulated kinases (ERKs) phosphorylate Lin28a protein to modulate p19 cell proliferation and differentiation. J Biol Chem. 2017;292:3970–3976. doi: 10.1074/jbc.C117.775122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takashima Y, Guo G, Loos R, Nichols J, Ficz G, Krueger F, Oxley D, Santos F, Clarke J, Mansfield W, Reik W, Bertone P, Smith A. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158:1254–1269. doi: 10.1016/j.cell.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garavello NM, Pena DA, Bonatto JM, Duarte ML, Costa-Junior HM, Schumacher RI, Forti FL, Schechtman D. Activation of protein kinase C delta by ψδRACK peptide promotes embryonic stem cell proliferation through ERK 1/2. J Proteomics. 2013;94:497–512. doi: 10.1016/j.jprot.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 48.Nii T, Marumoto T, Kawano H, Yamaguchi S, Liao J, Okada M, Sasaki E, Miura Y, Tani K. Analysis of essential pathways for self-renewal in common marmoset embryonic stem cells. FEBS Open Bio. 2014;4:213–219. doi: 10.1016/j.fob.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Balko JM, Schwarz LJ, Bhola NE, Kurupi R, Owens P, Miller TW, Gómez H, Cook RS, Arteaga CL. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer Res. 2013;73:6346–6358. doi: 10.1158/0008-5472.CAN-13-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang HY, Cheng Z, Malbon CC. Overexpression of mitogen-activated protein kinase phosphatases MKP1, MKP2 in human breast cancer. Cancer Lett. 2003;191:229–237. doi: 10.1016/s0304-3835(02)00612-2. [DOI] [PubMed] [Google Scholar]
- 51.Ma G, Pan Y, Zhou C, Sun R, Bai J, Liu P, Ren Y, He J. Mitogen-activated protein kinase phosphatase 1 is involved in tamoxifen resistance in MCF7 cells. Oncol Rep. 2015;34:2423–2430. doi: 10.3892/or.2015.4244. [DOI] [PubMed] [Google Scholar]
- 52.Wise R, Zolkiewska A. Metalloprotease-dependent activation of EGFR modulates CD44(+)/CD24(-) populations in triple negative breast cancer cells through the MEK/ERK pathway. Breast Cancer Res Treat. 2017;166:421–433. doi: 10.1007/s10549-017-4440-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Judd NP, Winkler AE, Murillo-Sauca O, Brotman JJ, Law JH, Lewis JS Jr, Dunn GP, Bui JD, Sunwoo JB, Uppaluri R. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res. 2012;72:365–374. doi: 10.1158/0008-5472.CAN-11-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang YQ, Wei XL, Liang YK, Chen WL, Zhang F, Bai JW, Qiu SQ, Du CW, Huang WH, Zhang GJ. Over-expressed twist associates with markers of epithelial mesenchymal transition and predicts poor prognosis in breast cancers via ERK and Akt activation. PLoS One. 2015;10:e0135851. doi: 10.1371/journal.pone.0135851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith BN, Burton LJ, Henderson V, Randle DD, Morton DJ, Smith BA, Taliaferro-Smith L, Nagappan P, Yates C, Zayzafoon M, Chung LW, Odero-Marah VA. Snail promotes epithelial mesenchymal transition in breast cancer cells in part via activation of nuclear ERK2. PLoS One. 2014;9:e104987. doi: 10.1371/journal.pone.0104987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dasgupta A, Sawant MA, Kavishwar G, Lavhale M, Sitasawad S. AECHL-1 targets breast cancer progression via inhibition of metastasis, prevention of EMt and suppression of cancer stem cell characteristics. Sci Rep. 2016;6:38045. doi: 10.1038/srep38045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu S, Wang Y, Sun L, Zhang Z, Jiang Z, Qin Z, Han H, Liu Z, Li X, Tang A, Gui Y, Cai Z, Zhou F. Decreased expression of dual-specificity phosphatase 9 is associated with poor prognosis in clear cell renal cell carcinoma. BMC Cancer. 2011;11:413. doi: 10.1186/1471-2407-11-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Y, Lagowski J, Sundholm A, Sundberg A, Kulesz-Martin M. Microtubule disruption and tumor suppression by mitogen-activated protein kinase phosphatase 4. Cancer Res. 2007;67:10711–10719. doi: 10.1158/0008-5472.CAN-07-1968. [DOI] [PubMed] [Google Scholar]
- 59.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, Hingorani SR, Zaks T, King C, Jacobetz MA, Wang L, Bronson RT, Orkin SH, DePinho RA, Jacks T. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 60.Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F, Brown Swigart L, Pham DM, Seo Y, Evan GI, Martins CP. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature. 2010;468:567–571. doi: 10.1038/nature09526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cho JA, Park H, Lim EH, Lee KW. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int J Oncol. 2012;40:130–138. doi: 10.3892/ijo.2011.1193. [DOI] [PubMed] [Google Scholar]