

Abstract
Cation-coupled chloride cotransporters (CCC) play a role in modulating intracellular chloride concentration ([Cl−]i) and cell volume. Cell shrinkage and cell swelling are accompanied by an increase or decrease in [Cl−]i, respectively. Cell shrinkage and a decrease in [Cl−]i increase the activity of NKCCs (Na-K-Cl cotransporters: NKCC1, NKCC2, and Na-Cl) and inhibit the activity of KCCs (K-Cl cotransporters: KCC1 to KCC4), wheras cell swelling and an increase in [Cl−]i activate KCCs and inhibit NKCCs; thus, it is unlikely that the same kinase is responsible for both effects. WNK1 and WNK4 are chloride-sensitive kinases that modulate the activity of CCC in response to changes in [Cl−]i. Here, we showed that WNK3, another member of the serine-threonine kinase WNK family with known effects on CCC, is not sensitive to [Cl−]i but can be regulated by changes in extracellular tonicity. In contrast, WNK4 is highly sensitive to [Cl−]i but is not regulated by changes in cell volume. The activity of WNK3 toward NaCl cotransporter is not affected by eliminating the chloride-binding site of WNK3, further confirming that the kinase is not sensitive to chloride. Chimeric WNK3/WNK4 proteins were produced, and analysis of the chimeras suggests that sequences within the WNK’s carboxy-terminal end may modulate the chloride affinity. We propose that WNK3 is a cell volume-sensitive kinase that translates changes in cell volume into phosphorylation of CCC.
Keywords: CCCs, cell volume, chloride, NCC, WNK3, WNK4
INTRODUCTION
The SLC12 family of solute carriers, known as the electroneutral cation-coupled chloride cotransporters (CCC), is divided into two major branches. The Na-coupled chloride branch (NKCCs) includes the thiazide-sensitive Na-Cl (NCC) and the bumetanide-sensitive Na-K-2Cl cotransporters NKCC1 and NKCC2; the K-coupled branch (KCCs) includes the chloride cotransporters KCC1 to KCC4. The coordinated activity of these transporters functions in the response to changes in cell volume by modulating intracellular chloride concentration ([Cl−]i) and transepithelial ion transport. For instance, with a regulatory volume increase (RVI), which occurs during cell shrinkage, or with a decrease in the [Cl−]i, NKCCs are activated and KCCs inhibited. In contrast, with a regulatory volume decrease (RVD), which occurs during cell swelling, or with an increase in the [Cl−]i, activation of the K-driven transporters is required along with the inhibition of the Na-driven cotransporters (4, 11).
The coordinated modulation of CCC activity is possible because phosphorylation/dephosphorylation of key serine/threonine residues promotes opposite effects in the Na-driven and K-driven branches. Phosphorylation activates Na-Cl cotransporters but inhibits K-Cl cotransporters, whereas dephosphorylation inhibits Na-Cl cotransporters and activates K-Cl cotransporters (2, 4). Accordingly, cell shrinkage or intracellular chloride depletion results in CCC phosphorylation, activating Na-driven members and inhibiting K-driven transporters. Conversely, cell swelling or increasing [Cl−]i results in dephosphorylation of CCCs, inhibiting Na-coupled transporters and activating K-driven cotransporters.
Recent works have shown that recognition of intracellular chloride depletion is communicated to the CCCs by a complex cascade of kinases and phosphatases (2, 23). The STE20-related proline/alanine-rich kinase (SPAK) and the oxidative stress responsive kinase (OSR1) are directly responsible for CCC phosphorylation; NCC, NKCC1, and NKCC2 are phosphorylated at key residues of the amino-terminal domain to increase their activity, and KCCs are phosphorylated in key residues of the carboxy-terminal domain to decrease their activity (9, 20). SPAK/OSR1 activity in turn is regulated by the with no lysine (WNK) kinases (2, 26). When autophosphorylated in the T-loop, the WNK kinases are able to activate SPAK/OSR1, which in turn phosphorylates the CCCs. Of the WNK kinase isoforms, WNK4 is the most sensitive to [Cl−]i, followed by WNK1 (5, 19, 24). Thus, WNK1 and WNK4 are currently considered chloride-sensitive kinases.
It is not clear how a chloride-sensitive kinase can modulate CCCs in response to changes in cell volume, given that they are expected to produce the opposite effect in [Cl−]i. For instance, cell shrinkage that activates NKCC1/NKCC2 and inhibits KCCs is expected to result in an increase in [Cl−]i, which would inhibit NKCCs and activate KCCs. Conversely, cell swelling that activates KCCs and inhibits NKCCs is expected to result in a decrease in [Cl−]i that should activate NKCCs and inhibit KCCs. Thus, changes in cell volume for regulation of CCCs must be translated by a kinase that is sensitive to cell volume rather than to [Cl−]i. In this regard, work from our laboratory revealed that WNK3 and its catalytically inactive version are able to bypass the tonicity requirements for CCC modulation (10, 14, 21), suggesting that WNK3 activity could be modulated by cell volume. In addition, Zhang et al. (27) have provided evidence based on functional kinomics that WNK3 plays a critical role in the neuronal response to changes in cell volume. These data suggest that WNK3 activity is not regulated by changes in [Cl−]i but by changes in cell volume.
In the present work, we analyzed the differential modulation of WNK3 and WNK4 by cell volume and [Cl−]i and began to elucidate the regions of the kinases involved in the sensing to these physiological variables.
MATERIALS AND METHODS
Clones and mutations.
The following previously described clones were used in this study: human L-WNK1 (6), human WNK2 (22), human c-Myc-tagged WNK3 cDNA clone containing exon 18a without exon 22 (isoform 3) and the corresponding human c-Myc-tagged catalytically inactive form WNK3-D294A (8), mouse HA-tagged WNK4 (6), rat Flag-tagged NCC, and mouse KCC4 (13, 16, 17). The following mutations were introduced in corresponding clones, using custom primers (Sigma) and site-directed mutagenesis: WNK3-L295F, WNK3-L297F, WNK3-L295F/L297F, WNK4-L319F, and WNK3-DA-L295F/L297F. To avoid unwanted mutations, in all cases, the mutant fragment was fully sequenced between two unique restriction sites, digested, and ligated back into the original wild-type cDNA.
Chimeric clone construction and in vitro cRNA translation.
Human WNK3 (hWNK3) and mouse WNK4 (mWNK4) were previously cloned into the pGH19 vector and have been sequenced and analyzed. Restriction site analysis was used to define potential bases where internal silent restriction sites could be introduced to be used in the construction of a chimeric WNK3/WNK4. To create WNK3/WNK4 chimeric constructs, KpnI, NsiI, and NotI internal silent restriction sites were introduced by site-directed mutagenesis (QuickChange II; Stratagene) of hWNK3 and mWNK4 in the linker sequence between the amino-terminal and kinase domains of the kinase and carboxy-terminal domain. Once we had the WNK3 and WNK4 mutants with the silent restriction sites, chimeric clones were constructed de novo by exchanging the coding sequence of the amino-terminal domain, the kinase domain, or the carboxy-terminal domain between the two kinases using appropriate restriction enzymes and DNA ligase to cut and paste sequences.
As a control in all experiments, we used the wild-type WNK3 (333) and WNK4 (444) constructs, in which the silent restriction sites KpnI, NsiI, and NotI were introduced without disturbing the functional WNK3 and WNK4 wild-type regulation of CCCs.
To prepare in vitro cRNA constructs for microinjection, WNK1 was linearized with the MluI restriction enzyme, WNK4 and chimeric WNK3/WNK4 were linearized at the 3′ end with XhoI, NCC and WNK3 were linearized with NotI, and KCC4 was linearized with NheI. All restriction enzymes were purchased from New England BioLabs (Carlsbad, CA), and sequences were transcribed with the T7 RNA polymerase mMESSAGE mMACHINE (Ambion, Austin, TX) transcription system. The transcriptional integrity of the products was confirmed on agarose gels, and the cRNA concentration was determined by the absorbance reading at 260 nm (DU 640; Beckman, Fullerton, CA). The cRNAs were stored in aliquots at −80°C until use.
Functional expression in Xenopus laevis oocytes.
The use of Xenopus frogs and this study were approved by the Institutional Animal Care and Use Committee of our institutions (Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán and Escuela de Medicina Universidad Panamericana) (protocol no. NMM-1428). Adult female Xenopus laevis frogs (NASCO; Fort Atkinson, MI) were kept in our animal facility under controlled light conditions, water conditions, and temperature (16°C). Oocytes were surgically collected after anesthesia was administered (0.17% tricaine immersion). Then, oocytes were incubated for 1 h in Ca2+-free ND96 medium (in mM: 2 KCl, 96 NaCl, 1 MgCl2, and 5 HEPES-Tris, pH 7.4, 200 mOsm/kg) with 2 mg/mL collagenase A, washed four times with regular ND96 200 mOsm/kg, manually defolliculated, and incubated overnight in ND96 at 16°C. Mature oocytes were injected with 50 nL of water or with 0.1–0.4 µg/µL in vitro-transcribed cRNA from the constructs described above. Oocytes were incubated in ND96 at 16°C supplemented with 2.5 mM sodium pyruvate and 5 mg/100 mL of gentamicin for 72 h before being used in transport assays or having their protein extracted for Western blot assay. The incubation medium was changed every 24 h. To determine the effect of the [Cl−]i on oocytes, cells were switched to Cl−-free ND96 for 16 h or to low-chloride hypotonic stress (LCHS) medium for 16 h before the uptake assay, as previously described (5, 18). To determine the effect of changes in extracellular osmolarity on oocytes, experiments were performed using a similar solution with three different osmolarities. We used ND50 medium (in mM: 10 KCl, 33 NaCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES; 100 mOsm/kg) to which sucrose was added to increase the osmolarity; 34.2 g/L was used to increase to 200 mOsm/kg, and 53.36 g/l was used to increase to 250 mOsm/kg. Thus, all three solutions were identical in ion content but unique in osmolarity.
Functional assays with CCCs and statistical analysis.
X. laevis oocytes were microinjected with NCC or KCC4 cRNA alone or in combination with one of the following: human WNK1 cRNA, human WNK2, wild-type or mutant human WNK3, wild-type or mutant mouse WNK4, and chimeric cRNA construct. After 72 h of incubation, thiazide-sensitive 22Na+ (for NCC) or Cl−-dependent 86Rb+ uptake (for KCCs) was determined. Briefly, NCC activity was assessed under isotonic (220 mOsm/kg H2O) or low-chloride hypotonic stress conditions (170 mOsm/kg H2O). For the assay in isotonic condition, oocytes were preincubated for 10 min in a chloride-free solution containing ouabain, amiloride, and bumetanide, followed by 60-min uptake in the presence of chloride and the cocktail drugs. We have previously shown that the 10 min of preincubation in chloride-free medium had no effect on NCC activity (15, 17). For the assay in low-chloride hypotonic solution, oocytes were preincubated for 10 min in the same solution with cocktail drugs, followed by 60 min in a NaCl-containing medium. KCC4 activity was assessed either in hypotonic conditions (110 mOsm/kg H2O) in which the cotransporter is maximally active or in isotonic conditions in which the cotransporter is inhibited. The following inhibitors were used during the incubation and uptake period, as needed: ouabain to block the Na-K-ATPase, amiloride to inhibit the Na+ channels, bumetanide to block the activity of the Na-K-2Cl cotransporter, and metolazone or other thiazides to inhibit the Na-Cl cotransporter. All uptake measurements were performed at 32°C. At the end of the uptake period, oocytes were dissolved in 10% SDS, and β-scintillation counting was used to determine the tracer activity for each oocyte. All experiments were performed at least three times with a minimum of 10 oocytes per condition. Statistical significance was defined as a two-tailed P ≤ 0.05, and results were reported as means ± SE. We set the control group uptake at 100%, and experimental groups were normalized accordingly. The significant differences between groups were tested by one-way ANOVA, and Bonferroni corrections were used for comparisons of multiple samples.
Antibodies.
The WNK3, WNK4, chimeras, and NCC protein expression were analyzed using monoclonal antibodies against c-Myc, HA, and Flag tags, as described in results. In some experiments and for some chimeras, we used the COOH-terminal WNK3 antibody obtained from the Medical Research Council Protein Phosphorylation Unit (MRC-PPU) at Dundee University (Dundee, UK). Phosphorylation of WNK3 or WNK4 was assessed using the phospho-antibody L-WNK1 at S382; SPAK phosphorylation was determined using the phosphor-SPAK-S373 (S670B). Both polyclonal antibodies were obtained from the MRC-PPU at Dundee University. The specificity of all used antibodies has been tested previously.
RESULTS
We first explored the different abilities of the four WNKs to sense a reduction in [Cl−]i by assessing the effect of each one on the activity of NCC. Oocytes were injected with NCC cRNA alone or in the presence of each WNK cRNA, and 3 days later, Na+ influx was assessed in control conditions (Fig. 1A) or after 16 h of incubation in low-chloride hypotonic stress (LCHS) solution (Fig. 1B). Our previous experience revealed that in control conditions the [Cl−]i in oocytes is ∼45 mM, and after 16 h of exposure to LCHS it is reduced to ∼20 mM, which is associated with increased phosphorylation of NCC and WNK4 (3, 5, 18). In Fig. 1A, the uptake observed in NCC cRNA-injected oocytes under control conditions was established as 100%, and the rest of the groups were normalized accordingly. WNK1 and WNK3 were able to increase activation of NCC to 331 ± 45% and 490 ± 94%, respectively, whereas both WNK2 and WNK4 had no effect on NCC [128 ± 25% and 149 ± 23%, respectively; P = not significant (NS) vs. NCC].
Fig. 1.

With no lysine kinase (WNK) 3 kinase is not sensitive to low-chloride hypotonic stress (LCHS). Thiazide-sensitive Na-Cl (NCC) activity was measured in X. laevis oocytes microinjected with NCC cRNA alone or together with each of the following kinase cRNAs: WNK1 (W1), WNK2 (W2), WNK3 (W3), or WNK4 (W4). A: uptake was measured in control conditions where the intracellular chloride concentration ([Cl−]i) was ∼45 mM (5). NCC activity in control conditions was set to 100%, and the rest of the groups were normalized accordingly. In control conditions, WNK1 and WNK3 were able to activate NCC to 331 ± 45% and 490 ± 94%, respectively, but no significant effect was mediated by W2 and W4 (128 ± 25% and 149 ± 23%, respectively). B: oocyte incubation in LCHS conditions for 16 h reduced the [Cl−]i by ∼22 mM (5). In this condition, NCC activity increased to 366% due to activation of endogenous WNKs, and this activation was subtracted from each group; thus, NCC activity under LCHS conditions is set to 100% (black bar). WNK1 increased NCC activity by an extra 127% over what was observed in control conditions, and WNK2 and WNK4 showed increased NCC activation of 215 ± 56% and 356 ± 55%, respectively. In contrast, in LCHS conditions, WNK3 induced NCC activity to a level that was similar to what was seen in control conditions, indicating that WNK3 is not further activated by LCHS conditions. Data are the means ± SE of 8 different experiments (*P = not significant vs. NCC).
As we have shown before, the activity of NCC is increased by exposure to LCHS from 100% (control condition) to 366 ± 25%. This is due to activation of an endogenous WNK, since we have previously shown that this increase is completely prevented by treatment with WNK463, which is a specific WNK inhibitor (3). In Fig. 1B, the increased uptake observed due to the endogenous activation of WNKs was subtracted from each group. Thus, uptake in NCC injected oocytes is set at 100%. Thus, following LCHS exposure (Fig. 1B, gray bars), WNK1 caused NCC activity to be increased to 458 ± 70%. Thus, WNK1 is able to increase NCC activity in control conditions to 331 ± 45% and further increase it by an extra 127% in LCHS conditions, suggesting that the activity of WNK1 is further increased after decreasing the [Cl−]i. In contrast, the level of NCC activity in the presence of WNK3 and LCHS was similar to the level observed in control conditions (490 ± 94%), indicating that WNK3 is able to activate NCC in control conditions, but no further activation by WNK3 was observed after LCHS exposure.
Our WNK2 and WNK4 observations suggest that these kinases are much more sensitive to [Cl−]i because, in control conditions, they exhibited no activating effect on NCC, whereas LCHS exposure in the presence of WNK2 or WNK4 resulted in the activity of NCC being significantly higher than it was in their absence (Fig. 1B). In oocytes injected with NCC cRNA alone and exposed to LCHS, NCC activity was set to 100 ± 25%, as previously described, whereas in the presence of WNK2 or WNK4 the NCC activity was 215 ± 56% and 356 ± 56% higher than NCC in LCHS conditions. These observations suggested that the ability of WNKs to sense [Cl−]i is as follows: WNK2 = WNK4 > WNK1 > WNK3. Furthermore, the high concentration of chloride in oocytes actually implies that WNK3 is not sensitive to [Cl−]i at all.
We next analyzed the effect of eliminating conserved leucine residues from subdomain I of WNK3; these residues are known to be critical for chloride binding in WNK1 and WNK4 (5, 19). As shown in Fig. 2, wild-type WNK4 had no effect on NCC (97.2 ± 11%), and the substitution of leucine 319 with phenylalanine in WNK4 (WNK4-L319F) resulted in a constitutively active kinase (NCC increase to 243 ± 22%, P < 0.01). This was not observed for WNK3. In the presence of wild-type WNK3, NCC activity was increased to 543 ± 50%, whereas in the presence of the mutant WNK3 the increase was similar: 653 ± 37% with WNK3-L295F, 585 ± 33% with WNK3-L297F, and 443 ± 37% with the double-mutant WNK3-L295/297F. Thus, eliminating the chloride-binding site in WNK3 had no effect on the activity of this kinase in relation to NCC. This observation is consistent with the results in Fig. 1, which show that the effect of WNK3 on NCC is fully present in control conditions and is not increased by intracellular chloride depletion. These data show that, as previously suggested (24), WNK4 is a chloride-sensitive kinase, but WNK3 is not.
Fig. 2.

With no lysine kinase (WNK) 3 is not as sensitive to the elimination of the chloride-binding site as WNK4 is. X. laevis oocytes were microinjected with thiazide-sensitive Na-Cl (NCC) cRNA alone, with each of the single mutants (WNK3-L295F or WNK3-L297F), with the double chloride-binding site L295F/L297F-WNK3 mutant, or with the single chloride-binding site mutant WNK4-L319F. As an uptake control, NCC was coinjected with wild-type human WNK3 or coinjected with wild-type mouse WNK4. Activity in the NCC-alone injection was arbitrarily set to 100% (black bar), and coinjection with wild-type kinases (WNK3 and WNK4) and mutants was normalized accordingly (open bars). The WNK3 with eliminated chloride-binding sites (L295F or L297F) and the double-mutant (L295F/L297F) did not yield results that were different from wild-type WNK3; however, the elimination of the chloride-binding site L319F increased NCC activity compared with wild-type WNK4 coinjection. Data are the means ± SE of 5 different experiments (*P < 0.01 WNK4 vs. WNK4-L319F).
The other major modulator of CCC activity and phosphorylation is cell volume, and previous data from our laboratory suggested that WNK3 could be involved in cell volume regulation, so we assessed the response of WNK3 and WNK4 to changes in extracellular tonicity. The phosphorylation of the canonical T loop threonine, which requires phosphorylation to activate the WNK kinases, was assessed using a phospho-specific antibody originally developed for the recognition of serine residue 382 of WNK1 (25). Because of the high conservation in this region between WNKs, the antibody cross-reacts and detects phosphorylation in any WNK. We thus used phosphorylation as a surrogate of activity. Oocytes injected exclusively with wild-type WNK3, wild-type WNK4, or mutant WNK4-L319F were incubated overnight in an isotonic solution containing 46 mM sodium chloride (with added sucrose to reach 200 mOsm/kg). The next day, the oocytes were switched for 1 h to hypotonic media containing 46 mM NaCl (150 mOsm/kg), isotonic media (with same solution with added sucrose to reach 200 mOsm/kg), or hypertonic solution (same solution but with added sucrose to reach 250 mOsm/kg). Thus, during this incubation, the [Cl−]i was not expected to change, since all groups were exposed to the same concentration of extracellular chloride. For WNK4, a group in LCHS solution was included to show phosphorylation under reduced [Cl−]i. As shown in Fig. 3A, phosphorylation of WNK3 was modulated by extracellular tonicity. Compared with isotonic conditions (lane 2), WNK3 phosphorylation was decreased by hypotonicity (lane 1) and increased by hypertonicity (lane 3). Comparative analysis of five different experiments revealed that these differences were statistically significant. In contrast, as shown in Fig. 3B, phosphorylation of WNK4 was similar in hypotonic, isotonic, or hypertonic conditions. The LCHS treatment groups show that WNK4 phosphorylation is achieved after intracellular chloride depletion. We reasoned that one possibility could be that the high-[Cl−]i environment of oocytes precludes WNK4 from being modulated by extracellular tonicity. Thus, we also assessed the effect of tonicity in the mutant WNK4-L319F, in which the ability to sense [Cl−]i is prevented by the elimination of the chloride-binding site. As shown in Fig. 3C, basal phosphorylation of WNK4-L319F is higher, as expected, but no change in phosphorylation was observed following changes in extracellular tonicity. Thus, these observations strongly suggest that WNK3 activity is modulated by cell volume and not by [Cl−]i, whereas WNK4 activity is modulated by [Cl−]i but not by cell volume.
Fig. 3.

With no lysine kinase (WNK) 3 activity is modulated by cell volume but not by intracellular chloride concentration ([Cl−]i), whereas WNK4 activity is modulated by [Cl−]i but not by cell volume. Representative Western blot assay (top) and densitometric analysis (bottom) are shown for experiments that used a phosphoantibody against p-Ser382-WNK1 (p-WNK) (25), which detects WNK3 and WNK4 phosphorylation at Ser308 and Ser379, respectively; phosphorylation of the proteins was used as surrogates of WNK activity. cMyc-WNK3, HA-WNK4, and β-actin levels were used as total protein loading control. Total proteins were extracted from X. laevis oocytes injected with wild-type hWNK3 (WNK3; A), wild-type mWNK4 (WNK4; B), or the chloride-binding motif mutant mWNK4-L319F (C) cRNAs; oocytes were previously incubated in hypotonic media (46 mM sodium chloride, 150 mOsm), isotonic media (46 mM sodium chloride, 200 mOsm), and hypertonic media (46 mM sodium chloride, 250 mOsm) for 1 h (see materials and methods). Low-chloride hypotonic stress (LCHS) incubation was used as control group for wild-type WNK4 (B) and mutant WNK4-L319F phosphorylation (C). As shown in A, WNK3 phosphorylation was modulated by extracellular tonicity, since all groups were exposed to the same concentration of extracellular chloride. B and C show no significant difference between tonicity conditions regarding WNK4 phosphorylation. Densitometric analysis used WNK phosphorylation/total protein under isotonic conditions as 100% (an arbitrary unit), and tonicity conditions were normalized accordingly. Data are the means ± SE of at least 4 different experiments (*P < 0.01 vs. isotonic bar).
The WNK kinases contain a short amino-terminal domain, followed by the kinase domain and a large, complex carboxy-terminal domain (Fig. 4). The degree of kinase domain identity between the four WNKs is very high (>80%), whereas the degree of identity for the amino-terminal and the carboxy-terminal domains is <20%. It is thus likely that sequences within the amino-terminal or carboxy-terminal domain are responsible for differences in modulation of WNK3 or WNK4 by changes in the [Cl−]i or the cell volume. Because WNKs are such large kinases, studying the behavior of the full-length proteins in simple assays has been impossible. In this regard, our results in Figs. 1–3 suggest that the differential activity of WNK3 and WNK4 toward NCC can be used as a surrogate to study the structure-function relationship in WNKs regarding the ability to sense chloride. Thus, to begin to explore potential regions or sequences that regulate WNK activation in response to [Cl−]i, we constructed chimeric proteins between WNK3 and WNK4. As shown in Fig. 4, to construct the chimeras, silent restriction sites were introduced into WNK3 and WNK4 sequences in the linker regions between the amino or the carboxy-terminal domains and the kinase domain. These constructs containing the new silent sites were sequenced and used as the wild-type WNK3 or WNK4 to ensure that no unwanted mutations were introduced. Then, by swapping the amino and/or carboxy-terminal domains between WNK3 and WNK4, we constructed six chimeras. For each chimera, the first letter denotes the origin of the amino-terminal domain, the second letter denotes the origin of the kinase domain, and the third letter denotes the origin of the carboxy-terminal domain. In addition, in Fig. 4, to further facilitate the data presentation, we use blue to indicate fragments belonging to WNK3 and red for those of WNK4.
Fig. 4.

Chimeric construction of with no lysine kinase (WNK) 3 and WNK4 kinases. A: schematic representation of amino-terminal (1st letter), kinase domain (2nd letter), and carboxy-terminal domain (3rd letter) WNK3 (blue) and WNK4 (red) kinases. The number below each domain shows the amino acid residues that conform to the domain. Vertical lines show the silent mutation introduced to generate the chimeric construction below. B: 6 combinations of WNK3/WNK4 domains were constructed using the silent restriction sites shown above.
Proteins were extracted from oocytes injected with wild-type WNK3 or WNK4 or with any of the chimeric cRNAs and were resolved in PAGE gels for Western blot analysis. All chimeric proteins were expressed at the expected size and were phosphorylated (Fig. 5A). Given that the effect of WNK3 and WNK4 on KCC4 is similar, that is, both kinases inhibit the activity of KCC4, we assessed the effect of all chimeras on KCC4 activity to demonstrate that the constructs were functional. The inhibitory effect of all chimeras in KCC4 was between 60 and 70%, similar to what was observed for wild-type WNK3 or WNK4 (data not shown).
Fig. 5.

Expression and functional characterization of with no lysine kinase (WNK) 3/WNK4 chimeric proteins in isotonic conditions. cRNA from WNK3/WNK4 chimeras (344, 334, 343, 433, 443, and 434) was expressed in X. laevis oocytes, and wild-type WNK3 (333) and WNK4 (444) cRNAs were used as references. A: Western blot data show the phosphorylation levels of the WNK3/WNK4 chimeras, as determined by the pWNK1-Ser382 (pWNK-S382) antibody (25) that recognizes WNK3 and WNK4 Ser308 and Ser379 phosphorylation, respectively (top). Total protein expression from WNK3 and chimeras 344 and 334 was determined using a cMyc antibody (amino-terminal tag), since chimeras 343, 433, and 443 were assessed by the WNK3 carboxy-terminal antibody, and 434 chimera and WNK4 detection was performed using HA antibody (carboxy-terminal tag) (middle). β-Actin expression was measured as an internal control (bottom). B: functional thiazide-sensitive Na-Cl (NCC) activity in X. laevis oocytes microinjected with NCC cRNA alone or in combination with each of WNK3 (333), WNK4 (444), or the chimeras (344, 443, and 343) in isotonic conditions showed that chimera 344, which contains the WNK4-carboxy-terminal domain, behaved as WNK4 did, and chimeras 443 and 343, which contain the WNK3-carboxy-terminal domain, activated NCC as WNK3 did. NCC activity alone was arbitrarily set to 100% (black bar), and coinjection with WNK3 (333) and WNK4 (444) and chimeras WNK3/WNK4 was normalized accordingly. C: functional expression showed that chimera 433, which contained the WNK3-carboxy-terminal domain, induced NCC activity as WNK3 did, since chimeras 334 and 434 decreased the NCC activity compared with WNK3, which induced activity; however, chimeras 334 and 434 could not decrease NCC activity as WNK4 did. For B and C, data are the means ± SE of at least 5 different experiments with 10 oocytes/experimental group [*P < 0.01 vs. NCC (black bar)].
The effect of chimeric WNKs on NCC is shown in Fig. 5, B and C. Chimeras that contain the WNK4 kinase domain are depicted in Fig. 5B. In this series of experiments, as expected, wild-type WNK4 had no effect on NCC activity, and wild-type WNK3 induced a significant increase in NCC activity, to ∼800%, compared with the control. The chimera containing the kinase and carboxy-terminal domains of WNK4 and the amino-terminal domain of WNK3 (344) behaved as wild-type WNK4. In contrast, the chimera containing the carboxy-terminal domain of WNK3 attached to the amino-terminal and kinase domains of WNK4 (443) induced a significant increase in NCC activity. A similar behavior was observed with chimera 343. On the other hand, as shown in Fig. 5C, the chimera with the amino-terminal domain of WNK4 and the kinase and carboxy-terminal domain of WNK3 (433) exhibited an NCC activating capacity that was similar to that of the wild-type WNK3. In contrast, in chimeras containing the carboxy-terminal domain of WNK4 (334 or 434), the NCC-activating capacity was significantly reduced, although the level of NCC activity was still significantly higher than that of the NCC control.
Finally, we assessed whether chloride sensitivity was involved in the activity changes in the chimeras. To this end, we used two approaches. First, we eliminated the chloride-binding site in each chimeric construct by introducing an L-F substitution, depending on the kinase domain of the chimera. Figure 6, A and B, shows results obtained from chimeras that contain the kinase domain of WNK3 or WNK4, respectively. Given that the chloride-binding site is located in the kinase domain, our initial hypothesis was that the sensitivity to chloride would reflect which kinase domain was present. For instance, containing the kinase domain of WNK4 would cause chloride sensitivity; thus, the L-F substitution would make the chimera more active. Conversely, the chimeras containing the kinase domain of WNK3 would be insensitive to chloride; that is, the L-F substitution would have no further effect on the chimera’s activity. However, to our surprise, only chimeras 434 (Fig. 6A) and 344 (Fig. 6B) slightly increased by the L-F substitution. Thus, the carboxy-terminal domain of WNK4 is able to modulate the chloride-sensing ability of the WNK3 kinase domain, and the carboxy-terminal domain of WNK3 eliminates the chloride-sensing ability of the WNK4 kinase domain.
Fig. 6.
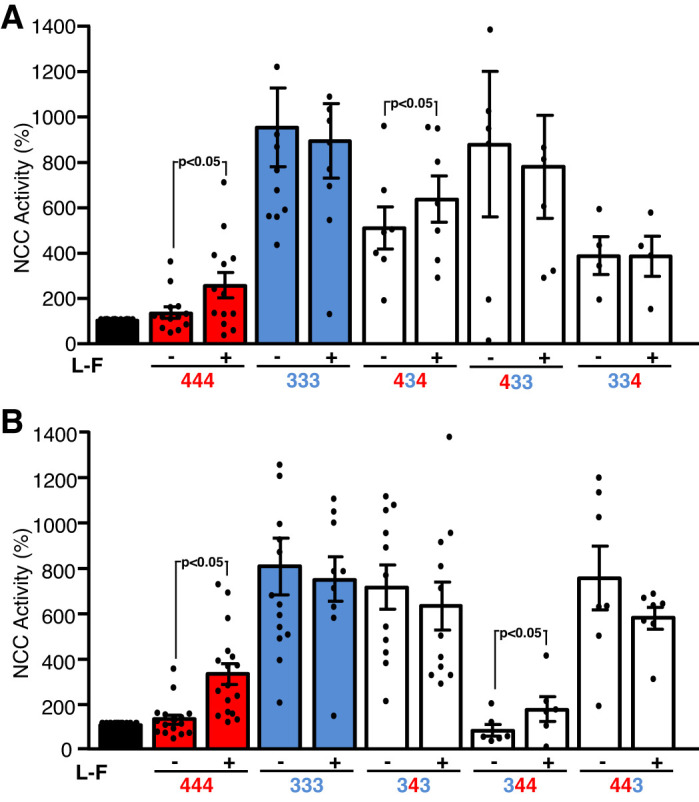
Effect of eliminating the chloride-binding site on the chimeras toward thiazide-sensitive Na-Cl (NCC). We assessed the effect of with no lysine kinase (WNK) 4 (444), WNK3 (333), and WNK3/WNK4 chimeras with or without the chloride-binding site (L-F: L295F-WNK3 kinase domain or L319F-WNK4 kinase domain) on NCC activity. As a control, we used wild-type WNK3 (333) and wild-type WNK4 (444) without (−) or with the elimination (+) of the chloride-binding site (LF). As we have previously shown, WNK4 is sensitive to chloride-binding site elimination (L-F), but WNK3 is not. A: chimeric proteins containing the WNK3 kinase domain (chimeras 434, 433, and 334). B: functional NCC activity induced by WNK3/WNK4 chimeras containing the WNK4 kinase domain (343, 344, and 443). In A, the chimera 434 is chloride sensitive, and in B, chimera 344 is also sensitive to chloride. Data are the means ± SE of at least 5 different experiments with 10 oocytes/experimental group.
Our data show that WNK3 carboxy-terminal domain confers constitutive activation of NCC that is no longer affected by eliminating the chloride-binding sites on the chimera. Thus, these chimeras were not analyzed further. Regarding the WNK4 carboxyl-terminal domain, the activity observed on the chimeras 334 and 434 was at a middle point between no activity (as WNK4) and full activity (as WNK3) (Fig. 5C), and we observed a slight increase with the L-F substitution (Fig. 6). We thus further analyzed whether these chimeras exhibit chloride sensitivity when exposed to LCHS. Unfortunately, during these experiments the functional expression of NCC was not working properly in the oocytes, so we were not able to use NCC as surrogate of WNKs or chimera activation. Instead, we analyzed the phosphorylation of SPAK/OSR1 in oocytes injected with corresponding cRNAs and exposed to the same process as in functional expression assays, that is, 2 or 3 days of incubation in ND96 after injection, followed by overnight incubation in control or LCHS and then by 1 h of incubation in the corresponding uptake solution (with exception of the tracer 22Na+). Figure 7A shows a representative blot and Fig. 7B the pooled results from three different experiments. As expected and consistent with Figs. 2 and 6, in oocytes injected with WNK3, SPAK/OSR1 phosphorylation was increased in control conditions, and no further increase was observed after LCHS, whereas in those injected with WNK4, no increase on phosphorylation was observed in control condition, but a significant increase was present after LCHS. Chimera 344 revealed neither an effect in control condition nor an increase after LCHS in SPAK/OSR1 phosphorylation. In contrast, chimeras 334 or 434 induced SPAK/OSR1 phosphorylation that was not further increased by LCHS.
Fig. 7.

Effect of with no lysine kinase (WNK) 4 carboxy-terminal containing chimeras on the STE20-related proline/alanine-rich kinase (SPAK)/oxidative stress-responsive kinase (OSR1) phosphorylation in oocytes exposed to control conditions or low-chloride hypotonic stress (LCHS). Total protein extracted from X. laevis oocytes microinjected with NCC and each of the chimeras 344, 334, and 434, as well as WNK3 (333) and WNK4 (444), was used for Western blot assay, using phospho-SPAK/OSR1 antibody (pSPAK). Oocytes microinjected were incubated in isotonic (Control) and 16-h exposure to LCHS. A: representative Western blot assay, in which SPAK/OSR1 phosphorylation was unaffected when 344 chimera was incubated in isotonic or LCHS. In contrast, chimeras 334 and 434 promoted an increase in SPAK/OSR1 phosphorylation that was not further increased when exposed to LCHS. As expected, coexpression with WNK3 in isotonic conditions induced SPAK/OSR1 phosphorylation that did not increase further after LCHS, whereas with WNK4, it did not increase phosphorylation in control condition, but it did after exposure to LCHS. B: densitometric analysis from 4 pooled experiments. Phosphorylation observed in isotonic conditions was set as 100% (as an arbitrary unit), and exposure to LCHS conditions was normalized accordingly. Data are the means ± SE of 4 different experiments.
DISCUSSION
The most important mechanisms for regulation of the cation-coupled chloride cotransporter activity appear to be counterintuitive. As represented in Fig. 8, the CCCs that are activated during cell shrinkage (NKCCs) are also inhibited when the [Cl−]i increases, which is expected to occur during cell shrinkage due to the efflux of water out of the cell. Thus, during cell shrinkage, one stimulus (cell volume) promotes activation of NKCCs and inhibition of KCCs despite the rise of [Cl−]i that would normally promote inhibition of NKCCs and activation of KCCs. A similar situation occurs during cell swelling; the activation of KCCs and inhibition of NKCCs is promoted despite the reduction in [Cl−]i that would be expected to produce the opposite effects. Because CCC activity is modulated by phosphorylation, a cell volume-sensitive kinase was expected to be responsible for this modulation. We hypothesized that such a kinase should be a chloride-insensitive kinase, unlike WNK1 or WNK4, given the movement of chloride during changes in cell volume discussed above.
Fig. 8.

Proposed model for with no lysine kinase (WNK) 3 and WNK4 sensitivity to cell volume and intracellular chloride concentration [Cl−]i toward cation-coupled chloride cotransporter (CCC) regulation. See text for explanation. KCC, K-Cl cotransporters; NKCC, Na-K-Cl cotransporters; P, phosphorylated state.
Recent work has clearly shown that WNK1 and WNK4 are chloride-sensitive kinases. Crystal structure information on the WNK1 kinase domain revealed that there is a chloride-binding site in subdomain I, where chloride binds directly into the catalytic site, stabilizing WNK1 in the inactive conformation (19). The WNK1 leucine residues 369 and 371 are critical for chloride binding, and substituting them for phenylalanine completely prevents the binding of an anion to the kinase, rendering WNK1 constitutively active. We have shown that WNK4 behaves in a similar fashion (5) and that the affinity of WNK4 for chloride is even higher than the affinity of WNK1 for chloride (24). Thus, WNK1 and WNK4 are clearly chloride-sensitive kinases (Fig. 8).
Regarding cell volume-mediated regulation of CCCs, studies from our laboratory demonstrated that the presence of WNK3 bypasses the tonicity requirements for CCC regulation. When coexpressed with WNK3, NKCC1 and NKCC2 are activated and remain activated, even when oocytes are exposed to a hypotonic solution that induces cell swelling and would be expected to abrogate their activity (14, 21). Similarly, when exposed to hypotonicity, which normally activates KCCs, the presence of WNK3 prevents such activation (10). Interestingly, the catalytically inactive form of WNK3 (WNK3-D294A) does the opposite. When coexpressed with WNK3-D294A, NKCC1 or NKCC2 activation is inhibited, even if cells are exposed to a hypertonic solution that would be expected to activate the transporters (14, 21). Similarly, in the presence of WNK3-D294A, KCCs are activated under isotonic conditions in which they are normally inactive. That is, extracellular hypotonicity is no longer required to activate KCCs (10). Thus, WNK3 makes CCCs behave as if the cells were exposed to hypertonicity, and WNK3-D294A makes CCCs behave as if cells were exposed to hypotonicity. Additionally, at least two different studies have also proposed that WNK3 is a kinase modulating cell volume (7, 27). These observations suggested that WNK3 activity is modulated by cell volume rather than by [Cl−]i.
Supporting this proposal, in the present study, we present evidence that the ability to sense [Cl−]i and the ability to sense cell volume is different among the WNK3 and WNK4 isoforms. When expressed in X. laevis oocytes that contain a relatively high [Cl−]i compared with many cells, the effect of WNKs on NCC varies tremendously. In one extreme, WNK2 and WNK4 cannot activate NCC in control conditions but are able to do so after exposure to LCHS, in which [Cl−]i is reduced (5). In a moderate response, WNK1, which is able to activate NCC in control conditions, is further stimulated by LCHS conditions. In the other extreme, WNK3 is able to activate NCC in control conditions, and no change is observed after LCHS exposure. Additionally, we observed that elimination of the putative chloride-binding sites in WNK3 does not change the WNK3-activating effect on NCC, indicating that even at the high [Cl−]i of oocytes, WNK3 is fully active toward NCC. Thus, whereas WNK2 and WNK4 are highly sensitive to chloride, WNK3 is not. Interestingly, an opposite effect occurs between WNK3 and WNK4 regarding changes in cell volume. WNK3 phosphorylation is modulated by cell volume, whereas WNK4 phosphorylation is not. During cell swelling, phosphorylation of WNK3 is decreased, and during cell shrinkage it is increased. The effect of cell volume on WNK3 agrees with previous observations that WNK3 promotes the activation of CCCs involved in RVI, such as NKCC1 or NKCC2, and inhibits those involved in RVD, such as KCCs. In this regard, a recent report (1) demonstrated that sucrose or the stronger osmolyte PGE400 affects the kinase activity of phospho-WNK3, strongly suggesting that macromolecular crowding, as is known to occur during cell shrinkage, promotes activation of WNK3. Unfortunately, WNK4 was not included in this study.
In our study, we used NCC as readout of WNK’s effect, rather than NKCC1, which is the primary transporter involved in RVI, because in our expression system the effect of WNK4 on NCC is much easier to observe than the effect on NKCC1, and the absence of an endogenous expression of NCC in oocytes makes the experiments cleaner than those to assess NKCC1, due to the endogenous expression of this transporter (12). Because WNK3 and WNK4 have been shown to be activators of both NKCC1 and NCC, through the SPAK pathway, we reasoned that either one would be good as a readout to analyze the effect of [Cl−]i or cell volume changes upon the activity or WNK3 or WNK4 toward the SLC12A cotransporters.
Together, these observations strongly suggest that, whereas WNK1 and WNK4 are the chloride-sensitive kinases that are communicating changes in [Cl−]i to the CCCs, WNK3 is a cell volume-sensitive kinase translating the changes in cell volume to the CCCs (Fig. 8). In this regard, it is important to clarify that our study of WNK3 is only regarding its relationship to CCCs, in which phosphorylation has been clearly demonstrated to modulate their activity. Whether WNK3 could mediate cell volume-induced regulation of other molecular players that participate in RVD and RVI is beyond the scope of this work.
In WNKs, the chloride-binding site is completely conserved, and the degree of identity in the kinase domain is very high, but the sensitivity for chloride varies tremendously; thus, it is reasonable to think that the amino and/or carboxy-terminal domains may play a role in modulating the affinity of each WNK for chloride. However, the only study designed to analyze the ability of WNKs to sense chloride was performed using only the kinase domains, since the expression of full-size WNKs in vitro was impossible (24). Taking advantage of the differential effects of WNK3 and WNK4 on NCC activity due to their difference in chloride affinity, we constructed a series of chimeric proteins by swapping the amino-terminal and carboxy-terminal domains between WNK3 and WNK4, thus producing proteins containing the kinase domain of WNK3 or WNK4, with the amino and/or carboxy-terminal domains of the opposite WNK. WNK3 and particularly the WNK4 kinase domain have the ability to sense chloride, and thus their activity toward NCC was clearly modulated by the carboxy-terminal domain. The WNK3 carboxy-terminal domain reduced the WNK4 kinase domain affinity for chloride, which was determined because chimeras 443 and 343 were able to activate NCC in control conditions, to a similar intensity as that shown with wild-type WNK3. This observation suggests that the WNK3 carboxyl-terminal domain can modulate chloride affinity in WNK4 kinase domain.
In contrast, the ability to activate NCC in chimeras containing the WNK3 kinase domain with the WNK4 carboxy-terminal domain was reduced but still remained significant. This could have two potential explanations. One is that the presence of WNK4 carboxyl-terminal domain induces some chloride affinity in the WNK3 kinase domain. The other is that in these chimeras the nonaffinity for WNK3 kinase domain for chloride persists, but the conformation of the resulting protein is far from ideal, and thus its capacity to activate NCC is lower. Our current data cannot clearly distinguish between both but suggest that the second is more likely. Although these chimeras 434 and 344 exhibited a slight but significant increase in the effect toward NCC by eliminating the chloride binding site in the kinase domain (which belongs to WNK3), their ability to induce SPAK/OSR1 phosphorylation is similar to wild-type WNK3, and no further effect was observed by LCHS, suggesting that activation of the chimera and SPAK/OSR1 is conserved, but the interaction with the SPAK-NCC complex is probably not ideal, and thus activation of NCC is partially precluded. Additional work is required to describe the precise mechanism that explains these observations.
Our study provides evidence that WNK3 and WNK4 sensitivities for chloride and cell volume changes are opposite. Thus we propose that whereas WNK4 is a chloride-sensitive kinase modulating the effect of intracellular chloride concentration upon the CCCs, WNK3 could be the cell volume-sensitive kinase translating the effect of changes in cell volume toward the CCCs.
GRANTS
This work was supported by CONACyT Grant No. S-8290, National Institute of Diabetes and Digestive and Kidney Diseases Grant DK051496 (to G.G.), and internal financing, Escuela de Medicina Universidad Panamericana (to D.P.A.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.P.-A., D.L.C.-P., A.M., K.L.-R., E.M., M.C.-B., and G.G. conceived and designed research; D.P.-A., D.L.C.-P., A.M., K.L.-R., E.M., E.H.-M., and N.V. performed experiments; D.P.-A., D.L.C.-P., A.M., E.M., E.H.-M., M.C.-B., N.V., and G.G. analyzed data; D.P.-A., D.L.C.-P., A.M., M.C.-B., and G.G. interpreted results of experiments; D.P.-A., A.M., and G.G. prepared figures; D.P.-A., A.M., and G.G. drafted manuscript; D.P.-A., D.L.C.-P., A.M., M.C.-B., and G.G. edited and revised manuscript; D.P.-A., D.L.C.-P., A.M., K.L.-R., E.M., E.H.-M., M.C.-B., N.V., and G.G. approved final version of manuscript.
REFERENCES
- 1.Akella R, Drozdz MA, Humphreys JM, Jiou J, Durbacz MZ, Mohammed ZJ, He H, Liwocha J, Sekulski K, Goldsmith EJ. A phosphorylated intermediate in the activation of WNK kinases. Biochemistry 59: 1747–1755, 2020. doi: 10.1021/acs.biochem.0c00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alessi DR, Zhang J, Khanna A, Hochdörfer T, Shang Y, Kahle KT. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal 7: re3, 2014. doi: 10.1126/scisignal.2005365. [DOI] [PubMed] [Google Scholar]
- 3.Argaiz ER, Chavez-Canales M, Ostrosky-Frid M, Rodríguez-Gama A, Vázquez N, Gonzalez-Rodriguez X, Garcia-Valdes J, Hadchouel J, Ellison D, Gamba G. Kidney-specific WNK1 isoform (KS-WNK1) is a potent activator of WNK4 and NCC. Am J Physiol Renal Physiol 315: F734–F745, 2018. doi: 10.1152/ajprenal.00145.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arroyo JP, Kahle KT, Gamba G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol Aspects Med 34: 288–298, 2013. doi: 10.1016/j.mam.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Bazúa-Valenti S, Chávez-Canales M, Rojas-Vega L, González-Rodríguez X, Vázquez N, Rodríguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, García-Valdés J, Hadchouel J, Gamba G. The effect of WNK4 on the Na+-Cl− cotransporter is modulated by intracellular chloride. J Am Soc Nephrol 26: 1781–1786, 2015. doi: 10.1681/ASN.2014050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chávez-Canales M, Zhang C, Soukaseum C, Moreno E, Pacheco-Alvarez D, Vidal-Petiot E, Castañeda-Bueno M, Vázquez N, Rojas-Vega L, Meermeier NP, Rogers S, Jeunemaitre X, Yang CL, Ellison DH, Gamba G, Hadchouel J. WNK-SPAK-NCC cascade revisited: WNK1 stimulates the activity of the Na-Cl cotransporter via SPAK, an effect antagonized by WNK4. Hypertension 64: 1047–1053, 2014. doi: 10.1161/HYPERTENSIONAHA.114.04036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cruz-Rangel S, Gamba G, Ramos-Mandujano G, Pasantes-Morales H. Influence of WNK3 on intracellular chloride concentration and volume regulation in HEK293 cells. Pflugers Arch 464: 317–330, 2012. doi: 10.1007/s00424-012-1137-4. [DOI] [PubMed] [Google Scholar]
- 8.Cruz-Rangel S, Melo Z, Vázquez N, Meade P, Bobadilla NA, Pasantes-Morales H, Gamba G, Mercado A. Similar effects of all WNK3 variants on SLC12 cotransporters. Am J Physiol Cell Physiol 301: C601–C608, 2011. doi: 10.1152/ajpcell.00070.2011. [DOI] [PubMed] [Google Scholar]
- 9.de los Heros P, Alessi DR, Gourlay R, Campbell DG, Deak M, Macartney TJ, Kahle KT, Zhang J. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl− co-transporters. Biochem J 458: 559–573, 2014. doi: 10.1042/BJ20131478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Los Heros P, Kahle KT, Rinehart J, Bobadilla NA, Vázquez N, San Cristobal P, Mount DB, Lifton RP, Hebert SC, Gamba G. WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc Natl Acad Sci USA 103: 1976–1981, 2006. doi: 10.1073/pnas.0510947103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev 85: 423–493, 2005. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- 12.Gamba G, Miyanoshita A, Lombardi M, Lytton J, Lee WS, Hediger MA, Hebert SC. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J Biol Chem 269: 17713–17722, 1994. [PubMed] [Google Scholar]
- 13.Garzón-Muvdi T, Pacheco-Alvarez D, Gagnon KB, Vázquez N, Ponce-Coria J, Moreno E, Delpire E, Gamba G. WNK4 kinase is a negative regulator of K+-Cl− cotransporters. Am J Physiol Renal Physiol 292: F1197–F1207, 2007. doi: 10.1152/ajprenal.00335.2006. [DOI] [PubMed] [Google Scholar]
- 14.Kahle KT, Rinehart J, de Los Heros P, Louvi A, Meade P, Vazquez N, Hebert SC, Gamba G, Gimenez I, Lifton RP. WNK3 modulates transport of Cl− in and out of cells: implications for control of cell volume and neuronal excitability. Proc Natl Acad Sci USA 102: 16783–16788, 2005. doi: 10.1073/pnas.0508307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mercado A, de Los Heros P, Melo Z, Chávez-Canales M, Murillo-de-Ozores AR, Moreno E, Bazúa-Valenti S, Vázquez N, Hadchouel J, Gamba G. With no lysine L-WNK1 isoforms are negative regulators of the K+-Cl− cotransporters. Am J Physiol Cell Physiol 311: C54–C66, 2016. doi: 10.1152/ajpcell.00193.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mercado A, Song L, Vazquez N, Mount DB, Gamba G. Functional comparison of the K+-Cl− cotransporters KCC1 and KCC4. J Biol Chem 275: 30326–30334, 2000. doi: 10.1074/jbc.M003112200. [DOI] [PubMed] [Google Scholar]
- 17.Moreno E, Plata C, Rodríguez-Gama A, Argaiz ER, Vázquez N, Leyva-Ríos K, Islas L, Cutler C, Pacheco-Alvarez D, Mercado A, Cariño-Cortés R, Castañeda-Bueno M, Gamba G. The european Eel NCCβ gene encodes a thiazide-resistant Na-Cl cotransporter. J Biol Chem 291: 22472–22481, 2016. doi: 10.1074/jbc.M116.742783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pacheco-Alvarez D, Cristóbal PS, Meade P, Moreno E, Vazquez N, Muñoz E, Díaz A, Juárez ME, Giménez I, Gamba G. The Na+:Cl− cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem 281: 28755–28763, 2006. doi: 10.1074/jbc.M603773200. [DOI] [PubMed] [Google Scholar]
- 19.Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7: ra41, 2014. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richardson C, Alessi DR. The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J Cell Sci 121: 3293–3304, 2008. doi: 10.1242/jcs.029223. [DOI] [PubMed] [Google Scholar]
- 21.Rinehart J, Kahle KT, de Los Heros P, Vazquez N, Meade P, Wilson FH, Hebert SC, Gimenez I, Gamba G, Lifton RP. WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl− cotransporters required for normal blood pressure homeostasis. Proc Natl Acad Sci USA 102: 16777–16782, 2005. doi: 10.1073/pnas.0508303102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rinehart J, Vázquez N, Kahle KT, Hodson CA, Ring AM, Gulcicek EE, Louvi A, Bobadilla NA, Gamba G, Lifton RP. WNK2 kinase is a novel regulator of essential neuronal cation-chloride cotransporters. J Biol Chem 286: 30171–30180, 2011. doi: 10.1074/jbc.M111.222893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shekarabi M, Zhang J, Khanna AR, Ellison DH, Delpire E, Kahle KT. WNK kinase signaling in ion homeostasis and human disease. Cell Metab 25: 285–299, 2017. doi: 10.1016/j.cmet.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int 89: 127–134, 2016. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thastrup JO, Rafiqi FH, Vitari AC, Pozo-Guisado E, Deak M, Mehellou Y, Alessi DR. SPAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem J 441: 325–337, 2012. doi: 10.1042/BJ20111879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391: 17–24, 2005. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Gao G, Begum G, Wang J, Khanna AR, Shmukler BE, Daubner GM, de Los Heros P, Davies P, Varghese J, Bhuiyan MI, Duan J, Zhang J, Duran D, Alper SL, Sun D, Elledge SJ, Alessi DR, Kahle KT. Functional kinomics establishes a critical node of volume-sensitive cation-Cl− cotransporter regulation in the mammalian brain. Sci Rep 6: 35986, 2016. doi: 10.1038/srep35986. [DOI] [PMC free article] [PubMed] [Google Scholar]