

Abstract
C57BL/6 mice are one of the most commonly used mouse strains in research, especially in kidney injury studies. However, C57BL/6 mice are resistant to chronic kidney disease-associated pathologies, particularly the development of glomerulosclerosis and interstitial fibrosis. Our laboratory and others developed a more clinically relevant dosing regimen of cisplatin (7 mg/kg cisplatin once a week for 4 wk and mice euthanized at day 24) that leads to the development of progressive kidney fibrosis in FVB/n mice. However, we found that treating C57BL/6 mice with this same dosing regimen does not result in kidney fibrosis. In this study, we demonstrated that increasing the dose of cisplatin to 9 mg/kg once a week for 4 wk is sufficient to consistently induce fibrosis in C57BL/6 mice while maintaining animal survival. In addition, we present that cohorts of C57BL/6 mice purchased from Jackson 1 yr apart and mice bred in-house display variability in renal outcomes following repeated low-dose cisplatin treatment. Indepth analyses of this intra-animal variability revealed C-C motif chemokine ligand 2 as a marker of cisplatin-induced kidney injury through correlation studies. In addition, significant immune cell infiltration was observed in the kidney after four doses of 9 mg/kg cisplatin, contrary to what has been previously reported. These results indicate that multiple strains of mice can be used with our repeated low-dose cisplatin model with dose optimization. Results also indicate that littermate control mice should be used with this model to account for population variability.
Keywords: acute kidney injury, C-C motif chemokine ligand 2, chronic kidney disease, cisplatin, fibrosis
INTRODUCTION
Cisplatin is a commonly used chemotherapeutic for many types of solid-organ cancers. Although it is very effective in killing cancer cells, it is hindered by its dose-limiting nephrotoxicity. Thirty percent of patients who receive cisplatin develop acute kidney injury (AKI), characterized by proximal tubule cell death, inflammation, and vascular injury (23, 26, 27). In the past, cisplatin-induced kidney injury has been modeled in mice by administering a single high dose of cisplatin. This model invokes a high level of tubule cell death and is lethal to mice within 3–4 days. Although this model can be useful for studying the short-term effects of cisplatin and AKI development, it does not allow for study of long-term renal function following treatment (36).
In the past decade, several studies have revealed the interconnectedness of AKI and chronic kidney disease (CKD), identifying AKI as a risk factor for CKD development (6–8). Studies have also identified a risk of long-term declines in renal function in pediatric patients treated with cisplatin who do not develop clinical AKI (40). These studies demonstrate the importance of studying the long-term effects of renal injuries. Our laboratory (33–35) and others (15, 19, 37) have used a repeated low-dose cisplatin model to observe AKI to CKD transitions following cisplatin treatment. In these studies, mice can survive up to 6 mo after cisplatin treatment and develop progressive renal fibrosis accompanied by declines in kidney function and chronic inflammation (35).
Our laboratory used FVB/n mice to characterize the renal response to repeated low-dose cisplatin treatment (34, 35). Mice were given four weekly doses of 7 mg/kg cisplatin. Following the fourth dose, mice developed moderate increases in blood urea nitrogen (BUN) and neutrophil gelatinase-associated lipocalin (NGAL), indicating subclinical kidney injury. Interestingly, mice also developed significant renal fibrosis, as measured by collagen deposition and α-smooth muscle actin (αSMA) expression. This fibrosis was accompanied by increased inflammatory cytokine mRNA expression and endothelial dysfunction. Kidney function continued to decline and fibrosis progressed up to 6 mo after cisplatin treatment, indicative of progressive CKD (35). We have since set out to optimize this repeated dosing model in different strains of mice.
C57BL/6 mice are used to generate many transgenic strains and are widely used in studies of kidney injury. However, these mice have been reported to be resistant to development of interstitial fibrosis and glomerulosclerosis in different models of CKD (30). When we treated C57BL/6 mice with 7 mg/kg cisplatin once a week for 4 wk, we did not observe development of fibrosis. Other studies have also demonstrated that C57BL/6 mice require a higher dose of cisplatin to develop pathologies indicative of CKD. However, this higher dosage can lead to decreased survival (3, 11, 15, 43).
The goal of this study was to determine if C57BL/6 mice can be used in our repeated low-dose cisplatin model with an increased dose of cisplatin without compromising animal survival and human relevance. In this study, we treated FVB/n and C57BL/6 mice with repeated weekly doses of 7 mg/kg and 9 mg/kg cisplatin for 4 wk. We observed that whereas FVB/n mice develop fibrosis and CKD-associated pathology with 7 mg/kg cisplatin, C57BL/6 mice require 9 mg/kg cisplatin to develop similar phenotypes. This increased dosage did not affect survival. Upon repeating studies with C57BL/6 mice, we also found that certain renal outcomes were variable. We observed a difference in structural damage and kidney function loss among C57BL/6 mice bred at and purchased from Jackson Laboratory 1 yr apart and C57BL/6 mice bred in-house. We analyzed this population variation and identified C-C motif chemokine ligand 2 (CCL2) as a marker of cisplatin-induced kidney injury. CCL2 is known to play a role in myeloid cell trafficking, and in accordance, we also observed an increase in infiltrating renal macrophages and monocytes in C57BL/6 mice following repeated doses of 9 mg/kg cisplatin. This study demonstrates that C57BL/6 mice can be used in the repeated low-dose cisplatin model and highlights the importance of using littermate control wild-type mice.
MATERIALS AND METHODS
Animals.
Eight-week-old male FVB/n and C57BL/6 mice were maintained on a 12:12-h light-dark cycle and provided food and water ad libitum. Animals were maintained under standard laboratory conditions. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Louisville (Protocol ID: 16593 and 19568) and followed the guidelines of the American Veterinary Medical Association. Briefly, mice were intraperitoneally injected with either 0.9% N saline vehicle or cisplatin at either 7 mg/kg or 9 mg/kg at 8:00 AM once a week for 4 wk and euthanized at day 24 (Supplemental Fig. S1; all Supplemental Material is available at https://doi.org/10.6084/m9.figshare.12658364.v1). Pharmaceutical-grade cisplatin purchased directly from the University of Louisville hospital pharmacy (1 mg/mL in 0.9% N saline from either Teva or Intas Pharmaceuticals) was used for all experiments. Mice were monitored for weight loss or evidence of high levels of discomfort/stress. Upon euthanasia, plasma was prepared and stored at −80°C. Kidneys were flash-frozen in liquid nitrogen or fixed in 10% neutral buffered formalin.
Blood urea nitrogen determination.
BUN was measured in the plasma of mice using a kit from AMS Diagnostics (80146, AMS Diagnostics) as per the manufacturer’s instructions and as previously published (35).
ELISAs.
ELISA for NGAL (DY1857, R&D Systems) was performed on mouse urine as previously published (35). ELISA for CCL2 (DY479-05, R&D Systems) was performed on mouse urine according to the manufacturer’s protocol.
Pathology scoring and analysis.
The morphological damage to the kidney was analyzed by light microscopy using sections stained with hematoxylin-eosin (H&E) and periodic acid-Schiff (PAS) stains and evaluating the entire kidney cross-section by a semiquantitative method. The following histological criteria were scored on a 0–4 scale: tubular necrosis, brush border loss, tubular dilatation, tubular cast formation, distal nephron damage, degeneration, and inflammation. Degeneration was scored in the proximal tubules that were not necrotic and included cell swelling, the formation of vacuoles, PAS-positive granules in the cytoplasm due to the damage to the membranes, and disruption of ion transport resulting in the accumulation of intracellular lipids and proteins or lipoproteins. Regeneration was scored in the tubules that underwent necrosis and were relined by new undifferentiated epithelial cells. The score was given depending on the extent of these changes and the state of differentiation of the epithelium. Inflammation was determined based on the density and the number of inflammatory cells throughout the kidney. These parameters were evaluated on a scale of 0–4: not present (0), mild (1), moderate (2), severe (3), and very severe (4).The scores were given based on the presence and extent of histologic damage using the following findings: 0 = damage is not present; 1 = damage is present only in and/or around individual tubules; 2 = damage is present in and/or around small group of tubules; 3 = damage is present confluently in the corticomedullary junction; and 4 = the damage extends to the outer cortex and it can reach the surface of the kidney.
Gene expression.
Total RNA was isolated from the kidney cortex, and cDNA was made as previously published (35). The following predesigned TaqMan primers (Life Technologies) were used: tumor necrosis factor-α (Tnfα) (Mm00443258_m1), interleukin-6 (Il-6) (Mm00446190_m1), C-X-C motif chemokine ligand 1 (Cxcl1) (Mm04207460_m1), Ccl2 (Mm00441242_m1), β2-microglobulin (B2m) (Mm00437762_m1), and arginase 1 (Arg-1) (Mm00475988_m1). The following self-designed primers were used: kidney injury molecule-1 (Kim-1) forward: AGATCCACACATGTACCAACATCAA and reverse: CAGTGCCATTCCAGTCTGGTTT; and inducible nitric oxide synthase (iNOS) forward: GAGATTGGAGGCCTTGTGTCA and reverse: TCAAGCACCTCCAGGAACGT. Quantitative RT-PCR was performed using iTaq Universal Probes Supermix (172–5134, Bio-Rad) or iTaq Universal SYBR Green Supermix (172–5124, Bio-Rad).
Protein isolation/quantification and Western blot analysis.
Protein isolation, quantification, and Western blot analysis were performed on the kidney cortex, as previously published (35), using 1:5,000 dilutions for primary antibodies, except for β-actin (1:10,000). For secondary antibodies, 1:20,000 dilution was used. Proteins of interest were detected by using a chemiluminescence substrate. The following antibodies were purchased from Cell Signaling: cleaved caspase-3 (no. 9664) and C/EBP homologous protein (CHOP; no. 2895). β-Actin antibody was purchased from Sigma (no. A2228).
Immunohistochemistry and Sirius Red/Fast Green staining.
αSMA immunohistochemistry for myofibroblasts and Sirius Red/Fast Green (SR/FG) staining for total collagen deposition were performed on paraffin-embedded kidney sections, as previously published (35). Quantification of positive staining was done using ImageJ to determine the mean intensity of positive staining. The optical density was calculated as follows: Log (255/mean intensity), as 255 is the max intensity for 8-bit images.
Flow cytometry.
Whole kidneys were homogenized into single-cell suspensions via mechanical disruption and enzymatic digestion with Liberase DL Research Grade (05466202001, Millipore/Sigma). After being passed through a 40-μm filter, cells were treated with ACK Lysing Buffer (A1042-01, Life Technologies) for 2 min to remove red blood cells. Cells were then suspended in PBS with 0.5% BSA, 0.01% sodium azide, and 2 mM EDTA. CD16/32 antibody (101321, BioLegend) was used to page Fc-gamma3 receptors. Cells were then stained with 10 μg/mL of CD45-PerCP (Cat. No. 103130, BioLegend), Ly6C-APC-Cy7 (Cat. No. 560596, BD Biosciences), F4/80-BV421 (Cat. No. 565411, BD Biosciences), and 7.5 μg/mL of CD11b-BV650 (Cat. No. 563402, BD Biosciences). After being stained, cells were permeabilized with the FoxP3/Transcription Factor Staining Buffer Set (Cat. No. 00-5523-00, Invitrogen). Intracellular staining was done with 10 μg/mL of CD206-PE. Flow cytometry was done using a BD LSRFortessa flow cytometer, collecting 1 million events per sample.
Statistical analysis.
Data are expressed as means ± SE for all experiments. Comparisons of normally distributed data sets were analyzed by either a one-way or two-way ANOVA, as appropriate, and group means were compared using Tukey’s post-hoc tests. Nominal data were analyzed with individual chi-square tests. Correlations were determined using a linear regression model. The criteria for statistical differences were as follows: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
RESULTS
Loss of kidney function is similar between FVB/n and C57BL/6 mice treated with repeated doses of cisplatin.
BUN is commonly used as a measure of kidney function, as levels of urea increase in the blood with loss of kidney function. We have previously shown that loss of kidney function as determined by BUN is minimal with our repeated dosing regimen of cisplatin (34, 35). When treated with four weekly doses of 7 mg/kg cisplatin, neither FVB/n nor C57BL/6 mice had a significant increase in BUN (P = 0.99 and 0.20, respectively). With repeated doses of 9 mg/kg cisplatin, BUN was significantly elevated in FVB/n mice (P = 0.045), but not in C57BL/6 mice (P = 0.99) (Fig. 1A). We also assessed levels of urinary NGAL, a sensitive kidney injury biomarker. When treated with repeated doses of either 7 mg/kg or 9 mg/kg cisplatin, NGAL levels increased comparably in both strains, but neither increase was statistically significant (Fig. 1B). Taken together, these data indicate that C57BL/6 and FVB/n mice sustain similar levels of kidney injury when treated with repeated, low doses of cisplatin. Thus, loss of kidney function is minimal and comparable between the two strains of male mice, characteristic of subclinical kidney injury.
Fig. 1.
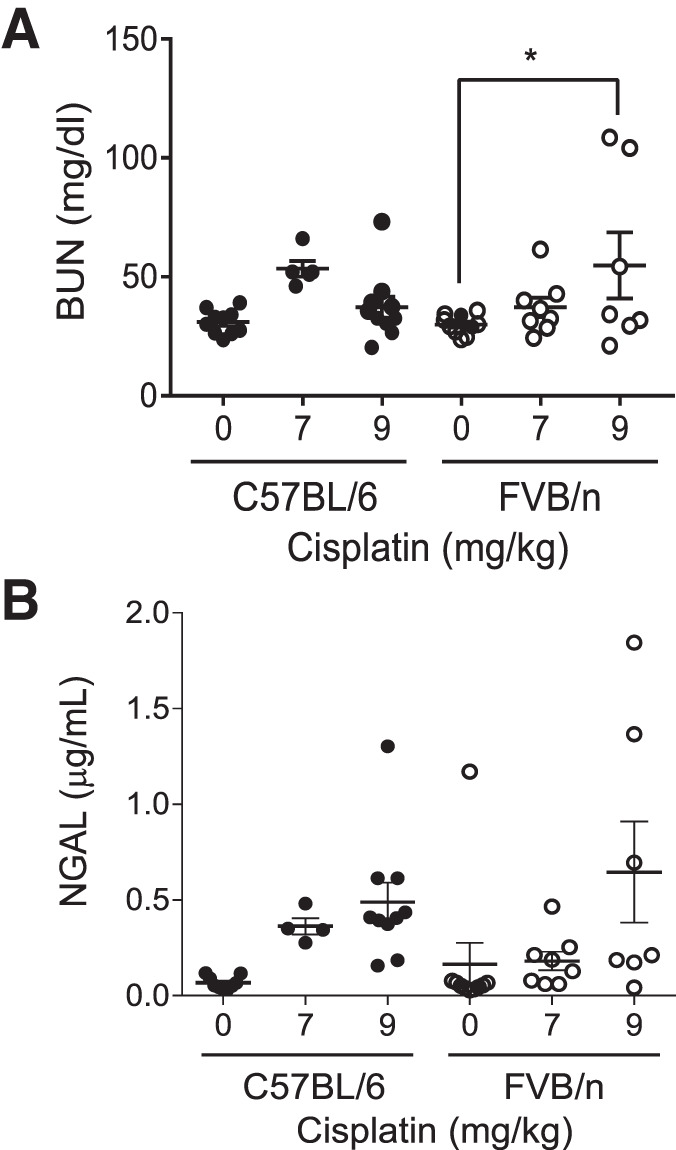
Kidney function and injury in FVB/n and C57BL/6 mice treated with repeated administration of cisplatin. A: blood urea nitrogen (BUN) levels were measured in plasma. B: neutrophil gelatinase-associated lipocalin (NGAL) levels were measured in urine. Statistical analysis was determined by two-way ANOVA followed by Tukey’s post hoc test. *P < 0.05.
A higher dose of cisplatin is required for the development of fibrosis in C57BL/6 mice than in FVB/n mice.
We have previously published that treating FVB/n mice with 7 mg/kg cisplatin once a week for 4 wk induces renal fibrosis and myofibroblast accumulation (35). Myofibroblasts are the main cell type responsible for collagen deposition and are identified by expression of αSMA. Immunohistochemistry staining for αSMA indicated that there were significantly more myofibroblasts present in FVB/n kidneys after repeated treatment with 7 mg/kg cisplatin (P = 0.03). Myofibroblasts were present in lower numbers when FVB/n mice were treated with repeated 9 mg/kg doses of cisplatin. In contrast, αSMA levels did not increase in C57BL/6 mice until the dose was increased to 9 mg/kg cisplatin, where levels were significantly elevated compared with both vehicle- and 7 mg/kg cisplatin-treated mice (P = 0.004 and 0.003, respectively) (Fig. 2, A and B). SR/FG staining revealed the same pattern. After repeated 7 mg/kg cisplatin treatment, FVB/n mice had their highest levels of collagen deposition, significantly elevated above both vehicle- and 9 mg/kg cisplatin-treated mice (P < 0.0001 and = 0.003, respectively). C57BL/6 mice did not have increased collagen deposition when treated with repeated 7 mg/kg doses of cisplatin (P > 0.99). When the weekly dose was increased to 9 mg/kg, however, collagen levels significantly increased (P = 0.0006) (Fig. 2, C and D). Thus, C57BL/6 mice require a higher dose of cisplatin for the development of interstitial fibrosis. In addition, FVB/n mice seem to lose their fibrotic phenotype when treated with a higher dose of cisplatin.
Fig. 2.

Development of renal fibrosis with repeated administration of cisplatin in C57BL/6 and FVB/n mice. A: α-smooth muscle actin (αSMA) immunohistochemistry for myofibroblasts. B: quantification of optical density of positive staining. C: Sirius Red/Fast Green (SR/FG) staining for total collagen deposition. D: quantification of optical density of positive staining. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Differences in apoptosis and endoplasmic reticulum stress in C57BL/6 and FVB/n mice when treated with cisplatin.
The main pathology associated with the single, high-dose model of cisplatin-induced kidney injury is cell death through both apoptotic and necrotic mechanisms (44, 45). However, we have previously shown with our repeated, 7 mg/kg dosing regimen of cisplatin that the kidneys of FVB/n mice do not have overt cell death (34). Thus, we wanted to determine if apoptosis was prevalent in C57BL/6 mice treated with either 7 mg/kg or 9 mg/kg cisplatin weekly (Fig. 3, A and D). When given repeated doses of 7 mg/kg cisplatin, neither FVB nor C57BL/6 mice displayed an increase in cleaved caspase 3 expression, an indicator of apoptosis. When the weekly dose was increased to 9 mg/kg cisplatin, both FVB/n and C57BL/6 mice expressed cleaved caspase 3, indicating that apoptosis occurs in both strains at this dose (Fig. 3, C and F). Interestingly, we found that CHOP, a marker for endoplasmic reticulum (ER) stress, was increased in a dose-dependent manner in C57BL/6 mice, but levels of CHOP were highest with repeated 7 mg/kg cisplatin treatments in FVB/n mice (Fig. 3, B and E).
Fig. 3.

Markers of cell stress and cell death with repeated administration of cisplatin in C57BL/6 and FVB/n mice. C/EBP homologous protein (CHOP), cleaved caspase 3 (CC3), and β-actin were measured in kidney cortex homogenates via Western blot in C57BL/6 mice (A) with quantification of CHOP (B) and CC3 (C) and in FVB/n mice (D) with quantification of CHOP (E) and CC3 (F). *P < 0.05, **P < 0.01.
C57BL/6 mice display variability in renal injury following repeated low-dose (9 mg/kg) cisplatin treatment.
One year after the previous experiments were performed, a second individual in the Siskind laboratory repeated the 9 mg/kg cisplatin dosing regimen of C57BL/6 mice with male mice that again were purchased from Jackson Laboratory (Table 1). Both purchased sets of mice had at least 3 wk of acclimation before the first dose of cisplatin was administered. Unexpectedly, the cisplatin-treated C57BL/6 mice purchased in 2018 had much greater weight loss than was previously observed (Fig. 4A). This presented problems for the use of this model, as 25% body weight loss was set as a humane end point. We believe dehydration is a major contributor to weight loss, as mice treated with cisplatin become less active and are likely drinking less water. We then treated C57BL/6 male mice bred in-house with repeated doses of 9 mg/kg cisplatin. Weight loss appeared to be following a similar trend, so we administered subcutaneous injections of 500 μL of 0.9% N saline one day before dose 3, two days after dose 3, and one day before dose 4. This kept most mice under the 25% body weight loss cutoff. We want to note that although saline reduced body weight loss, we still observed development of kidney injury and fibrosis, indicating that these outcomes are not a result of dehydration.
Table 1.
Experimental details of three populations of C57BL/6 mice treated with repeated administration of 9 mg/kg cisplatin
| C57BL/6 Experiment 1 | C57BL/6 Experiment 2 | C57BL/6 Experiment 3 | |
|---|---|---|---|
| Done by | Student 1 | Student 2 | Student 2 |
| Dose | 9 mg/kg | 9 mg/kg | 9 mg/kg |
| Sex | Male | Male | Male |
| Age | 8 wk | 10 wk | 8–10 wk |
| Source of mice | Jackson 5-8-17 (5 wk old) | Jackson 8-27-18 (4 wk old) | Bred in-house 2/2019 |
| Adjustment time | 3 wk | 6 wk | 8–10 wk |
| Numbers | 10 CIS, 5 VEH | 10 CIS, 5 VEH | 12 CIS, 5 VEH |
| Dosing regimen | Chronic, 8 AM | Chronic, 8 AM | Chronic, 8 AM |
CIS, cisplatin treated; VEH, vehicle treated.
Fig. 4.

Weight loss, kidney function, and injury in C57BL/6 mice treated with repeated administration of 9 mg/kg cisplatin. A: percent body weight loss. B: blood urea nitrogen (BUN) levels measured in plasma. C: neutrophil gelatinase-associated lipocalin (NGAL) levels measured in urine. Statistical analysis was determined by two-way ANOVA followed by Tukey’s post-hoc test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Interestingly, we also observed variability in BUN and NGAL levels among these three cohorts of C57BL/6 mice. Mice purchased from Jackson Laboratory in 2018 and mice bred in-house had significantly greater increases in BUN following cisplatin treatment than mice purchased from Jackson Laboratory in 2017 (P = <0.0001 and 0.002, respectively), indicating greater loss of renal function (Fig. 4B). Urinary NGAL levels of purchased mice were similarly elevated following treatment; however, in-house bred mice had significantly lower NGAL levels following 9 mg/kg cisplatin treatment compared with the 2017 and 2018 purchased groups (P = 0.03 and 0.009, respectively) (Fig. 4C).
C57BL/6 mice have variable structural damage, regeneration, and necrosis following repeated low-dose (9 mg/kg) cisplatin treatment.
Pathological assessment was performed on H&E- and PAS-stained sections from all three sets of C57BL/6 mice by the same pathologist in a parallel and blinded manner. Pathology scoring revealed variability in renal damage in C57BL/6 mice following repeated 9 mg/kg cisplatin treatment. Mice purchased in 2018 and in-house bred mice had higher levels of brush border loss, tubular casts, and tubular dilation compared with mice purchased in 2017 (Fig. 5, A–C). This suggests that mice purchased in 2017 had less structural damage when treated with repeated doses of 9 mg/kg cisplatin. On the other hand, widening of the interstitium was similar in mice purchased in 2017 and in-house bred mice, but it trended lower in mice purchased in 2018 (Fig. 5E). Inflammation was similarly elevated in mice purchased in 2017 and 2018, whereas it was higher in in-house bred mice (Fig. 5F). The greatest differences observed were large increases in tubular necrosis and regeneration displayed in the mice purchased in 2018 and in-house bred mice. The mice purchased in 2017 had almost no detectable necrosis or regeneration (Fig. 5, G and H). Representative images of pathology scoring are provided (Fig. 5, I and J).
Fig. 5.

Pathology scoring of tubular damage in C57BL/6 mice treated with repeated administration of 9 mg/kg cisplatin. Hematoxylin and eosin (H&E)-stained and periodic acid-Schiff (PAS)-stained sections were scored on a scale of 1–4 for severity of loss of brush border (A), tubular casts (B), tubular dilation (C), tubular degeneration (D), widening of the interstitium (E), inflammation (F), tubular necrosis (G), and tubular regeneration (H) by a mouse renal pathologist. Statistical analysis was determined by individual chi-square tests. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. I and J: representative H&E-stained sections. Reg, regeneration; c, cast; I, inflammatory cells; n, necrosis; Wi&I, widening of the interstitium and inflammatory cells.
C57BL/6 mice have differential inflammatory cytokine responses following repeated low-dose (9 mg/kg) cisplatin treatment.
Previously, we have characterized an increase in Tnfα, IL6, Cxcl1, and Ccl2 [also known as monocyte chemoattractant protein-1 (Mcp-1)] mRNA following repeated cisplatin treatment in FVB/n mice (34, 35). We also observed an increase in M2 macrophage marker Arg-1 accompanied by a decrease in M1 macrophage marker iNOS (35). In this study, we found that this response was consistent in C57BL/6 mice but varied in magnitude. C57BL/6 mice purchased in 2017 had a slightly greater increase in Tnfα and IL6 following treatment than those purchased in 2018 or the in-house bred mice, whereas C57BL/6 mice purchased in 2018 had the largest increase in Ccl2 levels (Fig. 6, A–C). In-house bred C57BL/6 mice had the greatest induction of Cxcl1, with levels significantly elevated above the treated 2017 and 2018 purchased groups (P = 0.0006 and 0.002, respectively) (Fig. 6D). Arg-1 levels were not elevated in either purchased set of cisplatin-treated C57BL/6 mice but showed a significant increase in in-house bred mice. Cisplatin-treated in-house bred mice had significantly elevated expression compared with both sets of cisplatin-treated purchased mice (P < 0.0001) (Fig. 6E). The decrease in iNOS expression was consistent among all treated groups (Fig. 6F). Overall, the inflammatory cytokine response was similar in each C57BL/6 group treated, with the exception of Cxcl1 and Arg-1 induction.
Fig. 6.

Inflammatory cytokine production in C57BL/6 mice treated with repeated administration of 9 mg/kg cisplatin. mRNA from the renal cortex was analyzed with quantitative RT-PCR to determine levels of tumor necrosis factor-α (Tnfα) (A), interleukin 6 (IL6) (B), C-C motif chemokine ligand 2 (Ccl2) (C), C-X-C motif chemokine ligand 1 (Cxcl1) (D), arginase 1 (Arg-1) (E), and inducible nitric oxide synthase (iNos) (F). Expression levels were normalized to β2-microglobulin. Statistical analysis was determined by two-way ANOVA followed by a Tukey’s post-hoc test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
C57BL/6 populations all develop renal fibrosis following repeated low-dose (9 mg/kg) cisplatin treatment.
Despite the differences observed in renal function and tubular injury, all C57BL/6 mice treated with 9 mg/kg cisplatin appeared to develop renal fibrosis, as assessed by SR/FG staining and αSMA immunohistochemistry. All C57BL/6 mice treated with repeated 9 mg/kg cisplatin had significant increases in collagen deposition in the kidney, as indicated by the increased red color in the SR/FG stains (Fig. 7, B and D). All C57BL/6 mice also had increased αSMA-positive cells following repeated 9 mg/kg cisplatin treatment (Fig. 7, A and C). Overall, these results indicate that although variation in renal function and tubular damage may be observed in different populations of C57BL/6 mice, renal collagen deposition and αSMA accumulation appear to be a consistent phenotype associated with the repeated dosing regimen of 9 mg/kg cisplatin in this strain.
Fig. 7.

Development of renal fibrosis with repeated administration of cisplatin in C57BL/6 mice. A: α-smooth muscle actin (αSMA) immunohistochemistry for myofibroblasts. B: Sirius Red/Fast Green (SR/FG) staining for total collagen deposition. Quantification of optical density of αSMA (C) and SR/FG positive staining (D). *P < 0.05, **P < 0.01, ***P < 0.001.
CCL2 is an indicator of injury following repeated low-dose (9 mg/kg) cisplatin treatment.
Owing to the variation observed in these studies with C57BL/6 mice, we set out to identify consistent correlations between different kidney injury parameters. We performed linear regressions comparing each quantitative data set collected for cisplatin-treated C57BL/6 mice. We found that among treated mice, Ccl2 mRNA expression in the kidney cortex was significantly correlated with body weight loss, BUN, and Kim-1 expression (P = 0.003, 0.02, and 0.0001, respectively) (Fig. 8, A–D). Cisplatin-treated mice with large increases in Ccl2 expression also had larger amounts of body weight loss and greater increases in BUN, NGAL, and Kim-1 expression, indicating worse kidney injury and decreased kidney function. In contrast, Tnfα, Il6, and Cxcl1 did not correlate with these markers of injury, indicating that this relationship is specific to Ccl2 and not general inflammation (Supplemental Fig. S2). These results suggest that Ccl2 may be useful in identifying cisplatin-induced kidney injury. These correlations also demonstrate that the variation in these experiments is likely due to different susceptibilities of individual mice when treated with cisplatin; mice with high levels of one injury indicator are likely to score high in other measures as well. In addition, urinary CCL2 has recently been identified as a potential biomarker in patients who have developed cisplatin-induced nephrotoxicity (25, 38). This prompted us to evaluate CCL2 protein levels in the urine of C57BL/6 mice treated with four doses of 9 mg/kg cisplatin. Although no CCL2 protein was detected in the urine of vehicle-treated mice, cisplatin-treated mice had significantly elevated levels of 7.8 ± 1.8 pg/mL in their urine (P = 0.0012) (Fig. 8E). These results highlight one factor that is similar in the kidney response to cisplatin among mice and humans.
Fig. 8.
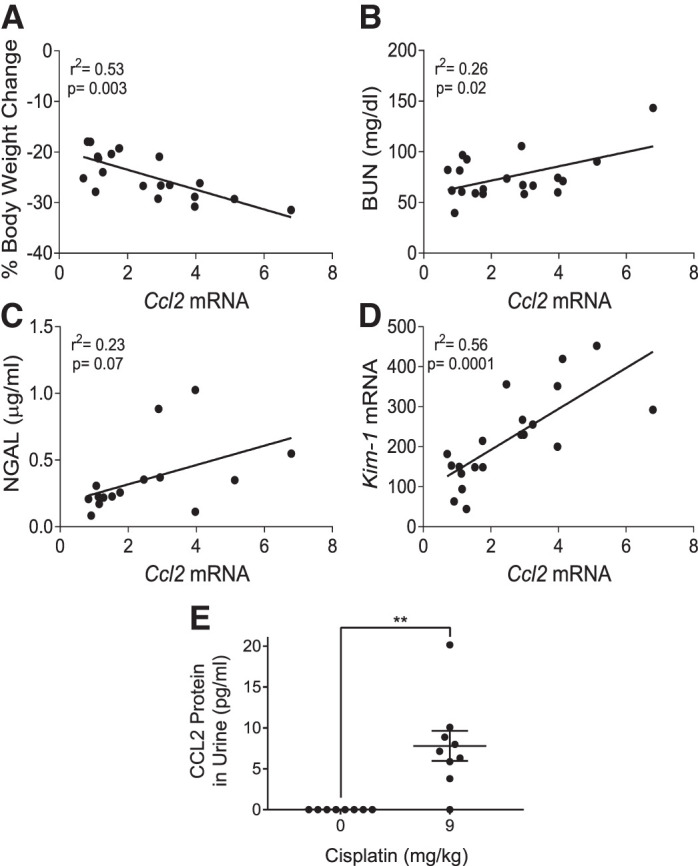
C-C motif chemokine ligand 2 (CCL2) expression in cisplatin-induced kidney injury. Correlations of Ccl2 mRNA expression with body weight loss (A), blood urea nitrogen (BUN) (B), neutrophil gelatinase-associated lipocalin (NGAL) (C), and kidney injury molecule-1 (Kim-1) mRNA (D) in C57BL/6 mice treated with repeated administration of 9 mg/kg cisplatin. Correlations were found using a linear regression model with significance determined as P < 0.05. D: urinary CCL2 in C57BL/6 mice treated with repeated administration of 9 mg/kg cisplatin. An ELISA was performed on 50 μL of undiluted urine from vehicle- and cisplatin-treated mice. Vehicle-treated mice had no detectable signal. Statistical analysis was determined by one-way ANOVA. **P < 0.01.
Renal immune cell infiltration is observed in C57BL/6 mice following repeated low-dose (9 mg/kg) cisplatin treatment.
CCL2 is known to play an important role in immune cell trafficking, specifically in myeloid cell recruitment (1). We hypothesized that the elevation of Ccl2 mRNA and urinary CCL2 observed in mice treated with cisplatin would be indicative of renal immune cell infiltration. We used the gating strategy shown in Supplemental Fig. S3 to evaluate the immune cells present in the kidney following cisplatin treatment. Contrary to what Black et al. (3) observed, we found a significant increase in the percentage of CD45+ immune cells in the kidney of cisplatin-treated mice (P < 0.0001) (Fig. 9A). We observed significantly increased populations of F4/80 hi and F4/80 lo macrophages (P = 0.01 and <0.0001, respectively) (Fig. 9, B and C). Further analysis also revealed a significant increase in F4/80+ CD206+ macrophages (P = 0.002), indicative of an “M2” phenotype (Fig. 9D). Finally, we observed a significant increase in both Ly6c hi and Ly6c lo monocytes (P < 0.0001 for both) (Fig. 9, E and F). These findings suggest that there is a significant immune response to cisplatin-induced renal damage in the repeated low-dose model. More studies are needed to assess whether the activity of these immune cells in the kidney plays a major role in promotion of fibrosis.
Fig. 9.

Flow cytometric analysis of renal macrophages and monocytes. Whole kidneys were homogenized and ~1 million cells were stained. One million events were collected from each sample. Analysis identified CD45+ immune cells (A), CD11b+ F4/80 hi resident macrophages (B), CD11b+ F4/80 lo infiltrating macrophages (C), F4/80+ CD206+ M2 macrophages (D), CD11b+ Ly6c hi inflammatory monocytes (E), and CD11b+ Ly6c lo resident monocytes (F). Populations are expressed as a percentage of the number of single cells counted. Statistical analysis was determined by one-way ANOVA. **P < 0.01 and ****P < 0.0001.
DISCUSSION
The development of mouse models of renal fibrosis is important for studying CKD. Previously, we developed a model of fibrosis that results from four weekly treatments of 7 mg/kg cisplatin in FVB/n mice. Fibrosis was characterized by increased collagen deposition via SR/FG staining and accumulation of myofibroblasts by αSMA immunohistochemistry. We also observed an increase in inflammatory cytokine mRNA expression and markers of endothelial dysfunction (35). C57BL/6 mice are well known to be resistant to development of fibrosis in other models, limiting their use in studies of CKD development (30). We observed here that administration of 7 mg/kg cisplatin in C57BL/6 did not result in fibrosis as seen in FVB/n mice. These results highlight that this repeated low-dose model of cisplatin-induced kidney injury is in fact a model of renal fibrosis and not AKI. The biological processes induced by the acute, high-dose model of cisplatin-induced kidney injury are different from what occurs in this repeated low-dose model.
Similar low-dosing regimens of cisplatin have indicated that with higher doses of cisplatin (9–15 mg/kg), C57BL/6 mice will develop mild tubulointerstitial fibrosis. Torres et al. (43) showed that two treatments of 15 mg/kg cisplatin 2 wk apart led to slight fibrosis and changes in glomeruli, although glomerulosclerosis was not present. In addition, Katagiri et al. (15) and Ravichandran et al. (31) have shown that administration of 10 mg/kg cisplatin once a week for 3 or 4 wk, respectively, results in mild fibrosis. We have found that 10 mg/kg cisplatin administered once a week for 4 wk increases frequency of animal death and leads to unacceptable levels of body weight loss (data not shown). Thus, the goal of our study was to optimize a dose of cisplatin in C57BL/6 mice that would induce the development of fibrosis with minimal animal death and <25% body weight loss.
In this study, we found that intraperitoneal administration of 9 mg/kg cisplatin once a week for 4 wk is sufficient to cause interstitial fibrosis in C57BL/6 mice. Administration of subcutaneous saline with this dosing regimen may also be used to prevent severe weight loss and does not hinder development of kidney injury or fibrosis. Similar studies by other groups have also indicated that repeated low doses of 8–9 mg/kg cisplatin induces fibrosis in C57BL/6 mice without compromising survival (3, 11). In addition, we observed marked differences between FVB/n and C57BL/6 mice in apoptosis induction and CHOP expression. We also observed variation in kidney function and tubular damage in different populations of C57BL/6 mice. Despite this variation, collagen deposition and αSMA accumulation with repeated 9 mg/kg cisplatin treatment were consistent.
It is well established that experimental AKI is met with high levels of inflammation and consequently high levels of both apoptotic and necrotic cell death. We have shown that treating FVB/n mice with 7 mg/kg cisplatin once a week for 4 wk results in little to no apoptosis or necrosis (35). In this study, both FVB/n and C57BL/6 mice expressed cleaved caspase 3 when treated with 9 mg/kg cisplatin, indicative of apoptosis. Surprisingly, C57BL/6 mice had apoptosis occurring with the development of fibrosis, whereas fibrosis developed with a lower dose in FVB/n mice and was less pronounced when there were higher levels of the cell death marker cleaved caspase 3. This may be key in explaining why development of a fibrosis model with cisplatin in C57BL/6 mice is often met with decreased overall survival.
ER stress is another major component of kidney injury. We found that expression of CHOP, an indicator of ER stress, corresponded to the development of fibrosis in both strains of mice. Although CHOP and ER stress are classically associated with apoptosis, these also play a role in renal fibrosis (22). Several studies have found that CHOP-deficient mice subjected to unilateral ureteral obstruction or ischemia reperfusion injury had lower levels of fibrosis, inflammatory cell infiltration, and lipid peroxidation compared with wild-type mice (20, 39, 47). Thus, CHOP is important for the progression of CKD and should be examined in the repeated low-dose cisplatin model.
A surprising outcome in this study was the loss of fibrotic phenotype in FVB/n mice given 9 mg/kg cisplatin. Our rationale for this result is based on the fact that cisplatin induces kidney injury on a spectrum from AKI to CKD pathologies. We hypothesize that at 9 mg/kg, FVB/n mice develop more of an AKI phenotype. These mice have higher levels of BUN, NGAL, structural damage, and cell death, indicating greater acute injury. We believe that this higher level of acute injury is causing injured cells to be cleared quickly by apoptosis or necrosis and preventing accumulation of senescent cells. CKD and fibrosis development are known to be promoted by processes of maladaptive repair, including cell cycle arrest of sublethally injured proximal tubule cells (9). Literature also suggests that apoptotic clearance of injured cells may prevent accumulation of senescent cells that may otherwise contribute to fibrosis development (2, 13, 29). Future studies should evaluate pathways of cell death activated at different cisplatin dosing levels as well as ER stress induction and cellular senescence. We also need to reevaluate the phenotype presented in FVB/n mice treated with 9 mg/kg cisplatin to assess if this higher dose increases group variability as in C57BL/6 mice.
Although 9 mg/kg cisplatin treatment consistently induces fibrosis in C57BL/6 mice, we did observe variability in renal function and structural damage. We used this variability to identify correlations among different injury parameters in our C57BL/6 populations. We analyzed correlations among quantitative data sets of cisplatin-treated mice and found that Ccl2 mRNA levels were significantly correlated with increased body weight loss, BUN, NGAL, and Kim-1 mRNA levels. These data suggest that variability in these markers corresponds to susceptibility for developing cisplatin-induced kidney injury, as mice that score high in one category are likely to score high in others. To further assess this correlation, we measured Ccl2 mRNA in FVB/n mice treated with 7 mg/kg and 9 mg/kg cisplatin. We observed a dose-dependent increase in Ccl2 mRNA levels (data not shown). Ccl2 mRNA was also increased more in FVB/n mice at 7 mg/kg than in C57BL/6 mice. This Ccl2 expression follows the patterns of kidney injury markers such as BUN and NGAL but not markers of fibrosis. This could suggest a disconnect between kidney injury and fibrosis development, with Ccl2 mRNA being more of an indicator of kidney injury than development of fibrosis.
We also observed a significant increase in urinary CCL2 protein in cisplatin-treated mice. Urinary CCL2 did not correlate with injury parameters or immune cell infiltration. Although we were able to detect an increase in urinary CCL2, the protein was present in such low amounts that we do not believe the ELISA used is sensitive enough to calculate exact quantities and may be the reason why a correlation was not observed. Clinically, urinary CCL2 has been identified as an indicator of cisplatin-induced kidney injury in patients with lung cancer treated with cisplatin (38). The presence of CCL2 in the urine of the mice in this study provides support to the argument that it could be used as a more sensitive biomarker of cisplatin-induced kidney injury than BUN or serum creatinine. These results also suggest that our repeated dosing regimen of cisplatin induces pathological processes that are similar to what occurs in humans.
The increase in CCL2 in this model could also hint at mechanisms promoting cisplatin-induced renal fibrosis. CCL2 is known to play a role in immune cell trafficking. It has been demonstrated that Ccl2 mRNA levels increase in different models of renal injury, indicating the CCL2 protein may play a role in orchestrating the development of renal fibrosis (32, 41). Kitagawa et al. (17) demonstrated that pharmacological inhibition and genetic knockdown of C-C motif chemokine receptor 2 (CCR2), the receptor of CCL2, protected mice from renal fibrosis in the unilateral ureteral obstruction model. They also observed a significant decrease in infiltrating F4/80 macrophages. Furuichi et al. (12) found similar results using the ischemia-reperfusion model, with CCR2-deficient mice showing resistance to injury and fibrosis. Pharmacological inhibition of CCL2 also provided protection in a model of diabetic nephropathy (4). These studies attribute the protective effects of CCR2 and CCL2 inhibition to modulation of immune response following renal injury.
In light of these studies, we evaluated renal immune cell infiltration following repeated low-dose (9 mg/kg) cisplatin treatment in C57BL/6 mice. Black et al. (3) recently published results indicating that repeated 9 mg/kg cisplatin doses in C57BL/6 mice did not lead to immune cell infiltration in the kidneys. Interestingly, this group also reported only mild levels of fibrosis. In contrast, we found a significant increase in renal CD45+ immune cells as well as macrophage and monocyte populations. This variability could be due to several factors. In discussion with this group, we have noted differences in the type of cisplatin given (pharmaceutical-grade predissolved cisplatin vs. the one purchased from a company as a powder and then dissolved in-house) and time of day of cisplatin injections. Administration of cisplatin at different times of day does alter its impact on kidney function and fibrosis (unpublished data in the Siskind laboratory). As demonstrated in this study, there is also inherent variability in different populations of C57BL/6 mice, which could explain the different responses. Despite this inherent variability, Fu et al. (11) recently published findings similar to ours using repeated 8 mg/kg cisplatin doses in C57BL/6 mice, indicating that there is reproducibility across some settings.
Macrophages have been studied in many different models of renal fibrosis. As in other organs, renal inflammation following an injury is a common precursor to fibrosis development (5, 24, 42). In models of ischemia-reperfusion, depletion of macrophages and monocytes with clodronate liposomes has been shown to attenuate renal fibrosis (10, 16, 21, 46). Similar results were also noted in models of unilateral ureteral obstruction (18, 28). The role of macrophages in CKD is complex owing to the diverse activity and heterogeneity of cell types (14). Some studies have suggested persistent “M2” macrophage activity could be a driver of renal fibrosis (16, 28). We hypothesize that the repeated nature of injury in our repeated dosing model likely leads to sustained renal inflammation and persistent repair-like activities with M2 polarization. Future studies will determine if macrophage subsets are major drivers of fibrosis development in this model.
Taken together, this study indicates the importance of choosing the correct mouse strain and dosing regimen for renal studies. We have observed that group variability, in terms of renal function and structural damage, is inherent in the cisplatin model of injury, but our repeated dosing regimen of cisplatin consistently induces fibrosis in C57BL/6 mice with an increased cisplatin dose of 9 mg/kg. Administration of subcutaneous saline may also be used to prevent weight loss. This dosing regimen also has a low rate of animal death. This study demonstrates that C57BL/6 mice are a viable option to use as a model of repeated low-dose cisplatin-induced fibrosis. We also demonstrate the importance of using littermate control mice in this model owing to population variability in renal function and tubular damage.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01DK124112 (to L.J.S.) and F31DK126400 (to S.M.S.) and National Cancer Institute Grant R01CA193220 (to L.J.B.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.M.S., C.N.S., L.J.B., and L.J.S. conceived and designed research; S.M.S. and C.N.S. performed experiments; S.M.S., C.N.S., A.K., D.S., and M.A.D. performed animal dissections; S.M.S., C.N.S., M.A.D., J.M., and G.B.O. analyzed data; S.M.S., C.N.S., and G.B.O. interpreted results of experiments; S.M.S. and C.N.S. prepared figures; S.M.S. drafted manuscript; S.M.S., M.A.D., L.J.B., and L.J.S. edited and revised manuscript; S.M.S., C.N.S., A.K., D.S., M.A.D., L.J.B., and L.J.S. approved final version of manuscript.
REFERENCES
- 1.Anders H-J, Vielhauer V, Schlöndorff D. Chemokines and chemokine receptors are involved in the resolution or progression of renal disease. Kidney Int 63: 401–415, 2003. doi: 10.1046/j.1523-1755.2003.00750.x. [DOI] [PubMed] [Google Scholar]
- 2.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol 2: 1303–1353, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Black LM, Lever JM, Traylor AM, Chen B, Yang Z, Esman SK, Jiang Y, Cutter GR, Boddu R, George JF, Agarwal A. Divergent effects of AKI to CKD models on inflammation and fibrosis. Am J Physiol Renal Physiol 315: F1107–F1118, 2018. doi: 10.1152/ajprenal.00179.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boels MGS, Koudijs A, Avramut MC, Sol WMPJ, Wang G, van Oeveren-Rietdijk AM, van Zonneveld AJ, de Boer HC, van der Vlag J, van Kooten C, Eulberg D, van den Berg BM, IJpelaar DHT, Rabelink TJ. Systemic monocyte chemotactic protein-1 inhibition modifies renal macrophages and restores glomerular endothelial glycocalyx and barrier function in diabetic nephropathy. Am J Pathol 187: 2430–2440, 2017. doi: 10.1016/j.ajpath.2017.07.020. [DOI] [PubMed] [Google Scholar]
- 5.Cao Q, Harris DCH, Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology (Bethesda) 30: 183–194, 2015. doi: 10.1152/physiol.00046.2014. [DOI] [PubMed] [Google Scholar]
- 6.Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int 79: 1361–1369, 2011. doi: 10.1038/ki.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med 371: 58–66, 2014. doi: 10.1056/NEJMra1214243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int 81: 442–448, 2012. doi: 10.1038/ki.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11: 264–276, 2015. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferenbach DA, Sheldrake TA, Dhaliwal K, Kipari TMJ, Marson LP, Kluth DC, Hughes J. Macrophage/monocyte depletion by clodronate, but not diphtheria toxin, improves renal ischemia/reperfusion injury in mice. Kidney Int 82: 928–933, 2012. doi: 10.1038/ki.2012.207. [DOI] [PubMed] [Google Scholar]
- 11.Fu Y, Cai J, Li F, Liu Z, Shu S, Wang Y, Liu Y, Tang C, Dong Z. Chronic effects of repeated low-dose cisplatin treatment in mouse kidneys and renal tubular cells. Am J Physiol Renal Physiol 317: F1582–F1592, 2019. doi: 10.1152/ajprenal.00385.2019. [DOI] [PubMed] [Google Scholar]
- 12.Furuichi K, Wada T, Iwata Y, Kitagawa K, Kobayashi K, Hashimoto H, Ishiwata Y, Asano M, Wang H, Matsushima K, Takeya M, Kuziel WA, Mukaida N, Yokoyama H. CCR2 signaling contributes to ischemia-reperfusion injury in kidney. J Am Soc Nephrol 14: 2503–2515, 2003. doi: 10.1097/01.ASN.0000089563.63641.A8. [DOI] [PubMed] [Google Scholar]
- 13.Gewin LS. Renal fibrosis: primacy of the proximal tubule. Matrix Biol 68-69: 248–262, 2018. doi: 10.1016/j.matbio.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guiteras R, Flaquer M, Cruzado JM. Macrophage in chronic kidney disease. Clin Kidney J 9: 765–771, 2016. doi: 10.1093/ckj/sfw096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katagiri D, Hamasaki Y, Doi K, Negishi K, Sugaya T, Nangaku M, Noiri E. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int 89: 374–385, 2016. doi: 10.1038/ki.2015.327. [DOI] [PubMed] [Google Scholar]
- 16.Kim M-G, Kim SC, Ko YS, Lee HY, Jo S-K, Cho W. The role of M2 macrophages in the progression of chronic kidney disease following acute kidney injury. PLoS One 10: e0143961, 2015. doi: 10.1371/journal.pone.0143961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitagawa K, Wada T, Furuichi K, Hashimoto H, Ishiwata Y, Asano M, Takeya M, Kuziel WA, Matsushima K, Mukaida N, Yokoyama H. pageade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol 165: 237–246, 2004. doi: 10.1016/S0002-9440(10)63292-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitamoto K, Machida Y, Uchida J, Izumi Y, Shiota M, Nakao T, Iwao H, Yukimura T, Nakatani T, Miura K. Effects of liposome clodronate on renal leukocyte populations and renal fibrosis in murine obstructive nephropathy. J Pharmacol Sci 111: 285–292, 2009. doi: 10.1254/jphs.09227FP. [DOI] [PubMed] [Google Scholar]
- 19.Landau SI, Guo X, Velazquez H, Torres R, Olson E, Garcia-Milian R, Moeckel GW, Desir GV, Safirstein R. Regulated necrosis and failed repair in cisplatin-induced chronic kidney disease. Kidney Int 95: 797–814, 2019. doi: 10.1016/j.kint.2018.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu SH, Wu C-T, Huang K-H, Wang C-C, Guan S-S, Chen L-P, Chiang C-K. C/EBP homologous protein (CHOP) deficiency ameliorates renal fibrosis in unilateral ureteral obstructive kidney disease. Oncotarget 7: 21900–21912, 2016. doi: 10.18632/oncotarget.7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu L, Faubel S, He Z, Andres Hernando A, Jani A, Kedl R, Edelstein CL. Depletion of macrophages and dendritic cells in ischemic acute kidney injury. Am J Nephrol 35: 181–190, 2012. doi: 10.1159/000335582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maekawa H, Inagi R. Stress Signal Network between hypoxia and ER stress in chronic kidney disease. Front Physiol 8: 74, 2017. doi: 10.3389/fphys.2017.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins (Basel) 2: 2490–2518, 2010. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nikolic-Paterson DJ, Wang S Lan HY. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int Suppl (2011) 4: 34–38, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishihara K, Masuda S, Shinke H, Ozawa A, Ichimura T, Yonezawa A, Nakagawa S, Inui K, Bonventre JV, Matsubara K. Urinary chemokine (C-C motif) ligand 2 (monocyte chemotactic protein-1) as a tubular injury marker for early detection of cisplatin-induced nephrotoxicity. Biochem Pharmacol 85: 570–582, 2013. doi: 10.1016/j.bcp.2012.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res Int 2014: 1, 2014. doi: 10.1155/2014/967826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73: 994–1007, 2008. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 28.Pan B, Liu G, Jiang Z, Zheng D. Regulation of renal fibrosis by macrophage polarization. Cell Physiol Biochem 35: 1062–1069, 2015. doi: 10.1159/000373932. [DOI] [PubMed] [Google Scholar]
- 29.Priante G, Gianesello L, Ceol M, Del Prete D, Anglani F. Cell death in the kidney. Int J Mol Sci 20: 3598, 2019. doi: 10.3390/ijms20143598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rabe M, Schaefer F. Non-transgenic mouse models of kidney disease. Nephron 133: 53–61, 2016. doi: 10.1159/000445171. [DOI] [PubMed] [Google Scholar]
- 31.Ravichandran K, Wang Q, Ozkok A, Jani A, Li H, He Z, Ljubanovic D, Weiser-Evans MC, Nemenoff RA, Edelstein CL. CD4 T cell knockout does not protect against kidney injury and worsens cancer. J Mol Med (Berl) 94: 443–455, 2016. doi: 10.1007/s00109-015-1366-z. [DOI] [PubMed] [Google Scholar]
- 32.Sahin H, Wasmuth HE. Chemokines in tissue fibrosis. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1832: 1041–1048, 2013. doi: 10.1016/j.bbadis.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 33.Sharp CN, Doll M, Dupre TV, Beverly LJ, Siskind LJ. Moderate aging does not exacerbate cisplatin-induced kidney injury or fibrosis despite altered inflammatory cytokine expression and immune cell infiltration. Am J Physiol Renal Physiol 316: F162–F172, 2019. doi: 10.1152/ajprenal.00463.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharp CN, Doll MA, Dupre TV, Shah PP, Subathra M, Siow D, Arteel GE, Megyesi J, Beverly LJ, Siskind LJ. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am J Physiol Renal Physiol 310: F560–F568, 2016. doi: 10.1152/ajprenal.00512.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharp CN, Doll MA, Megyesi J, Oropilla GB, Beverly LJ, Siskind LJ. Subclinical kidney injury induced by repeated cisplatin administration results in progressive chronic kidney disease. Am J Physiol Renal Physiol 315: F161–F172, 2018. doi: 10.1152/ajprenal.00636.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharp CN, Siskind LJ. Developing better mouse models to study cisplatin-induced kidney injury. Am J Physiol Renal Physiol 313: F835–F841, 2017. doi: 10.1152/ajprenal.00285.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi M, McMillan KL, Wu J, Gillings N, Flores B, Moe OW, Hu MC. Cisplatin nephrotoxicity as a model of chronic kidney disease. Lab Invest 98: 1105–1121, 2018. doi: 10.1038/s41374-018-0063-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shinke H, Masuda S, Togashi Y, Ikemi Y, Ozawa A, Sato T, Kim YH, Mishima M, Ichimura T, Bonventre JV, Matsubara K. Urinary kidney injury molecule-1 and monocyte chemotactic protein-1 are noninvasive biomarkers of cisplatin-induced nephrotoxicity in lung cancer patients. Cancer Chemother Pharmacol 76: 989–996, 2015. doi: 10.1007/s00280-015-2880-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shu S, Zhu J, Liu Z, Tang C, Cai J, Dong Z. Endoplasmic reticulum stress is activated in post-ischemic kidneys to promote chronic kidney disease. EBioMedicine 37: 269–280, 2018. doi: 10.1016/j.ebiom.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skinner R, Parry A, Price L, Cole M, Craft AW, Pearson AD. Persistent nephrotoxicity during 10-year follow-up after cisplatin or carboplatin treatment in childhood: relevance of age and dose as risk factors. Eur J Cancer 45: 3213–3219, 2009. doi: 10.1016/j.ejca.2009.06.032. [DOI] [PubMed] [Google Scholar]
- 41.Stroo I, Stokman G, Teske GJD, Raven A, Butter LM, Florquin S, Leemans JC. Chemokine expression in renal ischemia/reperfusion injury is most profound during the reparative phase. Int Immunol 22: 433–442, 2010. doi: 10.1093/intimm/dxq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang PM-K, Nikolic-Paterson DJ, Lan H-Y. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol 15: 144–158, 2019. doi: 10.1038/s41581-019-0110-2. [DOI] [PubMed] [Google Scholar]
- 43.Torres R, Velazquez H, Chang JJ, Levene MJ, Moeckel G, Desir GV, Safirstein R. Three-dimensional morphology by multiphoton microscopy with clearing in a model of cisplatin-induced CKD. J Am Soc Nephrol 27: 1102–1112, 2016. doi: 10.1681/ASN.2015010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Volarevic V, Djokovic B, Jankovic MG, Harrell CR, Fellabaum C, Djonov V, Arsenijevic N. Molecular mechanisms of cisplatin-induced nephrotoxicity: a balance on the knife edge between renoprotection and tumor toxicity. J Biomed Sci 26: 25, 2019. doi: 10.1186/s12929-019-0518-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, Wang Y, Huang Z, Ren J, Liu S, Chen X, Han J. A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol 26: 2647–2658, 2015. doi: 10.1681/ASN.2014080741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Q, Wang Y, Pei G, Deng X, Jiang H, Wu J, Zhou C, Guo Y, Yao Y, Zeng R, Xu G. Bone marrow-derived Ly6C- macrophages promote ischemia-induced chronic kidney disease. Cell Death Dis 10: 291, 2019. doi: 10.1038/s41419-019-1531-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang M, Guo Y, Fu H, Hu S, Pan J, Wang Y, Cheng J, Song J, Yu Q, Zhang S, Xu JF, Pei G, Xiang X, Yang P, Wang C-Y. Chop deficiency prevents UUO-induced renal fibrosis by attenuating fibrotic signals originated from Hmgb1/TLR4/NFκB/IL-1β signaling. Cell Death Dis 6: e1847, 2015. doi: 10.1038/cddis.2015.206. [DOI] [PMC free article] [PubMed] [Google Scholar]