

Keywords: acute kidney injury, chronic kidney disease, hypertension, renal perfusion, soluble epoxide hydrolase inhibitor
Abstract
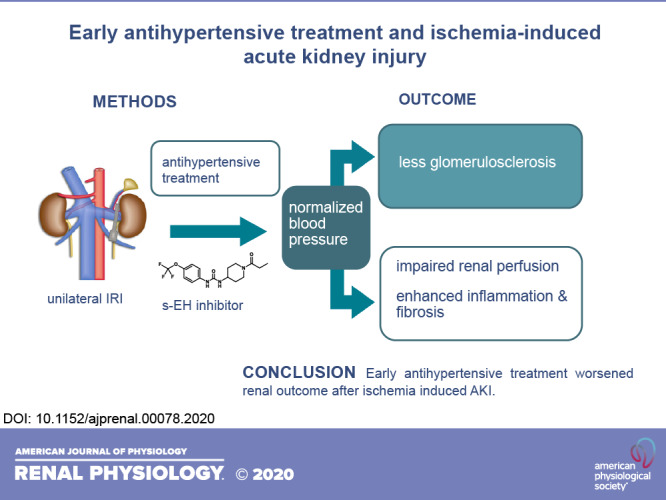
Acute kidney injury (AKI) frequently complicates major surgery and can be associated with hypertension and progress to chronic kidney disease, but reports on blood pressure normalization in AKI are conflicting. In the present study, we investigated the effects of an angiotensin-converting enzyme inhibitor, enalapril, and a soluble epoxide hydrolase inhibitor, 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl)urea (TPPU), on renal inflammation, fibrosis, and glomerulosclerosis in a mouse model of ischemia-reperfusion injury (IRI)-induced AKI. Male CD1 mice underwent unilateral IRI for 35 min. Blood pressure was measured by tail cuff, and mesangial matrix expansion was quantified on methenamine silver-stained sections. Renal perfusion was assessed by functional MRI in vehicle- and TPPU-treated mice. Immunohistochemistry was performed to study the severity of AKI and inflammation. Leukocyte subsets were analyzed by flow cytometry, and proinflammatory cytokines were analyzed by quantitative PCR. Plasma and tissue levels of TPPU and lipid mediators were analyzed by liquid chromatography mass spectrometry. IRI resulted in a blood pressure increase of 20 mmHg in the vehicle-treated group. TPPU and enalapril normalized blood pressure and reduced mesangial matrix expansion. However, inflammation and progressive renal fibrosis were severe in all groups. TPPU further reduced renal perfusion on days 1 and 14. In conclusion, early antihypertensive treatment worsened renal outcome after AKI by further reducing renal perfusion despite reduced glomerulosclerosis.
INTRODUCTION
Hypertension after acute kidney injury (AKI) is common (12) and may be a risk factor for chronic kidney disease (CKD) (5). Many patients at risk for AKI receive antihypertensive medication, but data on antihypertensive treatment in the acute phase of AKI are limited. In older patients needing cardiac surgery, risk factors such as hypertension, diabetes, and concomitant vascular disease can increase the AKI risk to 30%. Another patient group at high risk for AKI is recipients of solid organ transplantation. After lung transplantation, an incidence of AKI of 50–60% has been reported and is linked to CKD progression (30). Whether or not antihypertensive drugs, especially angiotensin-converting enzyme (ACE) inhibitors, should be withdrawn is a matter of debate (1, 4, 31). Epoxyeicosatrienoic acids (EETs) are synthesized from arachidonic acid (ARA) by cytochrome P-450, and this pathway offers several attractive novel drug targets (16, 17). EETs act as endothelium-derived hyperpolarizing factors on preglomerular vascular smooth muscle cells (3), increase renal blood flow (16), and exert vasodilatory, anti-inflammatory, antiapoptotic, and proangiogenic effects (15, 16, 21). The degradation of EETs to dihydroxyeicosatrienoic acids and other fatty acid epoxides to their corresponding diols is catalyzed by soluble epoxide hydrolase (sEH), which can be blocked by sEH inhibitors (sEHIs) (27). A potent and highly selective sEHI is 1-(1-propanoylpiperidin-4-yl)-3-[4-(trifluoromethoxy)phenyl]urea (TPPU) (24). TPPU has been shown to decrease reperfusion injury after focal cerebral ischemia (28). Furthermore, administration of sEHI reduced renal damage in ANG II-dependent hypertension (11, 29). We have recently shown that CD1 mice develop hypertension secondary to ischemia-induced AKI and developed progressive renal fibrosis and glomerulosclerosis within 2 wk of followup (9). Based on the previously reported beneficial effects of sEHI in models of cerebral ischemia and hypertension, we tested whether early antihypertensive therapy with TPPU compared with enalapril as an established antihypertensive drug attenuates AKI after renal ischemia-reperfusion injury (IRI) and improves renal outcome.
METHODS
Animals.
The Hannover Medical School Committee on Animal Research approved the present study (33.19-42502-04-14/1657 and 12/0916). The German guidelines are in accordance with National Institutes of Health guidelines for animal welfare. Adult male CD1 mice (30–35 g, 8–10 wk of age) were purchased from Charles River (Sulzfeld, Germany) and used for all experiments. Mice had free access to food (Altromin 1324 standard mouse diet) and domestic quality drinking water and were housed under conventional conditions in individually ventilated cages with a 10:14-h light-dark cycle. Animals were cared for in accordance with our institutional guidelines for experimental animals.
Treatment.
The sEH-inhibitor TPPU was administered once daily by gavage. Enalapril treatment via drinking water was calculated based on the medium amount of daily fluid intake (30 mg·kg−1·day−1). Vehicle treatment with PBS served as the control. All treatments were started 1 day before IRI and continued over the observation period of 2 wk. Vehicle treatment was done in n = 15 mice, and TPPU was administered to n = 7 IRI mice at a dose of 1 mg/kg and to n = 8 IRI mice and n = 4 sham mice at a dose of 10 mg/kg. Plasma levels of TPPU were monitored.
Renal IRI.
Mice were anesthetized with isoflurane (3% induction and 1.5% maintenance). Butorphanol (1 mg/kg sc) was given before surgery for analgesia. Renal IRI was induced by left renal pedicle clamping for 35 min (microaneurysm clip, Aesculap). Mice were monitored until fully awake. Physical condition was assessed daily and scored for general appearance and well-being. Organ retrieval was done in deep isoflurane anesthesia (4%), and total body perfusion with ice-cold PBS via the left ventricle caused circulatory arrest. IRI kidneys as well as contralateral kidneys were fixed in 4% paraformaldehyde or shock frozen. Inulin and p-aminohippurate clearance were measured surgically to analyze glomerular filtration rate and renal blood flow as described in the Supplemental Methods in the Supplemental Material (Supplemental Material is available online at https://doi.org/10.6084/m9.figshare.12047412.v1).
Functional MRI to measure renal perfusion.
Functional MRI was done before surgery at baseline and on days 1 and 14 after IRI using a 7-T small animal scanner (Bruker, Pharmascan) and a circular polarized volume coil (Bruker T10327V3). Mice were anesthetized by isoflurane inhalation. Respiration was monitored and kept between 30 and 60 breaths/min. Respiratory-triggered, fat-saturated T2-weighted sequences were acquired. Kidney volumes were determined by manual segmentation. Renal perfusion was measured without administration of contrast agent using an arterial spin labeling technique as previously described (13). Renal perfusion maps were calculated. Mean local perfusion in the renal cortex was quantified in the IRI kidney and contralateral kidney in milliliters per minute per 100 g kidney. Functional MRI experiments were done at baseline and on day 1 in n = 7 IRI vehicle-treated and IRI TPPU-treated mice and in n = 4 sham mice treated with TPPU. Baseline measurements were done before treatment and IRI. At day 14 after IRI, n = 4 mice/group were examined, respectively. For day 14, the untouched contralateral control kidney served as control.
Histology and immunohistochemistry.
Kidneys were harvested on days 2 and 14 after IRI. The middle part of the kidney was immediately fixed in 4% paraformaldehyde and embedded in paraffin. Two-micrometer paraffin sections were cut and stained with sirius red (collagen type I and type III deposition). Immunohistochemistry to investigate neutrophil infiltration (GR-1) and methenamine silver stain to assess mesangial matrix thickness was done according to standard diagnostic protocols (7) (see Supplemental Methods, available online at https://doi.org/10.6084/m9.figshare.12047412.v1). Interstitial collagen deposition in the cortex and outer medulla was quantified on sections stained with sirius red using ImageJ software (see Supplemental Methods, available online at https://doi.org/10.6084/m9.figshare.12047412.v1). Analysis was performed in a blinded manner using a Leica imaging microscope. Histology for renal morphology and immunohistochemistry was done in n = 4 mice/group at day 2 and n = 7–11 mice/group at day 14 after IRI.
Real-time quantitative PCR.
Quantitative PCR was done on an Lightcycler 420 II (Roche Diagnostics, Penzberg, Germany) using FastStart SYBR green chemistry as provided in the Supplemental Methods (available online at https://doi.org/10.6084/m9.figshare.12047412.v1). Gene-specific primers for connective tissue growth factor (CTGF; QT00096131), plasminogen activator inhibitor-1 (PAI-1; forward: 5′-ATGTTTAGTGCAACCCTGGC-3′ and reverse 5′-CTGCTCTTGGTCGGAAAGAC-3′), and IL-6 (QT00098875) were used. Results were normalized to hypoxanthine-guanine phosphoribosyltransferase expression (QT00166768). Quantification was carried out using qgene software.
ELISA to measure systemic chemokine (C-X-C motif) ligand 13 levels.
Serum chemokine (C-X-C motif) ligand 13 (CXCL13) levels in mice were measured by ELISA (Quantikine Mouse CXCL13/BLC/BCA-1, Immunoassay catalog no. MCX130) using the Tecan spectra mini ELISA reader (Tecan, Crailsheim, Germany). CXCL13 levels were quantified by comparison with internal CXCL13 standards.
Flow cytometry.
FACS analysis was performed in n = 4 mice/group at day 2 after IRI using the following anti-mouse antibodies: CD45 (clone 30-F11), CD49b (clone DX5), CD11c (clone N418), F4/80 (clone BM8), CD11b (clone M1/70), and fixable viability dye eFluor 506 (65-0866) from eBioscience (Santa Clara, CA) as well as Ly6-C (clone HK1.4) and Ly6G (clone 1A8) from Biolegend (San Diego, CA). The preparation protocol for renal tissue is provided in the Supplemental Methods (available online at https://doi.org/10.6084/m9.figshare.12047412.v1). For flow cytometry, a FACS CantoII (BD Biosciences) was used, and data analysis was done with Kaluza software 1.3 (Beckmann Coulter).
Liquid chromatography mass spectrometry.
Liquid chromatography coupled to mass spectrometry (LC-MS) was used to quantify epoxy fatty acid and dihydroxy fatty acid concentrations in plasma and drug concentrations in plasma and renal tissue at day 2 and day 14 in n = 4 animals/group. Quantification of renal tissue and plasma TPPU concentration was carried out using online solid phase extraction (SPE) LC-MS as previously described (26) using a QTRAP triple quadrupole MS (ABSciex, Darmstadt, Germany) (25, 32) or a Mircomass LC Quattro instrument (Waters, Eschborn, Germany) (33). The preparation of renal tissue and plasma for LC-MS is provided in the Supplemental Methods (available online at https://doi.org/10.6084/m9.figshare.12047412.v1). Plasma analysis of oxylipins (epoxy fatty acids and dihydroxy fatty acids) was carried out by means of LC-MS following offline SPE using the SepPak tC18 protocol as previously described (23).
Tail-cuff blood pressure measurement.
We are aware that radiotelemetry is the gold standard for blood pressure measurements in mice (20). However, the surgical interventions in our study made sensor placement (particularly in the abdomen) untenable. Carotid artery placement interfers with baroreflex regulation. Systolic blood pressure was measured with a noninvasive tail-cuff blood pressure measurement system (TSE BloodPressure 209000-series, TSE Systems) as provided in the Supplemental Methods (available online at https://doi.org/10.6084/m9.figshare.12047412.v1). Baseline systolic blood pressure was recorded in n = 28 mice before treatment initiation and in n = 7 mice in the vehicle-treated group and TPPU-treated group (1 mg/kg) at different time points after IRI.
Statistical analysis.
For statistical analysis, Graphpad prism software (version 5.0c, GraphPad Software, San Diego, CA) was used. For multiple comparisons, ANOVA with post hoc Tukey correction was applied. For comparison of two groups, Student’s unpaired t test was used. Linear regression analysis and Pearson correlation was done for TPPU tissue concentration and renal perfusion. Differences were considered significant at a P value of <0.05. Data are presented as means ± SE.
RESULTS
Antihypertensive treatment normalized blood pressure and attenuated glomerulosclerosis.
CD1 mice already had mild hypertension at baseline and showed further blood pressure elevation after unilateral IRI (+20 mmHg after 2 wk in the vehicle-treated group). Enalapril and TPPU normalized blood pressure during the 2-wk followup (Fig. 1E). Glomerulosclerosis can develop subsequent to hypertension, and a morphological correlate is mesangial matrix expansion. Therefore, mesangial matrix thickness was measured on silver-stained sections (Fig. 1, A–D). Mesangial matrix thickness was significantly increased in the vehicle-treated group 2 wk after IRI. In contrast, glomeruli of TPPU- and enalapril-treated mice showed significantly less expansion of the mesangial matrix than vehicle-treated mice. Notably, the contralateral untouched kidney showed no signs of glomerulosclerosis, although this kidney was also affected by systemic hypertension. Enalapril is known to reduce renal perfusion (10). Therefore, we focused on TPPU for further experiments on renal outcome by functional MRI, inflammation, and fibrosis.
Fig. 1.

Blood pressure and glomerulosclerosis. A−D: mesangial matrix thickness was measured at day 14 after ischemia-reperfusion injury (IRI) on methenamin silver-stained tissue samples. Mesangial matrix expansion was enhanced in hypertensive IRI vehicle-treated mice, whereas 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU)- and enalapril (enal)-treated IRI mice had reduced mesangial matrix expansion. The contralateral kidney served as the control and showed no signs of mesangial matrix expansion. Bar = 10 µm. E: the blood pressure elevation after IRI was attenuated by enalapril and TPPU treatment. F: quantification of mesangial matrix thickness. SBP, systolic blood pressure; d0, day 0; d14, day 14; veh, vehicle. *P < 0.05; **P < 0.01.
Inflammation after IRI.
In the early phase after IRI, neutrophils are the first leukocyte subsets infiltrating the kidney. Afterward, macrophages invade the tissue. By flow cytometry, we showed that Ly6G-positive neutrophils and activated Ly6c-positive macrophages were the main leukocyte populations at day 2 after IRI (Fig. 2, A–D). Contralateral untouched kidneys served as controls and had only minor neutrophil and macrophage infiltration similar to sham-operated mice treated with TPPU. GR-1-positive neutrophils were mainly localized in the outer medulla (Fig. 2, E–H). Proinflammatory cytokine expression in IRI kidneys for IL-6 was significantly upregulated in both vehicle- and TPPU-treated groups. In addition, the systemic inflammation marker CXCL13 was significantly increased at day 1 after IRI in the plasma of vehicle- and TPPU-treated mice (Fig. 2J). TPPU treatment did not attenuate early inflammation after IRI.
Fig. 2.

Inflammation at day 2 after ischemia-reperfusion injury (IRI). A−D: flow cytometry of whole kidney lysates showed enhanced neutrophil and macrophage infiltration in IRI kidneys compared with sham-operated control (con) kidneys after 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU) treatment. E−H: infiltrating GR-1-positive neutrophils were mainly localized in the outer medulla (GR-1 scoring: 0–4). Bar = 50 µm. I: IL-6 mRNA transcripts were significantly increased at day 2 after IRI in all groups. J: serum levels of proinflammatory chemokine (C-X-C motif) ligand 13 (S-CXCL13) were enhanced in all IRI mice without differences between groups. veh, vehicle; bl, baseline. *P < 0.05; **P < 0.01.
Kidney volume and CKD.
Kidney volume was measured by functional MRI longitudinally at baseline and on days 1 and 14 (Fig. 3, A–D). After unilateral IRI, the untouched contralateral kidney showed hypertrophy over time (13). To outweigh this effect, changes in kidney volume after IRI were expressed relatively to the untouched contralateral control kidney at day 1. In the chronic phase after IRI, severe kidney volume loss was detected in all groups as a correlate of CKD. Kidney wet weight was markedly reduced at 14 days after IRI compared with the contralateral control. This effect was even more pronounced in TPPU-treated compared with vehicle-treated IRI kidneys (Fig. 3K). In line with kidney shrinkage, sirius red-positive collagen deposition was enhanced in the IRI groups, indicating progressive renal fibrosis (Fig. 3, E−H). In addition, transcripts for CTGF and PAI-1, both downstream targets of profibrotic transforming growth factor (TGF)-β, were increased after 2 wk in IRI kidneys of both groups; contralateral control kidneys showed normal expression at day 14 (Fig. 3, I–J). TPPU treatment did not attenuate upregulation of profibrotic genes and did not protect from kidney volume loss and renal fibrosis.
Fig. 3.

Kidney volume and chronic kidney disease at day 14 (d14) after ischemia-reperfusion injury (IRI). A−D: morphometric MRI imaging was done to assess kidney volume longitudinally and showed severe volume loss at day 14 after IRI in vehicle (veh)- and 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU)-treated mice. E−H: tubulointerstitial fibrosis showed enhanced collagen deposition after IRI in all groups (all images were kept in the same magnification and the stack function was used). Quantification of fibrosis in the outer medulla (OM) is shown. I: connective tissue growth factor (CTGF) mRNA was significantly increased in IRI kidneys compared with contralateral controls without differences between treatment strategies. J: plasminogen activator inhibitor-1 (PAI-1) mRNA expression was even higher in TPPU-treated compared with vehicle-treated IRI kidneys. K: the kidney wet weight decrease was more pronounced in TPPU-treated IRI kidneys compared with vehicle-treated kidneys. *P < 0.05; **P < 0.01; ***P < 0.001.
TPPU treatment aggravated early renal perfusion impairment after IRI.
IRI caused substantial renal perfusion impairment, as measured by arterial spin labeling (Fig. 4, A–E). Surprisingly, TPPU treatment even aggravated renal perfusion impairment. Untouched contralateral kidneys served as controls and had perfusion values comparable to baseline perfusion and sham-operated mice treated with TPPU. At day 14, substantial renal perfusion impairment in IRI vehicle- and TPPU-treated mice was present (Fig. 4E). Tissue levels of TPPU were measured by LC-MS and were significantly lower in IRI kidneys compared with contralateral control kidneys at days 2 and 14 (Fig. 4H). Renal perfusion correlated with renal tissue levels of TPPU at day 14, and lower tissue levels were measured in less perfused kidneys (Fig. 4G). To assess renal function after IRI, the healthy contralateral kidney was removed at day 14, and inulin clearance was measured. Renal function was decreased to 20% from normal after IRI and was even further reduced after TPPU treatment (data are expressed as proportion of normal inulin clearance in healthy control mice, i.e., 150 mL/min). IRI resulted in poor renal function in vehicle-treated mice and was even worse in TPPU-treated mice with normal blood pressure (IRI vehicle: 22 ± 4% vs. IRI TPPU: 8 ± 2%; Fig. 4F).
Fig. 4.

1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU) aggravated renal perfusion impairment after ischemia-reperfusion injury (IRI). A−D: renal perfusion was measured by arterial spin labeling. The contralateral kidney at day 1 (d1) served as the control (con; B). Renal perfusion was significantly impaired at day 1 and also at day 14 (d14) after IRI. E: renal perfusion impairment was more profound after TPPU treatment. F: renal function was measured by inulin clearance after contralateral nephrectomy and was poor in vehicle (veh)-treated mice and even worse after blood pressure normalization by TPPU (relative inulin clearance was calculated as a proportion of normal). H and G: TPPU concentration in the renal tissue was lower in IRI kidneys compared with contralateral control kidneys (H) and low renal perfusion correlated with low tissue levels of TPPU (G). bl, baseline; d2, day 2. *P < 0.05; ***P < 0.001.
TPPU treatment affected oxylipin profiles.
Therapeutic plasma levels of TPPU were achieved, and TPPU treatment resulted in increased plasma epoxide-to-diol ratios at days 2 and 14 (Supplemental Fig. S1, available online at https://doi.org/10.6084/m9.figshare.12047247.v2). When we compared day 2 and day 14 under IRI and TPPU treatment, the epoxide-to-diol ratios were observed to be more strongly elevated at day 2 (Supplemental Fig. S1, available online at https://doi.org/10.6084/m9.figshare.12047247.v2). For example, this is shown for the epoxy arachidonic acid ratios of 11(12)-EpETrE to 11,12-DiHETrE and 14(15)-EpETrE to 14,15-DiHETrE, which were significantly increased under TPPU treatment at day 2, whereas this effect was less pronounced at day 14 (Supplemental Fig. S1, available online at https://doi.org/10.6084/m9.figshare.12047247.v2). For plasma epoxide-to-diol ratio measurements, sham-operated mice treated with TPPU served as the control.
DISCUSSION
We tested whether or not sEH inhibition with TPPU can normalize blood pressure elevation and attenuate renal damage in the CD1 mouse model of ischemia-induced AKI with progression to CKD. Systolic blood pressure, renal morphology, inflammation, perfusion impairment, and progression to CKD were investigated. We found that both TPPU and our positive control enalapril reduced blood pressure and attenuated glomerulosclerosis. However, aggravation of renal perfusion impairment caused enhanced inflammation and tubulointerstitial fibrosis after TPPU treatment. We have previously characterized CD1 mice in a model of renal IRI (9). CD1 mice developed marked hypertension after ischemia-induced AKI, which was associated with renal perfusion impairment and rarefication of the peritubular capillary network (9). Persistent inflammation and progression to CKD with development of glomerulosclerosis was also seen in CD1 mice after IRI (9). EETs have been protective in experimental models of ANG II-induced hypertension-mediated renal damage (35) and exerted anti-inflammatory (19, 34) as well as antifibrotic effects (18).
Blood pressure elevation was effectively normalized after TPPU or enalapril treatment. We showed that TPPU levels were in the therapeutic range and that target engagement could be demonstrated by oxylipin measurement. TPPU elevated plasma epoxide/diol levels effectively. While in the first days the epoxides of all polyunsaturated fatty acids were strongly elevated, the extent of change in the plasma epoxy-to-diol ratio decreased over the treatment period and reached statistically significance solely for the linoleic acid metabolites by day 14. This finding indicates a homeostatic adaption to the (sub)chronic sEH inhibition downregulation the formation of the highly potent epoxy-polyunsaturated fatty acid such as well-characterized ARA-derived EpETrEs (also known as EETs). This effect has been shown in a large number of studies evaluating the activity of sEH inhibitors showing that linoleic acid metabolites are the most sensitive biomarker effective chronic systemic sEH inhibition (22).
Despite the proven pharmacological effects of TPPU treatment on normalization of systemic blood pressure and severe kidney volume loss, progressive renal fibrosis was present in all IRI AKI groups. By comparing the outcome after enalapril and TPPU treatment following renal IRI, it became apparent that in CD1 mice, the TPPU-mediated elevation of EETs resulted in a similar ability to reduce hypertension as enalapril. Thus, the dominant mechanism of EET stabilization in the context of renal IRI seems to be a reduction of blood pressure, but further specific effects of EET metabolism inhibition on renal IRI cannot be excluded by this study and requires further investigation.
In previous experimental studies, TPPU reduced bleomycin-induced pulmonary fibrosis (36) and sEH deficiency attenuated renal fibrosis after unilateral ureter obstruction via reduced TGF-β expression (18). In our study, CTGF as a downstream target of TGF-β was increased following IRI and TPPU treatment. The reason for this discrepancy remains to be determined; however, one explanation could be that renal perfusion impairment may have led to a hypoxic microenvironment, promoting progressive renal fibrosis (2, 8). Indeed, we detected early renal perfusion impairment after IRI, which was associated with tubular capillary rarefication contributing to chronic hypoxia in a previous study (9). In the present study, we showed that TPPU treatment with normalization of blood pressure after IRI even aggravated the early renal perfusion impairment measured by functional MRI.
Since we chose a unilateral IRI model to overcome early mortality and study pathophysiological changes longitudinally, renal function could not be measured longitudinally throughout the entire followup. To outweigh this limitation, we measured renal function surgically by inulin clearance after nephrectomy of the untouched contralateral kidney at the end point after a 14-day followup. Renal function after IRI was poor in vehicle-treated hypertensive mice after IRI and even worse after blood pressure normalization with TPPU. This state of affairs correlated with the aggravation of renal perfusion impairment as has been shown in our previous study (13). One advantage of functional MRI techniques used in the present study is to be able to monitor renal perfusion longitudinally in the living animal under the influence of systemic blood pressure modification (13). Hypertension can lead to glomerulosclerosis and CKD. Mesangial matrix expansion as a sign of glomerulosclerosis was assessed and showed that antihypertensive treatment by TPPU and also enalapril attenuated mesangial matrix expansion and therefore glomerulosclerosis. The contralateral untouched control kidneys had no signs of glomerulosclerosis, although they were also affected by systemic blood pressure elevation.
Despite the beneficial effects on glomerulosclerosis, TPPU treatment failed to decrease inflammation and fibrosis after renal IRI. We started antihypertensive treatment 1 day before IRI induction and continued it over a period of 2 wk. In another study, Cheng et al. (6) showed that lorsartan reduced CKD development after contralateral nephrectomy and later renal IRI. In line with our observations, they showed that CD1 mice developed severe hypertension following IRI, but these authors started antihypertensive treatment 1 mo after AKI induction and continued it over a followup period of 5 mo. Interestingly, they showed that the development of tubulointerstitial fibrosis as well as the activation of profibrotic genes were attenuated by lorsartan treatment at this late time point. It is possible that later initiation of antihypertensive treatment in the context of AKI might avoid the more profound decrease of renal perfusion and thus be more beneficial in preventing CKD progression. By LC-MS, we could show that drug delivery to the kidney was impaired, which might be also a reason for poor effectivity of TPPU in the present study.
Taken together, TPPU was effective in controlling blood pressure elevation and preventing glomerulosclerosis after ischemia-induced AKI. On the other side, TPPU enhanced renal perfusion impairment and did not attenuate inflammation and progression to CKD. This study provides evidence that antihypertensive treatment might be detrimental in the early phase of ischemia-induced AKI. Therefore, timing and duration of antihypertensive treatment in the context of AKI requires careful consideration.
GRANTS
This work was supported in part by Deutsche Forschungsgemeinschaft (DFG) GU 613/1-1 and HU 2232/1-1 (to F.G.) and DFG Sche 1801 (to N.H.S.). Partial support was provided by National Institute of Environmental Health Sciences Grant R35ES030443-01 (RIVER Award) (to B.D.H.).
DISCLOSURES
B.D.H. founded EicOsis, which is developing sEHI as therapeutics.
AUTHOR CONTRIBUTIONS
F.G. conceived and designed research; R.G., K.D., B.H., A.T., S.R., R.C., S.H., M.-S.J., M.M., I.W., S.H.H., and F.G. performed experiments; R.G., K.D., B.H., A.T., S.H., M.-S.J., J.H.B., I.W., N.H.S., and F.G. analyzed data; R.G., K.D., B.H., A.T., S.H., M.-S.J., J.H.B., I.W., S.I., H.H., D.P., B.D.H., N.H.S., and F.G. interpreted results of experiments; R.G. prepared figures; R.G. drafted manuscript; K.D., F.C.L., D.P., S.H.H., B.D.H., N.H.S., and F.G. edited and revised manuscript; R.G., K.D., B.H., A.T., S.R., R.C., S.H., M.-S.J., J.H.B., M.M., I.W., S.I., H.H., F.C.L., D.P., S.H.H., B.D.H., N.H.S., and F.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Herle Chlebusch, Gabriele Bauerschaper, and Christian Bergen for excellent technical assistance.
REFERENCES
- 1.Bainey KR, Rahim S, Etherington K, Rokoss ML, Natarajan MK, Velianou JL, Brons S, Mehta SR; CAPTAIN Investigators . Effects of withdrawing vs continuing renin-angiotensin blockers on incidence of acute kidney injury in patients with renal insufficiency undergoing cardiac catheterization: results from the Angiotensin Converting Enzyme Inhibitor/Angiotensin Receptor Blocker and Contrast Induced Nephropathy in Patients Receiving Cardiac Catheterization (CAPTAIN) trial. Am Heart J 170: 110–116, 2015. doi: 10.1016/j.ahj.2015.04.019. [DOI] [PubMed] [Google Scholar]
- 2.Brezis M, Rosen S. Hypoxia of the renal medulla−its implications for disease. N Engl J Med 332: 647–655, 1995. doi: 10.1056/NEJM199503093321006. [DOI] [PubMed] [Google Scholar]
- 3.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res 78: 415–423, 1996. doi: 10.1161/01.RES.78.3.415. [DOI] [PubMed] [Google Scholar]
- 4.Chatzikyrkou C, Eichler J, Karch A, Clajus C, Scurt FG, Ramackers W, Lehner F, Menne J, Haller H, Mertens PR, Schiffer M. Short- and long-term effects of the use of RAAS blockers immediately after renal transplantation. Blood Press 26: 30–38, 2017. doi: 10.1080/08037051.2016.1182856. [DOI] [PubMed] [Google Scholar]
- 5.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med 371: 58–66, 2014. doi: 10.1056/NEJMra1214243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng SY, Chou YH, Liao FL, Lin CC, Chang FC, Liu CH, Huang TM, Lai CF, Lin YF, Wu VC, Chu TS, Wu MS, Lin SL. Losartan reduces ensuing chronic kidney disease and mortality after acute kidney injury. Sci Rep 6: 34265, 2016. doi: 10.1038/srep34265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehrenreich T, Espinosa T. Chromotrope silver methenamine stain of glomerular lesions. Am J Clin Pathol 56: 448–451, 1971. doi: 10.1093/ajcp/56.4.448. [DOI] [PubMed] [Google Scholar]
- 8.Fine LG, Orphanides C, Norman JT. Progressive renal disease: the chronic hypoxia hypothesis. Kidney Int Suppl 65: S74–S78, 1998. [PubMed] [Google Scholar]
- 9.Greite R, Thorenz A, Chen R, Jang MS, Rong S, Brownstein MJ, Tewes S, Wang L, Baniassad B, Kirsch T, Brasen JH, Lichtinghagen R, Meier M, Haller H, Hueper K, Gueler F. Renal ischemia reperfusion injury causes hypertension and renal perfusion impairment in the CD1 mice which promotes progressive renal fibrosis. Am J Physiol Renal Physiol 314: F881−F892, 2018. doi: 10.1152/ajprenal.00519.2016. [DOI] [PubMed] [Google Scholar]
- 10.Guidi E, Minetti EE, Cozzi MG. Acute and long-term effects of ACE inhibition on renal haemodynamics in glomerular and interstitial nephropathies. J Renin Angiotensin Aldosterone Syst 3: 40–45, 2002. doi: 10.3317/jraas.2002.007. [DOI] [PubMed] [Google Scholar]
- 11.Honetschlägerová Z, Kitada K, Husková Z, Sporková A, Kopkan L, Bürgelová M, Varcabová Š, Nishiyama A, Hwang SH, Hammock BD, Imig JD, Kramer HJ, Kujal P, Vernerová Z, Červenka L. Antihypertensive and renoprotective actions of soluble epoxide hydrolase inhibition in ANG II-dependent malignant hypertension are abolished by pretreatment with l-NAME. J Hypertens 31: 321–332, 2013. doi: 10.1097/HJH.0b013e32835b50aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu CY, Hsu RK, Yang J, Ordonez JD, Zheng S, Go AS. Elevated BP after AKI. J Am Soc Nephrol 27: 914–923, 2016. doi: 10.1681/ASN.2014111114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hueper K, Gutberlet M, Rong S, Hartung D, Mengel M, Lu X, Haller H, Wacker F, Meier M, Gueler F. Acute kidney injury: arterial spin labeling to monitor renal perfusion impairment in mice-comparison with histopathologic results and renal function. Radiology 270: 117–124, 2014. doi: 10.1148/radiol.13130367. [DOI] [PubMed] [Google Scholar]
- 15.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev 92: 101–130, 2012. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imig JD. Epoxyeicosatrienoic acids, hypertension, and kidney injury. Hypertension 65: 476–482, 2015. doi: 10.1161/HYPERTENSIONAHA.114.03585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imig JD. Prospective for cytochrome P450 epoxygenase cardiovascular and renal therapeutics. Pharmacol Ther 192: 1–19, 2018. doi: 10.1016/j.pharmthera.2018.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J, Imig JD, Yang J, Hammock BD, Padanilam BJ. Inhibition of soluble epoxide hydrolase prevents renal interstitial fibrosis and inflammation. Am J Physiol Renal Physiol 307: F971–F980, 2014. doi: 10.1152/ajprenal.00256.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu JY, Lin YP, Qiu H, Morisseau C, Rose TE, Hwang SH, Chiamvimonvat N, Hammock BD. Substituted phenyl groups improve the pharmacokinetic profile and anti-inflammatory effect of urea-based soluble epoxide hydrolase inhibitors in murine models. Eur J Pharm Sci 48: 619–627, 2013. doi: 10.1016/j.ejps.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luft FC. Men, mice, and blood pressure: telemetry? Kidney Int 96: 31–33, 2019. doi: 10.1016/j.kint.2018.12.033. [DOI] [PubMed] [Google Scholar]
- 21.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285: 1276–1279, 1999. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ostermann AI, Herbers J, Willenberg I, Chen R, Hwang SH, Greite R, Morisseau C, Gueler F, Hammock BD, Schebb NH. Oral treatment of rodents with soluble epoxide hydrolase inhibitor 1-(1-propanoylpiperidin-4-yl)-3-[4-(trifluoromethoxy)phenyl]urea (TPPU): resulting drug levels and modulation of oxylipin pattern. Prostaglandins Other Lipid Mediat 121: 131–137, 2015. doi: 10.1016/j.prostaglandins.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ostermann AI, Willenberg I, Schebb NH. Comparison of sample preparation methods for the quantitative analysis of eicosanoids and other oxylipins in plasma by means of LC-MS/MS. Anal Bioanal Chem 407: 1403–1414, 2015. doi: 10.1007/s00216-014-8377-4. [DOI] [PubMed] [Google Scholar]
- 24.Rose TE, Morisseau C, Liu JY, Inceoglu B, Jones PD, Sanborn JR, Hammock BD. 1-Aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: structure-activity relationships, pharmacokinetics, and reduction of inflammatory pain. J Med Chem 53: 7067–7075, 2010. doi: 10.1021/jm100691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schebb NH, Huby M, Morisseau C, Hwang SH, Hammock BD. Development of an online SPE-LC-MS-based assay using endogenous substrate for investigation of soluble epoxide hydrolase (sEH) inhibitors. Anal Bioanal Chem 400: 1359–1366, 2011. doi: 10.1007/s00216-011-4861-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schebb NH, Inceoglu B, Rose T, Wagner K, Hammock BD. Development of an ultra fast online-solid phase extraction (SPE) liquid chromatography electrospray tandem mass spectrometry (LC-ESI-MS/MS) based approach for the determination of drugs in pharmacokinetic studies. Anal Methods 3: 420–428, 2011. doi: 10.1039/C0AY00714E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res 43: 55–90, 2004. doi: 10.1016/S0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 28.Tu R, Armstrong J, Lee KSS, Hammock BD, Sapirstein A, Koehler RC. Soluble epoxide hydrolase inhibition decreases reperfusion injury after focal cerebral ischemia. Sci Rep 8: 5279, 2018. doi: 10.1038/s41598-018-23504-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ulu A, Harris TR, Morisseau C, Miyabe C, Inoue H, Schuster G, Dong H, Iosif AM, Liu JY, Weiss RH, Chiamvimonvat N, Imig JD, Hammock BD. Anti-inflammatory effects of ω-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin-II-dependent hypertension. J Cardiovasc Pharmacol 62: 285–297, 2013. doi: 10.1097/FJC.0b013e318298e460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wehbe E, Duncan AE, Dar G, Budev M, Stephany B. Recovery from AKI and short- and long-term outcomes after lung transplantation. Clin J Am Soc Nephrol 8: 19–25, 2013. doi: 10.2215/CJN.04800512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whiting P, Morden A, Tomlinson LA, Caskey F, Blakeman T, Tomson C, Stone T, Richards A, Savović J, Horwood J. What are the risks and benefits of temporarily discontinuing medications to prevent acute kidney injury? A systematic review and meta-analysis. BMJ Open 7: e012674, 2017. doi: 10.1136/bmjopen-2016-012674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willenberg I, Meschede AK, Schebb NH. Determining cyclooxygenase-2 activity in three different test systems utilizing online-solid phase extraction-liquid chromatography-mass spectrometry for parallel quantification of prostaglandin E2, D2 and thromboxane B2. J Chromatogr A 1391: 40–48, 2015. doi: 10.1016/j.chroma.2015.02.059. [DOI] [PubMed] [Google Scholar]
- 33.Willenberg I, von Elsner L, Steinberg P, Schebb NH. Development of an online-SPE-LC-MS method for the investigation of the intestinal absorption of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PHIP) and its bacterial metabolite PHIP-M1 in a Caco-2 Transwell system. Food Chem 166: 537–543, 2015. doi: 10.1016/j.foodchem.2014.06.038. [DOI] [PubMed] [Google Scholar]
- 34.Zhang G, Kodani S, Hammock BD. Stabilized epoxygenated fatty acids regulate inflammation, pain, angiogenesis and cancer. Prog Lipid Res 53: 108–123, 2014. doi: 10.1016/j.plipres.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol 15: 1244–1253, 2004. [PubMed] [Google Scholar]
- 36.Zhou Y, Yang J, Sun GY, Liu T, Duan JX, Zhou HF, Lee KS, Hammock BD, Fang X, Jiang JX, Guan CX. Soluble epoxide hydrolase inhibitor 1-trifluoromethoxyphenyl-3- (1-propionylpiperidin-4-yl) urea attenuates bleomycin-induced pulmonary fibrosis in mice. Cell Tissue Res 363: 399–409, 2016. doi: 10.1007/s00441-015-2262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]