

Abstract
The past two decades have witnessed a resurgence in neutrophil research, inspired in part by the discovery of neutrophil extracellular traps (NETs) and their myriad roles in health and disease. Within the lung, dysregulation of neutrophils and NETosis have been linked to an array of diseases including pneumonia, cystic fibrosis, acute respiratory distress syndrome (ARDS), chronic obstructive pulmonary disease (COPD), and severe asthma. However, our understanding of pathologic neutrophil responses in the lung remains incomplete. Two methodologic issues have contributed to this gap: first, an emphasis on studying neutrophils from blood rather than the lung and second, the technical difficulties of interrogating neutrophil responses in mice, which has largely restricted basic murine research to specialized laboratories. To address these limitations, we have developed a suite of techniques for studying neutrophil effector functions specifically in the mouse lung. These include ex vivo assays for phagocytosis and NETosis using bronchoalveolar neutrophils and in situ evaluation of NETosis in a murine model of pneumonia. Throughout, we have prioritized technical ease and robust, quantitative readouts. We hope these assays will help to standardize research on lung neutrophils and improve accessibility to this burgeoning field.
Keywords: bronchoalveolar neutrophils, neutrophil extracellular traps, phagocytosis, pneumonia, Pseudomonas aeruginosa
INTRODUCTION
Neutrophil research has undergone a renaissance during the last two decades. Long considered a unidimensional phagocyte designed to traffic into inflamed tissue, engulf extracellular pathogens, and then die, there is now growing appreciation of the neutrophil’s exquisite biological complexity. Evidence of phenotypic heterogeneity, bidirectional trafficking from tissues, contributions to adaptive immunity, and orchestration of tissue repair have transformed the field (10, 26). Perhaps most impactful was the discovery that neutrophils possess another mechanism of pathogen containment in addition to phagocytosis: formation of neutrophil extracellular traps (NETs) (4, 5). These webs of extruded chromatin, coated with citrullinated histones and cytotoxic granule proteins such as neutrophil elastase (NE), function to trap and kill pathogens during infection but also inflict collateral damage to the host during infectious and sterile inflammation (6, 31, 44).
The importance of NETs in the pathogenesis of lung disease has become increasingly clear (15, 32, 40). Indeed, NETosis and other pathological neutrophil responses have been implicated in pneumonia (25), acute respiratory distress syndrome (ARDS) (42), cystic fibrosis (24), severe asthma (41), and chronic obstructive pulmonary disease (COPD) (41). A common finding is the co-occurrence of excessive NETosis, which induces histotoxicity, and defective phagocytosis, which impairs pathogen clearance (8, 11, 17, 27, 28). Yet the mechanisms underlying these phenomena remain incompletely understood.
Untangling these and other fundamental questions in neutrophilic respiratory disease requires a robust, standardized, and widely accessible toolset for studying NETosis and phagocytosis within the lung. However, the majority of published methodology utilizes neutrophils from human blood. These cells offer crucial physiological insights into human immunity but poorly recapitulate the biology of neutrophils in the inflamed lung. Indeed, quiescent neutrophils in circulation must undergo several priming steps before they reach their fully activated state in the airspace, including transmigration into the tissue per se (3, 16, 19, 43). As such, bronchoalveolar neutrophils represent a more accurate model for studies of neutrophilic lung pathology.
A second drawback of human neutrophils is their genetic intractability, which derives from their short survival time in culture, precluding manipulation by siRNA and other gene silencing tools in vitro. In contrast, the study of neutrophils in genetically modified mouse models permits precise interrogation of gene function both in vivo and ex vivo. The use of murine systems also enables examination of neutrophils in well-established preclinical models of respiratory disease, such as pneumonia and asthma (23, 25). However, despite the many advantages of studying neutrophils within the murine lung, research in this area has been limited to specialized laboratories.
A primary reason for this is the technical difficulty of functional studies in murine neutrophils. Assessment of NET formation in vivo, for instance, requires codetection of DNA and NET-specific proteins such as NE or citrullinated histone H3 (H3Cit), either biochemically (via ELISA) or by colocalization (via microscopy). This is most commonly achieved using immunofluorescence microscopy, but the technique is challenging both to perform and quantitate (1, 9). In contrast, ELISA-based quantification of NETs in bronchoalveolar lavage (BAL) fluid represents a highly attractive alternative, especially because of its translatability to BAL samples from patients. However, few laboratories have reported specific techniques for detecting NETs in mouse BAL to date, and those described require costly reagents (Supplemental Table S1; all supplemental material is available at https://doi.org/10.6084/m9.figshare.12721736) (25, 33).
Measurement of phagocytosis, meanwhile, requires precise discrimination between substrate that is internalized and that which is adherent to the neutrophil’s exterior. Existing techniques solve this problem using relatively cumbersome, low-throughput approaches such as antibiotic protection (22) or sophisticated imaging technology that is not widely available (21). Therefore, a simple, accurate, and high-throughput phagocytosis assay in murine neutrophils remains in need.
In an effort to address these methodological gaps, we set out to establish a set of assays for interrogating NET formation and phagocytosis in murine neutrophils using models of bacterial pneumonia. Throughout, we have prioritized techniques that are quantitative, translatable, economical, and easily performed using widely available instrumentation.
METHODS
Murine pneumonia model.
Wild-type male and female C57BL/6 mice (Jackson Laboratories) were bred at Yale University under specific pathogen-free conditions. All animal studies were performed in accordance with Institutional Animal Care and Use Committee (IACUC)-approved protocols at Yale University. We used 8- to 12-wk-old age- and gender-matched mice for experiments after randomizing to control for litter effects. Growth of Pseudomonas aeruginosa strain PAO1, intrapulmonary instillation of P. aeruginosa lipopolysaccharide (LPS, cat. no. L9143, Sigma) and P. aeruginosa via oropharyngeal (OP) aspiration, and performance of BAL were performed as described in the Supplemental Material (see https://doi.org/10.6084/m9.figshare.12721736) (2, 14).
Ex vivo phagocytosis assay.
Synthesis of custom phagocytic substrates (pHrodo-PsA and fluor-PsA) from heat-killed P. aeruginosa was performed as described in the Supplemental Material. To collect neutrophils for phagocytosis assays, BAL fluid was harvested from pneumonic mice and centrifuged at 2,000 g for 2 min (speeds lower than 1,000 g resulted in significant loss of neutrophils) (12). The cell pellet was resuspended in DMEM/F-12 without phenol red (cat. no. 21041025, ThermoFisher), quantified using a coulter counter, and diluted to 7 × 105 cells/mL in DMEM/F-12. We added 75 μL of the cell suspension to wells of a nontreated 96-well flat bottom plate on ice. To each well, 30 μL of a suspension of phagocytic substrate was added at a final multiplicity of infection (MOI) of 40 for fluor-PsA and 80 for pHrodo-PsA. Plates were centrifuged at 2,000 g for 2 min to ensure contact between substrate and neutrophils and placed in a humidified incubator at 37°C with 5% CO2 for 90 min to allow phagocytosis to proceed. Negative control samples were incubated in parallel at 4°C to inhibit phagocytosis. After incubation, plates were immediately placed on ice. The nonadherent cell-containing supernatant was transferred to a nontreated U-bottom plate, 100 μL of PBS containing 10 mM EDTA was added to the adherent cells, and the plate was incubated for 20 min at 4°C. We detached cells from the plate by pipetting and gently scraping with the pipette tip and then transferred them to corresponding wells in the U-bottom plate. The plate was centrifuged at 2,000 g for 2 min, and the supernatant was discarded. The cells were resuspended in 50 μL FACS buffer (2% BSA in PBS) containing 1:100 Fc block (cat no. 123107, BioLegend,), incubated for 10 min on ice, washed, and stained with 50 μL of 1:100 Ly6G-APC (cat. no. 127613, BioLegend) for 30 min on ice. Finally, cells were washed, resuspended in FACS buffer, and analyzed on a tabletop flow cytometer (BD Accuri C6). Technical triplicates were used throughout.
Ex vivo NETosis assay.
BAL cells were resuspended in DMEM/F-12 at 1 × 106 white blood cells (WBCs)/mL, and 70 μL of cell suspension was added to each well of a black-wall, clear-bottom 96-well plate (cat. no. 3603, Corning). Sytox Orange (ThermoFisher) was added at a final concentration of 5 μM, and either live P. aeruginosa (100 MOI) or phorbol myristate acetate (PMA; 1 μM, cat. no. P1585, Sigma) was added to stimulate NETosis (total well volume of 100 μL). To control wells, saponin was added (1 μg/mL final concentration) to lyse cells and indicate maximal DNA release. Wells used for the assay were surrounded by a rim of wells filled with water to prevent evaporation. The plate was incubated with the lid on at 37°C without shaking, and fluorescence was measured through the clear bottom of the well every 10–15 min for 4 h using a fluorescence plate reader (540 nm excitation and 580 nm emission, Cytation 3, BioTek). Technical duplicates were used throughout.
In situ NETosis assay.
Semiquantitative ELISA-based detection of DNA-H3Cit complexes was performed as follows: 50 μL of a 2.5-μg/mL solution of murine monoclonal antibody against anti-double stranded DNA (cat. no. ab27156, Abcam) in PBS was added to each well of a high-binding 96-well plate (cat. no. 3369, Corning) and incubated overnight at 4°C. The plate was washed 3 times with 400 μL PBS per well, blocked with 200 μL of 1% BSA in PBS per well for 2 h at room temperature, and washed again. Then, 100 μL of BAL supernatant (collected by centrifuging BAL for 2 min at 2,000 g) was added to each well and incubated for 2 h at room temperature. Wells were washed twice, and 100 μL/well of a 1-μg/mL solution of anti-H3Cit detection antibody (cat. no. ab5103, Abcam) in PBS was added before incubation for 1 h at room temperature. After being washed twice, 100 μL/well of anti-rabbit horseradish peroxidase-conjugated antibody (1:1,000 dilution; cat. no. 7074, Cell Signaling Technology) was added and incubated for 1 h at room temperature. Three more washes were performed, 100 μL of TMB solution (cat. no. 34028, ThermoFisher) was added to each well, and the reaction was incubated for 10–20 min at room temperature on a rotary shaker, avoiding direct light. We added 50 μL of 2N HCl to each well to stop the reaction. The plate was then read immediately at 450 nm and 560 nm (a reduction measurement to correct for background) using a plate reader (Cytation 3, BioTek). In control experiments, the nuclease Benzonase (cat. no. E1014, Sigma) was used to digest the DNA backbone of NETs. As the concentration of Benzonase is reported by the manufacturer as ≥250 units/μL, values described herein are approximate. Technical singlicates were used due to a low intra-assay coefficient of variability.
Statistical analysis.
Statistical analysis, including calculation of IC50, was performed using Prism 8 (GraphPad Software, Inc). Results are presented as means ± standard deviation (SD), except where indicated. Data were analyzed using Student’s t test or one-way ANOVA as appropriate. Data shown are representative of multiple independent repetitions of the experiment. Statistical significance throughout is defined as P < 0.05.
RESULTS
Phagocytosis assay.
A primary challenge in assaying phagocytosis is discriminating between substrate internalized into phagosomes and that adherent to the cell exterior. To solve this problem, we employed the pH-sensitive dye, pHrodo, which fluoresces only in acidic environments such as the phagosome (Fig. 1). As P. aeruginosa is our primary pathogen of interest, we utilized this bacterium as a phagocytic substrate by covalently conjugating pHrodo-STP ester to heat-killed P. aeruginosa. This rapid and facile chemical reaction furnishes a large quantity of substrate for phagocytosis assays, referred to here as pHrodo-PsA.
Fig. 1.

Schematic overview of ex vivo phagocytosis assay. Pneumonia is induced in mice via oropharyngeal aspiration of Pseudomonas aeruginosa (PsA) or LPS. Bronchoalveolar lavage (BAL) cells are then collected and incubated with pHrodo-PsA ex vivo to evaluate phagocytosis. pHrodo-PsA only fluoresces within the acidified phagosome of neutrophils; adherent bacteria remain nonfluorescent. Upon flow cytometric analysis, neutrophils are identified via Ly6G staining and assessed for pHrodo-PsA positivity.
To test the performance of this custom phagocytic substrate, we elicited pneumonia in vivo using live P. aeruginosa, collected airway neutrophils via BAL, and incubated the BAL cells with opsonized pHrodo-PsA to induce phagocytosis (Supplemental Fig. S1). After 90 min, neutrophils were collected, identified via Ly6G positivity, and analyzed as shown in the gating strategy in Fig. 2A. Control samples were incubated at 4°C to inhibit phagocytosis and thereby identify the background fluorescent signal associated with adherent bacteria. As shown in Fig. 2B, the 4°C controls showed negligible fluorescence, whereas samples incubated at 37°C demonstrated robust uptake of pHrodo-PsA. These data were quantified in terms of the percentage of neutrophils positive for pHrodo (% positive, a binary readout) and mean fluorescence intensity (MFI), a continuous readout that reflects both the multiplicity of bacterial uptake and efficiency of phagosomal acidification.
Fig. 2.

Ex vivo phagocytosis assay. A: flow cytometry gating strategy for identifying bronchoalveolar lavage (BAL) neutrophils. Mice were infected with 2 × 105 Pseudomonas aeruginosa (PsA) and euthanized 16 h later to obtain BAL. From the total BAL cell population, a neutrophil gate was selected using scatter properties [forward scatter (FSC), side scatter (SSC)], singlets were identified, and Ly6G+ cells were used for analysis. B: uptake of pHrodo-PsA by neutrophils. Compared with negative controls incubated at 4°C, samples at 37°C showed uptake of pHrodo-PsA as indicated by a shift in FL-1 fluorescence. Phagocytosis was quantified by mean fluorescence intensity (MFI) and by the percentage of neutrophils positive for pHrodo-PsA (% phagocytosis). C: fluorescence images demonstrating uptake of pHrodo-PsA by LPS-elicited Ly6G+ neutrophils (top). Phagocytosis was inhibited by 10 μM cytochalasin D (Cyto D; bottom). D: raw flow cytometry traces of pHrodo-PsA uptake by neutrophils. Fluorescence of pHrodo-PsA alone, neutrophils alone, and pHrodo-PsA plus neutrophils at 4°C was negligible. At 37°C, phagocytosis was dose-dependently inhibited by cytochalasin D. Quantification of pHrodo-PsA uptake by MFI (E) and % phagocytosis (F). G: raw flow cytometry traces of fluor-PsA uptake by neutrophils. Unphagocytosed fluor-PsA was highly fluorescent, leading to a strong signal in 4°C negative controls due to substrate adherence. At 37°C, phagocytosis was inhibited by cytochalasin D. Quantification of fluor-PsA uptake by MFI (H) and % phagocytosis (I) showed diminished dynamic range compared with pHrodo-PsA. Error bars represent SE. MOI, multiplicity of infection.
Of note, we found that most neutrophils were contained within a population that was readily identifiable using scatter properties alone (Supplemental Fig. S2). Indeed, simply gating on this neutrophil subplot yielded results that were nearly indistinguishable from those obtained when gating on Ly6G+ cells (Supplemental Fig. S3). Therefore, the phagocytosis protocol presented here may be further streamlined by omitting Ly6G staining, allowing for one-color flow cytometry analysis.
Given the complex interactions between live P. aeruginosa and host immune cells during lung infection (including the inhibitory and cytotoxic effects of pseudomonal virulence factors on neutrophils), we next sought to simplify the pneumonia model (27). To this end, we utilized LPS to elicit pneumonia, reasoning that the resulting neutrophils would provide a more generalizable model for phagocytosis ex vivo than those in pseudomonal infection. In this LPS-stimulated pneumonia model, we used fluorescence microscopy to demonstrate uptake of pHrodo-PsA by Ly6G+ neutrophils ex vivo (Fig. 2C). We also demonstrated abrogation of uptake by cytochalasin D, an inhibitor of actin polymerization and therefore of phagocytosis (Fig. 2C). We then quantified uptake of pHrodo-PsA using flow cytometry. This showed a dose-dependent inhibition of phagocytosis by cytochalasin D (red traces, Fig. 2D) and robust increases in both MFI (65-fold increase) and percentage of positivity (90% difference) compared with the 4°C control (Fig. 2, E and F).
To further illustrate the specificity of pHrodo-PsA for phagocytosis over adherence, we synthesized a control substrate that does fluoresce outside the neutrophil: fluorescein-conjugated P. aeruginosa (fluor-PsA). The fluorescent properties of these two substrates are shown in the top traces in Fig. 2, D and G, respectively: unphagocytosed pHrodo-PsA shows negligible fluorescence whereas fluor-PsA is extremely bright. As a consequence, there is minimal signal when pHrodo-PsA is incubated with neutrophils at 4°C (Fig. 2D, third trace), but a strong signal from fluor-PsA under the same conditions (Fig. 2G, third trace). This fluorescence, which derives from adherent fluor-PsA, results in severely diminished dynamic range of the assay, with only a fivefold increase in MFI (Fig. 2H) and 47% increase in positive cells (Fig. 2I) upon incubating at 37°C.
In summary, these results describe straightforward synthesis of a pH-dependent fluorescent bacterial substrate, which enables highly specific assessment of phagocytosis by bronchoalveolar neutrophils using a flow cytometry-based assay marked by technical simplicity and expansive dynamic range.
Ex vivo NETosis assay.
Next, we sought to develop a method to quantify NET formation by bronchoalveolar neutrophils ex vivo. To do so, we adapted a plate-based fluorescence kinetic assay used previously to assess NETosis by blood neutrophils (18, 36). In this approach, neutrophils are incubated with a NETotic stimulus and a cell-impermeable DNA dye such as Sytox Orange, which fluoresces upon binding to extruded chromatin. As such, this assay is not specific for NET detection in sensu stricto, as it detects release of DNA, not complexes of DNA and NET proteins. Nevertheless, its ease and high throughput make it a valuable surrogate for studying NETosis in vitro.
To demonstrate the applicability of this technique to bronchoalveolar neutrophils, we stimulated BAL cells with live P. aeruginosa or PMA in the presence of Sytox Orange. Representative images at 2.5 h are shown in Fig. 3A. Notably, unstimulated neutrophils demonstrated uptake of Sytox dye at this timepoint, leading to modest but appreciable staining of their characteristic multilobulated nuclei. As reported previously, PMA induced nuclear decondensation and cloud-like DNA release (29), whereas P. aeruginosa elicited DNA extrusion in strand-like structures (25). Notably, P. aeruginosa was also stained by Sytox dye.
Fig. 3.

Ex vivo NETosis assay. Mice were administered LPS and euthanized 12 h later for bronchoalveolar lavage (BAL). A: fluorescent images of BAL neutrophils undergoing NETosis. Unstimulated neutrophils stained with Sytox Orange demonstrated characteristic multilobed nuclei. Neutrophils incubated with 1 μM phorbol myristate acetate (PMA) or 100 multiplicity of infection (MOI) live Pseudomonas aeruginosa (PsA) for 2.5 h formed abundant neutrophil extracellular traps (NETs), as indicated by extracellular DNA staining. B: kinetic fluorescence assay for quantifying NETosis from BAL neutrophils. Lysis with saponin defined maximal DNA release, whereas stimulation with PMA or P. aeruginosa stimulated NETosis. P. aeruginosa itself spontaneously released low levels of DNA. C: DNA release in response to PMA was dose-dependently inhibited by the NADPH oxidase (NOX) inhibitor, diphenyleneiodonium (DPI). Error bars represent SE.
The kinetics of DNA release by control and stimulated neutrophils are represented in Fig. 3B. Consistent with the images in Fig. 3A, both unstimulated cells and P. aeruginosa alone produce appreciable fluorescence. However, neutrophils incubated with NETotic stimuli release abundant DNA to generate substantially stronger signals (blue and green traces).
Next, we sought to confirm that the DNA release measured in this assay was dependent on reactive oxygen species (ROS) and therefore more likely reflective of NETosis than necrotic cell death. To this end, we tested whether NADPH oxidase (NOX) inhibition would abrogate DNA release and hence the fluorescent signal. PMA was used as a stimulus because it is known to trigger NETosis exclusively through the ROS-dependent suicidal pathway (34). As shown in Fig. 3C, the NOX inhibitor diphenyleneiodonium (DPI) indeed decreased DNA release stimulated by PMA in a dose-dependent manner, with an IC50 very similar to reported values (13). Taken together, these results demonstrate that kinetic assessment of DNA release from bronchoalveolar neutrophils represents a faithful surrogate for studying NETosis ex vivo.
In situ NETosis assay.
Lastly, we sought to develop a semiquantitative biochemical method to measure bronchoalveolar NET formation in vivo (Fig. 4). As a model, we used three different severities of pseudomonal pneumonia, reasoning that NET production would correlate with escalating severity, as indicated by increasing alveolar leukocytosis, hemorrhage, and damage (Fig. 5, A–C) (25). Indeed, commonly used surrogates for NETs, including NE and cell-free DNA, rose as expected (Fig. 5, D and E). Interestingly, we found that NE activity (as measured by cleavage of a fluorogenic NE substrate) also increased with severity of infection, but this was partly attributable to cleavage by an elastase expressed by P. aeruginosa itself (LasB, Supplemental Fig. S4) Thus, enzymatic assays for NE activity should be interpreted with caution in models of pneumonia elicited by elastase-secreting pathogens.
Fig. 4.
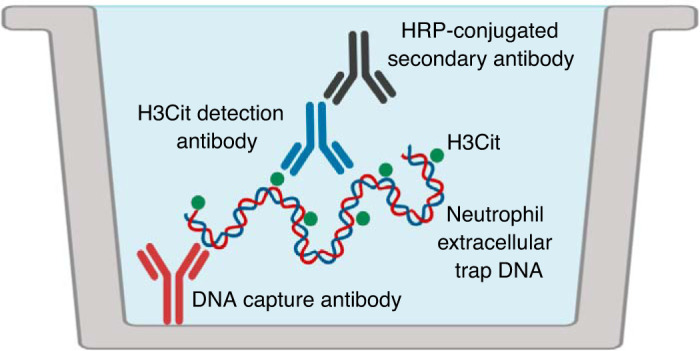
Schematic overview of in situ NETosis assay. Pneumonic bronchoalveolar lavage (BAL) is evaluated for neutrophil extracellular trap (NET) content using an ELISA-based method. This assay utilizes an anti-DNA capture antibody, which binds NETs, and an anti-citrullinated histone H3 (H3Cit) antibody to detect this NET-specific protein. A horseradish peroxidase (HRP)-conjugated secondary antibody against the H3Cit antibody is used to generate a measurable assay signal.
Fig. 5.

In situ NETosis assay. Mice were infected with three inocula of live Pseudomonas aeruginosa as indicated and euthanized 16 h later for bronchoalveolar lavage (BAL). Inflammation and lung damage scaled with severity of infection as revealed by BAL white blood cells (WBCs) (A), red blood cells (RBCs) (B), and total protein (C). Surrogates of neutrophil extracellular traps (NETs), including cell-free DNA (D, measured using PicoGreen) and neutrophil elastase (NE) (E, measured by ELISA) also increased with severity. Elevated DNA-citrullinated histone H3 (H3Cit) complexes (F, measured by ELISA) specifically demonstrated NETosis in situ. G: control experiments demonstrating specificity of DNA-H3Cit ELISA for NETs. PBS, pure genomic DNA (isolated from salmon sperm), and BAL from mice administered PBS intratracheally contained no detectable NETs (black bars, left). The ELISA signal was dependent on capture and detection antibody (black bars, right) and on DNA integrity, as shown via degradation with Benzonase at concentrations ranging from 5 × 10−1 to 5 × 10−5 U/μL (gray bars). H: PicoGreen lacks specificity for NETs, as it detects both NET-associated DNA and pure genomic DNA (second black bar from left). Ordinary one-way ANOVA was used to test for significance in A–F: ****P < 0.0001; *P < 0.05. Technical singlicates were used in G and H; data shown is representative of multiple repetitions of the experiment. AU, arbitrary units; CFUs, colony-forming units; OD, optical density.
To specifically detect NETs, we developed an ELISA based on capture of DNA and detection of DNA-associated H3Cit, a highly specific marker for NETosis (Fig. 4). As shown in Fig. 5F, DNA-H3Cit complexes were effectively quantified in BAL using this ELISA and correlated well with severity of pneumonia. To further validate this method, we demonstrated that the ELISA signal was dependent on the presence of capture and detection antibody and the integrity of the DNA backbone of NETs (confirmed by nuclease-mediated digestion of DNA, Fig. 5G). Unlike the more commonly used method of quantifying cell-free DNA by PicoGreen fluorescence (Fig. 5H), the DNA-H3Cit ELISA did not detect pure genomic DNA (Fig. 5G) and is therefore less likely to detect DNA released during non-NETotic forms of cell death (e.g., necrosis). Finally, of practical value, NETs were stable to freezing and thawing, with no decrement in signal by DNA-H3Cit ELISA (Supplemental Fig. S5).
DISCUSSION
In these studies, we have developed and characterized a set of techniques for quantifying neutrophil effector functions in murine models of pneumonia. Aiming to maximize accessibility of these assays to nonspecialized laboratories, we have emphasized technical ease and cost-effectiveness. Additionally, we have sought to ensure translatability to human studies by utilizing neutrophils from the bronchoalveolar compartment, which is easily sampled in patients by BAL.
Consistent with prior observations (25), the neutrophil fraction of WBCs in pneumonic BAL was around 90%, which is comparable to that achieved using density gradient centrifugation of mouse blood [86% reported in Siemsen et al. (35)]. Thus, a simple protocol of OP aspiration followed by BAL offers access to bronchoalveolar neutrophils that are directly amenable to downstream functional studies without the technically demanding step of purification. Notably, the flow cytometry-based phagocytosis assay described here further precludes the need for purification, as analysis is restricted to neutrophils by gating on Ly6G+ cells. It is important to recognize, however, that whole BAL contains additional cell types, including red blood cells (RBCs) and platelets (Fig. 1A, left), which are known to affect neutrophil function (7). For experiments requiring pure WBCs, RBCs and platelets may be removed using standard methodology, e.g., osmotic RBC lysis. Alternatively, Ly6G+ neutrophils may be purified using techniques such as magnetic-activated cell sorting (38).
The dose and timing of intrapulmonary delivery of inflammatory stimuli are important considerations. Here, we selected the 12-h timepoint to collect BAL after LPS instillation because it corresponds with the time of maximal neutrophil recruitment (33) and the 16-h timepoint after P. aeruginosa infection to allow adequate time for NETosis to proceed and yet minimize mouse morbidity (25). Utilizing milder doses and/or later timepoints, for instance, may allow examination of neutrophil function during the resolution phase of inflammation, at which point effector responses may be less robust. Thus, treatment dose and timing should be tailored according to experimental aims.
Throughout, we have highlighted the importance of studying neutrophils in the mouse to take advantage of disease models and genetic knockouts. We have also emphasized the use of bronchoalveolar cells, which best represent the phenotype of neutrophils during lung inflammation. However, it is important to note that these methods cannot completely replace studies using human neutrophils, which are necessary to confirm the relevance of preclinical findings to human health and disease (37). Similarly, they cannot replace the use of quiescent neutrophils isolated from blood or bone marrow in the interrogation of certain aspects of neutrophil biology, including transmigration, chemotaxis, and priming (3, 16, 43). As such, the techniques presented herein should be viewed as a complement to human blood neutrophil experiments, with particular relevance to the study of lung inflammation in situ.
The phagocytosis assay described here is high-throughput, robust in dynamic range owing to negligible background signal, and simple enough to perform on any benchtop flow cytometer. At most, two colors are needed, allowing for use of the fluorescein and Cy5/APC channels, which have almost no spectral overlap. Also, unlike existing flow cytometry-based techniques, which use pathogen-specific antibodies to identify adherent substrate (30), ours exploits an essential mechanism of phagocytosis (phagosomal acidification) to distinguish between substrate uptake and adherence. As such, our approach is not only simpler to perform and analyze, but also more physiologically representative of phagocytosis. Finally, the use of pHrodo-STP ester, a commercially available bioconjugatable dye, allows investigators to attach the compound to virtually any culturable microbe at low cost to create a customized substrate from their pathogen of interest. For experiments not requiring specific pathogens, ready-made pH-sensitive phagocytic substrates are available, although at higher cost (20). It should also be noted that studies on neutrophil interaction with live pathogens may require alternative strategies, such as the use of fluorescent-protein expressing strains.
As evidence for the role of NETs in lung disease grows, it is increasingly important that pulmonary researchers gain facility with NETosis assays and also recognize their limitations. For instance, numerous protocols have been established for visualizing NETs in fixed lung tissue by immunofluorescence microscopy, but image-based quantification remains a challenge (9). Immunoblotting for H3Cit within tissue homogenates offers a simple means of quantifying whole lung NET content, but it cannot distinguish between the vascular, interstitial, and bronchoalveolar compartments. Microscopy- and flow cytometry-based techniques have been developed for assessing active production of NETs from BAL neutrophils ex vivo, but these are technically demanding and do not reflect in situ NET formation within the airspace (25, 39, 45). Measuring cell-free DNA in BAL using fluorescent dyes (e.g., PicoGreen) represents a simple way of quantifying in situ NETosis, but it cannot distinguish NETs from necrotic DNA release. Alternatively, measurement of protein components within NETs, such as NE, may be used as a surrogate for in situ NETosis, either by ELISA or enzymatic assays. However, neither are specific, as ELISA-based detection of NE in BAL may indicate release from degranulating neutrophils, whereas enzymatic assays may detect activity of bacterial elastases such as pseudomonal LasB, as shown above (Supplemental Fig. S4).
The DNA-H3Cit ELISA described here solves many of these problems, enabling specific and quantitative detection of DNA-NET protein complexes from BAL. A second layer of specificity derives from targeting H3Cit, which is uniquely found in NETs. This assay of in situ NET formation may be complemented by our ex vivo assay (Fig. 3) to provide a powerful quantitative toolset for assessing NETosis in the murine lung. It should be noted, however, that generation of H3Cit via histone citrullination occurs only during peptidylarginine deiminase-4 (PAD4)-dependent NETosis. As such, assessment of PAD4-independent NETosis in situ may require the use of ELISAs that detect additional NET components, such as the NE-DNA ELISAs reported by Raderbecker et al. (33) and Lefrançais et al. (25) (Supplemental Table S1).
In summary, the techniques described here offer a facile means of interrogating NETosis in situ within the murine lung and obtaining pure, highly functional, and fully activated bronchoalveolar neutrophils for ex vivo assays of NETosis and phagocytosis. We hope that these methods will facilitate the study of neutrophil effector functions in mouse models, yielding further insights into these central mechanisms in lung immunity and disease.
GRANTS
This work was supported in part by National Heart, Lung, and Blood Institute Grant T32-HL007778 (to S.G.); Veterans Health Administration Health Services Research and Development Grant 1I01BX004661-01 (to C.S.D.); and National Heart, Lung, and Blood Institute Grant 5R01HL126094-05 (to C.S.D.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.G., Y.S., G.M.Y., and A.J.C. conceived and designed research; S.G., Y.S., G.M.Y., A.J.C., D.A.T., and T.M. performed experiments; S.G., Y.S., G.M.Y., R.H., A.J.C., D.A.T., and T.M. analyzed data; S.G., Y.S., G.M.Y., R.H., and A.J.C. interpreted results of experiments; S.G., Y.S., G.M.Y., and R.H. prepared figures; S.G., Y.S., G.M.Y., and A.J.C. drafted manuscript; S.G., Y.S., G.M.Y., R.H., A.J.C., D.A.T., T.M., L.S., and C.S.D.C. edited and revised manuscript; S.G., Y.S., G.M.Y., R.H., A.J.C., D.A.T., T.M., L.S., and C.S.D.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We appreciate Dr. Zahra Toossi’s critical reading of the manuscript.
REFERENCES
- 1.Abu Abed U, Brinkmann V. Immunofluorescence labelling of human and murine neutrophil extracellular traps in paraffin-embedded tissue. J Vis Exp 151: e60115, 2019. doi: 10.3791/60115. [DOI] [PubMed] [Google Scholar]
- 2.Allen IC. The utilization of oropharyngeal intratracheal PAMP administration and bronchoalveolar lavage to evaluate the host immune response in mice. J Vis Exp 86: 51391, 2014. doi: 10.3791/51391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borregaard N. Neutrophils, from marrow to microbes. Immunity 33: 657–670, 2010. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 4.Brinkmann V. Neutrophil extracellular traps in the second decade. J Innate Immun 10: 414–421, 2018. doi: 10.1159/000489829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535, 2004. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 6.Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 133: 2178–2185, 2019. doi: 10.1182/blood-2018-11-844530. [DOI] [PubMed] [Google Scholar]
- 7.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z, Looney MR. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest 122: 2661–2671, 2012. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conway Morris A, Kefala K, Wilkinson TS, Dhaliwal K, Farrell L, Walsh T, Mackenzie SJ, Reid H, Davidson DJ, Haslett C, Rossi AG, Sallenave JM, Simpson AJ. C5a mediates peripheral blood neutrophil dysfunction in critically ill patients. Am J Respir Crit Care Med 180: 19–28, 2009. doi: 10.1164/rccm.200812-1928OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Buhr N, von Köckritz-Blickwede M. How neutrophil extracellular traps become visible. J Immunol Res 2016: 4604713, 2016. doi: 10.1155/2016/4604713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deniset JF, Kubes P. Neutrophil heterogeneity: bona fide subsets or polarization states? J Leukoc Biol 103: 829–838, 2018. doi: 10.1002/JLB.3RI0917-361R. [DOI] [PubMed] [Google Scholar]
- 11.Donnelly LE, Barnes PJ. Defective phagocytosis in airways disease. Chest 141: 1055–1062, 2012. doi: 10.1378/chest.11-2348. [DOI] [PubMed] [Google Scholar]
- 12.Floyd M, Winn M, Cullen C, Sil P, Chassaing B, Yoo DG, Gewirtz AT, Goldberg JB, McCarter LL, Rada B. Swimming motility mediates the formation of neutrophil extracellular traps induced by flagellated Pseudomonas aeruginosa. PLoS Pathog 12: e1005987, 2016. doi: 10.1371/journal.ppat.1005987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gatto GJ Jr, Ao Z, Kearse MG, Zhou M, Morales CR, Daniels E, Bradley BT, Goserud MT, Goodman KB, Douglas SA, Harpel MR, Johns DG. NADPH oxidase-dependent and -independent mechanisms of reported inhibitors of reactive oxygen generation. J Enzyme Inhib Med Chem 28: 95–104, 2013. doi: 10.3109/14756366.2011.636360. [DOI] [PubMed] [Google Scholar]
- 14.Gautam S, Cohen AJ, Stahl Y, Valda Toro P, Young GM, Datta R, Yan X, Ristic NT, Bermejo SD, Sharma L, Restrepo M, Dela Cruz CS. Severe respiratory viral infection induces procalcitonin in the absence of bacterial pneumonia. In press. doi: 10.1136/thoraxjnl-2020-214896. [DOI] [PubMed] [Google Scholar]
- 15.Gautam S, Sharma L, Dela Cruz CS. Personalizing the management of pneumonia. Clin Chest Med 39: 871–900, 2018. doi: 10.1016/j.ccm.2018.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Giacalone VD, Margaroli C, Mall MA, Tirouvanziam R. Neutrophil adaptations upon recruitment to the lung: new concepts and implications for homeostasis and disease. Int J Mol Sci 21: 851, 2020. doi: 10.3390/ijms21030851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease: too little, too late or too much, too soon? Am J Respir Cell Mol Biol 48: 531–539, 2013. doi: 10.1165/rcmb.2012-0492TR. [DOI] [PubMed] [Google Scholar]
- 18.Hoppenbrouwers T, Autar AS, Sultan AR, Abraham TE, van Cappellen WA, Houtsmuller AB, van Wamel WJ, van Beusekom HM, van Neck JW, de Maat MP. In vitro induction of NETosis: comprehensive live imaging comparison and systematic review. PLoS One 12: e0176472, 2017. doi: 10.1371/journal.pone.0176472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itou T, Collins LV, Thorén FB, Dahlgren C, Karlsson A. Changes in activation states of murine polymorphonuclear leukocytes (PMN) during inflammation: a comparison of bone marrow and peritoneal exudate PMN. Clin Vaccine Immunol 13: 575–583, 2006. doi: 10.1128/CVI.13.5.575-583.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Juss JK, House D, Amour A, Begg M, Herre J, Storisteanu DML, Hoenderdos K, Bradley G, Lennon M, Summers C, Hessel EM, Condliffe A, Chilvers ER. Acute respiratory distress syndrome neutrophils have a distinct phenotype and are resistant to phosphoinositide 3-kinase inhibition. Am J Respir Crit Care Med 194: 961–973, 2016. doi: 10.1164/rccm.201509-1818OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karsten CB, Mehta N, Shin SA, Diefenbach TJ, Slein MD, Karpinski W, Irvine EB, Broge T, Suscovich TJ, Alter G. A versatile high-throughput assay to characterize antibody-mediated neutrophil phagocytosis. J Immunol Methods 471: 46–56, 2019. doi: 10.1016/j.jim.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JH, Chaurasia AK, Batool N, Ko KS, Kim KK. Alternative enzyme protection assay to overcome the drawbacks of the gentamicin protection assay for measuring entry and intracellular survival of Staphylococci. Infect Immun 87: e00119-19, 2019. doi: 10.1128/IAI.00119-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krishnamoorthy N, Douda DN, Brüggemann TR, Ricklefs I, Duvall MG, Abdulnour RE, Martinod K, Tavares L, Wang X, Cernadas M, Israel E, Mauger DT, Bleecker ER, Castro M, Erzurum SC, Gaston BM, Jarjour NN, Wenzel S, Dunican E, Fahy JV, Irimia D, Wagner DD, Levy BD; National Heart, Lung, and Blood Institute Severe Asthma Research Program-3 Investigators . Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci Immunol 3: eaao4747, 2018. doi: 10.1126/sciimmunol.aao4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Law SM, Gray RD. Neutrophil extracellular traps and the dysfunctional innate immune response of cystic fibrosis lung disease: a review. J Inflamm (Lond) 14: 29, 2017. doi: 10.1186/s12950-017-0176-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 3: e98178, 2018. doi: 10.1172/jci.insight.98178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, Catz SD. Neutrophils: new insights and open questions. Sci Immunol 3: eaat4579, 2018. doi: 10.1126/sciimmunol.aat4579. [DOI] [PubMed] [Google Scholar]
- 27.Malhotra S, Hayes D Jr, Wozniak DJ. Cystic fibrosis and Pseudomonas aeruginosa: the host-microbe interface. Clin Microbiol Rev 32: e00138–18, 2019. doi: 10.1128/CMR.00138-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manfredi AA, Ramirez GA, Rovere-Querini P, Maugeri N. The neutrophil’s choice: phagocytose vs make neutrophil extracellular traps. Front Immunol 9: 288, 2018. doi: 10.3389/fimmu.2018.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neubert E, Meyer D, Rocca F, Günay G, Kwaczala-Tessmann A, Grandke J, Senger-Sander S, Geisler C, Egner A, Schön MP, Erpenbeck L, Kruss S. Chromatin swelling drives neutrophil extracellular trap release. Nat Commun 9: 3767, 2018. doi: 10.1038/s41467-018-06263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nordenfelt P. Quantitative assessment of neutrophil phagocytosis using flow cytometry. Methods Mol Biol 1124: 279–289, 2014. doi: 10.1007/978-1-62703-845-4_18. [DOI] [PubMed] [Google Scholar]
- 31.Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18: 134–147, 2018. doi: 10.1038/nri.2017.105. [DOI] [PubMed] [Google Scholar]
- 32.Porto BN, Stein RT. Neutrophil extracellular traps in pulmonary diseases: too much of a good thing? Front Immunol 7: 311, 2016. doi: 10.3389/fimmu.2016.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radermecker C, Sabatel C, Vanwinge C, Ruscitti C, Maréchal P, Perin F, Schyns J, Rocks N, Toussaint M, Cataldo D, Johnston SL, Bureau F, Marichal T. Locally instructed CXCR4hi neutrophils trigger environment-driven allergic asthma through the release of neutrophil extracellular traps. Nat Immunol 20: 1444–1455, 2019. doi: 10.1038/s41590-019-0496-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, Noppen S, Delforge M, Willems J, Vandenabeele P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 21: 290–304, 2011. doi: 10.1038/cr.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siemsen DW, Malachowa N, Schepetkin IA, Whitney AR, Kirpotina LN, Lei B, Deleo FR, Quinn MT. Neutrophil isolation from nonhuman species. Methods Mol Biol 1124: 19–37, 2014. doi: 10.1007/978-1-62703-845-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sil P, Yoo DG, Floyd M, Gingerich A, Rada B. High throughput measurement of extracellular DNA release and quantitative NET formation in human neutrophils in vitro. J Vis Exp 2016: 2016. doi: 10.3791/52779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stackowicz J, Jönsson F, Reber LL. Mouse models and tools for the in vivo study of neutrophils. Front Immunol 10: 3130, 2020. doi: 10.3389/fimmu.2019.03130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swamydas M, Luo Y, Dorf ME, Lionakis MS. Isolation of mouse neutrophils. Curr Protoc Immunol 110: 3.20.1–3.20.15, 2015. doi: 10.1002/0471142735.im0320s110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tripathi JK, Sharma A, Sukumaran P, Sun Y, Mishra BB, Singh BB, Sharma J. Oxidant sensor cation channel TRPM2 regulates neutrophil extracellular trap formation and protects against pneumoseptic bacterial infection. FASEB J 32: 6848–6859, 2018. doi: 10.1096/fj.201800605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Twaddell SH, Baines KJ, Grainge C, Gibson PG. The emerging role of neutrophil extracellular traps in respiratory disease. Chest 156: 774–782, 2019. doi: 10.1016/j.chest.2019.06.012. [DOI] [PubMed] [Google Scholar]
- 41.Uddin M, Watz H, Malmgren A, Pedersen F. NETopathic inflammation in chronic obstructive pulmonary disease and severe asthma. Front Immunol 10: 47, 2019. doi: 10.3389/fimmu.2019.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vassallo A, Wood AJ, Subburayalu J, Summers C, Chilvers ER. The counter-intuitive role of the neutrophil in the acute respiratory distress syndrome. Br Med Bull 131: 43–55, 2019. doi: 10.1093/bmb/ldz024. [DOI] [PubMed] [Google Scholar]
- 43.Vogt KL, Summers C, Chilvers ER, Condliffe AM. Priming and de-priming of neutrophil responses in vitro and in vivo. Eur J Clin Invest 48, Suppl 2: e12967, 2018. doi: 10.1111/eci.12967. [DOI] [PubMed] [Google Scholar]
- 44.Wigerblad G, Kaplan MJ. NETs spread ever wider in rheumatic diseases. Nat Rev Rheumatol 16: 73–74, 2020. doi: 10.1038/s41584-019-0352-1. [DOI] [PubMed] [Google Scholar]
- 45.Zhao W, Fogg DK, Kaplan MJ. A novel image-based quantitative method for the characterization of NETosis. J Immunol Methods 423: 104–110, 2015. doi: 10.1016/j.jim.2015.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]