

Abstract
Balancers are rearranged chromosomes used in Drosophila melanogaster to maintain deleterious mutations in stable populations, preserve sets of linked genetic elements and construct complex experimental stocks. Here, we assess the phenotypes associated with breakpoint-induced mutations on commonly used third chromosome balancers and show remarkably few deleterious effects. We demonstrate that a breakpoint in p53 causes loss of radiation-induced apoptosis and a breakpoint in Fucosyltransferase A causes loss of fucosylation in nervous and intestinal tissue—the latter study providing new markers for intestinal cell identity and challenging previous conclusions about the regulation of fucosylation. We also describe thousands of potentially harmful mutations shared among X or third chromosome balancers, or unique to specific balancers, including an Ankyrin 2 mutation present on most TM3 balancers, and reiterate the risks of using balancers as experimental controls. We used long-read sequencing to confirm or refine the positions of two inversions with breakpoints lying in repetitive sequences and provide evidence that one of the inversions, In(2L)Cy, arose by ectopic recombination between foldback transposon insertions and the other, In(3R)C, cleanly separates subtelomeric and telomeric sequences and moves the subtelomeric sequences to an internal chromosome position. In addition, our characterization of In(3R)C shows that balancers may be polymorphic for terminal deletions. Finally, we present evidence that extremely distal mutations on balancers can add to the stability of stocks whose purpose is to maintain homologous chromosomes carrying mutations in distal genes. Overall, these studies add to our understanding of the structure, diversity and effectiveness of balancer chromosomes.
Keywords: balancer chromosomes, chromosomal inversions, p53, Ankyrin 2, Fucosyltransferase A
Balancer chromosomes occupy an important place in the Drosophila melanogaster genetic toolkit. Their extensive rearrangements function both to inhibit meiotic recombination and, when recombination does occur, prevent the recovery of recombinant chromosomes. Usually, the rearrangements that make up the balancer have two or three breakpoints that result in the inversion of chromosomal segments. The frequency of crossing over between a balancer and a normal-sequence homolog is low when the balancer breakpoints are closely linked, but, when the breakpoints are more distantly spaced, two-strand double crossovers can occur and exchange stretches of DNA. Likewise, single crossovers can occur distal to the distalmost breakpoints on chromosome arms if the breakpoints are not close enough to the telomeres. In addition, most balancers carry at least one recessive lethal or sterile mutation to prevent them from outcompeting homologous chromosomes in stock populations, and at least one dominant visible mutation with an easily scored phenotype so they may be tracked in crosses (Miller et al. 2019).
Balancers are used most often to maintain stable stocks carrying recessive lethal or sterile mutations and to assure that sets of alleles on homologous chromosomes remain linked together. They are also used in nearly all crosses that generate complex combinations of chromosomes. Because of their incredible usefulness, balancers have been tremendously important in the development of Drosophila melanogaster as a genetic model organism.
Despite the widespread use of balancers, the genomic positions of many of the breakpoints of the most commonly used balancers were identified only recently, with many of the breakpoints found to lie within protein-coding genes (Miller et al. 2016b, 2016a, 2018a; Ghavi-Helm et al. 2019). Miller et al. (2018a) assessed the phenotypic consequences of the breakpoints on second chromosome balancers by complementation testing the balancers against chromosomal deletions for breakpoint regions and showed that most breakpoints were not associated with severely deleterious phenotypes, but that the disruption of some genes by breakpoints caused recessive lethality or sterility. For example, the 22E breakpoint on the second chromosome balancer SM5 disrupts dachsous, resulting in lethality with escapers having shortened appendages, and the 22A breakpoint on SM1, SM5 and SM6a disrupts no individualized sperm resulting in male sterility.
In addition to identifying inversion breakpoints, sequencing the second chromosome balancers CyO, SM5 and SM6a identified potentially damaging missense, splice site and nonsense polymorphisms (Miller et al. 2018a). Some polymorphisms were shared by some, but not all of the balancers sequenced. For example, all SM5 and SM6a balancers sequenced have a splice-site mutation in asteroid, a gene involved in photoreceptor and wing development, that was not observed on any CyO balancers. Other polymorphisms appeared to be unique to the balancer from a single stock. For example, the SM6a balancer from one particular stock carries a nonsense mutation in the pickpocket 11 sodium channel gene.
Information on breakpoints and background polymorphisms on balancers is important because it can guide investigators in the choice of balancers for maintaining stocks with mutations in breakpoint-associated genes. It can also alert researchers to potential dose-dependent effects of heterozygous balancer-borne mutations and potential dominant interactions between mutations on balancers and other chromosomes. Moreover, breakpoint information has revealed previously unknown duplicated chromosomal segments such as the region containing 117 protein-coding genes present in two copies on all SM5 balancers (Miller et al. 2018a).
In this study, we use information from sequencing the third chromosome balancers TM3, TM6 and TM6B (Miller et al. 2016a) to assess the phenotypic effects of breakpoint-associated gene disruptions. We present a survey of the breakpoints on these balancers for severe deleterious phenotypes and demonstrate that breakpoints in the p53 and FucTA genes are associated with more subtle effects that may, nevertheless, be important in many experimental situations. Similar to the previous analysis of second chromosome balancers (Miller et al. 2018a), we identify sequence polymorphisms on X and third chromosome balancers that may affect protein structure or gene expression. We also provide a characterization of one specific background mutation in the Ankyrin 2 gene present on many TM3 balancers.
Previous characterizations of second and third chromosome balancers were unable to localize the breakpoints of two component inversions at single-nucleotide resolution because the breakpoints were associated with repetitive sequences (Miller et al. 2016a, 2018a). Here, we refine the genomic positions of the recently mapped In(3R)C inversion breakpoints (Ghavi-Helm et al. 2019) using long-read, single-molecule sequencing and show that the data provide evidence that balancers may carry terminal deletions. We similarly confirm the genomic positions of the In(2L)Cy breakpoints determined by the same investigators and show the inversion arose by ectopic recombination between foldback transposons. Finally, we use the genetic characterization of one particular CyO balancer to show that deleterious mutations at extremely distal positions can explain the unexpected stability of many stocks.
Materials and Methods
Complementation analyses
Fly crosses were made on standard medium, reared under routine conditions and evaluated by customary standards (details provided upon request). In general, complementation results were based on samples exceeding 50 progeny and sterility was judged in tests of >10 progeny. Genomic coordinates are given in terms of the Release 6 assembly. Table S1 provides a list of stocks used and our sources.
Apoptosis assay
As described previously (Qi and Calvi 2016), young adult females were conditioned on wet yeast for three days and then exposed to 40 Gy of gamma irradiation from a cesium source. Four hours after irradiation, ovaries were dissected and stained by the TUNEL method (In Situ Cell Death Detection Kit, TMR red, version 11 (12 156 792 910; Roche, Basel, Switzerland)) and DAPI. Percent of apoptotic cells was determined by scoring TUNEL staining of cells with DAPI-stained nuclei in flattened follicle surfaces, excluding deformed cells at follicle edges and the polar follicle cells that have been shown previously to undergo p53-independent, developmentally programmed cell death (Besse and Pret 2003; Mehrotra et al. 2008).
Antibody staining
Brains from adults aged 4–6 days were immunostained as described in Kahsai et al. (2010). Briefly, brains were dissected in sodium phosphate-buffered saline with 0.1% Triton X-100 (PBST), fixed in ice-cold 4% paraformaldehyde (Electron Microscopy Services) in sodium phosphate buffer for two hours, rinsed in PBST and incubated with affinity-purified Alexa Fluor 488-conjugated rabbit anti-Horseradish Peroxidase antibody (1:200 dilution) and the mouse nc82 monoclonal antibody (1:10) for 72 hr at 4°. An overnight secondary antibody incubation at 4° with Alexa Fluor 568-conjugated goat anti-mouse antibody (1:1000) was followed by a PBST rinse, a final wash in PBS and mounting in 2.5% (w/v) DABCO (1,4-Diazabicyclo[2.2.2]-octane, Aldrich). The nc82 antibody detects the protein Bruchpilot and provided a counterstain to define overall brain structure by highlighting presynaptic terminals (Rein et al. 1999; Wagh et al. 2006). Images were collected on a Leica SP5 scanning confocal microscope at the Indiana University Light Microscopy Imaging Center. At least 15 brains were analyzed for each experiment. Images that required comparisons were acquired using the same settings and processed simultaneously using Adobe Photoshop CC.
Adult intestines were immunostained as described in Buddika et al. (2020). Briefly, 5–7 day old females were dissected in ice-cold phosphate-buffered saline, and intestines were fixed with ice-cold 4% paraformaldehyde (Electron Microscopy Services) in sodium phosphate buffer for 45 min, rinsed in PBST, blocked with 0.5% bovine serum albumin in sodium phosphate buffer for 45 min and immunostained overnight with the primary rabbit anti-RFP (1:1000) and mouse anti-Prospero (1:100) antibodies in experiments verifying the M{mira-His2A.mCherry.HA} reporter. Alexa Fluor 568-conjugated goat anti-rabbit and Alexa Fluor 633-conjugated goat anti-mouse antibodies (1:1000) were used for secondary staining. In experiments to detect protein fucosylation, we used the same anti-RFP and anti-Pros primary antibodies in the primary incubation with Alexa Fluor 647-conjugated goat anti-HRP (1:500), Alexa Fluor 488-conjugated goat anti-mouse (1:1000) and Alexa Fluor 568-conjugated goat anti-rabbit (1:1000) antibodies in the secondary incubation. Antibodies for detecting Prospero were omitted in some experiments. After antibody incubations, samples were washed, counterstained with DAPI (1:10000) and mounted in Vectashield medium. Images were collected on a Leica SP8 scanning confocal microscope at the Indiana University Light Microscopy Imaging Center. Samples to be compared were acquired under the same settings and processed simultaneously using Adobe Photoshop CC. See Reagent Table (Table S1) for antibody sources.
Transgene construction
M{mira-His2A.mCherry.HA} expresses sequences encoding a nuclear-localized His2A histone protein fused to a catamer of four mCherry coding sequences and the sequence for a C-terminal hemagglutinin (HA) tag. The His2A coding sequence is shared by several His2A gene repeats in the histone gene cluster. This fusion transcript is expressed under the control of miranda gene regulatory sequences (2.6 kb upstream and 1.6 kb downstream) that were previously shown to drive expression in intestinal progenitor cells (Bardin et al. 2010). The progenitor plasmid was generated using Gateway MultiSite cloning (Shearin et al. 2014) using plasmids provided by Steve Stowers (Montana State University). Full construction details are available upon request. It was inserted into the M{3xP3-RFP.attP}ZH-2A landing site by Rainbow Transgenic Flies, Inc. (Camarillo CA). The loxP-flanked 3xP3-RFP cassette was subsequently removed from the progenitor insertion by Cre-mediated in vivo excision to leave only the miniwhite marker associated with the inserted sequence.
High molecular weight DNA extraction for Nanopore sequencing
High molecular weight DNA extractions were performed as previously described with slight modifications to improve DNA yield (Miller et al. 2018b). A total of 40 male and female flies from Bloomington stock 2475 (w*; T(2;3)apXa, apXa/In(2L)Cy, In(2R)Cy, DuoxCy; TM3, Sb1) were collected, starved for 48 hr, and frozen at –80° for 72 hr before extraction. Frozen flies were transferred to a 2 mL Kimble Dounce homogenizer with 1 mL of homogenization buffer (0.1 M NaCl, 30 mM Tris-HCl pH 8.0, 10 mM EDTA, 0.5% Triton X-100) and homogenized with 10 strokes of looser-fitting pestle A and then 10 strokes of tighter-fitting pestle B. This homogenate was transferred to a 1.5 mL Eppendorf tube. The Dounce homogenizer was rinsed with an additional 500 µL homogenization buffer, and this was combined with the rest of the homogenate. This tube was centrifuged at 2,000 G for five minutes to pellet fly material. The supernatant was discarded and the pellet was resuspended in 100 µL of homogenization buffer using a wide-bore tip.
A fresh 1.5 mL Eppendorf tube was prepared with 380 µL of lysis buffer (0.1 M Tris-HCl pH 8.0, 0.1 M NaCl, 20 mM EDTA), 10 µL of Proteinase K (20 mg/mL), 10 µL 10% w/v SDS, and 5 µL RNAse A (Sigma R6148-1.7mL). Using a wide-bore tip, resuspended homogenate was transferred to this tube and mixed by pipetting. Lysis occurred at 50° for 6 hr, with gentle swirling and inversion every 45–60 min. If visible clumps of homogenate could not be broken up by gentle mixing, the tube was shaken briefly to encourage mixing.
Lysate was extracted twice with an equal volume (∼600 µL) of phenol/chloroform/isoamyl alcohol (25:24:1, pH 8.0) in a single 2 mL 5PRIME phase lock gel light tube. The extraction was mixed on a platform rocker at medium speed for eight minutes and then centrifuged for eight minutes at 16,000 G. To maximize DNA purity, the aqueous (upper) phase was extracted twice in the same tube. After the second extraction, the aqueous phase was carefully decanted into a new 2 mL phase lock gel tube and extracted once with an equal volume (∼600 µL) of chloroform as described above. The aqueous phase was then decanted quickly into a 2 mL Eppendorf DNA LoBind tube. DNA was precipitated by adding 0.1 volume of NaOAc, gently swirling to mix, and then adding 2.1 volumes of absolute ethanol. Gentle mixing by inversion was performed until all shimmering strands were precipitated into a white, stringy clump of DNA.
The DNA clump was transferred into a fresh 1.5 mL DNA LoBind tube with a wide bore tip. Excess ethanol was removed by pipetting, and the DNA pellet washed twice with 200 µL of 70% ethanol without centrifugation. Another 200 µL of 70% ethanol was added to the tube and the DNA was pelleted by centrifugation at 2,000 G for two minutes. All ethanol was removed, and the DNA dried for 5–10 min to the immediate moment where the pellet became translucent. Then, 100µL of TE pH 8.0 was added and the tube incubated at 50° for one hour, briefly spun in a tabletop centrifuge, and then incubated at 4° for 48 hr. DNA was lightly sheared by gently pipetting ten times through a P1000 tip and incubated at 4° for an additional 48 hr.
Nanopore library preparation and sequencing
Nanopore libraries were prepared with the SQK-LSK109 Ligation Sequencing Kit with slight modifications to the standard protocol. To start the protocol, three micrograms of high molecular weight DNA were diluted with water to a total volume of 47.5 µL. The DNA repair and dA-tailing steps were then performed with a mixture of 47.5 µL sample, 3.5 µL FFPE DNA Repair Buffer, 2 µL FFPE DNA Repair Mix, 3.5 µL Ultra II End-prep Reaction Buffer, 3 µL Ultra II End-prep Enzyme Mix, and 0.5 µL 100x NAD+. This mixture was incubated in a 200 µL PCR tube at 20° for 60 min and 65° for 30 min in a thermal cycler, and then transferred to a 1.5 mL DNA LoBind tube. Because magnetic beads cause DNA clumping when the sample contains many large fragments, the sample was cleaned up without beads by adding an equal volume (60 µL) of precipitation buffer (9% w/v PEG 8000, 900 mM NaCl, 10 mM Tris-HCl pH 8.0), incubating for 15 min, and centrifuging for 30 min at 10,000 G. To wash the pellet, the supernatant was removed and 150 µL of SFB (from the kit) was added, and then centrifuged at 10,000 G for 2 min. The pellet was washed another time in this manner, and then immediately resuspended in 30 µL of 10 mM Tris-HCl pH 8.0. The sample was incubated at 50° for one hour, spun on a benchtop centrifuge briefly, and further resuspended at 4° for 48 hr.
For adapter ligation, half the volume recommended by the manufacturer’s protocol were used: 12.5 µL ligation buffer (LNB from the ligation kit), 5 µL T4 ligase, and 2.5 µL adapter mix (AMX from the ligation kit) was added to the 1.5 µL DNA LoBind tube containing the sample. After mixing, the sample was incubated for 30 min at room temperature. For the same reason as described previously, magnetic beads were omitted from the protocol. Since the ligation buffer causes DNA precipitation, the sample was centrifuged at 10,000 G for 30 min to pellet the DNA. To wash the pellet, the supernatant was removed and 150 µL LFB (from the ligation kit) added to the tube and centrifuged at 10,000 G for two minutes. The pellet was washed another time in this manner and immediately resuspended in 30 µL of 10 mM Tris-HCl pH 8.0. The sample was incubated at 40° for one hour, briefly spun down in a benchtop centrifuge, and stored at 4° for 48 hr. Small DNA fragments were removed from the prepared library using the Short Read Eliminator buffer (Circulomics) according to the manufacturer’s protocol, importantly, substituting LFB from the ligation kit or a 1:1 dilution of water and precipitation buffer instead of 70% ethanol to wash the pellet.
The adapter-ligated and size selected library was prepared for loading by quantifying with a Qubit fluorimeter and by transferring 350 ng (∼7.5 µL) of prepared library to a fresh 1.5 µL DNA LoBind tube. An equal volume (∼7.5 µL) of sequencing buffer (SQB from the ligation kit) was then added. Flush buffer (FB) from the EXP-FLP002 Flow Cell Priming Kit was added to a final volume of 70 µL. The library was loaded and sequenced according to manufacturer’s instructions. After 12 hr, the sequencing run was paused, and the flow cell was flushed with the EXP-WSH003 Flow Cell Wash Kit. A fresh library was loaded as described above and this procedure was performed one more time during the sequencing run.
Raw Nanopore reads were converted to FASTQ files using Guppy 3.2.4 in high-accuracy mode and all reads were aligned to release 6 of the D. melanogaster genome assembly using BWA MEM version 0.7.17-r1188 (Li and Durbin 2009).
Sequencing of the Df(2L)bhe chromosome
Three pools of males heterozygous for the Df(2L)bhe chromosome from Bloomington stock 3268 and the second chromosome from the genome reference strain (Bloomington stock 2057) were collected from three separate single-male crosses to take into account cryptic genetic changes that have accumulated in the reference strain (Gutzwiller et al. 2015). DNA for sequencing was prepared as described in Miller et al. (2018a). Libraries were sequenced in 150-bp paired-end mode on an Illumina NextSeq 500 sequencer. Illumina Real Time Analysis version 2.4.6 was used to demultiplex reads and generate FASTQ files. Alignment to version 6 of the Drosophila melanogaster genome assembly was performed with bwa version 0.7.15-r1140 (Li and Durbin 2009) and SNPs were called using SAMtools version 1.5 (Li et al. 2009).
Identification and analysis of shared and unique SNPs on X and third chromosome balancers
SNPs and indels for the X chromosome balancers FM7a and FM7c and the third chromosome balancer TM3 were obtained from previous alignments (Miller et al. 2016a, 2016b). Polymorphisms with VCF quality scores greater than 220 were identified using VCFtools version 0.0.15 (Danecek et al. 2011) and then merged using vcf-merge. VCF files were annotated using SnpEff version 4.3 (Cingolani et al. 2012) and annotated files were filtered using custom scripts.
Data availability
The accompanying tables contain complete complementation data. Stocks may be obtained from the Bloomington Drosophila Stock Center as indicated in the Reagent Table (Table S1). Basecalled Nanopore data are available from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/) under project number PRJNA623115. Data related to characterization of the Df(2L)bhe chromosome are available under project PRJNA623116. Original data underlying this manuscript can be accessed from the Stowers Original Data Repository at http://www.stowers.org/research/publications/LIBPB-1520. Custom scripts used for data analysis, including the genome assembly of Bloomington stock 2475, are available on GitHub (https://github.com/danrdanny/balancerPhenotypes). Supplemental material is available at figshare: https://doi.org/10.25387/g3.12996710.
Results
Recessive lethal, female-sterile and visible phenotypes associated with third chromosome balancer breakpoints
The molecular analysis of third chromosome balancers presented by Miller et al. (2016a) and summarized in Table S2 raised the question of how the disruption of genes by inversion breakpoints contributes to the homozygous lethality of the balancers. Some component inversions were known to be homozygous viable and fertile, but some inversion breakpoints had not, to our knowledge, been examined. As shown in Tables 1 and S3, we tested all TM3, TM6 and TM6B breakpoints for strong phenotypic effects by performing complementation tests with deficiencies spanning these breakpoints and scoring for lethality, female sterility or grossly abnormal morphology. For each test, we had independent control crosses to show the stocks were not compromised (Table S4).
Table 1. Assessing phenotypes associated with third chromosome balancer breakpoints.
| Inversiona | Balancer | Breakpointb | Genes disrupted | Deletion or mutation tested | Phenotypes | Related observations |
|---|---|---|---|---|---|---|
| In(3LR)P88 | TM6 | 61A6 | Between CG13485 & CG12483 | Df(3L)ED50002, Df(3L)ED4079 | Viable, female fertile | Df(3L)ED50002 & Df(3L)ED4079 are homozygous viable |
| 61B3 | Tudor-SN | Df(3L)BSC125, Df(3L)Exel6084 | Viable, female fertile | |||
| 89B14 | ss | ss1 | Viable, female fertile, bristle loss | ss mutation associated with In(3LR)P88 (Lindsley and Grell 1967; Morata and Lawrence 1978; Struhl 1982) | ||
| In(3LR)HR33 | TM6B | 61B2 | Between CG34453 & E(bx) | Df(3L)ED201, Df(3L)Exel6084 | Viable, female fertile | In(3LR)HR33 is homozygous viable (Ashburner 1972) |
| 87B3 | Between CG12256 & CG3916 | Df(3R)Exel7313, Df(3R)Exel6162 | Viable, female fertile | |||
| In(3L)P | TM6, TM6B | 63B9 | CG14964 | Df(3L)BSC671, Df(3L)BSC672c | Viable, weak female fertility | In(3L)P is homozygous viable and fertile (Payne 1918, 1924). |
| 72E1 | Between CG13042 & CR32160 | Df(3L)BSC560, Df(3L)BSC579 | Viable, female fertile | |||
| In(3LR)sep | TM3 | 65D3 | Between Prat2 & CG45413 | Df(3L)Exel6109, vvlsep | Viable, female fertile, vein defects | In(3LR)sep homozygotes and other vvl mutants show vein defects (Diaz-Benjumea and García-Bellido 1990) |
| 85F2 | Glut4EF | Df(3R)Exel6155, Df(3R)Exel6154, Glut4EFsep | Viable, female fertile, outheld wings | Glut4EF mutants have outheld wings (Yazdani et al. 2008) | ||
| In(3LR)TM3-3 | TM3 | 71B6 | FucTA | Df(3L)ED218, Df(3L)BSC837 | Viable, weakly female fertile | FucTA mutants viable (Yamamoto-Hino et al. 2010) |
| 94D10 | p53 | Df(3R)BSC803, Df(3R)ED6103 | Viable, female fertile | p53 mutants viable and fertile (Rong et al. 2002; Sogame et al. 2003; Xie and Golic 2004) | ||
| In(3LR)M6 | TM6, TM6B | 75D6 | CR43987 | Df(3L)BSC832, Df(3L)BSC416 | Viable, female fertile | Some TM6C chromosomes, which carry In(3LR)M6, are homozygous viable and fertile |
| 94A11 | CG13857 | Df(3R)BSC511, Df(3R)BSC685 | Viable, female fertile | |||
| In(3LR)TM3-2 | TM3 | 76B1–2 | CG32206 & possibly ms(3)76Ba | Df(3L)ED228 | Viable, female fertile, male fertile | |
| 92F4 | Lrrk | Df(3R)BSC141, Df(3R)BSC488 | Viable, female fertile, normal locomotion | Locomotory and female fertility loss in aging Lrrk mutants (Lee et al. 2007). We saw no overt phenotypes in young flies. | ||
| In(3LR)TM3-1 | TM3 | 79F3 | CG14459 | Df(3L)BSC451, Df(3L)ED230 | Viable, female fertile | |
| 100D1 | kek6 | Df(3R)ED6361, Df(3R)BSC505 | Viable, female fertile | |||
| In(3R)Hu | TM6B | 84B2 | Between CR44933 & Sodh-1 | Df(3R)Antp17 | Viable, female fertile, malformed tergites | In(3R)Hu is homozygous viable (Bridges and Brehme 1944). TkR86C mutations are homozygous viable and fertile (Asahina et al. 2014) |
| 84F8 | CR44318 | Df(3R)Exel6147 | Viable, female fertile | |||
| 86C5 | TkR86C | Df(3R)BSC529, Df(3R)Exel6159 | Viable, female fertile | |||
| In(3R)C | TM3, TM6, TM6B | 92E2 | Between CG4362 & CG42668 | Df(3R)ED6025, Df(3R)ED6027 | Viable, female fertile | In(3R)C is homozygous viable and fertile (Dexter 1914; Muller 1918; Bridges and Morgan 1923; Sturtevant 1926) |
| 100F2–3 | Distal to genes | Df(3R)ED50003, Df(3R)ED6361, Df(3R)ED6362d | Viable, female fertile |
The In(3LR)TM3-1, In(3LR)TM3-2 and In(3LR)TM3-3 inversions are named here for the first time.
Cytological bands predicted from sequence coordinates (Table S2) using FlyBase reference table except 100F2–3, where we cite polytene analysis (Morgan et al. 1937)
Fertility higher in TM6B crosses than TM6 crosses with both deletions.
P{RS3}CB-0686-3, the FRT-bearing progenitor of the distal Df(3R)ED6361 and Df(3R)ED6362 breakpoints, lies within the subtelomeric region (Phalke et al. 2009) at a position that may not be distal to the In(3R)C breakpoint.
We verified that some inversion breakpoints are associated with recessive visible phenotypes (Table 1). The 89C breakpoint of In(3LR)P88 lies in the fourth intron of spineless (ss), resulting in short bristles (Duncan et al. 1998). In(3LR)sep failed to complement alleles of ventral veins lacking (vvl) and deletions of the region of the 65D breakpoint, resulting in wing venation defects (Diaz-Benjumea and García-Bellido 1990; de Celis et al. 1995). Third chromosome balancers do not appear to contain mutations in the vvl coding region (Miller et al. 2016a), so disruption of vvl function by the In(3LR)sep breakpoint ∼130 kb downstream of vvl likely comes from perturbing long-distance regulation. The 85F breakpoint of In(3LR)sep disrupts Glut4EF, resulting in outspread wings (Yazdani et al. 2008; Miller et al. 2016a). Df(3R)Exel6154, which deletes only a subset of Glut4EF 5′ exons, showed phenotypes one would expect of a hypomorphic Glut4EF allele. Unlike flies with amorphic Glut4EF genotypes, which showed obvious wing spreading soon after eclosion and progressed to strong spreading within a week, flies with mutant genotypes involving Df(3R)Exel6154 showed infrequent and slight spreading in the first three days after eclosion and progressed to intermediate frequency and severity by one week (Tables 1 and S3).
We were surprised to find that none of the breakpoints were associated with complete lethality or female sterility (Table 1). We did not measure viability or fertility rates in ways that would allow us, in most cases, to identify intermediate levels, but the 71B breakpoint on TM3, which disrupts the FucTA gene, was associated with severely reduced female fecundity. The relative innocuousness of the breakpoints likely reflects the care taken in choosing preexisting progenitor inversions to assure that balancers would be as trouble free as possible and in designing screens for isolating new inversions that did not rely on lethal or sterile phenotypes. These results suggest that the recessive lethality of third chromosome balancers in most current stocks is attributable to spontaneous mutations that have accumulated since the balancers were generated, although it is possible that deleterious effects of multiple breakpoints contribute to recessive lethality.
A TM3 inversion breakpoint disrupting the p53 gene results in loss of apoptosis in response to irradiation
The 94D breakpoint of In(3LR)TM3-3 on TM3 lies within an intron of the p53 gene. The exons on either side of this breakpoint encode a protein region involved in DNA binding in all p53 isoforms, suggesting that the p53 allele on TM3 is amorphic. Previous studies have shown that homozygosity for p53 null alleles does not result in lethality or sterility in Drosophila (Rong et al. 2002; Sogame et al. 2003; Xie and Golic 2004), so viability and fertility in our complementation tests of TM3 and deletions of p53 (Table 1) was not surprising. Nevertheless, other studies have shown that apoptotic responses to DNA damage are defective in p53 mutants (Lee et al. 2003; Brodsky et al. 2004; Mehrotra et al. 2008; Qi and Calvi 2016).
To determine whether the TM3 breakpoint eliminates p53 activity, we examined the effect of the breakpoint on apoptosis in a convenient proliferative cell population, the mitotically dividing ovarian follicle cells that surround maturing egg chambers. Females with putative p53 loss-of-function genotypes were irradiated, aged four hours and assayed for apoptosis by TUNEL assay. Unlike wild-type controls, which showed TUNEL staining in ∼12% of dividing follicle cells, the cells from females where TM3 had been combined with a p53 deficiency (Df(3R)ED6103 or Df(3R)BSC803) or null mutation (p535A-1-41 or p53-ns) had much lower frequencies of TUNEL staining (<1% of cells, a level similar to unirradiated controls; Figure 1, Table S5).
Figure 1.

The TM3 breakpoint at 94D disrupts p53 apoptosis activity. Irradiation-induced apoptosis is seen as TUNEL staining in stage 1–5 follicle cells counterstained with DAPI of control females (e.g., y1 w67c23 homozygotes shown here). TUNEL staining is absent in follicle cells of females carrying TM3 combined with chromosomal deletions removing the p53 gene (e.g., TM3/Df(3R)ED6103 shown here) and females carrying TM3 and loss-of-function p53 alleles (e.g., TM3/p535A-1-4 shown here). TUNEL-stained cells in p53 mutants are polar follicle cells, which undergo p53-independent, developmentally programmed cell death. See Table S5 for all genotypes tested.
These results show that p53 function is indeed disrupted by the TM3 breakpoint and highlight the danger of balancing p53 mutations with TM3 and viewing the genotypes as “wild type”. A broader problem, however, is the effect of reducing p53 copy number on cellular processes. p53 is haploinsufficient with respect to the induction of apoptosis in response to telomere loss (Kurzhals et al. 2011) and may show haploinsufficiency with respect to other stress responses. Consequently, investigators should be careful not to use TM3 heterozygotes as “normal” controls in experiments that might involve p53-related processes.
A TM3 inversion breakpoint disrupts FucTA resulting in loss of immunostaining with antibodies against horseradish peroxidase
The 71B breakpoint of In(3LR)TM3-3 on TM3 lies within the 5′ UTR of one of two reported transcripts of the Fucosyltransferase A (FucTA) gene and likely results in partial loss of expression (Miller et al. 2016a). The FucTA enzyme catalyzes the attachment of fucose to N-linked glycans on a variety of proteins (Fabini et al. 2001). This modification is detected by antibodies raised against horseradish peroxidase (HRP) and is enriched in neural tissues of ecdysozoans including Drosophila (Jan and Jan 1982; Haase et al. 2001). Yamamoto-Hino et al. (2010) showed that labeled anti-HRP antibodies do not stain the nervous system of FucTAf03774 mutant larvae and Snow et al. (1987) demonstrated that neural staining is lost in homozygous TM3 embryos (though a few nonneural tissues show staining). As expected, when we combined TM3 from multiple stocks with either a deletion of the FucTA gene (Df(3L)BSC837) or FucTAf03774, we saw loss of anti-HRP staining in adult brains (Figure 2, Table S6).
Figure 2.

The TM3 breakpoint in FucTA is associated with loss of anti-HRP antibody staining in nervous tissue and intestinal epithelial cells. A. TM3/Sb1 flies were crossed to mutation- or deletion-bearing flies. Anti-HRP staining was not detected in adult brains when deletions of FucTA (Df(3L)BSC837 and Df(3L)Brd15) or a FucTA mutation (FucTAf03774) were combined with TM3, but it was seen in control crosses where a chromosome with no FucTA mutation (Sb1) was combined with the same deletions or mutation. Staining was seen with either TM3 or Sb1 combined with a Tollo deletion (Df(3L)BSC578) or mutations (Tollo1 and TolloMI11573). Anti-HRP staining shown in green; anti-BRP counterstaining shown in magenta to highlight neuropils. B. Crosses between mira-His2A.mCherry.HA; wgSp-1/CyO; TM3/TM6B females and males carrying the same deletions or mutations gave identical results for anti-HRP staining of intestinal epithelial cells where TM6B serves as the FucTA+ control (and only wgSp-1/+ progeny were scored). Anti-HRP staining shown in green; miranda-expressing progenitor and enteroendocrine cells shown in red; DAPI staining of nuclei shown in blue. C. The 71B breakpoint in TM3 lies within an alternative 5′ UTR of FucTA and ∼85 kb distal to Tollo. Df(3L)Brd15 deletes both genes while Df(3L)BSC837 disrupts only FucTA and Df(3L)BSC578 deletes only Tollo. FucTA-specific mutations and deletions failed to complement TM3 with respect to anti-HRP staining while Tollo-specific mutations and deletions complemented. A 412 transposon insertion in the 3′ UTR and a polymorphism leading to substitution of phenylalanine for leucine are present in Tollo in all TM3 chromosomes examined, but they may be neutral with respect to Tollo function.
To test whether FucTA mutations affect anti-HRP antibody staining in another tissue, we examined the intestine, where O’Brien et al. (2011) showed staining of progenitor cells (stem cells and enteroblasts). To facilitate this analysis, we generated a new cell marker containing regulatory sequences from the miranda gene previously shown to be expressed in progenitor cells (Bardin et al. 2010) and characterized its expression relative to known progenitor and enteroendocrine cell markers (Micchelli and Perrimon 2006). Figure S1 shows that it is expressed strongly in progenitor cells and weakly in some enteroendocrine cells in a pattern matching anti-HRP staining. As we saw in neurons, TM3 combined with either FucTA deletions or FucTAf03774 eliminated anti-HRP intestinal cell staining (Figure 2; Table S6). This observation highlights the utility of FucTA mutations and anti-HRP antibody staining for investigating the significance of protein fucosylation in intestinal cells, which remains largely unexplored.
The association of fucosylation defects with disruption of FucTA by a TM3 breakpoint would be straightforward were it not for previous studies suggesting that Tollo, a gene linked closely to FucTA, is disrupted on TM3 chromosomes and that Tollo regulates fucosylation. Seppo et al. (2003) mapped loss of embryonic nervous system anti-HRP staining on TM3 to the region of Df(3L)Brd15, showed that 412 transposon sequences exist within the 3′ UTR of Tollo on TM3 (Figure 2C), and speculated that the 412 sequences coincided with the TM3 breakpoint. Our long-read sequence showed that the 412 element is full length and not associated with an aberration breakpoint, but an examination of the short-read sequence data from Miller et al. (2016a) verified 412 termini in the same position (3L:15,241,752–15,241,755) in a broad sampling of TM3 chromosomes. We also saw that all sequenced TM3 chromosomes share a polymorphism in Tollo encoding a leucine-to-phenylalanine substitution at amino acid 851. We now know, however, that Df(3L)Brd15 deletes both FucTA and Tollo, and that fucosylation defects that have been attributed to Tollo mutation may be the result of FucTA mutation. We did not see immunostaining eliminated in adults carrying TM3 and Tollo mutations or Df(3L)BSC578, which deletes Tollo but leaves FucTA intact (Figure 2, Table S6), which is consistent with the results of Yagi et al. (2010), who saw immunostaining in embryos homozygous for the null Tollo59 mutation. We also saw immunostaining in FucTA heterozygotes when Tollo copy number was reduced (Table S6). While results suggest that the immunostaining defects are not attributable to Tollo disruption, Seppo et al. (2003) reported rescue of anti-HRP staining when they combined Df(3L)Brd and TM3 with transgenic constructs expressing Tollo, Baas et al. (2011) reported loss of immunostaining in flies homozygous for the TolloC5 null allele, and the position of the TM3 breakpoint in the 5′ UTR of an alternative FucTA transcript may allow plastic expression. Reconciling these observations is beyond the scope of this paper, but our studies indicate that the purported regulation of fucosylation by Tollo should be reevaluated.
Polymorphisms on FM7a, FM7c and TM3 balancers disrupt protein-coding genes
The second chromosome balancers SM5, CyO and SM6a have been shown to share mutations that affect protein-coding genes (Miller et al. 2018a). For example, all SM5 and SM6a balancers carry a C-to-T mutation in the uncharacterized gene CG12506, resulting in a premature translational stop. In addition, every sequenced balancer has unique mutations that are not seen on any other balancer chromosome, indicating that spontaneous mutations occur in stocks at an appreciable frequency. We therefore undertook a similar analysis of previously sequenced X and third chromosome balancers (Miller et al. 2016a, 2016b) to identify SNP and indel polymorphisms creating missense, splice-site, or nonsense mutations that are either shared or unique among the FM7a, FM7c or TM3 balancers (Table 2). We identified 1,536 mutations shared among all eight FM7a and FM7c balancers (Table S7), 339 mutations shared only among the three FM7a balancers sequenced (Table S8), 56 mutations shared only among all five FM7c balancers (Table S9) and 3,011 mutations shared among at least 15 of 17 TM3 balancers (Table S10). Altogether, we found 1,722 mutations unique to individual balancer chromosomes that likely affect protein function including 1,652 missense mutations, 25 stop mutations, and 45 splice-site mutations (Table S11). These statistics highlight a challenge in using balancers experimentally: specific balancers carry unique constellations of polymorphisms that may affect experimental outcomes and the interpretation of results.
Table 2. Number of SNP or indel mutations shared by multiple balancers or present on only one balancer.
| Balancer | Stocka | Stop mutations | Missense mutations | Splice-site mutations |
|---|---|---|---|---|
| Mutations on multiple balancers | ||||
| FM7a | All | 0 | 331 | 8 |
| FM7a and FM7c | All | 11 | 1503 | 22 |
| FM7c | All | 0 | 56 | 0 |
| TM3 | All | 7 | 2956 | 48 |
| Mutations on a single balancer | ||||
| FM7a | 785 | 0 | 7 | 0 |
| FM7a | 35522 | 0 | 6 | 0 |
| FM7a | 36489 | 0 | 17 | 1 |
| FM7c | 616 | 0 | 4 | 2 |
| FM7c | 3378 | 0 | 7 | 3 |
| FM7c | 5193 | 1 | 114 | 2 |
| FM7c | 23229 | 0 | 34 | 0 |
| FM7c | 36337 | 0 | 70 | 3 |
| TM3 | 120 | 1 | 50 | 3 |
| TM3 | 500 | 0 | 33 | 3 |
| TM3 | 504 | 3 | 265 | 1 |
| TM3 | 560 | 6 | 419 | 8 |
| TM3 | 1614 | 0 | 129 | 3 |
| TM3 | 1679 | 1 | 7 | 0 |
| TM3 | 2053 | 3 | 51 | 2 |
| TM3 | 2098 | 0 | 16 | 1 |
| TM3 | 2198 | 0 | 9 | 1 |
| TM3 | 2485 | 0 | 17 | 0 |
| TM3 | 3251 | 2 | 8 | 0 |
| TM3 | 5457 | 0 | 7 | 2 |
| TM3 | 8852 | 6 | 93 | 2 |
| TM3 | 9013 | 0 | 29 | 0 |
| TM3 | 22239 | 0 | 92 | 4 |
| TM3 | 24759 | 0 | 66 | 0 |
| TM3 | 38418 | 3 | 183 | 4 |
These numbers identify the original balancer stocks (Table S1) outcrossed to a common stock for sequencing (Miller et al. 2016a, 2016b).
An Ankyrin 2 mutation is present on Sb1-marked TM3 chromosomes
In a series of crosses involving mutations in polytene region 65D–F several years ago, we noticed that flies inheriting TM3 and the deletions Df(3R)RM5-1, Df(3R)RM5-2 or Df(3R)pbl-X1 were largely lethal, but escapers had unexpanded wings, disarranged bristles, small body size, improperly tanned cuticle, dark pigmentation, low female fecundity and a generally weak and sickly appearance (Table S12). The partial lethality of TM3 with Df(3R)RM5-1 and Df(3R)RM5-2 had been noted previously (Grasso et al. 1996). Our subsequent analysis of TM3 breakpoints with molecularly defined deletions (Table 1) showed that the 65D2–3 breakpoint of In(3LR)sep is not associated with these abnormal phenotypes and indicated that they were instead attributable to a mutation immediately proximal to the breakpoint. We mapped the mutation to a seventeen-gene interval based on Df(3R)RM5-1 deleting liquid facets through Ankyrin 2 (Ank2) (Koch et al. 2008). TM3 sequences (Miller et al. 2016a) showed the presence of a deletion within the last exon of most Ank2 transcripts that removes thirteen nucleotides (3L:7,658,562–7,658,574) and affects all but three Ank2 isoforms. A transposon insertion into the same exon, PBac{WH}Ank2f02001, produced the same phenotypes in combination with TM3 (Table S12), consistent with the report of Koch et al. (2008) that most Ank2 mutations could not be maintained in stock using TM3.
Interestingly, it appears from the TM3 sequences (Miller et al. 2016a) and complementation tests against Ank2 deletions and Ank2f02001 (Table S12) that all TM3 chromosomes marked with Sb1 (or chromosomes derived from them) carry the Ank2 mutation. The original version of TM3 lacking the Sb1 and Ser1 markers and a later version carrying only Ser1 that was the immediate progenitor of all Sb1-marked TM3 chromosomes (Tinderholt 1960; Miller et al. 2016a) lack the Ank2 mutation. It seems likely that the Ank2 mutation, which we call Ank2TM3, arose spontaneously on TM3 around the time Sb1 was introduced and has since been propagated to all Sb1-marked TM3 balancers.
The distal breakpoint of In(3R)C lies between subtelomeric heterochromatin and the telomeric transposon array
A previous analysis of third chromosome balancers (Miller et al. 2016a) was unable to determine the precise genomic positions of the In(3R)C breakpoints using short-read sequencing, because the distal breakpoint lies within repetitive sequence at the distal end of chromosome arm 3R. Recently, chromatin conformation capture data from TM3 heterozygotes was used to estimate the genomic position of the proximal 92E breakpoint to 3R:20,308,200 (Ghavi-Helm et al. 2019). Using this estimate as a guide, we isolated long Nanopore sequencing reads that spanned the proximal and distal In(3R)C breakpoints and performed a de novo genome assembly using Flye (version 2.7-b1585) with default parameters and the “–keep-haplotypes” flag (Kolmogorov et al. 2019).
We localized the proximal In(3R)C breakpoint to 3R:20,308,209–20,308,213 (the breakpoint contains a 5 bp duplication) in the intergenic region between CG4362 and CG42668 (Figure 3). The assembly spanning the proximal inversion end (shown as the A|C junction in Figure 3A) juxtaposed sequences distal to CG4362 to sequences distal to Map205, the distalmost gene on 3R. This assembly included the distalmost sequences of the reference genome assembly at 32,079,330 and 11,664 bp of novel sequence distal to it, which contained 1360, invader4 and copia transposons (Figure 3B, File S1). These transposons are commonly found in subtelomeric regions (Anderson et al. 2008; Mason and Villasante 2013).
Figure 3.
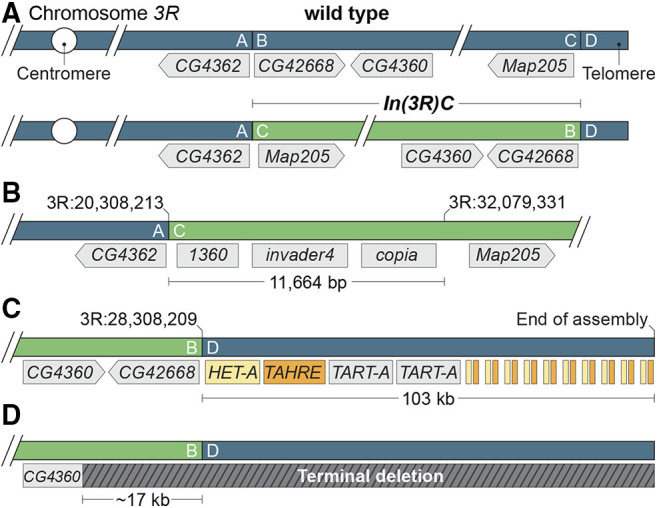
The structure of In(3R)C. A. The wild-type and inverted arrangement of the In(3R)C inversion breakpoints with neighboring genes labeled. The exact position of the distal (C|D) breakpoint was not previously known, but was suspected to lie within subtelomeric heterochromatin. B. Assembly of long sequencing reads revealed the molecular structure of the proximal (A|C) junction, with nearly 12 kb of sequence between the distal end of the reference 3R assembly at 32,079,331 and the distal break. This region contained three transposable elements that were, in all likelihood, originally positioned immediately proximal to the telomeric transposon repeats. C. The distal In(3R)C breakpoint fell immediately proximal to telomeric repeats. Our assembly of this region extended ∼103 kb distally from the distal (B|D) junction and contained the HeT-A, TAHRE, and TART-A elements expected for a telomeric region as well as repeated fragments of HeT-A and TART-A at the distal end of the assembly. The positions and number of elements shown distal to the second TART-A element are estimates. All elements, including the incomplete HeT-A and TAHRE fragments, are oriented with their 3′ ends toward the centromere. D. Having established the structure of In(3R)C, we interpret the ∼17 kb deletion of sequences immediately distal to 3R:20,308,209–20,308,213 observed by Ghavi-Helm et al. (2019) as evidence of a 3R terminal deletion specific to the TM3 chromosome they characterized.
The distal end of In(3R)C (shown as the B|D junction in Figure 3A) juxtaposes sequences immediately proximal to CG42668 to ∼103 kb of sequence composed of telomeric HeT-A, TAHRE, and TART-A transposons adjacent to repeated partial fragments of HeT-A and TAHRE (Figure 3C). To confirm this assembly, we identified individual reads that spanned the inversion end and extended up to 50 kb distally and found that they included the same HeT-A, TAHRE, and TART-A transposons. No individual read extended past the first TART-A element, so we could not confirm the presence of the second TART-A element or the subsequent HeT-A and TAHRE fragments with this approach. The repeated HeT-A and TAHRE fragments are similar in structure to partial duplications of HeT-A and TART elements observed by Levis et al. within telomeric sequences (Levis et al. 1993). The nongenic positions of the breakpoints largely explain why In(3R)C gives no overt phenotypes when homozygous (Dexter 1914; Muller 1918; Bridges and Morgan 1923; Sturtevant 1926) or combined with deletions for the regions of the breakpoints (Table 1). These complementation tests provided no evidence for position-effect suppression of genes juxtaposed to telomeric or subtelomeric sequences by In(3R)C.
Our results show that the distal In(3R)C breakpoint separates telomere-associated sequences (TAS) from telomeric sequences, and moves them from their usual subtelomeric position. While the two domains are juxtaposed on most chromosomes (Asif-Laidin et al. 2017), their relationship is unclear. Both express piRNAs to repress transposon activity, but the domains are regulated differently in germ line and somatic cells (Radion et al. 2018). TAS regions have heterochromatic properties that may be relevant to telomere function (Radion et al. 2018), but TAS are absent from some chromosomes (Asif-Laidin et al. 2017) and In(3R)C has a worldwide distribution in wild populations (Inoue and Igarashi 1994). These observations indicate that any interdependence of the two domains is complicated and suggest In(3R)C may prove valuable in exploring their interactions.
As discussed previously (Miller et al. 2016a, 2018a), meiotic recombination in chromosomal regions distal to the distalmost balancer breakpoint may be relatively high if the balancer breakpoint is positioned a large distance from the telomere, e.g., the 65D breakpoint of TM3. In this context, the distal 3R breakpoint of In(3R)C provides the “perfect” balancer end, because no genes lie distal to the breakpoint. Consequently, even extremely distal 3R mutations can be maintained with confidence in stocks utilizing the In(3R)C-containing balancers TM3, TM6, TM6B, TM6C, TM8 and TM9.
Interestingly, the chromatin conformation capture data of Ghavi-Helm et al. (2019) contained a ∼17-kb deletion immediately distal to the 92E breakpoint of In(3R)C (region B in Figure 3), which we did not see in our long-read sequencing data or on reanalysis of seventeen previously sequenced TM3 chromosomes. This observation indicates that the TM3 balancer they used in their studies contained a terminal deletion that removed the 3R tip and euchromatic sequences placed near the tip by the inversion event (Figure 3D). So, while CG42668 and the adjacent CG4360 gene were intact in the seventeen TM3 chromosomes sequenced previously and the TM3 chromosome sequenced in this study, they were deleted in the TM3 balancer analyzed by Ghavi-Helm and colleagues. We do not know if Het-A, TAHRE or TART elements have transposed to the truncated end of the balancer sequenced in their study. Unrecognized terminal deletions are not unusual in Drosophila. For example, cryptic 2L terminal deletions removing the l(2)gl tumor suppressor gene have bedeviled studies of growth control (Roegiers et al. 2009). These results show that terminal deletions are yet one more kind of genetic variation involving balancers that can affect experimental outcomes.
In(2L)Cy arose by ectopic recombination Between transposon insertions
Similar to In(3R)C discussed above, Miller et al. (2018a) were unable to localize the breakpoints of In(2L)Cy, a component of most second chromosome balancers, with single-nucleotide resolution using short sequence reads due to the presence of repetitive sequences at the breakpoints. Ghavi-Helm et al. (2019) used chromatin conformation capture data to estimate the positions of these breakpoints followed by paired-end sequencing to provide precise coordinates. To confirm their breakpoint mapping and provide more details about the inversion event, we sequenced DNA from flies carrying In(2L)Cy using long-read sequencing and identified multiple reads spanning both the distal and proximal In(2L)Cy breakpoints. We confirmed that the distal breakpoint lies at 2L:2,137,067–2,137,075 in the 3′ UTR of GlyP and the proximal breakpoint lies at 2L:12,704,649–12,704,657 in the intergenic region between CG5776 and spict (Figure 4A). These breakpoints have no seriously deleterious effects: In(2L)Cy has no overt phenotypic effects when homozygous (Sturtevant 1931) or when combined with deletions for the breakpoint regions (the deletions tested by Miller et al. (2018a) span the refined breakpoint positions reported here).
Figure 4.

In(2L)Cy was likely created by ectopic recombination between two foldback transposable elements. A. The distal In(2L)Cy breakpoint (A|B) lies in the 3′ UTR of GlyP and proximal breakpoint (C|D) lies between CG5776 and spict. Reference genome coordinates are shown. B. The general structure of the breakpoint-associated FB insertions. Both insertions have end sequences, spacer sequences and blocks of repeats in mirrored orientations flanking a middle region. Each block contains a single copy of five distinct short repeats, blocks are repeated tandemly a variable number of times, and each block terminates with a CTC motif. An additional 23 bp may be appended to a consensus end sequence. C. The distal (A|C) end of In(2L)Cy includes 2,418 bp of FB sequences, which contain an alternative end sequence and clusters of two and seven repeat blocks, but lack one spacer sequence. Consistent with a recombinant origin, the FB sequences are flanked by a 9 bp tandem site duplication from FB transposition into GlyP (shown as “A”) and another from FB transposition into the CG5776–spict region (“B”). D. The proximal (B|D) end includes 2,563 bp of FB sequences including two spacer sequences and clusters of two and eight repeat blocks. These FB sequences are flanked by the 9 bp duplicated sequences from FB transposition into GlyP (“A”) and the CG5776–spict region (“B”) expected for inversion via ectopic recombination. E. In(2L)Cy arose by recombination between progenitor FB insertions. The high similarity of the FB sequences at the In(2L)Cy ends and the presence of an alternative end sequence at the distal In(2L)Cy end suggests that an FB element within GlyP transposed to the CG5776–spict region and subsequently recombined with an FB element at the donor site.
From the de novo assembly produced by Flye and the sequences of individual long reads, we determined that each In(2L)Cy breakpoint is associated with an intact FB transposon that shows high conservation with FB elements characterized previously (Badal et al. 2006b). Each FB element carries inverted repeat end sequences 467 to 479 bp in length (Figure 4B) and the four end sequences showed ≥97% identity (File S1). Internal to the end sequences and flanking a middle region, the FB elements carry a single spacer region and a variable number of blocks of five 27–31 bp repeat sequences oriented in opposite directions. Both FB elements had two blocks of repeats distal to the middle region, but the distal FB element had seven blocks proximal to the middle region (Figure 4C) while the proximal FB element had eight (Figure 4D). All five repeats within all blocks showed high identity to consensus sequences and all repeat blocks were separated by single CTC motifs as expected (Badal et al. 2006b). The middle regions of FB elements often show some conservation, but the 321–322 bp middle regions of the In(2L)Cy breakpoint FB elements were essentially identical (99% identity) and contained three degenerated vestiges of two different 31-bp repeat sequences. No NOF transposons were present, even though they are often inserted into FB elements (Badal et al. 2006b, 2013).
That both breakpoints are associated with FB transposons strongly suggests that In(2L)Cy arose by ectopic recombination between transposon insertions, a major mode by which inversions arise in Drosophila populations (Ranz et al. 2007; Delprat et al. 2009; Reis et al. 2018; Orengo et al. 2019). The presence of end, spacer and block repeats makes FB and FB-like elements particularly recombinogenic (Cáceres et al. 1999; Casals et al. 2003; Moschetti et al. 2004; Badal et al. 2006a; Delprat et al. 2009) and the mirrored orientations of repeats allow single exchange events to produce inversions regardless of the relative orientations of FB insertions on progenitor chromosomes (Figure 4E). Because we do not know the sequences of the progenitor insertions, we cannot localize the sites of exchange within the FB elements.
The high sequence identity of the middle regions and the similarity in numbers and arrangements of repeat blocks make the two FB elements associated with In(2L)Cy look more like each other than any other FB element we have identified in any sequenced genome. This observation suggests that transposition of an FB element to a new site was followed by ectopic recombination with the donor site to invert the intervening chromosomal segment (Figure 4E). The difference in the number of repeat blocks (seven vs. eight), the absence of one spacer sequence and the minor differences in repeat sequences could easily have arisen, in the time since, by unequal sister chromatid exchange and spontaneous mutation—and larger changes, such as the repeat-mediated inversion of sequences within FB elements, are conceivable. In this scenario, the insertion in GlyP was likely the donor, because one FB end is distinctive: there are 23 bp of sequence between the conserved FB end sequence and the 9 bp tandem duplication generated upon FB insertion. This 23 bp sequence is also associated with an FB end in the reference genome sequence (FB{}nmoFB), suggesting that alternative end sequences may occasionally be used in transposition. The conserved end lying internal to the 23 bp sequence in the GlyP insertion would have been available for FB transposition to the CG5776–spict intergenic region. The alternative, that the progenitor insertions arose from independent transpositions of very closely related FB elements, is also possible. FB elements quite similar to those associated with In(2L)Cy do exist, even though they appear to be uncommon, e.g., there are two FB insertions in the reference genome sequence (FB{}CG34376FB and FB{}773) with identical middle regions (≥99% identity among the four FB insertions) even though they differ in the number and arrangement of repeat blocks.
Recessive lethal or sterile mutations in extremely distal positions add balancing power
Miller et al. (2018a) showed that CyO balances distal 2L mutations poorly because single crossovers occur distal to the distalmost breakpoint in 22D at notable frequency to give rise to nonbalancer chromosomes lacking distal mutations (Figure 5A). We were therefore puzzled by the unusual stability of a CyO stock carrying the broadhead mutation, which Kahsai and Cook (2018) showed is associated with a small terminal deletion called Df(2L)bhe. This mutation, which affects development of the anterior end of homozygous embryos (Nüsslein-Volhard et al. 1984; Tearle and Nüsslein-Volhard 1987), has been maintained stably in stock with CyO for approximately 40 years. Our short-read sequencing data from a sample of pooled flies showed that the position of the Df(2L)bhe breakpoint is variable—as is typical of chromosome ends not capped by a telomere (Biessmann and Mason 1988). The ends fell in the 2L:18,000–19,000 interval within l(2)gl. This deletion of the distalmost two genes lies next to a tandem duplication (2L:24,509–25,359) within the adjacent Ir21a gene (called Ir21abhe). We predicted that CyO in this stock carries a recessive lethal or sterile mutation in the region immediately proximal to Ir21a, so that single crossovers distal to 22D would result in lethal or sterile progeny and stock breakdown would be avoided (Figure 5B).
Figure 5.

A deleterious mutation can prevent stock breakdown when a crossover occurs distal to the distalmost balancer breakpoint. Panel A shows that a meiotic crossover distal to the CyO breakpoint in region 22D can result in the formation of mutation-free recombinant chromosomes, which will have a selective advantage in a stock population. In contrast, panel B shows that the addition of a recessive lethal or sterile mutation (shown here as a female-sterile mutation) to the balancer leads to elimination of the same recombinant chromosomes through routine population dynamics.
In crosses combining CyO from the Df(2L)bhe, Ir21abhe stock with a set of molecularly defined deletions encompassing 94% of the genes from the 2L telomere to the 22D breakpoint (Table S13), we found no recessive lethal or male-sterile mutations, but we identified a recessive female-sterile mutation (fs(2)21Ba1) in the 15-gene CG11374–ovm interval immediately proximal to Ir21a (Figure S2). In follow-up crosses, we narrowed the position of the mutation to the seven-gene region between net and CG3164. Sterile females had underdeveloped ovaries with only the occasional mature-looking egg chamber. The rare eggs they laid usually collapsed. We suspect the mutation arose spontaneously in the Df(2L)bhe, Ir21abhe stock, because the CyO chromosomes in 14 related stocks established in the Nüsslein-Volhard lab at roughly the same time lack the mutation (Table S13).
Any recombinant chromosomes arising from single crossovers in the region between fs(2)21Ba1 and the 22D breakpoint would be eliminated from the Df(2L)bhe, Ir21abhe/CyO, fs(2)21Ba1 stock. This observation suggests that distal deleterious mutations on balancers are the most likely mechanistic explanation for the unexpected stability of stocks that combine poor balancers of distal regions, such as CyO and TM3, with chromosomes carrying extremely distal mutations. Indeed, geneticists may have unwittingly selected for balancers with distal mutations through routine stockkeeping practices. In all likelihood, the suppression of meiotic recombination that occurs near telomeres (Hawley 1980; Comeron et al. 2012) and tight linkage work together to prevent crossovers between two distal trans-heterozygous mutations, such as Ir21abhe and fs(2)21Ba1, from being a significant risk for generating recombinant chromosomes lacking mutations that would lead to stock breakdown. Figure S3 explores the specific case of Ir21abhe–fs(2)21Ba1 recombination in detail.
Discussion
This is the fourth in a series of papers (Miller et al. 2016b, 2016a, 2018a) aimed at understanding the genomic structure of balancer chromosomes, demonstrating genetic variation among balancers, and illuminating the effectiveness of balancers in maintaining mutation-bearing chromosomes in stable stocks. These studies have shown that balancers, as arguably the most widely and frequently used tools in Drosophila genetics, are interesting both for their utility and the ways by which they inform us about biological processes.
Balancers owe their usefulness to the simple fact that the likelihood of a meiotic recombination event is reduced in the vicinity of aberration breakpoints. Multiple inversions both reduce crossing over between balancers and unrearranged chromosomes and prevent the recovery of most recombinant chromosomes that are formed—though rare, two-strand double crossovers within large regions not interrupted by breakpoints do result in the exchange of sequences (Miller et al. 2016b, 2016a, 2018a). Here, we provided molecular detail regarding two important component inversions that were challenging to characterize molecularly, In(3R)C and In(2L)Cy. Amusingly, In(3R)C was the last inversion on a major balancer to have its breakpoints defined at single-nucleotide resolution, but it was the first inversion discovered. Muller (1918) identified In(3R)C as a crossover suppressor (C = Crossover suppressor) on a chromosome isolated from a wild population. He showed that a recessive lethal mutation (l(3)a1) that arose spontaneously within In(3R)C allowed the recessive lethal mutation SerBd-1 on a homologous chromosome to be maintained in the first balanced stock. Schwartz (2008) provides an excellent discussion of the historic importance of this discovery and the critical role a clear understanding of inversions and balanced lethal systems played in disproving nonmendelian theories of inheritance and evolution. Coincidentally, the other inversion whose breakpoints were sequenced in this study, In(2L)Cy, is one of the two inversions on the second balancing chromosome described (Ward 1923). The characterization of this balancer was an important achievement and is an underappreciated contribution to the history of genetics by an early twentieth-century female scientist.
The precise breakpoints of In(3R)C and In(2L)Cy could not be determined by short-read sequencing in previous studies (Miller et al. 2016a, 2018a) because they are associated with repetitive genomic sequences. The chromatin conformation capture data of Ghavi-Helm et al. (2019) was key to narrowing the positions of the inversion ends so that long-read sequence runs spanning the breakpoints could be identified. Inversions isolated from wild Drosophila populations have arisen by two mechanisms: they were either induced by double-strand breaks followed by nonhomologous end-joining, as happened with In(3R)C, or by ectopic recombination between repetitive sequences, as happened with In(2L)Cy (Ranz et al. 2007; Delprat et al. 2009; Reis et al. 2018; Orengo et al. 2019). Curiously, we know of no inversions from wild populations generated by hybrid element insertion, the predominant mode by which inversions have been induced in experiments with P elements (Gray et al. 1996) and a mechanism by which one might expect inversions to arise after mobilization of P element-related foldback elements, such as those involved in generating In(2L)Cy.
Although the genomic locations of most breakpoints on third chromosome balancers had been determined previously (Miller et al. 2016a), we did not know the phenotypic consequences of these breaks, so we examined each breakpoint in complementation tests with deletions. Impressively, most of the breakpoints had no serious effects on viability. This means that the inversions were either homozygous viable and fertile when they were chosen as progenitor chromosomes or they were induced on top of preexisting inversions and kept for their minor effects. Only the In(3R)C breakpoint in FucTA resulted in severely reduced female fecundity in our tests. In fact, the most recently generated third chromosome balancer, TM6C, was homozygous viable and fertile when it was isolated, even though most TM6C chromosomes now carry secondary lethal mutations. We have not ascertained how investigators screened for the newly induced inversions on TM3, TM6, TM6B and TM6C without relying on overt phenotypes. We suspect they screened for changes in pairing-sensitive interallelic interactions.
We confirmed that breakpoints in he p53 and FucTA genes result in the loss-of-function phenotypes one might expect from disrupting these genes. The p53 gene is a well-studied tumor suppressor (Zhou 2019; Levine 2020; Tang et al. 2020) and FucTA affects nervous system development (Yamamoto-Hino et al. 2010). While the phenotypes we assayed—failures in apoptosis and protein fucosylation—are more subtle than lethality or abnormal morphological phenotypes, it is easy to see how even heterozygosity for these balancer-borne mutations might affect the interpretation of many experiments.
Likewise, we demonstrated that numerous missense, nonsense and splice-acceptor mutations are shared by balancers and that many others are unique to specific balancers—as one might expect for chromosomes sharing progenitors that have diverged over time. Indeed, balancers, because they undergo limited recombination, provide an excellent “fossil record” of sequence divergence by descent. The Ank2TM3 mutation we identified serves as a marker for a specific lineage of TM3 chromosomes, and other mutations undoubtedly mark different lineages. While we demonstrated that shared and unique mutations exist on balancers, we did not compare stocks established at a known time with the same balancer. Such an experiment would be interesting with respect to the rates at which mutations accumulate and balancers diverge under routine stock-rearing conditions.
In addition to documenting the accumulation of SNPs and indels, we saw evidence from characterizing In(3R)C that balancer chromosomes can be polymorphic for terminal deletions. Cryptic terminal deletions are common in stocks (Mason et al. 2004; Roegiers et al. 2009), so it is not surprising that some balancers are missing tip sequences. It is easy to imagine terminal deletions preventing the maintenance of distal mutations in stocks or enhancing the phenotypic effects of other mutations, so it is important to consider the potential impact of a terminal deletion when interpreting experimental results.
Finally, our studies provide evidence that distal deleterious mutations can improve the effectiveness of balancers in maintaining the integrity of distal regions on homologous chromosomes. While balancers with distalmost breakpoints positioned far from the telomere, such as CyO and TM3, usually allow the recovery of recombinant chromosomes from crossovers distal to the breakpoint, our observations indicate that balancers that carry distal deleterious mutations do not. While this mechanism provides a simple, straightforward and perhaps obvious explanation of the unexpected stability of stocks carrying distal mutations, we are not aware of another explicit demonstration.
This report adds detail to our understanding of the most frequently used balancers in Drosophila. We trust our observations improve the usefulness and increase the appreciation of these remarkable chromosomes.
Acknowledgments
We are grateful for the support and input of our colleagues at Indiana University and the Stowers Institute for Medical Research. We thank FlyBase (supported by NIH grant U41 HG000739 and MRC grant N030117/1), Drosophila Genomics and Genetic Resources at the Kyoto Institute of Technology, the Developmental Studies Hybridoma Bank (supported by NICHD and the University of Iowa) and the Indiana University Light Microscopy Imaging Center for their services; Steve Stowers for cloning vectors; Norbert Perrimon for the esg-lacZ stock; Bob Levis for helpful discussions; Angela L. Miller for editing and figure preparation; and Dmitri Petrov for supporting the Nanopore sequencing performed by B.Y.K. L.K. and K.R.C. were supported by NIH grant P40 OD018537. M.J.D. and B.R.C. were supported by NIH grant R01 GM113107. K.B. and N.S.S. were supported by NIH grant R01 GM124220. R.S.H. is an American Cancer Society Research Professor.
Footnotes
Supplemental material available at figshare: https://doi.org/10.25387/g3.12996710.
Communicating editor: B. Reed
Literature Cited
- Anderson J. A., Song Y. S., and Langley C. H., 2008. Molecular Population Genetics of Drosophila Subtelomeric DNA. Genetics 178: 477–487. 10.1534/genetics.107.083196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahina K., Watanabe K., Duistermars B. J., Hoopfer E., González C. R. et al. , 2014. Tachykinin-expressing neurons control male-specific aggressive arousal in Drosophila. Cell 156: 221–235. 10.1016/j.cell.2013.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., 1972. New Mutants Report. Drosoph. Inf. Serv. 49: 34. [Google Scholar]
- Asif-Laidin A., Delmarre V., Laurentie J., Miller W. J., Ronsseray S. et al. , 2017. Short and long-term evolutionary dynamics of subtelomeric piRNA clusters in Drosophila. DNA Res. 24: 459–472. 10.1093/dnares/dsx017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas S., Sharrow M., Kotu V., Middleton M., Nguyen K. et al. , 2011. Sugar-free frosting, a homolog of SAD kinase, drives neural-specific glycan expression in the Drosophila embryo. Development 138: 553–563. 10.1242/dev.055376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badal M., Portela A., Baldrich E., Marcos R., Cabré O. et al. , 2006a An FB-NOF mediated duplication of the white gene is responsible for the zeste 1 phenotype in some Drosophila melanogaster unstable strains. Mol. Genet. Genomics 275: 35 10.1007/s00438-005-0068-6 [DOI] [PubMed] [Google Scholar]
- Badal M., Portela A., Xamena N., and Cabré O., 2006b Molecular and bioinformatic analysis of the FB-NOF transposable element. Gene 371: 130–135. 10.1016/j.gene.2005.11.020 [DOI] [PubMed] [Google Scholar]
- Badal M., Xamena N., and Cabré O., 2013. FB-NOF is a non-autonomous transposable element, expressed in Drosophila melanogaster and present only in the melanogaster group. Gene 526: 459–463. 10.1016/j.gene.2013.04.082 [DOI] [PubMed] [Google Scholar]
- Bardin A. J., Perdigoto C. N., Southall T. D., Brand A. H., and Schweisguth F., 2010. Transcriptional control of stem cell maintenance in the Drosophila intestine. Development 137: 705–714. 10.1242/dev.039404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse F., and Pret A.-M., 2003. Apoptosis-mediated cell death within the ovarian polar cell lineage of Drosophila melanogaster. Development 130: 1017–1027. [DOI] [PubMed] [Google Scholar]
- Biessmann H., and Mason J. M., 1988. Progressive loss of DNA sequences from terminal chromosome deficiencies in Drosophila melanogaster. EMBO J. 7: 1081–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges C. B., and Brehme K. S., 1944. The Mutants of Drosophila Melanogaster Publs Carnegie Instn 552: vii + 257pp. [Google Scholar]
- Bridges C. B. and Morgan T. H., 1923. The third-chromosome group of mutant characters of Drosophila melanogaster Publs Carnegie Instn 327: 1–251. [Google Scholar]
- Brodsky M. H., Weinert B. T., Tsang G., Rong Y. S., McGinnis N. M. et al. , 2004. Drosophila melanogaster MNK/Chk2 and p53 Regulate Multiple DNA Repair and Apoptotic Pathways following DNA Damage. Mol. Cell. Biol. 24: 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buddika K., Ariyapala I. S., Hazuga M. A., Riffert D., and Sokol N. S., 2020. Canonical nucleators are dispensable for stress granule assembly in Drosophila intestinal progenitors. J. Cell Sci. 133: jcs243451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres M., Ranz J. M., Barbadilla A., Long M., and Ruiz A., 1999. Generation of a Widespread Drosophila Inversion by a Transposable Element. Science 285: 415–418. [DOI] [PubMed] [Google Scholar]
- Casals F., Cáceres M., and Ruiz A., 2003. The Foldback-like Transposon Galileo Is Involved in the Generation of Two Different Natural Chromosomal Inversions of Drosophila buzzatii. Mol. Biol. Evol. 20: 674–685. [DOI] [PubMed] [Google Scholar]
- de Celis J. F., Llimargas M., and Casanova J., 1995. Ventral veinless, the gene encoding the Cf1a transcription factor, links positional information and cell differentiation during embryonic and imaginal development in Drosophila melanogaster. Development 121: 3405–3416. [DOI] [PubMed] [Google Scholar]
- Cingolani P., Platts A., Wang L. L., Coon M., Nguyen T. et al. , 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron J. M., Ratnappan R., and Bailin S., 2012. The many landscapes of recombination in Drosophila melanogaster. PLoS Genet. 8: e1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P., Auton A., Abecasis G., Albers C. A., Banks E. et al. , 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delprat A., Negre B., Puig M., and Ruiz A., 2009. The transposon Galileo generates natural chromosomal inversions in Drosophila by ectopic recombination. PLoS One 4: e7883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexter J. S., 1914. The Analysis of a Case of Continuous Variation in Drosophila by a Study of Its Linkage Relations. Am. Nat. 48: 712–758. [Google Scholar]
- Diaz-Benjumea F. J., and García-Bellido A., 1990. Genetic analysis of the wing vein pattern of Drosophila. Roux’s Archives Dev Biology Official Organ Edbo 198: 336–354. [DOI] [PubMed] [Google Scholar]
- Duncan D. M., Burgess E. A., and Duncan I., 1998. Control of distal antennal identity and tarsal development in Drosophila by spineless-aristapedia, a homolog of the mammalian dioxin receptor. Genes Dev. 12: 1290–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabini G., Freilinger A., Altmann F., and Wilson I. B., 2001. Identification of core alpha 1,3-fucosylated glycans and cloning of the requisite fucosyltransferase cDNA from Drosophila melanogaster. Potential basis of the neural anti-horseadish peroxidase epitope. J. Biol. Chem. 276: 28058–28067. [DOI] [PubMed] [Google Scholar]
- Ghavi-Helm Y., Jankowski A., Meiers S., Viales R. R., Korbel J. O. et al. , 2019. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat. Genet. 51: 1272–1282. 10.1038/s41588-019-0462-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso, D. Bordne, and K. White, 1996 Deletions and lethals in the 66A region of Drosophila. Drosophila Information Service 77: 94–96.
- Gray Y. H., Tanaka M. M., and Sved J. A., 1996. P-element-induced recombination in Drosophila melanogaster: hybrid element insertion. Genetics 144: 1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzwiller F., Carmo C. R., Miller D. E., Rice D. W., Newton I. L. G. et al. , 2015. Dynamics of Wolbachia pipientis Gene Expression Across the Drosophila melanogaster Life Cycle. G3 (Bethesda) 5: 2843–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase A., Stern M., Wächtler K., and Bicker G., 2001. A tissue-specific marker of Ecdysozoa. Dev. Genes Evol. 211: 428–433. [DOI] [PubMed] [Google Scholar]
- Hawley R. S., 1980. Chromosomal sites necessary for normal levels of meiotic recombination in Drosophila melanogaster. I. Evidence for and mapping of the sites. Genetics 94: 625–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y., and Igarashi Y., 1994. On the category of naturally occurring inversions of Drosophila melanogaster. Jpn. J. Genet. 69: 105–118. [DOI] [PubMed] [Google Scholar]
- Jan L. Y., and Jan Y. N., 1982. Antibodies to horseradish peroxidase as specific neuronal markers in Drosophila and in grasshopper embryos. Proc. Natl. Acad. Sci. USA 79: 2700–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahsai L., and Cook K. R., 2018. Mapping Second Chromosome Mutations to Defined Genomic Regions in Drosophila melanogaster. G3 (Bethesda) 8: 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahsai L., Martin J.-R., and Winther A. M. E., 2010. Neuropeptides in the Drosophila central complex in modulation of locomotor behavior. J. Exp. Biol. 213: 2256–2265. [DOI] [PubMed] [Google Scholar]
- Koch I., Schwarz H., Beuchle D., Goellner B., Langegger M. et al. , 2008. Drosophila ankyrin 2 is required for synaptic stability. Neuron 58: 210–222. [DOI] [PubMed] [Google Scholar]
- Kolmogorov M., Yuan J., Lin Y., and Pevzner P. A., 2019. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37: 540–546. [DOI] [PubMed] [Google Scholar]
- Kurzhals R. L., Titen S. W. A., Xie H. B., and Golic K. G., 2011. Chk2 and p53 are haploinsufficient with dependent and independent functions to eliminate cells after telomere loss. PLoS Genet. 7: e1002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson J., Zhang J., and Rasmuson-Lestander A., 1996. Mutations in the Drosophila melanogaster gene encoding S-adenosylmethionine synthetase suppress position-effect variegation. Genetics 143: 887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. B., Kim W., Lee S., and Chung J., 2007. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem. Biophys. Res. Commun. 358: 534–539. [DOI] [PubMed] [Google Scholar]
- Lee J. H., Lee E., Park J., Kim E., Kim J. et al. , 2003. In vivo p53 function is indispensable for DNA damage-induced apoptotic signaling in Drosophila. FEBS Lett. 550: 5–10. [DOI] [PubMed] [Google Scholar]
- Levine A. J., 2020. P53 and The Immune Response: 40 Years of Exploration—A Plan for the Future. Int. J. Mol. Sci. 21: 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levis R. W., Ganesan R., Houtchens K., Tolar L. A., and Sheen F., 1993. Transposons in place of telomeric repeats at a Drosophila telomere. Cell 75: 1083–1093. [DOI] [PubMed] [Google Scholar]
- Li H., and Durbin R., 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J. et al. , 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley D. L., and Grell E. H., 1967. Genetic Variations of Drosophila melanogaster. Publs Carnegie Instn 627: 469pp.
- Mason J. M., Ransom J., and Konev A. Y., 2004. A Deficiency Screen for Dominant Suppressors of Telomeric Silencing in Drosophila. Genetics 168: 1353–1370. 10.1534/genetics.104.030676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason, J. M., and A. Villasante, 2013 Subtelomeres in Drosophila and Other Diptera. In: E. Louis and M. Becker (eds) Subtelomeres pp. 211–225. Springer, Berlin, Heidelberg. 10.1007/978-3-642-41566-1_12 [DOI]
- Mehrotra S., Maqbool S. B., Kolpakas A., Murnen K., and Calvi B. R., 2008. Endocycling cells do not apoptose in response to DNA rereplication genotoxic stress. Genes Dev. 22: 3158–3171. 10.1101/gad.1710208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micchelli C. A., and Perrimon N., 2006. Evidence that stem cells reside in the adult Drosophila midgut epithelium. Nature 439: 475–479. 10.1038/nature04371 [DOI] [PubMed] [Google Scholar]
- Miller D. E., Cook K. R., Arvanitakis A. V., and Hawley R. S., 2016a Third Chromosome Balancer Inversions Disrupt Protein-Coding Genes and Influence Distal Recombination Events in Drosophila melanogaster. G3 (Bethesda) 6: 1959–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D. E., Cook K. R., and Hawley R. S., 2019. The joy of balancers. PLoS Genet. 15: e1008421 10.1371/journal.pgen.1008421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D. E., Cook K. R., Hemenway E. A., Fang V., Miller A. L. et al. , 2018a The Molecular and Genetic Characterization of Second Chromosome Balancers in Drosophila melanogaster. G3 (Bethesda) 8: 1161–1171. 10.1534/g3.118.200021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D. E., Cook K. R., Kazemi N. Y., Smith C. B., Cockrell A. J. et al. , 2016b Rare recombination events generate sequence diversity among balancer chromosomes in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 113: E1352–E1361. 10.1073/pnas.1601232113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D. E., Staber C., Zeitlinger J., and Hawley R. S., 2018b Highly Contiguous Genome Assemblies of 15 Drosophila Species Generated Using Nanopore Sequencing. G3 (Bethesda) 8: 3131–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morata G., and Lawrence P. A., 1978. Anterior and posterior compartments in the head of Drosophila. Nature 274: 473–474. 10.1038/274473a0 [DOI] [PubMed] [Google Scholar]
- Morgan T., Bridges C., and Schultz J., 1937. Constitution of the germinal material in relation to heredity. Yearb. Carnegie Instn 36: 298–305. [Google Scholar]
- Moschetti R., Marsano R. M., Barsanti P., Caggese C., and Caizzi R., 2004. FB elements can promote exon shuffling: a promoter-less white allele can be reactivated by FB mediated transposition in Drosophila melanogaster. Mol. Genet. Genomics 271: 394–401. 10.1007/s00438-004-1007-7 [DOI] [PubMed] [Google Scholar]
- Muller H. J., 1918. Genetic Variability, Twin Hybrids and Constant Hybrids, in a Case of Balanced Lethal Factors. Genetics 3: 422–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nüsslein-Volhard C., Wieschaus E., and Kluding H., 1984. Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster: I. Zygotic loci on the second chromosome. Wihelm Roux Arch. Dev. Biol. 193: 267–282. 10.1007/BF00848156 [DOI] [PubMed] [Google Scholar]
- O’Brien L. E., Soliman S. S., Li X., and Bilder D., 2011. Altered modes of stem cell division drive adaptive intestinal growth. Cell 147: 603–614. 10.1016/j.cell.2011.08.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo D. J., Puerma E., and Aguadé M., 2019. The molecular characterization of fixed inversions breakpoints unveils the ancestral character of the Drosophila guanche chromosomal arrangements. Sci Rep-uk 9: 1706 10.1038/s41598-018-37121-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne F., 1918. An experiment to test the nature of the variations on which selection acts. Indiana Univ. Studies, 5: 1–45.
- Payne F., 1924. Crossover Modifiers in the Third Chromosome of Drosophila melanogaster. Genetics 9: 327–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phalke S., Nickel O., Walluscheck D., Hortig F., Onorati M. C. et al. , 2009. Retrotransposon silencing and telomere integrity in somatic cells of Drosophila depends on the cytosine-5 methyltransferase DNMT2. Nat. Genet. 41: 696–702. 10.1038/ng.360 [DOI] [PubMed] [Google Scholar]
- Qi S., and Calvi B. R., 2016. Different cell cycle modifications repress apoptosis at different steps independent of developmental signaling in Drosophila. Mol. Biol. Cell 27: 1885–1897. 10.1091/mbc.e16-03-0139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radion E., Morgunova V., Ryazansky S., Akulenko N., Lavrov S. et al. , 2018. Key role of piRNAs in telomeric chromatin maintenance and telomere nuclear positioning in Drosophila germline. Epigenetics Chromatin 11: 40 10.1186/s13072-018-0210-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranz J. M., Maurin D., Chan Y. S., von Grotthuss M., Hillier L. W. et al. , 2007. Principles of Genome Evolution in the Drosophila melanogaster Species Group. PLoS Biol. 5: e152 10.1371/journal.pbio.0050152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rein K., Zöckler M., and Heisenberg M., 1999. A quantitative three-dimensional model of the Drosophila optic lobes. Curr. Biol. 9: 93–96. 10.1016/S0960-9822(99)80021-9 [DOI] [PubMed] [Google Scholar]
- Reis M., Vieira C. P., Lata R., Posnien N., and Vieira J., 2018. Origin and consequences of chromosomal inversions in the virilis group of Drosophila. Genome Biol. Evol. 10: 3152–3166. 10.1093/gbe/evy239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roegiers F., Kavaler J., Tolwinski N., Chou Y.-T., Duan H. et al. , 2009. Frequent unanticipated alleles of lethal giant larvae in Drosophila second chromosome stocks. Genetics 182: 407–410. 10.1534/genetics.109.101808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y. S., Titen S. W., Xie H. B., Golic M. M., Bastiani M. et al. , 2002. Targeted mutagenesis by homologous recombination in D. melanogaster. Genes Dev. 16: 1568–1581. 10.1101/gad.986602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz J., 2008. In pursuit of the gene: from Darwin to DNA. Cambridge (MA): Harvard University Press. 10.4159/9780674043336 [DOI]
- Seppo A., Matani P., Sharrow M., and Tiemeyer M., 2003. Induction of neuron-specific glycosylation by Tollo/Toll-8, a Drosophila Toll-like receptor expressed in non-neural cells. Development 130: 1439–1448. 10.1242/dev.00347 [DOI] [PubMed] [Google Scholar]
- Shearin H. K., Macdonald I. S., Spector L. P., and Stowers R. S., 2014. Hexameric GFP and mCherry reporters for the Drosophila GAL4, Q, and LexA transcription systems. Genetics 196: 951–960. 10.1534/genetics.113.161141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow P., Patel N., Harrelson A., and Goodman C., 1987. Neural-specific carbohydrate moiety shared by many surface glycoproteins in Drosophila and grasshopper embryos. J. Neurosci. 7: 4137–4144. 10.1523/JNEUROSCI.07-12-04137.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogame N., Kim M., and Abrams J. M., 2003. Drosophila p53 preserves genomic stability by regulating cell death. Proc. Natl. Acad. Sci. USA 100: 4696–4701. 10.1073/pnas.0736384100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G., 1982. Spineless-aristapedia: a homeotic gene that does not control the development of specific compartments in Drosophila. Genetics 102: 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtevant A. H., 1926. A crossover reducer in Drosophila melanogaster due to inversion of a section of the third chromosome. Biol. Zent. Bl. 697: 702. [Google Scholar]
- Sturtevant A. H., 1931. Known and probable inverted sections of the autosomes of Drosophila melanogaster. Publications of the Carnegie Institution 421: 1–27. [Google Scholar]
- Tang Q., Su Z., Gu W., and Rustgi A. K., 2020. Mutant p53 on the Path to Metastasis. Trends Cancer 6: 62–73. 10.1016/j.trecan.2019.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tearle R., and Nüsslein-Volhard C., 1987. Tübingen mutants and stock list. Drosoph. Inf. Serv. 66: 209. [Google Scholar]
- Tinderholt V., 1960. New Mutants Report. Drosoph. Inf. Serv. 34: 53–54. [Google Scholar]
- Wagh D. A., Rasse T. M., Asan E., Hofbauer A., Schwenkert I. et al. , 2006. Bruchpilot, a Protein with Homology to ELKS/CAST, Is Required for Structural Integrity and Function of Synaptic Active Zones in Drosophila. Neuron 49: 833–844. 10.1016/j.neuron.2006.02.008 [DOI] [PubMed] [Google Scholar]
- Ward L., 1923. The Genetics of Curly Wing in Drosophila. Another Case of Balanced Lethal Factors. Genetics 8: 276–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H. B., and Golic K. G., 2004. Gene Deletions by Ends-In Targeting in Drosophila melanogaster. Genetics 168: 1477–1489. 10.1534/genetics.104.030882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi Y., Nishida Y., and Ip Y. T., 2010. Functional analysis of Toll-related genes in Drosophila. Dev. Growth Differ. 52: 771–783. 10.1111/j.1440-169X.2010.01213.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto-Hino M., Kanie Y., Awano W., Aoki-Kinoshita K. F., Yano H. et al. , 2010. Identification of genes required for neural-specific glycosylation using functional genomics. PLoS Genet. 6: e1001254 10.1371/journal.pgen.1001254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani U., Huang Z., and Terman J. R., 2008. The glucose transporter (GLUT4) enhancer factor is required for normal wing positioning in Drosophila. Genetics 178: 919–929. 10.1534/genetics.107.078030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., 2019. Advances in Experimental Medicine and Biology. Adv. Exp. Med. Biol. 1167: 105–112. 10.1007/978-3-030-23629-8_6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The accompanying tables contain complete complementation data. Stocks may be obtained from the Bloomington Drosophila Stock Center as indicated in the Reagent Table (Table S1). Basecalled Nanopore data are available from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/) under project number PRJNA623115. Data related to characterization of the Df(2L)bhe chromosome are available under project PRJNA623116. Original data underlying this manuscript can be accessed from the Stowers Original Data Repository at http://www.stowers.org/research/publications/LIBPB-1520. Custom scripts used for data analysis, including the genome assembly of Bloomington stock 2475, are available on GitHub (https://github.com/danrdanny/balancerPhenotypes). Supplemental material is available at figshare: https://doi.org/10.25387/g3.12996710.