

Abstract
An experiment was performed at the National Superconducting Cyclotron Laboratory using a 140 MeV/nucleon 48Ca beam and a flowing-water target to produce 47Ca for the first time with this production route. A production rate of 0.020 ± 0.004 47Ca nuclei per incoming beam particle was measured. An isotope harvesting system attached to the target was used to collect radioactive cationic products, including 47Ca, from the water on a cation-exchange resin. The 47Ca collected was purified using three separation methods optimized for this work: (1) DGA extraction chromatography resin with HNO3 and HCl, (2) AG MP-50 cation-exchange resin with an increasing concentration gradient of HCl, and (3) AG MP-50 cation-exchange resin with a methanolic HCl gradient. These methods resulted in ≥99 ± 2% separation yield of 47Ca with 100% radionuclidic purity within the limits of detection for HPGe measurements. Inductively coupled plasma-optical emission spectrometry (ICP-OES) was used to identify low levels of stable ions in the water of the isotope harvesting system during the irradiation and in the final purified solution of 47Ca. For the first time, this experiment demonstrated the feasibility of the production, collection, and purification of 47Ca through isotope harvesting for the generation of 47Sc for nuclear medicine applications.
Introduction
Several scandium isotopes have recently been identified as potential candidates for the theranostic treatment of metastatic cancers. Isotopes such as 43,44Sc are suitable for imaging with half-lives on the order of a few hours and low-energy positron emissions (43Sc: t1/2 = 3.891 h, Eavg,β = 476 keV and 44Sc: t1/2 = 3.97 h, Eavg,β = 632 keV).1−7 The ideal therapeutic partner for these diagnostic isotopes is 47Sc with a half-life of 3.35 days and a 100% low-energy β– emission (Eavg,β = 162 keV).8−11 Additionally, a low energy 159 keV γ ray accompanies this decay, allowing for visualization of the therapeutic dose with SPECT imaging.8
Though several routes have been explored, it has proven difficult to find a sustainable production method for 47Sc. Proton and neutron irradiations as well as photonuclear reactions on calcium and titanium targets have been explored (see Figure 1 for the relevant area in the chart of the nuclides).12−20 One obstacle encountered with these methods is the coproduction of other sufficiently long-lived scandium isotopes such as 44mSc (t1/2 = 2.44 days), 46Sc (t1/2 = 83.8 days), and 48Sc (t1/2 = 1.82 days). Among these contaminants, 46Sc and 48Sc emit high-energy and high-intensity γ rays (46Sc: 889 keV at 99.98%, 1121 keV at 99.99%; 48Sc: 984 and 1312 keV at 100%) that make them undesirable byproducts in terms of added radiation dose that does not contribute to therapy.21,22 To minimize these contaminants, enriched 46Ca, 48Ca, and 48Ti targets can be used. These low-abundance calcium isotopes are available only to a low level of enrichment and can be prohibitively expensive. Ti targets are particularly difficult to convert to and maintain in a form that is readily dissolved; this sensitivity limits irradiation intensities that can be used and extends the preparation time for the target material. Recycling methods must be used to recover enriched target material after each irradiation, reducing long-term cost of enriched targets but extending the processing time. Although some of these production routes show promise, a sustainable production route has not yet been fully developed.
Figure 1.
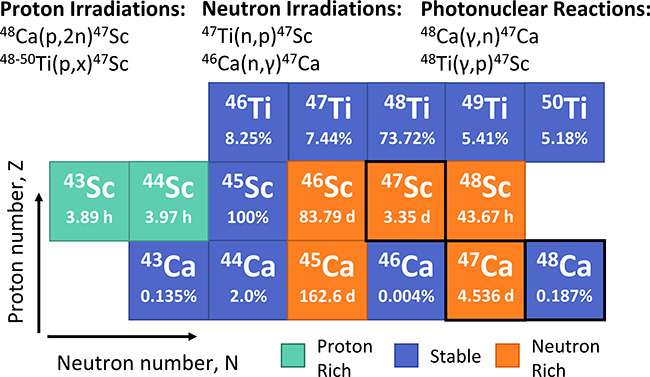
Key nuclei for the reactions discussed to produce 47Sc are shown. For stable nuclei, the natural abundance is given, and for radioactive nuclei, the half-life is given.
While work continues to find a long-term production route, an untapped supply of 47Sc exists at the National Superconducting Cyclotron Laboratory (NSCL) and will exist in the future at the Facility for Rare Isotope Beams (FRIB) when a primary 48Ca beam is used for a nuclear physics experiment.23 These facilities accelerate heavy (A ≥ 16), stable ion beams to approximately 50% the speed of light into thin targets. A small portion of the beam undergoes fragmentation reactions in which the stable beam is broken into smaller radioactive fragments, some of which are interesting for studies in nuclear physics. However, upwards of 90% of the accelerated stable beam at the NSCL goes unreacted at the target, is separated from the secondary exotic product beams, and is collected in a solid metal block known as a beam blocker. By replacing the current beam blocker with a flowing-water target (i.e., a metal shell with a hollow interior through which water flows), the radionuclides formed from this “left-over” beam can be accessed.24
Several previous experiments have been performed to test the feasibility of this isotope production method at the NSCL.24−28 These experiments range from collecting radioactive secondary beams in a water matrix to irradiating a prototype flowing-water target with a stable primary beam. This work has led to the development of a flowing-water target and extended water system for use in the beam blocker position at the NSCL (see Figure 2 for a simplified schematic and Figure S1 in the Supporting Information for a more detailed schematic).29 The water in this system serves as a cooling agent to remove heat from the target shell, as a target medium for isotope production where the accelerated beam reacts with the 16O and 1H nuclei in the water molecules, and as a transportation medium to move products from the target to collection components. Together, this method of production and collection of byproduct radionuclides at an accelerator facility is one example of isotope harvesting.23
Figure 2.
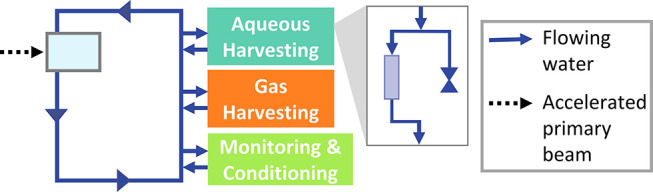
The harvesting system contains a main loop in which 37 ± 4 L of water are circulated from a reservoir at 10 L/min through a titanium alloy (Ti64) target shell (gray box with light blue interior). Subsystems off the main loop collect radioactive and stable ions from the water with ion-exchange resins, collect radioactive gases, and monitor and condition the water. The inset shows the flow of water through an ion-exchange resin and a valve-capped line for water sampling in the aqueous harvesting loop.
It is predicted that GBq (mCi equivalent) quantities of 47Ca can be produced during a typical 5 day experiment at the NSCL through fragmentation of the primary 48Ca beam in the water medium. Production of 47Ca allows for the generation of its daughter, 47Sc, without other scandium isotope impurities (see Figures 1 and 3). With a similar percentage of the unused beam and an almost 400 times increase in beam intensity for the 48Ca beam at FRIB, TBq (Ci equivalent) quantities of 47Ca are predicted to be formed in the beam dump during the regular operation of the facility when a 48Ca primary beam is used. This level of activity is more than enough to support research and preclinical work to determine the efficacy of 47Sc in the theranostic treatment of cancer. In these tests, it will be particularly important to compare the therapeutic application of 47Sc with that of the clinically available 177Lu. This work in turn could inspire or discourage further investments into finding a sustainable production method for clinical tests.
Figure 3.
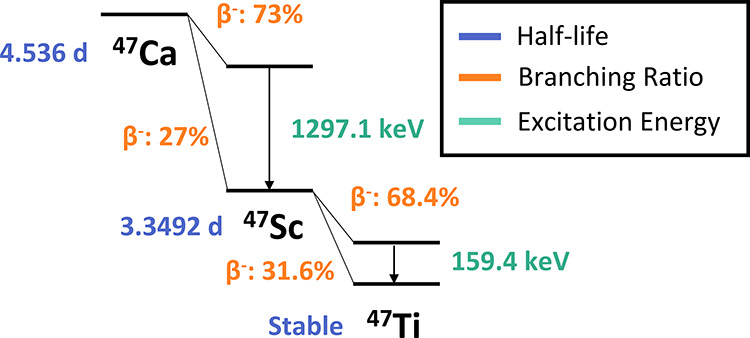
Radioactive decay scheme of 47Ca to 47Sc.11
In this study, a flowing-water target was irradiated for the first time with a 140 MeV/nucleon 48Ca beam to produce 47Ca. Results will be given for the measured production rate of 47Ca in this target, the collection and purification of the 47Ca produced, and the identification of stable contaminants in the isotope harvesting water during the irradiation. The radionuclidic purity and the level of stable ions in the purified samples from this work are used to predict the viability of using generated 47Sc for future radiolabeling experiments.
Materials and Methods
Materials
Reagents
Before the irradiation, the water in the isotope harvesting system was purified using mixed bed resins (McMaster-Carr, Filter media PVC water deionizer), resulting in a conductivity level of 250 nS/cm. Chemical processing of the products was performed with the following reagents: hydrochloric acid (VWR Chemicals, ACS grade, 36.5–38%), nitric acid (VWR Chemicals, ACS grade, 68–70%), methanol (Macron Fine Chemicals, anhydrous, ACS grade), and MilliQ water (Thermo Scientific MicroPure Ultrapure Water System, 18.2 MΩ cm).
Extraction Chromatography and Ion-Exchange Resins
DGA-exchange chromatography resin (N,N,N′,N′-tetra-n-octyldiglycolamide, normal resin, particle size 50–100 μm, TrisKem International) was dry loaded in a column and sequentially prerinsed with 20 mL of 5 M HCl, 5 M HNO3, and MilliQ water before use in the separations. Two cation-exchange resins (AG 50W-X8, mesh size 20–50, BioRad and AG MP-50, 100–200 mesh size, BioRad) were prepared in large quantities by rinsing with the following solutions twice: 50 mL of 2 M HCl, 50 mL of 4 M HCl, 50 mL of 6 M HCl, and 100 mL of MilliQ water. The rinsing steps described here were used to remove ionic impurities, especially metallic impurities, from the resins.
Column Construction
The columns used for collection and separation were made of rigid polycarbonate tubing (3/8″ OD, 1/4″ ID, McMaster-Carr, PN:9176T1) and push-to-connect fittings with a poly(butylene terephthalate) (PBT) body and stainless steel tube gripping clamps on both ends of the column (Pneumatic NITRA Union Reducer, 3/8″ to 1/4″). Two pieces of glass wool were used on each end of the column to stabilize the resin.
Instruments
Identification of stable ions was performed with an Agilent inductively coupled plasma-optical emission spectrometer (ICP-OES) (TCP700.) Identification and quantification of radionuclides were performed with an HPGe Canberra BEGe γ-ray Detector (BE2020). Energy and efficiency calibrations were previously performed with a 152Eu point source 50 cm from the detector face. Analysis of spectra was performed with Genie 2000 software (Mirion Technologies). The handling of nonstandard sample geometries is addressed in the Supporting Information.
48Ca Irradiation
A 140 MeV/nucleon 48Ca20+ beam was used to irradiate a flowing-water target over 8.5 h. Beam current measurements were recorded every second on average from the unsuppressed target, meaning the recorded values were proportional but not equivalent to the true beam current. Intermittently, an intercepting faraday cup was inserted into the beam to get an absolute measurement of the current. A linear trend between the true beam current values from the Faraday cup and the relative measurements on the target (see the Supporting Information) was used to calibrate the frequently recorded values from the target. After a short tuning period at an intensity of 0.26 pnA, the beam intensity was increased and maintained at an average of 0.92 pnA for about 5.1 h.
The irradiation was paused occasionally for samples of the system water to be withdrawn and ion-exchange resin beds to be removed and inserted (see Figure 2). According to the timeline in Figure 4, three 1 L water samples were taken during the irradiation, and a fourth sample was taken after the end of the irradiation (named Water Sample 1–4, respectively). After water sample 3 was withdrawn, a cation-exchange resin bed (AG 50W-X8, mesh size 20–50, H+ form, 1.5 g, 8.9 cm × 0.6 cm ID) was placed in the system with a water flow of approximately 180 mL/min over the resin bed. This cation-exchange resin bed was replaced with fresh resin beds after two more irradiation periods (resins 1, 2, and 3, respectively). Resin 3 was left in the system for 2 h after the irradiation ended with the flow rate increased to 500 mL/min for the second hour to increase the collection rate. Two more resin beds (resin 4 and 5, respectively) were put in the system in parallel the day after the irradiation and a final 1 L water sample was taken at the end of the collection effort (water sample 5). Altogether, activity was collected in five 1 L water samples and five cation-exchange resin beds that were used for further measurements and experiments.
Figure 4.
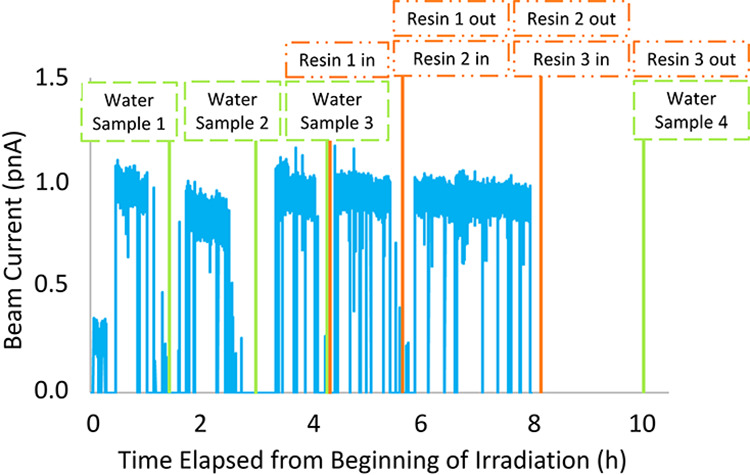
Beam current (blue), water sample collection (green), and resin bed changes (orange) are shown as a function of time. The beam current shown are the calibrated values of the current on the target.
Production of 47Ca
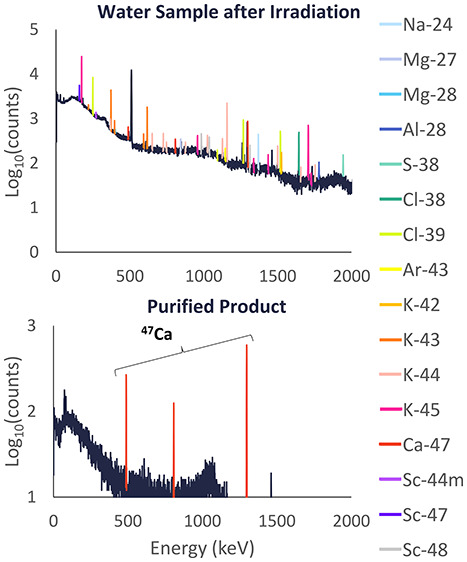
The activity of 47Ca found in the water samples and on the cation-exchange resin beds was measured with the HPGe detector. Analysis of these spectra was performed with Genie software to detect and integrate peaks, implement baseline corrections, and calculate efficiencies at each peak energy. This allowed for the detection and quantification of radionuclides based on their characteristic γ-ray energy emissions. Peaks from three characteristic γ rays were used to quantify the activity of 47Ca: 489.2 keV at 5.9 ± 1.2%, 807.9 keV at 5.9 ± 1.2%, and 1297.1 keV at 67 ± 13%.11
The total produced activity of 47Ca was estimated to be the sum of activity found in each water sample, on each of the collection ion-exchange resin beds, and in the water after collection with the resin beds. The activity remaining in the system water was approximated by measuring the activity in a water sample taken when the last column was removed from the system and scaling up this activity to account for the total remaining water volume of 33 ± 4 L. This large uncertainty in the water volume mainly resulted from difficulties in draining the water from all components and tubing in the system. Due to this 11% uncertainty in the total water volume of the system and 19–20% uncertainties in the branching ratios, the estimated activity has a large associated uncertainty. For future measurements of this production rate, other indirect methods of measurement will be used to determine the system water volume more accurately and these branching ratios will be remeasured to reduce the associated errors. More details on the production rate calculation are given in the Supporting Information.
The production rate of 47Ca in the flowing-water target of the isotope harvesting system has also been predicted using two simulation codes that predict the production rate of radionuclides in nuclear reactions: Particle and heavy ion transport code system (PHITS) and LISE++.30,31 For both estimates, a target was designed in the program to account for a 500 μm layer of Ti alloy followed by a water cavity. Additionally, both fragmentation reactions (at higher particle energies) and fusion evaporation reactions (at lower particle energies) with 16O and 1H were included in the production rate estimates.
Collection and Sample Processing
In addition to 47Ca, the following radionuclides were collected on the resins and identified with γ spectroscopy: 24Na, 27,28Mg, 42,43,44,45K, and 44m,47,48Sc. These activities were quantified using characteristic γ-rays observable above the limit of detection and with the branching ratios reported in the Evaluated Nuclear Structure Data File for each radionuclide (see Table S1 in the Supporting Information).6,7,11,21,22,32−36 The collection efficiency for 47Ca was calculated by comparing the total activity collected on the cation-exchange resin beds to the activity remaining in the system water, where the activity remaining in the water was approximated as described previously.
The 47Ca activity collected on five cation-exchange resin beds was removed with 50–70 mL of 3 M HNO3 per resin with a flow rate of 1.8–2.0 mL/min. This solution contained a mixture of cationic radionuclides from each resin and was separated into several fractions for use in three separation methods. Method 1 used the eluate directly as the load solution. Fractions used for methods 2 and 3 were evaporated to dryness on a rotary evaporator and reconstituted in 0.1 M HCl and 0.5 M HCl/90% methanol, respectively. See Figure 5 for a schematic overview of the sample processing.
Figure 5.
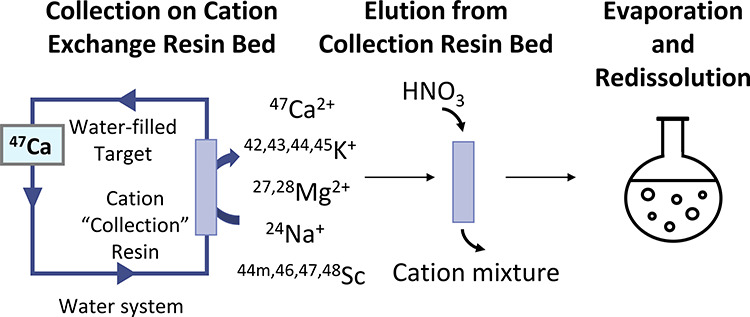
Sample processing was performed in this manner except for samples used with separation method 1, which were used directly after elution from the collection resin.
Purification of 47Ca
The following three methods were developed and optimized in this work to separate Ca from Na, Mg, K, Sc, and Fe. These elements took the form of 24Na, 27,28Mg, 42,43,44,45K, 44m,47,48Sc, and stable Fe during the experiment. After each separation, the columns were rinsed with water for storage.
Separation Method 1: DGA Resin with HCl and HNO3
A column of 1.07 g of DGA resin (dry packed, 7.2 cm × 0.6 cm ID) was preconditioned with 20 mL of 3 M HNO3 and a 10–20 mL sample of the collection column eluate in 3 M HNO3 was loaded at a flow rate of 1.8 mL/min. These conditions were chosen to elute many of the coproduced radionuclides such as 24Na, 28Mg, and 43,44K as well as stable Fe while retaining 47Ca on the resin. The pump rate was decreased to 1.3 mL/min and an additional 10–15 mL of 3 M HNO3 was used to rinse the column and ensure all Na, Mg, and K isotopes were entirely removed. Elution of 47Ca was carried out with approximately 20 mL of 3 M HCl to selectively remove 47Ca and leave the Sc isotopes adsorbed to the resin.
Separation Method 2: AG MP-50 with HCl
A column of 1.5 g AG MP-50 resin (slurry packed, 7.5 cm × 0.6 cm ID) was preconditioned with 0.1 M HCl. Evaporated fractions from the collection columns were redissolved in 20 mL of 0.1 M HCl. This concentration was used since Ca has a large distribution coefficient with AG MP-50 at this condition, allowing for the creation of a narrow 47Ca band during the loading step. This solution was loaded on the column and was followed by rinse steps of 10 mL of 0.1 M and 23–25 mL of 2 M HCl. Rinsing with these HCl concentrations allowed for the elution of coproduced radionuclides such as 7Be, 24Na, 28Mg, 43,44K, and stable Fe. Then, 10 mL of 5 M HCl was used to elute 47Ca while retaining the Sc isotopes on the column. This separation was carried out with a flow rate of 0.8 mL/min throughout.
Separation Method 3: AG MP-50 with Methanolic HCl
One gram of AG MP-50 resin (slurry packed, 5 cm × 0.6 cm ID) was preconditioned with 0.5 M HCl in 90% methanol. Evaporated fractions from the collection columns were reconstituted in 20 mL of 0.5 M HCl in 90% methanol and the solution was loaded on the column. Rinse solutions of 0.5 M HCl/90% methanol (20 mL, 1 mL/min), 2 M HCl/60% methanol (50 mL, 0.75 mL/min), and 2 M HCl/30% methanol (35 mL, 0.75 mL/min) were used in succession. The load and rinse solution of 0.5 M HCl/90% methanol was chosen due to the high distribution coefficient of Ca and the low distribution coefficient of Fe under these conditions. Therefore, stable Fe could be eluted in this step while forming a narrow 47Ca band on the column during loading. The two intermediate rinse steps were used to elute coproduced radionuclides such as 7Be, 24Na, 28Mg, and 43,44K. Specifically, the 2 M HCl/30% methanol rinse step was added during the optimization of this separation to allow for complete separation of the K isotopes from 47Ca. Elution of 47Ca from the column was performed with 15 mL of 4 M HCl at a flow rate of 1.25 mL/min. The high distribution coefficient of Sc for all rinse media used in this separation means all Sc isotopes remain on the column.
Separation Yield and Radionuclidic Purity
For each separation performed, the separation yield was calculated as the activity of 47Ca in the purified fractions divided by the total activity loaded for the separation. The radionuclidic purity was found as the activity of 47Ca in the purified fractions divided by the total activity in these fractions. Both the separation yield and radionuclidic purity of the purified 47Ca were found using the rate at which characteristic γ rays for each radionuclide were observed by the γ detector to avoid using the reported branching ratios and their large associated errors for 47Ca. This was made possible as all spectra were taken at 25 cm from the detector face and the yield and purity were calculated in terms of ratios, avoiding the need for absolute activities. Therefore, the only error considered in the γ ray rates measured was statistical counting errors. Since the separation yield and radionuclidic purity of the 47Ca processed with each of the separation methods were high (i.e., 100% separation yield and radionuclidic purity), the limit of detection for HPGe measurements of 47Ca in samples around the 47Ca elution peak and 43K for samples in the 47Ca elution peak were found. See the Supporting Information for more details.
Any activity of 45Ca that was present in the purified 47Ca fractions was not measurable due to the absence of reasonably intense γ-rays from this radionuclide (i.e., the only γ emission is 12.47 keV at an intensity of 3 × 10–6%). While this calcium isotope, if present, would follow 47Ca through all separation methods, it would also follow 47Ca through the previously published pseudo generator.15 Additionally, 45Ca decays slowly (t1/2 = 162.61 d) to stable 45Sc, which would not affect the radionuclidic purity and only minorly affect the specific activity of the final radiolabeling solution. Since 45Ca should not interfere with the generation of highly radiopure 47Sc in future work, the radionuclidic purity reported for 47Ca does not consider any 45Ca present in the purified product.
Stable Elemental Analysis
Samples from water samples 1–4 and the final purified 47Ca solution containing fractions from each separation performed were analyzed using a semiquantitative method on ICP-OES.37 This analysis was performed to identify and semiquantify stable ions above the limit of detection of the instrument with a one-point calibration semiquantification method for 69 elements. Among these elements were those that would indicate corrosion of the target or metal components in the system (e.g., Ti, V, Al, Fe, Ni, Cr) and elements that have been previously identified as common contaminants in the isotope harvesting system38 (e.g., Na, Mg, Ca, Si, Zn). More details on this method and sample preparation are given in the Supporting Information.
Results and Discussion
Production of 47Ca
The total activity of 47Ca measured and decay corrected to the end of the irradiation was 3.7 ± 0.7 MBq (100 ± 20 μCi). The errors considered in this measurement are from counting statistics, errors in reported γ-ray branching ratios, and an uncertainty in the total water volume of the system. By far, the dominant factor is the error in the reported branching ratios with 19–20% error for the three main γ rays. Table S2 in the Supporting Information gives the decay corrected activity to the end of the irradiation for 47Ca in each of the samples measured.
A production rate of 0.020 ± 0.004 47Ca nuclei produced per incoming 48Ca nuclei was measured. The reported error for this rate is solely from the uncertainty in the quantification of the total activity of 47Ca produced. The production rate can also be thought of as a 2.0 ± 0.4% conversion rate of beam particles to the desired nucleus, which is relatively high for a charged particle irradiation. In comparison, this rate is 10–20 times higher than that for 18F through the routine production route of 18O(p,n)18F.39
The predicted production rates using both PHITS and LISE++ are given in Table 1. These predictions are significantly lower than the production rate measured in this work. This difference demonstrates the importance of measuring the production rate of radionuclides in the isotope harvesting system as predictions have been found to be inaccurate as seen previously with a 40Ca beam experiment with a water target at the NSCL.38
Table 1. Predicted and Measured Production Rates of 47Ca in Isotope Harvesting Water Target with a 140 MeV/Nucleon 48Ca Beama.
| source of production rate | production rate (%) |
|---|---|
| experimentally measured | 2.0 ± 0.4 |
| predicted with PHITS | 1.19 |
| predicted with LISE++ | 1.03 |
The production rate is given as the percent of beam particles converted to 47Ca.
The measured production rate can be extrapolated to higher intensity irradiations anticipated at the NSCL and FRIB. This allows for more detailed safety and experimental planning for future isotope harvesting of 47Ca. Approximately 4.8 GBq (130 mCi) would be expected at the end of a 120 h irradiation with a 140 MeV/nucleon 80 pnA 48Ca beam, assuming 90% of the primary beam is directed to the isotope harvesting beam blocker. Since 47Ca would be produced as a byproduct of the NSCL experimental program, this estimate uses the average length of a nuclear physics experiment at the NSCL and the standard settings for the available 48Ca beam. Without any dedicated beamtime or additional use of enriched 48Ca, a significant supply of 47Ca could be produced for research purposes during normal NSCL operations.
The measured production rate can be extended to the 48Ca beam at FRIB as an underestimation of the potential production of 47Ca. At FRIB, the 48Ca beam that reaches the isotope harvesting beam dump will have an estimated energy of 189 MeV/nucleon. With a higher energy beam, a larger fraction of the beam particles undergoes fragmentation reactions, resulting in a higher production rate of 47Ca. These principles were supported using LISE++ (see the Supporting Information).30 While it has been noted that the absolute production rates predicted from LISE++ differ from the experimentally measured rate, this program can provide reliable relative trends. Using the production rate measured in this experiment, a 1 day irradiation of the isotope harvesting beam dump at FRIB full beam power (189 MeV/nucleon 30 pμA 48Ca beam, 86% primary beam transmission to beam dump) would produce >520 GBq (14 Ci) of 47Ca. As with the 47Ca produced at the NSCL, the 47Ca production at FRIB will occur simultaneously with the nuclear physics program as the unused primary beam from these experiments is stopped in an aqueous beam dump.40
Collection and Sample Processing
The efficiency with which 47Ca was collected from the system on resins 1–3 for about 5.5 h on the day of the irradiation was found to be 65 ± 1%. Two additional resin beds were used to increase the collected fraction, resulting in 82 ± 3% of the 47Ca collected on five resins. The errors considered for these efficiencies result from counting statistics and an uncertainty in the volume of water in the system. Offline experiments have demonstrated that increasing the flow through the ion-exchange resins can increase the overall collection rate. While there is a lower collection efficiency “per pass” of water, the high flow rate increases the frequency of water passing over the column.29 For future experiments in which the half-life of the radionuclide of interest is short, the flow rate through ion-exchange resins can be increased to expedite the collection process.
In addition to 47Ca, other cationic radionuclides were collected on the cation-exchange resin beds. The activities on these resins decay corrected to when they were removed from the system are given in Table 2. Short-lived radionuclides were not observed on resin 3 because it was removed hours after the irradiation ended. Therefore, the activities on resins 1 and 2 give an understanding of the radionuclides and their activities that would be encountered soon after an irradiation and the activities on resin 3 represent the activities that would be present after a 21 h cool-down period.
Table 2. Quantification of Radionuclides Collected on Cation-Exchange Resins 1–3a.
| activity
collected (kBq) |
activity
eluted (%) |
||||||
|---|---|---|---|---|---|---|---|
| nuclide | half-life | resin 1 | resin 2 | resin 3 | resin 1 | resin 2 | resin 3 |
| 7Be | 53.22 days | 12(2) | 21(2) | 100(22) | 100.0(2) | ||
| 24Na | 14.997 h | 92(4) | 250(10) | 290(10) | 100(1) | 100.0(7) | 100.0(1) |
| 27Mg | 9.458 month | 26(2) | |||||
| 28Mg | 20.915 h | 13(1) | 37(3) | 46(3) | 100(3) | 100(2) | 100.0(1) |
| 42K | 12.355 h | 280(8) | 740(50) | 990(70) | 100(2) | 100(1) | 100.0(1) |
| 43K | 22.3 h | 236(5) | 639(8) | 930(10) | 100.0(3) | 99.8(2) | 100.0(1) |
| 44K | 22.13 month | 650(100) | 1.0(2) × 103 | ||||
| 45K | 17.18 month | 470(30) | 560(40) | ||||
| 47Ca | 4.536 days | 300(60) | 880(170) | 1.2(2) × 103 | 99.8(7) | 93.9(4) | 92.42(1) |
| 44mSc | 58.61 h | 5.7(5) | 15.5(6) | 16.5(9) | 75(4) | 57(3) | 64.97(8) |
| 47Sc | 3.3492 days | 53(3) | 159(9) | 310(20) | 79(2) | 67(3) | 62.9(1) |
| 48Sc | 43.67 h | 30.3(6) | 85(2) | 85(2) | 77(1) | 60(1) | 64.98(4) |
| 46Sc | 83.79 days | 1.8(3) | 3.9(4) | 3.6(4) | 100(20) | 60(14) | 100.0(1) |
The activities collected with and eluted from resin beds 1–3 are reported with the uncertainty in parentheses afterwards. The activities eluted from resin beds 1–3 were achieved in 70, 62, and 55 mL, respectively. Additionally, the activity eluted for each radionuclide is given as a percentage of the activity removed from each column based on the measurements of each column before and after elution.
Using 3 M HNO3, an average of 96 ± 1% (n = 4) of the 47Ca collected on the cation-exchange resins was removed with 50–70 mL. The highest removal rate was observed when 70 mL were used to remove 99.8 ± 0.7% of the 47Ca from Resin 1 and the lowest was observed when 55 mL were used to remove 89 ± 2% from Resin 4. The sodium, magnesium, and potassium isotopes were entirely eluted from the collection resins, while the scandium isotopes were eluted to a lesser extent (Table 2). The first 50–55 mL of 3 M HNO3 used to remove 47Ca from each column were used in the separations. When more than this volume was used in the elution from the collection resins, the last few milliliters contained a low activity of 47Ca due to tailing elution behavior.
For the fractions evaporated to dryness for separation methods 2 and 3, the activity was reconstituted in the proper matrix for each load solution with a high yield: >99% for separation method 2 and >98% for separation method 3. Since no 47Ca was detected on the flasks after the transfer, these yields are lower limits found using the limit of detection in the γ spectra at the 47Ca characteristic energies.
In a wide range of oxidation potentials and pH values, calcium is expected to be in the divalent state in water. This simple chemistry allows for the 47Ca produced in this harvesting experiment to be easily removed from the water and processed in the laboratory with high yields. The ease of working with calcium is a major advantage to harvesting 47Ca for use in a 47Ca/47Sc radionuclide generator. Other elements that are easily hydrolyzed in near-neutral pH conditions, such as 48V and 88Zr, have also been produced through isotope harvesting.28 These radionuclides, with more complicated chemistries, have proven less easily collected and chemically modified offline.
Purification
Separation Methods
Each separation method was performed three times to confirm the elution profiles of all radionuclides involved and the separation yield and radionuclidic purity for 47Ca. A representative elution profile for the replicate with the most finely collected fractions for each method is given in Figure 6 with details of the fraction volumes and compositions given in Tables S4–S6 in the Supporting Information. The error bars in Figure 6 result only from statistical uncertainties in the detection method.
Figure 6.
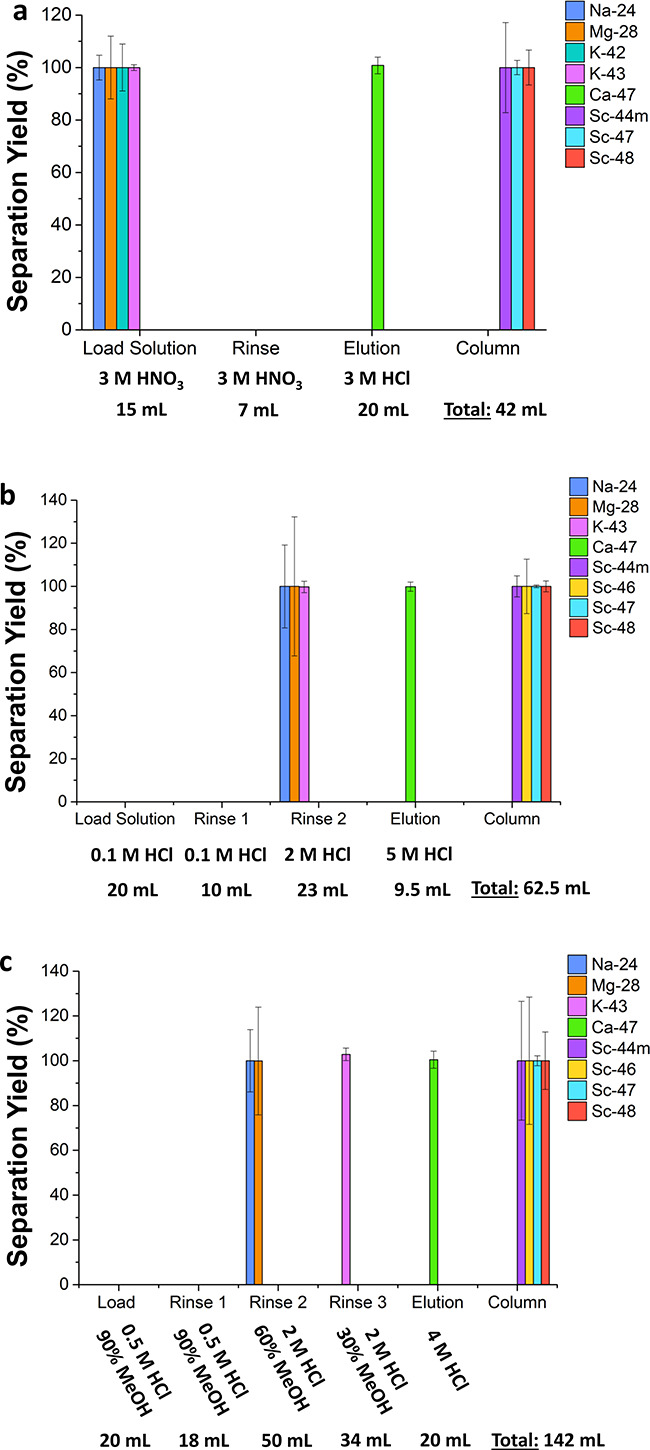
Separation profiles for methods 1–3 (1: DGA with HNO3/HCl, 2: AG MP-50 resin with an HCl gradient, and 3: AG MP-50 resin with HCl/methanol gradient) are shown in a–c, respectively. The liquid phase as well as its composition and volume are indicated along the x-axis for each separation method. The “column” label to the right side of each figure shows the species that remained on the column through the separation. The error bars are from counting statistics for one replicate of each method and not from deviations across replicates, with large errors resulting from low activities in the samples.
Depending on the time each separation was performed, a slightly different mix of radionuclides was identified due to their half-lives and production rates. For example, separation methods 2 and 3 required optimization, so 42K was not observed in the final elution profiles for either of these methods. As would be expected, 42K followed the elution pattern of 43K in all separations performed in this work, including the separations performed to optimize these two methods. Therefore, it can be confidently assumed that the elution profile for 42K is the same as that of 43K in the final protocols for methods 2 and 3. Conversely, 46Sc was not observed for any of the three replicates performed for separation method 1 due to the level of activity of other radionuclides at the time of separation and the relatively long half-life of 46Sc. However, this scandium isotope should behave identically to 44mSc, 47Sc, and 48Sc, and each of these isotopes were found exclusively on the DGA resin after the elution of 47Ca.
Two radionuclides appear during purification as daughters of isotopes produced during the irradiation: 28Al as the daughter of 28Mg and 44Sc as the daughter of 44mSc. The half-life of 28Al (t1/2 = 2.245 min) is so short that it has an apparent elution with 28Mg in all separations. Like all of the scandium isotopes observed, 44Sc remained on the column through each of these separations. Therefore, neither of these daughters affected the radionuclidic purity of the final 47Ca sample.
Separation Yield and Radionuclidic Purity
The average separation yields and radionuclidic purities across three replicates for 47Ca processed with these separation methods are quite high as shown in Table 3. The only separation that demonstrated less than 100% separation yield was one of the replicates for separation method 3, and even then, a 99 ± 2% average separation yield with high radionuclidic purity was demonstrated. The limit of detection (LOD) with HPGe measurements for 47Ca and 43K for the separation yield and radionuclidic purity, respectively, are shown to be within the statistical errors accounted for in the final values. Therefore, within the limits of detection, these methods resulted in 99–100% separation yield with 100% radionuclidic purity in the 47Ca recovered. This high purity indicates that any of the three methods would facilitate the generation of 47Sc with high radionuclidic purity for radiolabeling applications.
Table 3. Separation Yield and Radionuclidic Purity of 47Ca for Three Separation Methodsa.
| separation method | average separation yield (%) | LOD for 47Ca (%) | radionuclidic purity (%) | LOD for 43K (%) |
|---|---|---|---|---|
| 1 | 100 ± 1 | 0.9 | 100 ± 4 | 0.5 |
| 2 | 100 ± 2 | 0.5 | 100 ± 4 | 1 |
| 3 | 99 ± 2 | 0.8 | 100 ± 5 | 0.9 |
Separation method 1 used DGA resin with HNO3/HCl, 2 used AG MP-50 resin with an HCl gradient, and 3 used AG MP-50 resin with HCl/methanol gradient.
Comparing Separation Methods
Each of the three separation methods used in this work resulted in 10–20 mL of 3–5 M HCl containing essentially 100% of the 47Ca loaded on the column at 100% radionuclidic purity. This is a relatively small volume at an appropriate acid concentration for use as the loading solution in a previously published pseudo generator procedure to be used in future experiments.15 With these results, any of the three methods could be used for further work with harvested 47Ca. Once a method is selected, elution of 47Ca from the collection resin bed and sample processing will be tailored to the selected method to avoid an evaporation step in future experiments. For example, the collection resin bed could be processed with HCl instead of HNO3. The resulting solution could then be diluted to the correct load solution concentration for separation methods 2 and 3, avoiding the use of an evaporation step.
The advantages and disadvantages of these methods are found in practical considerations for routine future use: volume required, suitability of the final solution, and durability of the resin used. In terms of the volume required, it is likely that a small reduction in the early rinse stages and load solutions could be implemented with further optimization. Even with these optimizations, separation method 3 requires the highest volume to achieve the purification of 47Ca. Though each of the separation methods result in 47Ca in 3–5 M HCl, separation method 3 uses significant levels of methanol in all other rinse steps. This could potentially lead to small amounts of methanol in the final radiolabeling solution, which is not acceptable under current guidelines for radiopharmaceuticals. The TODGA extraction molecule used in DGA resin has been shown to be less durable than cation-exchange resins like AG MP-50 to the high levels of radiation expected at FRIB.41,42 Additionally, the surface tension interactions that immobilize TODGA on DGA resin are less strong than the covalent bonding in AG MP-50, leading to an increased risk of leaching extractant molecule from the resin after repeated use.43 Both durability issues could result in organic material from the DGA resin contaminating the final product and requiring an additional purification step. Finally, DGA resin is used in the 47Ca/47Sc generator that will be used in future experiments. Using one of the AG MP-50 resin methods, orthogonal resins can be used to potentially maximize the purification ability of the final procedure.
After these practical considerations, the most suitable separation method for future harvesting efforts with 47Ca is separation method 2. Adjustments can be made to this procedure based on future experimental results, including generator performance and radiolabeling efficiency. This separation method will be used in combination with the published 47Ca/47Sc generator15 in future experiments at the NSCL to determine the feasibility of generating 47Sc from harvested 47Ca for radiolabeling biological molecules.
Stable Elemental Analysis
The stable elemental analysis was performed on water samples 1–4 and the combined, purified solution of 47Ca from these separations as high levels of elements, such as iron, in the final purified solution may inhibit future radiolabeling experiments. This analysis identified only low levels of a few stable elements. In the total water volume of 35 L when water sample 3 was removed from the system, <750 μg of Ca, Mg, Na, Si, B, and Zn and <100 μg of Fe, Ni, and Cu were detected. The approximately 130 mL of purified 47Ca solution had concentrated levels of stable calcium (200–400 μg) as expected, lower levels of Mg, Na, Si, Fe, and Zn (<40 μg), and a very low level of Sc (<2 μg). More details on the semiquantification of these elements can be found in the Supporting Information.
Only small amounts of stable ions were found in the water samples, indicating that the system under these irradiation conditions did not contribute significant amounts of stable ions. This was a concern as a beam of energetic particles creates radiolysis products such as H+, OH–, HO•, H•, HO2̇•, and H2O2 as it deposits energy and stops in water. (More detail on radiolysis in the isotope harvesting system can be found in Domnanich et al.29) While most of these radiolysis products recombine rapidly, H2O2 is long-lived and can cause oxidative damage to metal components exposed to the system water. In this water system, a few stainless steel components are in contact with the system water and could become a source of Fe, Cr, and Ni. This stable element analysis demonstrates that under the irradiation conditions in this experiment, the mass of stable ions that accumulated in the water was low, and therefore, the corrosive effects of radiolysis in the water system were minimal.
The measured stable ion masses in the water system and in the purified 47Ca product demonstrate that the stable ion content is reduced through the purification steps. The fact that there are remaining stable ions in the purified sample is not surprising as reagent grade chemicals as opposed to Suprapur chemicals are used in each step of the purification process. It is anticipated that these levels of stable ions will be reduced in future experiments when higher purity chemicals are used. The only high level of ions observed in the purified fraction was stable calcium, which should follow 47Ca through the pseudo generator and not affect a purified 47Sc solution. These results indicate that stable elements in the water system and in the final purified 47Ca solution may not impede radiolabeling with 47Sc generated through isotope harvesting.
Conclusions
In this work, 47Ca was produced with a 48Ca beam on a flowing-water target for the first time. A relatively high 2.0 ± 0.4% beam conversion to 47Ca was measured and used to predict GBq to TBq 47Ca activities through isotope harvesting at the NSCL and FRIB. The 47Ca produced in this experiment was effectively collected on cation-exchange resin beds in the isotope harvesting water system, eluted from the collection resin beds, and purified with three different separation methods that were optimized in this work. These methods resulted in ≥99% separation yield for 47Ca with 100% radionuclidic purity. Together, the low levels of stable contaminants and the high radionuclidic purity in the purified 47Ca sample indicate that radiolabeling with 47Sc generated through isotope harvesting at the NSCL holds promising potential. Overall, the results from this experiment demonstrate for the first time the feasibility of producing, collecting, and purifying 47Ca for a 47Ca/47Sc generator through isotope harvesting.
Acknowledgments
The authors thank the A1900 staff and NSCL Operation team for providing the accelerated beam for this work. This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Nuclear Physics, DOE Isotope Program, under a funded proposal from DOE-FOA-0001588 (DE-SC0018637) and U.S. Department of Energy, Office of Science, Office of Nuclear Physics, under Contract No. DE-AC02-06CH11357. Additional support was provided by the U.S. Department of Energy NNSA SSGF Program (DE-NA0003864) and Michigan State University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c03020.
Nonstandard geometries; beam current calibration; production rate equation; table of nuclear data for quantification of radionuclides; explanation of LOD calculation; explanation of the semiquantification ICP-OES method; distribution of 47Ca activity; LISE++ support for the 47Ca production rate at FRIB; and results from semiquantification with ICP-OES (PDF)
Author Contributions
The manuscript was written through contributions and final approval of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Müller C.; Bunka M.; Reber J.; Fischer C.; Zhernosekov K.; Türler A.; Schibli R. Promises of Cyclotron-Produced 44Sc as a Diagnostic Match for Trivalent β--Emitters: In Vitro and in Vivo Study of A 44sc-Dota-Folate Conjugate. J. Nucl. Med. 2013, 54, 2168–2174. 10.2967/jnumed.113.123810. [DOI] [PubMed] [Google Scholar]
- van der Meulen N. P.; Bunka M.; Domnanich K. A.; Müller C.; Haller S.; Vermeulen C.; Türler A.; Schibli R. Cyclotron Production of 44Sc: From Bench to Bedside. Nucl. Med. Biol. 2015, 42, 745–751. 10.1016/j.nucmedbio.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Singh A.; Van Der Meulen N. P.; Müller C.; Klette I.; Kulkarni H. R.; Türler A.; Schibli R.; Baum R. P. First-in-Human PET/CT Imaging of Metastatic Neuroendocrine Neoplasms with Cyclotron-Produced 44Sc-DOTATOC: A Proof-of-Concept Study. Cancer Biother. Radiopharm. 2017, 32, 124–132. 10.1089/cbr.2016.2173. [DOI] [PubMed] [Google Scholar]
- Eppard E.; de la Fuente2 A.; Benešová M.; Khawar A.; Bundschuh R. A.; Gärtner F. C.; Kreppel B.; Kopka K.; Essler M.; Rösch F. Clinical Translation and First In-Human Use of [44Sc]Sc-PSMA-617 for Pet Imaging of Metastasized Castrate-Resistant Prostate Cancer. Theranostics 2017, 7, 4359. 10.7150/thno.20586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domnanich K. A.; Eichler R.; Müller C.; Jordi S.; Yakusheva V.; Braccini S.; Behe M.; Schibli R.; Türler A.; van der Meulen N. P. Production and Separation of 43Sc for Radiopharmaceutical Purposes. EJNMMI Radiopharm. Chem. 2017, 2, 14 10.1186/s41181-017-0033-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B.; Chen J. Nuclear Data Sheets for A = 43. Nucl. Data Sheets 2015, 126, 1–150. 10.1016/j.nds.2015.05.001. [DOI] [Google Scholar]
- Chen J.; Singh B.; Cameron J. A. Nuclear Data Sheets for A = 44. Nucl. Data Sheets 2011, 112, 2357–2495. 10.1016/j.nds.2011.08.005. [DOI] [Google Scholar]
- Müller C.; Bunka M.; Haller S.; Köster U.; Groehn V.; Bernhardt P.; Van Der Meulen N.; Türler A.; Schibli R. Promising Prospects for 44Sc-/47Sc-Based Theragnostics: Application of 47Sc for Radionuclide Tumor Therapy in Mice. J. Nucl. Med. 2014, 55, 1658–1664. 10.2967/jnumed.114.141614. [DOI] [PubMed] [Google Scholar]
- Müller C.; Domnanich K. A.; Umbricht C. A.; Van Der Meulen N. P. Scandium and Terbium Radionuclides for Radiotheranostics: Current State of Development towards Clinical Application. Br. J. Radiol. 2018, 91, 20180074 10.1259/bjr.20180074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siwowska K.; Guzik P.; Domnanich K. A.; Rodríguez J. M. M.; Bernhardt P.; Ponsard B.; Hasler R.; Borgna F.; Schibli R.; Köster U.; Van Der Meulen N. P.; Müller C. Therapeutic Potential of 47Sc in Comparison to 177Lu and 90Y: Preclinical Investigations. Pharmaceutics 2019, 11, 424 10.3390/pharmaceutics11080424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows T. W. Nuclear Data Sheets for A = 47. Nucl. Data Sheets 2007, 108, 923–1056. 10.1016/j.nds.2007.04.002. [DOI] [Google Scholar]
- Misiak R.; Walczak R.; Wąs B.; Bartyzel M.; Mietelski J. W.; Bilewicz A. 47Sc Production Development by Cyclotron Irradiation of 48Ca. J. Radioanal. Nucl. Chem. 2017, 313, 429–434. 10.1007/s10967-017-5321-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi M.; Kondo K. Preparation of Carrier-Free 47Sc by the 48Ti (γ,p) Reaction. Int. J. Appl. Radiat. Isot. 1977, 28, 463–468. 10.1016/0020-708X(77)90178-8. [DOI] [Google Scholar]
- Deilami-nezhad L.; Moghaddam-Banaem L.; Sadeghi M.; Asgari M. Production and Purification of Scandium-47: A Potential Radioisotope for Cancer Theranostics. Appl. Radiat. Isot. 2016, 118, 124–130. 10.1016/j.apradiso.2016.09.004. [DOI] [PubMed] [Google Scholar]
- Domnanich K. A.; Müller C.; Benešová M.; Dressler R.; Haller S.; Köster U.; Ponsard B.; Schibli R.; Türler A.; van der Meulen N. P. 47Sc as Useful β–-Emitter for the Radiotheragnostic Paradigm: A Comparative Study of Feasible Production Routes. EJNMMI Radiopharm. Chem. 2017, 2, 5 10.1186/s41181-017-0024-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane S.; Harris J. T.; Starovoitova V. N. 47Ca Production for 47Ca/47Sc Generator System Using Electron Linacs. Appl. Radiat. Isot. 2015, 97, 188–192. 10.1016/j.apradiso.2014.12.020. [DOI] [PubMed] [Google Scholar]
- Mamtimin M.; Harmon F.; Starovoitova V. N. Sc-47 Production from Titanium Targets Using Electron Linacs. Appl. Radiat. Isot. 2015, 102, 1–4. 10.1016/j.apradiso.2015.04.012. [DOI] [PubMed] [Google Scholar]
- Starovoitova V. N.; Cole P. L.; Grimm T. L. Accelerator-Based Photoproduction of Promising Beta-Emitters 67Cu and 47Sc. J. Radioanal. Nucl. Chem. 2015, 305, 127–132. 10.1007/s10967-015-4039-z. [DOI] [Google Scholar]
- Rotsch D. A.; Brown M. A.; Nolen J. A.; Brossard T.; Henning W. F.; Chemerisov S. D.; Gromov R. G.; Greene J. Electron Linear Accelerator Production and Purification of Scandium-47 from Titanium Dioxide Targets. Appl. Radiat. Isot. 2018, 131, 77–82. 10.1016/j.apradiso.2017.11.007. [DOI] [PubMed] [Google Scholar]
- Loveless C. S.; Radford L. L.; Ferran S. J.; Queern S. L.; Shepherd M. R.; Lapi S. E. Photonuclear Production, Chemistry, and in Vitro Evaluation of the Theranostic Radionuclide 47Sc. EJNMMI Res. 2019, 9, 42 10.1186/s13550-019-0515-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peker L. K. Nuclear Data Sheets Update for A = 46. Nucl. Data Sheets 1993, 68, 271–310. 10.1006/ndsh.1993.1009. [DOI] [Google Scholar]
- Burrows T. W. Nuclear Data Sheets for A = 48. Nucl. Data Sheets 2006, 107, 1747–1922. 10.1016/j.nds.2006.05.005. [DOI] [Google Scholar]
- Abel E. P.; Avilov M.; Ayres V.; Birnbaum E.; Bollen G.; Bonito G.; Bredeweg T.; Clause H.; Couture A.; DeVore J.; Dietrich M.; Ellison P.; Engle J.; Ferrieri R.; Fitzsimmons J.; Friedman M.; Georgobiani D.; Graves S.; Greene J.; Lapi S.; Loveless C. S.; Mastren T.; Martinez-Gomez C.; McGuinness S.; Mittig W.; Morrissey D.; Peaslee G.; Pellemoine F.; Robertson J. D.; Scielzo N.; Scott M.; Severin G.; Shaughnessy D.; Shusterman J.; Singh J.; Stoyer M.; Sutherlin L.; Visser A.; Wilkinson J. Isotope Harvesting at FRIB: Additional Opportunities for Scientific Discovery. J. Phys. G: Nucl. Part. Phys. 2019, 46, 100501 10.1088/1361-6471/ab26cc. [DOI] [Google Scholar]
- Abel E. P.; Clause H. K.; Severin G. W. Radiolysis and Radionuclide Production in a Flowing-Water Target during Fast 40Ca20+ Irradiation. Appl. Radiat. Isot. 2020, 158, 109049 10.1016/j.apradiso.2020.109049. [DOI] [PubMed] [Google Scholar]
- Pen A.; Mastren T.; Peaslee G. F.; Petrasky K.; Deyoung P. A.; Morrissey D. J.; Lapi S. E. Design and Construction of a Water Target System for Harvesting Radioisotopes at the National Superconducting Cyclotron Laboratory. Nucl. Instrum. Methods Phys. Res., Sect. A 2014, 747, 62–68. 10.1016/j.nima.2014.02.010. [DOI] [Google Scholar]
- Mastren T.; Pen A.; Peaslee G. F.; Wozniak N.; Loveless S.; Essenmacher S.; Sobotka L. G.; Morrissey D. J.; Lapi S. E. Feasibility of Isotope Harvesting at a Projectile Fragmentation Facility: 67Cu. Sci. Rep. 2014, 4, 6706 10.1038/srep06706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastren T.; Pen A.; Loveless S.; Marquez B. V.; Bollinger E.; Marois B.; Hubley N.; Brown K.; Morrissey D. J.; Peaslee G. F.; Lapi S. E. Harvesting 67Cu from the Collection of a Secondary Beam Cocktail at the National Superconducting Cyclotron Laboratory. Anal. Chem. 2015, 87, 10323–10329. 10.1021/acs.analchem.5b02322. [DOI] [PubMed] [Google Scholar]
- Loveless C. S.; Marois B. E.; Ferran S. J.; Wilkinson J. T.; Sutherlin L.; Severin G.; Shusterman J. A.; Scielzo N. D.; Stoyer M. A.; Morrissey D. J.; Robertson J. D.; Peaslee G. F.; Lapi S. E. Harvesting 48V at the National Superconducting Cyclotron Laboratory. Appl. Radiat. Isot. 2020, 157, 109023 10.1016/j.apradiso.2019.109023. [DOI] [PubMed] [Google Scholar]
- Domnanich K. A.; Abel E. P.; Clause H. K.; Kalman C.; Walker W.; Severin G. W. An Isotope Harvesting Beam Blocker for the National Superconducting Cyclotron Laboratory. Nucl. Instrum. Methods Phys. Res., Sect. A 2020, 959, 163526 10.1016/j.nima.2020.163526. [DOI] [Google Scholar]
- Tarasov O. B.; Bazin D. LISE++: Radioactive Beam Production with in-Flight Separators. Nucl. Instrum. Methods Phys. Res., Sect. B 2008, 266, 4657–4664. 10.1016/j.nimb.2008.05.110. [DOI] [Google Scholar]
- Sato T.; Iwamoto Y.; Hashimoto S.; Ogawa T.; Furuta T.; Abe S.; Kai T.; Tsai P.; Matsuda N.; Iwase H.; Shigyo N.; Sihver L.; Niita K. Features of Particle and Heavy Ion Transport Code System (PHITS) Version 3. 02. J. Nucl. Sci. Technol. 2018, 55, 684–690. 10.1080/00223131.2017.1419890. [DOI] [Google Scholar]
- Firestone R. B. Nuclear Data Sheets for A = 24. Nucl. Data Sheets 2007, 108, 2319–2392. 10.1016/j.nds.2007.10.001. [DOI] [Google Scholar]
- Shamsuzzoha Basunia M. Nuclear Data Sheets for A = 27. Nucl. Data Sheets 2011, 112, 1875–1948. 10.1016/j.nds.2011.08.001. [DOI] [Google Scholar]
- Basunia M. S. Nuclear Data Sheets for A = 28. Nucl. Data Sheets 2013, 114, 1189–1291. 10.1016/j.nds.2013.10.001. [DOI] [Google Scholar]
- Singh B.; Cameron J. A. Nuclear Data Sheets for A = 42. Nucl. Data Sheets 2001, 92, 1–145. 10.1006/ndsh.2001.0001. [DOI] [Google Scholar]
- Burrows T. W. Nuclear Data Sheets for A = 45. Nucl. Data Sheets 2008, 109, 171–296. 10.1016/j.nds.2007.12.002. [DOI] [Google Scholar]
- Neubauer K.; Thompson L. Semiquantitative Analysis in ICP-OES and ICP-MS. Spectroscopy 2011, 24–31. [Google Scholar]
- Abel E. P.; Clause H. K.; Severin G. W. Radiolysis and Radionuclide Production in a Flowing-Water Target during Fast 40Ca20+ Irradiation. Appl. Radiat. Isot. 2020, 158, 109049 10.1016/j.apradiso.2020.109049. [DOI] [PubMed] [Google Scholar]
- Charged Particle Cross-Section Database for Medical Radioisotope Production: Diagnostic Radioisotopes and Monitor Reactions IAEA-TECDOC-1211 Qaim S. M.; Tárkayáni F.; Capote R., Eds.; 2001, 292. 10.1016/S1041-6080(96)90013-8. [DOI]
- Avilov M.; Aaron A.; Amroussia A.; Bergez W.; Boehlert C.; Burgess T.; Carroll A.; Colin C.; Durantel F.; Ferrante P.; Fourmeau T.; Graves V.; Grygiel C.; Kramer J.; Mittig W.; Monnet I.; Patel H.; Pellemoine F.; Ronningen R.; Schein M. Thermal, Mechanical and Fluid Flow Aspects of the High Power Beam Dump for FRIB. Nucl. Instrum. Methods Phys. Res., Sect. B 2016, 376, 24–27. 10.1016/j.nimb.2016.02.068. [DOI] [Google Scholar]
- Gangwer T. E.; Goldstein M.; Pillay K. K. S.. Radiation Effects on Ion Exchange Resins, Technical Report, 1977.
- Sugo Y.; Izumi Y.; Yoshida Y.; Nishijima S.; Sasaki Y.; Kimura T.; Sekine T.; Kudo H. Influence of Diluent on Radiolysis of Amides in Organic Solution. Radiat. Phys. Chem. 2007, 76, 794–800. 10.1016/j.radphyschem.2006.05.008. [DOI] [Google Scholar]
- Horwitz E. P.; McAlister D. R.; Bond A. H.; Barrans J. E. Novel Extraction of Chromatographic Resins Based on Tetraalkyldiglycolamides: Characterization and Potential Applications. Solvent Extr. Ion Exch. 2005, 23, 319–344. 10.1081/SEI-200049898. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.