

The title compound, [systematic name: 5-(trifluoromethyl)pyridine-2-carboxylic acid monohydrate], C7H4F3NO2·H2O, is the acid hydrate of a pyridine with a carboxylic acid group and a trifluoromethyl substituent situated para to one another on the aromatic ring. The molecule forms a centrosymmetric water-bridged hydrogen-bonding dimer with graph-set notation  (12). The dimers are further linked into a two-dimensional sheet via two longer intermolecular hydrogen-bonding interactions between the second hydrogen atom on the bridging water molecule and both a pyridine nitrogen atom and carbonyl oxygen. The trifluoromethyl groups extend out the faces of the sheet providing for F⋯F and C—H⋯F contacts between the sheets, completing the three-dimensional packing.
(12). The dimers are further linked into a two-dimensional sheet via two longer intermolecular hydrogen-bonding interactions between the second hydrogen atom on the bridging water molecule and both a pyridine nitrogen atom and carbonyl oxygen. The trifluoromethyl groups extend out the faces of the sheet providing for F⋯F and C—H⋯F contacts between the sheets, completing the three-dimensional packing.
Keywords: crystal structure, hydrogen bonding, picolinic acid derivatives, trifluoromethyl group
Abstract
The title compound [systematic name: 5-(trifluoromethyl)pyridine-2-carboxylic acid monohydrate], C7H4F3NO2·H2O, is the acid hydrate of a pyridine with a carboxylic acid group and a trifluoromethyl substituent situated para to one another on the aromatic ring. The molecule forms a centrosymmetric water-bridged hydrogen-bonding dimer with graph-set notation R 4 4 (12). Hydrogen-bonding distances of 2.5219 (11) and 2.8213 (11) Å are observed between the donor carboxylic acid and the bridging water acceptor, and the donor water and carbonyl oxygen acceptor, respectively. The dimers are further linked into a two-dimensional sheet via two longer intermolecular hydrogen-bonding interactions between the second hydrogen atom on the bridging water molecule and both a pyridine nitrogen atom and carbonyl oxygen with distances of 3.1769 (11) and 2.8455 (11) Å, respectively. The trifluoromethyl groups extend out the faces of the sheet providing for F⋯F and C—H⋯F contacts between the sheets, completing the three-dimensional packing.
Chemical context
Picolinic acids, pyridine derivatives with a carboxylic acid substituent at the 2-position, are common bidentate chelating agents of metallic elements in the human body (Grant et al., 2009 ▸). The title compound, the hydrate of 5-(trifluoromethyl)-2-pyridinecarboxylic acid (I), commonly known as 5-(trifluoromethyl)picolinic acid, is a derivative of picolinic acid with potent chelating abilities and biological activities (Li et al., 2019 ▸). Its transition-metal complexes also exhibit outstanding photophysical and electrochemical properties that make them promising phosphorescent materials for OLEDs (Wei et al., 2016 ▸). The compound may be synthesized from a range of synthetic routes, one of which relies on the carboxylation reaction of 2-bromo-5-(trifluoromethyl)pyridine with butyllithium (Cottet et al., 2003 ▸).
Structural commentary
The structure of 5-(trifluoromethyl)picolinic acid (I) reveals that the crystalline material obtained from the supplier is a hydrate and confirms the position of the carboxylic acid group ortho to the pyridine nitrogen atom with trifluoromethyl substituent situated para to the acid group on the aromatic ring (Fig. 1 ▸). The two aromatic carbon–nitrogen bonds have bond lengths of N—C2 of 1.3397 (12) Å and N—C6 of 1.3387 (12) Å, shorter than the aromatic C—C bonds, which have an average bond length of 1.387 (3) Å, a wedge-type motif typical in pyridine structures (Montgomery et al., 2015 ▸). The aromatic carboxylic acid substituent has a C1—C2 bond length of 1.5081 (13) Å, similar to that of the trifluoromethyl substituent C5—C7 of 1.5019 (13) Å, and the C—F bond lengths of the trifluoromethyl group have an average bond length of 1.335 (4) Å. The carboxylic acid group is co-planar with the aromatic pyridine ring, with least-squares planes at an angle of 1.8 (2)°.
Figure 1.

A view of 5-(trifluoromethyl)picolinic acid (I) hydrate with the atom-numbering scheme. Displacement ellipsoids are shown at the 50% probability level.
Supramolecular features
The structure of 5-(trifluoromethyl)picolinic acid (I) reported is a hydrate (Fig. 1 ▸) exhibiting a water-linked two-dimensional hydrogen-bonding network. Four different hydrogen-bonding interactions are observed between the picolinic acid and water molecule, which acts as both a hydrogen-bonding donor and acceptor with the carboxylic acid group and pyridine nitrogen atom (Table 1 ▸).
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1⋯O1W | 0.92 (2) | 1.60 (2) | 2.5219 (11) | 174 (2) |
| O1W—H2W⋯O2i | 0.808 (19) | 2.038 (19) | 2.8213 (11) | 163.2 (17) |
| O1W—H1W⋯O2ii | 0.859 (19) | 2.615 (18) | 3.1769 (11) | 124.1 (13) |
| O1W—H1W⋯Nii | 0.859 (19) | 2.008 (18) | 2.8455 (11) | 164.5 (16) |
Symmetry codes: (i) 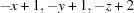 ; (ii)
; (ii)  .
.
The molecular packing in the solid state can be characterized by the 5-(trifluoromethyl)picolinic acid (I) hydrate asymmetric unit first forming a centrosymmetric water-bridged dimer unit with graph-set notation (12) (Fig. 2 ▸). The carboxylic acid hydrogen atom and the water oxygen form the stronger hydrogen bond, with an O1⋯O1W distance of 2.5219 (11) Å characterizing the O1—H1⋯O1W hydrogen bond, while the water hydrogen atom H2W bonds to the carbonyl oxygen atom with an O1W⋯O2i distance of 2.8213 (11) Å charaterizing the O1W-–H2W⋯O2i hydrogen bond [symmetry code: (i) −x + 1, −y + 1, −z + 2].
Figure 2.

A view of the intermolecular water-bridged hydrogen-bonding dimer in 5-(trifluoromethyl)picolinic acid (I) hydrate.
The water molecules, specifically using the other water hydrogen atom H1W, further bridge the dimer units together to form a pleated strip or tape motif that propagates along the crystallographic [010] direction (Fig. 3 ▸). The the O1W⋯Nii and O1W⋯O2ii distances of 3.1769 (11) and 2.8455 (11) Å, respectively, characterize the O1W—H1W⋯Nii and O1W—H1W⋯O2ii hydrogen bonds [symmetry code: (ii) −x + 1, y −  , −z +
, −z +  ]. The pleated nature of the strip exposes the H1W hydrogen atom of every other dimer to a pyridine nitrogen in the strip adjacent to it, forming a thick two-dimensional sheet (Fig. 4 ▸). The sheet can be considered a bilayer with a hydrophilic core due to the presence of water molecules and strong hydrogen bonding in the center and the more hydrophobic trifluoromethylaromatic groups extending to the faces of the sheet (Fig. 5 ▸).
]. The pleated nature of the strip exposes the H1W hydrogen atom of every other dimer to a pyridine nitrogen in the strip adjacent to it, forming a thick two-dimensional sheet (Fig. 4 ▸). The sheet can be considered a bilayer with a hydrophilic core due to the presence of water molecules and strong hydrogen bonding in the center and the more hydrophobic trifluoromethylaromatic groups extending to the faces of the sheet (Fig. 5 ▸).
Figure 3.

A view of a pleated strip formed between the water-bridged hydrogen-bonding dimers in 5-(trifluoromethyl)picolinic acid (I) hydrate.
Figure 4.

A view of the sheet hydrogen-bonding network in 5-(trifluoromethyl)picolinic acid (I) hydrate viewing the water-bridge hydrogen-bonding dimers end-on shows the hydrogen-bonding interactions between the pleated strips forming a two-dimensional sheet.
Figure 5.

An edge-on view of the two-dimensional sheet formed between the water-bridged hydrogen-bonding dimers in 5-(trifluoromethyl)picolinic acid (I) hydrate.
The sheets stack in the [100] direction (Fig. 6 ▸). The forces that guide the intermolecular interactions between neighboring sheets are van der Waals forces including F⋯F and C—H⋯F contacts. The shortest weak Caryl—H⋯F interaction, C4—H4A⋯F2 exhibits an H⋯F distance of 2.495 (1) Å. The most notable interaction is a dimeric F⋯F interaction between CF3 groups on neighboring sheets with an F1⋯F3 distance of 3.077 (1) Å, which is ∼0.15 Å longer than the sum of the van der Waals radii of fluorine (Bondi, 1964 ▸).
Figure 6.

A view of the stacking of the two-dimensional sheets in 5-(trifluoromethyl)picolinic acid (I) hydrate showing the trifluoromethylaromatic interactions at the interfaces of the sheets.
Database survey
Monocarboxylic derivatives of pyridine, pyridinecarboxylic acids, are also commonly known as picolinic acid, nicotinic acid, or isonicotinic acid when the carboxyl group resides at the 2-, 3-, or 4- position, respectively. The Cambridge Structural Database (Version 5.40, update of March 2020; Groom et al., 2016 ▸) contains no isomers of trifluoromethyl-substituted pyridinecarboxylic acids. The crystal structure of the base of the title compound, picolinic acid (PICOLA02), was shown to be 1:1 co-crystals of its neutral and zwitterionic forms, where the nitrogen atom can both be protonated and deprotonated (Hamazaki et al., 1998 ▸). The interactions form a zigzag chain by N—H⋯N and O—H⋯O intermolecular hydrogen bonding. A zwitterionic hydrogen-bonding motif can be found in substituted picolinic acid derivatives as well, such as 3-thioxo-2-pyridinecarboxylic acid (MPYDCX01; Bourne & Taylor, 1983 ▸).
A related solvated picolinic acid crystal structure can be found in the crystal structure of picolinic acid peroxosolvate (ANINES) which, while zwitterionic, exhibits a solvate-linked hydrogen-bonding pattern (Medvedev et al., 2013 ▸). In this structure, every hydrogen peroxide molecule links three picolinic acid molecules together with two hydrogen bonds between the H2O2 hydrogen atoms and two carboxylate groups, and an N—H⋯O hydrogen bond between the protonated pyridine nitrogen atom and one oxygen atom of the H2O2 molecule.
5-(Trifluoromethyl)-2-pyridinecarboxylic acid has been used as a monoanionic ligand in several metal complexes, including with CoII (VOVZOY; Li et al., 2019 ▸), CrIII (QEGWOR; Chai et al., 2017 ▸), MnII (ROKSIW; Wang et al., 2019 ▸), and IrIII [COKGAN and COKGIV (Sanner et al., 2019 ▸); GIZJOR (Hao et al., 2019 ▸)]. While the CoII and MnII complexes engage in intermolecular hydrogen bonding with metal-coordinated water molecules, the CrIII complex contains a water of solvation that facilitates the formation of a hydrogen-bonding network. In a fashion reminiscent of 5-(trifluoromethyl)picolinic acid (I) hydrate itself, [Cr(5-(trifluoromethyl)picolinate)2(H2O)2]NO3·H2O hydrogen bonds into thick two-dimensional sheets with the trifluoromethylaromatic groups extending to the faces of the sheets (Chai et al., 2017 ▸).
Synthesis and crystallization
5-(Trifluoromethyl)-2-pyridinecarboxylic acid (I, 95%) was purchased from Aldrich Chemical Company, USA, and was used as received.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms on carbon atoms were included in calculated positions and refined using a riding model with C—H = 0.95 Å and U iso(H) = 1.2U eq(C) of the aryl C-atoms. The position of the carboxylic acid and water hydrogen atoms were found in the difference map and refined freely.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | C7H4F3NO2·H2O |
| M r | 209.13 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 125 |
| a, b, c (Å) | 8.9213 (10), 10.0759 (12), 9.1010 (11) |
| β (°) | 99.983 (2) |
| V (Å3) | 805.70 (16) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.18 |
| Crystal size (mm) | 0.32 × 0.25 × 0.14 |
| Data collection | |
| Diffractometer | Bruker APEXII CCD |
| Absorption correction | Multi-scan (SADABS; Bruker, 2017 ▸) |
| T min, T max | 0.89, 0.98 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 19282, 2463, 2126 |
| R int | 0.026 |
| (sin θ/λ)max (Å−1) | 0.715 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.034, 0.103, 1.07 |
| No. of reflections | 2463 |
| No. of parameters | 139 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.50, −0.26 |
Analytical data
1H NMR (Bruker Avance III HD 400 MHz, DMSO d 6): δ 3.70 (br s, OH), 8.21 (d, 1H, Caryl H, Jortho = 8.0 Hz), 8.37 (dd, 1H, Caryl H, Jortho = 8.2 Hz, Jmeta = 2.0 Hz), 9.07 (s, 1H, Caryl H). 13C NMR (13C{1H}, 100.6 MHz, DMSO d 6): δ 123.23 (q, CF3, J C–F = 272.8 Hz), 124.73 (s, C aryl H), 127.30 (q, C arylCF3, J C–F = 32.7 Hz), 135.12 (q, C arylH, J C–F = 3.5 Hz), 146.24 (q, C arylH, J C–F = 3.8 Hz), 151.98 (s, C arylCOOH), 165.13 (s, COOH). 19F NMR (19F{1H}, 376.5 MHz, DMSO d6): δ −61.35 (s, 3F, CF3). IR (Thermo Nicolet iS50, FT–IR, KBr pellet, cm−1): 3469 (s br, O—H str), 3050 (s, Caryl—H str), 2849 (w), 2571 (w), 1961 (m), 1707 (s, C=O str), 1606 (s), 1582 (s), 1493 (s), 1440 (s), 1392 (s), 1328 (m), 1290 (m), 1251 (s), 1163 (m), 1126 (s), 1075 (s), 1023 (s), 948 (s), 878 (m), 864 (s), 806 (s), 760 (s), 704 (s), 643 (s), 524 (s). GC–MS (Agilent Technologies 7890A GC/5975C MS): M+ = 191 amu, corresponding to the anhydrous form, 5-(trifluoromethyl)pyridine-2-carboxylic acid (I), whose calculated exact molecular ion mass is 191.02 amu.
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989020013523/dj2015sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989020013523/dj2015Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989020013523/dj2015Isup3.cml
CCDC reference: 2036131
Additional supporting information: crystallographic information; 3D view; checkCIF report
supplementary crystallographic information
Crystal data
| C7H4F3NO2·H2O | F(000) = 424 |
| Mr = 209.13 | Dx = 1.724 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 8.9213 (10) Å | Cell parameters from 7786 reflections |
| b = 10.0759 (12) Å | θ = 3.0–30.2° |
| c = 9.1010 (11) Å | µ = 0.18 mm−1 |
| β = 99.983 (2)° | T = 125 K |
| V = 805.70 (16) Å3 | Block, colourless |
| Z = 4 | 0.32 × 0.25 × 0.14 mm |
Data collection
| Bruker APEXII CCD diffractometer | 2463 independent reflections |
| Radiation source: fine-focus sealed tube | 2126 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.026 |
| Detector resolution: 8.3333 pixels mm-1 | θmax = 30.5°, θmin = 2.3° |
| φ and ω scans | h = −12→12 |
| Absorption correction: multi-scan (SADABS; Bruker, 2017) | k = −14→14 |
| Tmin = 0.89, Tmax = 0.98 | l = −13→13 |
| 19282 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: dual |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.034 | Hydrogen site location: mixed |
| wR(F2) = 0.103 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.07 | w = 1/[σ2(Fo2) + (0.0559P)2 + 0.2301P] where P = (Fo2 + 2Fc2)/3 |
| 2463 reflections | (Δ/σ)max < 0.001 |
| 139 parameters | Δρmax = 0.50 e Å−3 |
| 0 restraints | Δρmin = −0.26 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| F1 | 1.02797 (8) | 0.44945 (10) | 0.19056 (8) | 0.0387 (2) | |
| F2 | 0.91945 (11) | 0.63911 (8) | 0.13936 (9) | 0.0398 (2) | |
| F3 | 0.80545 (8) | 0.45876 (7) | 0.06062 (7) | 0.02763 (17) | |
| O1 | 0.62864 (9) | 0.36976 (7) | 0.76271 (8) | 0.02126 (17) | |
| H1 | 0.572 (2) | 0.365 (2) | 0.838 (2) | 0.056 (6)* | |
| O2 | 0.57174 (9) | 0.58725 (7) | 0.74769 (8) | 0.02259 (17) | |
| N | 0.69746 (10) | 0.60686 (8) | 0.49521 (9) | 0.01815 (18) | |
| C1 | 0.62637 (11) | 0.48809 (9) | 0.70248 (10) | 0.01643 (18) | |
| C2 | 0.70005 (11) | 0.48977 (9) | 0.56536 (10) | 0.01547 (18) | |
| C3 | 0.76396 (12) | 0.37599 (9) | 0.51689 (11) | 0.01936 (19) | |
| H3A | 0.762813 | 0.29467 | 0.569519 | 0.023* | |
| C4 | 0.82968 (12) | 0.38369 (10) | 0.38964 (11) | 0.0205 (2) | |
| H4A | 0.87462 | 0.307748 | 0.353174 | 0.025* | |
| C5 | 0.82825 (11) | 0.50451 (10) | 0.31708 (11) | 0.01741 (19) | |
| C6 | 0.76044 (12) | 0.61375 (10) | 0.37220 (11) | 0.01917 (19) | |
| H6A | 0.758767 | 0.695976 | 0.32085 | 0.023* | |
| C7 | 0.89493 (12) | 0.51403 (10) | 0.17683 (11) | 0.0210 (2) | |
| O1W | 0.46520 (10) | 0.34230 (8) | 0.96069 (9) | 0.02433 (18) | |
| H2W | 0.469 (2) | 0.3734 (17) | 1.043 (2) | 0.040 (5)* | |
| H1W | 0.4241 (19) | 0.2651 (19) | 0.9607 (19) | 0.043 (5)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| F1 | 0.0238 (4) | 0.0669 (6) | 0.0280 (4) | 0.0091 (3) | 0.0118 (3) | −0.0011 (4) |
| F2 | 0.0630 (5) | 0.0278 (4) | 0.0368 (4) | −0.0179 (3) | 0.0317 (4) | −0.0058 (3) |
| F3 | 0.0315 (4) | 0.0352 (4) | 0.0167 (3) | −0.0061 (3) | 0.0055 (2) | −0.0055 (3) |
| O1 | 0.0303 (4) | 0.0167 (3) | 0.0190 (3) | 0.0011 (3) | 0.0104 (3) | 0.0034 (3) |
| O2 | 0.0328 (4) | 0.0185 (3) | 0.0189 (3) | 0.0026 (3) | 0.0112 (3) | −0.0006 (3) |
| N | 0.0240 (4) | 0.0154 (4) | 0.0161 (4) | 0.0013 (3) | 0.0066 (3) | 0.0005 (3) |
| C1 | 0.0192 (4) | 0.0165 (4) | 0.0135 (4) | −0.0014 (3) | 0.0027 (3) | 0.0004 (3) |
| C2 | 0.0180 (4) | 0.0150 (4) | 0.0136 (4) | −0.0004 (3) | 0.0031 (3) | −0.0006 (3) |
| C3 | 0.0252 (5) | 0.0151 (4) | 0.0188 (4) | 0.0012 (3) | 0.0066 (4) | 0.0006 (3) |
| C4 | 0.0248 (5) | 0.0174 (4) | 0.0207 (4) | 0.0015 (3) | 0.0078 (4) | −0.0033 (3) |
| C5 | 0.0182 (4) | 0.0197 (4) | 0.0153 (4) | −0.0028 (3) | 0.0056 (3) | −0.0031 (3) |
| C6 | 0.0252 (5) | 0.0167 (4) | 0.0168 (4) | −0.0006 (3) | 0.0070 (3) | 0.0006 (3) |
| C7 | 0.0218 (5) | 0.0234 (5) | 0.0194 (4) | −0.0044 (4) | 0.0079 (4) | −0.0036 (4) |
| O1W | 0.0376 (4) | 0.0188 (3) | 0.0198 (4) | −0.0073 (3) | 0.0140 (3) | −0.0031 (3) |
Geometric parameters (Å, º)
| F1—C7 | 1.3402 (13) | C3—C4 | 1.3879 (13) |
| F2—C7 | 1.3335 (13) | C3—H3A | 0.95 |
| F3—C7 | 1.3317 (12) | C4—C5 | 1.3840 (14) |
| O1—C1 | 1.3109 (11) | C4—H4A | 0.95 |
| O1—H1 | 0.92 (2) | C5—C6 | 1.3908 (13) |
| O2—C1 | 1.2143 (12) | C5—C7 | 1.5019 (13) |
| N—C6 | 1.3387 (12) | C6—H6A | 0.95 |
| N—C2 | 1.3397 (12) | O1W—H2W | 0.808 (19) |
| C1—C2 | 1.5081 (13) | O1W—H1W | 0.859 (19) |
| C2—C3 | 1.3861 (13) | ||
| C1—O1—H1 | 113.0 (13) | C4—C5—C6 | 119.52 (9) |
| C6—N—C2 | 117.98 (8) | C4—C5—C7 | 119.30 (9) |
| O2—C1—O1 | 125.78 (9) | C6—C5—C7 | 121.14 (9) |
| O2—C1—C2 | 121.95 (8) | N—C6—C5 | 122.17 (9) |
| O1—C1—C2 | 112.26 (8) | N—C6—H6A | 118.9 |
| N—C2—C3 | 123.42 (9) | C5—C6—H6A | 118.9 |
| N—C2—C1 | 115.48 (8) | F3—C7—F2 | 107.12 (9) |
| C3—C2—C1 | 121.09 (8) | F3—C7—F1 | 105.68 (8) |
| C2—C3—C4 | 118.41 (9) | F2—C7—F1 | 107.51 (9) |
| C2—C3—H3A | 120.8 | F3—C7—C5 | 112.15 (8) |
| C4—C3—H3A | 120.8 | F2—C7—C5 | 112.61 (8) |
| C5—C4—C3 | 118.49 (9) | F1—C7—C5 | 111.38 (9) |
| C5—C4—H4A | 120.8 | H2W—O1W—H1W | 107.4 (16) |
| C3—C4—H4A | 120.8 |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O1W | 0.92 (2) | 1.60 (2) | 2.5219 (11) | 174 (2) |
| O1W—H2W···O2i | 0.808 (19) | 2.038 (19) | 2.8213 (11) | 163.2 (17) |
| O1W—H1W···O2ii | 0.859 (19) | 2.615 (18) | 3.1769 (11) | 124.1 (13) |
| O1W—H1W···Nii | 0.859 (19) | 2.008 (18) | 2.8455 (11) | 164.5 (16) |
Symmetry codes: (i) −x+1, −y+1, −z+2; (ii) −x+1, y−1/2, −z+3/2.
Funding Statement
This work was funded by Vassar College grant . National Science Foundation grants 0521237 and 0911324. Salmon Fund and Olin College Fund of Vassar College grant .
References
- Bondi, A. (1964). J. Phys. Chem. 68, 441–451.
- Bourne, P. E. & Taylor, M. R. (1983). Acta Cryst. C39, 266–268.
- Bruker (2017). SAINT, SADABS and APEX2. Bruker AXS Inc., Madison, Wisconsin, USA.
- Chai, J., Liu, Y., Liu, B. & Yang, B. (2017). J. Mol. Struct. 1150, 307–315.
- Cottet, F., Marull, M., Lefebvre, O. & Schlosser, M. (2003). Eur. J. Org. Chem. pp. 1559–1568.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Grant, R. S., Coggan, S. E. & Smythe, G. A. (2009). Int. J. Tryptophan Res. 2, 71–79. [DOI] [PMC free article] [PubMed]
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Hamazaki, H., Hosomi, H., Takeda, S., Kataoka, H. & Ohba, S. (1998). Acta Cryst. C54, IUC9800049.
- Hao, H., Liu, X., Ge, X., Zhao, Y., Tian, X., Ren, T., Wang, Y., Zhao, C. & Liu, Z. (2019). J. Inorg. Biochem. 192, 52–61. [DOI] [PubMed]
- Li, B., Wang, J., Song, H., Wu, H., Chen, X. & Ma, X. (2019). J. Coord. Chem. 72, 2562–2573.
- Macrae, C. F., Sovago, I., Cottrell, S. J., Galek, P. T. A., McCabe, P., Pidcock, E., Platings, M., Shields, G. P., Stevens, J. S., Towler, M. & Wood, P. A. (2020). J. Appl. Cryst. 53, 226–235. [DOI] [PMC free article] [PubMed]
- Medvedev, A. G., Mikhailov, A. A., Prikhodchenko, P. V., Tripol’skaya, T. A., Lev, O. & Churakov, A. V. (2013). Russ. Chem. Bull. 62, 1871–1876.
- Montgomery, M. J., O’Connor, T. J. & Tanski, J. M. (2015). Acta Cryst. E71, 852–856. [DOI] [PMC free article] [PubMed]
- Sanner, R. D., Cherepy, N. J., Martinez, H. P., Pham, H. Q. & Young, V. G. (2019). Inorg. Chim. Acta, 496, 119040.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Wang, J., Li, B., Wu, H., Tian, X. & Ma, X. (2019). Jiegou Huaxue. 38, 1349–1355.
- Wei, X., Wang, S. & Wei, D. (2016). Huaxue Tongbao, 79, 947–951.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989020013523/dj2015sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989020013523/dj2015Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989020013523/dj2015Isup3.cml
CCDC reference: 2036131
Additional supporting information: crystallographic information; 3D view; checkCIF report