

Abstract
Zn2+ has been shown to have a wide range of modulatory effects on neuronal AMPARs. However, the mechanism of modulation is largely unknown. Here we show that Zn2+ inhibits GluA2(Q) homomeric receptors in an activity- and voltage-dependent manner, indicating a pore block mechanism. The rate of inhibition is slow, in the hundreds of milliseconds at millimolar Zn2+ concentrations; hence, the inhibition is only observed in the residual nondesensitizing currents. Consequently, the inhibition is higher for GluA2 receptors in complex with auxiliary subunits γ2 and γ8 where the residual activation is larger. The extent of inhibition is also dependent on charge at site 607, the site that undergoes RNA editing in GluA2 subunits replacing glutamine to arginine, with the percent inhibition being lower and IC50 being higher for the edited GluA2(R) relative to unedited GluA2(Q) and to GluA2(Q607E), a mutation observed in the genetic screen of a patient exhibiting developmental delays. We also show that Zn2+ inhibition is significant during rapid repetitive activity with pulses of millimolar concentrations of glutamate in both receptors expressed in HEK cells as well as in native receptors in cortical neurons of C57BL/6J mice of either sex, indicating a physiological relevance of this inhibition.
SIGNIFICANCE STATEMENT Zn2+ is present along with glutamate in synaptic vesicles and coreleased during synaptic transmission, modulating the postsynaptic ionotropic glutamate receptors. While Zn2+ inhibition of the NMDA subtype of the ionotropic glutamate receptors is well characterized, the mechanism of modulation of the AMPA subtype is much less known. Here we have systematically studied Zn2+ inhibition of AMPARs by varying calcium permeability, auxiliary subunits, and activation levels and show that Zn2+ inhibits AMPARs in an activity-dependent manner, opening up this pathway as a means to pharmacologically modulate the receptors.
Keywords: AMPA, electrophysiology, glutamate receptor, single channel
Introduction
Ionotropic glutamate receptors are the main excitatory receptors in the mammalian CNS. Glutamate binding to the extracellular domain of this receptor leads to the opening of cation-selective channels and subsequent desensitization. Overactivation of these receptors places metabolic stress on the neurons, resulting in neuronal injury, and underlies a wide range of conditions, such as ischemia, seizures, and amyotrophic lateral sclerosis (Kwak et al., 2010; Traynelis et al., 2010; Yuan et al., 2015; Iacobucci and Popescu, 2017). Ionotropic glutamate receptors are broadly classified into three classes: AMPARs, NMDARs, and kainate receptors. Early studies of excitotoxic mechanisms focused on the NMDARs as these are the primary calcium-permeable receptors among the three members of this family. However, despite the clear effect of NMDAR inhibitors in model systems, these have failed to show clear efficacy in therapeutic trials.
Recent studies have shed insight into the relevance and role of AMPARs in these pathologic and disease states. AMPARs are assembled by a combination of GluA1, GluA2, GluA3, and GluA4 subunits. In the adult brain, 99% of the GluA2 subunits undergo RNA editing that replaces a glutamine at site 607, which is at the tip of the reentrant pore-lining region of the receptor, with arginine (Wright and Vissel, 2012). The presence of RNA-edited GluA2, even in the heteromeric receptors, makes the receptors Ca2+-impermeable (Hollmann et al., 1991; Hume et al., 1991; Cull-Candy et al., 2006). However, a decrease in the editing of the GluA2 has been reported in several human neurologic conditions, such as Alzheimer's disease (Gaisler-Salomon et al., 2014), schizophrenia, Huntington's disease (Akbarian et al., 1995), amyotrophic lateral sclerosis (Kawahara et al., 2004), and ischemia (Peng et al., 2006), further implicating AMPARs in these pathologies. In line with these findings, a patient with de novo mutations at this site (Q607E/A) exhibited severe developmental delays (Salpietro et al., 2019). Thus, inhibitors with selectivity or higher inhibition toward the unedited or Q607E would have a potential to be used in these conditions.
Zn2+ has been previously shown to be endogenously released with glutamate at synapses and modulate the strength of synaptic AMPARs (Kalappa et al., 2015) and also shown to inhibit quisqualate-mediated signals in hippocampal neurons at 1 mm concentration (Mayer et al., 1989; Blakemore and Trombley, 2019). However, the specific mechanism of such inhibition has not been studied. Here we show that Zn2+ inhibits AMPARs in an activity-, auxiliary subunit-, and (Q/R) editing-dependent manner. For Zn2+ modulation of AMPARs in the presence of the auxiliary receptors, we have investigated γ2 and γ8 subunits, which are predominantly expressed in the brain. These auxiliary proteins not only play an essential role in anchoring and stabilizing AMPARs (Opazo et al., 2010; Jackson and Nicoll, 2011; Haering et al., 2014), but also stabilize the open channel active conformation of the receptor to varying degrees (for review, see Milstein and Nicoll, 2008). γ2 enhances agonist affinity and efficacy, slows deactivation and desensitization, and accelerates recovery from desensitization stabilizing the active open conformation (Tomita et al., 2005; Cho et al., 2007; MacLean and Bowie, 2011; MacLean et al., 2014; Carbone and Plested, 2016; Shaikh et al., 2016; Twomey et al., 2016). γ8 stabilizes the AMPAR active state even further, showing resensitization during the continued presence of glutamate (Kott et al., 2007; Suzuki et al., 2008; Kato et al., 2010; Jackson et al., 2011; Riva et al., 2017; Herguedas et al., 2019; Carrillo et al., 2020).
In addition to showing that Zn2+ exhibits activity-dependent block with higher inhibition toward the unedited GluR2(Q) and GluR2(E), we also show that Zn2+ inhibits AMPAR activation in mouse cortical neurons. These results suggest that this pathway could be further explored as a means for pharmacological modulation of the AMPARs.
Materials and Methods
Cell culture, mutagenesis, and transfection
HEK 293T cells were maintained in DMEM (GenDEPOT), supplemented with 10% FBS (GenDEPOT) and penicillin/streptomycin (Invitrogen). Mutations were introduced using standard site-directed mutagenesis and confirmed by sequencing (Genewiz). The tandem constructs for GluA2 with γ8 or γ2 were generated between the C-terminus of GluA2 and the N-terminus of γ8 or γ2 by a Gly-Ser linker using Gibson Assembly protocol.
HEK 293T cells at 40%-50% confluency were transfected using lipofectamine 2000 (Invitrogen) with one of the homomeric AMPAR subunits, GluA2(Q), GluA2(Q)/γ2, GluA2(Q)/γ8, GluA2(R), and GluA2(E) along with GFP. Cells were replated after 4-6 h at a low density in fresh media containing 30 μm NBQX. Whole-cell patch-clamp recordings were performed 24-48 h after transfection using 3-5 mΩ resistance fire-polished borosilicate glass pipettes filled with internal solution as follows: 135 mm CsF, 33 mm CsCl, 2 mm MgCl2, 1 mm CaCl2, 11 mm EGTA, and 10 mm HEPES, pH 7.4. The external solution was as follows: 150 mm NaCl, 1 mm CaCl2, and 10 mm HEPES, pH 7.4. The external solutions were locally applied to lifted cells using a LZ150M piezo translator (Burleigh Instruments) with voltage commands low-pass-filtered (eight-pole Bessel; Frequency Devices) at 100-200 Hz. All recordings were performed at room temperature with a holding potential of −60 mV using an Axopatch 200B amplifier (Molecular Devices), acquired at 10 kHz using pCLAMP10 software (Molecular Devices), and filtered online at 5 kHz.
Steady-state inhibition (Einhibition) was calculated using the following:
| (1) |
were IZinc is the steady-state current in the presence of zinc and Icontrol is the steady-state current in the absence of zinc.
Dose–response curves showing steady-state inhibition as a function of Zn2+ concentrations were fit to the Hill equation in Origin 9 and is shown in as follows:
| (2) |
Where Einhibition is steady-state inhibition as defined by Equation 1, Emin is the lowest value for steady-state inhibition, Emax is the maximum value for steady-state inhibition, IC50 is concentration of Zn2+ at half maximal inhibition, and C is concentration of Zn2+.
Single-channel recording
HEK 293T cells were plated in poly-D-lysine–coated 35 mm dishes and transfected 24 h later with one of the GluA2 constructs and GFP at a 1:1 ratio. Recordings were performed in the outside-out patch-clamp configuration, 8-24 h after transfection. Pipettes used had an 8-15 mΩ resistance. Buffers and solution concentrations were similar to those used for whole-cell recordings. Data were acquired at 50 kHz and low-pass filtered at 10 kHz (Axon 200B and Digidata 1550A; Molecular Devices). Pipette holding potential was −100 mV. Data were further filtered at 1 kHz. All recordings were idealized using the segmental k-means algorithm of QuB (Qin, 2004; Nicolai and Sachs, 2013). Recordings with a single level were used as a measure to ensure that the recordings were from single channels.
Animal experiments were conducted following the National Institutes of Health's Guide for the care and use of laboratory animals guidelines, and protocols were approved by the University of Texas Health Science Center at Houston. C57BL/6 mice (n = 10) 1-2 postnatal days of age were used. Animals were housed in an in-house animal facility with a 12:12 h light-dark cycle with food and water available ad libitum.
Neuron isolation
Cortical neurons from the PFC were prepared from early postnatal (P1-P2) C57BL/6 mice, either sex. The mice were decapitated, the skin and skull removed, and the brain placed in 4-5 ml ice-cold dissection solution (Earle's buffered salt solution, Invitrogen, 10 mm HEPES). Under a dissecting microscope, the PFC was removed and transferred to a solution containing 10 U/ml of papain (Worthington), 20 µl of DNase I (New England Biolabs, 2000 U/mg), and 500 μm APV (Abcam), and incubated in a 37°C water bath for 30 min. Every 10 min, the tissue was pipetted to dissociate into individual cells. The activity of papain was stopped by inactivated FBS for 2 min. The tissue was centrifuged and transferred to dissociation solution (B27/Neurobasal culture medium, Invitrogen, supplemented with 2 mm of L-glutamine, 0.2% penicillin/streptomycin, and 20 mm glucose). The neurons were mechanically dissociated from the tissue by gently sucking it back and forth with a flame-narrowed Pasteur pipette. The cell suspension was diluted with incubated feeding medium (B27/Neurobasal culture medium, Invitrogen, supplemented with 2 mm of L-glutamine, and 0.2% penicillin/streptomycin) and was plated on poly-L-lysine (Sigma Millipore)-coated glass coverslips and incubated in 5% CO2 at 37°C. Neurons were used 24 h after plating.
The neuron recordings were performed in whole-cell patch-clamp configuration, at room temperature. Pipettes, 8-15 mΩ, filled with internal solution as follows: 120 mm Cs-gluconate, 20 mm HEPES, 4 mm MgCl2, 10 mm EGTA, 0.4 mm GTP-Na, 4 mm ATP-Mg, and 5 mm phosphocreatine, pH 7.3. The external ACSF solution was as follows: 140 mm NaCl, 2.5 mm KCl, 2.5 mm CaCl2, 1 mm MgCl2, 1.25 mm NaH2PO4, 10 mm HEPES, 25 mm glucose, 50 mm APV, 10 μm bicuculline, and 1 mm TTX, pH 7.4.
mEPSC recordings
Spontaneous mEPSCs were recorded using neurons that were in culture for 14-21 d. For these recordings, pipettes with 8-15 mΩ were filled with internal solution as follows: 120 mm Cs-gluconate, 20 mm HEPES, 4 mm MgCl2, 10 mm EGTA, 0.4 mm GTP-Na, 4 mm ATP-Mg, and 5 mm phosphocreatine, adjusted to pH 7.3. The external buffer was as follows: 140 mm NaCl, 2.5 mm KCl, 2.5 mm CaCl2, 1 mm MgCl2, 1.25 mm NaH2PO4, 10 mm HEPES, 25 mm glucose, 10 μm bicuculline, and 1 mm TTX, adjusted to pH 7.4. mEPSC recordings were performed at room temperature with a holding potential of −80 mV using an Axopatch 200B amplifier (Molecular Devices), acquired at 50 kHz using pCLAMP10 software (Molecular Devices), and filtered online at 5 kHz. mEPSCs were analyzed using a mEPSC current-template search through Clampfit 10 software (Molecular Devices), with a detection threshold of −6 pA.
Statistics
At least three recordings were obtained for each condition studied from at least 3 different days. All electrophysiological data, except mEPSC recordings, were statistically analyzed using one-way ANOVA followed by pairwise comparisons using Bonferroni's multiple comparisons test. The mEPSC recordings were statistically analyzed using the Student´s paired t test. These tests were performed using GraphPad Prism software (GraphPad Software). For all tests, p < 0.05 was considered significant, and p < 0.001 was considered highly significant.
Results
Activity-dependent block of GluA2(Q) AMPARs
Whole-cell currents were recorded from HEK-293 cells expressing GluA2(Q) homomeric receptor in the absence (Fig. 1A) and the presence of auxiliary subunits (Fig. 1B,C). The auxiliary subunits γ2 and γ8 were chosen as they are commonly found to be coexpressed in AMPARs in the brain and modulate the receptors differently, with γ2 decreasing rates of desensitization and having a small fraction of nondesensitizing residual currents (Fig. 1B) and γ8 exhibiting resensitization with a larger fraction in open channel states in the continued presence of agonist (Fig. 1C). The whole-cell currents for GluA2(Q) receptors in the absence of auxiliary subunits with saturating concentration of agonist (10 mm glutamate) were not significantly altered when 1 mm Zn2+ was coapplied (Fig. 1A). However, the residual steady-state currents for GluA2(Q) were significantly inhibited in the presence of auxiliary subunits (Fig. 1B,C). The extent of inhibition was larger for GluA2(Q) in the presence of γ8 than in the case of GluA2(Q) in the presence of γ2. As the fraction of receptors open under residual conditions is more for GluA2(Q) in the presence of γ8 than in the case of GluA2(Q) in the presence of γ2, the higher inhibition current for GluA2(Q) in the presence of γ8 suggests that Zn2+ inhibits the open channel state of the receptor. Additionally, the lack of significant inhibition in the peak currents before desensitization suggests a slower rate for the block. To verify the open channel block and slower rate, we studied the inhibition of GluA2(Q) homomeric receptor by Zn2+ under conditions where steady-state currents are higher, by using saturating concentrations of agonist glutamate and 100 μm cyclothiazide, which reduces desensitization, (Fig. 1D) and also studied the time constants of Zn2+ inhibition at 1 mm Zn2+ (Fig. 1E,F). Under these conditions, 1 mm Zn2+ blocked the steady-state current nearly completely, consistent with an activity-dependent block by the Zn2+, and the time constants were 103 ± 23.4 ms for the on rate and 95 ± 17.7 ms for the off rate, indicating slow kinetics. Further, consistent with an open channel block, the extent of inhibition current was voltage-dependent with higher inhibition at more negative potentials (Fig. 1G–I). The dose dependence for the inhibition by Zn2+ showed that the IC50 values were also dependent on the fraction of receptors in the open channel state, with GluA2(Q) receptor in the presence of cyclothiazide showing the lowest IC50 of 460 ± 40 μm (Fig. 1J) and GluA2(Q) receptor in the presence of γ8 and γ2 having an IC50 of 520 ± 38 and 1754 ± 114 μm (Fig. 1J), respectively. The percent inhibition at the maximum concentrations of Zn2+ was higher for GluA2(Q) in the presence of CTZ, followed by GluA2(Q) in the presence of γ8, and then GluA2(Q) receptor in the presence of γ2. This suggests that the extent of inhibition is higher when the residual activation is higher.
Figure 1.
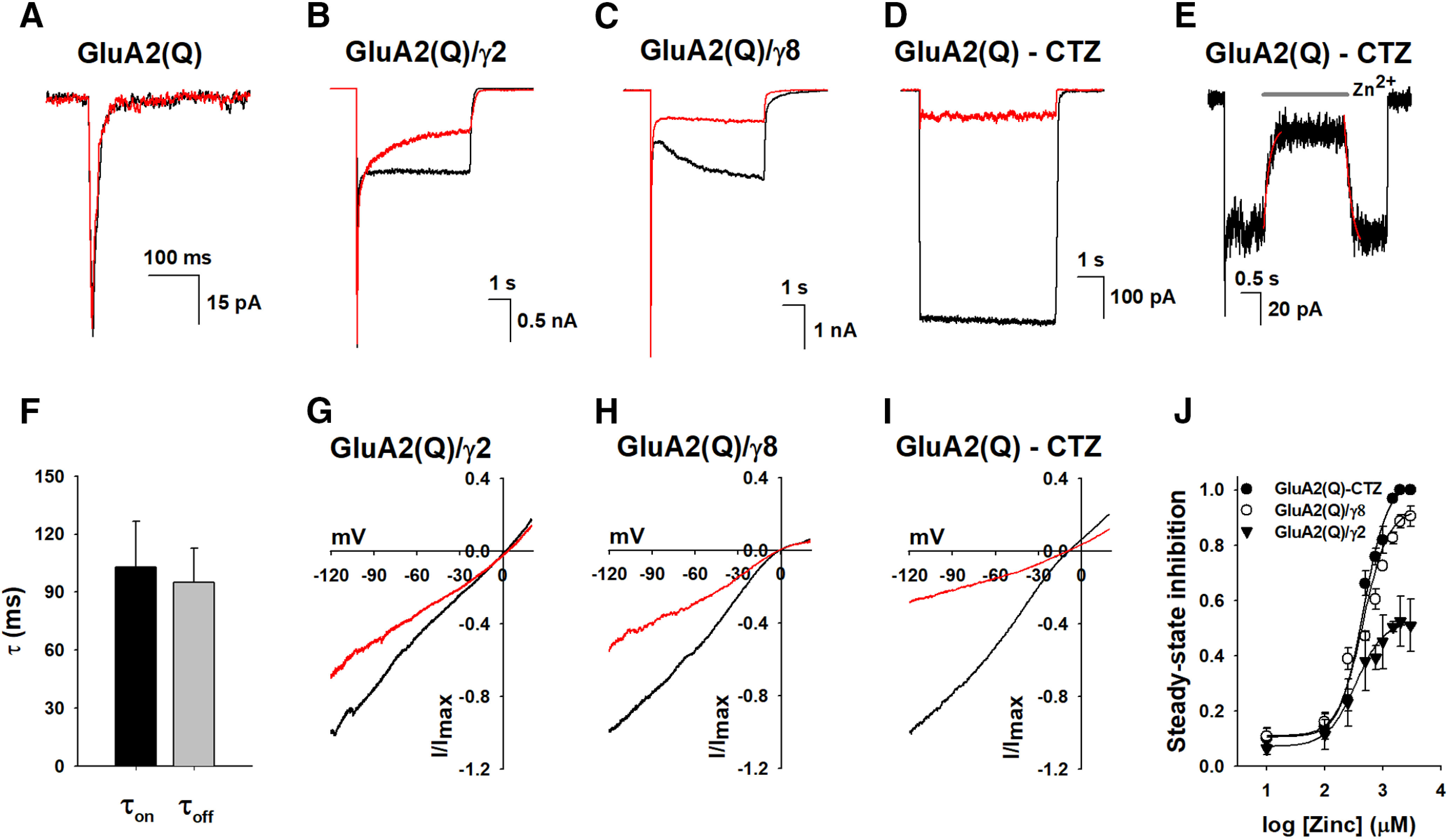
Zinc modulation of the AMPAR. Representative whole-cell recordings in response to 10 mm glutamate alone (black), or in the presence of 1 mm Zn2+ (red) for (A) GluA2(Q), (B) GluA2(Q)/γ2, (C) GluA2(Q)/γ8, and (D) GluA2(Q) in the presence of 100 μm cyclothiazide (CTZ). E, Time course of the inhibition and recovery by 1 mm Zn2+ in the presence of 10 mm glutamate and 100 μm CTZ. The On and Off phases were fitted to a single exponential function. Zn2+ application is marked by the gray bar on the top of the trace. F, Bar graph showing the time constant from the fits for the On and Off phases for Zn2+ inhibition. Voltage dependence curves (ramped from −120 to 20 mV) obtained using 10 mm glutamate alone, or in the presence of 1 mm Zn2+ for (G) GluA2 (Q)/γ2, (H) GluA2(Q)/γ8, and (I) GluA2(Q) in the presence of 100 μm CTZ. J, The dose-dependent inhibitory effects of Zn2+ on GluA2 (Q)/γ2 (▾), GluA2 (Q)/γ8 (○), and GluA2(Q) in the presence of 100 μm CTZ (●).
Dependence of Zn2+ inhibition on charge/editing at site 607
To determine whether the extent of Zn2+ inhibition is dependent on the charge at the site of editing, 607, we studied the unedited, GluA2(R), as well as a de novo mutation involved in developmental delay Q607E (GluA2(E)) (Fig. 2). Both of these proteins were studied in the context of γ2, the most common auxiliary subunit of the receptor; hence, all comparisons are done with respect to GluA2(Q) in the presence of γ2. The whole-cell currents of the GluA2(E) in the presence of γ2 showed a significantly higher level of residual steady-state currents relative to GluA2(Q) and GluA2(R) (Fig. 2A,B). The extent of inhibition current for the GluA2(E) in the presence of γ2 was significantly higher relative to that observed for GluA2(Q) in the presence of γ2, and significantly higher than that for GluA2(R) in the presence of γ2 (Fig. 2). The IC50 for Zn2+ inhibition of 980 ± 90 μm for GluA2(E) in the presence of γ2 (Fig. 2C) is significantly lower than the value of 2100 ± 150 μm for GluA2(R) in the presence of γ2 and 1754 ± 114 μm for GluA2(Q) in the presence of γ2 (Fig. 1J). The voltage dependence of the Zn2+ inhibition current was maintained for both GluA2(E) in the presence of γ2 and GluA2(R) in the presence of γ2 (Fig. 2D,E). The trends showing a higher extent of inhibition current and lower IC50 value for GluA2(E) relative to GluA2(Q) followed by GluA2(R) (Fig. 2F) suggest that charge at site 607 affects the extent of inhibition by Zn2+, with the difference between E to R being statistically significant.
Figure 2.

Zn2+ inhibition with site 607 mutants. Representative whole-cell recordings in response to 10 mm glutamate alone (black) or in the presence of 1 mm Zn2+ (red) for (A) GluA2(R)/γ2 and (B) GluA2(E)/γ2. C, The dose dependence of Zn2+ inhibition on GluA2(R)/γ2 (▾) and GluA2Q607E/γ2 (○). Voltage dependence curves (ramped from −120 to 20 mV) obtained using 10 mm glutamate alone, or in the presence of 1 mm Zn2+ for (D) GluA2(R)/γ2 and (E) GluA2(E)/γ2. F, Zinc inhibition of residual current in the presence of γ2 (left), and IC50 of zinc in the constructs of GluA2 coexpressed with γ2 (right). *p < 0.01. ***p < 0.001.
Frequency stimulation is affected by Zn2+
Given that Zn2+ inhibition current is significant in the physiological range of hundreds of micromolar to millimolar concentrations, we also studied Zn2+ inhibition using 1 ms pulses of 10 mm glutamate at 10 Hz frequency to mimic synaptic transmission. As expected based on the whole-cell recordings, GluA2(Q) currents are not inhibited significantly (8.5 ± 5%) by 1 mm Zn2+ (Fig. 3A,H). However, with 100 μm cyclothiazide, the current initially increased with frequency stimulation because cyclothiazide reduces desensitization, and these currents were inhibited by 73 ± 8.5% in the presence of 1 mm Zn2+ reaching a plateau at 15 s (Fig. 3B,H). GluA2(Q) in the presence of γ2 and γ8 was also inhibited by Zn2+: 52.5 ± 11% and 54 ± 5%, respectively (Fig. 3C,D). In the case of GluA2(R) in the presence of γ2, the inhibition current was similar to that of GluA2(Q) in the presence of γ2, showing an inhibition of 40.5 ± 11.5% (Fig. 3E), and the mutant GluA2(E) in the presence of γ2 showed a higher level of inhibition 60.5 ± 5% (Fig. 3F). The trends in the inhibition by Zn2+ for frequency stimulation are similar in all cases to the trends observed in the whole-cell currents, and further confirm that Zn2+ inhibition is observed under this stimulation. To determine whether Zn2+ inhibition plays a role in neuronal AMPARs, we also performed the experiments with mouse cortical neurons (Fig. 3G) and found that 1 mm Zn2+ showed inhibition as a function of time in the 10 Hz stimulation, showing an inhibition 18 ± 6%. The extent of inhibition was closer to that observed for GluA2(R) in the presence of γ2, consistent with the fact that heteromeric receptors containing GluA2(R) and γ2 are the most likely combination expected in these neurons.
Figure 3.
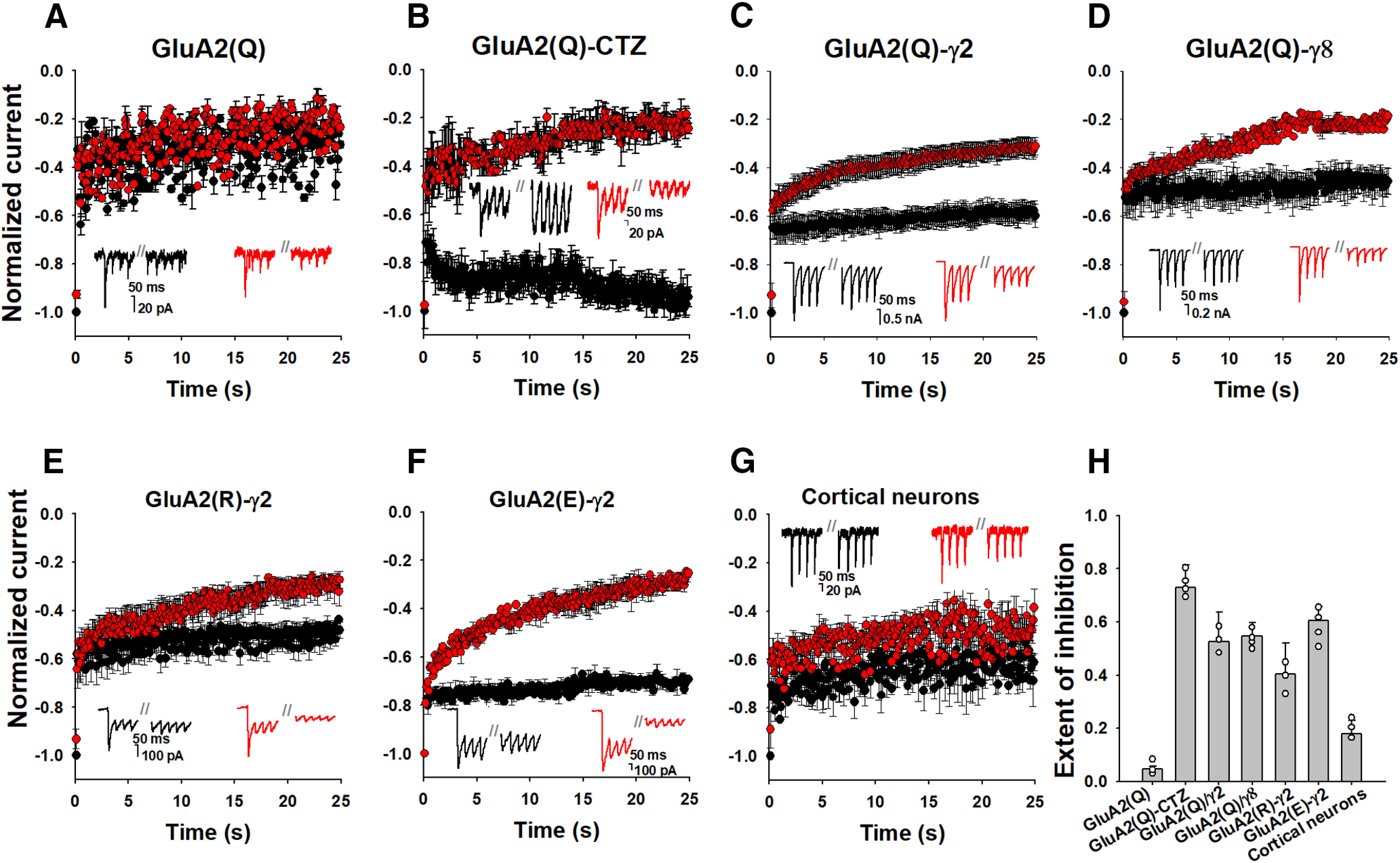
Effect of Zn2+ under 10 Hz frequency stimulation with 10 mm glutamate alone (black) or in the presence of 1 mm Zn2+ (red) for (A) GluA2(Q), (B) GluA2(Q)-CTZ, (C) GluA2(Q)/γ2, (D) GluA2(Q)/γ8, (E) GluA2(R)/γ2, and (F) GluA2(E)/γ2 recorded in the whole-cell configuration. Inset, Representative currents at the start and end of the train. G, Frequency stimulation in mouse cortical neurons, as in A. H, Histogram of Zn2+ inhibition for GluA2(Q) was 8.5 ± 5%, GluA2(Q)-CTZ 73 ± 8.5%, GluA2(Q)/γ2 52.5 ± 11%, GluA2(Q)/γ8 54 ± 5%, GluA2(R)-γ2 40.5 ± 11.5%, GluA2(E)-γ2 60.5 ± 5%, and cortical neurons was 18 ± 6%.
mEPSCs are inhibited by Zn2+
To test whether synaptic transmission was altered in the presence of Zn2+, we performed whole-cell voltage-clamp recordings in high cell density cortical neuronal culture. Spontaneous mEPSC recordings were in the presence of TTX, bicuculline, and Mg2+. As expected from these measurements, there is high variability in the mEPSCs. The mEPSCs showed a decrease in amplitude of spontaneous mEPSCs from 19.5 ± 1.5 to 15 ± 1 pA and a reduction in mEPSCs frequency from 32 ± 2.6 to 23.5 ± 2.8 (Fig. 4A,C) in the presence of 1 mm Zn2+. To evaluate whether endogenous Zn2+ affects the spontaneous mEPSC activity, we used the chelator ZX1, at a concentration of 100 μm (Pan et al., 2011). While a slight increase in the mean amplitude was noted for spontaneous mEPSCs, this was not statistically significant; however, the frequency was increased from 16 ± 3 to 27 ± 5 (Fig. 4B,D).
Figure 4.

Zinc inhibited the mEPSCs and ZX1 increased its frequency. Representative spontaneous mEPSCs, in control and (A) in the presence of 1 mm Zn2+, and (B) in the presence of ZX1 100 μm. Box plots of the amplitude and frequency activity of spontaneous mEPSC recordings, in control and (C) in the presence of 1 mm Zn2+, and (D) in the presence of ZX1 100 μm. Box plots represent median and interquartile range. Vertical lines indicate 5th and 95th percentiles. **p < 0.001.
Zinc inhibits the stabilized open state of GluA2(Q) AMPAR
We performed outside-out single-channel recordings of GluA2(Q) in the stabilized open state to further characterize the inhibition mechanism at the single-channel level. Consistent with previous reports (Kott et al., 2007; Suzuki et al., 2008; Jackson et al., 2011; Riva et al., 2017; Herguedas et al., 2019; Carrillo et al., 2020), GluA2(Q) in the presence of cyclothiazide and a saturating concentration of glutamate (10 mm) are predominantly open and populate the higher conductance levels (Fig. 5A). Similarly, GluA2(Q)/γ8 also populates the higher conductance levels, whereas GluA2(Q)/γ2 primarily populates the lower conductance states (Fig. 5B; Table 1). In the presence of Zn2+, the openings were brief and higher subconductance levels were not observed. This may suggest that Zn2+ predominantly blocks the higher subconductance levels. The single-channel recordings also show that the extent of inhibition under these conditions as measured by reduction in the open probability, 95 ± 2%, 86 ± 7%, and 44 ± 5%, for GluA2(Q) in presence of cyclothiazide, GluA2(Q) in the presence of γ8 and GluA2(Q) in the presence of γ2, respectively (Fig. 6), are similar to the inhibition as observed by whole-cell currents under similar conditions (Fig. 1J).
Figure 5.

Inhibition of GluA2 in the presence of CTZ and auxiliary proteins by Zn2+ in single-channel recordings. A, Single-channel currents recorded from GluA2(Q) in the presence of 100 μm cyclothiazide, GluA2(Q)/γ8, and GluA2(Q)/γ2 during continuous application of 10 mm glutamate alone (top) and in the presence 1 mm of Zn2+ (bottom). Openings are shown as downward deflections. B, Amplitude histograms of single-channel events from patches in the presence of glutamate alone (gray) and in the presence of Zn2+ (peach).
Table 1.
Conductance levels for single-channel recordingsa
| Conductance levels (pS) | GluA2(Q) + CTZ | GluA2(Q)/γ8 | GluA2(Q)/γ2 | |
|---|---|---|---|---|
| 1 | Control | 7.4 ± 0.5 (3.5 ± 0.4%) | 7 ± 0.7 (3 ± 0.6%) | 7 ± 0.4 (6.5 ± 0.9%) |
| Zinc | 11 ± 0.8 (73 ± 6%) | 7 ± 0.4 (12 ± 1%) | 10 ± 1 (74 ± 12.5%) | |
| 2 | Control | 15 ± 1 (9 ± 0.5%) | 14.6 ± 1.5 (7.5 ± 0.4%) | 13 ± 0.9 (48 ± 5%) |
| Zinc | 19 ± 1.1 (27 ± 4.5%) | 14 ± 0.9 (88 ± 11%) | 18.6 ± 1.5 (25 ± 3.5%) | |
| 3 | Control | 23.2 ± 2 (21 ± 2.6%) | 23 ± 1.2 (19 ± 2.7%) | 21 ± 1.7 (32 ± 3%) |
| Zinc | ------ | ------ | ------ | |
| 4 | Control | 33 ± 2.5 (70 ± 8.5%) | 35.4 ± 1.9 (68 ± 7%) | 31 ± 2.5 (12 ± 2%) |
| Zinc | ------ | ------ | ------ |
aPercentage of conductance levels is shown next to its value in parentheses. The error in the lifetimes is the SE of the fit. The table is related to Figure 5.
Figure 6.
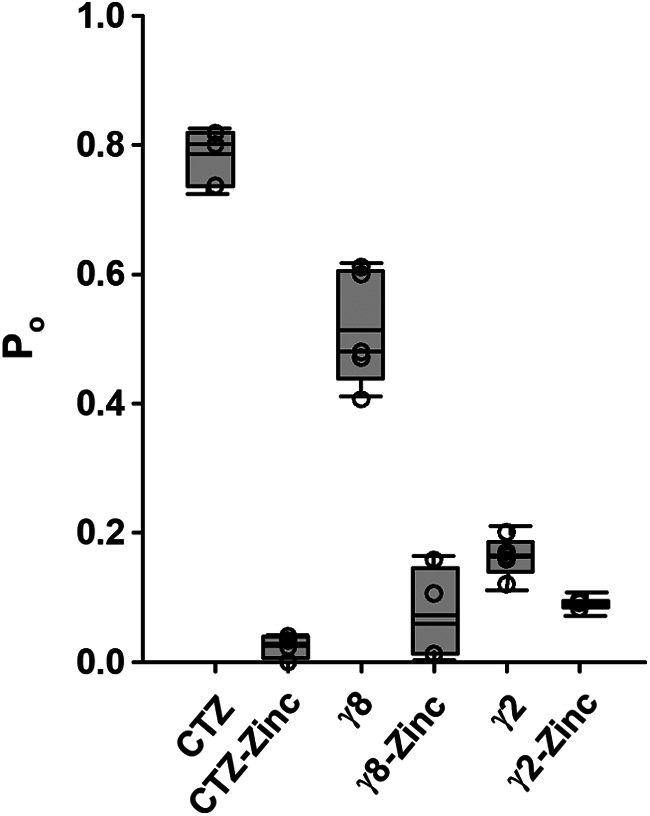
Zinc inhibition in single-channel recordings of GluA2(Q). Open probability (Po) of GluA2(Q) in the presence of CTZ, GluA2(Q)/γ8, and GluA2(Q)/γ2, in the absence and in the presence of 1 mm Zn2+.
Discussion
In the brain, free “chelatable” Zn2+ has been shown to be present at concentrations of ≥1 mm in glutamatergic synapses (Frederickson et al., 2000) and coreleased with glutamate during synaptic transmission (Qian and Noebels, 2005). Zn2+ modulates the function of ionotropic glutamate receptors, inhibiting the NMDA subtype of ionotropic glutamate receptors (for review, see Paoletti et al., 2009), and having a variable effect on the AMPA subtype of the ionotropic glutamate receptors (Mayer et al., 1989; Blakemore and Trombley, 2019). The inhibitory role of Zn2+ is best illustrated in studies with KO mice for ZnT3, the zinc transporter involved in maintaining vesicular synaptic Zn2+, which shows a marked increase in seizures and neuronal damage. While the mechanism of modulation of NMDARs by Zn2+ has been studied extensively, much less is known about the mechanism of modulation of AMPARs (Cole et al., 2000). The large variability in Zn2+ modulation of AMPARs, with some neurons showing no effect while others are inhibited to varying degrees, is one of the significant hurdles in understanding the mechanism. To address this, we have systematically studied Zn2+ modulation of AMPARs by varying the receptor properties of calcium permeability and also investigated the role of auxiliary proteins γ2 and γ8. Our data show that Zn2+ inhibition of AMPARs is slow, with a time constant of hundreds of milliseconds at millimolar concentrations of Zn2+. The kinetics of activation and desensitization for AMPARs (GluA2(Q)) is rapid (microseconds to tens of milliseconds); thus, the slow block by Zn2+ has minimal effect under these conditions. However, AMPARs in the presence of auxiliary proteins, such as γ2 and γ8, as well as edited and mutated forms of the receptor have slower kinetics and/or residual nondesensitized currents. Under these circumstances, Zn2+ is able to block the channel and inhibit the currents. Our data also show that the extent of inhibition correlates to the extent of residual nondesensitizing currents (activation) and is also dependent on the charge at site 607. These results along with the voltage dependence of the block suggest a channel block mechanism for the inhibition. Further, this block is significant under continual stimulations even in short pulses in both HEK-293 cells and neurons.
The requirement for the residual higher open probability as seen in the presence of γ2 and γ8 for significant inhibition by Zn2+ could underlie the large variability previously reported in primary neuronal recordings. Inhibition of AMPARs currents has been shown at millimolar concentrations of Zn2+ in hippocampal neurons (Mayer et al., 1989; Blakemore and Trombley, 2019); however, other studies that have shown no significant inhibition or even potentiation. For instance, Pan et al. (2011) reported that endogenous Zn2+ did not affect AMPARs at the mossy fiber CA3 synapses. The lack of an effect by Zn2+ could be because of the lack of γ8 at these synapses as shown by Fukaya et al. (2006), who found that mice with defective γ8 gene had no significant effect at the mossy fiber CA3, while having a large effect at other hippocampal synapses. Other studies have also shown potentiation at lower Zn2+ concentrations (micromolar concentration), and the mechanism of such potentiation is still not clear (Lin et al., 2001). However, even in these cases, addition of cyclothiazide to reduce desensitization eliminated this potentiation, possibly because of the inhibitory effects seen in our study compensating the potentiation from a yet unknown mechanism.
In conclusion, our studies suggest that Zn2+ should play a role in modulating AMPARs physiologically and also opens up the door for exploring this pathway as a means for pharmacological manipulation of AMPARs specifically targeting regions of the brain where there is overexpression of auxiliary proteins that prolong activation of the receptor.
Footnotes
The authors declare no competing financial interests.
This work was supported by National Institutes of Health Grant R35 GM122528 to V.J., American Heart Association Fellowship 18POST34030189 (National Institute of Health) to E.C., and 5R01GM115446 to A.M.A. We thank Ryan Durham for help editing the manuscript.
References
- Akbarian S, Smith MA, Jones EG (1995) Editing for an AMPA receptor subunit RNA in prefrontal cortex and striatum in Alzheimer's disease, Huntington's disease and schizophrenia. Brain Res 699:297–304. 10.1016/0006-8993(95)00922-D [DOI] [PubMed] [Google Scholar]
- Blakemore LJ, Trombley PQ (2019) Mechanisms of zinc modulation of olfactory bulb AMPA receptors. Neuroscience 410:160–175. 10.1016/j.neuroscience.2019.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone AL, Plested AJ (2016) Superactivation of AMPA receptors by auxiliary proteins. Nat Commun 7:10178. 10.1038/ncomms10178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo E, Shaikh SA, Berka V, Durham RJ, Litwin DB, Lee G, MacLean DM, Nowak LM, Jayaraman V (2020) Mechanism of modulation of AMPA receptors by TARP-gamma8. J Gen Physiol 152:e201912451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho CH, St-Gelais F, Zhang W, Tomita S, Howe JR (2007) Two families of TARP isoforms that have distinct effects on the kinetic properties of AMPA receptors and synaptic currents. Neuron 55:890–904. 10.1016/j.neuron.2007.08.024 [DOI] [PubMed] [Google Scholar]
- Cole TB, Robbins CA, Wenzel HJ, Schwartzkroin PA, Palmiter RD (2000) Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res 39:153–169. 10.1016/s0920-1211(99)00121-7 [DOI] [PubMed] [Google Scholar]
- Cull-Candy S, Kelly L, Farrant M (2006) Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol 16:288–297. 10.1016/j.conb.2006.05.012 [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Suh SW, Silva D, Frederickson CJ, Thompson RB (2000) Importance of zinc in the central nervous system: the zinc-containing neuron. J Nutr 130:1471S–1483S. 10.1093/jn/130.5.1471S [DOI] [PubMed] [Google Scholar]
- Fukaya M, Tsujita M, Yamazaki M, Kushiya E, Abe M, Akashi K, Natsume R, Kano M, Kamiya H, Watanabe M, Sakimura K (2006) Abundant distribution of TARP gamma-8 in synaptic and extrasynaptic surface of hippocampal neurons and its major role in AMPA receptor expression on spines and dendrites. Eur J Neurosci 24:2177–2190. 10.1111/j.1460-9568.2006.05081.x [DOI] [PubMed] [Google Scholar]
- Gaisler-Salomon I, Kravitz E, Feiler Y, Safran M, Biegon A, Amariglio N, Rechavi G (2014) Hippocampus-specific deficiency in RNA editing of GluA2 in Alzheimer's disease. Neurobiol Aging 35:1785–1791. 10.1016/j.neurobiolaging.2014.02.018 [DOI] [PubMed] [Google Scholar]
- Haering SC, Tapken D, Pahl S, Hollmann M (2014) Auxiliary subunits: shepherding AMPA receptors to the plasma membrane. Membranes (Basel) 4:469–490. 10.3390/membranes4030469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herguedas B, Watson JF, Ho H, Cais O, García-Nafría J, Greger IH (2019) Architecture of the heteromeric GluA1/2 AMPA receptor in complex with the auxiliary subunit TARP gamma8. Science 364:eaav9011 10.1126/science.aav9011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S (1991) Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science 252:851–853. 10.1126/science.1709304 [DOI] [PubMed] [Google Scholar]
- Hume RI, Dingledine R, Heinemann SF (1991) Identification of a site in glutamate receptor subunits that controls calcium permeability. Science 253:1028–1031. 10.1126/science.1653450 [DOI] [PubMed] [Google Scholar]
- Iacobucci GJ, Popescu GK (2017) NMDA receptors: linking physiological output to biophysical operation. Nat Rev Neurosci 18:236–249. 10.1038/nrn.2017.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, Nicoll RA (2011) Stargazin (TARP gamma-2) is required for compartment-specific AMPA receptor trafficking and synaptic plasticity in cerebellar stellate cells. J Neurosci 31:3939–3952. 10.1523/JNEUROSCI.5134-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, Milstein AD, Soto D, Farrant M, Cull-Candy SG, Nicoll RA (2011) Probing TARP modulation of AMPA receptor conductance with polyamine toxins. J Neurosci 31:7511–7520. 10.1523/JNEUROSCI.6688-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalappa BI, Anderson CT, Goldberg JM, Lippard SJ, Tzounopoulos T (2015) AMPA receptor inhibition by synaptically released zinc. Proc Natl Acad Sci USA 112:15749–15754. 10.1073/pnas.1512296112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato AS, Gill MB, Ho MT, Yu H, Tu Y, Siuda ER, Wang H, Qian YW, Nisenbaum ES, Tomita S, Bredt DS (2010) Hippocampal AMPA receptor gating controlled by both TARP and cornichon proteins. Neuron 68:1082–1096. 10.1016/j.neuron.2010.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S (2004) Glutamate receptors: RNA editing and death of motor neurons. Nature 427:801. 10.1038/427801a [DOI] [PubMed] [Google Scholar]
- Kott S, Werner M, Korber C, Hollmann M (2007) Electrophysiological properties of AMPA receptors are differentially modulated depending on the associated member of the TARP family. J Neurosci 27:3780–3789. 10.1523/JNEUROSCI.4185-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak S, Hideyama T, Yamashita T, Aizawa H (2010) AMPA receptor-mediated neuronal death in sporadic ALS. Neuropathology 30:182–188. 10.1111/j.1440-1789.2009.01090.x [DOI] [PubMed] [Google Scholar]
- Lin DD, Cohen AS, Coulter DA (2001) Zinc-induced augmentation of excitatory synaptic currents and glutamate receptor responses in hippocampal CA3 neurons. J Neurophysiol 85:1185–1196. 10.1152/jn.2001.85.3.1185 [DOI] [PubMed] [Google Scholar]
- MacLean DM, Bowie D (2011) Transmembrane AMPA receptor regulatory protein regulation of competitive antagonism: a problem of interpretation. J Physiol 589:5383–5390. 10.1113/jphysiol.2011.219485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean DM, Ramaswamy SS, Du M, Howe JR, Jayaraman V (2014) Stargazin promotes closure of the AMPA receptor ligand-binding domain. J Gen Physiol 144:503–512. 10.1085/jgp.201411287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Vyklicky L Jr, Westbrook GL (1989) Modulation of excitatory amino acid receptors by group IIB metal cations in cultured mouse hippocampal neurones. J Physiol 415:329–350. 10.1113/jphysiol.1989.sp017724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein AD, Nicoll RA (2008) Regulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol Sci 29:333–339. 10.1016/j.tips.2008.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai C, Sachs F (2013) Solving Ion Channel Kinetics with the QuB Software. Biophysical Reviews and Letters 08:1–21. [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D (2010) CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 67:239–252. 10.1016/j.neuron.2010.06.007 [DOI] [PubMed] [Google Scholar]
- Pan E, Zhang XA, Huang Z, Krezel A, Zhao M, Tinberg CE, Lippard SJ, McNamara JO (2011) Vesicular zinc promotes presynaptic and inhibits postsynaptic long-term potentiation of mossy fiber-CA3 synapse. Neuron 71:1116–1126. 10.1016/j.neuron.2011.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Vergnano AM, Barbour B, Casado M (2009) Zinc at glutamatergic synapses. Neuroscience 158:126–136. 10.1016/j.neuroscience.2008.01.061 [DOI] [PubMed] [Google Scholar]
- Peng PL, Zhong X, Tu W, Soundarapandian MM, Molner P, Zhu D, Lau L, Liu S, Liu F, Lu Y (2006) ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron 49:719–733. 10.1016/j.neuron.2006.01.025 [DOI] [PubMed] [Google Scholar]
- Qian J, Noebels JL (2005) Visualization of transmitter release with zinc fluorescence detection at the mouse hippocampal mossy fibre synapse. J Physiol 566:747–758. 10.1113/jphysiol.2005.089276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F. (2004) Restoration of Single-Channel Currents Using the Segmental k-Means Method Based on Hidden Markov Modeling Biophys. J 86:1488–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva I, Eibl C, Volkmer R, Carbone AL, Plested AJ (2017) Control of AMPA receptor activity by the extracellular loops of auxiliary proteins. Elife 6:e28680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpietro V, Dixon CL, Guo H, Bello OD, Vandrovcova J, Efthymiou S, Maroofian R, Heimer G, Burglen L, Valence S, Torti E, Hacke M, Rankin J, Tariq H, Colin E, Procaccio V, Striano P, Mankad K, Lieb A, Chen S, et al. (2019) AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat Commun 10:3094 10.1038/s41467-019-10910-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh SA, Dolino DM, Lee G, Chatterjee S, MacLean DM, Flatebo C, Landes CF, Jayaraman V (2016) Stargazin modulation of AMPA receptors. Cell Rep 17:328–335. 10.1016/j.celrep.2016.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki E, Kessler M, Arai AC (2008) The fast kinetics of AMPA GluR3 receptors is selectively modulated by the TARPs gamma 4 and gamma 8. Mol Cell Neurosci 38:117–123. 10.1016/j.mcn.2008.01.018 [DOI] [PubMed] [Google Scholar]
- Tomita S, Adesnik H, Sekiguchi M, Zhang W, Wada K, Howe JR, Nicoll RA, Bredt DS (2005) Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature 435:1052–1058. 10.1038/nature03624 [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496. 10.1124/pr.109.002451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Grassucci RA, Frank J, Sobolevsky AI (2016) Elucidation of AMPA receptor-stargazin complexes by cryo-electron microscopy. Science 353:83–86. 10.1126/science.aaf8411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright A, Vissel B (2012) The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Front Mol Neurosci 5:34. 10.3389/fnmol.2012.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Low CM, Moody OA, Jenkins A, Traynelis SF (2015) Ionotropic GABA and glutamate receptor mutations and human neurologic diseases. Mol Pharmacol 88:203–217. 10.1124/mol.115.097998 [DOI] [PMC free article] [PubMed] [Google Scholar]