

Abstract
Methyl-CpG binding protein 2 (MeCP2) is a nuclear protein critical for normal brain function, and both depletion and overexpression of MeCP2 lead to severe neurodevelopmental disease, Rett syndrome (RTT) and MECP2 multiplication disorder, respectively. However, the molecular mechanism by which abnormal MeCP2 dosage causes neuronal dysfunction remains unclear. As MeCP2 expression is nearly equivalent to that of core histones and because it binds DNA throughout the genome, one possible function of MeCP2 is to regulate the 3D structure of chromatin. Here, to examine whether and how MeCP2 levels impact chromatin structure, we used high-resolution confocal and electron microscopy and examined heterochromatic foci of neurons in mice. Using models of RTT and MECP2 triplication syndrome, we found that the heterochromatin structure was significantly affected by the alteration in MeCP2 levels. Analysis of mice expressing either MeCP2-R270X or MeCP2-G273X, which have nonsense mutations in the upstream and downstream regions of the AT-hook 2 domain, respectively, showed that the magnitude of heterochromatin changes was tightly correlated with the phenotypic severity. Postnatal alteration in MeCP2 levels also induced significant changes in the heterochromatin structure, which underscored importance of correct MeCP2 dosage in mature neurons. Finally, functional analysis of MeCP2-overexpressing mice showed that the behavioral and transcriptomic alterations in these mice correlated significantly with the MeCP2 levels and occurred in parallel with the heterochromatin changes. Taken together, our findings demonstrate the essential role of MeCP2 in regulating the 3D structure of neuronal chromatin, which may serve as a potential mechanism that drives pathogenesis of MeCP2-related disorders.
SIGNIFICANCE STATEMENT Neuronal function is critically dependent on methyl-CpG binding protein 2 (MeCP2), a nuclear protein abundantly expressed in neurons. The importance of MeCP2 is underscored by the severe childhood neurologic disorders, Rett syndrome (RTT) and MECP2 multiplication disorders, which are caused by depletion and overabundance of MeCP2, respectively. To clarify the molecular function of MeCP2 and to understand the pathogenesis of MECP2-related disorders, we performed detailed structural analyses of neuronal nuclei by using mouse models and high-resolution microscopy. We show that the level of MeCP2 critically regulates 3D structure of heterochromatic foci, and this is mediated in part by the AT-hook 2 domain of MeCP2. Our results demonstrate that one primary function of MeCP2 is to regulate chromatin structure.
Keywords: chromatin, imaging, MeCP2, neuron, Rett syndrome
Introduction
Methyl-CpG binding protein 2 (MeCP2) is a nuclear protein enriched in mature neurons and plays an essential role in maintaining normal gene expression (Lombardi et al., 2015). Proper neuronal function is dependent on the level of MeCP2, as both loss and gain of MeCP2 lead to severe neurodevelopmental disorders, Rett syndrome (RTT) and MECP2 multiplication disorders (Amir et al., 1999; Van Esch et al., 2005). These MECP2-related disorders are associated with many neurologic symptoms, which are recapitulated in mouse models either lacking or overexpressing MeCP2 (Guy et al., 2001; Collins et al., 2004). Importantly, neurologic deficit in the whole-body knock-out of Mecp2 is reproduced in mice which lack Mecp2 specifically in the brain (Chen et al., 2001). In addition, re-expression of MeCP2 in the postmitotic neurons of Mecp2-null mice restores their neurologic function (Guy et al., 2007). These findings demonstrate the importance of MeCP2 and show that understanding the physiological function of MeCP2 is the key to clarifying the pathogenesis of RTT and MECP2 multiplication disorders.
Extensive studies have aimed to clarify the molecular function of MeCP2, but what exactly MeCP2 does in vivo to regulate neuronal function remains elusive. MeCP2 has been shown to work as a transcriptional repressor by binding to methylated cytosines and recruiting co-repressor complexes (Lyst et al., 2013; Chen et al., 2015; Gabel et al., 2015; Lagger et al., 2017). However, deletion of Mecp2 in mice leads to both increased and decreased expression of MeCP2 bound genes (Chahrour et al., 2008; Ben-Shachar et al., 2009), arguing against a simple model where MeCP2 works as a repressor. Several studies have suggested that an additional function of MeCP2 is to modify the 3D structure of neuronal chromatin. First, a study in vitro showed that when purified MeCP2 is added to isolated nucleosomes, it induces changes in the beads-on-a-string structure of nucleosome arrays (Nikitina et al., 2007). Second, it was shown that MeCP2 deletion affects long-range chromosome interactions in the imprinted domains of neonatal mouse brains (Horike et al., 2005; Kernohan et al., 2014). Third, our group identified AT-hook like domains in MeCP2, which are homologous to those of high motility group AT-hook 1 (HMGA1). Because AT-hooks in HMGA1 can induce changes in the structural conformation of DNA, it has been speculated that the AT-hooks of MeCP2 have similar functions (Reeves, 2001; Baker et al., 2013). The AT-hook 2 domain is of particular interest, because it influences the onset of RTT-related symptoms (Baker et al., 2013). Fourth, work from a group using 3D FISH showed that the ectopic expression of MeCP2 induces clustering of pericentric heterochromatin in myoblasts (Brero et al., 2005). The same group also showed that cultured neurons lacking MeCP2 have increased number of heterochromatic foci (Bertulat et al., 2012). Finally, a study using array tomography analyzed heterochromatic foci in mouse brains and showed that MeCP2-negative neurons have foci with higher DAPI density, suggesting their heterochromatin is more compact than neurons expressing normal level of MeCP2 (Linhoff et al., 2015). While this work did not analyze the effect of MeCP2 overexpression or 3D shape of heterochromatic foci, the results suggested that one of the most important functions of MeCP2 could be to regulate chromatin structure.
Recently, we performed ChIP-seq on MeCP2 and the linker histone H1 in forebrain excitatory neurons and found that the two molecules occupy similar genomic regions while their distribution is independent of each other (Ito-Ishida et al., 2018). Because linker histone H1 is enriched in heterochromatin and induces structural changes on nucleosomes, that MeCP2 occupies similar genomic regions as linker histone H1 raises the possibility that MeCP2 may regulate chromatin structure. To address this possibility, here, we performed detailed structural analyses of heterochromatic foci in both MeCP2-depleted and MeCP2-overexpressing neurons using confocal and electron microscopy. We found that both depletion and overexpression of MeCP2 lead to different structural changes in heterochromatic foci, suggesting that MeCP2 levels regulate the architecture of neuronal chromatin.
Materials and Methods
Animals
Mice were housed in an AAALAS-certified Level three facility on a 14/10 h light/dark cycle. The mice were weaned at 21 d after birth and housed with three to five littermates per cage. Mecp2–null (Guy et al., 2001) and MECP2-Tg3 lines (Collins et al., 2004) were maintained in 129S6 and FVB background, respectively. Transgenic mice expressing either MeCP2-R270X or MeCP2-G273X were generated as previously described and were maintained in the FVB background (Baker et al., 2013). These mice were crossed with Mecp2+/− female mice, and male littermate mice were used for the analysis. Female mice were examined either at three months or three weeks of age, and male mice were examined at 8–10 weeks of age. For the immunostaining of histone modifications, we used symptomatic Mecp2+/− and Mecp2Tg3/+ female mice that were between four and 10 months old. All procedures to maintain and use these mice were approved by the Institutional Animal Care and Use Committee for Baylor College of Medicine and Affiliates and the Animal Resource Committee of Keio University.
Immunofluorescence
Brain sections were obtained as previously described (Ito-Ishida et al., 2015). The brains were fixed by transcardial perfusion of 4% paraformaldehyde (PFA) dissolved in PBS. Brains were removed, kept in 4% PFA overnight, cryoprotected in 25% sucrose solution, and frozen in optimal cutting temperature medium (O.C.T.). Sagittal sections were obtained using a Leica CM3050S cryostat at either 25- or 45-μm thickness. The slices were incubated in a PBS-buffered blocking solution containing 2% normal goat serum and 0.3% Triton X-100 for 1 h, followed by incubation in a primary antibody solution containing anti-MeCP2 (1:1000, rabbit monoclonal, D4F3, Cell Signaling, catalog #3456; 1:500, mouse monoclonal, mec-168, Sigma, #M6816), anti-parvalbumin (PV; 1:5000, mouse monoclonal, Swant, #PV235), anti-calmodulin-dependent protein kinase 2α (Camk2α; 1:1000, rabbit monoclonal, Abcam, #ab52476), or anti-GFP antibody (1:1000, chicken polyclonal, Abcam, #13970) overnight at 4°C. Immunostaining for histone methylation marks was performed after antigen retrieval by incubating the slices in sodium citrate buffer (10 mm sodium citrate and 0.05% Tween 20; pH 6.0) at 95°C for 5 min. The primary antibodies for histone methylation marks were anti-H3K27me3 (1:500, rabbit monoclonal, C36B11, Cell Signaling, catalog #9733), anti-H3K9me3 (1:500, rabbit polyclonal, Abcam, catalog #ab8898), anti-H4K20me2/3 (1:500, mouse monoclonal, 6F8-D9, Abcam, #ab78517), and anti-H3K4me3 (1:500, rabbit polyclonal, Abcam, #ab8580). After the overnight incubation with primary antibodies, slices were washed three times with PBS and were incubated with secondary antibodies conjugated with either Alexa Fluor 488, Alexa Fluor 555, or Alexa Fluor 633 (1:1000, Invitrogen) overnight at 4°C. After one wash wish PBS, PBS containing DAPI (2.5 µg/ml) was applied for 5 min, followed by two additional washes with PBS. To visualize nucleoli, we used Syto RNASelect green fluorescent cell stain (Invitrogen, #S32703), which was dissolved in PBS containing 0.3% Triton X-100 to the final concentration of 500 nm, and was applied overnight at 4°C, followed by the immunostaining as described above. Stained brain sections were imaged using a Leica LCS SP8 confocal microscope with 60× glycerol lens (N.A. 1.3).
Structural analysis of heterochromatic foci
Images were obtained to achieve the highest resolution following Nyquist rate: zoom 7 in 512 × 512 pixels or zoom 3.5 in 1024 × 1024 pixel, and serial z-sections at 0.13-µm thickness. In order to compare foci of Null-cells and Tg3-cells with wild-type (WT)-cells in the same brain sections, Null-cells or Tg3-cells were paired with their closest WT-cells and were imaged and analyzed using the same parameters. Quantitative analysis of DAPI-foci was performed using Image Pro Analyzer 7.0 (Media Cybernetics) and Imaris 8.3 (Bitplane) by following steps: (1) average intensity of DAPI in the euchromatic region within the MeCP2-positive (WT) nucleus was measured from three randomly selected areas, (2) the images were processed with 3 × 3 × 3 median filter, and (3) DAPI-foci were determined by using the surface tool of Imaris, using 2× intensity of average euchromatin intensity (obtained from step 1) as a threshold. Heterochromatic foci whose volume was >1000 voxels were included for analysis. Morphologic parameters, including average and maximum DAPI/MeCP2 intensity, ellipticity (oblate), and volume, were determined for each focus using the measurement tools in Imaris. Graphs for DAPI and MeCP2 intensity show the relative intensity normalized by the average intensity of the foci in either the respective cell (for WT-cells) or in the closest WT-cell (for Null-cells or Tg3-cells). If there were more than one DAPI-focus per cell, the results from all the foci within the nucleus were averaged to represent one cell. Results were obtained from seven to 12 cells per condition per mouse and were averaged to represent one animal. To quantify levels of histone methylation marks within the heterochromatic foci, DAPI images were used as described above to determine the presence of foci. Mean intensity of each histone mark was obtained from all the foci within the imaged area, which were grouped into either MeCP2-Null, Tg3, or WT foci based on the MeCP2 intensity. Values were averaged from two randomly selected regions to represent one animal.
Structural analysis of nucleoli
Images of nucleoli were obtained as above and analyzed using Image Pro Analyzer. The images were first processed twice with low pass filter. After adjustment of the intensity, DAPI images were subtracted from Syto RNASelect images to identify nucleoli which were defined as Syto RNASelect-positive and DAPI-negative region. The shape parameters of nucleoli, including irregularity (named “roundness” in the software) and area, were measured using “Automatic Counting” tool. Irregularity (i.e., roundness) was defined as (perimeter)2/(4π × area). The analyses were done on serial z-planes. The data from the plane with largest cross-sectional area was used to represent each nucleolus. When there was more than one nucleolus per cell, the results from all the nucleoli within one nucleus were averaged to represent one cell. Results were obtained from 10 cells per condition per mouse and were averaged to represent one animal.
Transmission electron microscopy (TEM)
The brains were fixed by transcardial perfusion of sodium cacodylate buffer containing 2% PFA and 2.5% glutaraldehyde. Hippocampal CA1 region was dissected under a dissection microscope and was postfixed overnight in the same cacodylate buffer. The samples were further fixed in aqueous 1% OsO4, and then dehydrated in an ethanol series followed by propylene oxide. The dehydrated samples were embedded in EMBED-812 resin (Electron Microscopy Sciences), sliced with a microtome, and the sections were stained with 1% uranyl acetate followed by lead citrate before imaging by TEM (1010; JEOL). The images were obtained at 800× magnification, processed by low-pass filter, and the electron-dense foci were automatically quantified using Image Pro Analyzer 7.0. The results were obtained from 8 images per mouse and all the identifiable nuclei in the images were analyzed to avoid bias. The numbers of nuclei and electron dense foci analyzed per mouse were 34–83 and 39–181, respectively.
Production and stereotaxic injection of adeno-associated virus (AAV)
Postnatal knock-down of MeCP2 was conducted using AAVs expressing miRNA against mouse Mecp2 (mirMecp2), which was designed using Block-IT RNAi designer kit (Invitrogen). Oligonucleotides containing mirMecp2 sequence (top, 5′−TGCTGTTCACCTGAACACCTTCTGATGTTTTGGCCACTGACTGACATCAGAAGGTTCAGGTGAA−3′; bottom, 5′−CCTGTTCACCTGAACCTTCTGATGTCAGTCAGTGGCCAAAACATCAGAAGGTGTTCAGGTGAAC−3′) were annealed and cloned into pCAGGS-GFP vector which induced GFP expression together with mirMecp2. Randomized sequence was used as a negative control (mirNega, top, 5′−TGCTGAAATGTACTGCGCGTGGAGACGTTTTGGCCACTGACTGACGTCTCCACGCAGTACATTT−3′; bottom, 5′−CCTGAAATGTACTGCGTGGAGACGTCAGTCAGTGGCCAAAACGTCTCCACGCGCAGTACATTTC−3′) and was processed similarly. Fragment containing either mirMecp2 or mirNega was digested from the pCAGGS-GFP-miRNA vector and was subcloned into pAAV-CAGGS-GFP vector. For the production of AAVs expressing human MECP2, PCR fragment from pDest-MECP2-GFP (Addgene, #48078) was inserted into a pAAV-hSyn-DIO-GFP vector. To produce AAV particles, the obtained pAAV-plasmids, pAAV-DJ (Cell Biolabs), and pHelper (AAV Helper-Free system, Agilent Technologies) were co-transfected into AAV293 cells (Agilent Technologies) with lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific) and incubated for 3 d in the presence of 1 μm tricostatin A. Virus particles were purified using AAV purification kit (VIRAPUR) and concentrated by filter units (Millipore Sigma). The titers of AAVs (vg/ml) were 1.3 × 1010 (AAV-mirMecp2), 3.4 × 1010 (AAV-mirNega), 6.8 × 1010 (AAV-DIO-huMECP2-GFP), and 3.9 × 1011 (AAV-DIO-GFP). AAV5-Camk2α-Cre was purchased from Addgene (ID: 105558, 6.8 × 1013 vg/ml). For the stereotaxic injection of AAVs, WT C57BL/J mice (six to seven weeks) were anesthetized with intraperitoneal injection of ketamine/xylazine and was placed on a stereotaxic instrument (Stoelting). Injection was performed using Nanoliter (World Precision Instrument) into bilateral PFC (AP 2.32, ML 0.4, 2.0–0.5 mm from pia, 0.6 µl each side) for the heterochromatic foci imaging. For the behavioral and transcriptional analysis, AAVs were injected into four sites (AP 2.0, ML 0.4, 2–0.5 mm from pia, 0.6 µl each side; AP 2.8, ML 0.4, 0.8 mm from pia, 0.4 µl each side) to achieve broader infection area.
Behavioral experiments
WT C57BL6J mice received AAV injection and were tested for behavior. We used equivalent number of both male and female mice. To perform open field (OF) test, mice were placed in 50 × 50 cm box for 15 min. Total distance traveled and time in open area were analyzed using custom software (Otsuka et al., 2016). Light-dark transition (LD) test was performed by placing mice in a 50 × 50 cm box separated into light and dark areas for 10 min. Fraction of time spent in the light area was measured using custom software.
Observational fear response was tested as described previously (Sakaguchi et al., 2018) with some modifications. Two identical chambers with grid floors were placed side-by-side in a sound-proof box. One chamber was used as an observation chamber and another as a shock chamber. Before the test day, mice were placed in the observation chamber and were habituated for 5 min. On the test day, mice were placed in the shock chamber and received brief prime shock (0.99 mA, 2 s, once) and were immediately returned to their home cages. Two hours after the prime shock, the mice were placed in the observation chamber and demonstrators (age and sex matched) were placed in the shock chamber. After 5 min of habituation phase, the demonstrators received repetitive foot shocks (0.99 mA, 2 s, 10-s interval, 24 times) for 4 min. The observers' freezing response was measured using automated analysis system (Image FZC, O'Hara) during the baseline (5 min), shock delivery (4 min), and after-shock phase (1 min).
mRNA-Seq from purified nuclei
WT C57BL6J mice (six to seven weeks) received injection of AAVs (AAV5-Camk2α-Cre and AAV(DJ)-DIO-huMECP2-GFP) into the bilateral PFC. Three weeks after the injection, tissue around injection sites was dissected in ice-cold PBS and snap frozen with liquid N2. Neuronal nuclei were purified using Iodixanol gradient method and sorted by FACS as described previously (Ito-Ishida et al., 2018). Anti-NeuN antibody (Clone A60, Chemicon), which was conjugated with Alexa Fluor 647 by Apex-labeling kit (Invitrogen), was used to label neuronal nuclei. Sorting was performed using Beckman MoFlo XDP system. Control samples, which had not received injection, was processed each time to determine background fluorescence and to set gating. NeuN-positive nuclei were sorted into four groups based on the GFP intensity and collected into RL buffer (Norgen) containing 1% mercaptoethanol and stored in −80°C until RNA isolation. We adjusted collection timing to obtain equivalent number of nuclei (15–50 × 103/sample) from each group within the same animal. Samples were collected from four mice and processed individually as biological replicates. Total RNA was purified using Single Cell RNA Purification kit (Norgen). RNA quality was determined by BioAnalyzer RNA6000 Pico kit (Illumina) and the RIN values ranged from 6.2 to 7.3. cDNA was synthesized from 1 ng of RNA using Smart-Seq HT kit (Takara). ERCC RNA Spike-in mix (Thermo Fisher Scientific) was included in the samples but was not used for analysis. Multiplexed library, which was prepared by Nextera XT DNA Library Prep kit (Illumina), was sent for 100-bp paired-end sequencing by HiSeq2500 platform at National Institute of Genetics (Shizuoka, Japan). Total number of reads per library was 28–37 M.
mRNA-Seq analysis
Paired-end reads from multiple lanes per sample were grouped together and quality control was performed using FastQC v0.11.8. Nextera mate pair adapters were trimmed by cutadapt v2.7 with options: “-a CTGTCTCTTATACACATCT -A CTGTCTCTTATACACATCT -m25.” Gene expressions were estimated by RSEM v1.3.1 using STAR v2.7.1a as the aligner. Trimmed reads were aligned to the Mus musculus genome Ensembl Version GRCh38.99 using the annotation gtf file obtained from Ensembl version GRCh38.99. In addition to the mouse genome, human MECP2-Cycle3GFP sequence was also used as the reference sequence. STAR indices were built by RSEM's rsem-prepare-reference. Read counts were calculated by RSEM's rsem-calculate-expression using parameters: “–paired-end–star–strandedness none–estimate-rspd.” Gene expression levels were quantified as transcripts per million mapped reads (TPM). We computed differential expression with the exact test method in the Bioconductor package edgeR v3.22.5. Four mice with the same conditions were grouped and GFP-low, GFP-med, and GFP-high groups were compared with the GFP-negative group. Genes with false discovery rate (FDR) ≦ 0.01 were classified as differentially expressed genes (DEGs). The results from mRNA-Seq analysis including total number of mapped reads and DEGs are summarized in Extended Data Fig. 12-1.
Results from mRNA-Seq analysis including the number of mapped reads and the list of DEGs. Download Figure 12-1, XLSX file (509.9KB, xlsx) .
Validation of mRNA-Seq by RT-qPCR
First-strand cDNA was synthesized from 5 ng RNA by using M-MLV reverse transcriptase (Life Technologies). Quantitative PCR was performed using Thunderbird SYBR qPCR Mix (Toyobo) and QuantStudio 5 (Applied Biosystems). The primer sets were designed to amplify target genes using UCSC genome browser and Primer 3. The reactions were conducted in duplicate and relative proportions of the cDNA were determined based on the threshold cycle (Ct). The results were averaged for each sample and normalized to the value of gapdh. Statistical analyses were performed using δ Ct values over gapdh (i.e., Ctgene − Ctgapdh). Following PCR primers were used:
Gapdh: forward 5′–ggagattgttgccatcaacga–3′, reverse 5′–tgaagacaccagtagactccacgac–3′;
Gipr: forward 5′–gggatcttggagagaccaca–3′, reverse 5′–gtcgtcagggacagggagta–3′;
Efnb3: forward 5′–gcttcttgtccttcctgtgc–3′, reverse 5′–ggggagcagttggtatgaaa–3′;
Pcdh9: forward 5′–cgaagagtacgcgaggtagg–3′, reverse 5′–tcagagttgggtgatggtga–3′;
Slc24a2: forward 5′–gactgccgaggaagaagatg–3′, reverse 5′–cggacgtcaggtaaggtgat–3′.
Experimental design and statistical analysis
Statistical tests for each assay were chosen based on their appropriateness for the assay. No statistical method was used to predetermine sample size but our sample sizes are similar to or exceed previous publications (Ito-Ishida et al., 2015; Linhoff et al., 2015). All statistical calculations were conducted using GraphPad Prism software with α level of 0.05. For experiments comparing Null-cells or Tg3-cells with WT-cells from the same set of mice, two-tailed paired t test was performed. The Gaussian distribution for the difference between the pairs was confirmed by the Shapiro–Wilk normality test. Paired t tests for MeCP2 and DAPI intensity were performed using average of the raw intensity data. Cumulative distribution was analyzed by Kolmogorov–Smirnov test. For the analysis comparing TEM images obtained from mice with different genotypes, the difference in variation was analyzed first by F test. As there was no difference between the groups, the datasets were analyzed by two-tailed t test. For experiments comparing multiple groups, data distribution was assumed to be normal and this was not formally tested. Variance was estimated by standard errors of mean. Multiple comparisons were corrected by either Tukey's (for one-way ANOVA) or Bonferroni's post hoc test (for two-way ANOVA). Statistical methods and values including all p values were described in figure legends.
Results
MeCP2 level determines structure of heterochromatic foci in hippocampal pyramidal cells
To address whether MeCP2 is required for maintaining chromatin structure in mature neurons, we examined the 3D architecture of neuronal heterochromatin-enriched foci in both MeCP2-negative (Null) and MeCP2-overexpressing (Tg3) cells by using Mecp2+/− and Mecp2Tg3/+ female mice. While heterochromatic foci in Null-cells have been previously analyzed (Linhoff et al., 2015), the impact of MeCP2 overexpression has not been studied. Because of random inactivation of X-chromosomes, Mecp2+/− females contain MeCP2-positive (WT) and MeCP2-negative (Null) neurons, allowing us to compare the heterochromatic foci in these two cellular genotypes under identical conditions. Because the extra copies of MECP2 in Mecp2Tg3/+ females are also on the X-chromosome, WT and Tg3 neurons could be compared in the same mice as well—importantly, the Tg3 allele models the human MECP2 triplication (Collins et al., 2004). We first looked at the nuclei of CA1 pyramidal cells because they have clear DAPI-intense foci, which are highly enriched in heterochromatin. We examined three-month-old female mice, because Mecp2+/− female mice develop significant neurologic features by this age (Samaco et al., 2013). Images of DAPI-foci were obtained by a confocal microscope at a Nyquist rate to achieve optimal resolution and were reconstructed using Imaris (Fig. 1; Movie 1). Neurons expressing a normal level of MeCP2 (WT-cells) could be clearly differentiated from either Null-cells or Tg3-cells by the immunofluorescent intensity of MeCP2 (Fig. 1A,D,G,J). When we analyzed heterochromatic foci of Mecp2+/− female mice, we found that the foci in Null-cells had higher mean DAPI intensity (N = 5 mice, p = 0.0162; Fig. 1B), which was consistent with the finding from the previous study (Linhoff et al., 2015). We further looked into the detailed structure of the foci and found that the shape of the foci in Null-cells was clearly different from WT-cells (Fig. 1A). Compared with the foci in WT-cells which had some irregularity on their surface, heterochromatic foci in Null-cells had smoother surface and simpler morphology, and their shape was closer to a simple sphere. We quantified the smoothness and irregularity of 3D structures by measuring their ellipticity index (Fig. 1C). This index represents how close an object is to a sphere assuming that an object is an oblate ellipsoid. The ellipticity index is zero when the object is a complete sphere. The value becomes higher as the object becomes flatter, with the maximum value being one. We found that the ellipticity index of foci in Null-cells was significantly lower than that in WT-cells, indicating that the morphology of the foci in Null-cells is closer to a simple sphere (N = 5 mice, p = 0.0008; Fig. 1C). The average volume of the foci was comparable between Null-cells and WT-cells (p = 0.1848; Fig. 1E), while the average number of foci per cell was mildly decreased in Null-cells (p = 0.0036; Fig. 1F).
Figure 1.
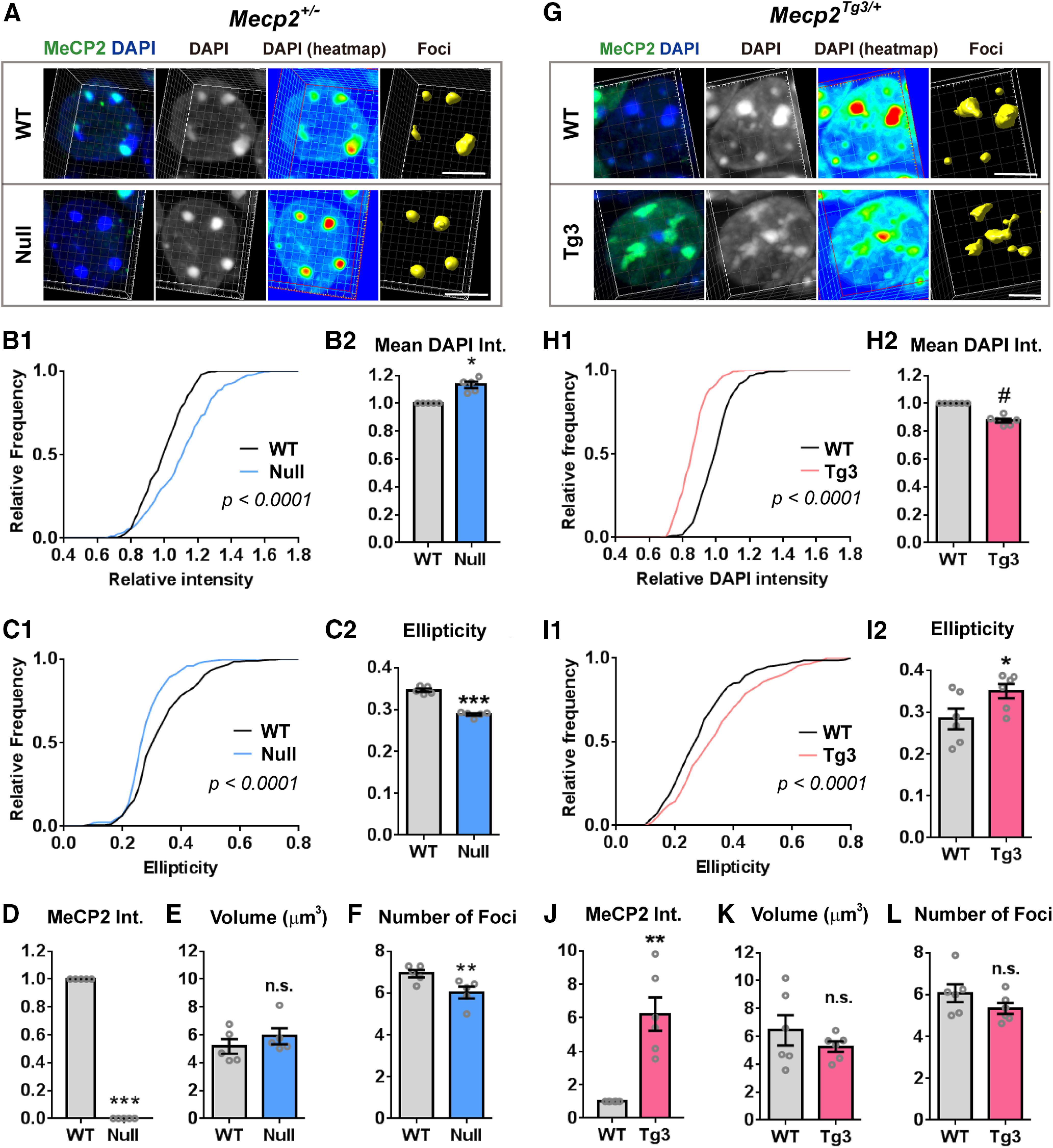
MeCP2 dosage regulates heterochromatic structure in CA1 pyramidal cells. A, CA1 pyramidal cells of Mecp2+/− female mice (three months old) were stained with DAPI (blue) and MeCP2 (green). 3D structure of the foci was constructed and analyzed by Imaris (yellow). Note the change in the DAPI intensity (heat map) and in the shape of foci in MeCP2-negative (Null) cells when compared with the MeCP2-positive (WT) cells. Scale bars: 5 µm. B–F, Results of the quantification from the images obtained in A. B, DAPI intensity (int.) in the heterochromatic foci was higher in Null-cells than in WT-cells. B1, Relative DAPI intensity of each focus was normalized to the mean DAPI intensity of the foci in WT-cell. The graph shows cumulative distribution of the normalized mean DAPI intensity. N = 271 foci from 5 mice (WT-cells) and 238 foci from 5 mice (Null-cells). Kolmogorov–Smirnov test; p < 0.0001. B2, Bar graph showing mean DAPI intensity averaged in each mouse. Mean DAPI intensity was higher in Null-cells. N = 5 mice. Two-tailed paired t test; p = 0.0162, t(4) = 3.995. C, Ellipticity in the foci was decreased in Null-cells. C1, Cumulative distribution of ellipticity in the foci was shifted toward left in Null-cells. N = 271 foci from 5 mice (WT-cells) and 238 foci from 5 mice (Null-cells). Kolmogorov–Smirnov test; p < 0.0001. C2, Bar graph showing ellipticity averaged across foci in each mouse. Ellipticity was lower in Null-cells than WT-cells. N = 5 mice. Two-tailed paired t test; p = 0.0008, t(4) = 9.137. D, Mean MeCP2 intensity was significantly reduced in Null-cells compared with WT-cells. N = 5 mice. Two-tailed paired t test; p = 0.0004, t(4) = 11.13. E, Average volume of heterochromatic foci was comparable between Null-cells and WT-cells. N = 5 mice, Two-tailed paired t test; p = 0.1848, t(4) = 1.6. F, Average number of heterochromatic foci per cell was reduced in Null-cells compared with WT-cells. N = 5 mice. Two-tailed paired t test; p = 0.0036, t(4) = 6.114. G, CA1 pyramidal cells of Mecp2Tg3/+ female mice (three months old) were stained and analyzed in the same way as in A. Scale bars: 5 µm. H–L, Quantified results from (G). Note the opposite changes in the parameters in Null-cells and Tg3-cells. H, DAPI intensity in the heterochromatic foci was reduced in Tg3-cells. H1, Relative DAPI intensity of each focus was normalized to the mean DAPI intensity of the foci in WT-cells. The graph shows cumulative distribution of the normalized mean DAPI intensity. N = 291 foci from 6 mice (WT-cells) and 256 foci from 6 mice (Tg3-cells). Kolmogorov–Smirnov test, p < 0.0001. H2, Bar graph showing mean DAPI intensity averaged in each mouse. Mean DAPI intensity was decreased in Tg3-cells. N = 6 mice. Two-tailed paired t test; p < 0.0001, t(5) = 16.92. I, Ellipticity in the foci was increased in Tg3-cells. I1, Cumulative distribution of ellipticity in each focus was shifted toward right in Tg3-cells. N = 291 foci from 6 mice (WT-cells) and 256 foci from 6 mice (Tg3-cells). Kolmogorov–Smirnov test; p < 0.0001. I2, Bar graph showing ellipticity averaged across foci in each mouse. Ellipticity was higher Tg3-cells than WT-cells. N = 6 mice. Two-tailed paired t test; p = 0.0422, t(5) = 2.711. J, Mean MeCP2 intensity (int.) was significantly higher in Tg3-cells than WT-cells. N = 6 mice. Two-tailed paired t test; p = 0.0037, t(5) = 5.114. K, Average volume of heterochromatic foci was comparable between Tg3-cells and WT-cells. N = 5 mice, Two-tailed paired t test; p = 0.2328, t(5) = 1.357. L, Average number of heterochromatic foci per cell was comparable between Tg3-cells and WT-cells. N = 5 mice. Two-tailed paired t test; p = 0.1478, t(5) = 1.711. Bars show average ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, # p < 0.0001; n.s., not significant.
Images of 3D-reconstructed nuclei. DAPI, MeCP2, and reconstructed heterochromatin foci are shown in gray, green, and yellow, respectively. Nuclei of CA1 pyramidal cells of a Mecp2+/− female mouse. MeCP2-negative (Null-) cell was observed by the side of a MeCP2-positive (WT-) cell. Heterochromatic foci of the Null-cell had increased DAPI intensity and simplified shape.
We also analyzed MeCP2-overexpressing cells (Tg3-cells) in the CA1 pyramidal layer of Mecp2Tg3/+ female mice using the same method (Fig. 1G; Movie 2). We found that the heterochromatic foci of Tg3-cells also had significantly different morphologic features when compared with the cells expressing one copy of Mecp2 (Fig. 1G). Interestingly, parameters which were altered in Null-cells were changed to the opposite direction in Tg3-cells (Fig. 1H,I). The mean intensity of DAPI within the heterochromatic foci was lower in Tg3-cells than that in WT-cells (N = 6 mice, p < 0.0001; Fig. 1H). The shape of the heterochromatic foci in Tg3-cells appeared more irregular, and the ellipticity index was higher than the foci of WT-cells (p = 0.0422; Fig. 1I). The average volume of foci (p = 0.2328; Fig. 1K) and the number of foci per cell (p = 0.1478; Fig. 1L) were not altered in Tg3-cells.
Images of 3D-reconstructed nuclei. DAPI, MeCP2, and reconstructed heterochromatin foci are shown in gray, green, and yellow, respectively. Nuclei of CA1 pyramidal cells of a Mecp2Tg3/+ female mouse. MeCP2-overexpressing (Tg3-) cells were observed by the side of a cell expressing single copy of Mecp2 (WT-cell). Heterochromatic foci of the Tg3-cells had decreased DAPI intensified and complex morphology.
MeCP2 level determines structure of heterochromatic foci in excitatory and inhibitory neurons of the cerebral cortex
Next, we asked whether MeCP2-dependent changes in heterochromatic foci were present in other neuronal cell types. To address this, we analyzed DAPI-enriched foci of excitatory and inhibitory neurons in the frontal cortex (Figs. 2, 3). We used the same method as for CA1 pyramidal neurons and analyzed the foci of Mecp2+/− and Mecp2Tg3/+ female brains (three months old) to compare Null-cells versus WT-cells and Tg3-cells versus WT-cells, respectively.
Figure 2.
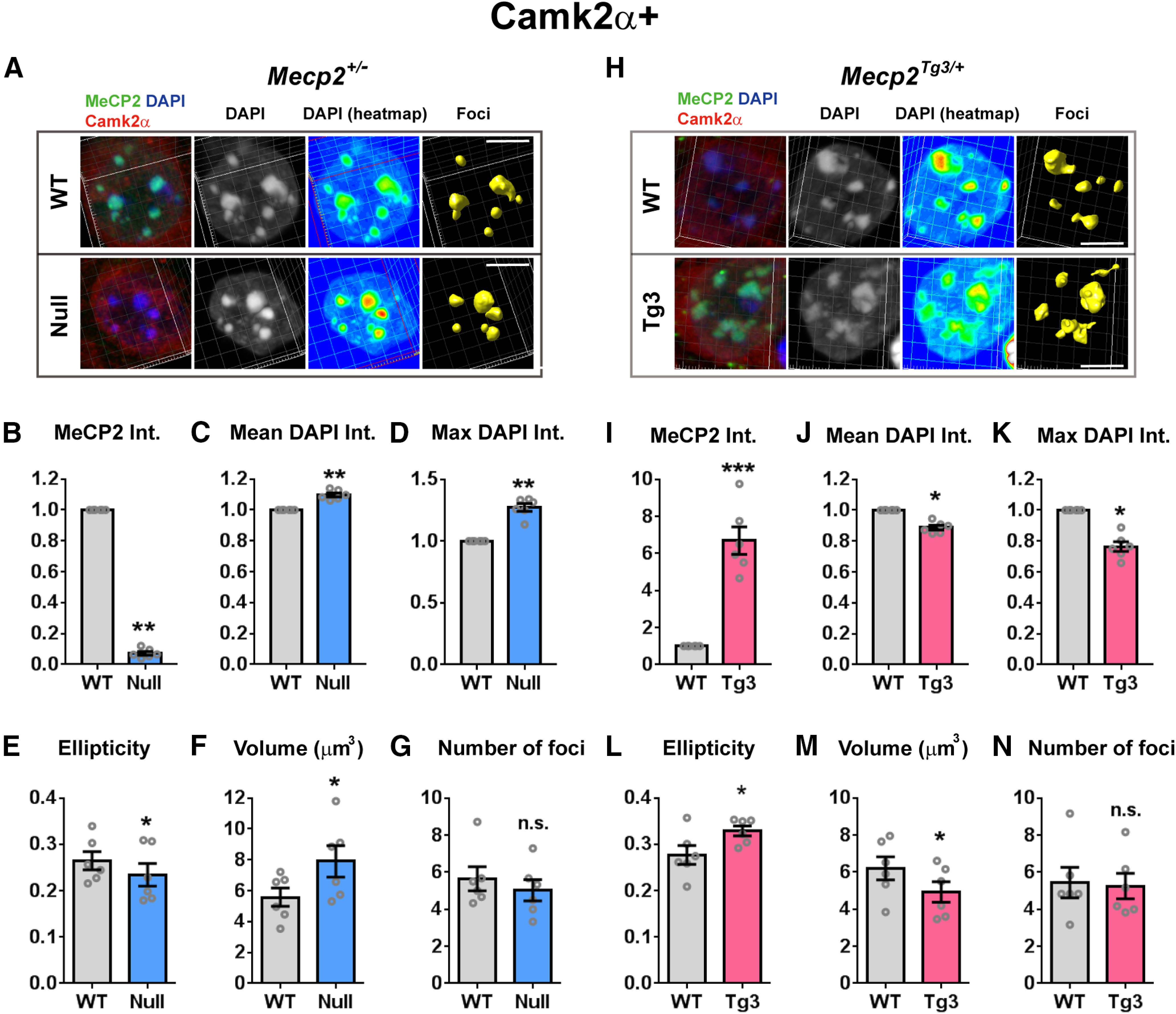
Impact of MeCP2 dosage on the heterochromatin structure of cortical excitatory cells. A, DAPI-foci in the cortical excitatory neurons of Mecp2+/− female mice (three months old). Neurons were stained for MeCP2 (green), DAPI (blue), and Camk2α (red). WT-cells and Null-cells were analyzed using the same method as in Figure 1. Scale bars: 5 µm. B–G, Quantified results from A. N = 6 mice. Two-tailed paired t test. B, Mean MeCP2 intensity (int.) was significantly reduced in Null-cells. p = 0.0032, t(5) = 5.299. C, Mean DAPI intensity was significantly increased in Null-cells. p = 0.0023, t(5) = 5.708. D, Maximum DAPI intensity within the heterochromatic foci was increased in Null-cells. p = 0.0016, t(5) = 6.157. E, Ellipticity of the heterochromatic foci was decreased in Null-cells. p = 0.0205, t(5) = 3.343. F, Average volume of foci was higher in Null-cells than in WT-cells. p = 0.0113, t(5) = 3.906. G, Average number of foci per nucleus was comparable between Null-cells and WT-cells. p = 0.0577, t(5) = 2.453. H, Heterochromatic foci in the cortical excitatory neurons of Mecp2Tg3/+ female mice (three months old). Neurons were stained for MeCP2 (green), DAPI (blue), and Camk2α (red). Scale bars: 5 µm. I–N, Quantified results from H. N = 6 mice. Two-tailed paired t test. I, Mean MeCP2 intensity was significantly increased in Tg3-cells. p = 0.0009, t(5) = 6.955. J, Mean DAPI intensity was significantly decreased in Tg3-cells. p = 0.0137, t(5) = 3.721. K, Maximum DAPI intensity within the heterochromatic foci was decreased in Tg3-cells. p = 0.0137, t(5) = 3.721. L, Ellipticity of the heterochromatic foci was increased in Tg3-cells. p = 0.0153, t(5) = 3.613. M, Average volume of foci was lower in Tg3-cells than in WT-cells. p = 0.0181, t(5) = 3.457. N, Average number of foci per nucleus was comparable between Tg3-cells and WT-cells. p = 0.5365, t(5) = 0.6632. Bars show average ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001; n.s., not significant.
Figure 3.
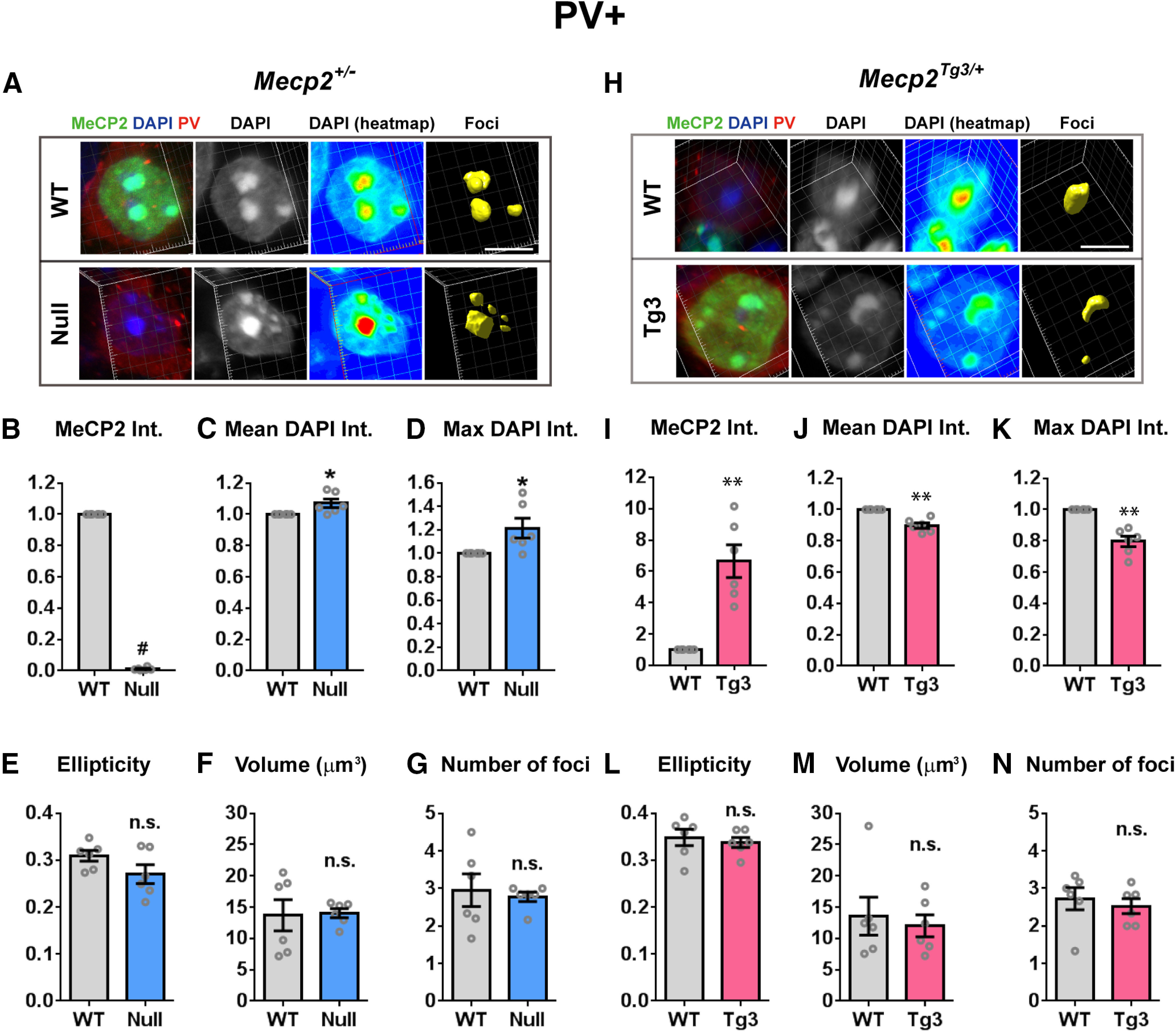
MeCP2-dependent structural changes of the heterochromatic foci in the cortical inhibitory neurons. A, DAPI-foci in the cortical PV-positive inhibitory neurons of Mecp2+/− female mice (three months old). Neurons were stained for MeCP2 (green), DAPI (blue), and PV (red). The images were analyzed using the same method as in Figures 1, 2. Scale bars: 5 µm. B–G, Quantified results from A. N = 6 mice. Two-tailed paired t test. B, Mean MeCP2 intensity (int.) was significantly reduced in Null-cells. p = 0.0006, t(5) = 7.771. C, Mean DAPI intensity was significantly increased in Null-cells. p = 0.0355, t(5) = 2.857. D, Maximum DAPI intensity within the heterochromatic foci was increased in Null-cells. p = 0.0474, t(5) = 2.615. E, Ellipticity of the heterochromatic foci was comparable between Null-cells and WT-cells. p = 0.1119, t(5) = 1.927. F, Average volume of foci was not changed in Null-cells. p = 0.9172, t(5) = 0.1093. G, Average number of foci per nucleus was comparable between Null-cells and WT-cells. p = 0.6920, t(5) = 0.42. H, DAPI-foci in the cortical PV-positive inhibitory neurons of Mecp2Tg3/ female mice (three months old). Scale bars: 5 µm. I–N, Quantified results from H. N = 6 mice. Two-tailed paired t test. I, Mean MeCP2 intensity was significantly higher in Tg3-cells. p = 0.0096, t(5) = 4.071. J, Mean DAPI intensity was significantly reduced in Tg3-cells. p = 0.0064, t(5) = 4.494. K, Maximum DAPI intensity within the heterochromatic foci was decreased in Tg3-cells. p = 0.0066, t(5) = 4.464. L, Ellipticity of the heterochromatic foci was comparable between Tg3-cells and WT-cells. p = 0.4481, t(5) = 0.8228. M, Average volume of foci was not changed in Tg3-cells. p = 0.5838, t(5) = 0.5853. N, Average number of foci per nucleus was comparable between Tg3-cells and WT-cells. p = 0.5219, t(5) = 0.6883. Bar graphs show average ± SEM; *p < 0.05, **p < 0.01, # p < 0.0001; n.s., not significant.
In order to identify excitatory neurons, slices from the frontal cortex of Mecp2+/− and Mecp2Tg3/+ female mice were labeled with anti-calcium/Camk2α antibody (Fig. 2; Movies 3, 4, 5, 6). The results from the structural analysis on the heterochromatic foci showed that the morphologic parameters of the foci were changed in both Null-cells and Tg3-cells in a similar way as in the foci of CA1 pyramidal cells (Fig. 2). Heterochromatic foci in Null-cells, which lacked MeCP2 (Fig. 2A,B), had significantly increased DAPI intensity [N = 6 mice, p = 0.0023 (Fig. 2C) and p = 0.0016 (Fig. 2D)] and reduced ellipticity when compared with WT-cells (p = 0.0205; Fig. 2E). In contrast, heterochromatic foci in Tg3-cells, which overexpressed MeCP2 (Fig. 2H,I), had reduced DAPI intensity [N = 6 mice, p = 0.0137 (Fig. 2J) and p = 0.0137 (Fig. 2K)] and increased ellipticity index compared with WT-cells (p = 0.0153; Fig. 2L). Interestingly, we found that the volume of heterochromatic foci was changed in the cortical Camk2α-positive cells, which was unlike the foci in CA1 pyramidal cells (Fig. 2F,M). The volume of foci was increased and decreased in Null-cells and Tg3-cells, respectively [p = 0.0113 for (Fig. 2F) and 0.0181 (Fig. 2M)]. The number of heterochromatic foci was not changed in either cell types (p = 0.0577, Fig. 2G; p = 0.5365, Fig. 2N). These results suggest that different cell types may respond differently to the alteration in MeCP2 dosage.
Images of 3D-reconstructed nuclei. DAPI, MeCP2, and reconstructed heterochromatin foci are shown in gray, green, and yellow, respectively. Nuclei of cortical pyramidal cells of a Mecp2+/− female mouse. MeCP2-positive (WT-) cell and MeCP2-negative (Null) cell from the same image were reconstructed and shown in separate movies (Movie 3, WT-cell; Movie 4, Null-cell). These images were processed using the same settings.
Images of 3D-reconstructed nuclei. DAPI, MeCP2, and reconstructed heterochromatin foci are shown in gray, green, and yellow, respectively. Nucleus of a cortical pyramidal cell of a Mecp2+/− female mouse. The movie shows a nucleus of MeCP2-negative (Null-) cell found in the proximity to the WT-cell shown in Movie 3.
Images of 3D-reconstructed nuclei. DAPI, MeCP2, and reconstructed heterochromatin foci are shown in gray, green, and yellow, respectively. Movie of a cortical pyramidal cell in a Mecp2Tg3/+ female mouse. Cell expressing single copy of Mecp2 (WT-cell) and a cell overexpressing MeCP2 (Tg3-cell) from the same image are shown in separate movies (Movie 5, WT-cell; Movie 6, Tg3-cell). These images were processed using the same settings.
Images of 3D-reconstructed nuclei. DAPI, MeCP2, and reconstructed heterochromatin foci are shown in gray, green, and yellow, respectively. Nucleus of a cortical pyramidal cell of a Mecp2Tg3/+ female mouse. The movie shows a nucleus of a MeCP2-overexpressing (Tg3-) cell found in the proximity to the WT-cell shown in Movie 5.
In order to visualize inhibitory cells, we stained slices with anti-PV antibody, because PV-positive inhibitory neurons constitute the largest population of inhibitory cells in the cortex (Fig. 3; Tremblay et al., 2016). Result from the morphologic analysis on the heterochromatic foci of PV-positive cells showed milder but still significant changes in foci when MeCP2 was either depleted or overexpressed (Fig. 3). In PV-positive cells, DAPI intensity of the heterochromatic foci was increased and decreased in Null-cells and Tg3-cells, respectively (N = 6 mice, p = 0.0355, p = 0.0474, p = 0.0064, and p = 0.0066, Fig. 3C,D,J,K). In contrast, the ellipticity index, the average foci volume, and the number of foci per cell were not changed in both Null-cells and Tg3-cells (Fig. 3E–G,L–N). Together with the results from the hippocampal and cortical excitatory cells, alteration in the DAPI intensity was consistently observed in the heterochromatic foci of all cell types and was consistently inverse in loss and gain of MeCP2. In contrast, the degree of shape changes in the heterochromatic foci varied between the neuronal cell types. These results suggest that MeCP2 dosage may differentially regulate heterochromatin structure depending on the cell types (excitatory vs inhibitory) and brain regions.
Electron microscopy reveals morphologic changes of subnuclear structures which is dependent on MeCP2 levels
To look into the details of the nuclear structural changes in MeCP2-depleted and MeCP2-overexpressed neurons, we used TEM and imaged subnuclear structures of CA1 pyramidal cells (Fig. 4). We analyzed electron-dense foci in neuronal nuclei, which are composed of condensed heterochromatin and nucleoli. TEM images from CA1 pyramidal cells were obtained from male Mecp2–/y, Mecp2Tg3/y, and their littermate Mecp2+/y (WT) mice (Fig. 4). We used eight- to nine-week-old mice when the majority of Mecp2–/y mice show clear neurologic deficits. As TEM requires intense analysis of each sample, we used three mice per genotype, but analyzed 34–83 nuclei per mouse to minimize variations. Consistent with the result from the confocal imaging of DAPI-foci (Fig. 1), we found that the shape of the electron-dense foci was significantly changed in both Mecp2–/y and Mecp2Tg3/y mice (Fig. 4). The electron-dense foci of Mecp2–/y mice appeared simpler in shape compared with the foci of Mecp2+/y mice (Fig. 4A). In contrast, the electron-dense foci of Mecp2Tg3/y mice appeared to have more irregularity and complexity (Fig. 4D). To evaluate the shape of electron-dense foci quantitatively, we measured “irregularity,” a parameter representing how close an object is to a circle. We found that the irregularity of the electron-dense foci was decreased in Mecp2–/y mice [N = 3 mice, two-way ANOVA, p = 0.002 (Fig. 4B), p < 0.001 (Fig. 4C)], while it was increased in Mecp2Tg3/y mice compared with their littermate WT mice [N = 3 mice, two-way ANOVA, p < 0.0001 (Fig. 4E), p = 0.0241 (Fig. 4F)].
Figure 4.
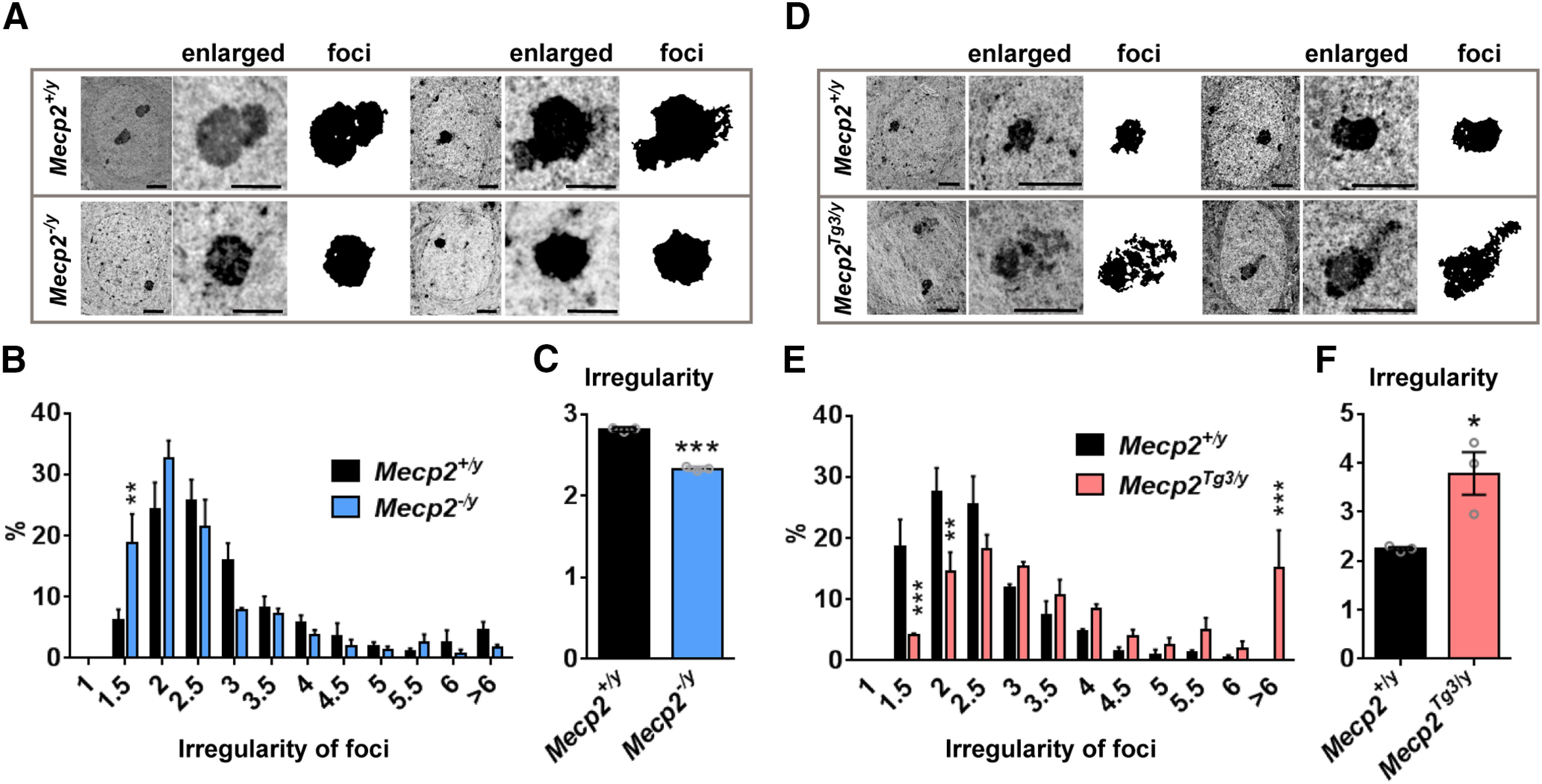
TEM analysis revealed subnuclear structures are altered in mice lacking and overexpressing MeCP2. A, TEM images of the nucleus in CA1 pyramidal cells obtained from Mecp2–/y and Mecp2+/y male mice (eight to nine weeks). Enlarged images show electron-dense foci. Scale bars: 2 µm (nucleus) and 1 µm (enlarged). B, C, Quantified result from A. B, Histogram showing the distribution for the irregularity of the electron-dense foci. The distribution was shifted toward left in Mecp2–/y mice. N = 3 mice. Two-way ANOVA (interaction), p = 0.0026, F(11,48) = 3.188. Bonferroni's multiple comparison test, p = 0.002 (irregularity = 1–1.5). C, Average irregularity of the electron-dense foci was significantly decreased in Mecp2–/y mice. N = 3 mice. Two-tailed t test, p < 0.0001, t(4) = 22.52. D, TEM images of the nucleus in CA1 pyramidal cells obtained from Mecp2Tg3/y and Mecp2+/y male mice (eight to nine weeks). Scale bars: 2 µm. E, F, Quantified results from E. E, Histogram showing the distribution of the irregularity of the electron-dense foci. The distribution was shifted toward right in Mecp2Tg3/y mice. N = 3 mice. Two-way ANOVA (interaction), p < 0.0001, F(11,44) = 5.535. Bonferroni's multiple comparison test, p = 0.0008 (irregularity = 1.0−1.5), 0.0029 (irregularity = 1.5–2.0), 0.0004 (irregularity > 6). F, Average irregularity of the electron-dense foci was significantly increased in Mecp2Tg3/y mice. N = 3 mice. Two-tailed t test, p = 0.0241, t(4) = 3.537. Bar graphs show average ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001.
Structural analysis of neuronal nucleoli and hepatocyte nuclei of Mecp2-deleted and overexpressing cells
Because heterochromatin is frequently associated with nucleoli (Manuelidis, 1984), it was possible that the alteration in electron-dense foci observed by TEM was caused by the changes in nucleoli. To address this, we visualized nucleoli of CA1 pyramidal cells and analyzed their shape using Mecp2+/− and Mecp2Tg3/+ female mice (three to four months old, N = 6 mice; Fig. 5). We used a fluorescent RNA-staining dye and confocal microscopy to image nucleoli and compared their shape between Null-cells, Tg3-cells, and WT-cells (Fig. 5). Overall, the shape of the nucleoli appeared similar in Null-cells, Tg3-cells, and WT-cells (Fig. 5). In order to evaluate their shape quantitatively, we obtained z-stack images from each nucleolus, and analyzed the image from a single plane in which the area of the nucleolus reached maximum. From each nucleolus, we measured irregularity, the same index that we used to analyze the electron-dense foci (Fig. 5). The irregularity was not changed in both Null-cells and Tg3-cells when compared with WT-cells (N = 6 mice, p = 0.3176 and p = 0.6960; Fig. 5B,F). These results suggested that the change in the irregularity of the electron-dense foci that was observed by TEM (Fig. 4) was because of changes in heterochromatin and not to changes in nucleoli. Interestingly, there was a subtle but significant decrease in the number of nucleoli per cell in Null-cells (p = 0.0046; Fig. 5C), while the maximum area of nucleoli was not changed (p = 0.0767; Fig. 5D). In contrast, while the number of nucleoli per cell was not changed in Tg3-cells (p = 0.5155; Fig. 5G), there was an increase in the size of nucleoli, as shown by the increase in the maximum area of nucleoli in Tg3-cells (p = 0.0390; Fig. 5H). These results indicated that, in addition to controlling the structure of heterochromatin, MeCP2 dosage may regulate the number and the size of neuronal nucleoli.
Figure 5.
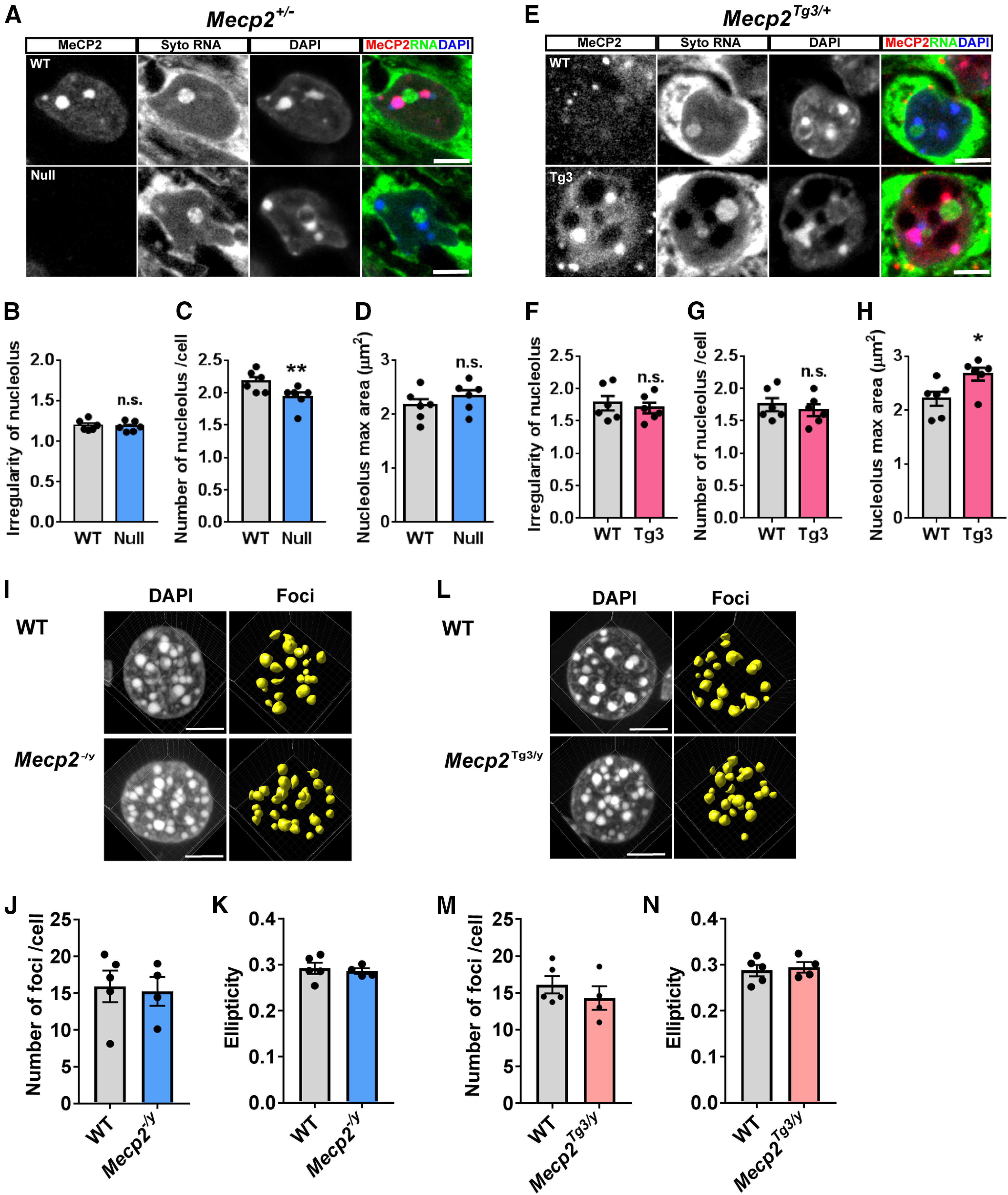
Structural analysis of nucleoli in CA1 pyramidal cells and analysis of DAPI foci in the hepatocytes in MeCP2-deleted and MeCP2-overexpressing cells. A, CA1 pyramidal cells in Mecp2+/− female mice (three months old) were stained with RNA-selective dye (Syto RNASelect, green), MeCP2 (red), and DAPI (blue). Nucleoli were determined as Syto RNASelect-positive and DAPI-negative regions. Scale bars: 5 µm. B–D, Quantified data from A. N = 6 mice. Two-tailed paired t test. B, The irregularity of nucleoli was comparable between Null-cells and WT-cells. p = 0.3176. t(5) = 1.11. C, The number of nucleoli per cell was mildly decreased in Null-cells. p = 0.0046. t(5) = 1.11. D, Maximum area of nucleoli was comparable between Null-cells and WT-cells. p = 0.0767. t(5) = 2.225. E, Nucleoli of CA1 pyramidal cells in Mecp2Tg3/+ female mice (three months old) were visualized using the same method as in A. F–H, Quantified data from A. N = 6 mice. Two-tailed paired t test. F, The irregularity of nucleoli was comparable between Tg3-cells and WT-cells. p = 0.1312. t(5) = 1.803. G, The number of nucleoli per cell was comparable between Tg3-cells and WT-cells. p = 0.5155. t(5) = 0.6993. H, The maximum area of nucleoli was higher in Tg3-cells than WT-cells. p = 0.0390. t(5) = 2.779. I, Heterochromatic foci in the hepatocytes of Mecp2–/y and WT male mice (eight to nine weeks old). N = 5 (WT) and 4 (Mecp2–/y) mice. J, The number of foci per cell was not changed in Mecp2–/y mice. Mann–Whitney U test, p = 0.9058. K, Ellipticity was not changed in Mecp2–/y mice. Two-tailed t test, p = 0.6792, t(7) = 0.4313. L, Heterochromatic foci in the hepatocytes of Mecp2Tg3/y and WT male mice (eight to nine weeks old). N = 5 (WT) and N = 4 (Mecp2Tg3/y) mice. M, The number of foci per cell was not changed in Mecp2Tg3/y mice. Mann–Whitney U test, p = 0.5556. N, Ellipticity was not changed in Mecp2Tg3/y mice. Two-tailed t test, p = 0.6960, t(7) = 0.4072. Bar graphs show average ± SEM; *p < 0.05, **p < 0.01; n.s., not significant.
We asked whether the structural changes of heterochromatic foci occur in non-neuronal cell types. We analyzed DAPI-stained nuclei in the hepatocytes of male Mecp2–/y and Mecp2Tg3/y mice and compared with those of littermate Mecp2+/y (WT) mice (N = 4–5 mice; Fig. 5I–N). The number of heterochromatic foci per nucleus and the average ellipticity of foci were not affected in both conditions. These results suggest that MeCP2 levels primarily affect heterochromatin in neurons.
Mild structural changes in heterochromatin in MeCP2-negative cells precede the onset of neurologic deficits
Symptoms in classical RTT patients develop only after they reach 6–18 months of age (Lombardi et al., 2015). This progression is mirrored in Mecp2+/− female mice which develop behavioral phenotypes as they age. To address whether the structural abnormalities of heterochromatin in MeCP2-negative cells occur before or after the onset of neurologic symptoms, we analyzed CA1 pyramidal cells of younger Mecp2+/− female mice at three weeks of age. The mice at this age did not show any behavioral deficit and appeared grossly similar to their littermate WT mice (Samaco et al., 2013). We used the same method to analyze DAPI-enriched heterochromatic foci of CA1 pyramidal neurons in the hippocampus and compared the structure of foci between Null-cells and WT-cells (Fig. 6). The results showed that the heterochromatic foci in Null-cells had higher mean DAPI intensity (N = 5 mice, p = 0.0023; Fig. 6B2) and a lower ellipticity index (p = 0.0012; Fig. 6C2) than those of WT-cells. The difference between Null-cells and WT-cells was small but statistically significant (Fig. 6B,C). There was no significant change in the average volume of foci or in the number of foci (Fig. 6E,F). These results indicate that the structural changes in heterochromatin occur before the onset of RTT-related symptoms in mice. In addition, these data also suggest that the heterochromatin changes in the Null-cells are likely caused directly by the absence of MeCP2 and not indirectly by the decline in the general health of neurons.
Figure 6.

Structural changes in MeCP2-negative cells are present in presymptomatic young Mecp2+/− female mice. A, Representative images of the heterochromatic foci in CA1 pyramidal cells of a three-week-old Mecp2+/− female mouse. There was a mild but significant difference in the DAPI intensity and shape between Null-cells and WT-cells. Scale bars: 5 µm. B–F, Quantitative results from images taken in A. B1, Cumulative distribution of the mean DAPI intensity for each heterochromatic focus. DAPI intensity for each focus was normalized to the average DAPI intensity in the foci of the nearest WT-cells. The distribution for Null-cells was slightly shifted toward right. N = 362 foci (WT-cells) and 392 foci (Null-cells). Kolmogorov–Smirnov test, p = 0.0137. B2, Average of mean DAPI intensity from five mice was mildly increased in Null-cells. N = 5 mice. Two-tailed paired t test, p = 0.0023, t(4) = 6.93. C1, Cumulative distribution of ellipticity for each heterochromatic focus showed mild shift toward left in Null-cells. N = 362 foci (WT-cells) and 392 foci (Null-cells). Kolmogorov–Smirnov test, p = 0.00102. C2, Average ellipticity for five mice was significantly lower in Null-cells than WT-cells. N = 5 mice. Two-tailed paired t test, p = 0.0012, t(4) = 8.292. D, The intensity of MeCP2 was significantly decreased in Null-cells. N = 5 mice. Two-tailed paired t test, p = 0.0082, t(4) = 4.874. E, Average volume of foci was comparable between Null-cells and WT-cells. N = 5 mice. Two-tailed paired t test, p = 0.3344, t(4) = 1.097. F, The number of foci per cell was not different between Null-cells and WT-cells. N = 5 mice. Two-tailed paired t test, p = 0.1057, t(4) = 2.083. Bar graphs show average ± SEM; **p < 0.01; n.s., not significant.
MeCP2 levels impact histone modifications within the heterochromatic foci
Heterochromatin contains distinct sets of histone modification such as H3 trimethylation at lysine 9 (H3K9me3), H3 trimethylation at lysine 27 (H3K27me3), and H4 trimethylation/dimethylation at lysine 20 (H4K20me2/3), while euchromatin typically contains H3 trimethylation at lysine 4 (H3K4me3). As the structure of heterochromatic foci was significantly dependent on MeCP2 levels, we asked whether the heterochromatin structural change was associated with any alteration in histone modifications. To address this, we performed immunostaining of hippocampal pyramidal cells in Mecp2+/− and Mecp2Tg3/+ female mice and quantified levels of histone modifications within the foci (Fig. 7). Average intensities of H3K27me3, H3K9me3, H4K20me2/3, and H3K4me3 were measured in MeCP2-deleted (Null) and MeCP2-overexpressing (Tg3) cells and the results were compared with those from cells expressing normal level of MeCP2 (WT-cells). Interestingly, we detected various changes in histone methylation marks in both Null-cells and Tg3-cells. In Null-cells, average intensities of H3K27me3 and H3K4me3 were increased [N = 6 mice, p = 0.0296 (Fig. 7A) and p = 0.0041 (Fig. 7D)]. Average intensity of H3K9me3 showed mild trend toward decrease but was not statistically significant (p = 0.0926; Fig. 7B). In Tg3-cells, average intensity of H3K4me3 was decreased (N = 6 mice, p = 0.0175; Fig. 7H). The intensity of H3K9me3 in Tg3-cells also showed trend toward increase but the change was not statistically significant (p = 0.0637; Fig. 7F). We detected no change in H4K20me2/3 in both Null-cells (p = 0.4856; Fig. 7C) and Tg3-cells (p = 0.3484; Fig. 7G). These results suggest that MeCP2 levels significantly impacts histone modifications such as H3K27me3, H3K4me3, and H3K9me3, which may further contribute to heterochromatin structure and transcription.
Figure 7.
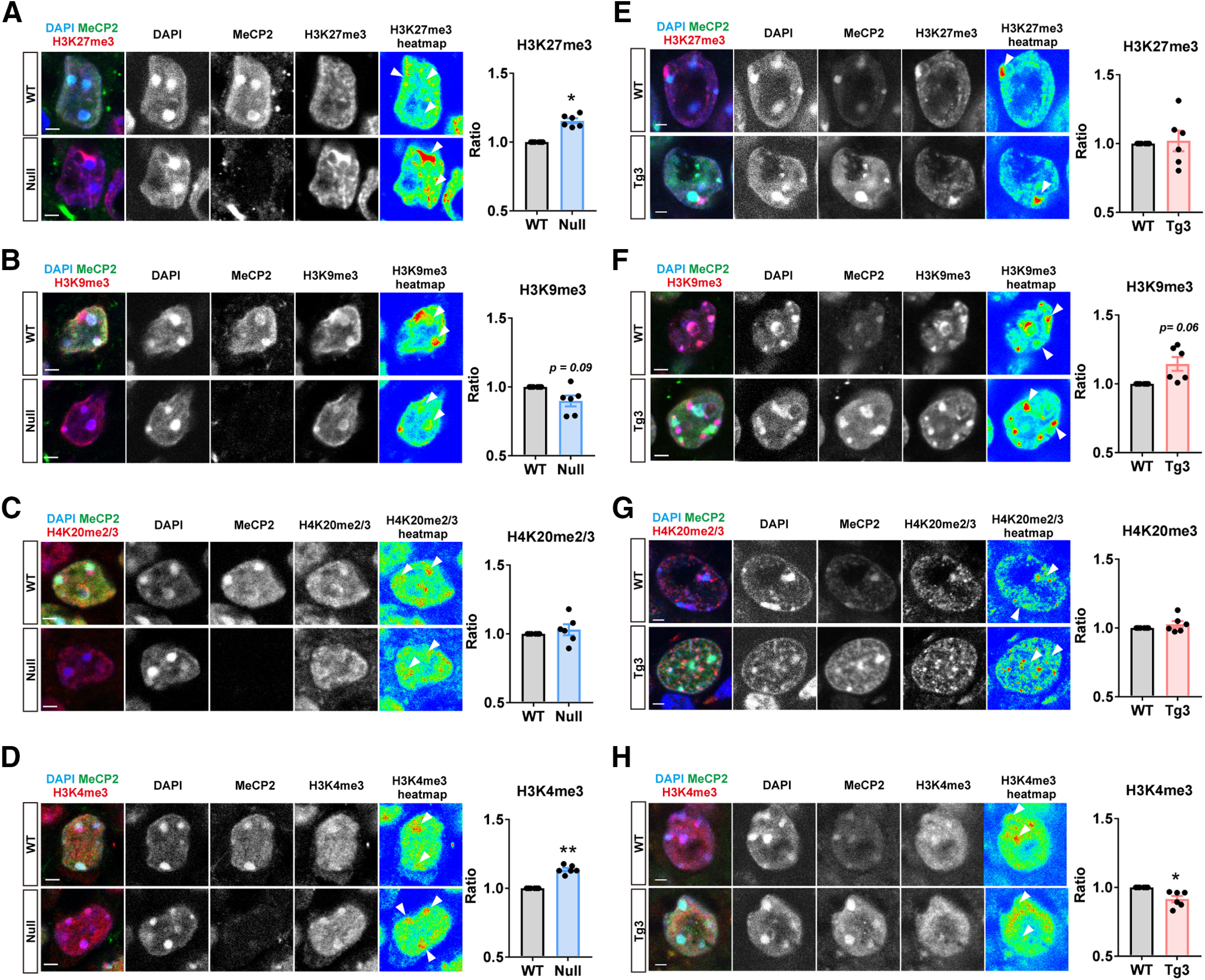
Levels of histone methylation marks in the heterochromatic foci of MeCP2-negative (Null) and MeCP2-overexpressing (Tg3) cells. Immunohistochemistry of CA1 pyramidal cells was performed using symptomatic Mecp2+/− (4–10 months old) and Mecp2Tg3/+ female mice (6–10 months old) to quantify H3K27me3 (A, E), H3K9me3 (B, F), H4K20me2/3 (C, G), and H3K4me3 (D, H) within the heterochromatic foci. Mean intensity of each histone modification mark was quantified by Imaris. The intensity was normalized to that of foci expressing normal level of MeCP2. MeCP2 deletion resulted in increased H3K27me3 (A) and H3K4me3 (D), while MeCP2 overexpression led to decreased H3K4me3 (H). H3K9me3 showed mild trend toward decrease in Null-cells and increase in Tg3-cells. Paired t test. N = 6 mice per genotype. A, H3K27me3 in Mecp2+/− mice. p = 0.0296, t(5) = 3.016. B, H3K9me3 in Mecp2+/− mice. p = 0.0926, t(5) = 2.075. C, H4K20me2/3 in Mecp2+/− mice. p = 0.4856, t(5) = 0.7525. D, H3K4me3 in Mecp2+/− mice. p = 0.0041, t(5) = 4.995. E, H3K27me3 in Mecp2Tg3/+ female mice. p = 0.8793, t(5) = 0.1598. F, H3K9me3 in Mecp2Tg3/+ mice. p = 0.0637, t(5) = 2.373. G, H4K20me2/3 in Mecp2Tg3/+ mice. p = 0.3484, t(5) = 1.034. H, H3K4me3 in Mecp2Tg3/+ mice. p = 0.0175, t(5) = 3.489. Arrowheads in the heatmaps indicate locations of the heterochromatic foci. Bar graphs show average ± SEM; *p < 0.05, **p < 0.01.
AT-hook 2 domain of MeCP2 contributes to the structural changes in heterochromatic foci
The results from the confocal and electron microscopy showed that MeCP2 is essential for the structural integrity of heterochromatin in neurons. In Null-cells, the DAPI intensity of the heterochromatic foci was increased and the irregularity of the electron-dense foci was decreased, suggesting that the heterochromatin became more compact when MeCP2 was depleted. Conversely, in Tg3-cells, DAPI intensity of the heterochromatic foci was reduced and the irregularity of the electron-dense foci was increased, suggesting that the heterochromatin became less condensed when MeCP2 was overexpressed.
To gain insight into the regions in MeCP2 that might impact chromatin structural changes, we asked whether the AT-hook 2 domain plays a role in effecting these changes. We focused on this domain because it determines disease onset of RTT (Baker et al., 2013), and the homologous domain in HMGA1 has been shown to induce chromatin structural changes (Reeves, 2010). MECP2-R270X and MECP2-G273X transgenic mice express MeCP2 with truncating mutations that either disrupt (R270X) or retain (G273X) the AT-hook 2 domain. We crossed these mice with Mecp2+/− female mice, and obtained male mice that express only one of these mutants and lack the WT allele (Mecp2–/y;MECP2-R270X, and Mecp2–/y;MECP2-G273X), mice that do not express MeCP2 (Mecp2–/y), and WT control mice at eight to nine weeks old. At this age, Mecp2–/y and Mecp2–/y;MECP2-R270X mice have clear RTT-related neurologic deficits, while Mecp2–/y;MECP2-G273X mice are still healthy and indistinguishable from WT mice. We imaged the heterochromatic foci of CA1 pyramidal neurons and analyzed their shape by Imaris. Because we used male mice for this experiment, it was necessary to compare cells obtained from different slices from different mice. Since this makes comparing DAPI intensity and related parameters difficult between samples, we analyzed only ellipticity and the number of foci per cell, which are not affected by the absolute DAPI intensity (Fig. 8). The genotype of each mouse was confirmed by immunostaining using an anti-MeCP2 antibody, which recognizes the C terminus of MeCP2 deleted in mutants, and an anti-GFP antibody, which detected the transgenic MeCP2 that was tagged with GFP (Fig. 8A). The results showed that the average ellipticity was significantly reduced in the foci of Mecp2–/y (Null) male mice when compared with that of WT mice [N = 7 (WT) and N = 4 mice (Null), p = 0.0013; Fig. 8B)]. This result was consistent with the result from Mecp2+/− female mice (Fig. 8). In Mecp2–/y; MECP2-R270X mice, the foci showed a reduction in ellipticity, which was comparable to the foci of Mecp2–/y mice [N = 7 (WT) and N = 4 mice (R270X), p = 0.0003 (WT vs R270X); Fig. 8B]. In contrast, ellipticity of the foci in Mecp2–/y; MECP2-G273X mice was not affected and was comparable to those of WT mice [N = 7 (WT) and N = 4 mice (G273X), p = 0.3267; Fig. 8B]. The number of foci per cell was similar in all four genotypes (Fig. 8C). We also analyzed older Mecp2–/y; MECP2-G273X mice at five months when the mice showed clear RTT-related symptoms (Fig. 8D–F). Interestingly, average ellipticity of the foci in these mice was significantly decreased [N = 4 (WT) and N = 6 (G273X) mice, p = 0.0026; Fig. 8E], whereas the number of foci remained unaffected (p = 0.6619; Fig. 8F). These results show that the structural changes in heterochromatin foci clearly correlated with the phenotypic severity of the RTT models. The presence of AT-hook 2 influences the onset of the changes in the heterochromatic foci, which became significant with the progression of RTT-related phenotypes.
Figure 8.

AT-hook 2 domain of MeCP2 influences onset of heterochromatic structural changes in CA1 pyramidal cells. A, Representative images of heterochromatic foci in CA1 pyramidal cells obtained from eight- to nine-week-old WT (Mecp2+/y, WT), Mecp2–/y (Null), Mecp2–/y;MECP2-R270X (R270X), and Mecp2–/y; MECP2-G273X (G273X) mice. The cells were stained with anti-GFP to confirm expression of mutant MeCP2 which were tagged with GFP, and anti-MeCP2 C terminus antibody to confirm depletion of endogenous MeCP2. Scale bars: 5 µm. B, C, Quantitative results from the images in A. N = 7 (WT), N = 4 (Null), N = 4 (R270X), and N = 4 mice (G273X). One-way ANOVA followed by Tukey's multiple comparison test. B, Ellipticity of the heterochromatic foci was decreased in Mecp2-null mice and in mice expressing MeCP2-R270X, while it was not changed in mice expressing MeCP2-G273X. One-way ANOVA, p = 0.0002, F(3,15) = 13.27. Tukey's multiple comparison tests; p = 0.0013 (WT vs Null), 0.0003 (WT vs R270X), and 0.3267 (WT vs G273X). C, Average number of heterochromatic foci per cell was comparable in all the genotypes analyzed. One-way ANOVA, p = 0.1523, F(3,15) = 2.034. D, Heterochromatic foci in the CA1 pyramidal cells of five-month-old G273X and WT mice. E, Ellipticity of the heterochromatic foci was decreased in five-month-old G273X mice. N = 4 (WT) and N = 6 (G273X) mice. Two-tailed t test. p = 0.0026, t(8) = 4.298. F, Average number of heterochromatic foci per cell was not altered. N = 4 (WT) and N = 6 (G273X) mice. Mann–Whitney U test. p = 0.6619. Bar graphs show average ± SEM; **p < 0.01; n.s., not significant.
AAV-induced knock-down and overexpression of MeCP2 induce distinct heterochromatin structural changes in cortical pyramidal cells
Next, we asked whether MeCP2 is necessary for the maintenance of heterochromatin structure in adult healthy neurons. To address this, AAV(DJ) vector introducing miRNA against Mecp2 together with GFP (AAV-mirMecp2) was injected into the prefrontal cortex (PFC) of adult WT mice (six to seven weeks) and the brains were collected five weeks after the injection. Brain slices were stained with DAPI and antibodies against MeCP2, GFP, and Camk2α to distinguish excitatory pyramidal cells (Fig. 9A). Heterochromatic foci were analyzed by Imaris, and the result was compared between AAV-infected GFP-positive cells and non-infected GFP-negative cells. The intensity of MeCP2 was significantly decreased in GFP-positive cells, confirming effective knock-down by mirMecp2 (N = 5 injected area from three mice, p = 0.0024; Fig. 9B). Interestingly, AAV-mirMecp2 did not induce any change in the average DAPI intensity (p = 0.6623; Fig. 9C), ellipticity (p = 0.6471; Fig. 9D), or volume (p = 0.1037; Fig. 9E), while all these parameters were significantly altered in Null-cells of Mecp2+/− mice (Fig. 2). In contrast, GFP-positive cells had significantly lower number of foci (p = 0.0085; Fig. 9F), while this change was not observed in Null-cells of Mecp2+/− mice (Fig. 2). As a control experiment, AAV(DJ) vector, which introduced miRNA containing randomized negative control sequence (AAV-mirNega), was injected into the PFC (Fig. 9G). Analysis of the heterochromatic foci showed no difference between GFP-positive and GFP-negative cells (Fig. 9H–L).
Figure 9.
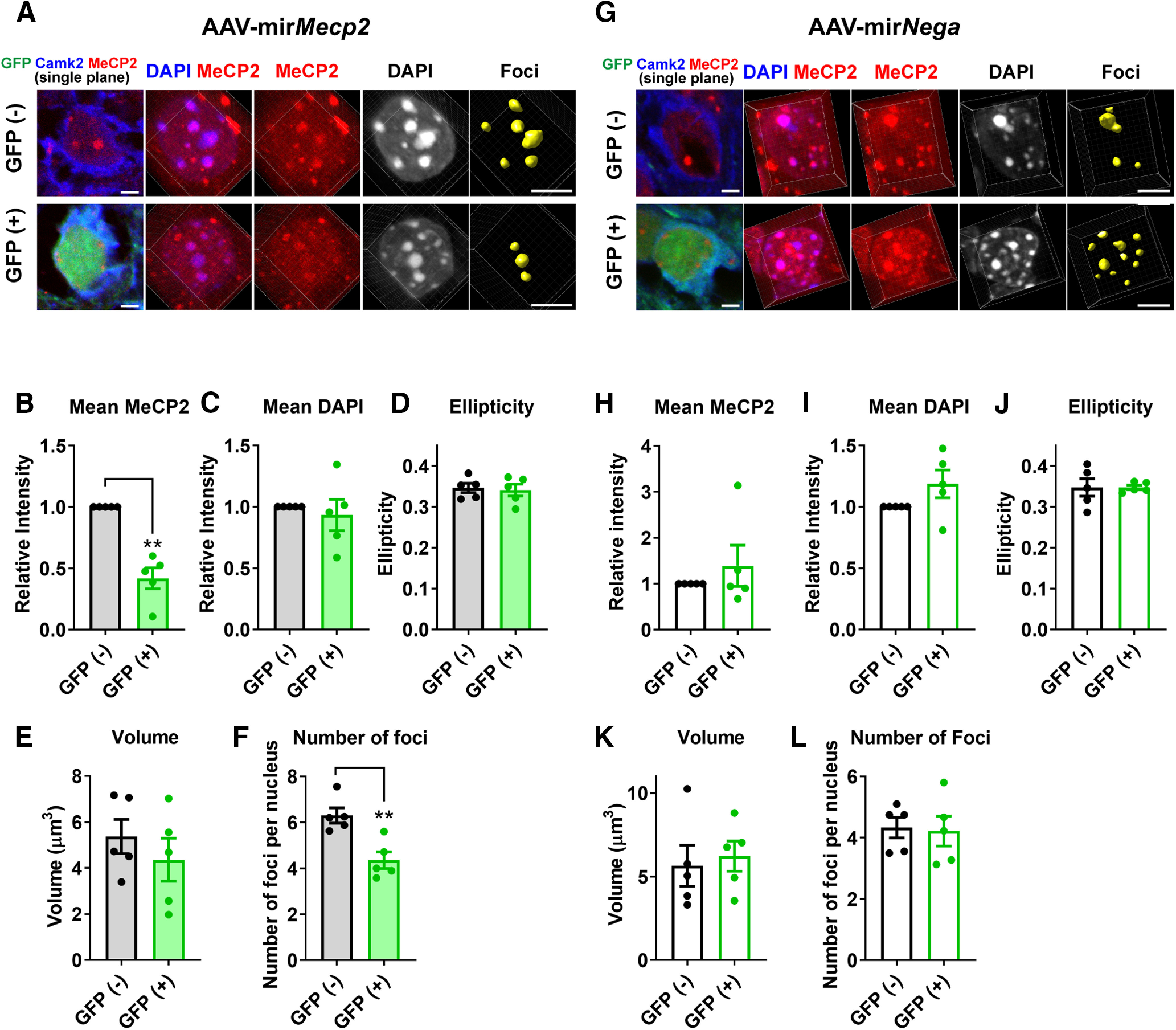
Knock-down of MeCP2 in the adult cortical pyramidal cells induces reduction in the number of heterochromatic foci. A, AAV(DJ)-vector inducing expression of Mecp2-targetting miRNA and GFP (AAV-mirMecp2) was injected into the PFC of WT mice (six to seven weeks). The mice were perfused five weeks after the injection, and the brain slices were stained with DAPI and antibodies for MeCP2, GFP, and Camk2α to mark pyramidal cells. Images are single plane images showing GFP and Camk2α signals (left panel) and 3D-reconstructed images from the same cells (right panels). Scale bars: 5 µm. B–F, Results from the quantitative analysis of DAPI foci. Reduction of MeCP2 was confirmed in GFP-positive cells (B, p = 0.0024, t(4) = 6.815). Average number of foci within the nucleus was decreased by Mecp2 knock-down (F, p = 0.0085, t(4) = 4.832). No changes were detected in the mean DAPI intensity (C, p = 0.6623, t(4) = 0.4708), ellipticity (D, p = 0.6471, t(4) = 0.4942), or volume (E, p = 0.1037, t(4) = 2.099). Paired t test. N = 5 injected area from 3 mice. G, AAV(DJ)-vector inducing expression of random sequence miRNA and GFP (AAV-mirNega) was injected into the PFC of WT mice (six to seven weeks), and the analysis was done in the same way as in A–F. H–L, Results from the quantitative analysis of DAPI foci. No change was detected in MeCP2 intensity (H, p = 0.6486, t(4) = 0.4919), DAPI intensity (I, p = 0.1705, t(4) = 1.669), ellipticity (J, p = 0.99, t(4) = 0.00018), number of foci (K, p = 0.8717, t(4) = 0.1722), or volume (L, p = 0.7768, t(4) = 0.3717). Paired t test. N = 5 injected area from 3 mice. Bar graphs show average ± SEM; **p < 0.01.
We also asked whether acute overexpression of MeCP2 in adult brains have any impact on the heterochromatic foci. To address this, MeCP2 overexpression was induced by injecting AAV5-Camk2α-Cre and AAV-DIO-human(hu)MECP2-GFP into the PFC of adult mice (Fig. 10A). Slices were collected three weeks after the injection and were stained with DAPI and antibodies against GFP and MeCP2. Cortical pyramidal cells expressed various levels of GFP, whose intensity strongly correlated with the intensity of MeCP2 (Fig. 10A). We found cells with higher MeCP2 intensity showed stronger structural changes in the heterochromatic foci. To evaluate this quantitatively, we grouped GFP-positive cells into two groups based on the GFP intensity. GFP-high group showed higher MeCP2 intensity when compared with GFP-low group (Fig. 10B). Mean DAPI intensity was decreased in both GFP-low and GFP-high groups, with higher statistical significance in the GFP-high group (N = 6 injected sites from three mice; Fig. 10C). While ellipticity was not affected by MeCP2 overexpression (ANOVA, p = 0.3881; Fig. 10D), maximum volume of foci was increased (Fig. 10E,F), and we observed largest increase in the GFP-high group [ANOVA, p = 0.0001; post hoc test, p = 0.0011 (GFP-negative vs GFP-high); Fig. 10F]. The number of foci showed mild trend toward increase but this was not statistically significant (ANOVA, p = 0.1416; Fig. 10G). These changes were again different from the observation in Mecp2Tg3/+ female mice, which showed decreased DAPI intensity, increased ellipticity, and reduced volume. Taken together, postnatal changes in MeCP2 levels in adult mice affected heterochromatic foci in a distinct manner from RTT-disease models.
Figure 10.

AAV-induced overexpression of MeCP2 caused heterochromatin changes in the adult cortical pyramidal cells. A, AAV(DJ)-DIO-huMECP2-GFP and AAV5-Camk2α-Cre were co-injected into the PFC of mature WT mice (six to seven weeks), and the brains were analyzed three weeks after the injection. The brain slices were stained with DAPI and antibodies against MeCP2 and GFP. The cells were grouped into GFP-negative, GFP-low, and GFP-high group based on the GFP intensity. Scale bars: 5 µm. B–G, Results from the quantitative analysis of DAPI foci. N = 6 injected sites from 3 mice. B, MeCP2 intensity was significantly increased with higher GFP expression. Friedman's ANOVA, p = 0.001; Dunn's multiple comparison test, p = 0.0011 (GFP-negative vs GFP-high). C, DAPI intensity within the foci was decreased in GFP-low and GFP-high cells. Friedman's ANOVA, p = 0.0055; Dunn's multiple comparison test, p = 0.0418 (GFP-negative vs GFP-low), p = 0.0078 (GFP-negative vs GFP-high). D, Ellipticity was not affected by MeCP2 expression. One-way ANOVA, p = 0.3881. E, Mean volume was not affected by MeCP2 overexpression. Friedman's ANOVA, p = 0.2522. F, Maximum volume of foci within each nucleus was significantly increased in GFP-high group. Friedman's ANOVA, p = 0.0001; Dunn's multiple comparison test, p = 0.1665 (GFP-negative vs GFP-low) and p = 0.0011 (GFP-negative vs GFP-high). G, Average number of foci was not changed. Friedman's ANOVA, p = 0.1416. Bar graphs show average ± SEM; *p < 0.05. **p < 0.01.
MeCP2 overexpression impacts cognitive behavior and the transcriptome
Our results so far revealed importance of MeCP2 dosages in the heterochromatin structure in neurons. However, its physiological significance remains an open question because the majority of heterochromatin is composed of repetitive elements whose function is largely unknown. To gain insight into this question, we asked whether any behavioral and transcriptional changes could be correlated to the changes in the heterochromatic foci (Figs. 11, 12). Behavioral analysis was performed in WT mice overexpressing MeCP2 in the bilateral PFC, which was induced by co-injection of AAV5-Camk2α-Cre and AAV(DJ)-DIO-huMECP2-GFP (Fig. 11). Control mice received injection of either only AAV5-Camk2α-Cre or AAV5-Camk2α-Cre together with AAV(DJ)-DIO-GFP. Activity and anxiety-related behavior was analyzed by OF test and LD test at two time points (Fig. 11A): two weeks after the injection when the heterochromatin changes were still unclear and three weeks after the injection when we observed clear changes in the heterochromatic foci (Fig. 10). While the results from two weeks did not show any difference (Fig. 11C), the results from three weeks showed mild changes in LD test: mice overexpressing MeCP2 showed trend toward increased time in light (N = 9–10 mice per condition, one-way ANOVA, p = 0.0475; Fig. 11D). We further analyzed cognitive performance at 3.5 weeks by observational fear response test, which has been shown to be dependent on PFC (N = 10–12 mice per condition; Fig. 11E; Sakaguchi et al., 2018). Mice were put in an observation chamber with transparent walls, which was placed side-by-side to a shock chamber in which demonstrator mice received repeated foot shocks. We analyzed freezing response in three phases: baseline, during shock, and after shock (Fig. 11E). The results showed that the mice overexpressing MeCP2 responded to the observed fear with increased freezing time at the after-shock phase [one-way ANOVA, p = 0.0260; post hoc test, p = 0.0295 (Cre:MeCP2-GFP vs Cre:GFP) and p = 0.0784 (Cre vs Cre:MeCP2-GFP); Fig. 11F] .
Figure 11.
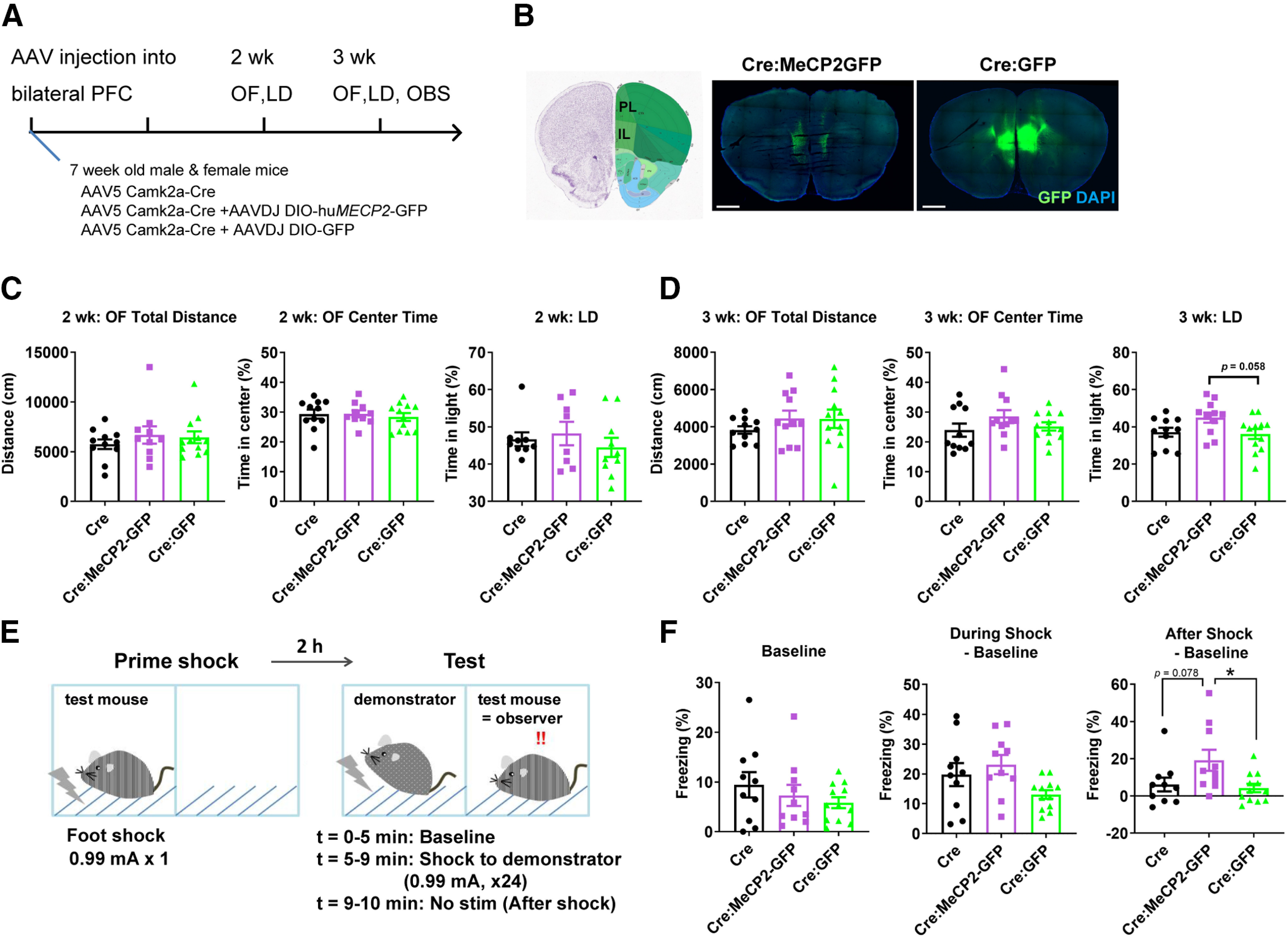
MeCP2 overexpression in the adult PFC induces altered response to observational fear. A, Experimental design. AAVs were injected into the bilateral PFC of WT mice at six to seven weeks. AAV5-Camk2α-Cre and AAV(DJ)-DIO-huMECP2-GFP were used to induce overexpression of MeCP2 in the cortical pyramidal cells (Cre:MeCP2-GFP). Control mice received injection of either AAV5-Camk2α-Cre only (Cre) or AAV5-Camk2α-Cre together with AAV(DJ)-DIO-GFP (Cre:GFP). LD test and OF activity test were performed two and three weeks after the injection. Observational fear response (OBS) was performed at 3.5 weeks. B, Images of coronal slices showing distribution of GFP around injection areas. PL, prelimbic. IL, infralimbic. Scale bars: 1 mm. C, Results from OF and LD tests two weeks after the injection. No change was found. One-way ANOVA. p = 0.5881, F(2,30) = 0.5403 (OF, total distance), p = 0.8259, F(2,30) = 0.1925 (OF, center time), p = 0.586, F(2,24) = 0.5465 (LD). N (Cre, Cre: MeCP2-GFP, Cre:GFP) = (11, 10, 12) and (9, 8, 10) mice for OF and LD, respectively. D, Results from OF and LD tests three weeks after the injection. Mice overexpressing MeCP2 (Cre: MeCP2-GFP) showed trend toward increased time in light in the LD test. One-way ANOVA. p = 0.4695, F(2,31) = 0.7749 (OF, total distance), p = 0.2468, F(2,31) = 1.464 (OF, center time), p = 0.0475, F(2,31) = 3.367 (LD). Tukey's multiple comparison test for LD, p = 0.0583 (Cre:MeCP2-GFP vs Cre:GFP) and p = 0.1085 (Cre vs Cre:MeCP2-GFP). N (Cre, Cre:MeCP2-GFP, Cre:GFP) = (11, 11, 12) mice for OF and LD. E, Experimental design for the OBS test. See method section for details. F, Results from OBS test. Mice overexpressing MeCP2 (Cre:MeCP2-GFP) responded to the observational fear with higher freezing time in the after-shock phase. There was no significant change in the freezing time at the baseline and during the shock phase. One-way ANOVA. p = 0.4203, F(2,29) = 0.8931 (baseline); p = 0.0521, F(2,29) = 3.278 (during shock); p = 0.0260, F(2,29) = 4.150 (after shock). Tukey's multiple comparison test for after shock, p = 0.0295 (Cre:MeCP2-GFP vs Cre:GFP), p = 0.0784 (Cre vs Cre:MeCP2-GFP), p = 0.9355 (Cre vs Cre:GFP). N = 10 (Cre), N = 10 (Cre:MeCP2-GFP), and N = 12 (Cre:GFP) mice. Bar graphs show average ± SEM; *p < 0.05.
Figure 12.

MeCP2 overexpression in the adult cortical neurons induces MeCP2 dosage-dependent transcriptional changes. A, Experimental design. AAV5-Camk2α-Cre and AAV(DJ)-DIO-huMECP2-GFP were used to induce overexpression of MeCP2 in the bilateral PFC of adult mice (six to seven weeks). PFC was dissected three weeks after the injection and neuronal nuclei labeled by anti-NeuN antibody were processed by FACS. The nuclei were sorted into four groups based on the GFP intensity and used for mRNA-Seq. Results from mRNA-Seq analysis including the number of mapped reads and the list of DEGs are summarized in Extended Data Figure 12-1. B, Images of the sorted nuclei. Scale bars: 5 µm. C–I, Results from mRNA-Seq analysis. DEGs were obtained using GFP-negative group as a control. C, Transcript levels of endogenous mouse Mecp2 (left), exogenous human MECP2 (middle), and total Mecp2 normalized to the control. Overexpression of human MECP2 did not cause significant changes in the endogenous mouse Mecp2 (left). Total Mecp2 reads correlated positively with the GFP expression levels. D, Number of DEGs in GFP-low, GFP-med, and GFP-high groups. Higher numbers of DEGs were detected with higher expression of MECP2. E, Heatmap showing log2FC for genes altered in both GFP-med and GFP-high groups. Absolute log2FC in GFP-high group was consistently higher than that in GFP-med group. F, G, Difference in the absolute log2FC between GFP-med and GFP-high groups were plotted in y-axis and the absolute log2FC for each gene in GFP-med group were plotted in x-axis. Values in y-axis were higher than zero in the majority of common DEGs in both upregulated genes (F) and downregulated genes (G). The fractions of these genes are indicated in red. H, I, Comparison of log2FC for common DEGs between GFP-med and GFP-high groups. The analysis was done in both upregulated genes (H) and downregulated genes (I). The results show that the absolute log2FC for each gene is consistently higher in GFP-high group than in GFP-med group. Wilcoxon matched pair signed rank test. p < 0.0001, N = 103 genes (H); p < 0.0001, N = 108 genes (I). J, Results from RT-qPCR for the subset of common DEGs with the most significant FDRs. Fold change was obtained by normalizing Ct of each gene with that of Gapdh. The magnitude of fold change was consistently higher in GFP-high group and clearly correlated with MECP2 expression levels. One-way ANOVA: p < 0.0001, F(2.427,7.281) = 106.1 (Gipr), p = 0.0011, F(1.971,5.912) = 27.24 (Efnb3), p = 0.0031, F(1.299,3.897) = 39.50 (Pcdh9), p = 0.0025, F(1.265,3.796) = 47.24 (Slc24a2). ANOVA was followed by post hoc Tukey's multiple comparison tests. Gipr: p = 0.0121 (negative vs med), p = 0.0036 (negative vs high), p = 0.0128 (low vs med), p = 0.0021 (low vs high), p = 0.0067 (med vs high). Efnb3: p = 0.0075 (negative vs high) and 0.0164 (low vs high). Pcdh9: p = 0.0239 (negative vs low), 0.0021 (negative vs med), and 0.0074 (negative vs high). Slc24a2: p = 0.0427 (negative vs low), p = 0.0014 (negative vs med), p = 0.0099 (negative vs high), p = 0.0016 (low vs med), and p = 0.0309 (low vs high). Bar graphs show average ± SEM; *p < 0.05, **p < 0.001, ****p < 0.0001.
Finally, we performed mRNA-Seq of MeCP2-overexpressing nuclei to correlate transcriptional changes with the alteration in heterochromatin structure. Neuronal nuclei were purified from PFC, which was collected three weeks after the injection of AAV5-Camk2α-Cre and AAV(DJ)-DIO-huMECP2-GFP (Fig. 12). Nuclei were labeled with anti-NeuN-Alexa Fluor 647 antibody and NeuN-positive nuclei were purified by FACS. Based on the GFP intensity, NeuN-positive nuclei were sorted into GFP-high, GFP-medium (med), GFP-low, and GFP-negative groups (Fig. 12A,B). Microscopic images of the collected nuclei confirmed effectiveness of sorting (Fig. 12B). Four mice were processed separately and were used as biological replicates. mRNA-Seq was performed from 16 samples in total and the result was analyzed using GFP-negative samples as control. First, we analyzed endogenous and exogenous Mecp2 levels by measuring reads of mouse and human Mecp2 (Fig. 12C). While mouse Mecp2 reads were comparable between groups, human MECP2 reads correlated positively with the GFP intensity (Fig. 12C). Total Mecp2 reads normalized to that of GFP-negative group was around 2×, 5×, and 25× in GFP-low, GFP-med, and GFP-high group, respectively (Fig. 12C). Next, we evaluated the number of DEGs in each group. We detected significantly higher number of DEGs in groups expressing higher MECP2 (Fig. 12D). The number of DEGs were 25, 302, and 2114, in GFP-low, GFP-med, and GFP-high groups, respectively (Fig. 12D). The number of upregulated and downregulated DEGs were similar in all three groups, and this result was consistent with previous literature showing bidirectional impact of MeCP2 in transcriptional regulation. The number of common DEGs altered in both GFP-med and GFP-high groups was 211, which corresponded to 70% of DEGs detected in GFP-med group (Fig. 12D). The fraction of upregulated and downregulated genes was again similar. We focused on these common DEGs and asked whether log2 fold-change (log2FC) correlated with the MeCP2 level. The absolute log2FC for each common DEG was consistently higher in GFP-high group than in GFP-med group (Fig. 12E). To validate this finding, we quantified difference of absolute log2FC between GFP-med and GFP-high group (Fig. 12F,G). The fraction of DEGs which showed higher absolute log2FC in GFP-high group was 66% and 84% in upregulated genes and downregulated genes, respectively (Fig. 12F,G). Paired comparison of log2FC in common DEGs between GFP-med and GFP-high groups showed absolute log2FC was significantly higher in GFP-high group for both upregulated genes (N = 103 genes, p < 0.0001; Fig. 12H) and downregulated genes (N = 108 genes, p < 0.0001; Fig. 12I). Taken together, the magnitude of transcriptional changes caused by MeCP2 overexpression was highly correlated with the MeCP2 level. This finding was confirmed by quantitative PCR analysis on the subsets of common DEGs with the most significant FDRs (Fig. 12J).
Discussion
In this study, we examined the effect of MeCP2 dosage on the structure of heterochromatic foci in neurons. Results obtained from confocal and electron microscopy showed that both depletion and overexpression of MeCP2 significantly affected the structure of heterochromatic foci. Loss of MeCP2 in Mecp2-null mouse lines resulted in foci with a simplified shape and smooth surface. In contrast, gain of MeCP2 in Mecp2-Tg3 mouse lines led to foci with diffuse and irregular morphology (Figs. 1–4). Because a mild but significant change in heterochromatic foci was present in the MeCP2-deleted neurons of the presymptomatic female Mecp2+/− mice (Fig. 6), MeCP2-dependent heterochromatin change may be upstream of subsequent changes that lead to abnormal neuronal function. Furthermore, results from MeCP2-R270X and MeCP2-G273X mice, which lacked and retained AT-hook 2 domain, respectively, showed that the heterochromatin structural changes correlated clearly with the severity of RTT-related symptoms (Fig. 8). We also analyzed the impact of MeCP2 knock-down and overexpression in the postnatal brains (Figs. 9, 10) and found that the changes occurred in a distinct manner from Mecp2-null or Mecp2-Tg3 mice. Finally, we analyzed physiological outcome of postnatal MeCP2-overexpression and found significant changes in behavior and transcriptome, which occurred in parallel with the heterochromatin changes (Figs. 11, 12). Notably, the magnitude of transcriptional alteration clearly correlated with MeCP2 levels (Fig. 12). Taken together, our results suggest that one of the essential functions of MeCP2 is to regulate 3D structure of heterochromatin, which could explain the gene expression changes that occur on changes in MeCP2 function or levels.
The nucleus is organized into distinct compartments, such as nucleoli, nuclear lamina, and pericentric chromocenters, which consist of heterochromatic AT-rich DNA that we visualized by DAPI as heterochromatic foci (Cremer and Cremer, 2010). Recent studies using cutting-edge imaging technology revealed that chromatin within the nucleus is spatially organized according to its chromatin state (Boettiger et al., 2016; Wang et al., 2016; Watson and Tsai, 2017). Transcriptionally suppressed heterochromatic regions are organized into tight clusters while actively transcribed euchromatic regions are spatially segregated from these areas, suggesting that such spatial compartmentalization of chromatin is essential for normal transcription (Boettiger et al., 2016). Because MeCP2 levels significantly affected the structure of heterochromatic foci, it is tempting to speculate that MeCP2 organizes spatial compartmentalization of chromatin within the neuronal nuclei and also regulates transcription. To test this hypothesis, it would be critical to examine the effect of MeCP2 depletion and overexpression on chromatin structure genome wide. It will be also crucial to examine whether the location or spatial organization of the MeCP2-regulated genes is altered in the nucleus of MeCP2-depleted and GFP-overexpressing cells. Application of techniques such as high-C to the repetitive elements and 3D FISH with advanced imaging techniques will be helpful to address these possibilities (Lieberman-Aiden et al., 2009; Rao et al., 2014; Schmitt et al., 2016).
How could MeCP2 alter the structure of heterochromatic foci? The results from mice expressing MeCP2-R270X and MeCP2-G273X showed that the AT-hook 2 domain was important in determining the onset of the heterochromatin structural changes (Fig. 8). The presence of AT-hook 2 must be critical for the function of MeCP2 because loss of this domain leads to severe neurologic symptoms both in humans and in mice (Villard, 2007; Baker et al., 2013). The importance of AT-hook 2 was further confirmed by a recent study which showed that expression of MeCP2, which is deprived of nuclear localization signal but retains AT-hook 2, significantly prolongs survival in mice (Lyst et al., 2018). Because DNA binding of MeCP2-R270X is preserved but weakened in heterochromatic repeat elements (Baker et al., 2013), the AT-hook 2 domain may be needed to stabilize the binding of MeCP2 to DNA. Alternatively, AT-hook 2 domain may mediate chromatin architectural change through directly affecting DNA conformation and nucleosome positioning. AT-hooks in Mecp2 are homologous to those of HMG proteins, which can bend, straighten or twist the double helix when bound to DNA (Reeves, 2001, 2010). A final consideration is the recruitment of α-thalassemia/mental retardation syndrome X linked (ATRX) to heterochromatic foci. ATRX is lost in the heterochromatic foci of both MeCP2-null and MeCP2-R270X-expressing mice and its loss correlated well with the disease progression (Nan et al., 2007; Baker et al., 2013). Whether ATRX loss from the heterochromatic foci is downstream or upstream of the structural changes observed in this study requires further investigation.
We detected MeCP2-dependent changes in heterochromatic foci in all the three neuronal populations we analyzed, which included CA1 excitatory cells, layer 2/3 cortical excitatory cells, and PV-positive cortical inhibitory cells (Figs. 1–3). Interestingly, while the changes in DAPI intensity were consistently observed in all the three groups of neurons, the degree of changes in the shape of heterochromatic foci varied by cell type. Cortical excitatory cells showed the most robust structural changes (Fig. 2) and CA1 pyramidal cells showed milder but significant changes in heterochromatin structure (Fig. 1). In contrast, heterochromatic foci of PV-positive inhibitory cells in the cortex showed changes in the DAPI intensity but did not show changes in the ellipticity when MeCP2 was either depleted or overexpressed (Fig. 2). Furthermore, when we induced MeCP2 knock-down or overexpression in the postnatal brains (Figs. 9, 10), the effect was distinct from the effect we observed in Mecp2-null or Mecp2-Tg3 mice. Previous studies using cultured neurons and myocytes reported changes that were quite different from our observations (Brero et al., 2005; Bertulat et al., 2012). These studies analyzed ectopic overexpression of MeCP2 and found reduction in the number of heterochromatic foci (Brero et al., 2005). In contrast, we performed AAV-induced MeCP2 overexpression in adult mice and observed decreased DAPI intensity and increased volume of foci. The effect of MeCP2 in the cultured cells may be mediated by a different mechanism from that of neurons in vivo, because the effect of MeCP2 in vitro was shown to be solely dependent on the methyl-binding domain (Brero et al., 2005). While the difference in the time course of MeCP2 expression changes may have contributed to the variations in results, the difference in the degree of the structural changes between the cell types and between maturation stages could be also explained by the difference in the DNA methylation pattern and MeCP2 distribution (Johnson et al., 2017; Stroud et al., 2017). Additionally, neuronal activity may also have impact on MeCP2 distribution. This is supported by a study which showed that increased activity in cortical neurons induced accumulation of MeCP2 into the heterochromatic foci (Ballas et al., 2005), whose size was also shown to be affected by the neuronal activity (Singleton et al., 2011). These possibilities need to be addressed in vivo, because the results may provide explanation for the difference in transcriptional changes observed in Mecp2-null mice between various cell types or between different brain regions (Chahrour et al., 2008; Ben-Shachar et al., 2009; Stroud et al., 2017).
In order to clarify the molecular pathogenesis of RTT and MECP2 multiplication disorders, it will be crucial to identify how loss and overexpression of MeCP2 lead to neuronal gene expression changes. In this study, we attempted to correlate transcriptional changes with the structural changes of heterochromatic foci by performing mRNA-Seq on neurons overexpressing MeCP2 (Fig. 12). Our results clearly demonstrated the impact of MeCP2 dosage on gene expression, as the magnitude of transcriptional changes correlated significantly with MeCP2 levels. However, causal relationships between the heterochromatic foci and gene transcription remain to be addressed. In addition to the heterochromatin structural changes that we showed in this study, multiple modes of functions have been suggested for MeCP2, which include transcriptional repression (Nan et al., 1997, 1998; Lyst et al., 2013), activation (Chahrour et al., 2008), splicing regulation (Young et al., 2005), global regulation of translational activity (Li et al., 2013), and histone modifications (Thambirajah et al., 2012; Lee et al., 2020). Indeed, we detected mild changes in the number and size of nucleoli in cells lacking and overexpressing MeCP2 (Fig. 5), which suggested that MeCP2 may modify translational activity. Results from immunostaining of heterochromatic foci in MeCP2-deleted and GFP-overexpressing cells showed that several types of histone methylation such as H3K27me3 and H3K4me3 were affected. However, it is unclear whether the observed changes in the nucleoli and histone methylation were directly caused by MeCP2 or indirectly as a result of the changes in heterochromatic foci. Since MeCP2 is distributed genome wide and binds to DNA in multiple contexts (Chen et al., 2015; Gabel et al., 2015), the effect of MeCP2 on transcriptional regulation is likely mediated by multiple mechanisms. Future studies aiming to understand the combinatorial effect of the multiple modes of MeCP2's function will be essential for determining how loss and overexpression of MeCP2 result in neurologic disorders.
Footnotes
This work was supported by National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS) Grants 5R01NS057819, Howard Hughes Medical Institute (to H.Y.Z.), the NIH/NINDS Grant R01NS089664 (to R.V.S.), the Japan Society for the Promotion of Science KAKENHI Grants 18H05139 and 16H06279 (Platform for Advanced Genome Science), the AMED-Prime Grants 19gm6310001h0001 and 20gm6310001h0001 (to A.I.-I.), and the Neurovisualization Core at the BCM Intellectual and Developmental Disabilities Research Center Grant NIH/1U54HD083092. We thank Ms. Lita Duraine and Dr. Hugo Bellen for their help with transmission electron microscopy. We also thank the Collaborative Research Resources (Keio University School of Medicine) for help with cell sorting.
The authors declare no competing financial interests.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188. 10.1038/13810 [DOI] [PubMed] [Google Scholar]
- Baker SA, Chen L, Wilkins AD, Yu P, Lichtarge O, Zoghbi HY (2013) An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell 152:984–996. 10.1016/j.cell.2013.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G (2005) REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121:645–657. 10.1016/j.cell.2005.03.013 [DOI] [PubMed] [Google Scholar]
- Ben-Shachar S, Chahrour M, Thaller C, Shaw CA, Zoghbi HY (2009) Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum Mol Genet 18:2431–2442. 10.1093/hmg/ddp181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertulat B, De Bonis ML, Della Ragione F, Lehmkuhl A, Milden M, Storm C, Jost KL, Scala S, Hendrich B, D'Esposito M, Cardoso MC (2012) MeCP2 dependent heterochromatin reorganization during neural differentiation of a novel Mecp2-deficient embryonic stem cell reporter line. PLoS One 7:e47848. 10.1371/journal.pone.0047848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu CT, Zhuang X (2016) Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529:418–422. 10.1038/nature16496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brero A, Easwaran HP, Nowak D, Grunewald I, Cremer T, Leonhardt H, Cardoso MC (2005) Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J Cell Biol 169:733–743. 10.1083/jcb.200502062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong STC, Qin J, Zoghbi HY (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320:1224–1229. 10.1126/science.1153252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen K, Lavery LA, Baker SA, Shaw CA, Li W, Zoghbi HY (2015) MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc Natl Acad Sci USA 112:5509–5514. 10.1073/pnas.1505909112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R (2001) Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet 27:327–331. 10.1038/85906 [DOI] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY (2004) Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet 13:2679–2689. 10.1093/hmg/ddh282 [DOI] [PubMed] [Google Scholar]
- Cremer T, Cremer M (2010) Chromosome territories. Cold Spring Harb Perspect Biol 2:a003889. 10.1101/cshperspect.a003889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, Greenberg ME (2015) Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522:89–93. 10.1038/nature14319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A (2007) Reversal of neurological defects in a mouse model of Rett syndrome. Science 315:1143–1147. 10.1126/science.1138389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A (2001) A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 27:322–326. 10.1038/85899 [DOI] [PubMed] [Google Scholar]
- Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T (2005) Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet 37:31–40. 10.1038/ng1491 [DOI] [PubMed] [Google Scholar]
- Ito-Ishida A, Ure K, Chen H, Swann JW, Zoghbi HY (2015) Loss of MeCP2 in parvalbumin-and somatostatin-expressing neurons in mice leads to distinct Rett syndrome-like phenotypes. Neuron 88:651–658. 10.1016/j.neuron.2015.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito-Ishida A, Yamalanchili HK, Shao Y, Baker SA, Heckman LD, Lavery LA, Kim J-Y, Lombardi LM, Sun Y, Liu Z, Zoghbi HY (2018) Genome-wide distribution of linker histone H1.0 is independent of MeCP2. Nat Neurosci 21:794–798. 10.1038/s41593-018-0155-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BS, Zhao YT, Fasolino M, Lamonica JM, Kim YJ, Georgakilas G, Wood KH, Bu D, Cui Y, Goffin D, Vahedi G, Kim TH, Zhou Z (2017) Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat Med 23:1203–1214. 10.1038/nm.4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernohan KD, Vernimmen D, Gloor GB, Bérubé NG (2014) Analysis of neonatal brain lacking ATRX or MeCP2 reveals changes in nucleosome density, CTCF binding and chromatin looping. Nucleic Acids Res 42:8356–8368. 10.1093/nar/gku564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagger S, Connelly JC, Schweikert G, Webb S, Selfridge J, Ramsahoye BH, Yu M, He C, Sanguinetti G, Sowers LC, Walkinshaw MD, Bird A (2017) MeCP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. PLoS Genet 13:e1006793. 10.1371/journal.pgen.1006793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Kim J, Yun JM, Ohn T, Gong Q (2020) MeCP2 regulates gene expression through recognition of H3K27me3. Nat Commun 11:3140. 10.1038/s41467-020-16907-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang H, Muffat J, Cheng AW, Orlando DA, Lovén J, Kwok S-M, Feldman DA, Bateup HS, Gao Q, Hockemeyer D, Mitalipova M, Lewis CA, Vander Heiden MG, Sur M, Young RA, Jaenisch R (2013) Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell 13:446–458. 10.1016/j.stem.2013.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326:289–293. 10.1126/science.1181369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhoff MW, Garg SK, Mandel G (2015) A high-resolution imaging approach to investigate chromatin architecture in complex tissues. Cell 163:246–255. 10.1016/j.cell.2015.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi LM, Baker SA, Zoghbi HY (2015) MECP2 disorders: from the clinic to mice and back. J Clin Invest 125:2914–2923. 10.1172/JCI78167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, Guy J, Kastan NR, Robinson ND, de Lima Alves F, Rappsilber J, Greenberg ME, Bird A (2013) Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci 16:898–902. 10.1038/nn.3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, Ekiert R, Guy J, Selfridge J, Koerner MV, Merusi C, De Sousa D, Bird A (2018) Affinity for DNA contributes to NLS independent nuclear localization of MeCP2. Cell Rep 24:2213–2220. 10.1016/j.celrep.2018.07.099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L. (1984) Different central nervous system cell types display distinct and nonrandom arrangements of satellite DNA sequences. Proc Natl Acad Sci USA 81:3123–3127. 10.1073/pnas.81.10.3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A (1997) MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88:471–481. 10.1016/S0092-8674(00)81887-5 [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393:386–389. 10.1038/30764 [DOI] [PubMed] [Google Scholar]
- Nan X, Hou J, Maclean A, Nasir J, Lafuente MJ, Shu X, Kriaucionis S, Bird A (2007) Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc Natl Acad Sci USA 104:2709–2714. 10.1073/pnas.0608056104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitina T, Shi X, Ghosh RP, Horowitz-Scherer RA, Hansen JC, Woodcock CL (2007) Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Mol Cell Biol 27:864–877. 10.1128/MCB.01593-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka S, Konno K, Abe M, Motohashi J, Kohda K, Sakimura K, Watanabe M, Yuzaki M (2016) Roles of Cbln1 in non-motor functions of mice. J Neurosci 36:11801–11816. 10.1523/JNEUROSCI.0322-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159:1665–1680. 10.1016/j.cell.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves R. (2001) Molecular biology of HMGA proteins: hubs of nuclear function. Gene 277:63–81. 10.1016/S0378-1119(01)00689-8 [DOI] [PubMed] [Google Scholar]
- Reeves R. (2010) Nuclear functions of the HMG proteins. Biochim Biophys Acta 1799:3–14. 10.1016/j.bbagrm.2009.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi T, Iwasaki S, Okada M, Okamoto K, Ikegaya Y (2018) Ethanol facilitates socially evoked memory recall in mice by recruiting pain-sensitive anterior cingulate cortical neurons. Nat Commun 9:3526. 10.1038/s41467-018-05894-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaco RC, McGraw CM, Ward CS, Sun Y, Neul JL, Zoghbi HY (2013) Female Mecp2(+/-) mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum Mol Genet 22:96–109. 10.1093/hmg/dds406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AD, Hu M, Ren B (2016) Genome-wide mapping and analysis of chromosome architecture. Nat Rev Mol Cell Biol 17:743–755. 10.1038/nrm.2016.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MK, Gonzales ML, Leung KN, Yasui DH, Schroeder DI, Dunaway K, LaSalle JM (2011) MeCP2 is required for global heterochromatic and nucleolar changes during activity-dependent neuronal maturation. Neurobiol Dis 43:190–200. 10.1016/j.nbd.2011.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Su SC, Hrvatin S, Greben AW, Renthal W, Boxer LD, Nagy MA, Hochbaum DR, Kinde B, Gabel HW, Greenberg ME (2017) Early-life gene expression in neurons modulates lasting epigenetic states. Cell 171:1151–1164.e16. 10.1016/j.cell.2017.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thambirajah AA, Ng MK, Frehlick LJ, Li A, Serpa JJ, Petrotchenko EV, Silva-Moreno B, Missiaen KK, Borchers CH, Adam Hall J, Mackie R, Lutz F, Gowen BE, Hendzel M, Georgel PT, Ausió J (2012) MeCP2 binds to nucleosome free (linker DNA) regions and to H3K9/H3K27 methylated nucleosomes in the brain. Nucleic Acids Res 40:2884–2897. 10.1093/nar/gkr1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay R, Lee S, Rudy B (2016) GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 91:260–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, Moraine C, Marynen P, Fryns JP, Froyen G (2005) Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 77:442–453. 10.1086/444549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villard L. (2007) MECP2 mutations in males. J Med Genet 44:417–423. 10.1136/jmg.2007.049452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Su JH, Beliveau BJ, Bintu B, Moffitt JR, Wu CT, Zhuang X (2016) Spatial organization of chromatin domains and compartments in single chromosomes. Science 353:598–602. 10.1126/science.aaf8084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson LA, Tsai LH (2017) In the loop: how chromatin topology links genome structure to function in mechanisms underlying learning and memory. Curr Opin Neurobiol 43:48–55. 10.1016/j.conb.2016.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF, Kang D, Richman R, Johnson JM, Berget S, Zoghbi HY (2005) Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci USA 102:17551–17558. 10.1073/pnas.0507856102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Results from mRNA-Seq analysis including the number of mapped reads and the list of DEGs. Download Figure 12-1, XLSX file (509.9KB, xlsx) .
Images of 3D-reconstructed nuclei. DAPI, MeCP2, and reconstructed heterochromatin foci are shown in gray, green, and yellow, respectively. Nuclei of CA1 pyramidal cells of a Mecp2+/− female mouse. MeCP2-negative (Null-) cell was observed by the side of a MeCP2-positive (WT-) cell. Heterochromatic foci of the Null-cell had increased DAPI intensity and simplified shape.
Images of 3D-reconstructed nuclei. DAPI, MeCP2, and reconstructed heterochromatin foci are shown in gray, green, and yellow, respectively. Nuclei of CA1 pyramidal cells of a Mecp2Tg3/+ female mouse. MeCP2-overexpressing (Tg3-) cells were observed by the side of a cell expressing single copy of Mecp2 (WT-cell). Heterochromatic foci of the Tg3-cells had decreased DAPI intensified and complex morphology.
Images of 3D-reconstructed nuclei. DAPI, MeCP2, and reconstructed heterochromatin foci are shown in gray, green, and yellow, respectively. Nuclei of cortical pyramidal cells of a Mecp2+/− female mouse. MeCP2-positive (WT-) cell and MeCP2-negative (Null) cell from the same image were reconstructed and shown in separate movies (Movie 3, WT-cell; Movie 4, Null-cell). These images were processed using the same settings.
Images of 3D-reconstructed nuclei. DAPI, MeCP2, and reconstructed heterochromatin foci are shown in gray, green, and yellow, respectively. Nucleus of a cortical pyramidal cell of a Mecp2+/− female mouse. The movie shows a nucleus of MeCP2-negative (Null-) cell found in the proximity to the WT-cell shown in Movie 3.
Images of 3D-reconstructed nuclei. DAPI, MeCP2, and reconstructed heterochromatin foci are shown in gray, green, and yellow, respectively. Movie of a cortical pyramidal cell in a Mecp2Tg3/+ female mouse. Cell expressing single copy of Mecp2 (WT-cell) and a cell overexpressing MeCP2 (Tg3-cell) from the same image are shown in separate movies (Movie 5, WT-cell; Movie 6, Tg3-cell). These images were processed using the same settings.
Images of 3D-reconstructed nuclei. DAPI, MeCP2, and reconstructed heterochromatin foci are shown in gray, green, and yellow, respectively. Nucleus of a cortical pyramidal cell of a Mecp2Tg3/+ female mouse. The movie shows a nucleus of a MeCP2-overexpressing (Tg3-) cell found in the proximity to the WT-cell shown in Movie 5.