

Abstract
The pathologic hallmark of Parkinson's disease is the accumulation of α-synuclein-containing Lewy bodies/neurites almost exclusively in neurons, and rarely in glial cells. However, emerging evidence suggests that glia such as astrocytes play an important role in the development of α-synuclein pathology. Using induced pluripotent stem-derived dopaminergic neurons and astrocytes from healthy subjects and patients carrying mutations in lysosomal ATP13A2, a monogenic form of synucleinopathy, we found that astrocytes rapidly internalized α-synuclein, and exhibited higher lysosomal degradation rates compared with neurons. Moreover, coculturing astrocytes and neurons led to decreased accumulation of α-synuclein in neurons and consequently diminished interneuronal transfer of α-synuclein. These protective functions of astrocytes were attenuated by ATP13A2 deficiency, suggesting that the loss of ATP13A2 function in astrocytes at least partially contributes to neuronal α-synuclein pathology. Together, our results highlight the importance of lysosomal function in astrocytes in the pathogenesis of synucleinopathies.
SIGNIFICANCE STATEMENT While most neurodegenerative disorders are characterized by the accumulation of aggregated mutant proteins exclusively in neurons, the contribution of glial cells in this process remains poorly explored. Here, we demonstrate that astrocytes contribute to the removal of extracellular α-synuclein and that disruption of this pathway caused by mutations in the Parkinson's disease-linked gene ATP13A2 result in α-synuclein accumulation in human dopaminergic neurons. We found that astrocytes also protect neurons from α-synuclein propagation, whereas ATP13A2 deficiency in astrocytes compromises this protective function. These results highlight astrocyte-mediated α-synuclein clearance as a potential therapeutic target in disorders characterized by the accumulation of α-synuclein, including Parkinson's disease.
Keywords: α-synuclein, astrocyte, Kufor–Rakeb syndrome, Parkinson's disease
Introduction
Parkinson's disease (PD), the most common neurodegenerative disease next to Alzheimer's disease, is characterized by the accumulation of protein aggregates, Lewy bodies and neurites, which are immunoreactive for α-synuclein (α-syn), a presynaptic protein associated with the pathogenesis of sporadic and familial PD (Wong and Krainc, 2017). Abnormally elevated α-syn levels are toxic to neurons, as α-syn locus duplication causes late-onset PD, while triplication leads to early-onset PD, suggesting dose-dependent α-syn-mediated neurotoxicity (Singleton et al., 2003; Chartier-Harlin et al., 2004; Ibáñez et al., 2004). Therefore, it is critical for neurons to maintain α-syn within a certain level, which is conducted by a complex cellular machinery, including exocytosis, which is mainly conducted by two pathways—exosomes and lysosomal exocytosis. Both of these pathways are impaired in Kufor–Rakeb syndrome (KRS; Tsunemi et al., 2014, 2019), which was originally described as a rare hereditary neurodegenerative disorder caused by loss-of-function mutations in ATP13A2 (Ramirez et al., 2006).
ATP13A2 encodes a lysosomal type 5 P-type ATPase that has been extensively studied, but the precise mechanism of ATP13A2-mediated neurodegeneration remains to be explored (Dehay et al., 2012; Usenovic et al., 2012; Kett et al., 2015; Bento et al., 2016; Lopes da Fonseca et al., 2016; van Veen et al., 2020). We and others have reported that mutant (Mut) ATP13A2 results in insufficient generation of intraluminal vesicles and consequently decreased the release of exosomes and α-syn into extracellular space (Kong et al., 2014; Tsunemi et al., 2014). ATP13A2 also regulates another exocytotic pathway, lysosomal exocytosis, in which lysosomes directly fuse with plasma membrane and release their contents (Tsunemi et al., 2019). Importantly, intracellular levels of α-syn are inversely correlated with the amount of α-syn released from neurons by either pathway, suggesting that secretory pathways, at least in part, regulate intracellular α-syn levels (Tsunemi et al., 2014, 2019). While α-syn secretion would be beneficial for host neurons, it may enhance the progression of PD pathology as extracellularly released α-syn may be subsequently taken up by neighboring neurons, mediating α-syn propagation.
Here, we showed that induced pluripotent stem cell (iPSC)-derived astrocytes as well as rat primary astrocytes exhibit highly active phagocytosis, endocytosis, and proteolysis, and play a critical role in removing extracellular α-syn. We further found that cocultured astrocytes prevent not only neuronal α-syn accumulation in iPSC-derived dopaminergic (DA) neurons, but also α-syn transfer between neurons, suggesting a protective role against the progression of PD pathology. Importantly, these astrocytic protective functions are partially impaired by ATP13A2 mutations, resulting in the increased accumulation and propagation of α-syn. Together, these results suggest that astrocytic protective functions against α-syn toxicity may be potential targets for developing therapeutic agents for KRS and other related synucleinopathies such as PD.
Materials and Methods
Cell culture
Human iPSCs were taken from four normal patients of either sex and two male KRS patients (155°C > T, Mut 1; 3176 T > G, 3253 delC, Mut 2) were cultured and reprogrammed as described previously (Tsunemi et al., 2019). Differentiation toward dopaminergic neurons was conducted following the protocol described previously (Mazzulli et al., 2016; Tsunemi et al., 2019). At 40 d after the initiation of differentiation, we infected lentiviruses depending on the experiments. Differentiation into astrocytes and maturation were performed following the manufacturer instructions (STEMCELL Tech). At 12 d after the generation of embryonic bodies, astrocyte differentiation was induced. At 21 d after differentiation, the medium was replaced with the Astrocyte Maturation Medium. The astrocytes at 35 d after differentiation were used for the experiments (Fig. 1A,B). For neuron–astrocyte coculture experiments, the coverslips for DA neurons were prepared following a published protocol with a few modifications (Kaech and Banker, 2006). The paraffin wax (Sigma-Aldrich) was applied to the 18 mm coverslips on Parafilm. After being coated with 5 mg/ml poly-d-Lysine overnight, the coverslips were washed three times in sterile water every 2 h and coated with 50 μg/ml laminin overnight. DA neurons at 21–30 d after the start of differentiation were plated on the coated coverslips at a density of 5 × 105 cells/coverslip. At day 40, neurons on a coverslip were placed in each well of 6-well dishes on the bottom of which differentiated astrocytes were plated at a density of 2 × 106 cells/well (Fig. 2). For the α-syn transfer experiment, control (Cont) and ATP13A2 Mut DA neurons that were separately plated at a density of 5 × 105 cells/coverslip were cocultured in a 10 cm dish (see Fig. 5A,B), on the bottom of which astrocytes were plated at a density of 1 × 107 cells/dish (see Fig. 5C–F) for 5–7 d. Then, each DA neuron on the coverslips and astrocytes on the bottom of the plates were separately harvested for immunoblotting. Rat primary neurons and astrocytes were cultured as described previously (Meberg and Miller, 2003; Al-Bader et al., 2011). Astrocyte-conditioned medium was generated by culturing astrocytes for 24 h. Rat primary cortical neurons were cultured in the medium containing Neurobasal medium with B27 Supplement (Thermo Fisher Scientific) and 10% astrocyte-conditioned medium for 24 h (Fig. 2E–H). Fluorescent-tagged α-syn (α-syn Alexa Fluor 555) was generated as described previously (Tsunemi et al., 2019).
Figure 1.

Astrocytes absorb different α-syn species from culture media. A, Astrocytes differentiated from iPSCs that were taken from two normal individuals (Cont 1 and Cont 2) and two patients with ATP13A2 mutations (Mut 1 and Mut 2). The cells were stained with astrocyte markers GFAP (top) or β-S100 (bottom). B, Immunoblot characterization of astrocytes with astrocyte markers including vimentin (top), GFAP (second from top), glutamine synthetase (second from bottom), and β-S100 (bottom). C–E, iPSC-derived astrocytes can absorb α-syn from the media. C, Representative images of α-syn immunofluorescence in Cont 1 astrocytes before (top) and after culturing in the media containing monomeric α-syn (middle) and oligomeric/fibrillar α-syn (bottom) for 24 h. GFAP is used for astrocytic marker. D, Quantification of α-syn fluorescence intensities normalized by a nuclear marker, 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) intensities in astrocytes (n = 3, *p = 0.001, **p = 0.015, one-way ANOVA with Tukey's post hoc test). E, The quantification of α-syn levels in astrocytes that were cultured in the normal media or in the media containing α-syn (conditioned media; n = 3, *p = 0.001, **p = 0.0011, one-way ANOVA with Tukey's post hoc test). F, Trypan blue assay with Cont 1 and Mut 1 astrocytes that were cultured in the media containing oligomeric/fibrillar α-syn for 24 h. G, Representative immunoblot for α-syn of lysates from astrocytes before (leftmost lane), after being cocultured with Cont 1 and 2 and Mut 1 DA neurons and DA neurons harboring α-syn triplication (lanes 2–5), and after being cultured in the leftover media of Cont 1 and 2 and Mut 1 DA neurons and DA neurons harboring α-syn triplication (lanes 6–9). H, Densitometric quantification is shown as the relative α-syn levels against GFAP in astrocytes (n = 3, *p = 0.003, **p = 0.042, ***p = 0.001, one-way ANOVA with Tukey's post hoc test). I, Representative immunoblot for α-syn of lysates from astrocytes before (leftmost lane), after being cocultured with Cont 1 and 2 and Mut 1 DA neurons and DA neurons harboring α-syn triplication that were cultured in the media containing oligomeric/fibrillar α-syn for 24 h (lanes 2–5) or cultured in the leftover media of Cont 1 and 2, and Mut 1 DA neurons and DA neurons harboring α-syn triplication after they were cultured in the media containing oligomeric/fibrillar α-syn for 24 h (lanes 6–9). J, Densitometric quantification is shown as the relative α-syn levels against GFAP in astrocytes (n = 3, *p = 0.001, **p = 0.0013, ***p = 0.003, one-way ANOVA with Tukey's post hoc test). Scale bars: A, C, and F, 50 µm. In all graphs, error bars indicate the SEM.
Figure 2.
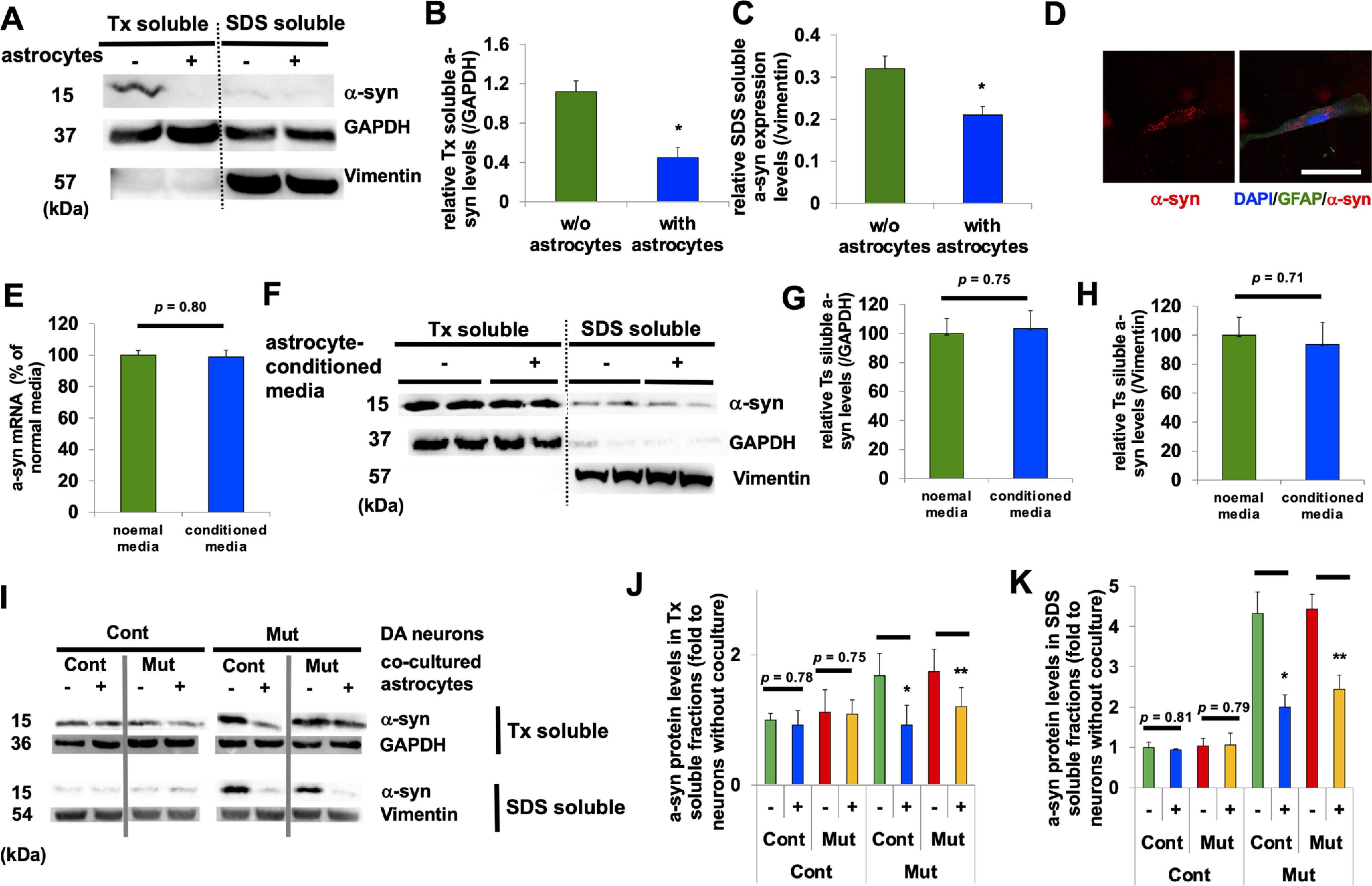
Coculturing of astrocytes and neurons lowers neuronal levels of α-syn. A, Representative immunoblot for α-syn, GAPDH, and vimentin in Triton-soluble (right two columns) and Sodium dodecyl sulfate (SDS)-soluble fractions (left two columns) from rat primary cortical neurons that were cultured without (each left column) or with (each right column) rat primary astrocytes. B, Densitometric quantification is shown as the relative α-syn levels against GAPDH in Triton-soluble fractions in cortical neurons (n = 3, *p = 0.021, Student's t test). C, Densitometric quantification is shown as the relative α-syn levels against vimentin in SDS-soluble fractions in cortical neurons (n = 3, *p = 0.035, Student's t test). D, A representative image of rat primary neuron that was cocultured with α-syn Alexa Fluor 555-containing astrocytes. E, Expression of α-syn mRNA in rat primary neurons before and after being cultured in astrocyte-conditioned media (n = 3, p = 0.80, Student's t test). F, Representative immunoblot for Triton-soluble (right four columns) and SDS-soluble fractions (left four columns) from rat primary cortical neurons before and after cultured in astrocyte-conditioned media. G, Densitometric quantification is shown as the relative α-syn levels against GAPDH in Triton-soluble fractions in rat primary cortical neurons (n = 3, p = 0.75, Student's t test). H, Densitometric quantification is shown as the relative α-syn levels against vimentin in SDS-soluble fractions in rat primary cortical neurons (n = 3, p = 0.71, Student's t test). I, Representative immunoblot for Triton-soluble fractions (top two lanes) and SDS-soluble fractions (bottom two lanes) from Cont (Cont 1; left four columns) and Mut (ATP13A2 Mut 1; right four columns) DA neurons that were cultured without (left) or with (right) Cont astrocytes. J, Densitometric quantification is shown as the relative α-syn levels against GAPDH in Triton-soluble fractions in DA neurons (n = 3, *p = 0.018, **p = 0.033, Student's t test). K, Densitometric quantification is shown as the relative α-syn levels against vimentin in SDS-soluble fractions in DA neurons (n = 3, *p = 0.005, **p = 0.009, Student's t test). In all graphs, error bars indicate SEM.
Figure 5.
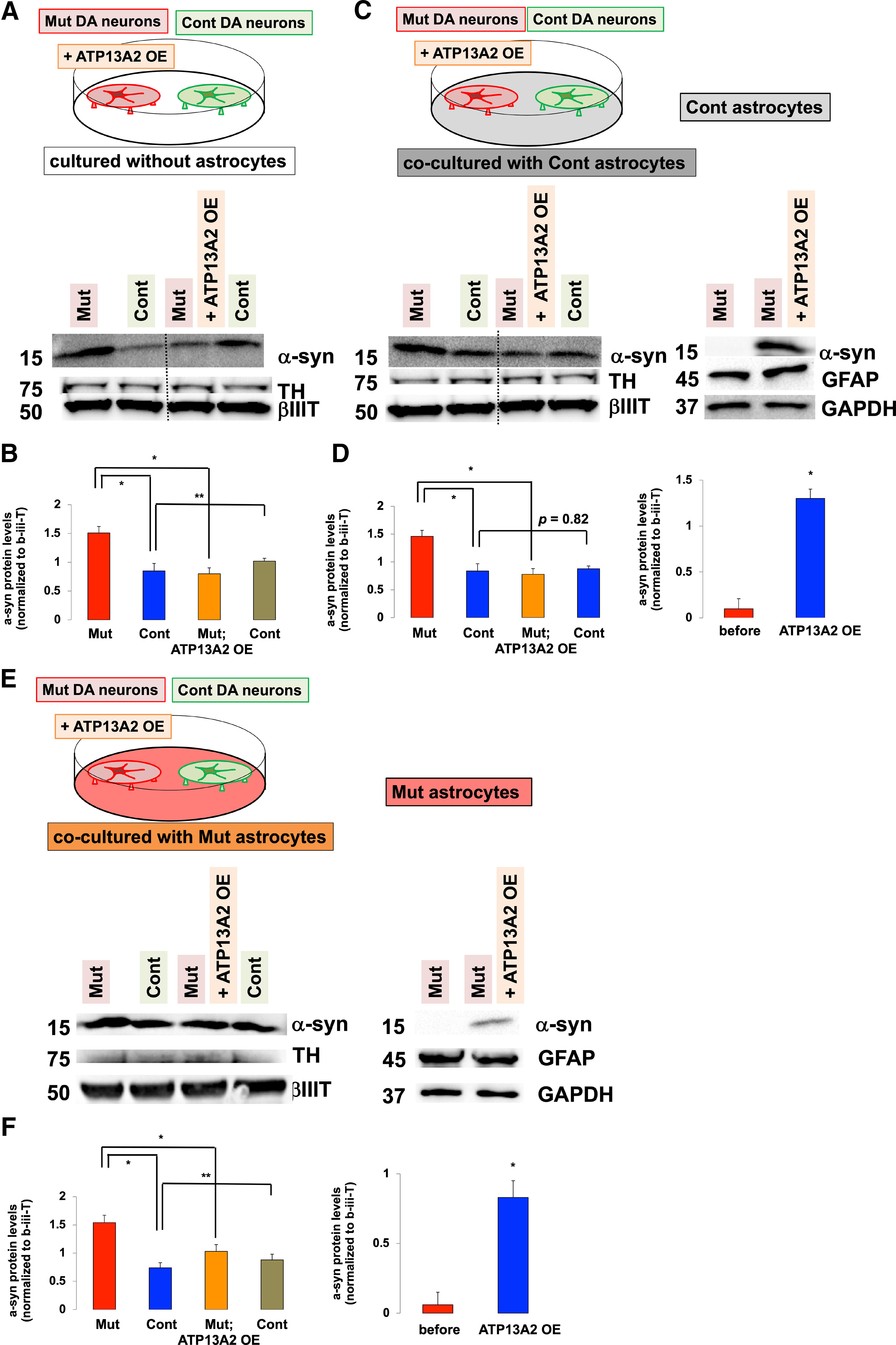
Coculturing neurons with astrocytes prevents α-syn transfer between neurons. A–D, The effect of coculturing astrocytes on the α-syn transmission between neurons. A, The α-syn transmission from Mut (ATP13A2 Mut 1) to Cont (Cont 1) DA neurons in the absence of coculturing astrocytes. Immunoblot analysis of α-syn levels in Mut and Cont DA neurons (left two columns) and α-syn levels in ATP13A2 overexpressing Mut and Cont DA neurons (right two columns). B, Densitometric quantification of α-syn levels normalized to β-III-tubulin in neurons that were not cocultured with astrocytes (n = 3, *p = 0.029, **p = 0.017, one-way ANOVA with Tukey's post hoc test). C, The α-syn transmission from Mut to Cont DA neurons in the presence of coculturing Cont astrocytes. Immunoblot analysis of α-syn levels in Mut and Cont neurons (first two columns) and α-syn levels in ATP13A2 overexpressing Mut and Cont DA neurons (next two columns) after being cocultured with Cont astrocytes. Immunoblot analysis of α-syn levels in Cont astrocytes cocultured with Mut and ATP13A2 overexpressing Mut DA neurons (right two columns). D, Densitometric quantification of α-syn levels normalized to β-III-tubulin in neurons in the presence of coculturing astrocytes (left graph; n = 3, *p = 0.027, one-way ANOVA with Tukey's post hoc test). Densitometric quantification of α-syn levels normalized to GFAP in Cont astrocytes (right graph; n = 3, *p = 0.002, Student's t test). E, F, The α-syn transmission from Mut to Cont DA neurons in the presence of coculturing Mut astrocytes. E, Immunoblot analysis of α-syn levels in Mut and Cont neurons (left two columns) and α-syn levels in ATP13A2 overexpressing Mut and Cont DA neurons (next two columns) after cocultured with Mut 1 astrocytes. Immunoblot analysis of α-syn levels in Mut astrocytes that were cocultured with Mut and ATP13A2 overexpressing Mut DA neurons (right two columns). F, Densitometric quantification of α-syn levels normalized to β-III-tubulin in neurons in the presence of coculturing Mut astrocytes (left graph; n = 3, *p = 0.017, **p = 0.022, one-way ANOVA with Tukey's post hoc test). Densitometric quantification of α-syn levels normalized to GFAP in Mut astrocytes (right graph; n = 3, *p = 0.003, Student's t test). Error bars indicate the SEM.
Immunocytochemistry
Immunocytochemical analysis was conducted as described previously (Tsunemi et al., 2014, 2019; Tsunemi and Krainc, 2014). Briefly, after fixation in 4% paraformaldehyde, the cells were permeabilized/blocked in PBS containing 0.1% saponin, 1% BSA, and 5% normal goat serum for 20 min. Specimens were then incubated with primary antibodies overnight, washed in PBS, and then incubated with Alexa Fluor-conjugated anti-rabbit or anti-mouse antibodies at 1:400 dilution for 1 h. Confocal imaging was conducted on the Zeiss LSM 880 confocal system with the Zeiss AX10 inverted microscope equipped with α Plan-APOCHROMAT 63× (1.4 numerical aperture) oil-immersion objective.
Electron microscopy
Electron microscopic analysis was conducted as described previously with modifications (Tsunemi et al., 2014). Briefly, the cells were fixed with 2.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, overnight. After being postfixed with 1% osmium tetroxide in phosphate buffer, the cells were dehydrated through a series of graded ethanol steps and embedded in Epon812 (Oken Shoji). The resin blocks were thin sectioned with Ultramicrotome UC6 (Leica). The sections were stained with uranyl acetate and lead citrate, and then analyzed with a transmission electron microscope (model HT7700, Hitachi).
Western blotting
Immunoblotting was conducted as described previously (Tsunemi et al., 2014, 2019; Tsunemi and Krainc, 2014). The antibodies used were anti-human S100 β (Abcam), anti-human glial fibrillary acidic protein (GFAP; Cell Signaling Technology), anti-human α-synuclein C-20 (Santa Cruz Biotechnology), anti-human α-synuclein 211 (Santa Cruz Biotechnology), anti-human glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Millipore), human vimentin (BD Biosciences), anti-human lysosome-associated membrane protein 1 (LAMP1; Santa Cruz Biotechnology), anti-human early endosome antigen 1 (EEA1; Cell Signaling Technology), anti-human epidermal growth factor receptor (EGFR; Cell Signaling Technology), anti-human CD63 (Developmental Studies Hybridoma Bank), anti-human Flotillin-1 (BD Biosciences), anti-human β-III tubulin (Covance), and anti-human TH (Millipore).
Exosome isolation and nanoparticle tracking analysis
Exosomes were purified as described previously (Tsunemi et al., 2014). Briefly, exosomes were collected from cell-conditioned media using a basic differential centrifugation method (200 × g for 5 min, 1200 × g for 10 min, and 16,500 × g for 30 min), followed by ultracentrifugation at 110,000 × g for 60 min. After washing in PBS, exosomes were collected by centrifugation at 110,000 × g for 60 min. Analysis of extracellular vesicles was conducted by the NanoSight LM10 system (NanoSight), configured with a 405 nm laser and a high-sensitivity digital camera system (OrcaFlash2.8, Hamamatsu C11440, NanoSight). Samples were administered and recorded for 1 min under sustained flow controlled by script control system equipped with a NanoSight syringe pump. Videos were analyzed by NTA software (version 2.3).
Lysosomal proteolysis in live neurons and lysosomal enzyme activity assays
EGFR degradation assay was conducted as described previously (Usenovic et al., 2012). Briefly, DA neurons or astrocytes were treated with 150 ng/ml human EGF (PreproTech) to stimulate endocytosis of EGFR. The levels of EGFR were monitored with anti-EGFR antibody. Long-lived protein degradation assays were performed by radioactive pulse-chase using tritium-labeled leucine (catalog #NET460A001MC, PerkinElmer) as previously described (Kaushik and Cuervo, 2009; Tsunemi et al., 2019). Glucocerebrosidase activity assays in lysosome-enriched P2 fractions isolated from DA neurons and astrocytes were performed using the artificial enzyme substrates 4MU-glucopyranoside (for GCase) and 4MU-sulfate potassium salt (for a-i-2-sulf) as described previously (Mazzulli et al., 2011; Tsunemi et al., 2019).
α-Synuclein detection
α-Synuclein oligomers/fibrils were formed as described previously (Mazzulli et al., 2011). Briefly, after α-syn monomers were incubated at 37°C for 10 d under continuous agitation of 1000 rpm, α-syn oligomers/fibrils were centrifuged at 10,000 × g for 30 min. The pellets were resuspended in PBS, and fibril formation was assessed by Thioflavin T spectroscopic assay and electron microscopic analysis. For analyzing α-synuclein internalization, 100 ng/ml α-synuclein oligomers/fibrils were added in the media for the indicated time, and DA neurons or astrocytes were washed four times in PBS and fixed in 4% paraformaldehyde.
Statistical analysis
All data were prepared for analysis with standard spreadsheet software (Excel, Microsoft). Statistical analysis was performed by one-way ANOVA with post hoc Tukey's test or Student's t test. All error bars represent the SEM in figures.
Results
Astrocytes can uptake α-synuclein secreted from DA neurons
We have reported that increased neuronal secretion of α-syn, via either exosomal secretion or lysosomal exocytosis, results in lower intracellular α-syn (Tsunemi et al., 2014, 2019). Secreted α-syn is taken up by glial cells such as astrocytes and causes inflammatory responses (Lee et al., 2010; Loria et al., 2017). Using astrocytes differentiated from control iPSCs (Fig. 1A,B), we found that under normal conditions, α-syn levels in astrocytes were very low (Fig. 1C, top, D, left). However, after culturing astrocytes in media containing α-syn monomers, astrocytic α-syn levels increased (Fig. 1C, middle, D, center), suggesting that astrocytes took up α-syn from the extracellular space, consistent with previous reports (Lee et al., 2010; Loria et al., 2017). Astrocytes were also able to take up α-syn oligomer/fibrils (Fig. 1C, bottom, D, right). This increase in α-syn levels in astrocytes was further confirmed by a sensitive ELISA (Tsunemi et al., 2014, 2019; Fig. 1E), and the uptake of α-syn was confirmed by the trypan blue assay to exclude the possibility that α-syn attached to the outside of plasma membranes (Fig. 1F; Tsunemi et al., 2019).
We next examined the relationship between the amounts of α-syn in the media and α-syn levels in astrocytes. While α-syn was not detectable in control astrocytes before coculturing with neurons (Fig. 1G,H, leftmost lane), it became detectable after 24 h of coculturing with DA neurons. (Fig. 1G,H, lanes 2–5). Interestingly, the increase in astrocytic α-syn was more pronounced when astrocytes were cocultured with control neurons compared with ATP13A2-mutant DA neurons, consistent with our prior observations that ATP13A2-mutant DA neurons secreted less α-syn than control neurons (Cont 1 and 2 vs ATP13A2 Mut; 0.73 ± 0.02, p = 0.042; Fig. 1G,H, lane 4; Tsunemi et al., 2014, 2019). In contrast, coculturing of astrocytes with neurons carrying α-syn triplication, which express higher levels of α-syn, resulted in dramatically increased uptake of α-syn into astrocytes (Cont 1 and 2 vs ATP13A2 Mut; 2.21 ± 0.03, p = 0.001; Fig. 1G,H, lane 5).
To exclude the possibility that α-syn directly transferred from neurons to astrocytes, we cultured astrocytes in cell media used for neuronal cultures, and found that α-syn levels in astrocytes were comparable to those cocultured with neurons, further suggesting that astrocytic α-syn originated from neuronally secreted α-syn (Fig. 1G,H, lanes 6–9). To examine whether astrocytes can uptake α-syn oligomers/fibrils secreted from neurons, we cultured astrocytes in media either containing α-syn oligomers/fibrils or cocultured with DA neurons. These experiments revealed that α-syn oligomers/fibrils were detected in astrocytes in the presence of control or mutant ATP13A2 neurons (Cont 1, Cont 2, Mut 1, and Mut 2) or DA neurons carrying SNCA triplications (Fig. 1I,J, leftmost lane; Fig. 1I,J, lanes 2–5). Similarly, astrocytic α-syn oligomers/fibrils were detected after culturing astrocytes with neuronal media (Fig. 1I,J, lanes 6–9). Together, these data demonstrate that astrocytes can take up different α-syn species secreted from DA neurons into the media.
Coculturing ATP13A2 neurons with astrocytes lowers levels of neuronal α-syn levels
Next, we examined how α-syn uptake by astrocytes affects α-syn levels in neurons in a steady-state condition. Using rat primary neurons and astrocytes (Meberg and Miller, 2003; Al-Bader et al., 2011), we found that both Triton-soluble (Fig. 2A, left two lanes, B) and SDS-soluble fractions of neuronal α-syn (Fig. 2A, right two lanes, C) were decreased after coculturing neurons with astrocytes, indicating that astrocyte-mediated α-syn uptake was able to lower intracellular α-syn levels in neurons. To further confirm that astrocytes uptake neuronal α-syn, we labeled synthetic α-syn fibrils with Alexa Fluor 555 (α-syn 555; Tsunemi et al., 2019). After DA neurons were exposed to α-syn 555 overnight and then cocultured with astrocytes, we were able to detect α-syn 555 in astrocytes, further demonstrating that astrocytic α-syn was of neuronal origin (Fig. 2D). To examine whether astrocytes generate and release some factors that could reduce α-syn expression in neurons, we analyzed the expression and protein levels of neuronal α-syn before and after culturing neurons with astrocyte-derived conditioned media. We did not find any differences in either α-syn expression (Fig. 2E) or α-syn protein levels (Fig. 2F–H) before and after culturing neurons with astrocyte-derived conditioned media. These results suggest that it is unlikely that astrocytes send some sort of a signal that would lower neuronal α-syn levels. To examine the effect of ATP13A2 on neuronal α-syn levels, iPSC-derived DA neurons and astrocytes were used. We found that α-syn levels in control DA neurons did not change when these neurons were cocultured with control or mutant astrocytes (Fig. 2I, left four lanes, J,K, left four lanes). In contrast, levels of α-syn in ATP13A2-mutant DA neurons were significantly reduced when the neurons were cocultured with control or mutant astrocytes (Fig. 2I, right four lanes, J,K, right four lanes). Importantly, the reduction of α-syn levels in ATP13A2-mutant neurons was more profound when the neurons were cocultured with control compared with mutant ATP13A2 astrocytes (Fig. 2J,K, right four lanes). Thus, astrocytes offer an important mechanism for regulating neuronal α-syn levels by taking up secreted α-syn, but this regulation is less efficient in mutant ATP13A2 astrocytes.
Proteolysis and degradation of α-syn is more efficient in astrocytes compared with neurons
Our data so far indicate that astrocytes can uptake secreted α-syn and therefore protect neurons from accumulation of α-syn. Based on these results, we hypothesized that α-syn gets efficiently degraded in astrocytes. To test this hypothesis, we first measured the degradation of EGFR, which is expressed on the plasma membrane, but is endocytosed and degraded in lysosomes on binding to EGF (Usenovic et al., 2012). These experiments revealed significantly increased average EGFR degradation rates in both control and PARK 9 mutant astrocytes compared with DA neurons (Fig. 3A,B). However, ATP13A2 deficiency because of mutations decreased EGFR degradation rates in either DA neurons or astrocytes (Fig. 3A,B), suggesting that ATP13A2 mutations impair the endocytic pathway in both cell types.
Figure 3.
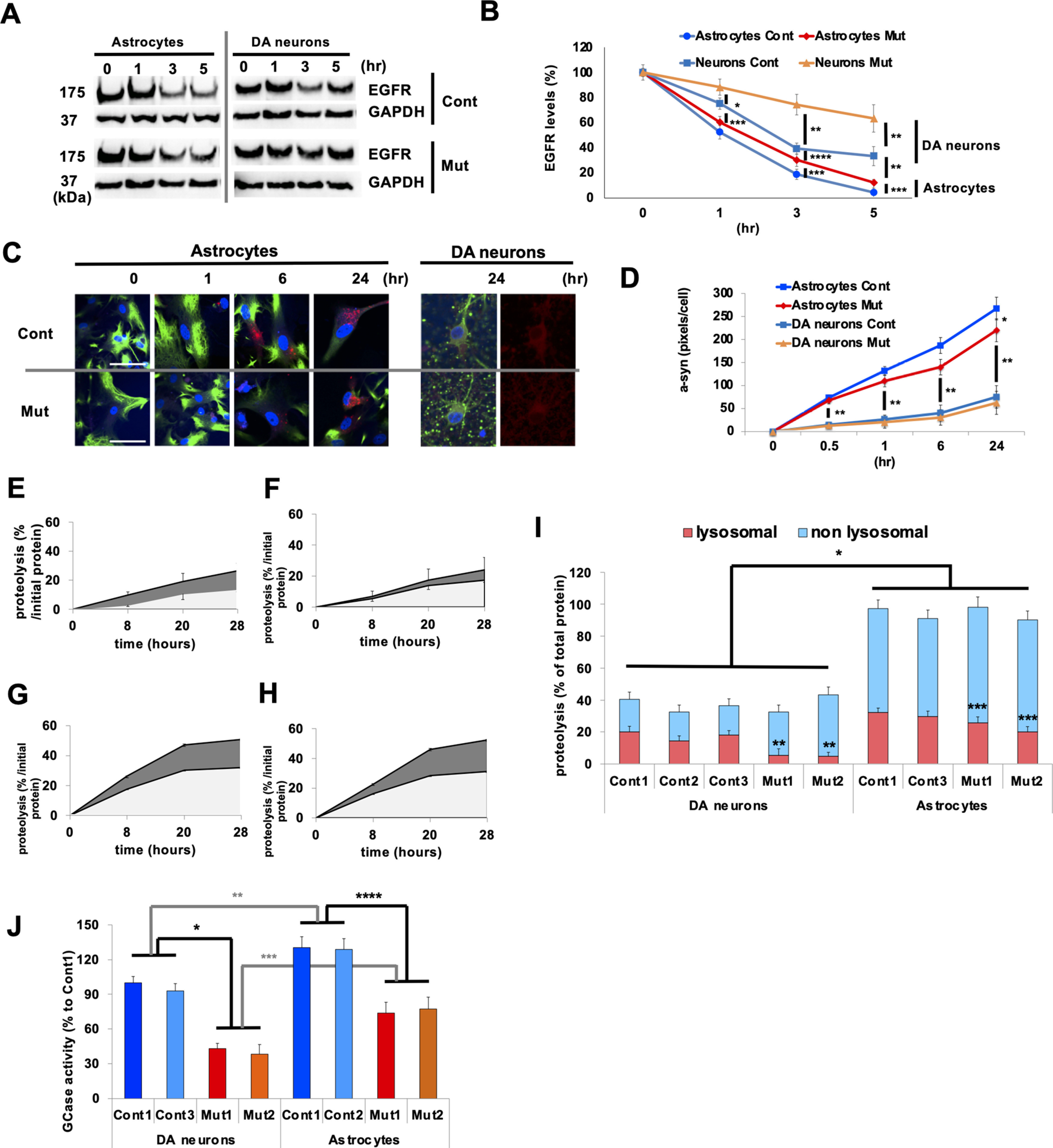
Astrocytes exhibit higher rates of lysosomal proteolysis compared with neurons. A, Representative immunoblot for EGFR degradation in iPSC-derived Cont (Cont 1) and Mut (Mut 1) astrocytes and Cont and Mut DA neurons that was followed for 5 h. B, Levels of EGFR were normalized to GAPDH levels and expressed as percentage levels of the initial time point (T0; n = 6, *p = 0.037, **p = 0.01, ***p = 0.015, ****p = 0.041, one-way ANOVA with Tukey's post hoc test). C, Representative images of α-syn internalization assay in Cont (Cont 1) and Mut (ATP13A2 Mut1) astrocytes and DA neurons. At 0, 1, 6, and 24 h after incubation with α-syn oligomers/fibrils, the astrocytes and DA neurons were fixed and stained with GFAP with DAPI and b-iii-T with DAPI, respectively. D, Analysis of α-syn internalization assay in astrocytes and DA neurons (n = 3, *p = 0.037, **p = 0.0014, one-way ANOVA with Tukey's post hoc test). E–I, Lysosomal proteolysis of Cont (E) and Mut (F) DA neurons and Cont (G) and Mut (H) astrocytes is calculated by subtracting lysosomal inhibitors (2.5 mm NH4Cl and 50 μm leupeptin) sensitive to proteolysis from total proteolysis at 8, 20, and 28 h after chase. I, Lysosomal (red) and nonlysosomal (light blue) proteolysis in Cont 1–3, Mut 1, and 2 DA neurons (left four columns) and in Cont 1 and 3 and Mut 1 and 2 astrocytes (right four columns; n = 3, *p = 0.009, **p = 0.022, ***p = 0.039, one-way ANOVA with Tukey's post hoc test). J, Glucocerebrosidase (GCase) activities in lysosome-enriched fractions extracted from Cont 1 and 3, Mut 1, and 2 DA neurons (left four columns) and in Cont 1 and 2, Mut 1 and 2 astrocytes (right four columns; n = 3, *p = 0.009, **p = 0.037, ***p = 0.027, ****p = 0.019, one-way ANOVA with Tukey's post hoc test). In all graphs, error bars indicate SEM.
We then measured α-syn internalization (Fig. 3C,D) by exposing the cells to oligomeric/fibrillar human α-syn (Tremblay et al., 2019). We found that oligomeric/fibrillar α-syn internalization was faster in astrocytes than neurons, demonstrating a higher rate of endocytic activity (Fig. 3D), whereas ATP13A2 deficiency decreased the α-syn internalization rates in astrocytes. Next, we compared the proteolytic capacity in control (Fig. 3E) and ATP13A2-mutated DA neurons (Fig. 3F) and control (Fig. 3G) and ATP13A2-mutated astrocytes (Fig. 3H) by pulse-chase analysis (Tsunemi and Krainc, 2014; Tsunemi et al., 2019). The results showed that lysosomal proteolysis was significantly higher in astrocytes than in neurons (Fig. 3I), whereas ATP13A2 deficiency leads to decreased lysosomal proteolysis in both DA neurons and astrocytes (Fig. 3I). The activity of glucocerebrosidase in lysosome-enriched fractions in astrocytes was higher than those in DA neurons (Fig. 3J; p = 0.037), further supporting the notion of higher proteolytic capacity in astrocytes.
To begin exploring the reasons for the increased proteolytic capacity of astrocytes, we compared endolysosomal protein levels and found that astrocytes contain increased levels of both EEA1, an early endosomal protein, and LAMP1, a lysosomal protein, compared with neurons (Fig. 4A–C). ATP13A2 deficiency resulted in increased EEA1 and LAMP1 levels both in astrocytes and neurons (Fig. 4B,C). Using TEM to quantify electron-dense vesicles (EDVs), we found that EDVs were abundant in astrocytes (Fig. 4D,E; Cont astrocytes vs Cont DA neurons, p = 0.017), whereas ATP13A2 deficiency resulted in a further increase of EDVs both in astrocytes and neurons. These results were confirmed by immunocytochemistry, showing that astrocytes contained more LAMP1-positive vesicles than neurons (Cont astrocytes vs Cont DA neurons, p = 0.009), while ATP13A2 deficiency resulted in increased LAMP1-positive vesicles and EDVs both in astrocytes and neurons (Fig. 4F,G). Together, these results suggest that both endocytic activities and lysosomal proteolytic activities were higher in astrocytes compared with neurons, possibly because of the higher density of endolysosomes in astrocytes.
Figure 4.
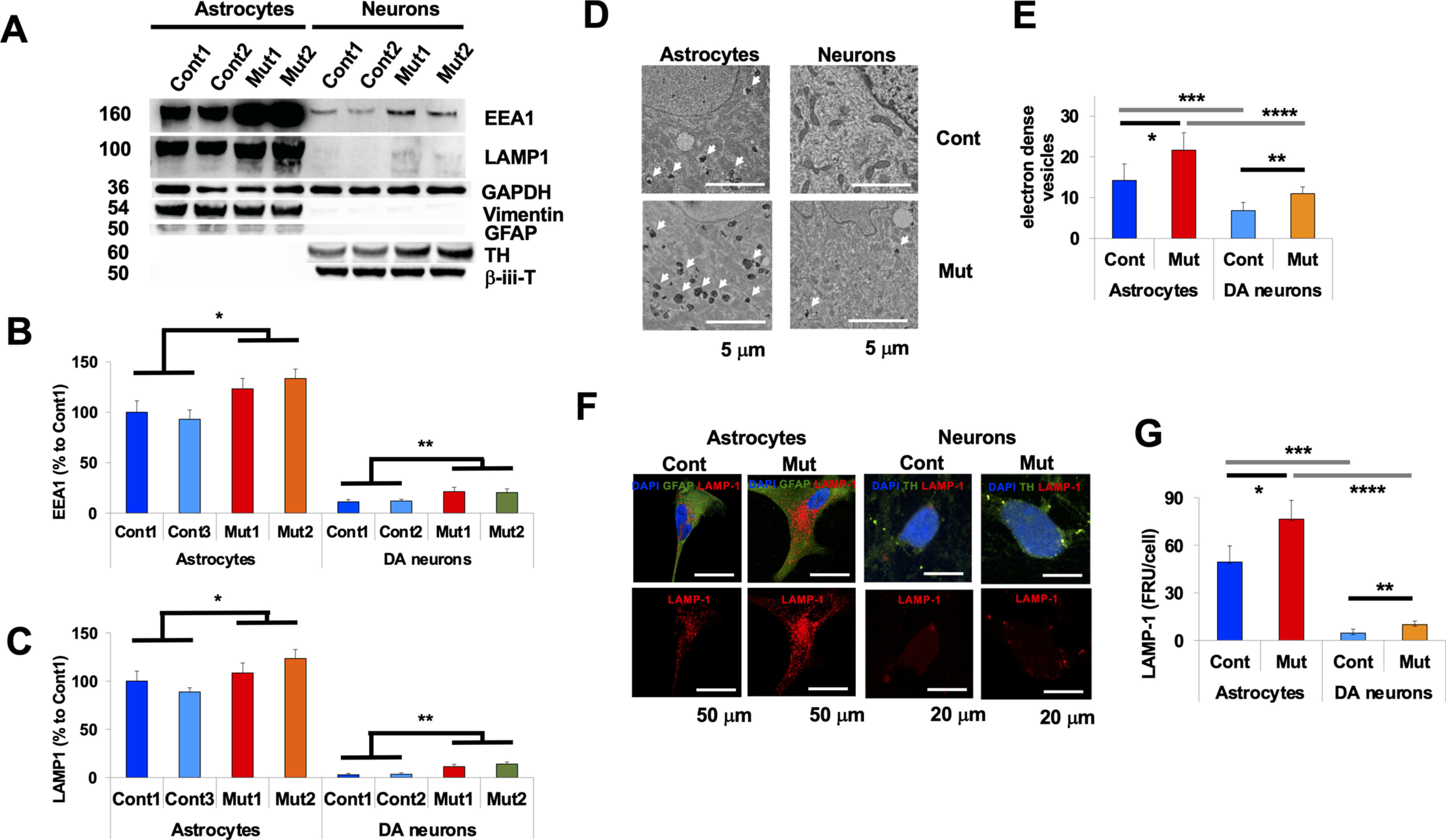
Higher endocytic activity in astrocytes compared with neurons. A–C, Immunoblot analysis for endosomal protein levels in astrocytes and neurons. A, Representative immunoblot for EEA1 (top), LAMP1 (second from top), GAPDH (middle), vimentin (second from bottom), and GFAP (bottom) of lysates from Cont 1 and 2, Mut 1 and 2 DA neurons (left four columns) and Cont 1 and 2 and Mut 1 and 2 astrocytes (right four columns). B, Densitometric quantification is shown as the relative EEA1 levels against GAPDH in Cont 1 and 2 and Mut 1 and 2 astrocytes (left four columns), and in Cont 1 and 2 and Mut 1 and 2 DA neurons (right four columns; n = 3, *p = 0.031, **p = 0.027, one-way ANOVA with Tukey's post hoc test). C, Densitometric quantification is shown as the relative LAMP1 levels against GAPDH in Cont 1 and 2 and Mut 1 and 2 astrocytes (left four columns), and Cont 1 and 2 and Mut 1 and 2 DA neurons (right four columns; n = 3, *p = 0.039, **p = 0.027, one-way ANOVA with Tukey's post hoc test). D, Representative EM images of electron-dense vesicles in Cont (Cont 1; top left) and Mut (ATP13A2 Mut 1; bottom left) astrocytes and in Cont (top right) and Mut (bottom right) DA neurons. Scale bar, 5 µm. E, Quantification analysis of the number of electron-dense vesicles from Cont and Mut astrocytes (left two columns) and Cont and Mut DA neurons (right two columns; n = 10–20, *p = 0.033, **p = 0.027, ***p = 0.017, ****p = 0.021, one-way ANOVA with Tukey's post hoc test). F, Representative images of LAMP1-positive vesicles in Cont and Mut astrocytes and Cont and Mut DA neurons. Scale bars: astrocytes, 50 µm; neurons, 10 µm. G, Quantification analysis of the number of LAMP1-positive vesicles from Cont and Mut astrocytes (left two columns) and Cont and Mut DA neurons (right two columns; n = 10-20, *p = 0.039, **p = 0.033, ***p = 0.009, ****p = 0.013, one-way ANOVA with Tukey's post hoc test). In all graphs, error bars indicate the SEM.
Coculturing astrocytes and neurons decreases transfer of α-syn between neurons
Based on these results, we hypothesized that the secretion of α-syn from neurons, followed by uptake and degradation of α-syn by astrocytes leads to less α-syn available for neuron-to-neuron propagation. To this end, we used a triple coculture system in which mutant and control DA neurons were cultured on the coverslips and astrocytes were cultured on the bottom of a dish, allowing us to analyze the changes in α-syn levels in each type of cell separately. We tested whether the presence of astrocytes affected neuron-to-neuron α-syn transmission (Fig. 5). In the absence of astrocytes, lentivirus-mediated expression of wild-type ATP13A2 in mutant DA neurons led to an decrease in α-syn levels in mutant DA neurons because of increased exocytosis (Tsunemi et al., 2014, 2019; Fig. 5A,B, first and third lanes from left) and an increase in α-syn levels in control DA neurons (Fig. 5A,B, second and forth lanes from left), showing that α-syn secreted from mutant DA neurons was taken up by control DA neurons. Then we examined the effect of coculturing astrocytes on this α-syn transfer between neurons (Fig. 5C–F). Similarly, lentivirus-mediated expression of wild-type ATP13A2 in mutant DA neurons increased α-syn secretion and resulted in reduced intracellular α-syn levels (Fig. 5C,D, first and third lanes from left). While α-syn was expressed in control astrocytes at very low levels, the secreted α-syn was taken up by astrocytes (Fig. 5C,D, right two lanes); therefore, α-syn levels remained unchanged in cocultured control DA neurons (Fig. 5C,D, second and fourth lanes from left). These results suggested that the presence of astrocytes protects healthy neurons from extracellular α-syn.
We next examined whether ATP13A2 deficiency in astrocytes affects this neuronal α-syn transmission. Indeed, α-syn levels increased in control DA neurons, suggesting that reduced uptake and degradation of α-syn in ATP13A2-mutated astrocytes resulted in increased α-syn transmission between DA neurons (Fig. 5E,F, first and third lanes from left). Together, our data demonstrated that astrocytes have the capacity to clear extracellular α-syn more efficiently than neurons, whereas ATP13A2 deficiency partially disrupts this protective role of astrocytes, potentially contributing to PD pathology.
Discussion
Recent studies using ATP13A2-mutant DA neurons demonstrated that decreased exosomal or lysosomal secretion of α-syn results in neuronal accumulation of α-syn, whereas enhancing these pathways leads to decreased levels of neuronal α-syn (Tsunemi et al., 2014, 2019; Filippini et al., 2019). However, the fate of secreted α-syn has not been explored in detail. In this study, we focused on the role of astrocytes and ATP13A2 in the regulation of α-syn secreted from neurons.
Astrocytes are known to provide metabolic and trophic support to neurons, and more recent studies also suggest that astrocytes play important roles in modulating neurotransmission, cell signaling, inflammation, synapse modulation, and metabolite and electrolyte homeostasis (Bélanger et al., 2011). For example, glutamate transporters (EAAT1 and EAAT2) and Kir4.1 are able to clear excess extracellular glutamate and potassium, respectively, released from neurons (Ben Haim and Rowitch, 2017); whereas, their malfunction results in aberrant neuronal excitability, leading to epilepsy (Barker-Haliski and White, 2015). In addition, astrocytes can uptake misfolded toxic protein aggregates including huntingtin (Shin et al., 2005), amyloid-β (Pihlaja et al., 2008; Xiao et al., 2014), and α-syn (Lee et al., 2010; Bliederhaeuser et al., 2016; Loria et al., 2017). In this study, we observed astrocyte-mediated clearance of neuronal α-syn (Figs. 1, 2), revealing an important role of astrocytes in regulation of neuronal α-syn. Interestingly, the astrocytic uptake was decreased by the presence of ATP13A2 deficiency (Fig. 3C,D), leading to increased α-syn levels in DA neurons (Fig. 2D,F). Astrocyte-mediated neuroprotection was also recently observed in another PD-associated mutation, LRRK2 G2019S (di Domenico et al., 2019). In addition, the astrocyte-specific overexpression of the redox-sensitive transcriptional factor erythroid 1 related factor 2 (Nrf2) leads to decreased α-syn levels in neurons from mice expressing mutant α-synA53T (Gan et al., 2012). These results suggest a significant involvement of astrocytes in the regulation of α-syn in PD. Of note, astrocytes that uptake mutant proteins may also become reactive astrocytes, which exhibit neurotoxicity (Shin et al., 2005) or induce inflammation (Lee et al., 2010). Indeed, a lack of ATP13A2 induces activation of nod‐like receptor protein 3 (NLRP3) inflammasome to produce excess IL‐1β from astrocytes, whereas the overexpression of ATP13A2 can reverse it (Qiao et al., 2016). However, our results favor a neuroprotective function for astrocytes through the removal of α-syn, as astrocyte-mediated α-syn clearance mitigates its accumulation in neurons, suggesting an important astrocytic regulation of neuronal protein homeostasis (Figs. 2, 3). Importantly, this neuroprotective α-syn clearance is reduced in ATP13A2-mutant astrocytes, contributing to α-syn accumulation in DA neurons (Fig. 2D,F).
We found that α-syn was abundantly expressed in neurons, but rarely in astrocytes (Fig. 1), which is consistent with previous reports (Zhang et al., 2014). In line with previous studies (Loria et al., 2017), we found that phagocytosis and the endosomal pathway were more active in astrocytes compared with neurons. Indeed, internalization and recycling of α-syn were more active in astrocytes compared with neurons (Fig. 3C,D), but were reduced in both cell types in the presence of ATP13A2 deficiency (Fig. 3C,D). However, α-syn internalization was still more efficient in mutant astrocytes compared with neurons, indicating that extracellular α-syn is more likely taken up by astrocytes even in the presence of ATP13A2 mutations. While α-syn is degraded through multiple pathways (Wong and Krainc, 2017), recent evidence emphasized the importance of lysosomal degradation of α-syn in neurons as defects in lysosomal hydrolases have been linked to several genetic forms of Parkinson's disease (Ramirez et al., 2006; Mazzulli et al., 2011). We found that lysosomal degradation was more active in astrocytes compared with neurons (Fig. 3E–I), suggesting that astrocytes have better capacity to digest lipids, amino acids, complex sugars, and proteins including α-syn (Loria et al., 2017). Interestingly, α-syn accumulation was observed in astrocytes in patients with sporadic PD, but with a lower frequency compared with neurons, possibly because of higher proteolysis in astrocytes (Wakabayashi et al., 2000). Moreover, PD-associated mutations in GBA1 also resulted in decreased lysosomal function in iPSC-derived astrocytes, raising the possibility that dysfunctional astrocytic proteolysis contributes to neuronal α-syn accumulation in PD (Aflaki et al., 2020).
Recent studies showed that α-syn can be transmitted from neuron to neuron, and α-syn pathology may spread in a “prion-like” manner (Guo and Lee, 2014), but the role of astrocytic ATP13A2 in modulating this transmission was previously not known. While the precise propagation mechanisms remain to be determined, initial studies in cultured cells demonstrated that α-syn can be transmitted via exosome secretion (Danzer et al., 2012), whereas later studies using microfluidic chambers revealed that α-syn can be released from axon terminals, suggesting synaptic propagation (Brahic et al., 2016; Mao et al., 2016). Studies in mouse brains have further documented that α-syn first spreads to anatomically connected lesions, and subsequently to lesions without direct connections, suggesting that both forms of transmissions may occur but at different rates (Luk et al., 2012). While some studies proposed direct transfer via tunnel nanotubes (Rostami et al., 2017), most previous results suggest that α-syn can be released and transmitted by multiple pathways including exosomal secretion and lysosomal exocytosis (Tsunemi et al., 2014, 2019). Our coculturing experiments demonstrated that astrocytes play a protective role in α-syn propagation by preventing indirect α-syn transfer, consistent with the previous report (Loria et al., 2017; Fig. 5A–D). We found that ATP13A2-deficiency impaired this protective role of astrocytes, leading to increased α-syn transfer between neurons (Fig. 5E,F).
In conclusion, using iPSC-derived astrocytes and neurons, we showed that intraneuronal α-syn levels were, at least in part, regulated by astrocytic uptake and lysosomal degradation. Impairment of these pathways by familial PD-associated ATP13A2 mutations contributed to increased accumulation and propagation of α-syn in DA neurons. Although patients with ATP13A2 mutations present with parkinsonism, supranuclear gaze palsy, and dementia, they also respond well to treatment with levodopa, suggesting a degeneration of nigral DA neurons that is typically seen in PD. In addition, Atp13a2 mouse models and DA neurons differentiated from patient-derived iPSCs all demonstrated increased levels of α-syn, as commonly seen in PD (Schultheis et al., 2013; Tsunemi et al., 2019). Importantly, cognitive decline and dementia develop in most patients with sporadic PD and in patients with genetic forms of PD (Kalia and Lang, 2015), and patients with sporadic PD have been also shown to have altered ATP13A2 in the nigra (Ramirez et al., 2006; Dehay et al., 2012; Ramonet et al., 2012; Murphy et al., 2013), suggesting that our studies of ATP13A2 may be more broadly informative for PD and other synucleinopathies. Therefore, targeting astrocytic clearance of α-syn may be an important therapeutic strategy for ameliorating α-syn-mediated pathology not only for KRS but also other synucleinopathies.
Footnotes
D.K. is the Founder and Scientific Advisory Board Chair of Lysosomal Therapeutics Inc. and Vanqua Bio; serves on the scientific advisory boards of The Silverstein Foundation, Intellia Therapeutics, and Prevail Therapeutics; and is a Venture Partner at OrbiMed. The authors declare no other competing financial interests.
This work is supported by National Institutes of Health Grant R37-NS-096241 (to D.K.); Japan Society for the Promotion of Science KAKENHI Grant 18K07510 and Juntendo University Research Institute for Environmental and Gender-Specific Medicine (to T.T.); Strategic Research Foundation Grant-in-Aid for Private Universities (to T.T., Y.I., K.I., S.S., W.A., and N.H.); and the Rare/Intractable Disease Project of the Japan from AMED Grants JP17ek0109244 and JP20ek0109429 (to T.T., K.I., S.S., W.A., and N.H.).
References
- Aflaki E, Stubblefield BK, McGlinchey RP, McMahon B, Ory DS, Sidransky E (2020) A characterization of Gaucher iPS-derived astrocytes: potential implications for Parkinson's disease. Neurobiol Dis 134:104647 10.1016/j.nbd.2019.104647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Bader MD, Malatiali SA, Redzic ZB (2011) Expression of estrogen receptor alpha and beta in rat astrocytes in primary culture: effects of hypoxia and glucose deprivation. Physiol Res 60:951–960. 10.33549/physiolres.932167 [DOI] [PubMed] [Google Scholar]
- Barker-Haliski M, White HS (2015) Glutamatergic Mechanisms Associated with Seizures and Epilepsy. Cold Spring Harb Perspect Med 5:a022863 10.1101/cshperspect.a022863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti PJ (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14:724–738. 10.1016/j.cmet.2011.08.016 [DOI] [PubMed] [Google Scholar]
- Ben Haim L, Rowitch DH (2017) Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci 18:31–41. 10.1038/nrn.2016.159 [DOI] [PubMed] [Google Scholar]
- Bento CF, Ashkenazi A, Jimenez-Sanchez M, Rubinsztein DC (2016) The Parkinson's disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat Commun 7:11803 10.1038/ncomms11803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliederhaeuser C, Grozdanov V, Speidel A, Zondler L, Ruf WP, Bayer H, Kiechle M, Feiler MS, Freischmidt A, Brenner D, Witting A, Hengerer B, Fändrich M, Ludolph AC, Weishaupt JH, Gillardon F, Danzer KM (2016) Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol 131:379–391. 10.1007/s00401-015-1504-2 [DOI] [PubMed] [Google Scholar]
- Brahic M, Bousset L, Bieri G, Melki R, Gitler AD (2016) Axonal transport and secretion of fibrillar forms of α-synuclein, Aβ42 peptide and HTTExon 1. Acta Neuropathol 131:539–548. 10.1007/s00401-016-1538-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin M-C, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A (2004) α-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364:1167–1169. 10.1016/S0140-6736(04)17103-1 [DOI] [PubMed] [Google Scholar]
- Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ (2012) Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener 7:42 10.1186/1750-1326-7-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E (2012) Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 109:9611–9616. 10.1073/pnas.1112368109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Domenico A, Carola G, Calatayud C, Pons-Espinal M, Muñoz JP, Richaud-Patin Y, Fernandez-Carasa I, Gut M, Faella A, Parameswaran J, Soriano J, Ferrer I, Tolosa E, Zorzano A, Cuervo AM, Raya A, Consiglio A (2019) Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson's disease. Stem Cell Rep 12:213–229. 10.1016/j.stemcr.2018.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini A, Gennarelli M, Russo I (2019) α-synuclein and glia in Parkinson's disease: a beneficial or a detrimental duet for the endo-lysosomal system? Cell Mol Neurobiol 39:161–168. 10.1007/s10571-019-00649-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Vargas MR, Johnson DA, Johnson JA (2012) Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J Neurosci 32:17775–17787. 10.1523/JNEUROSCI.3049-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Lee VM (2014) Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 20:130–138. 10.1038/nm.3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Pollak P, Agid Y, Dürr A, Brice A (2004) Causal relation between α-synuclein locus duplication and familial Parkinson's disease. Lancet 364:1169–1171. 10.1016/S0140-6736(04)17104-3 [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G (2006) Culturing hippocampal neurons. Nat Protoc 1:2406–2415. 10.1038/nprot.2006.356 [DOI] [PubMed] [Google Scholar]
- Kalia LV, Lang AE (2015) Parkinson's disease. Lancet 386:896–912. 10.1016/S0140-6736(14)61393-3 [DOI] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM (2009) Methods to monitor chaperone-mediated autophagy. Methods Enzymol 452:297–324. 10.1016/S0076-6879(08)03619-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kett LR, Stiller B, Bernath MM, Tasset I, Blesa J, Jackson-Lewis V, Chan RB, Zhou B, Di Paolo G, Przedborski S, Cuervo AM, Dauer WT (2015) α-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J Neurosci 35:5724–5742. 10.1523/JNEUROSCI.0632-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, Farghaian H, Cole AR, Lay PA, Sue CM, Cooper AA (2014) Parkinson's disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-synuclein externalization via exosomes. Hum Mol Genet 23:2816–2833. 10.1093/hmg/ddu099 [DOI] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ (2010) Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 285:9262–9272. 10.1074/jbc.M109.081125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes da Fonseca T, Pinho R, Outeiro TF (2016) A familial ATP13A2 mutation enhances alpha-synuclein aggregation and promotes cell death. Hum Mol Genet 25:2959–2971. 10.1093/hmg/ddw147 [DOI] [PubMed] [Google Scholar]
- Loria F, Vargas JY, Bousset L, Syan S, Salles A, Melki R, Zurzolo C (2017) α-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol 134:789–808. 10.1007/s00401-017-1746-2 [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM (2012) Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. 10.1126/science.1227157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Ou MT, Karuppagounder SS, Kam T-I, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin J-H, Kang HC, Zhang J, Xu J, Chen R, Park H, Andrabi SA, Kang SU, Goncalves RA, Liang Y, Zhang S, Qi C, Lam S, Keiler JA, Tyson J, Kim D, Panicker N, Yun SP, Workman CJ, Vignali DAA, Dawson VL, Ko HS, Dawson TM (2016) Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353:aah3374 10.1126/science.aah3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D (2011) Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146:37–52. 10.1016/j.cell.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D (2016) α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A 113:1931–1936. 10.1073/pnas.1520335113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meberg PJ, Miller MW (2003) Culturing hippocampal and cortical neurons. Methods Cell Biol 71:111–127. 10.1016/s0091-679x(03)01007-0 [DOI] [PubMed] [Google Scholar]
- Murphy KE, Cottle L, Gysbers AM, Cooper AA, Halliday GM (2013) ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol Commun 1:11 10.1186/2051-5960-1-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlaja R, Koistinaho J, Malm T, Sikkilä H, Vainio S, Koistinaho M (2008) Transplanted astrocytes internalize deposited β-amyloid peptides in a transgenic mouse model of Alzheimer's disease. Glia 56:154–163. 10.1002/glia.20599 [DOI] [PubMed] [Google Scholar]
- Qiao C, Yin N, Gu HY, Zhu JL, Ding JH, Lu M, Hu G (2016) Atp13a2 deficiency aggravates astrocyte-mediated neuroinflammation via NLRP3 inflammasome activation. CNS Neurosci Ther 22:451–460. 10.1111/cns.12514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38:1184–1191. 10.1038/ng1884 [DOI] [PubMed] [Google Scholar]
- Ramonet D, Podhajska A, Stafa K, Sonnay S, Trancikova A, Tsika E, Pletnikova O, Troncoso JC, Glauser L, Moore DJ (2012) PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum Mol Genet 21:1725–1743. 10.1093/hmg/ddr606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostami J, Holmqvist S, Lindström V, Sigvardson J, Westermark GT, Ingelsson M, Bergström J, Roybon L, Erlandsson A (2017) Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J Neurosci 37:11835–11853. 10.1523/JNEUROSCI.0983-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, Dickson DW, Mazzulli JR, Bardgett ME, Haik KL, Ekhator O, Chava AK, Howard J, Gannon M, Hoffman E, Chen Y, Prasad V, Linn SC, Tamargo RJ, Westbroek W, et al. (2013) Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited α-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet 22:2067–2082. 10.1093/hmg/ddt057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ (2005) Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol 171:1001–1012. 10.1083/jcb.200508072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. (2003) α-Synuclein locus triplication causes Parkinson's disease. Science 302:841 10.1126/science.1090278 [DOI] [PubMed] [Google Scholar]
- Tremblay ME, Cookson MR, Civiero L (2019) Glial phagocytic clearance in Parkinson's disease. Mol Neurodegener 14:16. 10.1186/s13024-019-0314-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Krainc D (2014) Zn(2)(+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet 23:2791–2801. 10.1093/hmg/ddt572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Hamada K, Krainc D (2014) ATP13A2/PARK9 regulates secretion of exosomes and α-synuclein. J Neurosci 34:15281–15287. 10.1523/JNEUROSCI.1629-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Perez-Rosello T, Ishiguro Y, Yoroisaka A, Jeon S, Hamada K, Rammonhan M, Wong YC, Xie Z, Akamatsu W, Mazzulli JR, Surmeier DJ, Hattori N, Krainc D (2019) Increased lysosomal exocytosis induced by lysosomal Ca(2+) channel agonists protects human dopaminergic neurons from α-synuclein toxicity. J Neurosci 39:5760–5772. 10.1523/JNEUROSCI.3085-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D (2012) Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci 32:4240–4246. 10.1523/JNEUROSCI.5575-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veen S, Martin S, Van den Haute C, Benoy V, Lyons J, Vanhoutte R, Kahler JP, Decuypere JP, Gelders G, Lambie E, Zielich J, Swinnen JV, Annaert W, Agostinis P, Ghesquière B, Verhelst S, Baekelandt V, Eggermont J, Vangheluwe P (2020) ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 578:419–424. 10.1038/s41586-020-1968-7 [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Hayashi S, Yoshimoto M, Kudo H, Takahashi H (2000) NACP/α-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson's disease brains. Acta Neuropathol 99:14–20. 10.1007/PL00007400 [DOI] [PubMed] [Google Scholar]
- Wong YC, Krainc D (2017) α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 23:1–13. 10.1038/nm.4269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Burchett JM, Schuler DR, Cirrito JR, Diwan A, Lee JM (2014) Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J Neurosci 34:9607–9620. 10.1523/JNEUROSCI.3788-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]