

Abstract
The second messenger cAMP is an important determinant of synaptic plasticity that is associated with enhanced neurotransmitter release. Long-term potentiation (LTP) at parallel fiber (PF)–Purkinje cell (PC) synapses depends on a Ca2+-induced increase in presynaptic cAMP that is mediated by Ca2+-sensitive adenylyl cyclases. However, the upstream signaling and the downstream targets of cAMP involved in these events remain poorly understood. It is unclear whether cAMP generated by β-adrenergic receptors (βARs) is required for PF–PC LTP, although noradrenergic varicosities are apposed in PF–PC contacts. Guanine nucleotide exchange proteins directly activated by cAMP [Epac proteins (Epac 1-2)] are alternative cAMP targets to protein kinase A (PKA) and Epac2 is abundant in the cerebellum. However, whether Epac proteins participate in PF–PC LTP is not known. Immunoelectron microscopy demonstrated that βARs are expressed in PF boutons. Moreover, activation of these receptors through their agonist isoproterenol potentiated synaptic transmission in cerebellar slices from mice of either sex, an effect that was insensitive to the PKA inhibitors (H-89, KT270) but that was blocked by the Epac inhibitor ESI 05. Interestingly, prior activation of these βARs occluded PF–PC LTP, while the β1AR antagonist metoprolol blocked PF–PC LTP, which was also absent in Epac2−/− mice. PF–PC LTP is associated with an increase in the size of the readily releasable pool (RRP) of synaptic vesicles, consistent with the isoproterenol-induced increase in vesicle docking in cerebellar slices. Thus, the βAR-mediated modulation of the release machinery and the subsequent increase in the size of the RRP contributes to PF–PC LTP.
SIGNIFICANCE STATEMENT G-protein-coupled receptors modulate the release machinery, causing long-lasting changes in synaptic transmission that influence synaptic plasticity. Nevertheless, the mechanisms underlying synaptic responses to β-adrenergic receptor (βAR) activation remain poorly understood. An increase in the number of synaptic vesicles primed for exocytosis accounts for the potentiation of neurotransmitter release driven by βARs. This effect is not mediated by the canonical protein kinase A pathway but rather, through direct activation of the guanine nucleotide exchange protein Epac by cAMP. Interestingly, this βAR signaling via Epac is involved in long term potentiation at cerebellar granule cell-to-Purkinje cell synapses. Thus, the pharmacological activation of βARs modulates synaptic plasticity and opens therapeutic opportunities to control this phenomenon.
Keywords: adrenergic receptors, Epac2 KO, neurotransmitter release, parallel fiber LTP, release machinery, RRP size
Introduction
The second messenger cAMP is an important determinant of presynaptic function, with increases in cAMP potentiating glutamate release (Herrero and Sánchez-Prieto, 1996; Millán et al., 2002) and synaptic transmission (Chavez-Noriega and Stevens, 1994; Chen and Regehr, 1997; Sakaba and Neher, 2001; Kaneko and Takahashi, 2004). Presynaptic forms of synaptic plasticity are also driven by cAMP, which involves enhanced neurotransmitter release (Salin et al., 1996; Villacres et al., 1998; Castillo et al., 2002). The mechanism by which cAMP potentiates neurotransmitter release does not affect Ca2+ influx or the action potential waveforms (Chen and Regehr, 1997; Kaneko and Takahashi, 2004), yet it does modify the release machinery. The readily releasable pool (RRP) of synaptic vesicles (SVs) is made up of docked and primed vesicles, with these latter vesicles in a state where Ca2+ influx triggers their fusion. The availability of cAMP heightens the size and release probability (Pr) of the RRP (Chen and Regehr, 1997; Sakaba and Neher, 2001; Kaneko and Takahashi, 2004; Yao and Sakaba, 2010), although the source of this cAMP remains to be established. Significantly, β-adrenergic receptors (βARs) are expressed in axons that establish asymmetric glutamatergic synapses (Ferrero et al., 2013), and they can activate adenylyl cyclase and generate cAMP, which through its downstream signaling may in turn potentiate glutamate release (Herrero and Sánchez-Prieto, 1996; Millán et al., 2002; Ferrero et al., 2016), synaptic transmission, and plasticity (Huang et al., 1996; Huang and Hsu, 2006).
Long-term potentiation (LTP) at parallel fiber (PF)–Purkinje cell (PC) synapses depends on a Ca2+-induced increase in presynaptic cAMP (Salin et al., 1996), as mediated by Ca2+-sensitive adenylyl cyclases (Storm et al., 1998). However, the upstream and downstream signaling elements involved in this presynaptic form of plasticity are poorly understood. Indeed, while βARs participate in presynaptic and cAMP-dependent LTP in the cerebral cortex (Huang and Hsu, 2006), and noradrenergic varicosities are seen in apposition at PF–PC contacts (Abbott and Sotelo, 2000), it remains unclear whether these receptors are required for PF–PC LTP. The active zone (AZ) Rab-3-interacting protein (RIM1α) might represent a downstream target of cAMP-dependent protein kinase A (PKA) in PF–PC LTP (Linden and Ahn, 1999; Castillo et al., 2002; Lonart et al., 2003), although this is yet to be confirmed (Kaeser et al., 2008; Kintscher et al., 2013). By contrast, the guanine nucleotide exchange proteins directly activated by cAMP (Epac) proteins (Epac1-2) are alternative cAMP targets to PKA. Indeed, Epac 2 is abundant in the cerebellum (Kawasaki et al., 1998), and its deletion impairs presynaptic hippocampal mossy fiber cAMP-dependent LTP (Fernandes et al., 2015).
We found that βAR activation enhanced the amplitude of EPSCs at PF–PC synapses by a presynaptic mechanism that diminished the paired-pulse ratio (PPR), while increasing the frequency of asynchronous quantal events. These effects were consistent with the immunoelectron microscopy identification of β1ARs at PF boutons. βAR synaptic potentiation was insensitive to PKA inhibitors (H-89, KT270), yet it was blocked by the Epac inhibitor ESI 05 and was mimicked by the Epac activator 8pCPT. Moreover, this potentiation was associated with an increase in SV docking. Interestingly, presynaptic PF–PC LTP was associated with an increase in the size of the RRP of SVs, while it was absent in the presence of the βAR1 antagonist metoprolol and in cerebellar slices from Epac2-null mice. Thus, βARs appear to activate a physiologically relevant signaling pathway that involves cAMP- and Epac2-dependent modulation of the SV release machinery required for presynaptic PF LTP.
Materials and Methods
Electrophysiology
All procedures for the handling of animals were performed in accordance with European Commission guidelines (2010/63/EU), and they were approved by the Animal Research Committee at the Complutense University. Wild-type (WT) and Epac2 knock-out (KO) mice (22–30 d of age), of either sex, were anesthetized with isoflurane (1.5–2% in a mixture of 80% synthetic air/20% oxygen), and they were then killed by decapitation. Parasagittal vibratome slices of the cerebellum (325 µm thick; model VT 1200S, Leica) were maintained in ice-cold Ringer's solution, and recordings at PF–PC synapses were obtained as described previously (Salin et al., 1996; Castillo et al., 2002). The slices were kept in a holding chamber containing Ringer's solution for at least 1 h and then transferred to a superfusion chamber to record in superfusion medium equilibrated with 95% O2/5% CO2 and containing the following: 119 mm NaCl, 2.5 mm KCl, 1.3 mm MgSO4, 2.5 mm CaCl2, 26 mm NaHCO3, 1 mm NaH2PO4, 10 mm glucose, and 100 μm picrotoxin (to block GABAA receptors). In some experiments, 0.5 mm ascorbic acid was added to the superfusion medium to avoid the oxidation of norepinephrine. The flow rate was established at 1 ml/min and recordings were obtained at 25°C maintained using a temperature controller (model TC-324C, Warner Instruments).
Theta capillaries (2–5 µm tip) filled with Ringer's solution were used for bipolar stimulation. The electrodes were connected to a stimulator (model S38, GRASS) through an isolation unit and placed near the pial surface of the molecular layer to stimulate the PF input to PCs. Stimuli (<50 µA, 100 µs) were delivered at 0.05 Hz, with paired pulses administered 80 ms apart to obtain the PPR as EPSC2/EPSC1.
Whole-cell recordings from individual PCs were obtained with a PC-ONE amplifier (Dagan Corporation) under voltage-clamp conditions, and the membrane potential was held at −70 mV to record glutamatergic-evoked EPSCs. Signals were fed to a Pentium-based PC through a DigiData1322A interface board (Axon Instruments), and the pCLAMP version 10.2 software (Molecular Devices) was used to generate the stimuli, as well as for data display, acquisition, storage, and analysis. Patch pipettes were pulled from thin-walled borosilicate glass capillary tubes (outer diameter, 1.5 mm) on a P-97 puller (Sutter Instrument), and the pipettes (3–4 MΩ) were filled with an internal solution containing the following: 122.5 mm cesium gluconate, 10 mm HEPES, 10 mm BAPTA, 2 mm Mg-ATP, 8 mm NaCl, and 5 mm QX-314-Br, pH 7.3, adjusted with CsOH to an osmolarity of 290 mOsm. Series and input resistances were monitored throughout the experiment using a −5 mV pulse, and the recordings were considered stable when the series and input resistances, resting membrane potential, and stimulus artifact duration did not change >20%. Cells that did not meet these criteria were discarded in all of the experiments. To avoid irreversible effects of agonists/antagonist/inhibitors or LTP protocols, only one neuron per slice was analyzed.
The presence of the Ca2+ chelator BAPTA in the recording pipette (Safo and Regehr, 2005) may prevent the generation of endocannabinoids by PCs (Kreitzer and Regehr, 2001; Maejima et al., 2001) and the postsynaptic PC LTP mediated by cAMP, also impeding the Epac-dependent increase in GluA3-containing AMPA receptor (AMPAR) permeability (Gutierrez-Castellanos et al., 2017).
To measure PF asynchronous EPSCs (aEPSCs), CaCl2 was replaced with 2.5 mm SrCl2, the stimuli were adjusted to yield a EPSC amplitude between 150 and 250 pA, and they were delivered every 20 s. Asynchronous release associated with each stimulus was estimated after 20 ms and over 500 ms. The asynchronous events associated with the last six stimuli of a 5 min period were quantified in the basal condition and after 10 min in the presence of isoproterenol (100 μm).
To induce LTP, a tetanic train of 100 stimuli was delivered at 10 Hz at least 15 min after the beginning of the recording to allow sufficient time for the diffusion of BAPTA into the dendritic tree. The baseline PF-evoked EPSC amplitude was maintained at <300 pA to avoid sodium spikes that escaped the voltage clamp, particularly during and after tetanization. The size of the RRP was estimated as described previously (Schneggenburger et al., 1999; Gioia et al., 2016). To minimize the variation in these estimates, the stimulus intensity before LTP induction was adjusted to yield EPSC amplitudes between 150 and 200 pA. Thus, 30 min after applying the 10 Hz train to induce presynaptic LTP, a further tetanic train was applied (100 stimuli at 40 Hz). The cumulative EPSC amplitudes during this train were plotted, and the y-intercept extended from the linear part of the curve (over times >1.5 s when the cumulative amplitude curve reached steady state), and it was used to estimate the RRP size. Moreover, the slope of this fitted line was used to estimate the RRP replenishment rate (all of these data were plotted and fitted using OriginLab 8 software). Finally, the mean probability of vesicle release (pves) was obtained by dividing the average of six consecutive EPSCs before the 40 Hz train by the estimated RRP size in each cell.
Immunoelectron microscopy
Immunohistochemistry for electron microscopy (EM) was conducted using the pre-embedding Immunogold method, as described previously (Luján et al., 1996). Three adult C57BL/6 mice [postnatal day 60 (P60)] were anaesthetized and perfused transcardially with ice-cold fixative containing 4% paraformaldehyde, 0.05% glutaraldehyde, and 15% (v/v) saturated picric acid diluted to 0.1 m phosphate buffer (PB), pH 7.4. After perfusion, the brain of the animal was removed, washed thoroughly in 0.1 m PB, and 60-μm-thick coronal vibratome sections were obtained (model V1000, Leica). The free-floating sections were incubated in 10% (v/v) normal goat serum (NGS) diluted in Tris-buffered saline (TBS) and then with goat β1AR antibodies (Sigma-Aldrich) at a final protein concentration of 3–5 μg/ml in TBS containing 1% (v/v) NGS. After several washes in TBS, the sections were incubated with 1.4 nm gold coupled rabbit anti-goat IgG (Nanoprobes). The sections were postfixed in 1% (v/v) glutaraldehyde and washed in double-distilled water before performing silver enhancement of the gold particles with an HQ Silver Kit (Nanoprobes). The sections were then treated with osmium tetroxide (1% in 0.1 m PB), block-stained with uranyl acetate, dehydrated in a graded series of ethanol and flat-embedded on glass slides in Durcupan (Fluka) resin. Regions of interest were cut at a thickness of 70–90 nm on an ultramicrotome (Reichert Ultracut E, Leica) and collected on single-slot Pioloform-coated copper grids. Staining was performed using drops of 1% aqueous uranyl acetate followed by Reynolds's lead citrate stain, and the ultrastructure of the tissue was analyzed on a JEM 1010 electron microscope (JEOL).
Quantification of adrenergic receptors
To establish the relative abundance of the β1AR subunits in the molecular layer of the cerebellum, we quantified their immunolabeling in 60 μm coronal sections processed for pre-embedding Immunogold immunohistochemistry following a procedure similar to that used previously (Luján et al., 1996). Briefly, three tissue samples were obtained from three different animals and embedded in blocks. To minimize false negatives, ultrathin sections were cut close to the surface of each block. We estimated the quality of immunolabeling by selecting areas with optimal gold labeling at approximately the same distance from the cut surface. Randomly selected areas were then photographed from the selected ultrathin sections at a final magnification of 45,000×, and the Immunogold labeling was quantified in sampling areas of each cortex totaling ∼1500 μm2. Immunoparticles for individual β1AR subunits at the plasma membrane of axon terminals were counted in each sampling area, analyzing the neuropile of the molecular layer containing postsynaptic and presynaptic compartments. In terms of the presynaptic compartments, only axon terminals (PF terminals) establishing synaptic contacts with dendritic spines of PCs were analyzed. A total of 285 gold particles were counted at the axon terminals immunostained for β1ARs. Subsequently, the proportion of immunoparticles for the β1AR subunits at the AZ and in the extrasynaptic plasma membrane of axon terminals was calculated, as well as the proportion of postsynaptic versus presynaptic elements stained for β1AR.
Controls
To determine the specificity of the methods used in the immunoelectron microscopy studies, the primary antibody was either omitted or replaced with 5% (v/v) of normal serum from the species of the primary antibody. No specific labeling was observed in these conditions. Labeling was also compared with that obtained for calretinin and calbindin, and only antibodies against β1AR consistently labeled the plasma membrane.
Synaptic vesicle distribution by electron microscopy
Parasagittal slices (325 µm thick) from the cerebellum were obtained as described above for the electrophysiology experiments and transferred to an immersion recording chamber. The slices were superfused at 1 ml/min with gassed Ringer's solution containing picrotoxin (100 μm), and, in some cases, the slices were also treated for 10 min with isoproterenol (100 μm). The slices from WT and Epac2 KO mice were fixed immediately afterward by immersion for 45 min at 37°C in 3.5% glutaraldehyde in 0.1 m PB, pH 7.4, and they were then left in glutaraldehyde solution for 30 min at room temperature before storing them for 20 h at 4°C. The slices were then rinsed six times with large volumes of 0.1 m PB, and the cerebellum was dissected out and postfixed for 1 h at room temperature in 1% OsO4-1.5% K3Fe(CN)6. After dehydrating through a graded series of ethanol (30, 50, 70, 80, 90, 95, and 100%), the samples were embedded using the SPURR embedding kit (TAAB). Ultrathin ultramicrotome sections (70–80 nm thick; model EM UC6 Ultramicrotome, Leica Microsystems) were routinely stained with uranyl acetate and lead citrate, and images were obtained on a JEM 1010 transmission electron microscope (JEOL). Randomly chosen areas of the cerebellar molecular layer were photographed at 80,000× magnification, and only asymmetric synapses with clearly identifiable electron-dense postsynaptic densities were analyzed with ImageJ software. The total number of SVs per synaptic terminal was determined, and the distance between the outer layer of the vesicle and the inner layer of the AZ was measured. These data were distributed in 10 nm bins, and the SVs up to a maximum of 10 nm from the AZ were considered as docked vesicles. The SV data were also were distributed in 5 nm bins to distinguish fully primed and tightly docked SVs (0–5 nm) from loosely docked SVs (5–10 nm). All the data were analyzed blind to the genotype and treatment.
Experimental design and statistical analysis
The data were analyzed using Instat or OriginPro 8.0 software, applying an unpaired two-tailed Student's t test. When the variances of the populations were differed significantly, a Welch's test was applied, and, when more than two groups were compared, an ANOVA with a Bonferroni's post hoc test was used. The data are represented as the mean ± SEM: *p < 0.05, **p < 0.01, ***p < 0.001. Differences were considered statistically significant at p < 0.05 with a confidence limit of 95%.
Results
Norepinephrine and isoproterenol potentiate synaptic transmission at PF–PC synapses through a presynaptic mechanism involving β1ARs
Noradrenergic innervation of the cerebellum is important to modulate its activity, and, as such, noradrenergic fibers are distributed diffusely throughout the cerebellar cortex. Indeed, noradrenergic varicosities are opposed to dendritic profiles that largely belong to PCs (Abbott and Sotelo, 2000). When we tested whether noradrenaline modulates synaptic transmission at PF–PC synapses, we found it increased the EPSC amplitude at these synapses from a basal state of 100.9 ± 3.4% to 194.1 ± 11.2% (n = 10; p < 0.0001, Welch's test; Fig. 1A,B). This potentiation was prevented by the specific β1AR antagonist metoprolol (from 97.9 ± 7.7 to 101.9 ± 4.1%; n = 4; p = 0.6627, Student's t test; Fig. 1A,B). The potentiation of EPSC amplitudes by norepinephrine is associated with a decrease in the EPSC2/EPSC1 PPR from a value of 1.72 ± 0.05 under basal conditions to 1.41 ± 0.02 on exposure to norepinephrine (p = 0.0001, Welch's test; Fig. 1C,D). Hence, the noradrenaline-induced increase in EPSC amplitude is the result of presynaptic β1AR activation, which enhances glutamate release.
Figure 1.

Norepinephrine potentiates synaptic transmission at PF–PC synapses. EPSCs were recorded from Purkinje cells after 0.05 Hz parallel fiber stimulation. A, Norepinephrine (10 min, 100 μm) induced a sustained increase in the EPSC amplitude that was sensitive to the β1AR antagonist metoprolol (60 μm, 30 min). B, Quantification of the changes in EPSC amplitude. The data were measured 40 min after norepinephrine application (2) in the absence (n = 10 cells/10 slices/5 mice, t = 7.963, df = 10) or the presence of metoprolol (n = 4 cells/4 slices/2 mice, t = 0.4585, df = 6) compared with the values before norepinephrine application (1). C, Changes in the PPR (EPSC2/EPSC1) induced by norepinephrine in the different conditions, quantified 40 min after treatment and relative to the respective basal values. D, Quantification of the changes in the PPR induced by norepinephrine (2) in the absence (t = 7.757, df = 11) or presence of metoprolol (t = 0.6868, df = 3). EPSC sample traces (A, C) represent the mean of six consecutive EPSCs at 0.05 Hz taken before (1) and 40 min after (2) treatment. Calibration: 50 pA and 15 ms. These data represent the mean ± SEM.
The βAR agonist isoproterenol also increases the amplitude of EPSCs at PF–PC synapses (from a basal state of 101.9 ± 4.2% to 202.6 ± 13.6%; n = 13; p < 0.0001, Welch's test; Fig. 2A,B), and this potentiation was prevented by the specific β1AR antagonist metoprolol (from 99.7 ± 5.9% to 96.1 ± 5.6%; n = 11; p = 0.6628, Student's t test; Fig. 2A,B). To exclude the activation of postsynaptic βARs (Lippiello et al., 2015), we substituted GTP for the nonhydrolyzable GTPγS in the recording pipette, although this did not prevent isoproterenol from increasing the EPSC amplitudes (from 104.4 ± 4.1% to 187.1 ± 13.2%; n = 11; p < 0.0001, Welch's test; Fig. 2A,B). Thus, the enhancement to EPSC amplitudes provoked by isoproterenol is not mediated by the activation of postsynaptic βARs at PF–PC synapses. This potentiation of the EPSC amplitudes by isoproterenol is associated with a decrease in the EPSC2/EPSC1 PPR from a basal value of 1.71 ± 0.09 to 1.38 ± 0.05; p = 0.0049, Welch's test; Fig. 2C,D). Hence, the isoproterenol-induced increase of EPSC amplitude is the result of presynaptic β1AR activation enhancing glutamate release.
Figure 2.

Isoproterenol potentiates synaptic transmission at PF–PC synapses by a presynaptic mechanism involving β1-adrenergic receptors. EPSCs were recorded from Purkinje cells after 0.05 Hz parallel fiber stimulation. A, The βAR agonist isoproterenol (100 μm, 10 min) induced a sustained increase in the EPSC amplitude that was sensitive to the specific β1AR antagonist metoprolol (60 μm, 30 min), yet was insensitive to the presence of GTPγS (1 mm, 15 min) in the recording pipette. B, Quantification of the changes in EPSC amplitude induced by isoproterenol. The data were measured 40 min after isoproterenol application (2) in the absence (n = 13 cells/13 slices, 6 mice, t = 7.075, df = 14) or the presence (n = 11 cells/11 slices/5 mice, t = 0.4426, df = 20) of metoprolol or GTP-γ-S (n = 11 cells/11slices/5 mice, t = 5.983, df = 11), and relative to the values before isoproterenol application (1). C, Changes in the PPR (EPSC2/EPSC1) induced by isoproterenol in the different conditions. D, Quantification of the changes in the PPR induced by isoproterenol (2) in the absence (t = 3.205, df = 18) and presence (t = 0.53, df = 20) of GTPγS (t = 3.221, df = 11) or metoprolol, and relative to the respective basal values (1). EPSC sample traces (A, C) represent the mean of six consecutive EPSCs at 0.05 Hz taken before and 40 min after treatment. Calibration: 50 pA and 15 ms. The data represent the mean ± SEM.
Subcellular localization of β1ARs in the presynaptic compartments of the cerebellum
The distribution of β1ARs in the presynaptic compartment of PF–PC synapses was assessed in the cerebellar molecular layer by examining the immunoparticles detected in electron micrographs. β1AR immunoparticles were observed along the plasma membrane (arrowheads) of PF terminals (Fig. 3A–C, pf), establishing excitatory synapses with dendritic spines (Fig. 3A–C, s) on PCs. Less often, immunoparticles for β1AR were also observed at postsynaptic sites along the plasma membrane (arrows) of PC dendritic spines (Fig. 3A–C, s) and shafts (Fig. 3A–C, Den). The distribution of β1ARs was also studied in electron micrographs of the molecular layer of the cerebellum stained for tyrosine hydroxylase (TH) using an immunoperoxidase reaction. β1ARs were observed along the plasma membrane (Fig. 3D,E, arrowheads) of PF terminals (Fig. 3D,E, pf), always close to fibers immunostained for TH (Fig. 3D,E, white asterisks). Quantification of the β1AR immunoparticles (n = 502 from three preparations) in the molecular layer of the cerebellum indicates that they were more frequent in presynaptic compartments (57.0 ± 0.9%; p = 0.0004, Student's t test), and that they were more frequent at the AZ of axon terminals (66.4 ± 1.1%; p < 0.0001, Student's t test) than at extrasynaptic sites (33.6 ± 1.1%; Fig. 3F). Thus, β1ARs are clearly present in PF–PC synapses.
Figure 3.

Subcellular localization of the β1-adrenergic receptors in the presynaptic compartments of the cerebellum. A–C, Electron micrographs of the molecular layer of the cerebellum showing pre-embedding Immunogold staining for β1AR. Immunoparticles for β1AR were observed along the plasma membrane (arrowheads) of parallel fiber terminals (pf) establishing excitatory synapses with dendritic spines (s) of PCs. Less frequently, immunoparticles for β1AR were also observed at postsynaptic sites along the plasma membrane (arrows) of dendritic spines (s) and the shafts (Den) of PCs. D, E, Electron micrographs of the molecular layer of the cerebellum showing β1AR immunoparticles and the immunoperoxidase reaction for TH detected using a dual-labeling pre-embedding method. β1AR immunoparticles were observed along the plasma membrane (arrowheads) of pf terminals, always close to fibers immunopositive for TH (filled with the peroxidase reaction end product, white asterisks). F, Quantitative analysis showing the percentage of β1AR immunoparticles in the molecular layer of the cerebellum. Immunoparticles (502) for β1ARs were more frequently observed in presynaptic compartments (57.0 ± 0.9%) and within axon terminals than in the postsynaptic compartments (43.0 ± 0.9%, t = 10.999, df = 4). Moreover, they were more frequently found in the active zone (66.4 ± 1.1%, t = 21.085, df = 4) than at extrasynaptic sites (33.6 ± 1.1%). Scale bars: A, D, E, 500 nm; B, C, 200 nm. The data represent the mean ± SEM.
Isoproterenol potentiation of synaptic transmission at PF–PC synapses involves Epac rather than PKA-dependent signaling
The downstream elements involved in the cAMP signaling associated with βAR-induced potentiation remain controversial, involving either PKA-dependent mechanisms (Herrero and Sánchez-Prieto, 1996; Huang and Hsu, 2006) or PKA-independent mechanisms (Huang and Hsu, 2006; Gekel and Neher, 2008; Ferrero et al., 2013; Fernandes et al., 2015). In a set of experiments independent from those presented above (Fig. 2A,B), we showed that the isoproterenol-induced increase in EPSC amplitude at PF–PC synapses (from 100.8 ± 4.1% to 201.8 ± 15.4%; n = 11; p < 0.0001, Welch's test) was not altered by the PKA inhibitors H-89 (from 99.8 ± 4.4% to 206.5 ± 11.4%; n = 11; p = 0.0004, Welch's test) or KT5720 (from 98.9 ± 4.5% to 200.0 ± 7.7%; n = 11; p < 0.0001, Welch's test; Fig. 4A,B). Some presynaptic effects of βARs are mediated by cAMP acting through Epac proteins (Kaneko and Takahashi, 2004; Huang and Hsu, 2006; Gekel and Neher, 2008; Yang et al., 2012; Ferrero et al., 2013; Ramírez-Franco et al., 2014; Fernandes et al., 2015; Alonso et al., 2017). Indeed, when we assessed the effect of the selective Epac2 inhibitor ESI 05 that binds to the interface formed between the two unique cAMP binding domains of Epac2 (Tsalkova et al., 2012), there was a clear impairment of the isoproterenol-induced increase in EPSC amplitude (from 101.3 ± 4.8% to 99.3 ± 3.5%; n = 10; p = 0.7125, Welch's test; Fig. 4A,B). Conversely, the Epac activator 8pCPT potentiated synaptic transmission (from 104.3 ± 4.6% to 204.3 ± 12.7%; n = 12; p = 0.0003, Welch's test; Fig. 4A,B), which also involved a presynaptic mode of action as this potentiation was associated with a decrease in the EPSC2/EPSC1 PPR (basal: 1.61 ± 0.03 and 1.43 ± 0.03 after 8pCPT; n = 12; p = 0.0004, Student's t test; Fig. 4C,D). Thus, Epac proteins are downstream targets of cAMP in the potentiation of PF–PC synaptic transmission by βARs.
Figure 4.

Isoproterenol potentiation of synaptic transmission at PF–PC synapses involves Epac but not PKA-dependent signaling. A, The βAR agonist isoproterenol (100 μm, 10 min) induced a sustained increase in the EPSC amplitude that was insensitive to the PKA inhibitors H-89 (10 μm, 30 min) and KT-5720 (2 μm, 30 min). However, this response was absent in the presence of the Epac2 inhibitor ESI 05 (10 μm, 30 min), and it was mimicked by the Epac activator 8pCPT (50 μm, 10 min). B, Quantification of the changes in EPSC amplitude measured 40 min after isoproterenol/8pCPT application (2). Isoproterenol in the absence (n = 11 cells/11 slices, 5 mice, t = 6.344, df = 11) or in the presence of H-89 (n = 11 cells/11 slices/5 mice, t = 4.496, df = 11), KT-5720 (n = 11 cells/11slices/5 mice, t = 5.945, df = 11), or ESI 05 (n = 10 cells/10 slices/4 mice, t = 0.3760, df = 11), or 8pCPT alone (n = 12 cells/12 slices/5 mice, t = 4.850 df = 13). All the data were compared with the respective values before isoproterenol or 8pCPT application (1). C, Changes in the PPR (EPSC2/EPSC1) induced by isoproterenol or 8pCPT in the different conditions. D, Quantification of the changes in the PPR induced by 8pCPT (t = 4.230, df = 21) and isoproterenol (2) in the absence (t = 2.219, df = 20) or presence of H-89 (t = 4.941, df = 18), KT-5720 (t = 2.133, df = 8) or ESI 05 (t = 1.28, df = 18), and relative to the respective basal values (1). EPSC sample traces (A, C) represent the mean of six consecutive EPSCs at 0.05 Hz taken before and 40 min after treatment. Calibration: 50 pA and 15 ms. The data represent the mean ± SEM.
Isoproterenol and 8pCPT enhance the frequency but not the amplitude of asynchronous release events
The presynaptic origin of the isoproterenol-induced potentiation can be assessed by measuring the changes in the frequency but not the amplitude of miniature EPSCs (mEPSCs). However, as it is not possible to distinguish between PF-mEPSCs from climbing fiber mEPSCs (Carey and Regehr, 2009), we measured the amplitude and frequency of PF-mEPSCs in the presence of Sr2+, which induces an asynchronous release of vesicles (i.e., aEPSCs; Goda and Stevens, 1994; Xu-Friedman and Regehr, 1999; Carey and Regehr, 2009). Under these conditions, aEPSCs can be attributed to PF stimulation and in the presence of Sr2+, delayed aEPSCs were associated with each stimulus in addition to the evoked eEPSCs because of synchronous release. Isoproterenol increased eEPSC amplitude relative to the controls (207.7 ± 14.1 and 332.9 ± 24.6 pA; n = 10; p < 0.0006, Welch's test; Fig. 5A,B) and the aEPSC mean frequency (10.0 ± 1.1 and 14.5 ± 1.5 Hz; n = 10; p = 0.0264, Student's t test; Fig. 5C,D). Moreover, while it changed the aEPSC frequency distribution (p < 0.0001, Kolmogorov–Smirnov test; Fig. 5E), it did not affect the aEPSC mean amplitude (9.50 ± 0.20 pA, n = 301; and 9.80 ± 0.16 pA, n = 435; p = 0.2385, Student's t test; Fig. 5F) or its amplitude distribution (p = 0.0799, Kolmogorov–Smirnov test; Fig. 5G). Similarly, 8pCPT enhanced the eEPSC mean amplitude (198.2 ± 10.8 and 340.2 ± 25.5 pA; n = 10; p < 0.0003, Welch's test; Fig. 5H,I), and it increased the mean frequency of aEPSCs (9.30 ± 0.6 and 16.2 ± 1.3 Hz; n = 10; p = 0.0003, Welch's test; Fig. 5J,K). This Epac activator also modified the aEPSC frequency distribution (p < 0.0001, Kolmogorov–Smirnov test; Fig. 5L), yet it did not change the aEPSC mean amplitude (9.89 ± 0.28, n = 280; and 9.96 ± 0.17 pA, n = 485; p = 0.8309, Welch's test; Fig. 5M), although it did slightly modify the cumulative probability distribution (p = 0.0132, Kolmogorov–Smirnov test; Fig. 5N). This latter phenomenon was probably related to the effect of Epac on the open-channel probability of postsynaptic AMPARs in PCs (Gutierrez-Castellanos et al., 2017). These experiments indicate that isoproterenol and 8pCPT potentiate synaptic transmission largely through their presynaptic effects.
Figure 5.

Isoproterenol and 8pCPT enhance the frequency but not the amplitude of the asynchronous release events. A, H, The effects of isoproterenol (100 μm, 10 min) and 8pCPT (50 μm, 10 min) on eEPSCs recorded in the presence of Sr2+ (2.5 mm). C, J, Individual traces showing asynchronous release events in control conditions (black) and after isoproterenol (C, red) or 8pCPT treatment (J, red). B, I, Quantification of the effects of isoproterenol (n = 10 cells/10 slices/8 mice, t = 4.416, df = 14) and 8pCPT (n = 10 cells/10 slices/8 mice, t = 5.128. df = 12) on eEPSC amplitude. D–G, K–N, Quantification of the changes in aEPSC frequency and amplitude induced by isoproterenol (D–G) and 8pCPT (K–N). The aEPSC frequency was enhanced by isoproterenol (D, E: n = 10 cells/10 slices/8 mice, t = 2.419, df = 18) and 8pCPT (K, L: n = 10 cells/10 slices/7 mice, t = 5.073, df = 12), without changing the aEPSC amplitude in isoproterenol (F, G: n = 10 cells/10 slices/7 mice, t = 1.180, df = 734) and 8pCPT (M, N: n = 10 cells/10 slices/7 mice, t = 0.2137, df = 484). Calibration: A, H, 100 pA and 50 ms; C, J, 25 pA and 50 ms. O, Effects of isoproterenol and 8pCPT on eEPSC kinetics in the presence of Sr2+ analyzed in scaled representations. P, The decay time (ms) was not altered by isoproterenol (t = 0.1109, df = 18) or by 8pCPT (t = 0.8003, df = 18). Q, The decay slope pA/ms was not modified by isoproterenol (t = 1.033, df = 18) or by 8pCPT (t = 1.51, df = 18). R, The decay tau (ms) was not changed by isoproterenol (t = 1.628, df = 11) or by 8pCPT (t = 0.818, df = 13). S–U, Effect of isoproterenol and 8pCPT on eEPSC kinetics in the presence of Ca2+. S, The decay time was not altered by isoproterenol (t = 0.6143, df = 24) or by 8pCPT (t = 0.3143 df = 22). T, The decay slope (pA/ms) was not changed by isoproterenol (t = 1.033, df = 24) or by 8pCPT (t = 0.2965, df = 22). U, The decay tau (ms) was not modified by isoproterenol (t = 1.164, df = 24) or by 8pCPT (t = 0.4317, df = 22). Replacing Ca2+ with Sr2+ increased the control decay time (t = 2.267, df = 12) and control decay tau (t = 3.409, df = 21). Calibration: O, 100 pA and 50 ms. The data represent the mean ± SEM.
The effects of isoproterenol and 8pCPT on eEPSC kinetics in the presence of Sr2+ were analyzed in scaled representations (Fig. 5O). Neither isoproterenol nor 8pCPT modified the decay time (in milliseconds; p = 0.9129 and p = 0.4340, respectively, Student's t test; Fig. 5P), the decay slope (in picoamperes per millisecond; p = 0.3102 and p = 0.1481, respectively, Student's t test; Fig. 5Q), or the tau of decay (in milliseconds; p = 0.1319 and p = 0.4281, Welch's test; Fig. 5R). The effects of isoproterenol and 8pCPT on eEPSC kinetics were also analyzed in the presence of Ca2+, with no effect of isoproterenol or 8pCPT evident on the following parameters: the decay time (in milliseconds; p = 0.545, Student's t test and p = 0.7563, respectively, Student's t test; Fig. 5S), the decay slope (in picoamperes per millisecond; p = 0.3117 and p = 0.8483, respectively, Student's t test; Fig. 5T), or the tau of decay (in milliseconds; p = 0.2558 and p = 0.4317, respectively, Student's t test; Fig. 5U). Although replacing Ca2+ with Sr2+ increased the control decay time (p = 0.0427, Welch's test; Fig. 5P) and the control tau of decay (p = 0.0026, Student's t test; Fig. 5R).
β1ARs and Epac are required for PF–PC LTP
We tested whether βAR signaling through Epac proteins participates in presynaptically expressed forms of LTP. At cerebellar granule cell-to-PC synapses, LTP depends on a Ca2+-mediated increase in presynaptic cAMP (Salin et al., 1996), and it requires the AZ protein RIM1α to enhance transmitter release (Castillo et al., 2002; Lonart et al., 2003). Stimulation at 10 Hz for 10 s induced PF–PC LTP in naive cerebellar slices (from 99.7 ± 6.7% to 149.2 ± 17.7%; n = 16; p = 0.0013, Welch's test; Fig. 6A,B), and the presynaptic origin of this LTP was evident through the decrease in the PPR (from 1.79 ± 0.08 to 1.51 ± 0.04; n = 16; p = 0.0049, Welch's test; Fig. 6E,F). Although Rp-8-CPT-cAMPS has been used to assess the involvement of PKA in PF–PC LTP, this cAMP analog also inhibits Epac proteins (Chen et al., 2014). Thus, we tested the contribution of βARs to PF–PC LTP using the broad βAR antagonist propranolol, which prevented PF–PC LTP (from 98.9 ± 4.8% to 96.5 ± 5.4%; n = 12; p = 0.7429, Student's t test; Fig. 6A,B). PF–PC LTP was also prevented by the specific β1AR antagonist metoprolol (from to 102.7 ± 3.5% to 98.3 ± 5.1%; n = 10; p = 0.4860, Student's t test; Fig. 6A,B) as did the selective Epac2 inhibitor ESI 05 (from 103.6 ± 5.1% to 96.3 ± 6.9%; n = 11; p= 0.405, Student's t test; Fig. 6C,D). By contrast, PF–PC LTP was insensitive to the PKA inhibitors H-89 (from 103.1 ± 3.8% to 161.2 ± 19.0%; n = 13; p = 0.0111, Welch's test) and KT5720 (from 103.8 ± 5.5% to 179.5 ± 16.4%; n = 10; p = 0.0014, Welch's test; Fig. 6C,D). Thus, PF–PC LTP requires βAR signaling and the activity of Epac proteins.
Figure 6.
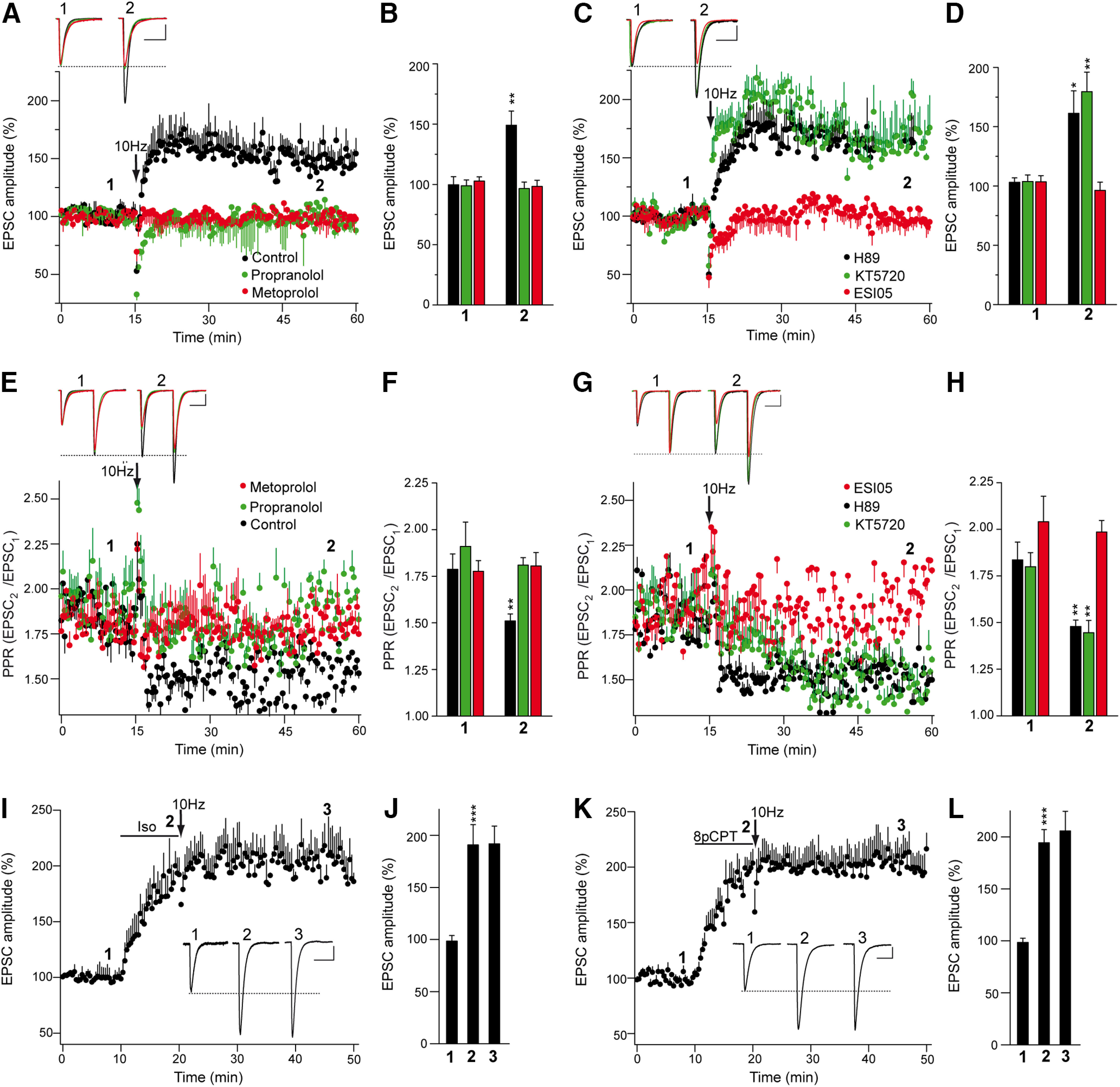
βAR and Epac activation is required for PF–PC LTP. A, C, A 10 Hz 10 s stimulation induced a sustained increase in EPSC amplitude in control and in KT-5720-treated (2 μm, 30 min prior 10 Hz stimulation) or H-89-treated (10 μm, 30 min) slices. Similar increases were absent in slices treated with the Epac inhibitor ESI 05 (10 μm, 30 min) or with the βAR antagonists propranolol (100 μm, 30 min) or metoprolol (60 μm, 30 min). B, D, Quantification of the changes in EPSC amplitude measured 40 min after stimulation (2) in control slices (n = 16 cells/16 slices/14 mice, t = 3.671, df = 23), propranolol-treated slices (n = 12 cells/12 slices/8 mice, t = 0.3322, df = 22), metoprolol-treated slices (n = 10 cells/10 slices/5 mice, t = 0.7113, df = 18), H-89-treated slices (n = 13 cells/13 slices/6 mice, t = 2.999, df = 12), KT-5720-treated slices (n = 10 cells/10 slices/7 mice, t = 4352, df = 10), and ESI 05-treated slices (n = 11 cells/11 slices/9 mice, t = 0.8508, df = 20) relative to the respective values before stimulation (1). E, G, Changes in the PPR (EPSC2/EPSC1) induced by 10 Hz stimulation in the conditions shown in A and C, respectively. F, H, Quantification of the changes in PPR 40 min after a 10 Hz stimulation in control [t = 3.13, df = 22 compared with (1)], and in the presence of propranolol [t = 0.7352, df = 13 compared with (1)], metoprolol [t = 0.2169, df = 18 compared with (1)], H-89 [t = 3448, df = 14 compared with (1)], KT-5720 [t = 3.387, df = 18, compared with (1)], or ESI 05 [t = 3.3283, df= 11, compared with (1)]. EPSC sample traces (A, C, E, G) represent the mean of six consecutive EPSCs at 0.05 Hz taken before and 40 min after 10 Hz stimulation. Calibration: 50 pA and 15 ms. I, K, The βAR agonist isoproterenol (10 min, 100 μm; I) and Epac activator 8pCPT (50 μm, 10 min; K) induced a sustained increase in EPSC amplitude that occludes the 10 Hz induced LTP. J, L, Quantification of the changes in EPSC amplitude induced by isoproterenol (J) and 8pCPT (L) followed by 10 Hz stimulation. The data were measured 10 min after isoproterenol/8pCPT application (2) [n = 12 cells/12 slice/6 mice, t = 453.5, df = 12 and n = 13 cells/13 slices/6 mice, t = 7.186, df = 14, respectively, compared with (1)]. The data were also measured 30 min after 10 Hz stimulation (3) [t = 0.04,229, df = 22 and t = 0.5006, df = 24, respectively, compared with (2)]. EPSC sample traces (I, K) represent the mean of six consecutive EPSCs at 0.05 Hz taken before (1) and 10 min after (2) isoproterenol/8pCPT treatment, and 30 min after 10 Hz stimulation (3). The data represent the mean ± SEM.
Further evidence that βARs participate in PF–PC LTP was obtained in occlusion experiments. The βAR agonist isoproterenol increased EPSC amplitude (from 98.5 ± 5.4% to 190.7 ± 19.6%; n = 12; p = 0.0007, Welch's test; Fig. 6I,J), yet a subsequent 10 Hz stimulation failed to further potentiate synaptic transmission (from 190.7 ± 19.6% to 191.8 ± 17.1%; p = 0.9666, Student's t test; Fig. 6I,J). Likewise, the Epac agonist 8pCPT induced a sustained increase in EPSC amplitude (from 98.4 ± 4.1% to 194.3 ± 12.7%; n = 13; p < 0.0001, Welch's test; Fig. 6K,L), which similarly occluded potentiation by a subsequent 10 Hz stimulation (from 194.3 ± 12.7% to 205.7 ± 18.9%; n = 13; p = 0.6212, Student's t test; Fig. 6K,L). Thus, prior activation of βARs or Epac proteins occluded tetanus-induced PF–PC LTP, suggesting that Epac-dependent signaling by βARs is involved in PF–PC LTP.
PF–PC LTP is absent in Epac2−/− mice, in which isoproterenol fails to enhance SV docking
The presynaptic action of Epac at PFs is consistent with the abundance of Epac2 in the cerebellum (Kawasaki et al., 1998), and, thus, we studied synaptic transmission in Epac2−/− mice to further assess the role of Epac2 in PF–PC LTP (Shibasaki et al., 2007). PF LTP was absent in cerebellar slices from Epac2−/− mice (from 100.6 ± 4.1% to 98.7 ± 2.0%; n = 15; p = 0.6812, Welch's test), whereas a 10 Hz stimulation for 10 s did induce PF–PC LTP in WT slices (from 101.3 ± 5.0% to 169.1 ± 15.6%; n = 13; p = 0.0025, Welch's test; Fig. 7A,B). PF LTP was associated with a decrease in the EPSC2/EPSC1 PPR (from 1.80 ± 0.07 to 1.52 ± 0.05; p = 0.0023 Student's t test; Fig. 7C), a phenomenon not observed in Epac2−/− slices (1.75 ± 0.06 and 1.73 ± 0.08; p = 0.8429, Student's t test).
Figure 7.
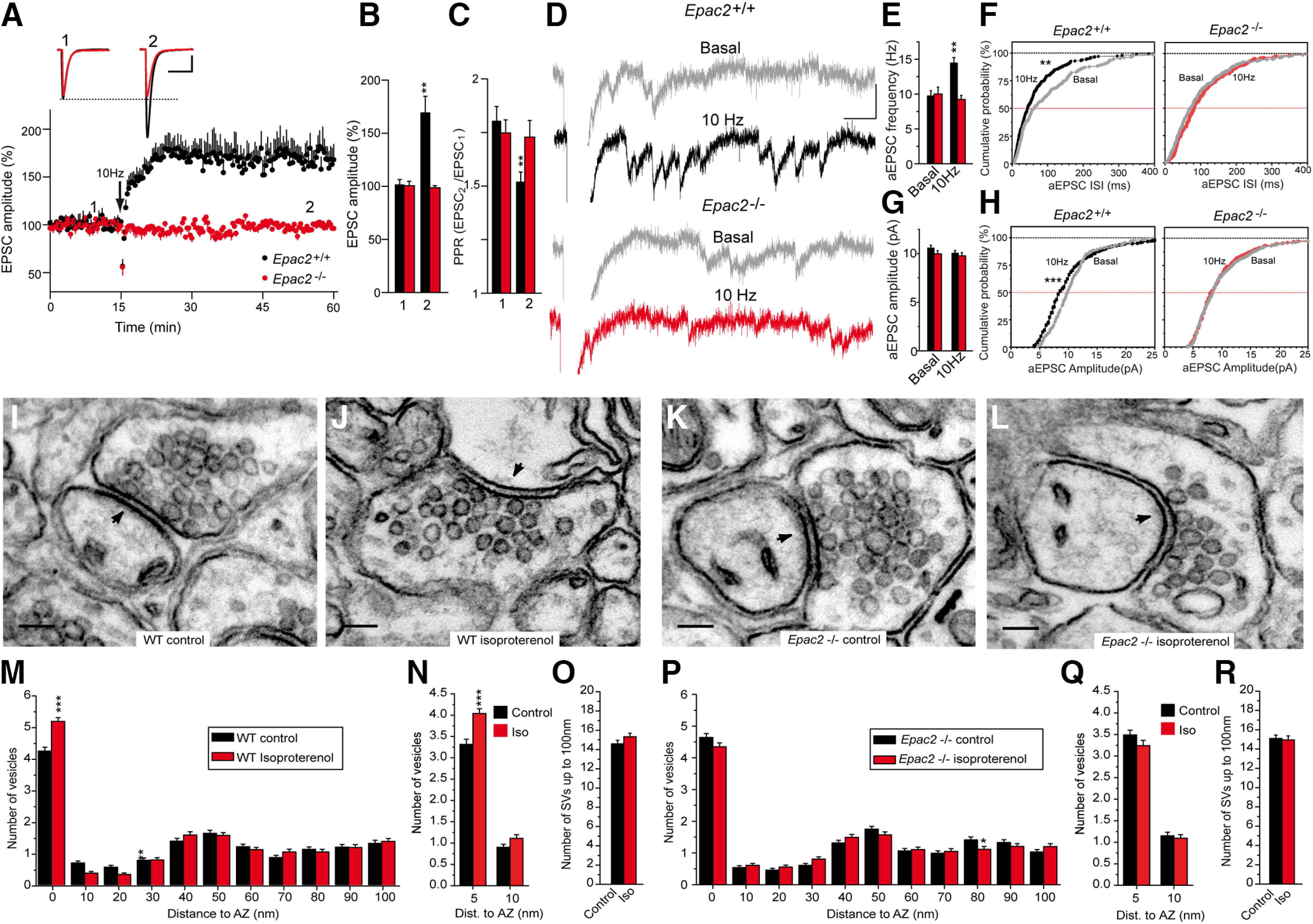
Epac2−/− slices lack PF–PC LTP, and they display no isoproterenol induced increase in SV docking. A, A 10 Hz, 10 s stimulation induced a sustained increase in the EPSC amplitude in WT cerebellar slices that was absent in slices from Epac2−/− littermates. B, Quantification of the changes in EPSC amplitude measured 40 min after stimulation (2) in slices from WT (n = 13 cells/13 slices/6 mice, t = 3.745, df = 13) or Epac2−/− mice (n = 15 cells/15 slices/8 mice, t = 0.4169, df = 15) compared with the values before stimulation (1). C, Quantification of the changes in the PPR measured 40 min after stimulation (2) in slices from WT (t = 3.403, df = 24) and Epac2−/− mice (t = 0.2000, df = 28) compared with the values before stimulation (1). D, The aEPSCs induced when replacing Ca2+ with Sr2+ (2.5 mm) 30 min after 10 Hz stimulation. After 5 min, individual traces showing aEPSCs were analyzed (2 min) in basal (gray) and stimulated WT (black) or Epac2−/− (red) slices. E, F, Changes in aEPSC frequency induced by 10 Hz stimulation in WT (t = 4.321, df = 11) and Epac2−/− (t = 0.6774, df = 14) slices. G, H, Changes in aEPSC amplitude induced by 10 Hz stimulation in WT (t = 1.225, df = 424) and Epac2−/− (t = 0.4063, df = 459) slices. I, J, Isoproterenol (100 μm, 10 min) increases SV docking in WT mice. M, Quantification of the isoproterenol induced an increase in SV docking in WT mice. N, Isoproterenol increases the number of SVs 0–5 nm from the AZ membrane without changing the SVs 5–10 nm from the AZ membrane in WT slices. O, Isoproterenol treatment did not change the total number of SVs up to 100 µm from the AZ in WT slices. K, L, Isoproterenol treatment fails to increase SV docking in Epac2−/− mice. P, Quantification of the isoproterenol-induced increase in SV docking in Epac2−/− slices. Q, Isoproterenol did not change the number of SVs within 5 or 5–10 nm from the AZ membrane in Epac2−/− slices. R, Isoproterenol treatment did not change the total number of SVs up to 100 µm from the AZ membrane in Epac 2−/− slices. Calibration: A, 50 pA and 15 ms; D, 20 pA and 40 ms. Scale bars: I–L, 100 nm. The data represent the mean ± SEM.
The presynaptic origin of PF LTP was further assessed by replacing Ca2+ with Sr2+ 30 min after a 10 Hz stimulation. In the presence of Sr2+, LTP increased the mean frequency of aEPSCs (9.72 ± 0.76, n = 6; and 14.43 ± 0.77 Hz, n = 7; p = 0.0012, Student's t test; Fig. 7D,E) and altered the aEPSC frequency distribution (p = 0.0011, Kolmogorov–Smirnov; Fig. 7F) in WT but not in Epac2−/− slices [10.0 ± 1.00, n = 8, and 9.21 ± 0.60 Hz, n = 8, p = 0.5039 Student's t test (Fig. 7E); and p = 0.2539, Kolmogorov–Smirnov test (Fig. 7F)]. By contrast, there were no changes in the aEPSC mean amplitude in WT slices undergoing LTP under these conditions (10.55 ± 0.31, n = 175; and 10.03 ± 0.29 Hz, n = 303; p = 0.2213, Welch's test; Fig. 7D,G), although there was a mild reduction in the aEPSC amplitude in the cumulative probability plots (p = 0.0008, Kolmogorov–Smirnov test; Fig. 7H). Similarly, there was no change in aEPSC mean amplitude during LTP in Epac2−/− slices (9.96 ± 0.33, n = 241; and 9.76 ± 0.33 pA, n = 220; p = 0.6847, Student's t test; Fig. 7G) or in the amplitude distribution of aEPSC (p = 0.949, Kolmogorov–Smirnov test; Fig. 7H). Thus, PF LTP largely involved changes in aEPSC frequency, providing further evidence of its presynaptic origin.
We determined to what extent the lack of PF–PC LTP in Epac2−/− mice was a result of changes in vesicle docking assessed by EM. Chemical LTP was induced by incubating cerebellar slices with isoproterenol (100 μm) for 10 min, and the SV distribution was then determined in fixed slices by EM. Isoproterenol-enhanced SV docking in WT slices [from 4.21 ± 0.12 (n = 182) to 5.15 ± 0.12] synaptic vesicles/AZ profile (n = 168; p < 0.001, ANOVA Bonferroni's test; Fig. 7I,J,M). The absence of PF–PC LTP was associated with a failure of isoproterenol to increase SV docking in Epac2−/− slices, which ranged from to 4.35 ± 0.12 synaptic vesicles/AZ profile (n = 160) in the presence of isoproterenol to 4.64 ± 0.13 synaptic vesicles/AZ profile (n = 180) in its absence (p > 0.05, ANOVA with Bonferroni's test; Fig. 7K,L,P).
Docked and primed SVs are heterogeneous, and the following two subpopulations can be distinguished: loosely docked SVs located 8 nm from the AZ membrane because of partial zippering of SNARE complexes; and fully primed and tightly docked SVs in which SNARE complex zippering has progressed further and, therefore, these SVs are closer to the AZ membrane (<5 nm; Fernández-Busnadiego et al., 2013; Imig et al., 2014; Neher and Brose, 2018). To distinguish loosely from tightly docked and primed SVs, we studied the distribution of SVs within a distance of 5 nm and 5–10 nm from the AZ membrane. Isoproterenol increased the number of SVs within 5 nm from the AZ membrane in WT slices [from 3.31 ± 0.12 (n = 182) to 4.04 ± 0.11 (n = 168)] synaptic vesicles/AZ profile (p < 0.001, ANOVA Bonferroni's test; Fig. 7N) without affecting the number of SVs located 5–10 nm from the AZ membrane (from 0.90 ± 0.07 to 1.11 ± 0.08 synaptic vesicles/AZ profile; p >0.05, ANOVA with Bonferroni's test; Fig. 7N). Conversely, no change in the number of SVs <5 nm from the AZ membrane was observed in Epac2−/− slices under control conditions relative to the WT slices (3.49 ± 0.11; n = 180; p > 0.05, ANOVA with Bonferroni's test; Fig. 7Q), and, moreover, isoproterenol failed to increase this parameter (3.24 ± 0.12; n = 160; p > 0.05, ANOVA with Bonferroni's test; Fig. 7Q). Similarly, isoproterenol did not alter the SVs 5–10 nm from the AZ membrane in Epac2−/− slices (1.15 ± 0.08, n = 180; and 1.09 ± 0.08, n = 160; p > 0.05, ANOVA with Bonferroni's test; Fig. 7Q). Thus, the absence of Epac2 prevents isoproterenol-induced increase in tightly docked and fully primed SVs. In addition, isoproterenol did not alter the total number of SVs within 100 µm from the AZ plasma membrane in WT or Epac2−/− slices (Fig. 7O,R). Together, these results indicate that the increase in neurotransmitter release during PF–PC LTP requires the isoproterenol-induced Epac2-dependent recruitment of SVs to the plasma membrane AZ.
PF–PC LTP is associated with an increase in the RRP size that is absent in Epac2−/− slices
The number of docked vesicles is strongly correlated to the size of the RRP of SVs (Rosenmund and Stevens, 1996; Schikorski and Stevens, 2001). Since PF–PC LTP is associated with SV docking, we tested whether PF–PC LTP also remodels the size of the SV pools, in particular, increasing the RRP size. The RRP size was calculated from the cumulative amplitude plots as the y-intercept from a linear fit of the steady-state values attained during high-frequency trains (Schneggenburger et al., 1999; Gioia et al., 2016). Thus, slices were stimulated at 40 Hz for 2.5 s in basal conditions or 30 min after LTP induction. In WT slices, the RRP size increased after LTP induction from 4347.4 ± 513.8 pA (n = 18) to 8586.9 ± 848.3 pA (n = 21; p = 0.012, Student's t test; Fig. 8A–D). The 10 Hz stimulation protocol failed to induce LTP in Epac2−/− slices (Figs. 7A,B, 8G), and it did not increase the RRP size (control, 4006.4 ± 481.2 pA, n = 21; after the 10 Hz protocol, 3544.5 ± 711.3 pA, n = 24; p = 0.5937, Welch's test; Fig. 8G–J). Moreover, LTP did not change the replenishment rate (in picograms per millisecond) in WT slices (1.64 ± 0.47 and 1.60 ± 0.4, in control and LTP conditions, respectively; p = 0.9490, Student's t test; Fig. 8E) or Epac2−/− slices (1.21 ± 0.27 and 1.47 ± 0.38, in control and LTP, respectively; p = 0.5801, Welch's test; Fig. 8K). PF–PC LTP mildly decreased the pves in WT slices (0.035 ± 0.004 and 0.025 ± 0.002, in control and LTP, respectively; p = 0.0345, Welch's test; Fig. 8F), yet the small decrease in pves was overcompensated for by the large increase in the RRP size, leading to an increase in the Pr, as evident by the decrease in PPR during PF–PC LTP (Fig. 6E,F). However, the pves did not change in Epac2−/− slices (0.029 ± 0.003 and 0.034 ± 0.005, in control and LTP, respectively; p = 0.397, Welch's test; Fig. 8L). Thus, βAR-mediated signaling through the Epac2 protein increases the size of the RRP of SVs and contributes to PF–PC LTP at cerebellar synapses.
Figure 8.
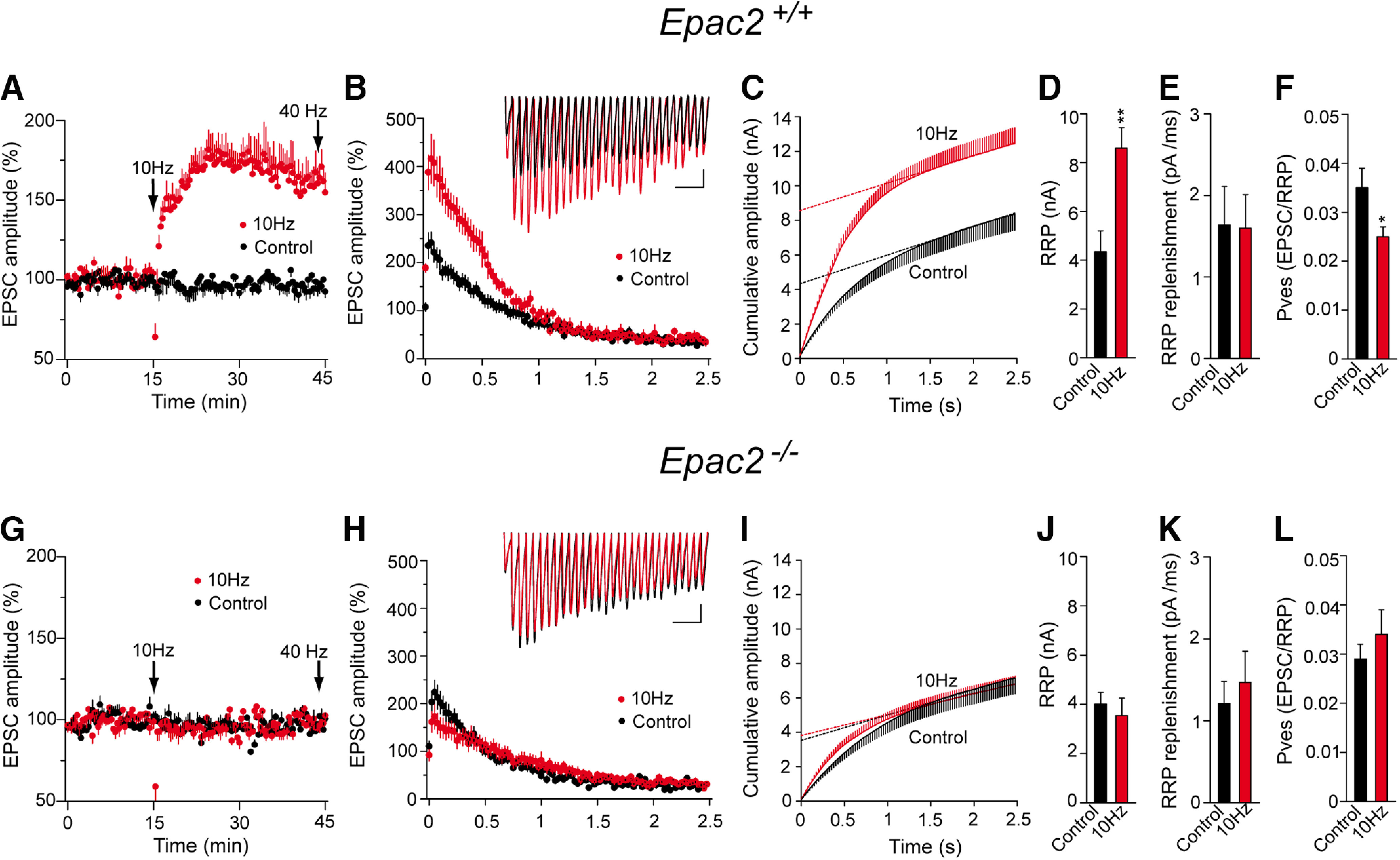
PF–PC LTP is associated with an increase in the RRP size that does not occur in Epac2−/− mice. A, G, The RRP size was measured after a train of 100 pulses at 40 Hz applied 30 min after LTP induction, as well as in control (unstimulated) slices from both WT and Epac2−/− mice (littermates). B, H, EPSC amplitude during the 40 Hz train, the traces showing individual examples. Calibration: 100 pA and 100 ms. C, I, Cumulative EPSC amplitudes in control and LTP-induced cells. The RRP size was calculated from the cumulative amplitude plots as the y-intercept from a linear fit of the steady-state level attained during the high-frequency train. D, J, Quantification of the changes in the RRP size induced by a 10 Hz stimulus in slices from WT (D: control slices, n = 18 cells/18 slices/8 mice, 10 Hz stimulated slices, n = 21 cells/21 slices/9 mice, t = 3.506, df = 37) and Epac2−/− mice (J: control slices, n = 21 cells/21 slices/10 mice, and 10 Hz stimulates slices, t = 0.5379, df = 39). E, K, Quantification of the replenishment rate (pA/ms) in slices from WT mice (E: t = 0.064, df = 37) and Epac2−/− mice (K: t = 0.5578, df = 40). F, L, Quantification of the pves in slices from WT mice (F: t = 2.236, df = 25) or Epac2−/− mice (L: t = 0.8575, df = 37). The data represent the mean ± SEM.
Discussion
We found here that isoproterenol increases synaptic transmission at PF–PC synapses by a presynaptic mechanism, whereby β1ARs expressed in PF synaptic boutons generate cAMP and activate Epac, enhancing SV docking. Interestingly, presynaptic PF–PC LTP is blocked by the selective β1AR antagonist metoprolol and it is not detected in cerebellar slices from Epac2−/− mice. Furthermore, the increase in the RRP size when synapses are potentiated is not observed in Epac2−/− slices. Thus, βARs appear to activate physiologically relevant signaling in presynaptic PF–PC LTP that involves cAMP- and Epac2-dependent modulation of the release machinery.
PF–PC LTP requires an increase in presynaptic cAMP generated by Ca2+-dependent adenylyl cyclase 1 (AC1; Storm et al., 1998). Since PF–PC LTP requires βAR activation, it would appear that AC1 activation by Ca2+ alone is insufficient to induce this LTP and that Gs stimulation is also required, consistent with the stimulation of all AC isoforms by Gsα (Sadana and Dessauer, 2009), and with the notion that AC1 is synergistically stimulated by Ca2+ and Gs-coupled receptors (Wayman et al., 1994). The expression of β1ARs in PF boutons near noradrenergic varicosities makes it likely that the PF stimulation that induces PF–PC LTP recruits noradrenergic fibers to release noradrenaline, which in turn activates β-adrenergic heteroreceptors at PF boutons, enhancing glutamate release. Noradrenergic fibers are distributed throughout the cerebellar cortex, forming varicosities closely apposed to PF–PC contacts on PC dendrites (Bloom et al., 1971; Abbott and Sotelo, 2000). The finding that βAR activation is required for PF–PC LTP is consistent with the presynaptic actions of these receptors at other synapses, as follows: (1) βARs increase cAMP and potentiate glutamate release in cerebrocortical synaptosomes (Herrero and Sánchez-Prieto, 1996; Millán et al., 2002; Ferrero et al., 2013, 2016); (2) βAR activation brings SVs closer to the plasma membrane (Ferrero et al., 2013) and prevents the presynaptic silencing induced by cannabinoid type 1 receptors in cerebellar granule cells (Alonso et al., 2017); (3) βARs potentiate synaptic transmission through a presynaptic mechanism (Parfitt et al., 1992; Huang et al., 1996; Huang and Hsu, 2006; Ji et al., 2008); and (4) βARs facilitate the induction of other cAMP-dependent forms of presynaptic LTP (Wójtowicz et al., 2010; Ul Haq et al., 2016). Thus, βAR activation recapitulates forskolin-induced enhancement of synaptic transmission (Chavez-Noriega and Stevens, 1994; Chen and Regehr, 1997; Sakaba and Neher, 2001; Kaneko and Takahashi, 2004), and these receptors constitute part of the physiologically relevant signaling involved in the activation of cAMP-dependent endogenous pathways that potentiate neurotransmitter release (Ferrero et al., 2013, 2016). Furthermore, noradrenaline enhances PC excitability in vivo because of an increase in the excitatory input from PFs (Guo et al., 2016).
Our pharmacological data with PKA inhibitors (H-89 and KT-5720) do not support a role of PKA in PF–PC LTP, while both pharmacological Epac2 inhibition (ESI 05) and activation (8pCPT), as well as genetic depletion of Epac2, strongly support the requirement of Epac2 for PF–PC LTP. It is important to note that several studies of PKA and LTP used synapses that developed in vitro, such as cultured granular neurons (Linden and Ahn, 1999; Lonart et al., 2003), and that the functional properties of these synapses are likely to be different from synapses in mature slices. In addition, PF–PC LTP in cerebellar slices was blocked by the PKA inhibitor Rp-8-CPT-cAMPS (Salin et al., 1996), a compound that also inhibits Epac proteins (Zieba et al., 2011; Chen et al., 2014). The search for PKA substrates involved in PF–PC LTP has also produced conflicting results. In one study, RIM1α phosphorylation by PKA at Ser-413 was seen to be essential for LTP in paired recordings of cerebellar granule cells and PCs in culture (Lonart et al., 2003). However, this conclusion has been questioned as PF–PC LTP remains intact in knock-in mice carrying a Ser-413 mutated to alanine (Kaeser et al., 2008). Finally, Epac2 plays an essential role in other forms of cAMP-dependent LTP expressed presynaptically as the increase in neurotransmitter release is associated with the hippocampal mossy fiber LTP (Fernandes et al., 2015).
Our data support a key role for Epac2 in the βAR potentiation of neurotransmitter release and presynaptic PF–PC LTP. Epac activation increases evoked EPSC amplitude and occludes 10 Hz induced PF–PC LTP. In addition, PF–PC LTP is blocked by the Epac2-specific inhibitor ESI 05 and is not detected in Epac2−/− KO mice. A presynaptic effect of Epac at PFs is consistent with the abundance of Epac2 in the cerebellum (Kawasaki et al., 1998). Epac proteins mediate many responses to presynaptic cAMP in non-neuronal (Ozaki et al., 2000) and neuronal systems, including neurotransmitter release (Kaneko and Takahashi, 2004; Gekel and Neher, 2008; Yang et al., 2012; Ferrero et al., 2013, 2016), SV recruitment (Sakaba and Neher, 2003; Ramírez-Franco et al., 2014; Alonso et al., 2017), and presynaptic plasticity (Fernandes et al., 2015; Tong et al., 2017). The priming activity of Munc13 (Ma et al., 2011) plays a key role in SV docking through assembly of the Munc13–Rim1–Rab3 complex (Dulubova et al., 2005) and SNARE complex. Munc13-1 forms supramolecular self-assemblies that serve as independent release sites (Sakamoto et al., 2018) through which phospholipase C (PLC)-coupled receptors (Bauer et al., 2007; Martín et al., 2010) enhance the RRP size (Martín et al., 2018). βARs and Epac also activate PLC, translocate Munc13-1, enhance Rab3A–RIM1α interaction, and increase SV docking (Ferrero et al., 2013). However, how a Gs-coupled receptor like βAR and a cAMP target like Epac activate PLC and Munc13-1 proteins is unlikely to be straightforward. Epac proteins are guanine nucleotide exchange factors for the small GTPases Rap1 and Rap2 (Kawasaki et al., 1998), which can bind to and activate PLC-ε in HEK293 cells (Schmidt et al., 2001). Significantly, PLC-ε is expressed in the brain (Thomas et al., 1991; Kelley et al., 2001), and diacylglycerol (DAG) can increase SV docking and priming through the activation of Munc13 proteins (Rhee et al., 2002).
We found that βARs strongly potentiate synaptic transmission and that this effect is associated with an increase in docked SVs. Specifically, βARs enhance the docking of SVs 0–5 nm from AZ membrane, SVs that are tightly docked and fully primed, and thus, release ready (Fernández-Busnadiego et al., 2013; Imig et al., 2014; Neher and Brose, 2018; Maus et al., 2020). The docked SVs are closely correlated with the RRP size (Rosenmund and Stevens, 1996; Schikorski and Stevens, 2001), although not all docked vesicles are released and some undocked vesicles contribute to the RRP (Kaeser and Regehr, 2017). EM also has some limitations as it provides a static image of a section of the synapse and chemical fixation may alter the SV distribution (Maus et al., 2020). It is possible that the strong potentiation of synaptic transmission mediated by βARs is not fully accounted for by the weak effect on docking and that a postdocking effect enhancing Ca2+-dependent fusion could also be involved. Thus, forskolin increases the Ca2+ sensitivity of transmitter release induced with weak Ca2+-uncaging flashes (∼0.5–1 μm Ca2+; Yao and Sakaba, 2010). A change in Ca2+-dependent fusion would be consistent with an increase in the Pr, as seen by the decrease in the PPR. Indeed, the decrease in PPR mediated by noradrenaline, isoproterenol, 8pCPT, and 10 Hz LTP seen here should not only be regarded as an indication of a presynaptic change but also, as an increase in the initial Pr (Branco and Staras, 2009). Noradrenaline also modulates release by increasing the Ca2+ sensitivity of vesicle fusion (Yawo, 1996, 1999). Thus, βAR-mediated potentiation of neurotransmitter release involves a docking effect, and it may also involve a post-docking component (Chen and Regehr, 1997; Kaneko and Takahashi, 2004). However, the decrease in the mean pves during PF–PC LTP suggests a limited influence of a change in the Ca2+ sensitivity of release in the potentiation of synaptic transmission by βARs. It is important to note that pves is very variable, even among synaptic boutons originating from the same axon in cultured neurons (Ariel et al., 2013). More specifically, the nanoscale molecular topography of PF–PC synapses indicates that the high heterogeneity in pves is because of the variable number of voltage-dependent calcium channels within close proximity to the Ca2+ sensor (Rebola et al., 2019). Moreover, this variability may be even higher if silent synapses are recruited during LTP (Jörntell and Ekerot, 2002). The small decrease in pves during PF–PC LTP would indicate that new docked SVs are less efficiently coupled to Ca2+ influx than the already docked ones. PF axon terminals coexpress Munc13-1 and Munc13-3, which play a similar role in the SV cycle but with an unequal contribution to Pr and RRP dynamics (Augustin et al., 2001; Chen et al., 2013). Munc13-3-deficient synapses have decreased Pr (Augustin et al., 2001; Ishiyama et al., 2014), and this isoform works as a developmentally upregulated mediator of tight coupling at PF–PC synapses (Kusch et al., 2018). Thus, a decrease in pves during PF–PC LTP could be possible if the new docked SVs are primed by a smaller Munc13-3/Munc13-1 proportion than the previously docked SVs. Importantly, the decrease in pves provoked by PF–PC LTP is compatible with an increase in the Pr because the large increase in the RRP size compensates for the small decrease in pves.
In conclusion, βARs enhance the docking and priming of SVs through βAR/cAMP/Epac signaling, thereby providing a link between βARs and the endogenous pathways that potentiate neurotransmitter release. Hence, our findings reveal a new mechanism driven by presynaptic receptors relevant to PF–PC LTP.
Footnotes
This work was supported by the Ministerio de Economía, Industria y Competitividad (Grant BFU 2013-43163R to J.S.-P.; Grant BFU 2012-32105 to M.T.) and the Ministerio de Ciencia e Innovación (Grant BFU2017-83292-R), the “Instituto de Salud Carlos III” (Grants RD12/0014 and RD16/0019), and the Community of Madrid (Grant B2017/BMB-3699) to J.S.-P. and M.T. This work was also supported by Grant SBPLY/17/180501/000229 from the Junta de Comunidades de Castilla-La Mancha to R.L. We thank Prof. Susumu Seino (Kobe University, Kobe, Japan) for providing the Epac2 KO mice. We also thank María Luisa García and Miriam González at the electron microscopy facility of the Universidad Complutense de Madrid for technical support; María del Carmen Zamora for excellent technical assistance; and Dr. Mark Sefton for editorial assistance.
The authors declare no competing financial interests.
References
- Abbott LC, Sotelo C (2000) Ultrastructural analysis of catecholaminergic innervation in weaver and normal mouse cerebellar cortices. J Comp Neurol 426:316–329. [DOI] [PubMed] [Google Scholar]
- Alonso B, Bartolomé-Martín D, Ferrero JJ, Ramírez-Franco J, Torres M, Sánchez-Prieto J (2017) CB1 receptors down-regulate a cAMP/Epac2/PLC pathway to silence the nerve terminals of cerebellar granule cells. J Neurochem 142:350–361. 10.1111/jnc.14059 [DOI] [PubMed] [Google Scholar]
- Ariel P, Hoppa MB, Ryan TA (2013) Intrinsic variability in Pv, RRP size, Ca(2+) channel repertoire, and presynaptic potentiation in individual synaptic boutons. Front Synaptic Neurosci 4:9. 10.3389/fnsyn.2012.00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin I, Korte S, Rickmann M, Kretzschmar HA, Súdhof TC, Herms JW, Brose N (2001) The cerebellum-specific Munc13 isoform Munc13-3 regulates cerebellar synaptic transmission and motor learning. J Neurosci 21:10–17. 10.1523/JNEUROSCI.21-01-00010.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CS, Woolley RJ, Teschemacher AG, Seward EP (2007) Potentiation of exocytosis by phospholipase C-coupled G-protein-coupled receptors requires the priming protein Munc13-1. J Neurosci 27:212–219. 10.1523/JNEUROSCI.4201-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom FE, Hoffer BJ, Siggins GR (1971) Studies on norepinephrine-containing afferents to Purkinje cells of art cerebellum. I. Localization of the fibers and their synapses. Brain Res 25:501–521. 10.1016/0006-8993(71)90457-4 [DOI] [PubMed] [Google Scholar]
- Branco T, Staras K (2009) The probability of neurotransmitter release: variability and feedback control at single synapses. Nat Rev Neurosci 10:373–383. 10.1038/nrn2634 [DOI] [PubMed] [Google Scholar]
- Carey MR, Regehr WG (2009) Noradrenergic control of associative synaptic plasticity by selective modulation of instructive signals. Neuron 62:112–122. 10.1016/j.neuron.2009.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC (2002) RIM1alpha is required for presynaptic long-term potentiation. Nature 415:327–330. 10.1038/415327a [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Stevens CF (1994) Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci 14:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG (1997) The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci 17:8687–8694. 10.1523/JNEUROSCI.17-22-08687.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Wild C, Zhou X, Ye N, Cheng X, Zhou J (2014) Recent advances in the discovery of small molecules targeting exchange proteins directly activated by cAMP (EPAC). J Med Chem 57:3651–3665. 10.1021/jm401425e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Cooper B, Kalla S, Varoqueaux F, Young SM Jr (2013) The Munc13 proteins differentially regulate readily releasable pool dynamics and calcium-dependent recovery at a central synapse. J Neurosci 33:8336–8351. 10.1523/JNEUROSCI.5128-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Lou X, Lu J, Huryeva I, Alam A, Schneggenburger R, Südhof TC, Rizo J (2005) A Munc13/RIM/Rab3 tripartite complex: from priming to plasticity? EMBO J 24:2839–2850. 10.1038/sj.emboj.7600753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes HB, Riordan S, Nomura T, Remmers CL, Kraniotis S, Marshall JJ, Kukreja L, Vassar R, Contractor A (2015) Epac2 mediates cAMP-dependent potentiation of neurotransmission in the hippocampus. J Neurosci 35:6544–6553. 10.1523/JNEUROSCI.0314-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Busnadiego R, Asano S, Oprisoreanu A-M, Sakata E, Doengi M, Kochovski Z, Zürner M, Stein V, Schoch S, Baumeister W, Lucić V (2013) Cryo-electron tomography reveals a critical role of RIM1α in synaptic vesicle tethering. J Cell Biol 201:725–740. 10.1083/jcb.201206063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero JJ, Alvarez AM, Ramírez-Franco J, Godino MC, Bartolomé-Martín D, Aguado C, Torres M, Luján R, Ciruela F, Sánchez-Prieto J (2013) β-Adrenergic receptors activate exchange protein directly activated by cAMP (Epac), translocate Munc13-1, and enhance the Rab3A-RIM1α interaction to potentiate glutamate release at cerebrocortical nerve terminals. J Biol Chem 288:31370–31385. 10.1074/jbc.M113.463877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero JJ, Ramírez-Franco J, Martín R, Bartolomé-Martín D, Torres M, Sánchez-Prieto J (2016) Cross-talk between metabotropic glutamate receptor 7 and beta adrenergic receptor signaling at cerebrocortical nerve terminals. Neuropharmacology 101:412–425. 10.1016/j.neuropharm.2015.07.025 [DOI] [PubMed] [Google Scholar]
- Gekel I, Neher E (2008) Application of an Epac activator enhances neurotransmitter release at excitatory central synapses. J Neurosci 28:7991–8002. 10.1523/JNEUROSCI.0268-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioia DA, Alexander NJ, McCool BA (2016) Differential expression of Munc13-2 produces unique synaptic phenotypes in the basolateral amygdala of C57BL/6J and DBA/2J mice. J Neurosci 36:10964–10977. 10.1523/JNEUROSCI.1785-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda Y, Stevens CF (1994) Two components of transmitter release at a central synapse. Proc Natl Acad Sci U S A 91:12942–12946. 10.1073/pnas.91.26.12942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, Feng J-Y, Li J, Ding N, Li Y-J, Qiu D-L, Piao R-L, Chu C-P (2016) Effects of norepinephrine on spontaneous firing activity of cerebellar Purkinje cells in vivo in mice. Neurosci Lett 629:262–266. 10.1016/j.neulet.2016.06.058 [DOI] [PubMed] [Google Scholar]
- Gutierrez-Castellanos N, Da Silva-Matos CM, Zhou K, Canto CB, Renner MC, Koene LMC, Ozyildirim O, Sprengel R, Kessels HW, de Zeeuw CI (2017) Motor learning requires Purkinje cell synaptic potentiation through activation of AMPA-receptor subunit GluA3. Neuron 93:409–424. 10.1016/j.neuron.2016.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero I, Sánchez-Prieto J (1996) cAMP-dependent facilitation of glutamate release by beta-adrenergic receptors in cerebrocortical nerve terminals. J Biol Chem 271:30554–30560. 10.1074/jbc.271.48.30554 [DOI] [PubMed] [Google Scholar]
- Huang CC, Hsu KS (2006) Presynaptic mechanism underlying cAMP-induced synaptic potentiation in medial prefrontal cortex pyramidal neurons. Mol Pharmacol 69:846–856. 10.1124/mol.105.018093 [DOI] [PubMed] [Google Scholar]
- Huang CC, Hsu KS, Gean PW (1996) Isoproterenol potentiates synaptic transmission primarily by enhancing presynaptic calcium channels influx via P- and/or Q-type calcium channels in the rat amygdala. J Neurosci 16:1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig C, Min SW, Krinner S, Arancillo M, Rosenmund C, Südhof TC, Rhee JS, Brose N, Cooper BH (2014) The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron 84:416–431. 10.1016/j.neuron.2014.10.009 [DOI] [PubMed] [Google Scholar]
- Ishiyama S, Schmidt H, Cooper BH, Brose N, Eilers J (2014) Munc13-3 superprimes synaptic vesicles at granule cell-to-basket cell synapses in the mouse cerebellum. J Neurosci 34:14687–14696. 10.1523/JNEUROSCI.2060-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X-H, Cao X-H, Zhang C-L, Feng Z-J, Zhang X-H, Ma L, Li B-M (2008) Pre- and postsynaptic β-adrenergic activation enhances excitatory synaptic transmission in layer V/VI pyramidal neurons of the medial prefrontal cortex of rats. Cereb Cortex 18:1506–1520. 10.1093/cercor/bhm177 [DOI] [PubMed] [Google Scholar]
- Jörntell H, Ekerot CF (2002) Reciprocal bidirectional plasticity of parallel fiber receptive fields in cerebellar Purkinje cells and their afferent interneurons. Neuron 34:797–806. 10.1016/s0896-6273(02)00713-4 [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Regehr WG (2017) The readily releasable pool of synaptic vesicles. Curr Opin Neurobiol 43:63–70. 10.1016/j.conb.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Kwon HB, Blundell J, Chevaleyre V, Morishita W, Malenka RC, Powell CM, Castillo PE, Südhof TC (2008) RIM1alpha phosphorylation at serine-413 by protein kinase A is not required for presynaptic long-term plasticity or learning. Proc Natl Acad Sci U S A 105:14680–14685. 10.1073/pnas.0806679105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Takahashi T (2004) Presynaptic mechanism underlying cAMP-dependent synaptic potentiation. J Neurosci 24:5202–5208. 10.1523/JNEUROSCI.0999-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM (1998) A family of cAMP-binding proteins that directly activate Rap. Science 282:2275–2279. 10.1126/science.282.5397.2275 [DOI] [PubMed] [Google Scholar]
- Kelley GG, Reks SE, Ondrako JM, Smrcka AV (2001) Phospholipase C (epsilon): a novel Ras effector. EMBO J 20:743–754. 10.1093/emboj/20.4.743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintscher M, Wozny C, Johenning FW, Schmitz D, Breustedt J (2013) Role of RIM1α in short- and long-term synaptic plasticity at cerebellar parallel fibres. Nat Commun 4:2392. 10.1038/ncomms3392 [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727. 10.1016/s0896-6273(01)00246-x [DOI] [PubMed] [Google Scholar]
- Kusch V, Bornschein G, Loreth D, Bank J, Jordan J, Baur D, Watanabe M, Kulik A, Heckmann M, Eilers J, Schmidt H (2018) Munc13-3 is required for the developmental localization of Ca 2+ channels to active zones and the nanopositioning of Ca v 2.1 near release sensors. Cell Rep 22:1965–1975. 10.1016/j.celrep.2018.02.010 [DOI] [PubMed] [Google Scholar]
- Linden DJ, Ahn S (1999) Activation of presynaptic cAMP-dependent protein kinase is required for induction of cerebellar long-term potentiation. J Neurosci 19:10221–10227. 10.1523/JNEUROSCI.19-23-10221.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippiello P, Hoxha E, Volpicelli F, Lo Duca G, Tempia F, Miniaci MC (2015) Noradrenergic modulation of parallel fiber-Purkinje cell synapse. Neuropharmacology 89:33–42. 10.1016/j.neuropharm.2014.08.016 [DOI] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Südhof TC, Linden DJ (2003) Phosphorylation of RIM1α by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell 115:49–60. 10.1016/s0092-8674(03)00727-x [DOI] [PubMed] [Google Scholar]
- Luján R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P (1996) Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci 8:1488–1500. 10.1111/j.1460-9568.1996.tb01611.x [DOI] [PubMed] [Google Scholar]
- Ma C, Li W, Xu Y, Rizo J (2011) Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat Struct Mol Biol 18:542–549. 10.1038/nsmb.2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31:463–475. 10.1016/s0896-6273(01)00375-0 [DOI] [PubMed] [Google Scholar]
- Martín R, Durroux T, Ciruela F, Torres M, Pin JP, Sánchez-Prieto J (2010) The metabotropic glutamate receptor mGlu7 activates phospholipase C, translocates munc-13-1 protein, and potentiates glutamate release at cerebrocortical nerve terminals. J Biol Chem 285:17907–17917. 10.1074/jbc.M109.080838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín R, Ferrero JJ, Collado-Alsina A, Aguado C, Luján R, Torres M, Sánchez-Prieto J (2018) Bidirectional modulation of glutamatergic synaptic transmission by metabotropic glutamate type 7 receptors at Schaffer collateral-CA1 hippocampal synapses. J Physiol 596:921–940. 10.1113/JP275371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus L, Lee CK, Altas B, Sertel SM, Weyand K, Rizzoli SO, Rhee JS, Brose N, Imig C, Cooper BH (2020) Ultrastructural correlates of presynaptic functional heterogeneity in hippocampal synapses. Cell Rep 30:3632–3643. 10.1016/j.celrep.2020.02.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millán C, Luján R, Shigemoto R, Sánchez-Prieto J (2002) Subtype-specific expression of group III metabotropic glutamate receptors and Ca2+ channels in single nerve terminals. J Biol Chem 277:47796–47803. 10.1074/jbc.M207531200 [DOI] [PubMed] [Google Scholar]
- Neher E, Brose N (2018) Dynamically primed synaptic vesicle states: key to understand synaptic short-term plasticity. Neuron 100:1283–1291. 10.1016/j.neuron.2018.11.024 [DOI] [PubMed] [Google Scholar]
- Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S (2000) cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol 2:805–811. 10.1038/35041046 [DOI] [PubMed] [Google Scholar]
- Parfitt KD, Doze VA, Madison DV, Browning MD (1992) Isoproterenol increases the phosphorylation of the synapsins and increases synaptic transmission in dentate gyrus, but not in area CA1, of the hippocampus. Hippocampus 2:59–64. 10.1002/hipo.450020108 [DOI] [PubMed] [Google Scholar]
- Ramírez-Franco J, Bartolomé-Martín D, Alonso B, Torres M, Sánchez-Prieto J (2014) Cannabinoid type 1 receptors transiently silence glutamatergic nerve terminals of cultured cerebellar granule cells. PLoS One 9:e88594. 10.1371/journal.pone.0088594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebola N, Reva M, Kirizs T, Szoboszlay M, Lőrincz A, Moneron G, Nusser Z, DiGregorio DA (2019) Distinct nanoscale calcium channel and synaptic vesicle topographies contribute to the diversity of synaptic function. Neuron 104:693–710. 10.1016/j.neuron.2019.08.014 [DOI] [PubMed] [Google Scholar]
- Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Südhof TC, Takahashi M, Rosenmund C, Brose N (2002) β phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell 108:121–133. 10.1016/S0092-8674(01)00635-3 [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16:1197–1207. 10.1016/s0896-6273(00)80146-4 [DOI] [PubMed] [Google Scholar]
- Sadana R, Dessauer C (2009) Physiological roles for G protein-regulated adenylyl cyclase isoform: insights from knockout and overexpression studies. Neurosignals 17:5–22. 10.1159/000166277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safo PK, Regehr WG (2005) Endocannabinoids control the induction of cerebellar LTD. Neuron 48:647–659. 10.1016/j.neuron.2005.09.020 [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E (2001) Preferential potentiation of fast-releasing synaptic vesicles by cAMP at the calyx of Held. Proc Natl Acad Sci U S A 98:331–336. 10.1073/pnas.021541098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E (2003) Direct modulation of synaptic vesicle priming by GABA(B) receptor activation at a glutamatergic synapse. Nature 424:775–778. 10.1038/nature01859 [DOI] [PubMed] [Google Scholar]
- Sakamoto H, Ariyoshi T, Kimpara N, Sugao K, Taiko I, Takikawa K, Asanuma D, Namiki S, Hirose K (2018) Synaptic weight set by Munc13-1 supramolecular assemblies. Nat Neurosci 21:41–49. 10.1038/s41593-017-0041-9 [DOI] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA (1996) Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron 16:797–803. 10.1016/s0896-6273(00)80099-9 [DOI] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF (2001) Morphological correlates of functionally defined synaptic vesicle populations. Nat Neurosci 4:391–395. 10.1038/86042 [DOI] [PubMed] [Google Scholar]
- Schmidt M, Evellin S, Weernink PA, von Dorp F, Rehmann H, Lomasney JW, Jakobs KH (2001) A new phospholipase-C-calcium signalling pathway mediated by cyclic AMP and a Rap GTPase. Nat Cell Biol 3:1020–1024. 10.1038/ncb1101-1020 [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E (1999) Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23:399–409. 10.1016/S0896-6273(00)80789-8 [DOI] [PubMed] [Google Scholar]
- Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, Zhang C, Tamamoto A, Satoh T, Miyazaki J-I, Seino S (2007) Essential role of Epac/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci U S A 104:19333–19338. 10.1073/pnas.0707054104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm DR, Hansel C, Hacker B, Parent A, Linden DJ (1998) Impaired cerebellar long-term potentiation in type I adenylyl cyclase mutant mice. Neuron 20:1199–1210. 10.1016/s0896-6273(00)80500-0 [DOI] [PubMed] [Google Scholar]
- Thomas GM, Geny B, Cockcroft S (1991) Identification of a novel cytosolic polyphosphoinositide-specific phospholipase C (PLC-86) as the major G-protein regulated enzyme. EMBO J 10:2507–2512. 10.1002/j.1460-2075.1991.tb07790.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Liu X, Vickstrom C, Li Y, Yu L, Lu Y, Smrcka AV, Liu QS (2017) The Epac-phospholipase Cε pathway regulates endocannabinoid signaling and cocaine-induced disinhibition of ventral tegmental area dopamine neurons. J Neurosci 37:3030–3044. 10.1523/JNEUROSCI.2810-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL Jr, Cheng X (2012) Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci U S A 109:18613–18618. 10.1073/pnas.1210209109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul Haq R, Anderson M, Liotta A, Shafiq M, Sherkheli MA, Heinemann U (2016) Pretreatment with β-adrenergic receptor agonists facilitates induction of LTP and sharp wave ripple complexes in rodent hippocampus. Hippocampus 26:1486–1492. 10.1002/hipo.22665 [DOI] [PubMed] [Google Scholar]
- Villacres EC, Wong ST, Chavkin C, Storm DR (1998) Type I adenylyl cyclase mutant mice have impaired mossy fiber long-term potentiation. J Neurosci 18:3186–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Wu Z, Kindsvogel W, Prichard L, Storm DR (1994) Synergistic activation of the wild type adenylyl cyclase by Ca2+ by Gs-coupled receptors in vivo. J Biol Chem 269:25400–25405. [PubMed] [Google Scholar]
- Wójtowicz AM, Fidzinski P, Heinemann U, Behr J (2010) Beta-adrenergic receptor activation induces long-lasting potentiation in burst-spiking but not regular-spiking cells at CA1 subiculum synapses. Neuroscience 171:367–372. 10.1016/j.neuroscience.2010.09.028 [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG (1999) Presynaptic strontium dynamics and synaptic transmission. Biophys J 76:2029–2042. 10.1016/S0006-3495(99)77360-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shu X, Liu D, Shang Y, Wu Y, Pei L, Xu X, Tian Q, Zhang J, Qian K, Wang YX, Petralia RS, Tu W, Zhu LQ, Wang JZ, Lu Y (2012) EPAC null mutation impairs learning and social interactions via aberrant regulation of miR-124 and Zif268 translation. Neuron 73:774–788. 10.1016/j.neuron.2012.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Sakaba T (2010) cAMP modulates intracellular Ca2+ sensitivity of fast-releasing synaptic vesicles at the calyx of Held synapse. J Neurophysiol 104:3250–3260. 10.1152/jn.00685.2010 [DOI] [PubMed] [Google Scholar]
- Yawo H. (1996) Noradrenaline modulates neurotransmitter release by enhancing the Ca2+ sensitivity of exocytosis in the chick ciliary presynaptic terminal. J Physiol 493:385–391. 10.1113/jphysiol.1996.sp021390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawo H. (1999) Involvement of cGMP-dependent protein kinase in adrenergic potentiation of transmitter release from the Calyx-type presynaptic terminal. J Neurosci 19:5293–5300. 10.1523/JNEUROSCI.19-13-05293.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieba BJ, Artamonov MV, Jin L, Momotani K, Ho R, Franke AS, Neppl RL, Stevenson AS, Khromov AS, Chrzanowska-Wodnicka M, Somlyo AV (2011) The cAMP-responsive Rap1 guanine nucleotide exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity. J Biol Chem 286:16681–16692. 10.1074/jbc.M110.205062 [DOI] [PMC free article] [PubMed] [Google Scholar]