

Abstract
African great apes harbor at least twelve Plasmodium species, some of which have served as a source of human infection. It is now well-established that Plasmodium falciparum emerged following the transmission of a gorilla parasite, perhaps within the last 10,000 years, while Plasmodium vivax emerged from an ancient parasite lineage that infected humans and apes in Africa before the spread of the Duffy-negative mutation eliminated the parasite from humans there. Both human parasites have greatly reduced genetic diversity and a relative excess of nonsynonymous mutations, consistent with severe genetic bottlenecks followed by rapid population expansion. A newly uncovered Plasmodium species widespread in chimpanzees, gorillas and bonobos places the origin of Plasmodium malariae in Africa. Here, we review what is known about the origins and evolutionary history of all human-infective Plasmodium species, the time and circumstances of their emergence, and the diversity, host specificity and zoonotic potential of their ape counterparts.
Keywords: malaria, Plasmodium, chimpanzee, gorilla, evolution, cross species transmission, interspecies gene transfer
1. HUMAN-INFECTIVE PLASMODIUM SPECIES
Malaria is a vector-borne disease of humans caused by apicomplexan parasites in the genus Plasmodium. With more than 220 million clinical cases and over 400,000 deaths in 2018, malaria remains a major public health threat that affects over 3 billion people in tropical and subtropical regions worldwide (3). Although mammals, birds and reptiles carry more than 250 named (and many more as yet unnamed) Plasmodium species, there are only five parasites that are known to be transmitted by Anopheles mosquitoes from one human host to another. These include Plasmodium falciparum, the most prevalent and virulent human parasite, which causes the great majority (>90%) of malaria-related deaths, especially in sub-Saharan Africa (1, 122); Plasmodium vivax, which is geographically more widespread than P. falciparum and responsible for about half of all malaria-related morbidity outside of Africa (8); Plasmodium malariae, which is common throughout the tropics, but less virulent than P. falciparum or P. vivax, and frequently found in multi-species infections (31, 91, 125); and Plasmodium ovale wallikeri and Plasmodium ovale curtisi, two sympatric sibling species, which are present in Africa and parts of Asia but not South America, and like P. malariae, only rarely cause disease (126).
In addition to these traditional human Plasmodium species, there are several primate parasites that are capable of causing malaria in humans (111). The clinically most relevant is Plasmodium knowlesi, which is a parasite of long-tailed (Macaca fascicularis) and pigtailed (Macaca nemestrina) macaques that zoonotically infects humans throughout Southeast Asia (121, 137). Although this emerging pathogen does not seem to be transmitted between humans, it is the most common cause of human malaria in Malaysia (133). Two other macaque parasites, Plasmodium cynomolgi and Plasmodium inui, are also capable of infecting humans, but do so only rarely and mainly cause asymptomatic infections (30, 62, 127). In South America, Plasmodium simium infects several New World monkey species, but is restricted to southeastern Brazil (15, 40). This parasite emerged in primates of the Atlantic Forest following the introduction of P. vivax, but has more recently been observed to cause zoonotic outbreaks in human communities where P. vivax has long been eradicated (15). Finally, Plasmodium brasilianum, which infects New World monkeys throughout Central and South America, has also been implicated as a zoonotic parasite (40). However, P. brasilianum and P. malariae are morphologically, biologically and genetically near-identical, suggesting that they represent members of the same Plasmodium species (67).
While the primate reservoirs of the zoonotic malarias are well documented, the origins of the human-specific Plasmodium species have been the subject of much debate (74). One hypothesis, popular until about 10 years ago, suggested that humans and chimpanzees acquired P. falciparum-like infections from their common ancestor and that these parasites had co-evolved with their respective host species for millions of years (44, 60). In contrast, P. vivax was believed to have emerged in Southeast Asia following the transmission of a macaque parasite to humans (46, 88). However, the discovery of parasites closely related to P. falciparum and P. vivax in wild-living chimpanzees (Pan troglodytes), bonobos (Pan paniscus) and western gorillas (Gorilla gorilla) has contradicted these theories, indicating that both human pathogens emerged much more recently from parasites infecting African apes (74, 113). Although the origins of P. malariae, P. ovale curtisi and P. ovale wallikeri have not yet been fully deciphered, all of these also have closely related chimpanzee, bonobo and/or gorilla counterparts (64, 70, 72). Given the propensity of primate parasites to switch host species, the success of malaria control and elimination efforts depends, at least in part, on understanding when and how the human-infective Plasmodium species evolved, and what conditions prompted their emergence.
2. FOUR DIVERGENT PRIMATE PLASMODIUM LINEAGES
Plasmodium parasites were first identified in African apes about 100 years ago, when researchers studying human malaria also examined blood from wild-caught chimpanzees and gorillas (4, 114). Three morphologically distinct forms were identified, two of which were indistinguishable from P. falciparum and P. malariae, and named P. reichenowi and P. rodhaini, respectively. The third resembled P. vivax and P. ovale, and was named P. schwetzi (28). While P. rodhaini and P. schwetzi parasites were never genetically characterized, one strain of P. reichenowi isolated from a chimpanzee in 1968 has been maintained (28). DNA sequences from this parasite confirmed that it was closely related to, but distinct from, P. falciparum (44–45). Because P. falciparum differed considerably from the other human parasites in both its biology and morphology, it was placed (with P. reichenowi) in a separate taxon, Laverania, which was eventually recognized as a distinct subgenus within Plasmodium (16, 50).
In the past decade, studies of both captive and wild-living apes have uncovered numerous additional parasites (42–43, 64, 66, 70–72, 94, 109), indicating the existence of at least seven distinct Laverania species in addition to P. falciparum. Ape samples also yielded sequences closely related to P. malariae, P. vivax and P. ovale (64, 70), consistent with the descriptions of P. rodhaini and P. schwetzi in these animals. However, analysis of the biology and life cycle of the newly discovered parasites has been difficult due to the endangered species status of their hosts. As an alternative, recent efforts have focused on generating genome sequences of Laverania (97–98, 124), P. vivax-like (52, 75) and P. malariae-like (120) parasites from both chimpanzee and gorilla samples.
Figure 1 depicts the relationships among mammalian Plasmodium species derived from their nuclear genomes. The topology differs from mitochondrial gene phylogenies in the order of divergence among the major lineages (55, 66, 74). Most previous analyses have been consistent in identifying Laverania as the most divergent mammalian parasite lineage. Mitochondrial DNA phylogenies then show a split between rodent and all other primate parasite species (28, 66, 74), consistent with the existing taxonomy, which places rodent parasites in one subgenus, Vinckeia, and all non-Laverania primate parasites in the Plasmodium subgenus (50). However, trees of nuclear (120) and apicoplast (5) sequences cluster the rodent parasites with the P. ovale lineage (Figure 1), making the Plasmodium subgenus paraphyletic. For consistency, each of the four major non-Laverania lineages should thus be placed in a separate subgenus. Since P. malariae is the type species of the Plasmodium subgenus (50), the lineages comprised of P. ovale species, and of P. vivax and related monkey parasite species, should each be given a new subgenus name. This may also apply to a clade of divergent parasites recently identified in lemurs, which has not yet been fully characterized (69, 99).
Figure 1.

Evolutionary relationships of primate and rodent Plasmodium species inferred from whole genome sequences. A maximum-likelihood tree of deduced protein sequences from 1,693 orthologous genes is shown. Two strains of P. praefalciparum are included to illustrate the position of P. falciparum within the radiation of this species. The G03 sequence was obtained by SNP calling of published reads using P. falciparum for reference (97). Human parasites are shown in red, with the zoonotic parasite P. knowlesi indicated by a red asterisk. The tree was rooted using the bird parasites P. gallinaceum and P. relictum as outgroups. Sequence data and gene annotations were derived from published genome assemblies (6, 12, 25, 58, 75, 96–98, 102–103, 120) and PlasmoDB. All relationships were supported by 100% of bootstrap replicates except for the position of P. cynomolgi. The scale bar indicates amino acid substitutions per site.
3. ORIGIN OF PLASMODIUM FALCIPARUM
3.1. Laverania species and their host tropism
Initially, the Laverania subgenus was thought to consist of only P. falciparum and P. reichenowi. However, beginning in 2009 additional Plasmodium species were discovered, first by characterizing blood or tissue samples of sanctuary apes (42, 64, 94, 109, 116), and later by amplifying parasite sequences from fecal samples collected non-invasively from chimpanzees, bonobos and gorillas across their ranges in sub-Saharan Africa (70–73, 109) (Figure 2). Phylogenetic analyses of mitochondrial, apicoplast and nuclear gene fragments identified several well-supported clades, which in addition to P. falciparum indicated the existence of seven ape Laverania species (Figure 3). These include P. reichenowi, P. gaboni and P. billcollinsi from chimpanzees, P. lomamiensis from bonobos, and P. praefalciparum, P. adleri and P. blacklocki from western gorillas. One additional species, P. billbrayi, was proposed on the basis of subdivisions in mitochondrial phylogenies (66), but this classification was not supported by analyses of nuclear loci (73). Except for P. lomamiensis, the distinctiveness of each of these ape Laverania species has been confirmed by whole genome analyses (97–98, 124).
Figure 2.

Geographic distribution and host association of ape Plasmodium parasites in sub-Saharan Africa. The distribution of (a) Laverania, and (b) P. vivax is shown, as determined by PCR amplification of parasite sequences from blood and fecal samples of captive and wild apes as well as mosquitoes. Field sites are shown in relation to the ranges of the four subspecies of the common chimpanzee (P. t. verus, black, upper left inset; P. t. ellioti, purple; P. t. troglodytes, magenta; P. t. schweinfurthii, blue), Cross River (G. g. diehli, white stripe), western lowland (G. g. gorilla, red stripe), and eastern lowland (G. b. graueri, yellow stripe) gorillas, and bonobos (P. paniscus, orange) (20). Sites where ape malaria was detected are highlighted in yellow, aqua and red indicating that chimpanzees, gorillas or both were infected, respectively. Circles, diamonds, and hexagons identify locations where fecal samples were collected from chimpanzees, gorillas, or both species, respectively. Squares indicate bonobo sites, with black stripes indicating one site where P. lomamiensis was detected in only one of 69 fecal samples (72). Triangles and asterisks denote ape sanctuaries and mosquito collection sites, respectively. At two sites, blood and tissue samples were obtained from injured or deceased chimpanzees habituated to human observers (39, 64). The star denotes the location where a European forester became infected with ape P. vivax (110). Data were compiled from published studies (10, 14, 39, 52, 64, 70–74, 79, 93, 97, 104, 110).
Figure 3.
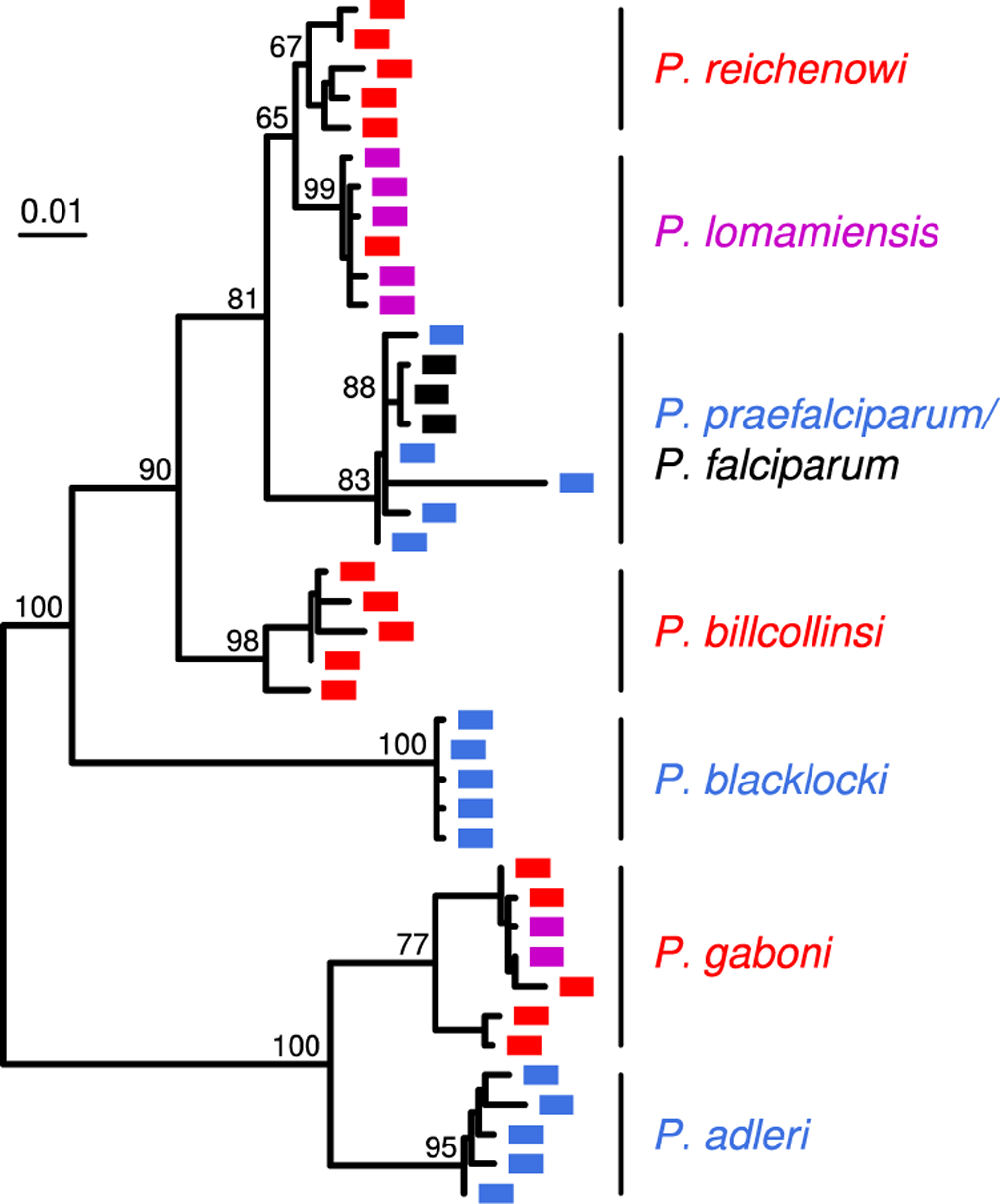
Evolutionary relationships of Laverania species. A maximum likelihood tree (midpoint rooted) of mitochondrial (cytb) parasite sequences (956 bp) from chimpanzee (red), gorilla (blue), bonobo (purple) and human (black) samples is shown. Sequences were selected from published data (63–64, 66, 70, 72–73, 94, 124) to illustrate the diversity within each species (percent bootstrap values are shown for inter-species branches). The scale bar indicates nucleotide substitutions per site.
Laverania infections are common and wide-spread in all four chimpanzee subspecies as well as western lowland gorillas (G. g. gorilla), with fecal detection rates ranging between 22% and 40% (Figure 2). However, the true prevalence of Laverania infections is probably close to 100% in many wild communities, since studies in humans have shown that only a fraction of blood-infected individuals also shed parasite DNA into their stool (76). Interestingly, bonobos were initially thought to lack Laverania parasites (70). However, a study of over 1,500 bonobo fecal samples from 11 field sites showed that bonobos are, in fact, commonly infected with two Laverania species, albeit only at a single location in the eastern most part of their range. In the Lomami River basin, bonobos were found to harbor the P. gaboni variant that is highly prevalent in neighboring eastern chimpanzees (Figure 2a), as well as a new species, P. lomamiensis, that appears to be largely bonobo-specific (Figure 3). Except for a single positive sample at a second site (hatched in Figure 2a), all other bonobo communities were Plasmodium-negative. Studies of parasite seasonality as well as bonobo population structure, plant consumption and fecal microbiome composition failed to provide an explanation for this geographic restriction (72), pointing to other factors, such as a paucity of transmitting vectors or the spread of a malaria protective mutation as potential reasons. Interestingly, eastern gorillas (G. beringei) also appear to lack Laverania infections (70), although fewer samples and sites have been tested (Figure 2). It will be important to determine whether the factors that restrict Laverania infections in bonobos also do so in eastern gorillas.
While Laverania parasites exhibit strict host specificity in the wild (70), captive chimpanzees are susceptible to the gorilla parasites P. praefalciparum and P. adleri, while captive gorillas can harbor the chimpanzee parasite P. reichenowi (93). Indeed, the P. praefalciparum reference genome (G01) was generated from a blood stage infected chimpanzee (97). However, after the screening of more than 3,000 fecal samples with highly sensitive PCR methods, P. praefalciparum, P. adleri and P. blacklocki have never been detected in wild chimpanzees, while P. reichenowi, P. gaboni and P. billcollinsi have never been detected in wild gorillas. This is despite the fact that transmitting Anopheles vectors are highly promiscuous, both with respect to the ape parasites that they carry as well as their feeding behavior, which is not limited to any one particular host (10, 79, 104). Similarly, captive bonobos and chimpanzees (albeit not gorillas) frequently become infected with P. falciparum, but the human parasite has never been detected in wild apes. Thus, there are strong isolating mechanisms in wild settings that are not recapitulated in captivity. Interestingly, the same degree of host specificity does not apply to Laverania parasites infecting wild chimpanzees and bonobos, presumably because these hosts are extremely closely related. In addition to P. gaboni, bonobos can harbor P. reichenowi, while eastern chimpanzees are susceptible to P. lomamiensis, although the latter cross-infections are exceedingly rare (72). The host specificity of Laverania in wild apes appears to be determined by multiple barriers at the vector-host, vector-parasite and/or host-parasite interface.
3.2. Laverania evolution
All P. falciparum strains lie within the radiation of a genetically much more diverse parasite species found only in wild gorillas (70). This makes it clear that P. falciparum arose from a recent zoonotic transmission, and the gorilla parasite has been named P. praefalciparum in recognition of this (113).
In the past, all attempts to infer dates of divergence among Plasmodium species had to either assume that these parasites evolved at the same rate as other eukaryotes, or that certain parasites co-speciated with their hosts. Thus, it was posited that P. falciparum and P. reichenowi had co-diverged with the ancestors of humans and chimpanzees, around 6–7 million years ago (44, 60). While the discovery of the gorilla origin of P. falciparum has refuted this hypothesis, the ancestors of P. praefalciparum and P. reichenowi could still have co-diverged with the ancestors of gorillas and chimpanzees (24), around 8–9 million years ago. This revised co-speciation hypothesis is supported by two observations. First, the bonobo-specific species, P. lomamiensis, is closely related to P. reichenowi (Figure 3), and together with P. praefalciparum these form a triad of parasites whose relationships reflect those of their hosts (72). Second, the genetic distance between P. gaboni and P. adleri is similar to that between P. praefalciparum and P. reichenowi (Figure 3) implying that, within each chimpanzee and gorilla parasite pair, divergence occurred at about the same time (97). The simplest explanation of this coincidence is that both splits reflect the divergence of their hosts. Under this scenario, the common ancestor of the entire Laverania clade might have existed ~25–30 million years ago. Since humans are also apes, this co-divergence hypothesis requires that our ancestors lost their Laverania, which is not without precedent given the curious absence of infection in most bonobos (72). The inactivation of the gene that produces N-glycolylneuraminic acid in human ancestors (discussed below) after their divergence from the chimpanzee lineage (26) probably impaired the ability of Laverania to infect human erythrocytes (82).
An alternative approach to dating the Laverania phylogeny is to combine estimates of the mutation rate per replication cycle (13, 27) with the numbers of replication cycles a parasite is likely to undergo in a year (97, 124). Such a molecular clock likely provides a good estimate of the short term rate of P. falciparum evolution. However, using this rate to extrapolate back to species divergence dates yields timescales that seem unrealistically short. For example, it has been suggested that the common ancestor of P. reichenowi and P. praefalciparum (and P. falciparum) existed ~190,000 years ago, and that the last common ancestor of the entire Laverania clade existed only about one million years ago (97). If the co-divergence hypothesis is true, the longer term rates of evolution must be at least one order of magnitude slower than the short term rate of nucleotide changes. Discrepancies of this kind, where long term rates of evolution seem much slower than short term rates, are becoming apparent in many contexts (57, 84).
Although genome-wide analyses have confirmed that Laverania represent distinct, non-recombining species, two examples of gene transfer have been documented (Figure 4). The first involves the transmission of an 8 kb segment of chromosome 4 from an ancestor of P. adleri to an ancestor of P. praefalciparum (Figure 4a). This region is of interest because it contains four genes, two of which encode the reticulocyte-binding-like homologous protein 5 (RH5) and the cysteine-rich protective antigen (CyRPA) (124), which together form part of a multi-protein complex that mediates the binding of the parasite to red blood cells (135). The second gene transfer appears to be the result of an ancient introgression event that involved the common ancestor of P. reichenowi and P. lomamiensis and an ancestor of P. billcollinsi (Figure 4b). This event resulted in the transfer of as many as 24 genes on six different chromosomes, most of which appear to encode proteins that are exported into the erythrocyte (107). Although rare, gene transfers between Laverania species may have played an important role in the evolution of their host interactions.
Figure 4.

Evidence of gene transfer between Laverania species. (a) A maximum likelihood tree of rh5 gene sequences (827 bp) is shown, indicating a gene transfer from an ancestor of P. adleri to the ancestor of P. praefalciparum (97, 124). P. lomamiensis sequences were obtained by limiting dilution PCR as described (72) (GenBank accession numbers MN175633 – MN175635); other sequences were taken from published data (97, 106, 124). (b) A maximum likelihood tree of fikk9.6 gene sequences (800 bp) indicating an ancient introgression event that involved an ancestor of P. reichenowi and P. lomamiensis and an ancestor of P. billcollinsi (107). Parasite sequences derived from chimpanzee, gorilla, bonobo and human samples are shown in red, blue, purple and black, respectively, except for the P. praefalciparum reference sequence (indicated by asterisk), which is shown in blue for consistency but was obtained from a captive chimpanzee (97). Bootstrap values are shown for inter-species branches. The scale bars indicate nucleotide substitutions per site.
3.3. Emergence of P. falciparum
It has long been speculated that P. falciparum has unusually low genetic diversity and that this reflects a recent severe genetic bottleneck (117), but the reason(s) for such a bottleneck were unclear. It is now apparent that the genomes of global P. falciparum strains exhibit levels of nucleotide diversity that are about an order of magnitude lower than what is observed in ape Laverania species (97, 124). This difference could reflect an extreme bottleneck resulting from a single gorilla-to-human transmission event. Since P. praefalciparum has not been found in humans, even in individuals who live in close proximity to infected apes (41, 81, 93, 123, 134), the transfer likely involved parasites with a unique set of mutations that rendered them infectious to humans. It has been argued that a single transmission scenario is inconsistent with the presence of a small number of “dimorphic” genes (e.g., msp1) in P. falciparum, where two highly divergent alleles must have originated from different gorilla parasites (97). However, most apes are multiply infected (70). Thus, a single transmission event could have involved a genetically diverse set of sporozoites, such that any mutations required for success in the new host could, through recombination, be present in different genetic backgrounds. Immediately after the transmission event, random genetic drift would have led to the loss of most genetic diversity, but strong selection could have maintained both haplotypes at a limited number of dimorphic loci.
Long-term molecular clocks based on the assumption of host-parasite co-divergence (60), or mammalian evolutionary rates (92), place the last common ancestor of P. falciparum at around 200,000 years ago. However, the short-term molecular clock for P. falciparum described above is consistent with a common ancestor within the last 5,000–10,000 years (74, 124). This shorter timescale is consistent with the fact that human mutations conferring resistance to falciparum malaria, such as mutant alleles at the loci encoding β-globin and glucose-6-phosphate dehydrogenase, are generally estimated to have arisen within the last 10,000 years (56). The shorter timescale is also consistent with the suggestion that the hunter-gather lifestyle of humans prior to that period was likely not conducive to maintaining Laverania infections (22).
An alternative scenario has recently been proposed by Otto and colleagues (97). They depicted P. falciparum and P. praefalciparum as diverged clades, even though for many genes the human parasites fall within the radiation of the gorilla parasites, indicating that the two species have not undergone complete lineage sorting. They used a Bayesian coalescent based approach (53), calibrated with the short-term molecular clock, and estimated that the gorilla and human parasites started to diverge around 50,000 years ago, but continued to undergo gene flow, from P. praefalciparum to P. falciparum. However, given the expected large asymmetry in effective population size between the two species, this scenario would be difficult to distiguish from one where there was a much more recent origin of P. falciparum (with no subsequent gene flow) from a large and possibly geographically structured ancestral P. praefalciparum population, especially if the host switch involved a strong bottleneck. Indeed, the estimated extent of post-divergence gene flow for genic sequences, which are less troublesome to align, was not significantly greater than zero (97).
This alternative (older) divergence scenario also leaves a number of unanswered questions. Given that modern humans emerged in Africa at least 200,000 years ago (18), it is unclear what occurred 50,000 years ago to suddenly allow P. praefalciparum to infect humans as well as gorillas? And if genetic exchange between the two parasites continued for thousands of years subsequently, why is there not more genetic diversity in the human parasite given that selection for human adaptive alleles, even at a number of different loci, would not be expected to homogenize a very large fraction of the genome? We suggest that the dearth of genetic diversity across the P. falciparum genome more likely reflects a severe bottleneck, resulting from a single transmission of a limited number of parasites, and that the timing of that event was precipitated by a change in human demography within the last 10,000 years.
3.4. Adaptation of P. falciparum to the human host
The gene encoding cytidine monophospho-N-acetylneuraminic acid hydroxylase (CMAH), which converts N-acetylneuraminic acid (Neu5Ac) to N-glcolylneuraminic acid (Neu5Gc), has been inactivated on the lineage leading to humans, but remains intact in African apes (26). Consequently, sialoglycoproteins on human erythrocytes contain only Neu5Ac, whereas those on ape erythrocytes contain both Neu5Gc and Neu5Ac (89). Sialoglycoproteins act as receptors for Laverania erythrocyte binding like (EBL) proteins. Interestingly, all known strains of P. falciparum contain a nucleotide deletion within the gene that encodes the erythrocyte binding-like protein 165 (EBA165), whereas this gene is intact in all other Laverania species (108). Correction of the inactivating mutation in P. falciparum yielded viable parasites. However, transcription of the restored eba165 gene was silenced, suggesting that expression of EBA165 is incompatible with parasite growth in human erythrocytes (108). Moreover, neither the restored P. falciparum EBA165, nor the wildtype P. reichenowi EBA165, were able to utilize Neu5Ac (108). Since erythrocyte invasion involves multiple, partially redundant receptor-ligand interactions (9, 34), an eba165 inactivating mutation in an ape parasite that used this protein as a dominant invasion ligand would be expected to switch to a subdominant invasion ligand. The erythrocyte binding-like protein 175 (EBA175) of P. falciparum, which binds glycophorin A (95, 129), is a major invasion ligand that interacts with both Neu5Ac and Neu5Gc containing sialoglycoproteins (95, 132). Thus, a switch from EBA165 to EBA175 could have increased the likelihood of transmission and/or enhanced parasite growth in humans (108). A similar loss of invasion ligands changed the host preference of P. knowlesi during in vitro adaptation to human red blood cells (38, 87).
The genes encoding two essential invasion ligands, CyRPA and RH5, lie within a short region of chromosome 4 that was transferred from an ancestor of P. adleri to the ancestor of P. praefalciparum, prior to its transfer to humans (124). CyRPA and RH5 form part of a multi-protein complex that enables RH5 to bind the host protein basigin (48, 135), and this is the only receptor-ligand interaction that is essential for erythrocyte invasion by P. falciparum (36). RH5 binds basigin in a host-specific manner (132, 136), and the rh5 gene has been under positive selection during the diversification of the Laverania (106). However, since all known strains of P. praefalciparum carry the P. adleri-derived allele (Figure 4a), this gene transfer alone cannot explain the cross-species transmission event.
To examine the role of RH5 in the origin of P. falciparum, a recent study investigated the ability of various chimpanzee and gorilla Laverania RH5 proteins to bind to ape and human basigin (49). RH5 proteins from three chimpanzee Laverania species bound chimpanzee, but not gorilla, basigin, while RH5 proteins from two gorilla Laverania species bound gorilla, but not chimpanzee, basigin. Remarkably, however, the RH5 proteins of both P. adleri and P. gaboni, the gorilla and chimpanzee parasites that form the Laverania clade most distant from P. falciparum (Figure 1), also bound human basigin, whereas those from P. reichenowi and P. billcollinsi did not (49). As expected, RH5 from P. praefalciparum, which is closely related to that from P. adleri (Figure 4a), also bound human basigin. However, the fact that P. reichenowi RH5 failed to bind human basigin suggests that, prior to the gene transfer, P. praefalciparum RH5 would not have bound human basigin either. Thus, the horizontal gene transfer appears to have been an absolute prerequisite for the subsequent jump into humans. However, the protein binding data alone do not explain why P. adleri and P. gaboni were not transmitted to humans. Moreover, the identification of one amino acid substitution that abrogates the binding of P. falciparum RH5 to gorilla basigin (49) may explain why gorillas are not susceptible to P. falciparum infection, but does not explain why P. praefalciparum does not normally infect humans. In the context of the discussion of the emergence of P. falciparum, the RH5-basigin binding data do not seem to provide evidence favouring one scenario over the other.
4. ORIGIN OF HUMAN PLASMODIUM VIVAX
4.1. P. vivax-related parasites in non-human primates
PCR testing of blood and fecal samples of captive and wild-living apes has identified parasites that are very closely related to human P. vivax (71). In humans, P. vivax grows to lower blood titers than P. falciparum due to its preference for immature red blood cells (83, 90) and this results in less parasite DNA shedding into fecal samples. Thus, the prevalence rates of P. vivax in wild ape populations are likely much higher than the observed fecal PCR detection rates of 4% to 8% (71). Indeed, P. vivax has been detected in all four chimpanzee subspecies, bonobos, western and eastern gorillas as well as several forest Anopheles species (Figure 2b), indicating that transmission rates are of sufficient intensity to maintain wide-spread endemic infection (64, 71–72, 110). Although New World monkeys are susceptible to P. vivax (15, 19, 40), PCR testing of nearly 1,000 blood samples from 16 African monkey species failed to detect P. vivax infection (71).
Molecular epidemiological studies of ape P. vivax also revealed the existence of a related Plasmodium species, termed P. carteri, in wild apes (74). This species appears to be very rare, having been detected thus far in only two wild chimpanzees sampled at different locations in Cameroon. Phylogenetic analyses of a number of mitochondrial, apicoplast and nuclear gene fragments revealed that P. carteri represents the closest known relative of P. vivax, which is significant because it places the last common ancestor of these two Plasmodium species in Africa (74).
4.2. Evolutionary history of human and ape P. vivax
A phylogenetic network of all published ape P. vivax genomes, together with a global sample of human P. vivax strains (52, 61, 75), reveals that the ape parasites are about 10-fold more diverse than the human strains and fall into two divergent lineages (Figure 5a). For different genes, the relationship of ape and human P. vivax strains appears to depend on the number of sequences analyzed, as well as the closeness of the outgroup (75). Phylogenetic analyses of individual gene sequences yielded two different topologies. For one third of genes, a tree rooted with P. cynomolgi placed human and ape P. vivax as sister clades (not shown). However, for the remaining two thirds, the same analysis placed human P. vivax within the radiation of the ape parasites (Figure 5b). Since ape and human parasites have long been geographically separated, it is likely that the discrepant topologies are an indication of ongoing lineage sorting in the absence of introgression (52, 75).
Figure 5.

Evolutionary relationships of ape and human P. vivax strains. (a) A phylogenetic network based on pairwise distances of coding sequences from all published ape and selected human P. vivax genomes. Sequences from Gilabert and colleagues (triangles) were used as published (Pvl01) or obtained from sequencing reads following the SNP calling procedure described by the authors (52), with the dominant alleles (by read count) used at heterozygous positions. Other genome-wide ape P. vivax sequences from Loy and colleagues (circles) and human P. vivax sequences (black) from Hupalo and colleagues have been described (61, 75). The network is based on 79,492 nucleotide positions from 280 genes with data included from all samples. Ape P. vivax sequences obtained from chimpanzees in Cameroon, Gabon and Cote d’Ivoire are shown in red (52, 75) and one mosquito-derived genome sequence from Gabon is shown in orange (52). (b) A maximum likelihood tree of partial sequences (405 bp) from one nuclear gene (PVP01_1418300) illustrates an example of human P. vivax falling within the ape P. vivax radiation. The tree is rooted with P. cynomolgi. Human (black), and chimpanzee (red), gorilla (blue) and mosquito (orange) derived ape P. vivax sequences are as in (a), except for the addition of an additional chimpanzee-derived (Pvl06) parasite (52). Parasite sequences were generated from ape blood and fecal samples described (52, 75). Bootstrap values >70% are shown. The scale bars indicate nucleotide substitutions per site.
Ape P. vivax appears to circulate freely between chimpanzees, bonobos and gorillas, and thus lacks the host specificity that characterizes Laverania parasites (71–72). Indeed, ape P. vivax has been shown to infect and cause disease in a Duffy-positive European forester (110), and human P. vivax has been transmitted to wild-living monkeys in southeastern Brazil, generating P. simium (15, 19, 40). Thus, while the cross-species transmission of an ape-specific parasite cannot be ruled out, it seems more likely that human P. vivax emerged from an ancient stock of parasites that infected both apes and humans in Africa, until the spread of the Duffy negative mutation eliminated the parasite from humans there (71, 74). Interestingly, the Duffy-negative mutation remains at a very high frequency across much of central Africa, in a geographic area that overlaps current and past ranges of apes (59), suggesting that the possibility of infection by ape P. vivax has maintained selection pressure for the mutant allele. Under this scenario, global human P. vivax strains represent a lineage that escaped out of Africa, undergoing a genetic bottleneck in the process. This may have occurred when modern humans first left Africa ~75,000 year ago (101), or as part of more recent migration events. Following its egress from Africa, P. vivax initially spread to Asia and Europe, and most likely from Europe into the Americas (21, 51, 131). Strains of P. vivax now infecting humans in Madagascar and East Africa appear to reflect reintroductions from Asia (37).
It has been argued that current data are insufficient to rule out an Asian origin of human P. vivax (52, 110). However, this claim ignores the existence of P. carteri as the closest relative of P. vivax (Figure 5b), which indicates a long history of this lineage in Africa (74). Furthermore, an important feature of recent nuclear (Figure 1) and apicoplast (5) phylogenies is the position of P. vivax as an outgroup to the clade of Plasmodium species that infect primates in Southeast Asia. The conclusion that P. vivax originated in Southeast Asia (46, 88) has always been at odds with the very high frequency of the protective Duffy negative mutation in central Africa (85–86), but not Asia (59). However, the position of P. gonderi, from African monkeys, at the base of this lineage, followed by P.vivax diverging before the radiation of the Asian monkey parasites, is consistent with this entire lineage having emerged in Africa, with migration to Asia occurring subsequent to the divergence of the P. vivax ancestor. This revised understanding of the phylogenetic position of P. vivax also renders redundant speculations of which Asian primate parasite is its closest relative (128), since it is now clear that P. vivax is equally distantly related to all of them (Figure 1).
4.3. No evidence of host-specific adaptations of P. vivax
Genome analyses have shown that human P. vivax strains contain inactivating mutations in three genes that encode reticulocyte binding proteins (rbp2d, rbp2e and rbp3), while chimpanzee and gorilla P. vivax strains encode intact orthologs (75). When the binding capacity of two of these proteins (RBPe and RBP3) to human and ape red blood cells was investigated, the results failed to yield evidence of host-specific barriers to red blood cell invasion (75). Thus, the maintenance of intact rbp2e and rbp3 genes in ape P. vivax cannot be explained by invoking an interaction with a host-specific erythrocyte receptor.
Conversely, two reported “ape-specific adaptations” of human P. vivax, invoked as supporting an Asian origin (52), are based on errors. The reported pseudogenization of a cytoadherence-linked asexual gene (clag) in chimpanzee, but not human, P. vivax genomes reflects a mis-annotation, while the gene that encodes the Duffy binding protein 2 (dbp2), which was reportedly lost in two chimpanzee reference strains (52), is intact in other ape P. vivax genomes (75).
5. PATTERNS OF POLYMORPHISM IN P. FALCIPARUM AND P. VIVAX
Both P. falciparum and P. vivax have greatly reduced levels of genetic diversity compared to their ape-infecting relatives (Figure 6a). Comparing gene sequences, the average pairwise nucleotide diversity among P. falciparum strains from across the world is about 0.0005 per site (124). Considering only 4-fold degenerate sites within genes, where all nucleotide changes are synonymous and thus likely to be effectively neutral, the value is about 0.0008 per site. When multiple P.reichenowi and P. gaboni genome sequences became available, it was found that both of these values were about 8–12 times higher (124), and a similarly higher nucleotide diversity was subsequently identified in the other ape Laverania species (97). Nucleotide diversity among global strains of human P. vivax is nearly twice as high as in P. falciparum (92), with values of 0.0009 and 0.0012 for genes and 4-fold degenerate sites, respectively, but again values in ape P. vivax are about an order of magnitude higher than that (75). These results indicate that both human parasites have passed through relatively recent genetic bottlenecks, although as discussed above the particular events involved were likely different.
Figure 6.
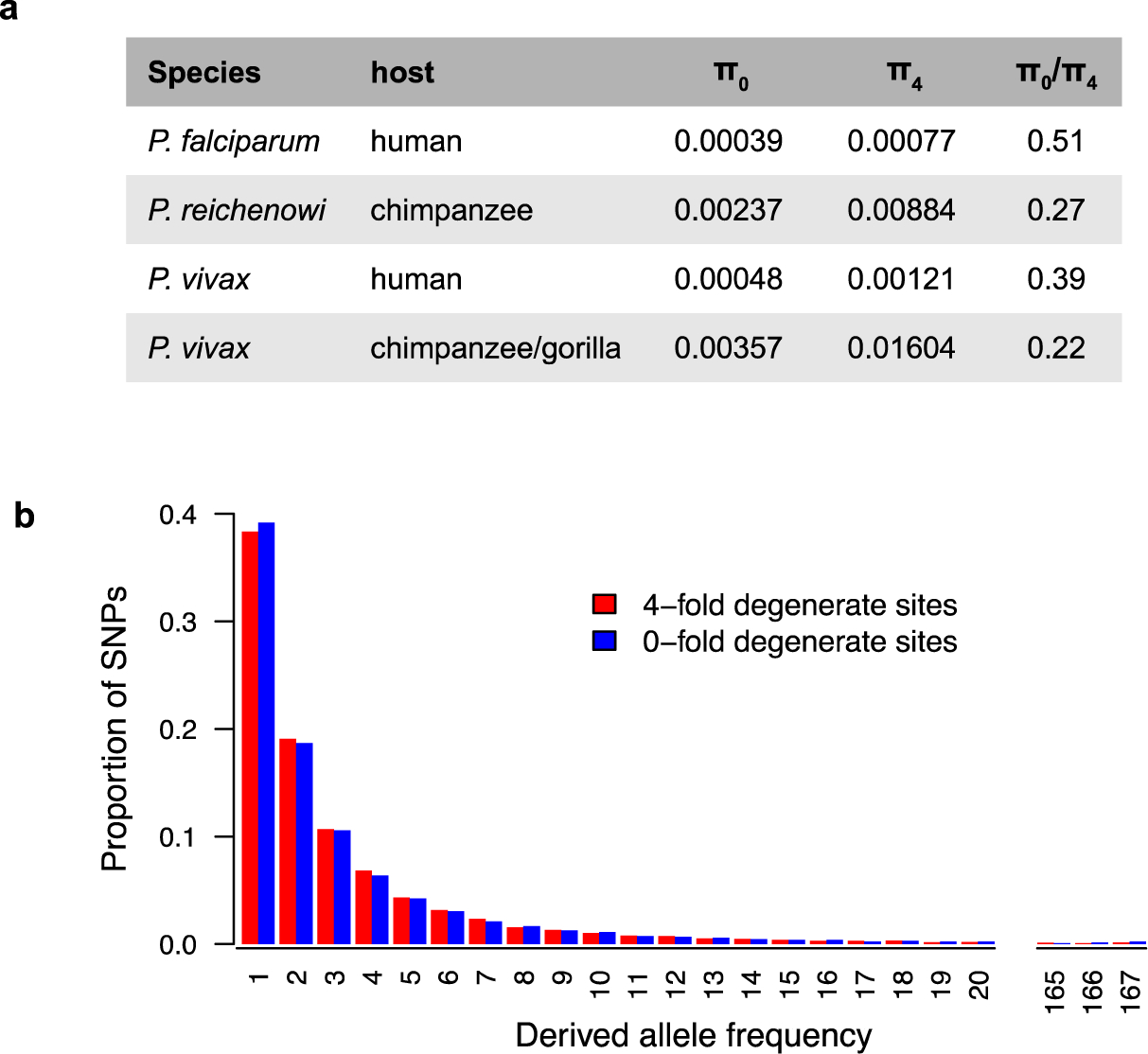
Excess of nonsynonymous polymorphisms in human versus ape Plasmodium parasites. (a) The mean pairwise diversity at zero-fold (π0) and four-fold (π4) degenerate sites is shown for P. falciparum and P. reichenowi as well as human and ape P. vivax derived from published data (2, 75, 105, 124) Both human parasites have much higher π0/π4 ratios than their ape counterparts, indicating a relative excess of nonsynonymous polymorphisms. (b) Site frequency spectra of polymorphisms at zero-fold degenerate (blue) and four-fold degenerate (red) sites in human P. vivax show almost identical patterns, indicating relaxed selection on nonsynonymous polymorphisms. Data were derived from 197 human P. vivax strains from southeast Asia (105) and processed as described (75).
When nucleotide differences are divided into nonsynonymous (NS) and synonymous (S) changes, i.e., those that do, or do not, change the encoded protein sequence, it is apparent that the nature of the genetic variation also differs between human and ape parasites. Among strains of human P. vivax the ratio of NS:S changes is about 1.4, whereas among ape P. vivax strains the ratio is only half of that (75). When expressed as the ratio of diversity at 0-fold degenerate sites (which can only change nonsynonymously) to diversity at 4-fold degenerate sites, the value is again about two times higher among human than ape P. vivax strains (Figure 6a). This suggests that selection against nonsynonymous mutations has been much less effective in the human parasite. Synonymous mutations should be able to spread through the population under random genetic drift, but selection against nonsynonymous mutations should constrain their spread. As a consequence, the site frequency spectrum (SFS) for polymorphisms at 4-fold degenerate should reflect the past demography of the population, whereas the SFS for 0-fold degenerate sites typically has a much larger excess of singletons, i.e., sites where the mutation is found only once in the population (65). Remarkably, the SFSs for polymorphisms at 0-fold and 4-fold degenerate sites are almost identical in human P. vivax (Figure 6b), suggesting that these nonsynonymous mutations are segregating as if effectively neutral. This most likely reflects a rapid expansion of the human P. vivax population, subsequent to its escape from Africa.
An earlier examination of the nature of polymorphism among 25 P. falciparum isoaltes from Senegal found the same high ratio of NS/S and the similarity between the SFSs for synonymous and nonsynonymous polymorphisms (24). It was suggested that this could reflect the unusual life cycle of P. falciparum, where transmission through the mosquito vector leads to recurrent population bottlenecks (23). However, the fact that the NS/S ratio is much lower in ape Laverania rules out the life cycle as the cause. It was noted that the SFS for synonymous polymorphisms was consistent with a major population expansion (24), and again this may explain the reduced selection against nonsynonymous mutations. This genome-wide excess of nonsynonymous mutations in human malaria parasites has consequences for identifying genes under selection (106).
6. OTHER HUMAN MALARIA SPECIES WITH RELATIVES IN WILD APES
6.1. African origin of P. malariae
P. malariae in humans has long been known to closely resemble P. rodhaini in African apes and P. brasilianum in New World monkeys, but the exact relationships of these parasites have remained unclear (29, 31). Although never genetically characterized, P. rodhaini was transmitted from chimpanzees to humans by blood inoculation, while P. malariae was transmitted from humans to chimpanzees by the same route (17, 29, 118–119). Similarly, P. brasilianum was transmitted from New World monkeys to humans by infective mosquitoes and transferred back to monkeys via infected blood (29, 33). Sequence analysis of a limited number of nuclear loci showed that P. brasilianum and P. malariae are genetically near-identical (67, 120). Together, these findings have raised the possibility that P. malariae, P. rodhaini and P. brasilianum represent members of the same species (29, 67, 112).
Initial field studies of wild apes revealed that chimpanzees harbor P. malariae-related parasites, but only few mitochondrial (cytb) and nuclear (18S rRNA) gene sequences were recovered (64, 70). Analysis of additional sequences from captive (42, 54, 66, 93) and free-ranging apes (64, 70, 76), as well as humans, has identified three distinct parasite lineages (Figure 7). Interestingly, all human-derived sequences fall in just one of these lineages (M1), which also includes P. brasilianum. M1 sequences were also identified in captive chimpanzees and bonobos, probably as a result of infection with human parasites (66, 76). In contrast, P. malariae-like parasites from free-ranging apes are more diverse (76) and form what appears to be a distinct (M1-like) lineage (Figure 7).
Figure 7.

Evolutionary relationships of ape and human P. malariae parasites. A maximum likelihood tree (midpoint rooted) of published (42, 54, 64, 66, 70, 75, 93) and newly derived (asterisks; GenBank accession numbers MN175636–MN175639) mitochondrial (cytb) sequences (576 bp) from chimpanzee (red), gorilla (blue), bonobo (purple), and mosquito (orange) samples as well as P. brasilianum (grey) and representative human P. malariae (black) strains is shown. The phylogeny depicts three lineages of P. malariae and P malariae-related strains (labelled at the side), one of which (M2) is likely to represent a new species tentatively named P. africanum sp. nov. All human P. malariae sequences fall into the M1 lineage together with P. brasilianum and parasite sequences from some captive apes. In trees of longer (2 kb) mitochondrial (76) and nuclear (120) loci (not shown), parasites from wild chimpanzees and gorillas form a distinct M1-like lineage. It is currently unknown whether wild apes also harbor M1 parasites, and whether M1 and M1-like parasites form two sister clades or one falls into the radiation of the other. Bootstrap values greater than 70% are denoted. The scale bar indicates nucleotide substitutions per site.
Genome sequences have been obtained for two P. malariae-like chimpanzee parasites (PmlGA01 and PmlGA02) from a wildlife reserve in Gabon (120). Although closely related to human P. malariae, the chimpanzee parasites were far more divergent from the human parasites (~1%) than these were from each other (~0.03%). Moreover, the two chimpanzee parasites exhibited ~0.7% nucleotide sequence diversity, although this distance is likely inflated since the read library of PmlGA02 contains large numbers of contaminating P. reichenowi and P. vivax reads. Based on their divergence from the human parasites, the two chimpanzee parasites represent members of the M1-like lineage. It remains to be determined whether M1 and M1-like parasites form sister clades, or whether one of these lineages falls within the radiation of the other.
Fecal-based screening of wild apes also identified a more divergent parasite lineage that forms a sister clade to M1 and M1-like parasites (Figure 7). Members of this M2 lineage were identified in free-ranging chimpanzees, gorillas and bonobos (70, 72) as well as in A. vinckei mosquitoes captured at two locations in Gabon (79). Indeed, all of 10 Anopheles-derived P. malariae-related sequences were of the M2 genotype (Figure 7), suggesting active transmission in wild ape populations. Although only a few of these parasites have so far been detected, members of this Plasmodium lineage do not appear to be host specific (Figure 7). Moreover, they have a wide geographic distribution, ranging from Cameroon, Gabon and the Republic of Congo to the eastern parts of the Democratic Republic of the Congo (70, 72, 79). Given their distance from the M1 and M1-like lineages in mitochondrial (Figure 7) and nuclear (not shown) phylogenies, it appears that the M2 parasites represent a distinct Plasmodium species. The finding of this new Plasmodium species in chimpanzees, bonobos and gorillas places the deeper ancestry of the P. malariae lineage in Africa. We propose to name the new species (M2) Plasmodium africanum sp. nov. to highlight its widespread distribution among African great apes. The African origin of P. malariae makes it clear that P. brasilianum represents an anthroponosis that was acquired by New World monkeys from P. malariae-infected humans who migrated from Africa (67, 112).
The finding of M1 parasites in captive apes provides further evidence that ape, human and monkey versions of P. malariae represent members of the same species (67, 112). What remains unknown is whether P. malariae-like (M1-like) parasites from wild apes represent a distinct species and if so, whether they have the potential to infect humans. Given the results of the P. rodhaini transmission studies, one would expect P. malariae-like parasites to circulate freely between humans and apes (17, 29, 31, 118–119). However, it is unknown whether M1, M1-like and/or M2 parasites were transmitted in these experiments. Although two M1-like genomes have been sequenced, introgression between human and chimpanzee parasites has not been examined (120). Genome-wide analysis of additional M1-like and M2 parasites will be necessary to determine the number of P. malariae-related species and their host specificities.
6.2. Ape-infecting relatives of P. ovale wallikeri and P. ovale curtisi
PCR testing of blood and fecal samples from captive and free-ranging apes also uncovered close relatives of P. ovale curtisi and P. ovale wallikeri (43, 64, 70, 80–81). However, despite screening thousands of fecal samples, P. ovale curtisi-like parasites have so far only been identified in five wild chimpanzees from Cote d’Ivoire (64), Cameroon (70), and the Republic of Congo (70), and one wild bonobo from the Democratic Republic of the Congo (72), while P. ovale wallikeri-like parasites have only been detected in two wild gorillas from Cameroon (70) and the Central African Republic (80–81), respectively. In each case, the gene sequences amplified were identical or near-identical to the corresponding sequences from human parasites.
To explain the existence of two sympatric P. ovale species in humans, it has been suggested that they were acquired from a single primate source on two separate occasions, spaced as many as a million years apart (125–126). However, given that wild apes harbor near-identical parasites, it seems more likely that P. ovale curtisi and P. ovale wallikeri already evolved as separate species prior to their introduction into humans. Existing data would suggest that humans acquired P. ovale curtisi from chimpanzees, while P. ovale wallikeri was transmitted from gorillas. However, since both parasite species have been identified in captive chimpanzees (43), it is not yet clear whether they really have a restricted host range in the wild. Interestingly, epidemiological studies conducted over 50 years ago showed that the areas with the highest P. ovale prevalence in humans coincided with the natural range of African apes, suggesting circulation of these parasites between man and ape (68, 77). It is thus possible that ape and human versions of P. ovale curtisi and P. ovale wallikeri represent members of the same Plasmodium species.
7. HUMAN ZOONOTIC RISK
7.1. Anopheles vectors of ape Plasmodium parasites
Although identifying the mosquito vectors that transmit Plasmodium parasites among apes is critical to gauge human zoonotic risk, few such studies have been conducted (7, 79, 104, 110). Initial analyses of whole mosquito DNA identified P. praefalciparum in Anopheles moucheti and ape P. vivax in A. moucheti and A. vinckei (104, 110), while subsequent more comprehensive studies identified A. vinckei, A. moucheti and A. marshallii as competent vectors of ape Laverania and non-Laverania parasites (79). Analyses of whole mosquitoes and dissected salivary glands showed that A. vinckei was commonly infected with both chimpanzee and gorilla derived Laverania parasites as well as ape P. vivax and P. africanum (the newly described species related to P. malariae), while A. moucheti and A. marshallii were mostly infected with ape P. vivax (79). Importantly, landing catches showed that all three of these Anopheles species feed on humans (79). A more recent study of forest Anopheles trapped near sanctuary chimpanzees confirmed their generalistic host preference, although none of the mostly A. obscurus mosquitoes was positive for ape Plasmodium parasites (7). Together these data indicate that forest mosquitoes are opportunistic in their feeding behavior and could thus serve as bridge vectors for human infection, as has been demonstrated for P. knowlesi (35, 121) and other Plasmodium parasites infecting wild macaques (78).
7.2. Human zoonotic risk of ape Laverania infections
Extensive molecular epidemiological studies have shown that Laverania infected apes do not currently serve as a reservoir of human infection. PCR screening of more than 5,000 humans living or working in close proximity to infected apes in Cameroon, Gabon and the Central African Republic, including individuals who observe habituated gorillas in their natural habitat, ruled out blood infection with ape Laverania parasites (41, 81, 93, 123). Moreover, PCR testing of fecal samples from over 500 Cameroonian forest dwellers failed to show evidence of abortive liver infection with ape Plasmodium parasites (76). These data, together with results from experimental transmission studies of P. reichenowi a century ago (11), indicate that ape Laverania parasites do not normally infect humans. However, since it is unknown how often and under what circumstance humans are bitten by mosquitoes that carry ape Laverania sporozoites, it is also unclear whether existing transmission barriers are indeed near-insurmountable or only rarely tested.
7.3. Human zoonotic risk of ape non-Laverania infections
In contrast to Laverania species, non-Laverania parasites have a much less restricted host tropism and may thus pose a greater zoonotic risk. Ape P. vivax infects chimpanzees, bonobos and gorillas in the wild (64, 71–72), and has been identified in a Duffy positive human traveler (110), while human P. vivax has been found in a confiscated orangutan (115) and (as P. simium) in several free-ranging New World monkey species (15, 19, 40). Similarly, human P. malariae has been found in captive chimpanzees and bonobos (64, 76) and (as P. brasilianum) in free-ranging New World monkey species (40, 67). Finally, “P. schwetzi”, which may have contained P. vivax-, P. ovale curtisi- and/or P. ovale wallikeri-like parasites, was experimentally transmitted from apes to humans by infective mosquitoes and blood inoculation on multiple occasions (29, 32, 118–119). Thus, current evidence indicates that non-Laverania parasites have a promiscuous host tropism, with documented bi-directional transmission between humans and African apes as well as humans and New World monkeys.
Among the non-Laverania parasites, ape P. vivax is of particular interest, since its zoonotic and disease causing potential has been documented (110). P. vivax is increasingly detected in humans in Africa, including in Duffy-negative individuals (130). It will thus be important to determine whether any of these cases reflect zoonotic transmissions from apes. Ape and human P. vivax strains may also be able to infect the same hosts and undergo genetic exchange. Since such recombination could generate biologically more versatile parasites, it will be important to search for signs of introgression in areas where an influx of Duffy positive humans through commerce and travel coincides with forest encroachment.
8. CONCLUSIONS
African apes have long been known to harbor different Plasmodium species, but their numbers, evolutionary relationships, geographic distribution, prevalence, and mammalian host and vector associations have only recently been determined. Non-invasive fecal-based methods have identified at least 12 Plasmodium species in wild-living apes, two of which have given rise to P. falciparum and P vivax (70–71, 75, 97). Moreover, ape parasites may well be responsible for the emergence of P. malariae, P. ovale curtisi and P. ovale wallikeri. Although orangutans and gibbons are also naturally Plasmodium infected, the infecting parasites appear to be of Asian macaque origin (100, 115).
It remains to be seen whether any of the newly identified ape Plasmodium species poses a zoonotic risk. Consideration of such risks could be important, especially for non-Laverania parasites, at a time when efforts to reduce and eliminate malaria in human populations are intensifying (47). With increasing human activities in natural environments the risk of zoonoses increases as exemplified by malaria outbreaks of P. knowlesi in Southeast Asia and P. simium in southeastern Brazil. The lack of in vitro culture systems poses a significant challenge to the functional characterization of ape Plasmodium parasites, but whole genome sequencing represents an important step towards understanding their biology (75, 97–98, 120, 124). Such analyses have already revealed a number of unexpected findings, such as horizontal transfer and adaptive processes of key invasion ligands (97, 107–108, 124). However, additional parasite genome sequences from wild apes, in particular of P. malariae-like and P. ovale-like parasites, will be needed to ascertain their relationship to their human counterparts. In general, new insight into the biology of ape malaria parasites will inform malaria eradication efforts by identifying potential zoonotic threats (47).
ACKNOWLEDGEMENTS
We thank Weimin Liu for artwork, Ameena Al-Amin for manuscript preparation, and Richard Carter and Konrad Lohse for discussion. This work was supported by grants from the National Institutes of Health R01 AI 091595, R01 AI 120810, R01 AI 050529 and P30 AI 045008.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.2014. Severe malaria. Trop Med Int Health 19 Suppl:7–131. World Health Organ. [DOI] [PubMed] [Google Scholar]
- 2.Malar. Genom. Epidemiol. Netw. 2016. Pf3k pilot data release 5. MalariaGEN https://www.malariagen.net/data/pf3k-5
- 3.World Health Organ. 2019. World Malaria Report 2019, Geneva: World Health Organ. [Google Scholar]
- 4.Adler S 1923. Malaria in chimpanzees in Sierra Leone. Annals of Tropical Medicine & Parasitology 17:13–8 [Google Scholar]
- 5.Arisue N, Hashimoto T, Kawai S, Honma H, Kume K, Horii T. 2019. Apicoplast phylogeny reveals the position of Plasmodium vivax basal to the Asian primate malaria parasite clade. Sci Rep 9:7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auburn S, Bohme U, Steinbiss S, Trimarsanto H, Hostetler J, et al. 2016. A new Plasmodium vivax reference sequence with improved assembly of the subtelomeres reveals an abundance of pir genes. Wellcome Open Res 1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bakker JW, Loy DE, Takken W, Hahn BH, Verhulst NO. 2019. Attraction of mosquitoes to primate odours and implications for zoonotic Plasmodium transmission. Med Vet Entomol, in press doi: 10.1111/mve.12402 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Battle KE, Lucas TCD, Nguyen M, Howes RE, Nandi AK, et al. 2019. Mapping the global endemicity and clinical burden of Plasmodium vivax, 2000–17: a spatial and temporal modelling study. Lancet 394:332–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baum J, Maier AG, Good RT, Simpson KM, Cowman AF. 2005. Invasion by P. falciparum merozoites suggests a hierarchy of molecular interactions. PLoS Pathog 1:e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bitome-Essono PY, Ollomo B, Arnathau C, Durand P, Mokoudoum ND, et al. 2017. Tracking zoonotic pathogens using blood-sucking flies as ‘flying syringes’. Elife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blacklock B, and Adler S. . 1922. A parasite resembling Plasmodium falciparum in a chimpanzee. Ann. Trop. Med. Parasitol XVI:99–106 [Google Scholar]
- 12.Bohme U, Otto TD, Cotton JA, Steinbiss S, Sanders M, et al. 2018. Complete avian malaria parasite genomes reveal features associated with lineage-specific evolution in birds and mammals. Genome Res 28:547–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bopp SE, Manary MJ, Bright AT, Johnston GL, Dharia NV, et al. 2013. Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Genet 9:e1003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boundenga L, Ollomo B, Rougeron V, Mouele LY, Mve-Ondo B, et al. 2015. Diversity of malaria parasites in great apes in Gabon. Malar J 14:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brasil P, Zalis MG, de Pina-Costa A, Siqueira AM, Junior CB, et al. 2017. Outbreak of human malaria caused by Plasmodium simium in the Atlantic Forest in Rio de Janeiro: a molecular epidemiological investigation. Lancet Glob Health 5:e1038–e46 [DOI] [PubMed] [Google Scholar]
- 16.Bray RS. 1958. Studies on malaria in chimpanzees. VI. Laverania falciparum. Am J Trop Med Hyg 7:20–4 [DOI] [PubMed] [Google Scholar]
- 17.Bray RS. 1960. Studies on malaria in chimpanzees. VIII. The experimental transmission and pre-erythrocytic phase of Plasmodium malariae, with a note on the host-range of the parasite. Am J Trop Med Hyg 9:455–65 [DOI] [PubMed] [Google Scholar]
- 18.Brown FH, McDougall I, Fleagle JG. 2012. Correlation of the KHS Tuff of the Kibish Formation to volcanic ash layers at other sites, and the age of early Homo sapiens (Omo I and Omo II). J Hum Evol 63:577–85 [DOI] [PubMed] [Google Scholar]
- 19.Buery JC, Rodrigues PT, Natal L, Salla LC, Loss AC, et al. 2017. Mitochondrial genome of Plasmodium vivax/simium detected in an endemic region for malaria in the Atlantic Forest of Espirito Santo state, Brazil: do mosquitoes, simians and humans harbour the same parasite? Malar J 16:437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caldecott JO, and Miles L. 2005. World Atlas of Great Apes and their Conservation. Berkley, California, USA: University of California Press [Google Scholar]
- 21.Carter R 2003. Speculations on the origins of Plasmodium vivax malaria. Trends Parasitol 19:214–9 [DOI] [PubMed] [Google Scholar]
- 22.Carter R, Mendis KN. 2002. Evolutionary and historical aspects of the burden of malaria. Clin Microbiol Rev 15:564–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang HH, Moss EL, Park DJ, Ndiaye D, Mboup S, et al. 2013. Malaria life cycle intensifies both natural selection and random genetic drift. Proc Natl Acad Sci U S A 110:20129–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang HH, Park DJ, Galinsky KJ, Schaffner SF, Ndiaye D, et al. 2012. Genomic sequencing of Plasmodium falciparum malaria parasites from Senegal reveals the demographic history of the population. Mol Biol Evol 29:3427–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chien JT, Pakala SB, Geraldo JA, Lapp SA, Humphrey JC, et al. 2016. High-Quality genome assembly and annotation for Plasmodium coatneyi, generated using single-molecule real-time PacBio technology. Genome Announc 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chou HH, Takematsu H, Diaz S, Iber J, Nickerson E, et al. 1998. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc Natl Acad Sci U S A 95:11751–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Claessens A, Hamilton WL, Kekre M, Otto TD, Faizullabhoy A, et al. 2014. Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of var genes during mitosis. PLoS Genet 10:e1004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coatney GRCWE, Warren M, Contacos PG 1971. The Primate Malarias. Washington, DC: US Government Printing Office [Google Scholar]
- 29.Coatney GR. 1971. The simian malarias: zoonoses, anthroponoses, or both? Am J Trop Med Hyg 20:795–803 [DOI] [PubMed] [Google Scholar]
- 30.Coatney GR, Chin W, Contacos PG, King HK. 1966. Plasmodium inui, a quartan-type malaria parasite of Old World monkeys transmissible to man. J Parasitol 52:660–3 [PubMed] [Google Scholar]
- 31.Collins WE, Jeffery GM. 2007. Plasmodium malariae: parasite and disease. Clin Microbiol Rev 20:579–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Contacos PG, Coatney GR, Orihel TC, Collins WE, Chin W, Jeter MH. 1970. Transmission of Plasmodium schwetzi from the chimpanzee to man by mosquito bite. Am J Trop Med Hyg 19:190–5 [DOI] [PubMed] [Google Scholar]
- 33.Contacos PG, Lunn JS, Coatney GR, Kilpatrick JW, Jones FE. 1963. Quartan-type malaria parasite of New World monkeys transmissible to man. Science 142:676. [DOI] [PubMed] [Google Scholar]
- 34.Cowman AF, Tonkin CJ, Tham WH, Duraisingh MT. 2017. The Molecular basis of erythrocyte invasion by malaria parasites. Cell Host Microbe 22:232–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cox-Singh J, Davis TM, Lee KS, Shamsul SS, Matusop A, et al. 2008. Plasmodium knowlesi malaria in humans is widely distributed and potentially life threatening. Clin Infect Dis 46:165–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crosnier C, Bustamante LY, Bartholdson SJ, Bei AK, Theron M, et al. 2011. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature 480:534–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Culleton R, Coban C, Zeyrek FY, Cravo P, Kaneko A, et al. 2011. The origins of African Plasmodium vivax; insights from mitochondrial genome sequencing. PLoS One 6:e29137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dankwa S, Lim C, Bei AK, Jiang RH, Abshire JR, et al. 2016. Ancient human sialic acid variant restricts an emerging zoonotic malaria parasite. Nat Commun 7:11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Nys HM, Calvignac-Spencer S, Thiesen U, Boesch C, Wittig RM, et al. 2013. Age-related effects on malaria parasite infection in wild chimpanzees. Biol Lett 9:20121160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deane LM. 1992. Simian malaria in Brazil. Mem Inst Oswaldo Cruz 87 Suppl 3:1–20 [DOI] [PubMed] [Google Scholar]
- 41.Delicat-Loembet L, Rougeron V, Ollomo B, Arnathau C, Roche B, et al. 2015. No evidence for ape Plasmodium infections in humans in Gabon. PLoS One 10:e0126933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duval L, Fourment M, Nerrienet E, Rousset D, Sadeuh SA, et al. 2010. African apes as reservoirs of Plasmodium falciparum and the origin and diversification of the Laverania subgenus. Proc Natl Acad Sci U S A 107:10561–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duval L, Nerrienet E, Rousset D, Sadeuh Mba SA, Houze S, et al. 2009. Chimpanzee malaria parasites related to Plasmodium ovale in Africa. PLoS One 4:e5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Escalante AA, Ayala FJ. 1994. Phylogeny of the malarial genus Plasmodium, derived from rRNA gene sequences. Proc Natl Acad Sci U S A 91:11373–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Escalante AA, Barrio E, Ayala FJ. 1995. Evolutionary origin of human and primate malarias: evidence from the circumsporozoite protein gene. Mol Biol Evol 12:616–26 [DOI] [PubMed] [Google Scholar]
- 46.Escalante AA, Cornejo OE, Freeland DE, Poe AC, Durrego E, et al. 2005. A monkey’s tale: the origin of Plasmodium vivax as a human malaria parasite. Proc Natl Acad Sci U S A 102:1980–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feachem RGA, Chen I, Akbari O, Bertozzi-Villa A, Bhatt S, et al. 2019. Malaria eradication within a generation: ambitious, achievable, and necessary. Lancet 394:1056–112 [DOI] [PubMed] [Google Scholar]
- 48.Galaway F, Drought LG, Fala M, Cross N, Kemp AC, et al. 2017. P113 is a merozoite surface protein that binds the N terminus of Plasmodium falciparum RH5. Nat Commun 8:14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galaway F, Yu R, Constantinou A, Prugnolle F, Wright GJ. 2019. Resurrection of the ancestral RH5 invasion ligand provides a molecular explanation for the origin of P. falciparum malaria in humans. PLoS Biol 17:e3000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garnham PCC. 1964. The subgenera of Plasmodium in mammals. Annales de la Societe Belge de Medecine Tropicale 44:267–72 [PubMed] [Google Scholar]
- 51.Gelabert P, Sandoval-Velasco M, Olalde I, Fregel R, Rieux A, et al. 2016. Mitochondrial DNA from the eradicated European Plasmodium vivax and P. falciparum from 70-year-old slides from the Ebro Delta in Spain. Proc Natl Acad Sci U S A 113:11495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilabert A, Otto TD, Rutledge GG, Franzon B, Ollomo B, et al. 2018. Plasmodium vivax-like genome sequences shed new insights into Plasmodium vivax biology and evolution. PLoS Biol 16:e2006035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gronau I, Hubisz MJ, Gulko B, Danko CG, Siepel A. 2011. Bayesian inference of ancient human demography from individual genome sequences. Nat Genet 43:1031–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hayakawa T, Arisue N, Udono T, Hirai H, Sattabongkot J, et al. 2009. Identification of Plasmodium malariae, a human malaria parasite, in imported chimpanzees. PLoS One 4:e7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayakawa T, Culleton R, Otani H, Horii T, Tanabe K. 2008. Big bang in the evolution of extant malaria parasites. Mol Biol Evol 25:2233–9 [DOI] [PubMed] [Google Scholar]
- 56.Hedrick PW. 2011. Population genetics of malaria resistance in humans. Heredity (London) 107:283–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ho SY, Duchene S, Molak M, Shapiro B. 2015. Time-dependent estimates of molecular evolutionary rates: evidence and causes. Mol Ecol 24:6007–12 [DOI] [PubMed] [Google Scholar]
- 58.Honma H, Kawai S, Motooka D, Nakamura S, Tougan T, et al. 2017. Draft genome sequence of Plasmodium gonderi, a malaria parasite of African Old World monkeys. Genome Announc 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Howes RE, Patil AP, Piel FB, Nyangiri OA, Kabaria CW, et al. 2011. The global distribution of the Duffy blood group. Nat Commun 2:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hughes AL, Verra F. 2010. Malaria parasite sequences from chimpanzee support the co-speciation hypothesis for the origin of virulent human malaria (Plasmodium falciparum). Mol Phylogenet Evol 57:135–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hupalo DN, Luo Z, Melnikov A, Sutton PL, Rogov P, et al. 2016. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat Genet 48:953–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imwong M, Madmanee W, Suwannasin K, Kunasol C, Peto TJ, et al. 2019. Asymptomatic natural human infections with the simian malaria parasites Plasmodium cynomolgi and Plasmodium knowlesi. J Infect Dis 219:695–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Joy DA, Feng X, Mu J, Furuya T, Chotivanich K, et al. 2003. Early origin and recent expansion of Plasmodium falciparum. Science 300:318–21 [DOI] [PubMed] [Google Scholar]
- 64.Kaiser M, Lowa A, Ulrich M, Ellerbrok H, Goffe AS, et al. 2010. Wild chimpanzees infected with five Plasmodium species. Emerg Infect Dis 16:1956–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keightley PD, Campos JL, Booker TR, Charlesworth B. 2016. Inferring the frequency spectrum of derived variants to quantify adaptive molecular evolution in protein-coding genes of Drosophila melanogaster. Genetics 203:975–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krief S, Escalante AA, Pacheco MA, Mugisha L, Andre C, et al. 2010. On the diversity of malaria parasites in African apes and the origin of Plasmodium falciparum from bonobos. PLoS Pathog 6:e1000765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lalremruata A, Magris M, Vivas-Martinez S, Koehler M, Esen M, et al. 2015. Natural infection of Plasmodium brasilianum in humans: Man and monkey share quartan malaria parasites in the Venezuelan Amazon. EBioMedicine 2:1186–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Languillon J 1957. Carte epidemiologique du paludisme au Cameroun. Bull. Soc. Path. Exot 50:585–600 [PubMed] [Google Scholar]
- 69.Larsen PA, Hayes CE, Williams CV, Junge RE, Razafindramanana J, et al. 2016. Blood transcriptomes reveal novel parasitic zoonoses circulating in Madagascar’s lemurs. Biol Lett 12:20150829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu W, Li Y, Learn GH, Rudicell RS, Robertson JD, et al. 2010. Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature 467:420–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu W, Li Y, Shaw KS, Learn GH, Plenderleith LJ, et al. 2014. African origin of the malaria parasite Plasmodium vivax. Nat Commun 5:3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu W, Sherrill-Mix S, Learn GH, Scully EJ, Li Y, et al. 2017. Wild bonobos host geographically restricted malaria parasites including a putative new Laverania species. Nat Commun 8:1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu W, Sundararaman SA, Loy DE, Learn GH, Li Y, et al. 2016. Multigenomic delineation of Plasmodium species of the Laverania subgenus infecting wild-living chimpanzees and gorillas. Genome Biol Evol 8:1929–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Loy DE, Liu W, Li Y, Learn GH, Plenderleith LJ, et al. 2017. Out of Africa: origins and evolution of the human malaria parasites Plasmodium falciparum and Plasmodium vivax. Int J Parasitol 47:87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Loy DE, Plenderleith LJ, Sundararaman SA, Liu W, Gruszczyk J, et al. 2018. Evolutionary history of human Plasmodium vivax revealed by genome-wide analyses of related ape parasites. Proc Natl Acad Sci U S A 115:E8450–E9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loy DE, Rubel MA, Avitto AN, Liu W, Li Y, et al. 2018. Investigating zoonotic infection barriers to ape Plasmodium parasites using faecal DNA analysis. Int J Parasitol 48:531–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lysenko AJ, Beljaev AE. 1969. An analysis of the geographical distribution of Plasmodium ovale. Bull World Health Organ 40:383–94 [PMC free article] [PubMed] [Google Scholar]
- 78.Maeno Y 2017. Molecular epidemiology of mosquitoes for the transmission of forest malaria in south-central Vietnam. Trop Med Health 45:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Makanga B, Yangari P, Rahola N, Rougeron V, Elguero E, et al. 2016. Ape malaria transmission and potential for ape-to-human transfers in Africa. Proc Natl Acad Sci U S A 113:5329–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mapua MI, Fuehrer HP, Petrzelkova KJ, Todd A, Noedl H, et al. 2018. Plasmodium ovale wallikeri in western lowland gorillas and humans, Central African Republic. Emerg Infect Dis 24:1581–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mapua MI, Qablan MA, Pomajbikova K, Petrzelkova KJ, Huzova Z, et al. 2015. Ecology of malaria infections in western lowland gorillas inhabiting Dzanga Sangha protected areas, Central African Republic. Parasitology 142:890–900 [DOI] [PubMed] [Google Scholar]
- 82.Martin MJ, Rayner JC, Gagneux P, Barnwell JW, Varki A. 2005. Evolution of human-chimpanzee differences in malaria susceptibility: relationship to human genetic loss of N-glycolylneuraminic acid. Proc Natl Acad Sci U S A 102:12819–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McQueen PG, McKenzie FE. 2006. Competition for red blood cells can enhance Plasmodium vivax parasitemia in mixed-species malaria infections. Am J Trop Med Hyg 75:112–25 [PMC free article] [PubMed] [Google Scholar]
- 84.Membrebe JV, Suchard MA, Rambaut A, Baele G, Lemey P. 2019. Bayesian inference of evolutionary histories under time-dependent substitution rates. Mol Biol Evol 36:1793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mendis K, Sina BJ, Marchesini P, Carter R. 2001. The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg 64:97–106 [DOI] [PubMed] [Google Scholar]
- 86.Miller LH, Mason SJ, Clyde DF, McGinniss MH. 1976. The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N Engl J Med 295:302–4 [DOI] [PubMed] [Google Scholar]
- 87.Moon RW, Sharaf H, Hastings CH, Ho YS, Nair MB, et al. 2016. Normocyte-binding protein required for human erythrocyte invasion by the zoonotic malaria parasite Plasmodium knowlesi. Proc Natl Acad Sci U S A 113:7231–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mu J, Joy DA, Duan J, Huang Y, Carlton J, et al. 2005. Host switch leads to emergence of Plasmodium vivax malaria in humans. Mol Biol Evol 22:1686–93 [DOI] [PubMed] [Google Scholar]
- 89.Muchmore EA, Diaz S, Varki A. 1998. A structural difference between the cell surfaces of humans and the great apes. Am J Phys Anthropol 107:187–98 [DOI] [PubMed] [Google Scholar]
- 90.Mueller I, Galinski MR, Baird JK, Carlton JM, Kochar DK, et al. 2009. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect Dis 9:555–66 [DOI] [PubMed] [Google Scholar]
- 91.Mueller I, Zimmerman PA, Reeder JC. 2007. Plasmodium malariae and Plasmodium ovale -- the “bashful” malaria parasites. Trends Parasitol 23:278–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Neafsey DE, Galinsky K, Jiang RH, Young L, Sykes SM, et al. 2012. The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet 44:1046–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ngoubangoye B, Boundenga L, Arnathau C, Mombo IM, Durand P, et al. 2016. The host specificity of ape malaria parasites can be broken in confined environments. Int J Parasitol 46:737–44 [DOI] [PubMed] [Google Scholar]
- 94.Ollomo B, Durand P, Prugnolle F, Douzery E, Arnathau C, et al. 2009. A new malaria agent in African hominids. PLoS Pathog 5:e1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Orlandi PA, Klotz FW, Haynes JD. 1992. A malaria invasion receptor, the 175-kilodalton erythrocyte binding antigen of Plasmodium falciparum recognizes the terminal Neu5Ac (alpha 2–3) Gal-sequences of glycophorin A. J Cell Biol 116:901–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Otto TD, Bohme U, Jackson AP, Hunt M, Franke-Fayard B, et al. 2014. A comprehensive evaluation of rodent malaria parasite genomes and gene expression. BMC Biol 12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Otto TD, Gilabert A, Crellen T, Bohme U, Arnathau C, et al. 2018. Genomes of all known members of a Plasmodium subgenus reveal paths to virulent human malaria. Nat Microbiol 3:687–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Otto TD, Rayner JC, Bohme U, Pain A, Spottiswoode N, et al. 2014. Genome sequencing of chimpanzee malaria parasites reveals possible pathways of adaptation to human hosts. Nat Commun 5:4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pacheco MA, Battistuzzi FU, Junge RE, Cornejo OE, Williams CV, et al. 2011. Timing the origin of human malarias: the lemur puzzle. BMC Evol Biol 11:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pacheco MA, Reid MJ, Schillaci MA, Lowenberger CA, Galdikas BM, et al. 2012. The origin of malarial parasites in orangutans. PLoS One 7:e34990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pagani L, Lawson DJ, Jagoda E, Morseburg A, Eriksson A, et al. 2016. Genomic analyses inform on migration events during the peopling of Eurasia. Nature 538:238–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pain A, Bohme U, Berry AE, Mungall K, Finn RD, et al. 2008. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature 455:799–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pasini EM, Bohme U, Rutledge GG, Voorberg-Van der Wel A, Sanders M, et al. 2017. An improved Plasmodium cynomolgi genome assembly reveals an unexpected methyltransferase gene expansion. Wellcome Open Res 2:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paupy C, Makanga B, Ollomo B, Rahola N, Durand P, et al. 2013. Anopheles moucheti and Anopheles vinckei are candidate vectors of ape Plasmodium parasites, including Plasmodium praefalciparum in Gabon. PLoS One 8:e57294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pearson RD, Amato R, Auburn S, Miotto O, Almagro-Garcia J, et al. 2016. Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nat Genet 48:959–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Plenderleith LJ, Liu W, MacLean OA, Li Y, Loy DE, et al. 2018. Adaptive evolution of RH5 in ape Plasmodium species of the Laverania subgenus. MBio 9:e02237–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Plenderleith LJ, Weimin L, Learn GH, Loy DE, Speede S, et al. 2019. Ancient introgression between two ape malaria parasite species. Genome Biol Evol 11:3269–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Proto WR, Siegel SV, Dankwa S, Liu W, Kemp A, et al. 2019. Adaptation of Plasmodium falciparum to humans involved the loss of an ape-specific erythrocyte invasion ligand. Nat Commun, 10:4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Prugnolle F, Durand P, Neel C, Ollomo B, Ayala FJ, et al. 2010. African great apes are natural hosts of multiple related malaria species, including Plasmodium falciparum. Proc Natl Acad Sci U S A 107:1458–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Prugnolle F, Rougeron V, Becquart P, Berry A, Makanga B, et al. 2013. Diversity, host switching and evolution of Plasmodium vivax infecting African great apes. Proc Natl Acad Sci U S A 110:8123–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ramasamy R 2014. Zoonotic malaria - global overview and research and policy needs. Front Public Health 2:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rayner JC. 2015. Plasmodium malariae malaria: From monkey to man? EBioMedicine 2:1023–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rayner JC, Liu W, Peeters M, Sharp PM, Hahn BH. 2011. A plethora of Plasmodium species in wild apes: a source of human infection? Trends Parasitol 27:222–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Reichenow E 1920. Über das Vorkommen der Malariaparasiten des Menschen bei den Afrikanischen Menschenaffen. Centralbl. f. Bakt. I. Abt 85:207–16 [Google Scholar]
- 115.Reid MJ, Ursic R, Cooper D, Nazzari H, Griffiths M, et al. 2006. Transmission of human and macaque Plasmodium spp. to ex-captive orangutans in Kalimantan, Indonesia. Emerg Infect Dis 12:1902–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rich SM, Leendertz FH, Xu G, LeBreton M, Djoko CF, et al. 2009. The origin of malignant malaria. Proc Natl Acad Sci U S A 106:14902–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rich SM, Licht MC, Hudson RR, Ayala FJ. 1998. Malaria’s Eve: evidence of a recent population bottleneck throughout the world populations of Plasmodium falciparum. Proc Natl Acad Sci U S A 95:4425–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rodhain J 1940. Les plasmodium des anthropoids de l’Afriqe centrale et leurs relations avec les plasmodiums humains. Ann. Soc. Belg. Med. Trop 35:69–73 [Google Scholar]
- 119.Rodhain JD R 1943. L’infection a Plasmodium malariae du chimpanze chez l’homme. Etude d’une premiere souche isolee de l’anthropoide Pan satyrus verus. Ann. Soc. Belg. Med. Trop 23:19–46 [Google Scholar]
- 120.Rutledge GG, Bohme U, Sanders M, Reid AJ, Cotton JA, et al. 2017. Plasmodium malariae and P. ovale genomes provide insights into malaria parasite evolution. Nature 542:101–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Singh B, Kim Sung L, Matusop A, Radhakrishnan A, Shamsul SS, et al. 2004. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet 363:1017–24 [DOI] [PubMed] [Google Scholar]
- 122.Snow RW, Sartorius B, Kyalo D, Maina J, Amratia P, et al. 2017. The prevalence of Plasmodium falciparum in sub-Saharan Africa since 1900. Nature 550:515–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sundararaman SA, Liu W, Keele BF, Learn GH, Bittinger K, et al. 2013. Plasmodium falciparum-like parasites infecting wild apes in southern Cameroon do not represent a recurrent source of human malaria. Proc Natl Acad Sci U S A 110:7020–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sundararaman SA, Plenderleith LJ, Liu W, Loy DE, Learn GH, et al. 2016. Genomes of cryptic chimpanzee Plasmodium species reveal key evolutionary events leading to human malaria. Nat Commun 7:11078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sutherland CJ. 2016. Persistent parasitism: The adaptive biology of malariae and ovale malaria. Trends Parasitol 32:808–19 [DOI] [PubMed] [Google Scholar]
- 126.Sutherland CJ, Tanomsing N, Nolder D, Oguike M, Jennison C, et al. 2010. Two nonrecombining sympatric forms of the human malaria parasite Plasmodium ovale occur globally. J Infect Dis 201:1544–50 [DOI] [PubMed] [Google Scholar]
- 127.Ta TH, Hisam S, Lanza M, Jiram AI, Ismail N, Rubio JM. 2014. First case of a naturally acquired human infection with Plasmodium cynomolgi. Malar J 13:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tachibana S, Sullivan SA, Kawai S, Nakamura S, Kim HR, et al. 2012. Plasmodium cynomolgi genome sequences provide insight into Plasmodium vivax and the monkey malaria clade. Nat Genet 44:1051–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Triglia T, Thompson JK, Cowman AF. 2001. An EBA175 homologue which is transcribed but not translated in erythrocytic stages of Plasmodium falciparum. Mol Biochem Parasitol 116:55–63 [DOI] [PubMed] [Google Scholar]
- 130.Twohig KA, Pfeffer DA, Baird JK, Price RN, Zimmerman PA, et al. 2019. Growing evidence of Plasmodium vivax across malaria-endemic Africa. PLoS Negl Trop Dis 13:e0007140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.van Dorp L, Gelabert P, Rieux A, de Manuel M, de-Dios T, et al. 2019. Plasmodium vivax malaria viewed through the lens of an eradicated European strain. Mol Biol Evol, in press doi: 10.1093/molbev/msz264 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wanaguru M, Liu W, Hahn BH, Rayner JC, Wright GJ. 2013. RH5-Basigin interaction plays a major role in the host tropism of Plasmodium falciparum. Proc Natl Acad Sci U S A 110:20735–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.White NJ. 2008. Plasmodium knowlesi: the fifth human malaria parasite. Clin Infect Dis 46:172–3 [DOI] [PubMed] [Google Scholar]
- 134.Woldearegai TG, Lalremruata A, Nguyen TT, Gmeiner M, Veletzky L, et al. 2019. Characterization of Plasmodium infections among inhabitants of rural areas in Gabon. Sci Rep 9:9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wong W, Huang R, Menant S, Hong C, Sandow JJ, et al. 2019. Structure of Plasmodium falciparum Rh5-CyRPA-Ripr invasion complex. Nature 565:118–21 [DOI] [PubMed] [Google Scholar]
- 136.Wright KE, Hjerrild KA, Bartlett J, Douglas AD, Jin J, et al. 2014. Structure of malaria invasion protein RH5 with erythrocyte basigin and blocking antibodies. Nature 515:427–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zaw MT, Lin Z. 2019. Human Plasmodium knowlesi infections in South-East Asian countries. J Microbiol Immunol Infect 52:679–84 [DOI] [PubMed] [Google Scholar]