

Abstract
Objective
To understand the role of gut microbiome in influencing the pathogenesis of neuromyelitis optica spectrum disorders (NMOSDs) among patients of south Indian origin.
Methods
In this case-control study, stool and blood samples were collected from 39 patients with NMOSD, including 17 with aquaporin 4 IgG antibodies (AQP4+) and 36 matched controls. 16S ribosomal RNA (rRNA) sequencing was used to investigate the gut microbiome. Peripheral CD4+ T cells were sorted in 12 healthy controls, and in 12 patients with AQP4+ NMOSD, RNA was extracted and immune gene expression was analyzed using the NanoString nCounter human immunology kit code set.
Results
Microbiota community structure (beta diversity) differed between patients with AQP4+ NMOSD and healthy controls (p < 0.001, pairwise PERMANOVA test). Linear discriminatory analysis effect size identified several members of the microbiota that were altered in patients with NMOSD, including an increase in Clostridium bolteae (effect size 4.23, p 0.00007). C bolteae was significantly more prevalent (p = 0.02) among patients with AQP4-IgG+ NMOSD (n = 8/17 subjects) compared with seronegative patients (n = 3/22) and was absent among healthy stool samples. C bolteae has a highly conserved glycerol uptake facilitator and related aquaporin protein (p59-71) that shares sequence homology with AQP4 peptide (p92-104), positioned within an immunodominant (AQP4 specific) T-cell epitope (p91-110). Presence of C bolteae correlated with expression of inflammatory genes associated with both innate and adaptive immunities and particularly involved in plasma cell differentiation, B cell chemotaxis, and Th17 activation.
Conclusion
Our study described elevated levels of C bolteae associated with AQP4+ NMOSD among Indian patients. It is possible that this organism may be causally related to the immunopathogenesis of this disease in susceptible individuals.
Neuromyelitis optica spectrum disorders1 (NMOSDs) are a group of autoimmune inflammatory diseases that predominantly target optic nerve and spinal cord. A disease-specific serum autoantibody (aquaporin-4 (AQP4) IgG) targets AQP4, which is an important water channel protein in the CNS.2 The resulting cascade of events include activation of compliment produced locally by astrocytes, increased blood-brain permeability and a massive influx of leukocytes (particularly neutrophils and eosinophils), and death of astrocytes, oligodendrocytes, and neurons.3 An interleukin 6 (IL-6)–dependent B cell subpopulation plays an important role in NMOSD pathogenesis, transforming into AQP4-IgG synthesizing plasmablasts during an acute relapse. Hence, NMOSD is primarily considered to be a humorally mediated autoimmune disorder. Evidence also points to a pivotal role for T lymphocytes in NMOSD immunopathogenesis.4,5 AQP4-IgG belongs to the IgG1 subclass, which is T cell dependent for its activity. Experimental models of NMOSD with passive transfer of AQP4-IgG requires a T cell–mediated inflammation to be concurrently present. Among T lymphocytes, T-helper cell 17 (Th17) cells and interleukin 17 (IL-17) secreting T cells are increased in the sera of patients in acute relapse6, and NMOSD lesions have elevated levels of IL-17, suggesting a prominent role for Th17 cells.7 Recently, AQP4-specific T cells were identified in patients with NMOSD that showed a Th17 bias.8
Globally, demographic data suggest that NMOSD, particularly AQP4-IgG–associated NMOSD, is likely to be more prevalent among non-White populations.9 Although genetic association suggests implication of human leucocyte antigen Class 11 molecules,10 very little is understood about the environmental factors related to this disease. The intestinal microbiota is an important environmental factor that can play a role in the initiation and progression of autoimmune disorders. Few studies have looked at the association of gut microbiome with NMOSD. Cree et al.11 found a strong association between AQP4-IgG–associated NMOSD and Clostridium perfringens, a Gram-positive commensal in the gut. This study suggested that aquaporin-4–specific T cells cross-react with an adenosine triphosphate–binding cassette (ABC) transporter of C perfringens that shares sequence homology with AQP4.8,12 Another study of Chinese patients living in Asia showed an abundance of Streptococcus associated with AQP4-IgG–positive NMOSD.13 Our study investigated the gut microbiome among Indian patients with NMOSD to explore the possibility that it may provide a greater understanding of the pathophysiology of disease among our patients.
Methods
Seventeen AQP4-IgG–positive patients (AQP4+), 22 AQP4-IgG–negative patients (AQP4−), and 36 healthy volunteers matched for age and body mass index were included from our registry (table 1). All patients were in remission at the time of sample collection, had AQP4 IgG status confirmed by live cell-based assay, and had given stool and blood samples. One AQP4-IgG+ patient and 6 seronegative patients were not on treatment at the time of sample collection. All others were receiving immunosuppressants. Patients and controls had not been on probiotics or antibiotics in the 3 months preceding stool collection. Histories of bowl surgery, inflammatory bowel disease, and other autoimmune disorders were excluded. Vegetarian status among patients and healthy volunteers was ascertained.
Table 1.
Demographic and clinical features of the study population

Sample collection
Stool samples were collected in containers provided to patients. They were delivered in person or by a relative on the same day of collection over a median period of 3 hours (range: 1–5 hours). Stool samples were then immediately frozen at −80°C until DNA extraction. Peripheral blood mononuclear cells were isolated using ficoll hypaque gradient centrifugation and stored in liquid nitrogen until use.
16S rRNA sequencing of the gut microbiota
DNA was extracted from stool using QIAamp® DNA Stool extraction kit as per the manufacturer's instructions. DNA was transported in dry ice from Nitte University to Brigham and Women's Hospital, Boston, USA, for further analysis. Samples were normalized, and bacterial 16S ribosomal RNA (rRNA) gene was amplified using primers for amplification of V4 region as per protocols described in the Earth Microbiome Project.14 Samples were sequenced by paired end 250 base pair reads at the Harvard Medical School Biopolymer facility using the Illumina MiSeq platform. FastQC program (www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to evaluate sequence quality. We used Qualitative Insights into Microbial Ecology, an open source bioinformatics platform, for downstream analysis.15 Sequences were demultiplexed and denoised using Divisive Amplicon Denoising Algorithm 2. Taxonomy was assigned using reference database SILVA (arb-silva.de). In addition, we used the EZ-Taxon (EzTaxon.org) to identify some species that could not be annotated by SILVA. Alpha diversity was calculated using Faith's phylogenetic diversity (PD), observed operational taxonomic unit (OTU), and Shannon diversity metrics. Beta diversity was calculated using weighted and unweighted UniFrac distances.16 Relative abundance of individual species was calculated as the relative frequency in each sample/sum of relative frequency in all samples tested (n = 75) and expressed as percentage.
We used linear discriminant analysis (LDA) effect size (LEfSe) on the Galaxy Browser, huttenhower.sph.harvard.edu/galaxy, to detect compositional changes in the microbiome in patients with NMOSD and controls.17 Accordingly, a nonparametric factorial Wilcoxon rank-sum test was used initially to detect bacterial taxa with significant (α set at 0.01) differential abundance in patients with NMOSD vs healthy controls. Then, estimate of the effect size of each differentially abundant feature was calculated using LDA, with a cutoff greater than 3.
Gene expression studies and correlation with microbiome
Gene expression studies were performed at Nitte University. In brief, CD4 T cells were sorted from PBMC of patients with AQP4-IgG+ NMOSD (N = 12) and healthy controls (N = 12) using the Miltenyi Biotec positive selection kits. Later, mRNA extraction was performed using RNeasy mini kits (QIAGEN) and stored at −80°C until further analysis. All 24 samples were subjected to immune gene expression using a NanoString array (nCounter, Gene expression code set, human immunology kit). One patient's sample failed to meet quality standards and was removed from the analysis. Data were normalized using nSolver Analysis software (nanostring.com/products/analysis-software/nsolver). Significant differences between patient and control samples were analyzed by t tests, and p-values were adjusted for multiple comparisons using Benjamini-Hochberg correction. Correlation between microbiome abundance and immune gene expression was determined using Spearman Rho correlation (IBM SPSS statistics 20). Kyoto encyclopedia of the genes and genomes (KEGGs) pathway analysis was performed using Enrichr web server18 (amp.pharm.mssm.edu/Enrichr), and the top canonical pathways that had an activation score >1.5/−1.5 were reported.
Sequence homology and phylogenetic placement
Sequence homology between AQP4 and other proteins were addressed using the Basic Local Alignment Search Tool (BLAST) from the National Center for Biotechnology Information (NCBI) and Clustal 2.1 multiple sequence alignment tool from European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI). Phylogenetic placement of 16S rRNA sequences was completed using Phylogeny.fr(phylogeny.fr).
Standard protocol approvals, registrations, and patient consents
This study was approved by the institutional ethics committees at Nitte University (India) and Brigham and Women's Hospital (USA). It was conducted in compliance with the Declaration of Helsinki and national regulations. A written informed consent was obtained from all participants involved in the study.
Data availability
The 16S rRNA sequencing data along with deidentified metadata have been deposited at DRYAD (Pandit, Lekha [2020], 16S rRNA sequences of gut microbiome and meta data, Dryad, Dataset, doi.org/10.5061/dryad.jsxksn075).
Results
Microbiota community structure differs in NMO patients
There were 39 patients with NMOSD (17 were AQP4+) and 36 healthy controls. Details of the study population are outlined in table 1. Measures of alpha and beta diversities were calculated to determine the overall differences in the microbial community among patients and controls. Alpha diversity (microbial diversity within samples) was similar between all patients with NMOSD and healthy controls, including Faith PD and Shannon metric diversity. The difference in beta diversity (measurement of microbial community structure between groups) was determined using unweighted and weighted UniFrac metric. The 3-dimensional principal component analysis of weighted UniFrac distances of 16S rRNA genes showed a shift in the microbiota community structure between AQP4+ patients and healthy controls (figure 1A), which was significant using the pairwise PERMANOVA analysis (adjusted p value = 0.002) (figure 1B). Beta diversity also differed by diet (vegetarian/nonvegetarian), showing a trend for significance (pairwise PERMANOVA, adjusted p value = 0.05).
Figure 1. Beta diversity between AQP4+ and healthy controls.

(A) 3D principal component analysis (PCoA) of weighted UniFrac distances of 16S rRNA genes showing significant beta diversity between AQP4+ patients (green) and healthy controls (red). (B) Results of pairwise PERMANOVA analysis showing a significant difference in beta diversity (p < 0.001, pairwise PERMANOVA test) between patients with AQP4-IgG+ NMOSD and healthy controls. NMOSD = neuromyelitis optica spectrum disorder.
Compositional differences in the gut microbiome in patients with NMOSD
At the phylum level, Bacteroidetes, Firmicutes, and Proteobacteria were dominant in both patients with NMOSD and healthy controls (data not shown), with smaller contributions from Tenericutes and Actinobacteria. A taxa graph (figure 2A) shows an overview of the differential abundance of microbiota at the species level among patients with NMOSD and healthy controls. Circular cladograms (figure 2, B and C) reporting the LEfSe results showed significant changes from the phylum to genus levels between the 2 groups. At the phylum level, Proteobacteria was significant in patients with AQP4+ NMOSD and Verrucomicrobia in patients with AQP4-NMOSD. At the genus level, Clostridium, Megamonas, Streptococcus, Enterobacteriaceae, and Bilophila were prominent in patients with AQP4+ NMOSD, whereas Bacteroides, Megamonas, Phascolarctobacterium, and Akkermansia were prominent in patients with AQP4-NMOSD.
Figure 2. Differential bacterial composition at species level between patients and controls.

(A) Taxa graph showing bacterial composition at the species level—differential abundance (>0.1% in any sample group) of microbes among patients with NMOSD and healthy controls. (B) Cladogram based on LEfSe of bacterial communities showing differentially abundant microbes between patients with AQP4+ NMOSD and controls and (C) showing differential abundance between patients with AQP4- NMOSD and controls. LEfSe = Linear discriminatory analysis effect size; NMOSD = neuromyelitis optica spectrum disorder.
At the species level, there was a significant difference in microbial abundance between patients with AQP4+ NMOSD and controls (table 2) and patients with AQP4-NMOSD and controls (table 3). Microbial populations with an LDA > 3 and adjusted p value < 0.01 were considered significant. Among patients with AQP4+ NMOSD, Clostridium bolteae (p = 0.00007) and Flavonifractor plautii (p = 0.00008) were the top hits, whereas Megamonas funiformis (p = 0.00003) and Clostridium ramosum (p = 0.0001) were the same for patients with AQP4-NMOSD. Some organisms were significantly abundant among patients with both seropositive and seronegative NMOSD and included M funiformis, Phascolarctobacterium faecium, F plautii, and C ramosum.
Table 2.
Differentially abundant microbes in patients with AQP4+ NMOSD compared with healthy controls

Table 3.
Differentially abundant microbes in patients with AQP4- NMOSD compared with healthy controls

Lachnospiraceae was one of the significantly altered taxa. At the amplicon sequence variant level (ASV), which is akin to the strain level, we found that Lachnospiraceae comprised 4 unique sequence variants, namely, ASV_d5b22a367668019c66d346e758f7a1ee, ASV_f90fe5d03a1c66fbae34d7261fb134d9, ASV_aa2eeeb505ac76273e49ca4ac4c973c1, and ASV_5796684ced9efadf36da73862ae01df3. The maximum percentage prevalence of Lachnoclostridium was 8.44%, 5.10%, and 2.74%, respectively, for patients with AQP4+ NMOSD, patients with seronegative NMOSD, and healthy controls, respectively (figure 3A). Using the EZ-taxon database, we identified ASV_d5b22a367668019c66d346e758f7a1ee as C bolteae, with a 100% homology to the type strain WAL1631T (ATCC (T)-BAA-613T, CCUG (T)-46953T. Total abundance of C bolteae in patients with AQP4+ NMOSD (4.06%) was more than double (figure 3, B and C) than that in patients with AQP4-NMOSD (2.04%). Clostridium bolteae was absent among control samples. It was detected in 47.1% (8/17) of patients with AQP4+ NMOSD and 13.6% (3/22) patients with AQP4-NMOSD (p 0.02). It is worth noting that the single untreated patient in the AQP4-IgG+ group and 2 of 6 untreated patients in the seronegative group harbored C bolteae. The remaining 3 non annotated species were evenly distributed among seropositive and negative patients but was significantly more abundant among patients (p < 0.001) than healthy controls. Clostridium bolteae showed phylogenetic dissimilarity from the other Lachnoclostridium species isolated in this study (figure e-1, links.lww.com/NXI/A334).
Figure 3. Distribution of Lachnoclostridium species among patients and controls.
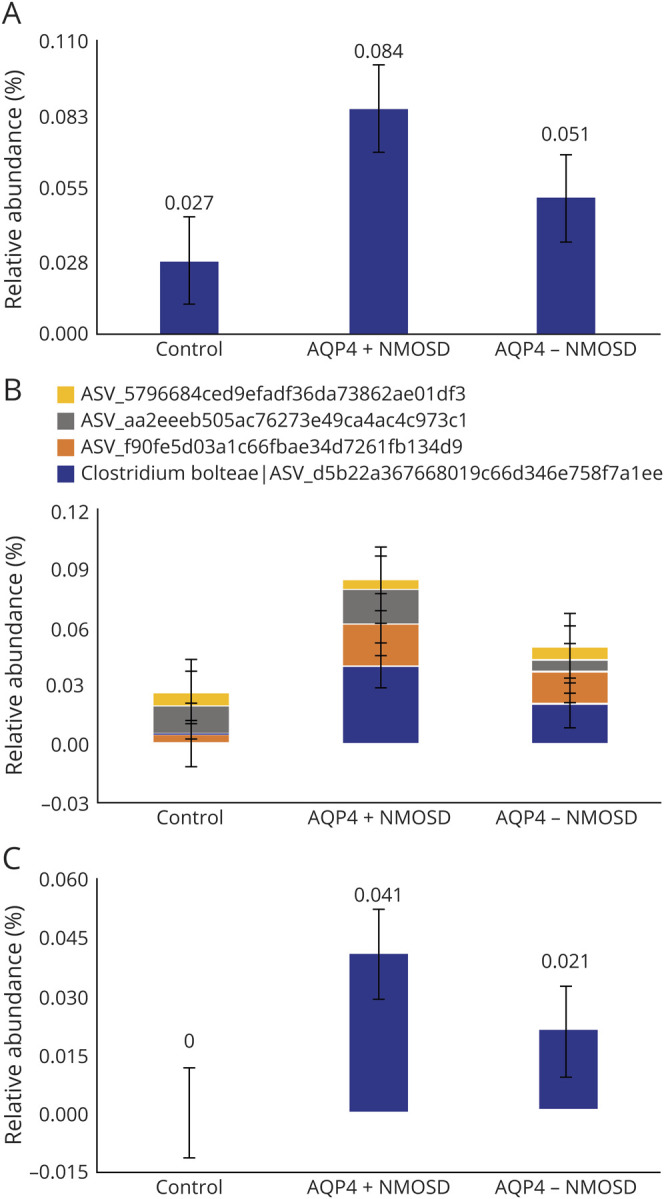
Lachnoclostridium distribution among (A) patients and control, (B) distribution of Lachnoclostridium species, and (C) distribution of Clostridum bolteae/d5b22a367668019c66d346e758f7a1ee among patients and controls.
C. bolteae has partial protein homology to AQP4
Sequence homology analysis (figure 4, A and B) showed that C bolteae peptide 59–71 (NCBI protein reference—Taxid ID208479) aligned with AQP4 peptide (p) 92–104. Topology (uniport.org) shows that AQP4 peptide sequence 92–104 is predominantly cytoplasmic (figure e-2, links.lww.com/NXI/A335) and likely to be highly reactive to AQP4 specific T cells.18 We identified a 62% homology (62% identity, 76% positive, and 0% gap) in the 13 aminoacid sequence 59–71 that falls within the highly conserved domain—glycerol uptake facilitator and related aquaporins (GlpF). It belongs to the major intrinsic proteins (MIP) responsible for carbohydrate transport and metabolism in the microbial cell.
Figure 4. Determination of sequence homology between AQP4 peptides and C. bolteae.

Sequence homology between AQP4 P92-104 and C. bolteae P59-71 peptides determined using (A) a basic local alignment tool (National Center for Biotechnology Information). (B) CLUSTAL 2.1 (multiple sequence alignment tool from European Molecular Biology Laboratory-European Bioinformatics Institute [EMBL-EBI]).
Gene expression studies in peripheral CD4+ lymphocytes
The NanoString Immunology Panel was used to measure the expression of 568 immune-related genes from peripheral blood derived CD4+ T cells from a subset of patients with AQP4+ NMOSD (n = 11) and healthy controls (n = 12). Because C bolteae was significantly elevated in AQP4-IgG+ patients, samples were selected based on the presence (n = 5) or absence of the same (n = 6). Demographic features of both groups were comparable (table e-1, links.lww.com/NXI/A337). The top list of genes upregulated (figure e-3, links.lww.com/NXI/A336) included those related to both innate and adaptive immunities. The presence of C bolteae correlated positively (Rho ≥ 0.5, p < 0.05) with transcriptomes of CD4+T cells implicated in inflammatory response, particularly CD70, IFI35 (interferon induced protein 35), MAPK11 (mitogen activated protein kinase 11), TAP2 (transporter 2), CCR10 (chemokine receptor 10), KLRAP1 (killer cell lectin like receptor A1), PML (promyelocyte leukemia) and IKBE (NF-kappa-B inhibitor epsilon gene)(figure 5A, table e-2, links.lww.com/NXI/A337). There was, in addition, a significant difference in gene upregulation among patients with AQP4+ NMOSD who harbored F plautii (KLRAP1 and PML) P faecium (MAPK11), and Bacteroides ovatus (MAPK11). Conversely, Prevotella, Bifidobacterium, and Ruminococcus showed a negative correlation for the same genes among patients and was similar to results in healthy controls. The KEGG pathway analysis (Enrichr)19 showed upregulation of pathways relevant to immunopathogenesis of AQP4-IgG–associated NMOSD (figure 5B). These included TH1, Th2, and Th17 differentiations, TLR1, TNF-alpha, and NFKB signaling pathways. Pathways projected to be upregulated in this study also shared similarity with other B cell mediated disorders such as inflammatory bowel disease and rheumatoid arthritis (RA).
Figure 5. Correlation between microbiota abundance and immune gene expression.

(A) Heat map showing correlation (Spearman Rho) between microbiota abundance and immune gene expression in patients with AQP4+ NMOSD (n-11) and healthy controls (n = 12). (B) KEGG pathway analysis (Enrichr*). Top list of pathways upregulated by the set of genes identified in the nanostring analysis were sorted by a combined score (log of p value obtained by the Fisher exact test multiplied by a Z score of the deviation from expected rank). Input genes are the rows, and cells in the matrix indicate whether a gene is associated with a term. KEGG = Kyoto encyclopedia of genes and genome; NMOSD = neuromyelitis optica spectrum disorder
Discussion
We have for the first time studied gut microbiome associations in Indian patients with NMOSD, using high throughput sequencing of 16S rRNA. There were significant alterations in beta diversity in gut microbiota among patients with NMOSD when compared with healthy controls and particularly in the AQP4+ NMOSD cohort. The microbial composition at the genus level showed over expression of Lachnoclostridium species (Class Clostridia and family Lachnospiraceae) in patients with NMOSD as opposed to healthy controls. Among them C bolteae, a gram positive, spore-forming bacterium was detected exclusively in patients, but not in healthy controls. There was also a significant difference in the relative prevalence (p = 0.02) when patients were stratified based on the presence/absence of AQP4-IgG. Because most patients with NMOSD were on treatment with immunosuppressants, hypothetically, it is possible that treatment favored microbial overgrowth in patients. However, the single untreated patient in the AQP4-IgG+ and 2 others in the seronegative group also harbored C bolteae.
Profiling of peripheral CD4+ T cell transcriptomes showed a positive correlation with this species for most upregulated genes in this study, and particularly relevant to gut dysbiosis and NMOSD pathogenesis. The latter included CD70, a tumor necrosis factor family ligand that binds to its receptor CD27 expressed on memory B cells and promotes plasma cell differentiation and immunoglobulin secretion.20 Gut microbiota are known to activate the differentiation of lamina propria cells, particularly CD70 into Th17.21 Another upregulated gene in our study was MAPK11. MAPK signaling pathway activation is known to be associated with gut dysbiosis.22 It is notable that CCR10 plays a role in the induction of mucosal humoral immunity. When stimulated by toll-like receptor 2 (TLR2), it is expressed in circulating B cells resulting in increased chemotaxis and B cell homing to the gastrointestinal tract.23 Other genes linked to innate immunity that were over expressed included PML, KLRAP1, and IFI35. IKBE gene, an NF-kB regulator involved in IL-6 gene transcription, and TAP2 gene have been previously linked with susceptibility to RA.24 In summary, pathways upregulating B cell activation, plasma cell differentiation, B cell chemotaxis, and Th17 activation were implicated. Further support came in the form of KEGG pathway analysis (Enrichr) that projected upregulation of pathways relevant to immunopathogenesis of AQP4-IgG-associated NMOSD. These included Th1, Th2, and Th17 differentiation, TLR, TNF-alpha, IL17, and NFKB signaling pathways. These pathways are also common to other B cell–mediated disorders such as inflammatory bowel disease and RA (figure 5B).
In our study, Lachoclostridium species, particularly C bolteae, was the most prominent microbe associated with gut dysbiosis among patients with AQP4+ NMOSD. Clostridium bolteae has gained attention in recent years because of its close association with autism spectrum disorders (ASD). The capsular polysaccharide of C bolteae, comprising disaccharide repeating blocks of mannose and rhamnose units, has been identified to be immunogenic in animal models.25 In children with ASD, C bolteae association is known to cause the upregulation of inflammatory cytokines (particularly IL-6 and IL-17).26,27 A study of gut microbiome in children with food allergy (an autoimmune mediated inflammatory state) has shown increased C bolteae when compared with nonallergic siblings and healthy controls.28
There was abundance of other microbiota in patients with NMOSD, which have been previously implicated in disease states, which could have additionally contributed to gut dysbiosis among our patients. These include Flavonifractor, which was recently found over expressed in gut dysbiosis associated with pemphigous vulgaris—an autoimmune disorder affecting the skin.29 Flavonifractor and Lachnoclostrium species (unidentified) have been associated with gut dysbiosis in ASD among Chinese children.26 There was a reduction in the abundance of Prevotella species among patients, which is similar to alterations in patients with MS.30 Prevotella histicola can ameliorate disease severity in experimental autoimmune encephalomyelitis, the animal model for MS.31 However, Prevotella copri is elevated in new onset RA patients and can drive inflammation when transferred to mice.32 Thus, further study on species-specific effects of Prevotella is warranted in NMOSD. Prevotella is the common species among vegetarians living in India and was most prominent in our control sample.33 Bifidobacterium species was reduced in patients, and this has been previously reported with another autoimmune disorder namely Crohn's disease.34 Clostridium bolteae was also increased in a small number of seronegative patients with NMOSD. A previous study has shown that C perfringens was not only associated with patients with AQP4+ NMOSD but was seen in a small number of MS patients also.11 It is possible that proinflammatory Clostridia species may be causally linked with more than one autoimmune disease.
The results of our study suggest that C bolteae plays a central in the immunopathogenesis of patients with AQP4+ NMOSD among our cohort of Indian patients, and we have identified 2 potential mechanisms. First, high levels of C bolteae may contribute to NMOSD by increasing Th17 and other inflammatory immune-mediated responses. It is well known that Clostridium species, which are prominent anerobes in the human intestinal tract, can influence the balance between Th17 and regulatory T cells.35,36
Second, the sequence similarity between C bolteae peptide 59-71 and AQP4 peptide 92-104 may have implications in disease pathogenesis. Several extracellular, membranous, and cytoplasmic AQP4 epitope determinants have been identified (figure e-2, links.lww.com/NXI/A335).8,18 Matsuya et al.18 have shown that AQP4-specific T cells exhibit significant reactivity to 2 intracellular AQP4 epitopes, one of which was AQP4p91-110 that overlaps with peptide sequences identified in our study (the other wasp11-40). In experimental settings, AQP4-specific T cells, which were CD4+ and MHC I I restricted, showed a similar response to this epitope in C57BL/6 mice and in SJL/J mice.37 Interestingly, in a separate study, B cell antigenic linear epitopes of AQP4 protein were found to be same as the T cell epitopes mentioned above.38 This response to AQP4 may be due to immunologic memory acquired through past infection with pathogens that share molecular similarity with AQP4.39 A potential link between infections and NMOSD pathogenesis has been studied before. An E coli–derived aquaporin-Z has been implicated in NMOSD in previous studies.40 In a more recent study, an immunodominant AQP4 T cell epitope was identified (p63-76), which showed 90% homology with p217-216, within a conserved ABC transporter protein from strains of C perfringens5 expressed abundantly in patients with NMOSD.11 In our study, another member of the clostridial class, namely, C bolteae p59-71, showed partial sequence homology with a recognized AQP4 specific T cell epitope within AQP4 p91-110. The aligned protein sequence corresponded to a multispecies aquaporin family protein, a member of the major intrinsic protein (MIP) superfamily ubiquitously present in eukaryotes, bacteria, and archaea. It is therefore possible that molecular mimicry, the mechanism by which exogenous agents, including plant, bacterial, and viral proteins, may trigger immune responses against self or nonself antigens contributes to the autoimmune response against AQP4 in NMOSD.
There are several limitations to our study, including the small sample size and the lack of sufficient number of untreated patients available for the analysis. Furthermore, evidence for a direct role for C bolteae in NMOSD pathogenesis needs to be established through invitro studies, which include the localization of the immunodominant peptide within the intracellular T cell–specific AQP4 epitope that we identified historically. Animal studies that involve colonization of germ-free mice with fecal samples from patients with NMOSD and individual bacterial species such as C bolteae may help to further strengthen the association between the gut microbiome and NMOSD.
Acknowledgment
L. Pandit was a Fulbright Nehru scholar at Harvard Medical School during part of the study and was partially supported by the United States–India Educational Foundation (USIEF).
Glossary
- ASD
autism spectrum disorder
- ASV
amplicon sequence variant level
- KEGG
Kyoto encyclopedia of genes and genome
- LDA
linear discriminant analysis
- LEfSe
Linear discriminatory analysis effect size
- NCBI
National Center for Biotechnology Information
- NMOSD
neuromyelitis optica spectrum disorder
- PD
phylogenetic diversity
Appendix. Authors

Study funding
No targeted funding reported.
Disclosure
L. Pandit, L. Cox, C. Malli, A. D'Cunha, T. Rooney, H. Lokhande, V. Willocq, and S. Saxena report no conflicts of interest. T. Chitnis reports consulting fees from Biogen. Idec, Novartis, Sanofi, Bayer, Celgene, Genentech Grants from Novartis, Octane Bioscience, EMD Serono, Verily Life Sciences; all outside the submitted work. Go to Neurology.org/NN for full disclosures.
References
- 1.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of Neuromyelitis optica: distinction from multiplesclerosis. Lancet 2004;364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 3.Jassiak ZM, Lyszczarz AK, Michalak S, Kozubski W. The immunology of neuromyelitis optica—current knowledge, clinical implications, controversies and future perspectives. Int J Mol Sci 2016;17:273: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol 2012;11:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol 2009;66:630–643. [DOI] [PubMed] [Google Scholar]
- 6.Wang HH, Dai YQ, Qiu W, et al. Interleukin-17-secretingTcellsinneuromyelitisopticaandmultiplesclerosisduringrelapse. J. Clin Neurosci 2011;18:1313–1317. [DOI] [PubMed] [Google Scholar]
- 7.Uzawa A, Mori M, Arai K, et al. Cytokine and chemokine profiles in neuromyelitis optica: significance of interleukin-6. Mult Scler 2010;16:1443–1452. [DOI] [PubMed] [Google Scholar]
- 8.Varrin- Doyer M, Spencer CM, Topphoff SU, et al. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol 2012;72:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pandit L, Asgari N, Apiwattanakul M, et al. Demographic and clinical features of neuromyelitis optica: a review. Mult Scler 2015;21:845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Estrada K, Whelan C, Zhao F. A whole-genome sequence study identifies genetic risk factors for neuromyelitis optica. Nat Comm 2018;9:1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cree BA, Spencer CM, Varrin-Doyer M, Baranzini SE, Zamvil SS. Gut Microbiome Analysis in neuromyelitis optica reveals overabundance of Clostridium perfringens. Ann Neurol 2016;80:443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zamvill SS, Spencer CM, Baranzini S, Cree BA. The gut microbiome in neuromyelitis optica. Neurotherapeutics 2018;15:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong J, Qiu W, Zeng Q, et al. Lack of short-chain fatty acids and overgrowth of opportunistic pathogens define dysbiosis of neuromyelitis optica spectrum disorders: a Chinese pilotstudy. Mult Scler J 2018;25:1316–1325. [DOI] [PubMed] [Google Scholar]
- 14.Caporaso J, Lauber C, Walters W, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISMEJ 2012;6:1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolven E, Rideout JR, Dilon MR, et al. QIIME 2: reproducible, interactive, scalable and extensible microbiome data science. PeerJ Preprints 6:e27295v2 10.7287/peerj.preprints.27295v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. App Env Microbiol 2005;71:8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011;12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuya N, Komori M, Nomura K, et al. Increased T-cell immunity against aquaporin-4 and proteolipid protein in neuromyelitis optica. Int Immunol 2011;23:565–573. [DOI] [PubMed] [Google Scholar]
- 19.Chen EY,Tan CM,Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Sci Rep 2016;6:33566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaw J, Wang YH, Ito T, Arima K, Liu YJ. Plasmacytoid dendritic cells regulate B-cell growth and differentiation via CD70. Blood 2010;115:3051–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atarashi K, Nishimura J, Shima T, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature 2008;455:808–812. [DOI] [PubMed] [Google Scholar]
- 22.Smith PK, Masilamani M, Li XM, Sampson HA. The false alarm hypothesis: food allergy is associated with high dietary advanced glycation end-products and proglycating dietary sugars that mimic alarmins. J Allergy Clin Immunol 2017;39:429–437. [DOI] [PubMed] [Google Scholar]
- 23.Liang Y, Hasturk H, Elliot J, et al. Toll-like receptor 2 induces mucosal homing receptor expression and IgA production by human B cells. Clin Immunol 2011;138:33–40. [DOI] [PubMed] [Google Scholar]
- 24.Myouzen K, Kochi Y, Okada Y, et al. Functional variants in NFKBIE and RTKN2 involved in activation of the NF-κB pathway are associated with rheumatoid arthritis in Japanese. PLoS Genet 2012;8:e1002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fattorusso A, Di Genova L, Dell'Isola GB, Mencaroni E, Esposito S. Autism spectrum disorders and the gut microbiota. Nutrients 2019;11:E521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma B, Liang J, Dai M, et al. Altered gut microbiotain Chinese children with autism spectrum disorders. Front Cell Infect Microbiol 2019;9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kourosh A, Luna RA, Balderas M, et al. Fecal microbiome signatures are different in food-allergic children compared to siblings and healthy children. Pediatr Allergy Immunol 2018;29:545–554. [DOI] [PubMed] [Google Scholar]
- 28.Peguegnat B, Monteiro MA. Carbohydrate scaffolds for the study of the autism associated bacterium, Clostridium bolteae. Curr Med Chem 2019;26:6341–6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.SenHuang S, Mao J, Zhou L, et al. The imbalance of gut microbiota and its correlation withplasma inflammatory cytokines in pemphigus vulgaris patients. Scand J Immunol 2019;18:e12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jangi S, Gandhi R, Cox LM, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun 2016;7:12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mangalam A, Shahi SK, Luckey D, et al. Human gut-derived commensal bacteria suppress CNS inflammatory and demyelinating disease. Cell Rep 2017;20:1269–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013;2:e01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhakan DB, Maji A, Sharma AK, et al. The unique composition of Indian gut microbiome,gene catalogue, and associated fecal metabolome deciphered using multi-omics approaches. GigaScience 2019;8:giz004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang W, Chen L, Zhou R, et al. Increased Proportions of Bifidobacterium and the Lactobacillus Group and loss of Butyrate-Producing Bacteria in Inflammatory Bowel Disease. J Clin Microbiol 2014;52:398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atarashi K, Tanoue T, Oshima K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the humanmicrobiota. Nature 2013;500:232–236. [DOI] [PubMed] [Google Scholar]
- 36.Atarashi K, Tanoue T, Ando M, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 2015;163:367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nelson PA, Khodadoust M, Prodhomme T, et al. Immunodominant T cell determinats of aquaporin-4, the autoantigen associated with neuromyelitis optica. PLoS One 2010;5:e1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kampylafka EI, Routsias JG, Alexopoulos H, et al. Fine specificities of antibodies against AQP4: epitope mapping reveals intracellular epitopes. J Autoimmun 2011;36:221. [DOI] [PubMed] [Google Scholar]
- 39.Benoist C, Mathis D. Autoimmunity provoked by infection: how good is the case for T cellepitope mimicry? Nat Immunol 2001;2:797. [DOI] [PubMed] [Google Scholar]
- 40.Ren Z, Wang Y, Duan T, et al. Cross-immunoreactivity between bacterial aquaporin-Z and human aquaporin-4: potential relevance to neuromyelitis optica. J Immunol 2012;189:4602–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The 16S rRNA sequencing data along with deidentified metadata have been deposited at DRYAD (Pandit, Lekha [2020], 16S rRNA sequences of gut microbiome and meta data, Dryad, Dataset, doi.org/10.5061/dryad.jsxksn075).