

Abstract
Objective
To determine whether the punctuated administration of low-dose rituximab, temporally linked to B-cell hyperrepopulation (defined when the return of CD19+ B cells approximates 40%–50% of baseline levels as measured before alemtuzumab treatment inception), can mitigate alemtuzumab-associated secondary autoimmunity.
Methods
In this hypothesis-driven pilot study, 10 patients received low-dose rituximab (50–150 mg/m2), a chimeric anti-CD20 monoclonal antibody, after either their first or second cycles of alemtuzumab. These patients were then routinely assessed for the development of autoimmune disorders and safety signals related to the use of dual monoclonal antibody therapy.
Results
Five patients received at least 1 IV infusion of low-dose rituximab, following alemtuzumab therapy, with a mean follow-up of 41 months. None of the 5 patients developed secondary autoimmune disorders. An additional 5 patients with follow-up over less than 24 months received at least 1 infusion of low-dose rituximab treatment following alemtuzumab treatment. No secondary autoimmune diseases were observed.
Conclusions
An anti-CD20 “whack-a-mole” B-cell depletion strategy may serve to mitigate alemtuzumab-associated secondary autoimmunity in MS by reducing the imbalance in B- and T-cell regulatory networks during immune reconstitution. We believe that these observations warrant further investigation.
Classification of evidence
This study provides Class IV evidence that for people with MS, low-dose rituximab following alemtuzumab treatment decreases the risk of alemtuzumab-associated secondary autoimmune diseases.
Alemtuzumab, a humanized anti-CD52 monoclonal antibody that depletes circulating B and T lymphocytes, is approved in the United States and Europe for the treatment of MS.1,2 Patients who receive alemtuzumab have an estimated 60% rate of attaining No Evidence of Disease Activity status, which is defined by no new clinical relapses, disease progression, or new MRI activity in a 5-year follow-up period.3,4 Antibody-mediated secondary autoimmune disease in patients with MS treated with alemtuzumab approaches an incidence of 40%–50% in prolonged follow-up, with a peak incidence by the third year following treatment initiation and waning incidence thereafter.5–16
The main adverse effect of alemtuzumab is the development of predominantly antibody-mediated secondary autoimmune disorders. The most common secondary autoimmune disorder is antibody-mediated thyroid disease; with autoimmune hyperthyroidism being the most common and exceeding those developing hypothyroidism.5,6 Other antibody-mediated autoimmune diseases have been reported, including idiopathic thrombocytopenic purpura, anti–glomerular basement membrane (GBM) disease, neutropenia, hemolytic anemia, and vitiligo, among others. T cell–mediated autoimmunity and granulomatous inflammatory diseases (principally sarcoidosis) occur at a considerably lower incidence.1–16 An increased risk of opportunistic infections continues to be an important and potentially serious complication of all cell-depleting disease-modifying treatment strategies, although there are a number of systematic risk-mitigating strategies.
Cooperation between B cells and T cells is required for B-cell differentiation and mature antibody formation, and yet it is now well established that following alemtuzumab disease-modifying therapy for MS, that there is a marked discordance in B vs T lymphocyte reconstitution kinetics; with the former being detected earlier and in considerably greater proportion, using objective methods for characterizing peripheral blood mononuclear cells.
Some evidence suggests that lymphocyte repopulation patterns, in patients treated with alemtuzumab, are not necessarily associated with the risk of developing secondary autoimmune diseases.16,17 Instead, a compromise in the integrity of cellular regulatory networks, corroborated stochastically by diminution in the regulatory signature ratios (e.g., the clonal frequency of regulatory T cells (Tregs) to TH-17 proinflammatory cells), could influence the functional thresholds that determine the ignition of dynamic immune response oscillations and their disposition toward activation vs anergy.11 Furthermore, reduced thymopoiesis can result in the restricted heterogeneity in the T-cell receptor repertoire, creating conditions that can predispose to a heightened risk of secondary autoimmunity.18 Therefore, the discrepancy between humoral and cellular immune networks appears to be beyond the simplistic stochastic considerations.
The kinetic disparities in the development, release, and recirculation of B and T lymphocytes may have implications for the coordinate-regulatory mechanisms, which represent the immune basis for self-tolerance, and the corresponding molecular check-point verification strategies, which are imperative for ensuring the perpetual fidelity to discriminate between self and non-self (i.e., tolerance and its durability in response to challenges fundamental to its integrity, and with time, especially with advancing age and the emergence of the increasingly recognized property of immune senescence).
We hypothesize that anti-CD20 B-cell depletion, punctually administered and temporally coinciding with the precocious B-cell hyperrepopulation, may represent a viable strategy for mitigating the risk of alemtuzumab-associated secondary autoimmunity.
Here, we report a strategic approach, along with pilot observations, suggesting that the risk of secondary autoimmunity can potentially be mitigated when low-dose anti-CD20 therapy is administered during B-cell repopulation (i.e., what is referred to as a “whack-a-mole” strategy19–23) following alemtuzumab therapy.
Methods
The study was approved by the Investigational Review Board of the Dell Medical School at the University of Texas at Austin. All patients consented for the off-label use of rituximab.
Our primary research question was to ascertain whether the punctuated administration of low-dose rituximab, temporally linked with the discordant B-cell hyperrepopulation (when the return of the CD19+ cells approximated 40%–50% of baseline measures examined before alemtuzumab therapy intervention), represents an effective strategy for mitigating alemtuzumab-associated secondary autoimmunity (Class IV evidence).
We examined 2 small cohorts of 5 patients each. The first cohort (see table 1 for demographics), which was originally treated with alemtuzumab, received at least 1 infusion of low-dose rituximab (range 1 to >3 doses; 50–150 mg/m2) after B-cell reconstitution began. Follow-up of these individuals occurred for more than 24 months. All patients who received alemtuzumab for treatment of MS were eligible. Patients were not selected on the basis of prior immunotherapy. Rituximab is an anti-CD20 monoclonal antibody therapeutic. In addition to CD20, CD19 is also a pan B-cell marker. Anti-CD20 therapeutic monoclonal antibody interferes with the recognition of the CD20 antigen. As such, the CD19+ B-cell level, as measured by flow cytometry, was used as a proxy to measure extent of B-cell depletion following anti-CD20 therapy.24
Table 1.
Clinical characteristics and results of patients with long-term follow-up
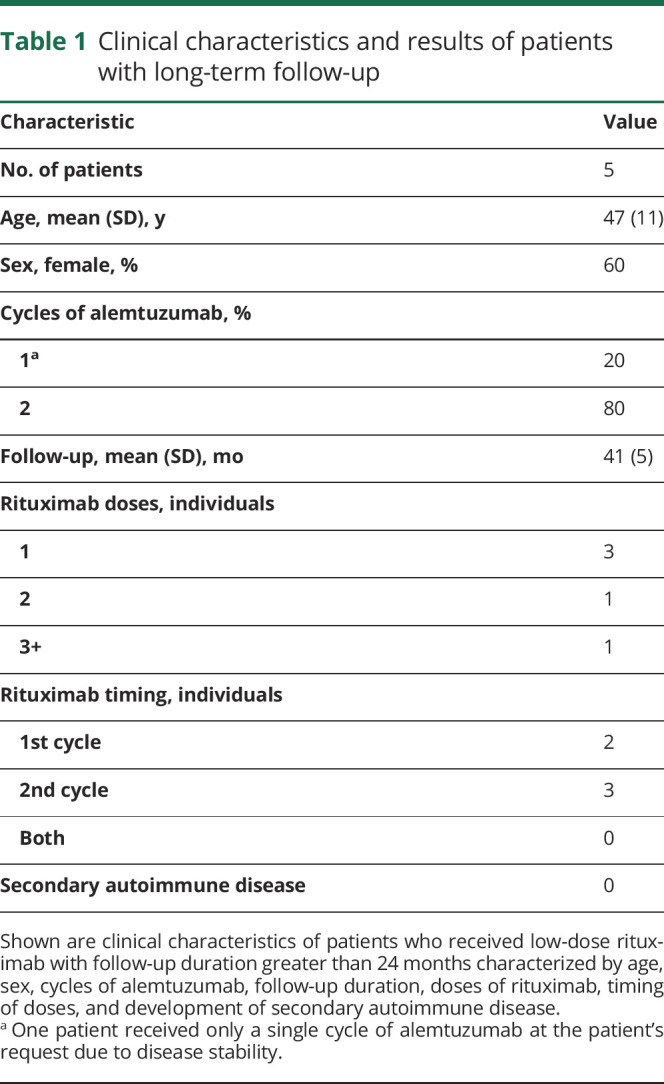
Alemtuzumab-treated patients were scheduled to receive rituximab when CD19+ B cells repopulated to 40%–50% of their baseline levels. Baseline B-cell frequency was determined before initiation of alemtuzumab treatments by performing lymphocyte subset analysis on 3–4 weekly blood draws. Rituximab dosing was predicated on insurance coverage. Some dosages were delayed due to difficulties obtaining insurance approval.
In addition to monthly monitoring of T- and B-cell (i.e., CD3+ and CD19+) lymphocyte subsets, we also measured thyroid function, urinalysis, and complete blood counts at baseline and monthly thereafter. This was performed in accordance with the Risk Evaluation and Mitigation Strategies program for alemtuzumab to identify evidence of autoimmune thyroiditis, anti-GBM antibody disease, or immune thrombocytopenic purpura.
The second cohort of 5 patients (demographics are shown in table 2), which was originally treated with alemtuzumab, received at least 1 infusion of low-dose rituximab after B-cell reconstitution to 40%–50% of baseline. Follow-up of these individuals occurred for less than 24 months. Lymphocyte subsets, thyroid function, urinalysis, and complete blood counts were measured at baseline and monthly thereafter, as described above.
Table 2.
Clinical characteristics and results of patients with short-term follow-up
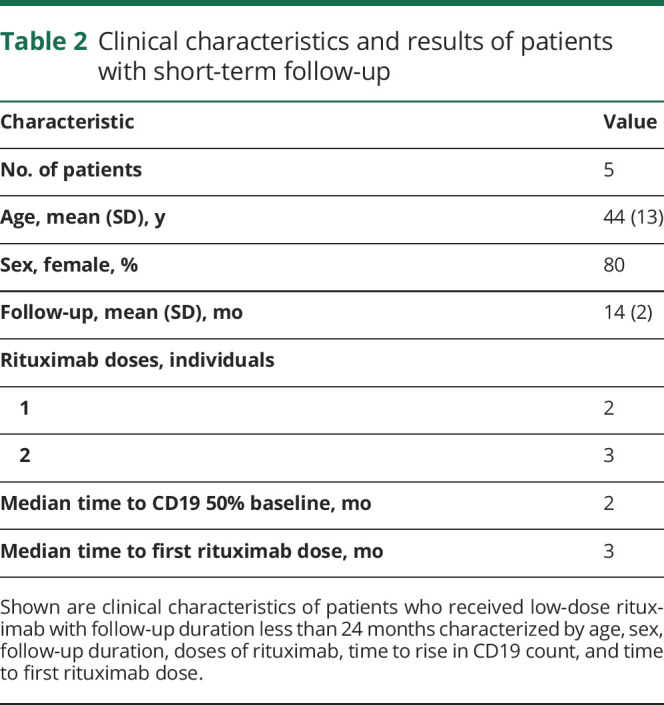
Given the low number of patients in this retrospective case series, and variations in protocol, we did not have adequate power to perform statistical tests to determine the efficacy of anti-CD20 therapy in preventing alemtuzumab-mediated secondary autoimmunity.
Data availability
Deidentified data will be shared with other investigators following requests made to the corresponding author.
Results
Of the 5 patients with follow-up greater than 24 months (table 1) who received low-dose rituximab infusions following alemtuzumab, 1 patient received 2 rituximab infusions, and 1 patient received greater than 3 rituximab infusions due to prolonged T-cell suppression. Two of the patients received rituximab after a single cycle of alemtuzumab, and 3 of the patients received rituximab after 2 cycles of alemtuzumab. None of the patients received 3 cycles of alemtuzumab, given evidence of clinical remission, and radiographic stabilization, as confirmed on follow-up MRI investigations.
For all 5 patients, the mean follow-up duration from the initial dose of alemtuzumab was 41 months (range 34–48 months). None of the patients developed secondary autoimmune complications as defined by the Comparison of Alemtuzumab and Rebif Efficacy in Multiple Sclerosis (CARE-MS) I and II trials.3,4 Based on the results of CARE-MS I, we anticipated that 30%–40% of patients with long-term follow-up would have developed autoimmune thyroid disease.3,4
Of the 5 additional patients with a follow-up duration of less than 24 months (table 2) who received low-dose rituximab treatment following alemtuzumab, 2 patients received 1 infusion of rituximab, and 3 patients received 2 infusions of rituximab. The median time to 40%–50% recovery of the CD19+ cell count was 2 months post-alemtuzumab, and the median time to the first rituximab dose administration was 3 months post-alemtuzumab.
In this second cohort, 1 patient developed transient neutropenia with an absolute neutrophil count (ANC) <500/μL 2 months after the second dose of 100 mg of rituximab. This patient was treated with granulocyte colony-stimulating factor and reconstituted the ANC within 1 month. Another patient developed a urinary tract infection, and an additional patient developed an upper respiratory tract infection after receiving low-dose rituximab, while both B- and T-cell lymphocytes were suppressed.
Late-onset neutropenia is a well-described phenomenon after rituximab infusion, which has been considered to be related to transient autoimmunity.25 Alternately, neutropenia has also been documented in postmarketing surveillance of alemtuzumab-treated patients, albeit at a much lower incident rate when compared with the frequency of the most common alemtuzumab-induced secondary autoimmune conditions, particularly thyroiditis (e.g., Graves disease and Hashimoto thyroiditis).3,4
All patients treated in both patient cohorts were on prophylactic valacyclovir or acyclovir for 24 consecutive months, including during treatment with low-dose rituximab, given high rates of herpes reactivation in the clinical trials.
Discussion
Alemtuzumab treatment in MS is associated with an early hyperrepopulation of B cells that emerge between 3 and 6 months following treatment, which precedes the repopulation of T cells that occurs between 12 and 24 months following treatment. A fundamental discordance in the return of humoral and cellular networks is likely to play a role, at least in part, in the incidence of the adverse event most strikingly associated with alemtuzumab treatment; secondary autoimmunity.26–29
T-cell help is required for B-cell differentiation, isotype switching, and antibody secretion. The interaction between B cells and T cells is bidirectional. As such, anti-CD20 B-cell depletion in MS and experimental autoimmune encephalitis (EAE) promotes T-cell immune modulation characterized by a significant reduction in proinflammatory Th1 and Th17 cells, which likely reflects a decrease in B-cell antigen-presenting cell function.26 In this context, selective deficiency of major histocompatibility complex (MHC) Class II expression by B cells in mice eliminates both susceptibility to EAE and the expansion of Th1 and Th17 cells.29
In the absence of T-cell help, B cells are rendered unable to differentiate between the antigen presentation of self- vs non–self-epitopes, at least in part secondary to a considerably lower threshold for presentation of low-magnitude antigens, when compared with macrophages or dendritic cells. Furthermore, anti-CD20 treatment of MS and EAE promotes T-cell immune modulation characterized by a significant reduction in the proinflammatory network, at least in part, mediated by Th17.26–29 Conspicuously, B cell–specific MHC Class II–deficient mice are rendered resistant to the development of EAE.28
Secondary humoral autoimmunity resulting in hyper- or hypothyroidism, thrombocytopenia or renal disease are well known risks following alemtuzumab treatment of MS.5,6 We hypothesize that anti-CD20 B-cell depletion after alemtuzumab administration may mitigate the risk of secondary autoimmunity and have investigated this possibility. Our initial results suggest that anti-CD20 B-cell depletion is safe and potentially efficacious. However, given the small sample size, and unrandomized and unblinded cohort, more rigorous studies need to be pursued before making any declaration regarding efficacy.
Several hypotheses attempt to delineate the cause of secondary autoimmune disease associated with alemtuzumab MS disease-modifying therapy. Unlike traditional B-cell return after anti-CD20 therapies, B-cell repopulation occurs rapidly following alemtuzumab treatment and is associated with hyperrepopulation of immature and mature B cells above baseline levels.30 Following alemtuzumab treatment, B-cell repopulation occurs well in advance of T-cell repopulation (∼3–6 months compared with ∼12–24 months, respectively), which may compromise the counterbalancing of humoral and cellular mechanisms that serve to maintain immune regulation.
Notwithstanding these findings, secondary autoimmune disease after alemtuzumab treatment, exhibits a long latency before presentation, that is well beyond the observed B-cell hyperrepopulation, and in fact coincides more closely with T-cell repopulation; as autoantibody production is likely CD4 T-cell dependent.31
T-cell regeneration after depletion in adults is predominantly achieved by thymic-independent homeostatic peripheral expansion. This process, unlike T-cell regeneration in children, which occurs primarily in the thymus, is associated with decreased self-tolerance, a potential molecular scaffolding on which autoimmunity can result.31
The goal of the temporally associated administration of low-dose rituximab is not exclusively restricted to delaying the return of the CD19+ B-cell population or an attempt to solely synchronize the reconstitution of B- and T-cell subsets. Data from clinical trials that have analyzed the kinetics of humoral and cellular subsets reveal no evidence of correlation that dichotomizes the risk predilection for alemtuzumab-mediated secondary autoimmunity.16 Future work should also characterize the effect of rituximab on a unique and proinflammatory mononuclear cell, which coexpresses both CD20 and CD3 (i.e., CD20+ T cells and chimeric or C cells), which may serve to promote secondary autoimmunity, and would be expected to be deleted along with CD20+ B cells during our administration of low-dose rituximab.
A unique member of the immune system's mononuclear cell pool is the CD20+ T cell (i.e., a chimeric lymphocyte or C cell; figure 1), which includes both CD4+ and CD8+ cells, with the latter being the predominant of this cell type. Its T-cell origin has been confirmed via the detection of CD3 encoded mRNA via reverse transcriptase-polymerase chain reaction (RT-PCR).32,33 The distribution of these cells is broad and includes the primary and secondary lymphoid tissue, thymus, bone marrow, lymph node, adenoids, liver, and cerebrospinal fluid (CSF). Conspicuously, at resting conditions, CD20+ T cells exhibit augmented release of cytokines vs CD20− T cells. Specifically, they release interferon γ, interleukin (IL) 1 β, IL-2, IL-4, IL-8, IL-10, transforming growth factor β, tumor necrosis factor α, and c-c chemokine.34
Figure 1. A model for the mitigation of secondary autoimmunity in patients with MS post-alemtuzumab.

In the upper portion of the figure, we illustrate alemtuzumab treatment and its effect on elimination of anti–self-lymphocytes by 2 principal mechanisms, clearance through the reticular endothelial system (RES; on the left) and assembly of the membrane attack complex (MAC) facilitating the passage of C9, which compromises cell membrane integrity, thereby culminating in cytotoxicity and cell death. The lower middle illustration emphasizes post-alemtuzumab treatment-induced mobilization of bone marrow stem cells, reconstituting the immune system. B cells undergo a discordant hyperrepopulation approaching pretreatment baseline numbers, 3–6 months following alemtuzumab treatment, whereas T-cell reconstitution is considerably delayed (12–24 months). Hypothetically, the highly discordant hyperrepopulation of B cells plays a role in the development of secondary autoimmunity. Post-alemtuzumab treatment, circulating mononuclear cells are principally CD3+-naive T cells, CD19+ CD20+-naive B cells, as well as a unique population of cells that express both the B-cell antigen CD20 and the T-cell antigen CD3. These latter cells we refer to as Chimeric lymphocytes (C cells), and they are known to express proinflammatory cytokines, which in the absence of adequate T-cell help can foment the production of anti–self-antibodies. This can initiate corresponding autoantibody-mediated disorders, such as those designated by green labels in the figure, as well as to T cells capable of orchestrating cellular mechanisms of autoimmunity (designated by purple labels). To the lower left of the lower diagram, one can see an enrichment in the CD3+ CD20+ C cells simultaneous with the discordant B-cell hyperrepopulation (generally emerging approximately 3–6 months following alemtuzumab treatment) in the context of a deficiency in the clonal frequency of regulatory T cells (Tregs) (i.e., identified as CD4+CD25hiFoxP3+). In the absence of T-cell help, such B cells are rendered incapable of discriminating between self and foreign epitopes to both other B cells, thereby coordinating the development of antibodies as well as to T cells leading to T-cell receptor maturation, with elaboration of cytokine and chemokine expression profiles commensurate to foment cellular autoimmune mechanisms. The central portion of the diagram serves to illustrate the potential diversity of antibody-mediated secondary autoimmune states (green line paths). We also see activation of T cells, which can mediate secondary autoimmunity (purple line paths), and granulomatous inflammatory disease, most specifically sarcoidosis. Monitoring monthly lymphocyte subsets facilitates detection of the discordant B-cell hyperrepopulation phase of bone marrow mononuclear cell mobilization after alemtuzumab therapy and thereby provides for the discrete and temporally punctuated administration of an anti-CD20 “whack-a-mole” strategy for mitigating secondary autoimmunity. Specifically, we administered low-dose rituximab to orchestrate the deletion of both CD20+ B cells in conjunction with the CD20+CD3+ C cells, along with their B-cell activation capabilities via the elaboration of proinflammatory cytokine and chemokine cascades. The net effect is seen approximately 4 months later (in the upper circle on the right lower side of the diagram) when there is a marked diminution of both effector/memory B cells and autoimmunity-inducing C cells, with a corresponding expansion in the clonal frequency of Tregs.
Under conditions of immune stimulation, there is an escalation in cytokine production in CD20+ T cells.32 Furthermore, enhanced expression of IL-17 is also characteristic of this small population of lymphocytes.33 In patients with relapsing-remitting and primary progressive MS vs healthy controls, there is an increased frequency of CD20+T cells in peripheral blood.35
In those with MS, there is an increased clonal frequency of such cells in CSF when compared with the analysis of peripheral blood mononuclear cells, and the magnitude of that clonal frequency is correlated with clinical disability in patients with MS as measured by the long-validated Expanded Disability Status Scale score.36 Also, CD20+ T cells are identified within chronic white matter plaque lesions derived from the MS brain.36 In drastic contrast to the small proportion of such cells from the peripheral blood of controls, the CD20+ T-cell fraction in patients with MS represents a striking 18.4% of all CD20+ cells including those mononuclear cells that are CD19+ B cells.37
B-cell hyperrepopulation, accompanied by a reduction of T-cell help, establishes circumstances whereby antigen presentation coordinated by B cells, renders them unable to differentiate between self (whereby antigen presentation of autoantigens stereotypically produces immune anergy), and foreign epitopes (which appropriately culminates in immune activation). Alternately, the B-cell depletion also comes with the downregulation of inflammatory cytokines, secondary to B-cell induction of T-cell activity and the eventual cytokine release syndrome.
A detailed figure (figure 1) delineates our hypothetical constellation of features proposed as the basis for a low-dose anti-CD20 whack-a-mole strategy aimed at mitigating secondary autoimmunity.
If our proposed risk mitigation strategy were to be used more broadly, for feasibility, there would need to be a straightforward way of timing rituximab infusions. As such, figure 2 illustrates a schematic for a potential protocol using a low-dose anti-CD20 (rituximab) whack-a-mole secondary autoimmunity mitigation strategy post-alemtuzumab treatment and its effect on B-cell repopulation. Specifically, we timed low-dose rituximab infusion with a CD19+ B-cell recovery of ∼50% of normal baseline. We chose this as a relatively easy marker to measure and to ensure that patients would receive rituximab synchronous to the B-cell hyperrepopulation phase of post–alemtuzumab-induced bone marrow mononuclear cell mobilization.
Figure 2. Low-dose anti-CD20 therapy post-alemtuzumab schematic illustrating a potential pilot protocol.

This schematic depicts normal CD4+ and CD19+ cell reconstitution,9 as well as the theoretical CD19+ reconstitution with our proposed low-dose rituximab protocol. Low-dose rituximab (e.g., 100 mg) is given subsequent to the detection of the precociously discordant B-cell hyperrepopulation phase of bone marrow mononuclear cell mobilization following alemtuzumab treatment; an intervention which attenuates promiscuous antigen presentation by B cells, capable of activating anti–self-humoral and cellular networks, while also serving to promote a more synchronized repopulation across the bone marrow mobilization of mononuclear cell heterogeneity (i.e., achieving a more balanced return of B and T lymphocyte populations; with the added dividend of amplifying the clonal frequency of Tregs; and observation akin to tolerance induction in patients with MS).
Alternatively, rituximab could be given at a fixed interval, such as 3 or 4 months after alemtuzumab infusion, a common, albeit not consistent, duration for the bone marrow mobilization and peripheral repopulation of B cells. However, if B-cell hyperrepopulation is delayed (e.g., until 4–6 months), a fixed timing for whack-a-mole administration of low-dose anti-CD20 therapy could predispose a significant proportion of patients to harbor a temporal discordance between such treatment and its intended targets, consequently producing an ineffective depletion of both CD20+ B cells and CD20+/CD3+ T cells (likely resulting in a potential type II error in efficacy analyses).
Given the heterogeneity in the kinetics of B-cell repopulation following alemtuzumab therapy, coupled with the known median half-life of rituximab at 22 days (with a range of 6.1–52 days, as per package insert), we strongly advise for monthly lymphocyte subset analysis to precisely synchronize administration of a rituximab low-dose anti-CD20 whack-a-mole therapy strategy to buffer against the discordant B-cell hyperrepopulation in the absence of adequate T-cell help. In addition, this strategy will attenuate the reemergence of the proinflammatory CD20+ T cells. It remains to be seen if a single dose of rituximab will be sufficient or whether multiple whack-a-mole cycles will be required for a therapeutic effect of preventing alemtuzumab-associated secondary autoimmunity.
It is of interest that anti-CD20 therapy achieves about a 99% loss of peripheral blood B lymphocytes, without affecting plasma cells or plasmablasts, the principal antibody-secreting cells, which are devoid of cell surface CD20 expression. In keeping with this observation, anti-CD20 therapy appears to exert no impact on CSF antibody indices such as oligoclonal banding, increased immuno gamma globulin (IgG) index, and/or an elevation in IgG synthesis rates, characteristic abnormalities identified in approximately 90% of patients with MS, when using isoelectric focusing techniques. Alternately, the peripheral loss of B cells does correspond to a marked reduction in the development of both T1 gadolinium-enhancing and combined unique MS plaque lesions.38,39
In a recent study, the third-generation, glycoengineered chimeric anti-CD20 therapy, ublituximab, was shown to reduce both effector and memory CD8 T-cell populations, while also augmenting the clonal frequency of naive CD8+ T cells.38 One of the most conspicuous observations was the identification of a population of lymphocytes with cell surface expression of both CD20+ and CD3+ (those designated as chimeric or C cells in our hypothetical model; figure 1), with further characterization revealing that such cells exhibit features in keeping with memory CD8+ T cells.40
The functionally chimeric lymphocytes produce proinflammatory mediators, which may play a key role in the promiscuous antigen presentation of self-motifs during the discordant hyperrepopulation of B cells following alemtuzumab treatment (figure 1). Lovett-Racke et al. categorically characterized both the cellular identification and cytokine/chemokine expression profiles using flow cytometric techniques from 47 patients with MS treated with ublituximab and confirmed that such treatment resulted in significantly attenuated expression of proinflammatory cytokines, reduced antigen presentation, a shift from the effector/memory phenotype to repopulation of naive lymphocytes, and importantly augmentation in the clonal frequency of regulatory T cells (Tregs).33
A primary immune defect in patients with MS is a deficiency in Tregs. Efficacy in treating the disorder, at least in part, involves reconstitution of the immune network's regulatory capabilities through the expansion of Tregs, specifically identified as CD4+CD25hiFoxP3+.28,38 The observations from this investigation suggest that anti-CD20 therapy is capable of provoking a skewing or immune deviation that may be secondary, perhaps at least in part, to the deletion of CD20+/CD3+ C cells, thereby resulting in reduced B cell–mediated degenerate antigen presentation of self-motifs, along with a broadening in the clonal frequency of Tregs, all of which may be germane to our understanding of how we can use alemtuzumab while actively reducing (vis a vis with the application of a ‘whack-a-mole’ cell depletion strategy) the incidence of secondary autoimmunity (figures 1 and 2).33
We successfully used low-dose rituximab 100 mg (∼50–75 mg/m2) for patients with neuromyelitis optica spectrum disorder (NMOSD) who could not afford access to higher doses of rituximab. A 100-mg infusion depletes the CD19 population of B cells to less than 2% for an average of 99 days (in essence, B-cell suppression is highly titratable, analogous to immunologic labetalol, the latter commonly used as a highly titratable agent for management of hypertensive crises, such as hypertensive encephalopathy, where loss of cerebrovascular autoregulation precludes the employment of long-acting agents, which could result in a protracted compromise in cerebral blood flow dynamics, predisposing to a hypoxic-ischemic insult) in patients with NMOSD compared with 184 days for a 1,000 mg dose of rituximab.41
Low-dose rituximab is a more attractive alternative to standard doses of 1,000 mg or 375 mg/m2. First, doses of rituximab as low as 1 mg/m2 suppress B cells by 97% in healthy controls, and B cells remain suppressed to 75% of baseline levels at 12 weeks after infusion.42,43
Second, low-dose anti-CD20 therapy does not result in prolonged B-cell depletion, so as not to interfere with repeat cycles of alemtuzumab therapy.41
Third, low-dose rituximab may facilitate greater control of B-cell repopulation and improved temporal synchronization with T-cell repopulation.
Last, reduced dosing magnitude reduces the duration of immunosuppression compared with conventional doses of rituximab and may eventually be shown in evidence-based studies to demonstrate both safety and cost advantages if few complications are identified with the low-dose regimen.
A more robust clinical trial with a primary outcome of the development of secondary autoimmunity would not likely require a large number of participants, given the incidence rate of 40% of autoimmune thyroid disease in patients with MS who have received alemtuzumab.3,4 Hence, we would estimate an incidence of 40% in the control group of alemtuzumab-only treated patients and with an assumption of an incidence of 10% in patients who would receive adjunct rituximab. If we assign the probability of a type I error of 5% and a power of 80%, a trial would need approximately 80 patients total (40 in each arm), accounting for drop-outs.
In addition to our small sample size, and an unrandomized, and unblended cohort, many physicians prescribe steroids with rituximab infusion (methylprednisolone 250 mg before each and every alemtuzumab infusion in our center) to prevent infusion reactions. It is possible that the potential positive effects of secondary autoimmunity prevention attributed to rituximab are instead, or at least in part, due to steroid usage. In addition, treatment may simply delay secondary autoimmunity rather than preventing it, and longer periods of ascertainment will be required to assess for this prospect.
An alternate explanation concerning the impact of rituximab on secondary autoimmunity is that some individuals may respond differently to low-dose anti-CD20 therapy due to the effects of the FCGR3A polymorphism.42 We do not know the effect of the polymorphism on low-dose rituximab kinetics; however, binding affinity may play a larger role in low-dose rituximab compared with conventional doses, given the lower absolute concentration of rituximab. Such issues should be carefully considered in the design of a larger trial, particularly given potential effects of such factors on response characteristics as well as on study cost.
As the field of neurology shifts from a model of step escalation to induction therapy for the treatment of MS, strategies that are aimed at mitigating the risks of potent disease modifying therapies are urgently needed.43
Only through the scientific method of subjecting our hypothesis to the rigors of a controlled, adequately blinded, prospective and adequately powered clinical trial can we confirm or refute the hypothesis-driven question; can the application of low-dose anti-CD20 therapy, temporally synchronized with objective confirmation of B-cell hyperrepopulation, serve to adequately reduce or even prevent alemtuzumab-associated secondary autoimmunity?
Furthermore, the addition of anti-CD20 therapy could potentially confer benefits on immune deviation that would exert added efficacy in synergy with the alemtuzumab-mediated mechanisms of action in the establishment of disease remission, while constituting augmented and durable regulatory properties in keeping with those achieved during the critical period that we now recognize collectively as self-tolerance.
Buffeted by the broad dissemination of bone marrow mobilization of stem cells, following alemtuzumab treatment may promote the process of restoration and neurologic functional reconstitution. Recent evidence has demonstrated that an emerging biomarker for assessing tissue damage in the CNS of patients with MS, the neurofilament light chain, is reduced most markedly following the administration of alemtuzumab when compared with other disease-modifying therapies and that such levels remain reduced to the greatest magnitude over time.44
It was once thought that a proposition such as repair and functional recovery from MS was wholly enigmatic and unlikely. However, we have arrived at a point in the history of our understanding of the disorder's pathobiology, in concert with the corresponding derangements in immune regulatory influences, and how to remediate them. We believe our work to represent but a single small step toward this constellation of goals that are so important to our deserving patients, their families, our community, and those of us who serve on those teams who care for and about them.
Acknowledgment
The authors thank Mr. Jason Ooi and Dr. Matthew Parsons for their transformation and production of Elliot and Teresa Frohman's formulation of the figure 1 medical illustration, which showcases alemtuzumab's mechanism of action, the pathobiological underpinnings for treatment-associated secondary autoimmunity, and the hypothetical mechanisms germane to our “whack-a-mole” B-cell deletion strategy aimed at mitigating alemtuzumab-associated autoimmunity in MS.
Glossary
- ANC
absolute neutrophil count
- EAE
experimental autoimmune encephalitis
- GBM
glomerular basement membrane
- NMOSD
neuromyelitis optica spectrum disorder
- Tregs
regulatory T cells
Appendix. Authors

Study funding
The National Multiple Sclerosis Society 5-Year Institutional Fellowship Training Grant: Awarded to: T.C. Frohman and E.M. Frohman. Fellows funded: E. Meltzer and R.A. Cruz (coauthors). MS and Neuroimmunology Center. The Dell Medical School at The University of Texas at Austin.
Disclosure
E. Meltzer served as a consultant for Genzyme and received honoraria from Novartis. S. Campbell and B. Ehrenfeld have nothing to disclose. L. Steinman is on the Editorial Boards of the Proceedings of the National Academy of Sciences and the Journal of Neuroimmunology. He has served on the Editorial Board of the Journal of Immunology and International Immunology. He has served as a member of grant review committees for the NIH and the National MS Society. He has served, or serves, as a consultant and received honoraria from Atara Biotherapeutics, Atreca, Biogen-Idec, Celgene, Centocor, Coherus, EMD-Serono, Genzyme, Johnson and Johnson, Novartis, Roche/Genentech, Teva Pharmaceuticals Inc., and TG Therapeutics. He has served on the Data Safety Monitoring Board for TG Therapeutics. He serves on the Board of Directors of Tolerion and Chairs the Scientific Advisory Board for Atreca. Currently, L. Steinman receives research grant support from the NIH and Atara Biotherapeutics. M.S. Parsons has nothing to disclose. S.S. Zamvil is Deputy Editor of Neurology, Neuroimmunology and Neuroinflammation and is an Associate Editor for Frontiers in Immunology and Frontiers in Neurology. He serves on the Advisory Committee for the American Congress on Treatment and Research in Multiple Sclerosis (ACTRIMS) and is a standing member of the research grant review committee for the National Multiple Sclerosis Society (NMSS). He has served on the Editorial Board of the Journal of Clinical Investigation, The Journal of Immunology and The Journal of Neurological Sciences and has been a charter member of the grant review committee for the NIH Clinical Neuroimmunology and Brain Tumors (CNBT). He has served, or serves, as a consultant and received honoraria from Alexion, Biogen-Idec, EMD-Serono, Genzyme, Novartis, Roche/Genentech, and Teva Pharmaceuticals Inc. and has served on Data Safety Monitoring Boards for Lilly, BioMS, Teva, and Opexa Therapeutics. Currently, S.S. Zamvil receives research grant support from the NIH, NMSS, Weill Institute, Race to Erase MS, and the Maisin Foundation. T.C. Frohman has received advisory board fees from Alexion. E.M. Frohman has received speaker honoraria from Genzyme, Novartis, Alexion, and Acorda. Go to Neurology.org/NN for full disclosures.
References
- 1.Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet 2012;380:1829–1839. [DOI] [PubMed] [Google Scholar]
- 2.Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet 2012;380:1819–1828. [DOI] [PubMed] [Google Scholar]
- 3.Coles AJ, Cohen JA, Fox EJ, et al. Alemtuzumab CARE-MS II 5-year follow-up. Neurology 2017;89:1117–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Havrdova E, Arnold DL, Cohen JA, et al. Alemtuzumab CARE-MS I 5-year follow-up. Neurology 2017;89:1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarvepalli D, Rashid MU, Ullah W, Zafar Y, Khan M. Idiopathic thrombocytopenic purpura: a rare syndrome with alemtuzumab, review of monitoring protocol. Cureus 2019;11:e5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruck T, Schulte-Mecklenbeck A, Pfeuffer S, et al. Pretreatment anti-thyroid autoantibodies indicate increased risk for thyroid autoimmunity secondary to alemtuzumab: a prospective cohort study. EBioMedicine 2019;46:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruck T, Pfeuffer S, Schulte-Mecklenbeck A, et al. Vitiligo after alemtuzumab treatment: secondary autoimmunity is not all about B cells. Neurology 2018;91:e2233–e2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zimmermann J, Buhl T, Müller M. Alopecia universalis following alemtuzumab treatment in multiple sclerosis: a barely recognized manifestation of secondary autoimmunity-report of a case and review of the literature. Front Neurol 2017;8:569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rotondi M, Molteni M, Leporati P, Capelli V, Marinò M, Chiovato L. Autoimmune thyroid diseases in patients treated with alemtuzumab for multiple sclerosis: an example of selective anti-TSH-receptor immune response. Front Endocrinol (Lausanne) 2017;8:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costelloe L, Jones J, Coles A. Secondary autoimmune diseases following alemtuzumab therapy for multiple sclerosis. Expert Rev Neurother 2012;12:335–341. [DOI] [PubMed] [Google Scholar]
- 11.Jones JL, Phuah CL, Cox AL, et al. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H). J Clin Invest 2009;119:2052–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torino F, Barnabei A, Paragliola R, Baldelli R, Appetecchia M, Corsello SM. Thyroid dysfunction as an unintended side effect of anticancer drugs. Thyroid 2013;23:1345–1366. [DOI] [PubMed] [Google Scholar]
- 13.Azzopardi L, Thompson SA, Harding KE, et al. Predicting autoimmunity after alemtuzumab treatment of multiple sclerosis. J Neurol Neurosurg Psychiatry 2014;85:795–798. [DOI] [PubMed] [Google Scholar]
- 14.Trinh T, Haridas AS, Sullivan TJ. Ocular findings in alemtuzumab (Campath-1H)-induced thyroid eye disease. Ophthalmic Plast Reconstr Surg 2016;32:e128–e129. [DOI] [PubMed] [Google Scholar]
- 15.von Kutzleben S, Pryce G, Giovannoni G, Baker D. Depletion of CD52-positive cells inhibits the development of central nervous system autoimmune disease, but deletes an immune-tolerance promoting CD8 T-cell population. Implications for secondary autoimmunity of alemtuzumab in multiple sclerosis. Immunology 2017;150:444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiendl H, Carraro M, Comi G, et al. Lymphocyte pharmacodynamics are not associated with autoimmunity or efficacy after alemtuzumab. Neurol Neuroimmunol Neuroinflamm 2019;7:1–10. doi:10.1212/NXI.0000000000000635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kousin-Ezewu O, Azzopardi L, Parker RA, et al. Accelerated lymphocyte recovery after alemtuzumab does not predict multiple sclerosis activity. Neurology 2014;82:2158–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones J, Thompson S, Loh P, et al. Human autoimmunity after lymphocyte depletion is caused by homeostatic T-cell proliferation. Proc Natl Acad Sci U S A 2013;110:20200–20205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costa DB, Kobayashi SS. Whacking a mole-cule: clinical activity and mechanisms of resistance to third generation EGFR inhibitors in EGFR mutated lung cancers with EGFR-T790M. Transl Lung Cancer Res 2015;6:809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shin J, MacCarthy T. Antagonistic coevolution drives whack-a-mole sensitivity in gene regulatory networks. PLoS Comput Biol 2015;11:e1004432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki K. Whack-a-mole strategy for multifocal ground glass opacities of the lung. J Thorac Dis 2017;9(suppl 3):S201–S207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swanson DS, Harrison CJ. Playing "Whack-a-Mole" with pneumococcal serotype eradication. Pediatrics 2017;140:e20172034. [DOI] [PubMed] [Google Scholar]
- 23.Meštrović T. A microbial game of whack-a-mole: clinical case series of the urethral uncloaking phenomenon caused by Corynebacterium glucuronolyticum in men treated for Chlamydia trachomatis urethritis. Infection 2019;47:121–124. [DOI] [PubMed] [Google Scholar]
- 24.Gelfand JM, Cree BAC, Hauser SL. Ocrelizumab and other CD20+ B-cell-depleting therapies in multiple sclerosis. Neurotherapeutics 2017;14:835–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Voog E, Morschhauser F, Solal-Céligny P. Neutropenia in patients treated with rituximab. N Engl J Med 2003;348:2691–2694; discussion 2691–2694. [DOI] [PubMed] [Google Scholar]
- 26.Bar-Or A, Fawaz L, Fan B, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol 2010;67:452–461. [DOI] [PubMed] [Google Scholar]
- 27.Weber MS, Prod'homme T, Patarroyo JC, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol 2010;68:369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monson NL, Cravens P, Hussain R, et al. Rituximab therapy reduces organ-specific T cell responses and ameliorates experimental autoimmune encephalomyelitis. PLoS One 2011;6:e17103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molnarfi N, Schulze-Topphoff U, Weber MS, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med 2013;210:2921–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baker D, Herrod SS, Alvarez-Gonzalez C, Giovannoni G, Schmierer K. Interpreting lymphocyte reconstitution data from the pivotal phase 3 trials of alemtuzumab. JAMA Neurol 2017;74:961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krupica T, Fry TJ, Mackall CL. Autoimmunity during lymphopenia: a two-hit model. Clin Immunol 2006;120:121–128. [DOI] [PubMed] [Google Scholar]
- 32.Wilk E, Witte T, Marquardt N, et al. Depletion of functionally active CD20+ T cells by rituximab treatment. Arthritis Rheum 2009;60:3563–3571. [DOI] [PubMed] [Google Scholar]
- 33.Schuh E, Berer K, Mulazzani M, et al. Features of human CD3+CD20+ T cells. J Immunol 2016;197:1111–1117. [DOI] [PubMed] [Google Scholar]
- 34.Gingele S, Kripuletz T, Jacobs R. Role of CD20+ T cells in multiple sclerosis:implications for treatment with ocrelizumab. Neural Regen Res 2020;15:663–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palanichamy A, Jahn S, Nickles D, et al. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J Immunol 2014;193:580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holley JE, Bremer E, Kendall AC, et al. CD20+ inflammatory T-cells are present in blood and brain of multiple sclerosis patients and can be selectively targeted for apoptotic elimination. Mult Scler Relat Disord 2014;3:650–685. [DOI] [PubMed] [Google Scholar]
- 37.von Essen MR, Ammitzbøll C, Hansen RH, et al. Proinflammatory CD20+ T cells in the pathogenesis of multiple sclerosis. Brain 2019;142:120–132. [DOI] [PubMed] [Google Scholar]
- 38.Lovett-Racke AE, Gormley M, Liu Y, et al. B cell depletion with ublituximab reshapes the T cell profile in multiple sclerosis patients. J Neuroimmunol 2019;332:187–197. [DOI] [PubMed] [Google Scholar]
- 39.Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol 2006;180:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naismith RT, Piccio L, Lyons JA, et al. Rituximab add-on therapy for breakthrough relapsing multiple sclerosis: a 52-week phase II trial. Neurology 2010;74:1860–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenberg BM, Graves D, Remington G, et al. Rituximab dosing and monitoring strategies in neuromyelitis optica patients: creating strategies for therapeutic success. Mult Scler 2012;18:1022–1026. [DOI] [PubMed] [Google Scholar]
- 42.Kim SH, Jeong IH, Hyun JW, et al. Treatment outcomes with rituximab in 100 patients with neuromyelitis optica: influence of FCGR3A polymorphisms on the therapeutic response to rituximab. JAMA Neurol 2015;72:989–995. [DOI] [PubMed] [Google Scholar]
- 43.Steinman L. Induction of new autoimmune diseases after alemtuzumab therapy for multiple sclerosis: learning from adversity. JAMA Neurol 2017;74:907–908. [DOI] [PubMed] [Google Scholar]
- 44.Delcoigne B, Manouchehrinia A, Barro C, et al. Blood neurofilament light levels segregate treatment effects in multiple sclerosis. Neurology 2020;94:e1201–e1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified data will be shared with other investigators following requests made to the corresponding author.