

Abstract
Squamate reptiles exhibit high variation in their phenotypic traits and geographical distributions and are therefore fascinating taxa for evolutionary and ecological research. However, genomic resources are very limited for this group of species, consequently inhibiting research efforts. To address this gap, we assembled a high-quality genome of the common lizard, Zootoca vivipara (Lacertidae), using a combination of high coverage Illumina (shotgun and mate-pair) and PacBio sequencing data, coupled with RNAseq data and genetic linkage map generation. The 1.46-Gb genome assembly has a scaffold N50 of 11.52 Mb with N50 contig size of 220.4 kb and only 2.96% gaps. A BUSCO analysis indicates that 97.7% of the single-copy Tetrapoda orthologs were recovered in the assembly. In total, 19,829 gene models were annotated to the genome using a combination of ab initio and homology-based methods. To improve the chromosome-level assembly, we generated a high-density linkage map from wild-caught families and developed a novel analytical pipeline to accommodate multiple paternity and unknown father genotypes. We successfully anchored and oriented almost 90% of the genome on 19 linkage groups. This annotated and oriented chromosome-level reference genome represents a valuable resource to facilitate evolutionary studies in squamate reptiles.
Keywords: linkage map, lizard genome, multiple paternity, reptile genomics, Lacertidae, squamates
Significance
We generated a high quality, chromosome-level, annotated and oriented reference genome for the Eurasian common lizard, Zootoca vivipara. This species is the most broadly geographically distributed terrestrial squamate and has lineages that are either oviparous or viviparous. Therefore, this genome is a critical resource for advancing research on evolution, ecology, physiology, reproduction, and development.
Introduction
Squamate reptiles are one of the largest group of vertebrates, with >10,000 species distributed worldwide. They have evolved extraordinary complex biological traits, such as live-bearing (Blackburn 2006; Pyron and Burbrink 2014), parthenogenesis (Neaves and Baumann 2011), and chromosomal variation (Deakin and Ezaz 2014). However, the lack of high-quality squamate genome assemblies has slowed research on understanding some of their hallmark adaptive traits.
Among squamates, the family Lacertidae, distributed across Eurasia and Africa, is the most species rich group of reptiles in Europe. Lacertids have adapted to various environments, from hot deserts to the coldest areas colonized by any reptile (Garcia-Porta et al. 2019), vary in traits such as coloration, including “paper-rock-scissors” strategies (Sinervo et al. 2007), and reproductive mode, including parthenogenic and live-bearing species (Neaves and Baumann 2011; Sites et al. 2011). One of these live-bearer—or viviparous—species is the Eurasian common lizard, Zootoca vivipara, a fascinating ecological and evolutionary model. It has the broadest natural range and the most northern distribution among terrestrial reptiles (Herczeg et al. 2003; Garcia-Porta et al. 2019). Inhabiting a range of altitudes, it has become a model for terrestrial ectotherm response to climate change and proximate stresses (Bestion et al. 2015, 2017; Dupoué et al. 2017, 2018). Several major intraspecific lineages have a divergence time of maximally ∼6 Myr (Cornetti et al. 2014; Horreo et al. 2018), and these strikingly include differing reproductive modes (viviparous and oviparous), associated life history traits, and reproductive physiologies (Foucart et al. 2014; Recknagel and Elmer 2019). Although sex determination and chromosomes differ across squamates (Pennell et al. 2018), the karyotype is generally conserved across lacertids (Rovatsos et al. 2016); however, Z. vivipara seems to be an exception showing variation in sex chromosome structure across lineages (Kupriyanova et al. 2008). However, to date no reference genome has been available.
We combined high-coverage Illumina-derived sequencing with multilayer PacBio and RNA-seq-based scaffolding to generate a high-quality genome assembly of the Eurasian common lizard, Z. vivipara (Lacertidae). An available genome of this lizard will facilitate studies of parity mode evolution, chromosomal architecture of sex determination, and environmental adaptations exhibited by this and other squamate reptiles.
Materials and Methods
Genome Biological Sample
The reference genome was constructed using a wild-caught adult female (heterogametic sex) collected from the Isle of Cumbrae, Scotland (permission of Scottish Natural Heritage 64972). This represents an exemplar from the Western Viviparous lineage (Surget-Groba et al. 2006; Recknagel et al. 2018), with a karyotype of n = 17 autosomes and Z1Z2W sex determination (Odierna et al. 1998; Kupriyanova et al. 2008). Euthanization followed Home Office protocols.
DNA Sequencing and Quality Control
For Illumina sequencing, high molecular weight DNA was extracted from tail tissue with the Dneasy Blood and Tissue Kit (Qiagen) following the manufacturer’s protocol with additional Riboshredder and phenol–chloroform clean-up. A TruSeq PCR-free library with 350 bp insert size was generated by Edinburgh Genomics for one lane of Illumina HiSeqX sequencing. Nextera mate-pair libraries of 3–5 and 8–12 kb were generated by Liverpool Centre for Genomic Research and sequenced on one lane of HiSeq4000 at Edinburgh Genomics.
For PacBio sequencing, we used a standard phenol–chloroform isolation method (Sambrook and Russell 2006) with minimal shaking. A 20-kb insert library was generated by the Centre for Genomic Research (NBAF Liverpool) and sequenced with four cells on a PacBio Sequel.
Raw reads were checked using FastQC v0.11.5 (Andrews 2015) and trimmed using Trimmomatic v036 (Bolger et al. 2014). We applied a read error correction using QuorUM v.1.1.0 (Marçais et al. 2015) to the short-insert size (350 bp) paired-ends [PEs]. Nextera junction adapters in the long insert size mate-pairs (3–5, 8–12 kb) were removed with NxTrim v0.4.1 (O’Connell et al. 2015).
RNA-Sequencing
Total RNA was extracted from RNAlater-preserved tissue (intestine, lungs, liver, muscle) using PureLink RNA Mini Kits (Life Technologies, Carlsbad, CA) following the protocol by Gunter et al. (2013). Libraries were prepared for each tissue separately with the Illumina TruSeq Total Stranded RNA-seq protocol by Edinburgh Genomics and sequenced on one lane of an Illumina HiSeq4000 (150 bp PEs).
Genome Assembly
Genome size was estimated using the k-mer distribution method of SGA v0.10.15 (Simpson 2014). Contigs were assembled using the Platanus v1.2.4 assembler (Kajitani et al. 2014). Initial scaffolding was performed using the platanus scaffold command with all the reads excluding by-product PE and SE from mate-pair libraries. Next, the resulting scaffolds were rescaffolded with the PacBio long reads (at least 1,000 bp long to reduce the chimera rate) and the 8–12 kb mate pairs using the OPERA-LG v2.0.6 software (Gao et al. 2016) and a k-mer size = 50.
The scaffolds outputted by OPERA-LG were additionally scaffolded using AGOUTI v0.3.3 (Zhang et al. 2016), which uses RNA-seq data and splicing information. To apply the AGOUTI algorithm, we first identified coding sequences in the draft genome using AUGUSTUS v3.3 (Stanke et al. 2004) and mapped the RNA-seq reads to the genome with the BWA v0.7.15-r1140 mem algorithm (Li and Durbin 2009).
At the final stage of the assembly, we closed gaps using the GapCloser v1.12 module in SoapDenovo2 (Luo et al. 2012) with all the available Illumina reads and then with the PBJelly v15.8.24 (English et al. 2012) long-read based algorithm. PacBio reads were error-corrected using Canu v1.5 (Koren et al. 2017) prior to the assembly. The overall assembly process is depicted in figure 1a.
Fig. 1.
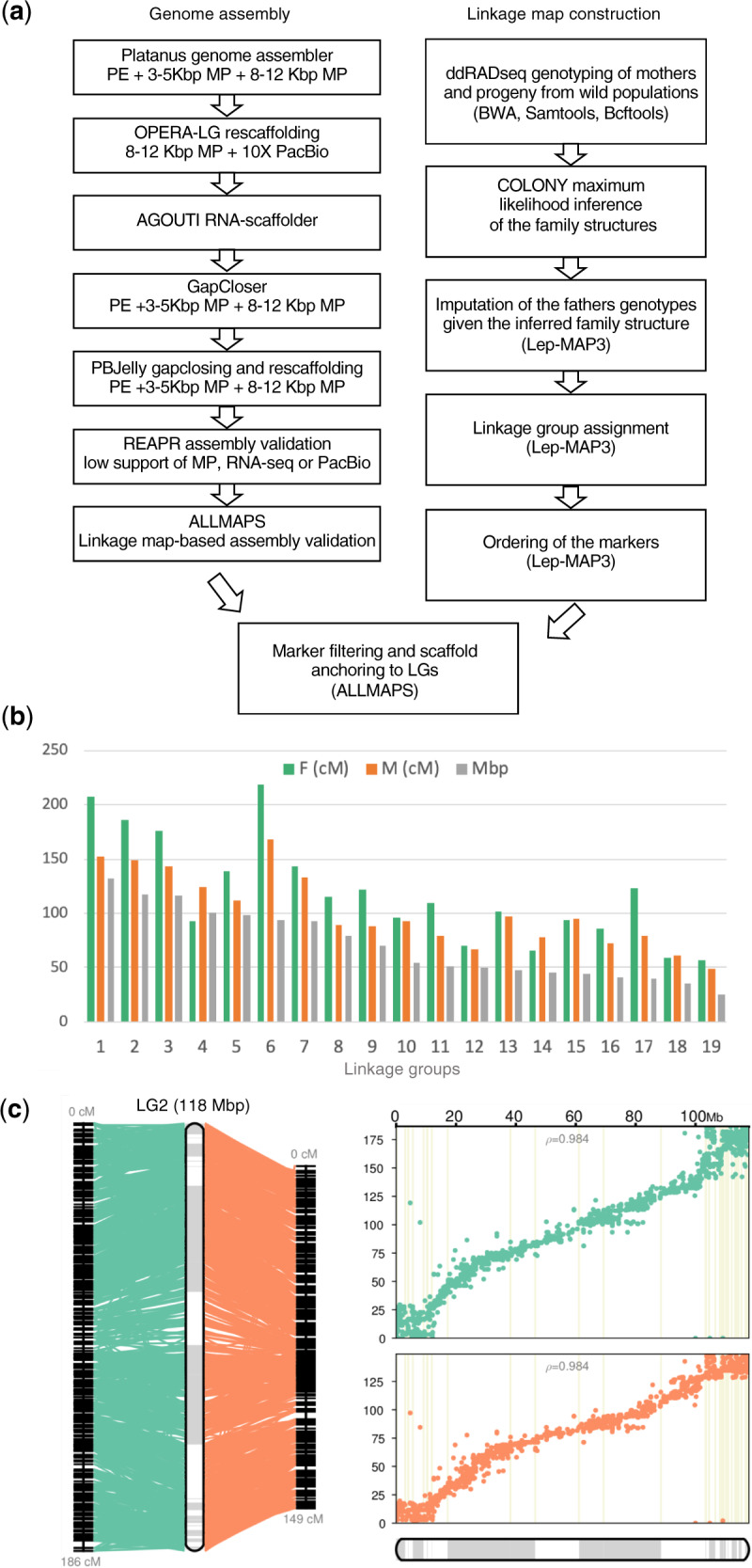
The genome assembly and linkage map construction for the common lizard, Zootoca vivipara. (a) The genome assembly and linkage map generation pipelines used in the study. (b) The length of the male (M), female (F), and consensus linkage groups. (c) An example of Linkage Group 2 based on the male (orange) and female (green) genetic maps. Pearson correlation coefficients between the physical (X axis) and genetic (Y axis) distances are indicated. Gray and white bars represent scaffolds.
Linkage Map Construction
In total, 205 individuals from 20 families of known mothers and progeny but without paternal data were sampled nonlethally from the Gailtal region in Austria (permission from Bezirkshauptmannschaft Hermagor HE3-NS-959/2013). Individuals were sampled from the Central Viviparous II and Eastern Oviparous lineages, at a site where some admixture occurs (McLennan et al. 2019). DNA was extracted from tail clip tissue using the Dneasy Blood and Tissue Kit. Genomic libraries were constructed using double-digest restriction-site associated DNA sequencing following methods in Recknagel et al. (2018). Libraries were sequenced at Edinburgh Genomics on two lanes of Illumina HiSeq4000 with 150-bp PE reads. Reads were aligned to the genomic scaffolds and SNPs were called using bcftools (Li 2011) with successive family assignment (see supplementary text, Supplementary Material online).
Next, we used Lep-MAP3 v0.2 (Rastas 2017) to convert VCF files and produce the male and female linkage maps. At the final stage of linkage map construction, we arranged scaffolds into linkage groups with ALLMAPS (Tang et al. 2015), using both the male and female linkage maps simultaneously (supplementary text, Supplementary Material online).
Assembly QC and Validation
We used REAPR v1.0.18 (Hunt et al. 2013) with mate-pairs and fragment coverage of PacBio and RNA-seq reads to validate the assembly. Finally, the assembly was validated and integrated with the linkage map (supplementary text, Supplementary Material online).
Genome Annotation
To annotate we employed homology-based (GeMoMa 1.4.2; Keilwagen et al. 2016), ab initio prediction (AUGUSTUS v3.3; Stanke et al. 2006), and RNA-seq-based methods (StringTie v1.3.1c; Pertea et al. 2015), which were combined using EVidenceModeler v1.1.1 (Haas et al. 2008) during two stages. First, the consensus gene-models were calculated and extracted. Then, the consensus proteins were blasted against the Swiss-Prot (Boutet et al. 2007) database using DIAMOND v0.9.13 (Buchfink et al. 2015) and genes without any homology to the database were excluded (supplementary text, Supplementary Material online).
Comparative Analysis
We identified single-copy orthologues from 16 published squamate genomes using Orthofinder (Emms and Kelly 2015). A phylogenetic tree of the aligned protein sequences was constructed in RAxML v.8.2.9 (Stamatakis 2014). Whole-genome alignment of the Z. vivipara assembly was performed against masked Podarcis muralis and Crotalus viridis genome assemblies using LASTZ (Harris 2007) (supplementary text, Supplementary Material online).
Results and Discussion
Genome Assembly
After read filtering and correction, we received 343M shotgun PE reads, 78M reads from 3- to 5-kb mate-pair libraries, and 53M reads from 8- to 12-kb mate-pair libraries from the short-read Illumina sequencing data (supplementary table S1, Supplementary Material online). These data were used to build contigs and scaffolds with the Platanus assembler along with 102M PE and 164M SE reads that were a by-product after trimming and filtering the PE and mate-pairs (only for contig assembly, supplementary table S1, Supplementary Material online).
The first scaffolds produced with Platanus had N50 metrics equal to 5.35 Mb and consisted of 366.9k contigs with a N50 of 5.23 kb (supplementary table S2, Supplementary Material online). After rescaffolding the assembly with Opera-LG using PacBio data (1.7 million reads) and the 8–12 kb mate-pairs, we doubled the N50 scaffold length to 12.52 Mb. Subsequent rescaffolding with the RNA-seq information on splicing events and gap closing using short reads increased the N50 contig size to 83.4 kb. Finally, gap closing with long PacBio reads allowed us to additionally increase the contig length distribution size to achieve an N50 of 220.4 kb.
The REAPR pipeline customized with additional PacBio and RNA-seq data allowed us to identify 1,733 likely erroneous joins between contigs, mostly at the ends of scaffolds that were then further broken. In sum, the assembled scaffolds were high quality and highly contiguous, benefiting from the combination of data types.
Linkage Map Construction and Scaffold Anchoring
We used linkage mapping as an established approach for chromosome-level assembly and which provides additional information on recombination rates, physical to genetic distances, and sex-specific recombination (Fierst 2015). Sequencing for linkage maps generated 643M clean PE reads that were used, representing 20 families of mothers and offspring. Through a stringent probabilistic estimation of family structure and parent assignment, we found widespread multiple paternities. Specifically, four families had a single father whereas all others had from two to four fathers, with mean of 3.7 progeny per half-sib family (supplementary table S5, Supplementary Material online). This agrees with other research suggesting multiple paternity is abundant in common lizards (Laloi et al. 2004; Fitze et al. 2005).
We retained 109,640 high-quality biallelic SNPs and used them for imputing the missing genotypes of fathers in a probabilistic framework. The high genomic diversity made imputation efficient due to a large number of highly polymorphic SNPs with heterozygous positions. At the first stage of linkage map construction, 17,210 markers were assigned to 19 linkage groups (from 395 to 1,648 markers per LG, LOD score = 10.7), in agreement with the Z. vivipara karyotype with 17 autosomes and the Z and W sex chromosomes (2n = 36 chromosomes including ZZ/Zw sex chromosomes) specific for these lineages (Kupriyanova et al. 2014). At the next step, an additional 7,177 markers were assigned to these LGs with a minimal LOD score of 9. Finally, we had 1.27 and 1.24 markers per cM for the male (1,929.24 cM) and female (2,263.13 cM) linkage maps with 2,487 and 2,845 unique points, respectively (supplementary table S3, Supplementary Material online). The relatively low rate (21%, 24,387/109,640) of linkage-informative, high-quality SNPs that were assigned to the final linkage map can partially be attributed to the imputation of father genotypes and our stringent criteria for inclusion (supplementary text, Supplementary Material online).
We anchored 91.2% and oriented 89.5% of the assembly using the linkage map (supplementary table S4, Supplementary Material online). The physical size of linkage groups varied from 24.9 to 131.77 Mb and physical positions of markers strongly correlated with the linkage-based positions on the map (fig. 1b and c). The average resolution of the male and female linkage maps was 0.67 and 0.59 Mb per cM, respectively (supplementary table S3, Supplementary Material online).
At the final stage we identified and broke 30 intrascaffold regions that showed signs of misassembly according to the linkage map data. After this validation step, the formal assembly quality metrics slightly reduced (scaffold N50 by 1.23–11.52 Mb), but still indicated a high level of assembly contiguity (supplementary table S2, Supplementary Material online). Therefore, given that these are all within the same species, a relatively stable autosomal karyotype was known (Odierna et al. 2001), very few scaffolds were reassembled by linkage map information, and the physical and genetic distances are concordant (fig. 1), the use of multiple lineages did not have significant consequences on the chromosome-level assembly. However, future lineage-specific assemblies would be valuable and informative.
To further quantify the quality of the final scaffolds, we estimated the number of recovered Tetrapoda single-copy orthologues (BUSCO) in the assembled genome. We found that 94% of orthologues were completely assembled (with 1.3% of them being duplicated), 3.7% were fragmented, and 2.3% of the 3,950 benchmarked genes were missed. This metric is comparable to other recently assembled high quality genomes (Andrade et al. 2019; Suryamohan et al. 2020) and indicates that the assembly was of high quality with only minor parts of the genome being fragmented.
Genome Annotation
Homology-based GeMoMa allowed us to identify 21,187 high quality gene-models with strong homology to chicken, Japanese gecko, and anole lizard genomes. The ab initio AUGUSTUS pipeline identified 15,637 gene-models which were finally combined using EVidenceModeler with 28,473 RNA-seq-based Transdecoder and GeMoMa gene models. After filtering out genes without any detected homology to the Swiss-Prot database, we received a final set of 19,829 protein-coding gene models. This is slightly lower than the three other lacertid species sequenced (Kolora et al. 2018; Andrade et al. 2019) and other squamates (Eckalbar et al. 2013; Suryamohan et al. 2020) which used less stringent filtering criteria but comparable to NCBI annotated genomes (19,431 protein coding genes for Anolis carolinensis; 19,535 for Gekko japonicus; and 18,971 for Pogona vitticeps).
Genome Size
The estimated genome size of Z. vivipara was ≈1.345 Gb based on SGA k-mer distribution analysis, agreeing with earlier flow-cytometry based reports (1.035–1.515 Gb) (Vinogradov 1998). The final assembly length, including all linkage groups and unanchored scaffolds, was 1.46 Gb.
Comparative Analysis
This Z. vivipara genome is one of six chromosome-level assemblies of the species-rich squamates to date. The hybrid assembly strategy we employed allowed us to achieve superior contig and scaffold size to Illumina-only squamate assemblies: 220.4 kb for contig and 11.54 Mb for scaffold N50 size in Z. vivipara versus O. gracilis genome with 42.8 kb contig and 1.27 Mb size as an example of one of the best Illumina-only squamate genome assemblies (Song et al. 2015). The contiguity of these Z. vivipara contigs is comparable to the other hybrid assembly-based genomes (Kolora et al. 2018) but is less than assemblies mainly generated by long-reads (Andrade et al. 2019). Our maximum-likelihood analysis resolved a phylogeny with Z. vivipara in a clade together with the recently assembled wall lizard (P. muralis) and two other Lacerta species (fig. 2a). The lacertid clade was, as expected (Irisarri et al. 2017), deeply divergent from other groups. Whole-genome alignment between Z. vivipara and the <40 Ma divergent (Garcia-Porta et al. 2019) P. muralis genome (Andrade et al. 2019) demonstrated a high level of synteny. The divergence time between common lizards and the rattlesnake, C. viridis, is >160 Ma (Pyron and Burbrink 2014), but nonetheless synteny was broadly conserved (fig. 2b). However, synteny analyses also demonstrated dynamic genome rearrangements between these distant lineages and many inter and intrachromosomal changes. In summary, the Z. vivipara genome shows high levels of contiguity and synteny with other squamate species, decreasing in synteny with divergence time.
Fig. 2.

Zootoca vivipara genome assembly in the context of other Squamata genomes. (a) Maximum-likelihood tree based on the 269 single-copy orthologs from available Squamata genomes. (b) Synteny between Z. vivipara chromosome-level assembly and snake (Crotalus viridis) and closely related wall lizard genome (Podarcis muralis).
Conclusions
Here, we report a chromosome-level genome assembly for the wide-ranging, cold-adapted and reproductively bimodal common lizard, Z. vivipara. The final assembly contains 19 linkage groups with almost 90% of the genome anchored and oriented, and assembly length is 1.46 Gb. We annotated 19,829 protein-coding genes and inferred high quality BUSCO metrics, with 97.7% of Tetrapoda-specific single-copy orthologues recovered (only 3.7% fragmented). We applied a novel linkage mapping approach from multiple families with absent paternal information and multiple paternity structure, which could be applied to other sexually reproducing systems in which one parent and sibs are known but the other parent must be imputed. This genome assembly will be a useful resource for a wide range of studies on the fascinating evolutionary diversity of squamate reptiles.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We are grateful for the help in library preparation and RNA extraction to Aileen Adams and Elizabeth Kilbride; NBAF Edinburgh (especially Karim Gharbi and Helen Gunter) and NBAF Liverpool for advice, library preparation, and sequencing; and Scottish Natural Heritage and Bezirkshauptmannschaft Hermagor for collection permits. This research was supported by the Natural Environment Research Council (NE/N003942/1, NBAF964, NBAF1018).
Contributor Information
Andrey A Yurchenko, Institute of Biodiversity, Animal Health and Comparative Medicine, College of Medical, Veterinary and Life Sciences, University of Glasgow, United Kingdom.
Hans Recknagel, Institute of Biodiversity, Animal Health and Comparative Medicine, College of Medical, Veterinary and Life Sciences, University of Glasgow, United Kingdom.
Kathryn R Elmer, Institute of Biodiversity, Animal Health and Comparative Medicine, College of Medical, Veterinary and Life Sciences, University of Glasgow, United Kingdom.
Data deposition: This project has been deposited at NCBI under the accessions PRJNA610958 (genome assembly and raw sequencing data) and PRJNA626507 (linkage map raw data), and Glasgow University Research Repository http://researchdata.gla.ac.uk/1005 (gene annotation).
Literature Cited
- Andrade P, et al. 2019. Regulatory changes in pterin and carotenoid genes underlie balanced color polymorphisms in the wall lizard. Proc Natl Acad Sci USA. 116(12):5633–5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2015. FASTQC a quality control tool for high throughput sequence data. Babraham Inst. http://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed January 1, 2019. [Google Scholar]
- Bestion E, et al. 2017. Climate warming reduces gut microbiota diversity in a vertebrate ectotherm. Nat Ecol Evol. 1:161. [DOI] [PubMed] [Google Scholar]
- Bestion E, Teyssier A, Richard M, Clobert J, Cote J. 2015. Live fast, die young: experimental evidence of population extinction risk due to climate change. PLOS Biol. 13(10):e1002281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn DG. 2006. Squamate reptiles as model organisms for the evolution of viviparity. Herpetol Monogr. 20(1):131. [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet E, Lieberherr D, Tognolli M, Schneider M, Bairoch A. 2007. UniProtKB/Swiss-Prot. Methods Mol Biol. 406:89–112. [DOI] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 12(1):59–60. [DOI] [PubMed] [Google Scholar]
- Cornetti L, Menegon M, Giovine G, Heulin B, Vernesi C. 2014. Mitochondrial and nuclear DNA survey of Zootoca vivipara across the eastern Italian Alps: evolutionary relationships, historical demography and conservation implications. PLoS One 9(1):e85912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin JE, Ezaz T. 2014. Tracing the evolution of amniote chromosomes. Chromosoma 123(3):201–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupoué A, et al. 2017. Shorter telomeres precede population extinction in wild lizards. Sci Rep. 7(1):16976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupoué A, et al. 2018. Reduction in baseline corticosterone secretion correlates with climate warming and drying across wild lizard populations. J Anim Ecol. 87(5):1331–1341. [DOI] [PubMed] [Google Scholar]
- Eckalbar WL, et al. 2013. Genome reannotation of the lizard Anolis carolinensis based on 14 adult and embryonic deep transcriptomes. BMC Genomics 14(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms DM, Kelly S. 2015. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16(1):157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English AC, et al. 2012. Mind the gap: upgrading genomes with pacific biosciences RS long-read sequencing technology. PLoS One 7(11):e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierst JL. 2015. Using linkage maps to correct and scaffold de novo genome assemblies: methods, challenges, and computational tools. Front Genet. 6:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitze PS, Le Galliard J-F, Federici P, Richard M, Clobert J. 2005. Conflict over multiple-partner mating between males and females of the polygynandrous common lizards. Evolution 59(11):2451–2459. [PubMed] [Google Scholar]
- Foucart T, Lourdais O, DeNardo DF, Heulin B. 2014. Influence of reproductive mode on metabolic costs of reproduction: insight from the bimodal lizard Zootoca vivipara. J Exp Biol. 217(22):4049–4056. [DOI] [PubMed] [Google Scholar]
- Gao S, Bertrand D, Chia BKH, Nagarajan N. 2016. OPERA-LG: efficient and exact scaffolding of large, repeat-rich eukaryotic genomes with performance guarantees. Genome Biol. 17(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Porta J, et al. 2019. Environmental temperatures shape thermal physiology as well as diversification and genome-wide substitution rates in lizards. Nat Commun. 10(1):4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter HM, et al. 2013. Shaping development through mechanical strain: the transcriptional basis of diet-induced phenotypic plasticity in a cichlid fish. Mol Ecol. 22(17):4516–4531. [DOI] [PubMed] [Google Scholar]
- Haas BJ, et al. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9(1):R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS. 2007. Improved pairwise alignment of genomic DNA [PhD thesis]. Pennsylvania State University.
- Herczeg G, Kovács T, Hettyey A, Merilä J. 2003. To thermoconform or thermoregulate? An assessment of thermoregulation opportunities for the lizard Zootoca vivipara in the subarctic. Polar Biol. 26(7):486–490. [Google Scholar]
- Horreo JL, et al. 2018. Phylogeography, evolutionary history and effects of glaciations in a species (Zootoca vivipara) inhabiting multiple biogeographic regions. J Biogeogr. 45(7):1616–1627. [Google Scholar]
- Hunt M, et al. 2013. REAPR: a universal tool for genome assembly evaluation. Genome Biol. 14(5):R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irisarri I, et al. 2017. Phylotranscriptomic consolidation of the jawed vertebrate timetree. Nat Ecol Evol. 1(9):1370–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajitani R, et al. 2014. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24(8):1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilwagen J, et al. 2016. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44(9):e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolora SRR, et al. 2018. Divergent evolution in the genomes of closely related lacertids, Lacerta viridis and L. bilineata, and implications for speciation. Gigascience 8:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, et al. 2017. Canu: scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 27(5):722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupriyanova L, Kuksin A, Odierna G. 2008. Karyotype, chromosome structure, reproductive modalities of three Southern Eurasian populations of the common lacertid lizard, Zootoca vivipara (Jacquin, 1787). Acta Herpetol. 3:99–106. [Google Scholar]
- Kupriyanova L, Niskanen M, Oksanen TA. 2014. Karyotype dispersal of the common lizard Zootoca vivipara (Lichtenstein, 1823) in eastern and northeastern Fennoscandia. Memo Soc pro Fauna Flora Fenn. 90:83–90. [Google Scholar]
- Laloi D, Richard M, Lecomte J, Massot M, Clobert J. 2004. Multiple paternity in clutches of common lizard Lacerta vivipara: data from microsatellite markers. Mol Ecol. 13(3):719–723. [DOI] [PubMed] [Google Scholar]
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27(21):2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1(1):2047–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais G, Yorke JA, Zimin A. 2015. QuorUM: an error corrector for Illumina reads. PLoS One 10(6):e0130821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan D, Recknagel H, Elmer KR, Monaghan P. 2019. Distinct telomere differences within a reproductively bimodal common lizard population. Funct Ecol. 33(10):1917–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neaves WB, Baumann P. 2011. Unisexual reproduction among vertebrates. Trends Genet. 27(3):81–88. [DOI] [PubMed] [Google Scholar]
- O’Connell J, et al. 2015. NxTrim: optimized trimming of Illumina mate pair reads. Bioinformatics 31(12):2035–2037. [DOI] [PubMed] [Google Scholar]
- Odierna G, et al. 1998. Progressive differentiation of the W sex-chromosome between oviparous and viviparous populations of Zootoca vivipara (Reptilia, Lacertidae). Ital J Zool. 65(3):295–302. [Google Scholar]
- Odierna G, et al. 2001. Evolutionary and biogeographical implications of the karyological variations in the oviparous and viviparous forms of the lizard Lacerta (Zootoca) vivipara. Ecography 24(3):332–340. [Google Scholar]
- Pennell MW, Mank JE, Peichel CL. 2018. Transitions in sex determination and sex chromosomes across vertebrate species. Mol Ecol. 27(19):3950–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, et al. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 33(3):290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyron RA, Burbrink FT. 2014. Early origin of viviparity and multiple reversions to oviparity in squamate reptiles. Ecol Lett. 17(1):13–21. [DOI] [PubMed] [Google Scholar]
- Rastas P. 2017. Lep-MAP3: robust linkage mapping even for low-coverage whole genome sequencing data. Bioinformatics 33(23):3726–3732. [DOI] [PubMed] [Google Scholar]
- Recknagel H, Elmer KR. 2019. Differential reproductive investment in co-occurring oviparous and viviparous common lizards (Zootoca vivipara) and implications for life-history trade-offs with viviparity. Oecologia 190(1):85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recknagel H, Kamenos NA, Elmer KR. 2018. Common lizards break Dollo’s law of irreversibility: genome-wide phylogenomics support a single origin of viviparity and re-evolution of oviparity. Mol Phylogenet Evol. 127:579–588. [DOI] [PubMed] [Google Scholar]
- Rovatsos M, et al. 2016. Conservation of sex chromosomes in lacertid lizards. Mol Ecol. 25(13):3120–3126. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. 2006. Purification of nucleic acids by extraction with phenol:chloroform. Cold Spring Harb Protoc. 2006:pdb-prot4455. [DOI] [PubMed] [Google Scholar]
- Simpson JT. 2014. Exploring genome characteristics and sequence quality without a reference. Bioinformatics 30(9):1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinervo B, et al. 2007. Models of density‐dependent genic selection and a new rock‐paper‐scissors social system. Am Nat. 170(5):663–680. [DOI] [PubMed] [Google Scholar]
- Sites JW, Reeder TW, Wiens JJ. 2011. Phylogenetic insights on evolutionary novelties in lizards and snakes: sex, birth, bodies, niches, and venom. Annu Rev Ecol Evol Syst. 42(1):227–244. [Google Scholar]
- Song B, et al. 2015. A genome draft of the legless anguid lizard, Ophisaurus gracilis. GigaScience 4(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, et al. 2006. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34(Web Server):W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Steinkamp R, Waack S, Morgenstern B. 2004. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32(Web Server):W309–W312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surget-Groba Y, et al. 2006. Multiple origins of viviparity, or reversal from viviparity to oviparity? The European common lizard (Zootoca vivipara, Lacertidae) and the evolution of parity. Biol J Linn Soc. 87(1):1–11. [Google Scholar]
- Suryamohan K, et al. 2020. The Indian cobra reference genome and transcriptome enables comprehensive identification of venom toxins. Nat Genet. 52(1):106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, et al. 2015. ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biol. 16(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov AE. 1998. Genome size and GC-percent in vertebrates as determined by flow cytometry: the triangular relationship. Cytometry 31(2):100–109. [DOI] [PubMed] [Google Scholar]
- Zhang SV, Zhuo L, Hahn MW. 2016. AGOUTI: improving genome assembly and annotation using transcriptome data. GigaScience 5(1):s13742–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.