

Abstract
DNA polymerase (dpol) β has served as a model for structural, kinetic, and computational characterization of the DNA synthesis reaction. The laboratory directed by Samuel H. Wilson has utilized a multifunctional approach to analyze the function of this enzyme at the biological, chemical, and molecular levels for nearly 50 years. Over this time, it has become evident that correlating static crystallographic structures of dpol β with solution kinetic measurements is a daunting task. However, aided by computational and spectroscopic approaches, novel and unexpected insights have emerged. While dpols generally insert wrong nucleotides with similar poor efficiencies, their capacity to insert the right nucleotide depends on the identity of the dpol. Accordingly, the ability to choose right from wrong depends on the efficiency of right, rather than wrong, nucleotide insertion. Structures of dpol β in various liganded forms published by the Wilson laboratory, and others, have provided molecular insights into the molecular attributes that hasten correct nucleotide insertion and deter incorrect nucleotide insertion. Computational approaches have bridged the gap between structures of intermediate complexes and provided insights into this basic and essential chemical reaction.
Keywords: DNA polymerase β, DNA synthesis, Fidelity, Genome integrity, Structure
Introduction
High fidelity DNA synthesis is an essential biological reaction that ensures genomic stability during cellular replication and DNA repair. Mammalian cells express at least 16 DNA polymerases that participate in a variety of specialized DNA synthesis transactions. DNA polymerase (dpol) β is the smallest eukaryotic dpol and has been extensively characterized at the biochemical, cellular, kinetic, and structural levels. Several overviews of its structural [1] and biological [2] features have recently been published. An overview of the contribution of the Wilson group to the structural characterization of dpol β appears in this issue [3].
Over the years, the laboratory directed by Samuel H. Wilson has been particularly interested in correlating dpol activity with biological and molecular mechanisms. This includes defining the structural attributes that modulate apparent enzyme activity and function through systematic modification of the enzyme and/or its ligands. The Wilson laboratory has a long history in kinetically and structurally characterizing several base excision DNA repair enzymes, but especially dpol β.
DNA synthesis roadmap
DNA polymerase β contributes two enzymatic reactions that modify both the 5´- and 3´-termini in a single-nucleotide gap during base excision DNA repair. This brief overview focuses on its DNA synthesis activity. A general kinetic scheme employed by dpols is outlined in Fig. 1A. Not depicted in this scheme, but discussed briefly below, dpols also require at least two magnesiums ions. Important aspects of this scheme are an ordered binding of substrates (DNA followed by dNTP; steps 1 and 2, respectively) and product release (PPi followed by DNA dissociation; steps 6 and 7, respectively). The chemical step (step 4) is surrounded by conformational changes(s) before (step 3) and after (step 5) chemistry that impact observed kinetic parameters. The identity of these conformational changes is not known, but is believed to be associated with the transition between open and closed forms of the enzyme. DNA polymerases undergo large subdomain motions and subtle conformational adjustments that hasten or deter the chemical steps (i.e., forward and reverse chemical steps; k4 and k−4, respectively). Following chemistry, the ternary product complex undergoes a conformational change that drives the reaction forward [4]. At this point, the extended product (DNA+1) is released (single-nucleotide insertion) or serves as substrate DNA for another round of nucleotide insertion following translocation (i.e., processive DNA synthesis). Alternatively, dpols with an associated proofreading exonuclease have the opportunity to excise the newly formed primer terminus prior to translocation. DNA polymerase β lacks a proofreading activity.
Fig. 1.

Mechanistic features of the dpol β nucleotidyl transferase reaction. (A) Over-simplified dpol reaction pathway. After binding DNA (step 1), the enzyme binds dNTP (step 2) leading to formation of a ternary complex. The ternary complex undergoes a conformational change that repositions the incoming dNTP (step 3) to hasten (correct nucleotide) or deter (incorrect nucleotide) insertion (step 4). A post-chemistry conformational change (step 5) facilitates PPi release (step 6). At this point, the DNA product has three fates: continued polymerization (dashed arrow), pyrophosphorolysis (reversal of steps 6–4), or DNA dissociation (step 7). The active and inactive enzyme (E) forms are colored green and red, respectively. (B) The pH-dependence of correct nucleotide insertion expressed as % of the maximal observed rate, kmax, indicates a single-ionization with a pKa of 8.1 [9, 10]. Thus, at pH 7.4, kpol,app is <20% of kmax. (C) A discrimination plot illustrating the catalytic efficiencies for dCTP (green) and dATP (red) insertion opposite tempting G and 8oxoG [28]. Discrimination, or fidelity, is proportional to the distance between the catalytic efficiencies for right and wrong nucleotides.
While activity measurements are central to deciphering an enzyme’s biological role, as well as providing mechanistic details, they need to be interpreted carefully. Historically, activity measurements attempted to define the rate-limiting step for the forward DNA synthesis reaction. For nearly 20 years, the rate-limiting step was thought to be a poorly-defined conformational change prior to chemistry. However, it was subsequently shown that the large subdomain motion occurring upon dNTP binding was considerably more rapid than the observed rate of nucleotide insertion [5, 6]. Although this pre-chemistry step is not rate limiting, it modulates insertion by controlling the enzyme’s forward commitment thereby affecting the concentration of active ternary substrate complex [6, 7]. The apparent nucleotide binding affinity (Kd,app) and observed insertion rate (kpol,app) determined by single-turnover analysis do not correspond to intrinsic kinetic parameters even though the chemical step is the slowest step in the forward direction (Fig. 1A). This is due to the reversal of the pre-chemistry conformational change (k−3) that influences the concentration of the productive ternary complex. Accordingly, the kinetic parameters Kd,app and kpol,app are a composite of several rate constants. Two steps (steps 2 and 3) influence the apparent binding affinity for the incoming nucleotide, and the magnitude of kpol,app is sensitive to step 4 (highlighted with a gray shaded box, Fig. 1A) as well as the pre- and post-chemistry steps. Kinetic simulation and fitting software have made it possible to extract intrinsic kinetic rate constants with an appropriate model from kinetic studies [8].
The 3´-hydroxyl of the DNA terminus of an active ternary substrate complex must be deprotonated to form the attacking oxyanion. The pKa of O3´ has been measured to be ~8.1 (Fig. 1B) [9, 10]. Site-directed mutagenesis of an active site metal coordinating ligand (Asp256) indicates that the catalytic metal (metal A) lowers the pKa of O3´ ~3 pH units. Since most dpol activity measurements are performed at around pH 7.4, the majority of the DNA termini are protonated so that measurements at physiological pH significantly underestimate the rate of the insertion step (k4). Accordingly, measured changes in kpol,app at pH 7.4 with an altered enzyme or with different substrates may be due to changes in the pKa of O3´ and not due to changes in k4.
DNA polymerase nucleotide insertion discrimination or fidelity is determined by how well competing nucleotides are inserted (i.e., ratio of their catalytic efficiencies, (kcat/Km)correct/(kcat/Km)incorrect or (kpol.app/Kd,app)correct/(kpol.app/Kd,app)incorrect). In a discrimination plot (Fig. 1C), the distance between the plotted catalytic efficiencies (short horizontal lines) represents fidelity or discrimination (i.e., 1/fidelity). This plot visually exposes the reason for an altered fidelity between different enzymes or substrates. For example, the dramatic loss in nucleotide discrimination observed with a templating 8-oxo-guanine (8oxoG), a highly mutagenic lesion, is primarily because of an increase in the efficiency of dATP insertion compared to the natural guanine (G) (Fig. 1C). This is due to the preferential syn-conformation of 8oxoG that presents its Hoogsteen edge to readily hydrogen bond with dATP [11].
DNA gap binding
The two enzymatic activities contributed by dpol β modify the 5´- and 3´-termini in a single-nucleotide gap during base excision DNA repair [2]. These activities reside on two domains: an 8-kDa amino-terminal lyase domain and a 31-kDa carboxyl-terminal polymerase domain (Fig. 2A). The polymerase domain is typically composed of three functionally distinct subdomains. The catalytic (C-) subdomain coordinates two divalent metal cations that facilitate DNA synthesis, whereas the other two subdomains are spatially situated on opposite sides of the C-subdomain and participate in substrate binding. DNA polymerase β prefers single-nucleotide gapped DNA that partially occludes the dNTP binding pocket (Fig. 2B). While the DNA major groove of the nascent base pair is solvent accessible, its minor groove can interact with several residues of the N-subdomain [12].
Fig. 2.

Domain organization of dpol β and single-nucleotide gapped DNA binding. The lyase and polymerase domains are colored gray and light purple, respectively. To highlight the intrinsic function of the polymerase subdomains of dpol β, a functionally based nomenclature is employed. The polymerase domain is composed of the C- (Catalytic), D- (Duplex DNA binding), and N-subdomains (Nascent base pair binding) (Fig. 2A). These correspond to the palm, thumb, and fingers subdomains of the right-handed replicative dpols, respectively. (B) Surface representation of dpol β using the same color scheme as in panel A (PDB ID 2FMS) [17]. The DNA is shown in a ladder representation with a green template strand and yellow primer and downstream DNA strands. The 5´-phosphate on the downstream strand is indicated and is surrounded by lysine residues of the lyase domain (blue molecular surface). The downstream duplex DNA is bent ~90° as it exits the polymerase active site so that the dpol can interact with the nascent base pair. The trajectory of the template strand is indicated. (C) The view is similar to panel B, but the surface has been removed. The two HhH motifs of dpol β contributed by the lyase domain and D-subdomain of the polymerase subdomain are illustrated. The templating nucleotide (t0) and correct incoming nucleotide analogue (dUMPNPP) are shown. The active site Mg2+ are shown as small green spheres.
In the one-nucleotide gapped DNA structure, the 5´-phosphate in the gap is hydrogen bonded to several lysine side chains in the lyase domain (Fig. 2B) [13]. The 5´-phosphate or 5´-deoxyribose phosphate moieties, key base excision repair intermediates, target dpol β to the downstream side of gapped DNA. The lyase domain also includes a Helix-hairpin-Helix (HhH) motif (residues 55–79) that interacts with the DNA backbone in a sequence independent manner (Fig. 2C). A second HhH motif (residues 92–118) in the D-subdomain interacts with the DNA backbone of upstream duplex DNA. Thus, the two HhH motifs make DNA backbone interactions with each end of the incised DNA strand thereby stabilizing the pronounced bend observed in the gapped DNA structure (Fig. 2B). This architectural feature positions the nascent base pair between the primer terminus base pair and α-helix N of the N-subdomain (Fig. 3) [14].
Fig. 3.

Molecular organization of the dpol β substrate and product ternary complexes. The two upper images are cartoon representations of a pre- (PDB ID 2FMS) [17] and post- (PDB ID 4KLI) [21] catalytic crystallographic ternary complexes (bottom images) for a correct nucleotide insertion. These ternary complex structures highlight key attributes [(a)–(d), see text for details) necessary to efficiently insert a deoxynucleoside triphosphate. The DNA major groove is closest to the viewer and is solvent exposed. The large colored arrows show the trajectory of the upstream (gray) and downstream (yellow) duplex DNA. The bases of the templating (coding) and incoming nucleotide are yellow, and those of the primer terminus base pair are gray. Metal and active site coordination are illustrated with dotted lines and Watson-Crick hydrogen bonds are green lines. Key active site residues are identified in the lower left panel. Magnesium and sodium ions are shown as green and purple spheres, respectively.
Chemistry
Following rapid lyase chemistry [15], dpol β catalyzes the nucleotidyl transferase reaction. Based on the exonuclease mechanism of Klenow fragment, a ‘two-metal ion’ mechanism for nucleotidyl transfer was proposed [16]. The ternary substrate complex of dpol β is consistent with this generally accepted mechanism (Fig. 3) [17]. The catalytic metal (metal A) lowers the pKa of O3´ of the growing primer terminus while the second nucleotide binding metal (metal B) coordinates the triphosphate moiety, hastening binding of the incoming nucleotide. Additionally, metal B assists PPi dissociation. Both metals are believed to stabilize the proposed penta-coordinated transition state of the nucleotidyl transferase reaction. The chemical mechanism proceeds by an in-line nucleophilic attack of the Mg2+-activated primer O3´– on the α-phosphate of the incoming nucleotide, leading to a penta-coordinated, bipyramidal α-phosphate transition state. The transition state is resolved by release of PPi from the opposite side of the attacking oxyanion, resulting in stereo-chemical inversion about the α-phosphorus of the newly incorporated nucleotide. This reaction is reversible, so that PPi and DNA can generate dNTP and a DNA primer strand that is one nucleotide shorter; i.e., pyrophosphorolysis. Detailed structural and kinetic characterization of the reverse reaction indicates that the reverse chemical step is not directly kinetically accessible; i.e., is limited by conformational change(s) that must occur to generate an active ternary complex [4].
Fidelity checkpoints
DNA polymerases must select the correct nucleotide from a pool of structurally similar molecules to faithfully copy the template strand during replication and repair. Kinetic analysis of single-nucleotide insertion demonstrates that dpol β efficiently inserts the correct nucleotide and inefficiently inserts the incorrect nucleotide (Fig. 1C) (fidelity ~105). A kinetic survey of single-nucleotide insertion efficiencies by dpols from various biological sources and polymerase families indicates that the divergent fidelities exhibited by these dpols is primarily due to their ability to insert the correct nucleotide [18]. Thus, high fidelity enzymes efficiently insert the correct nucleotide and inefficiently insert the wrong nucleotide. In contrast, low fidelity enzymes insert both the correct and incorrect nucleotide poorly. With this perspective, a molecular understanding of fidelity requires identifying dpol strategies that hasten correct insertion and deter insertion of the wrong nucleotide. Crystallographic structures of intermediate complexes during correct nucleotide binding and insertion have provided molecular details into how this is accomplished (Fig. 4).
Fig. 4.
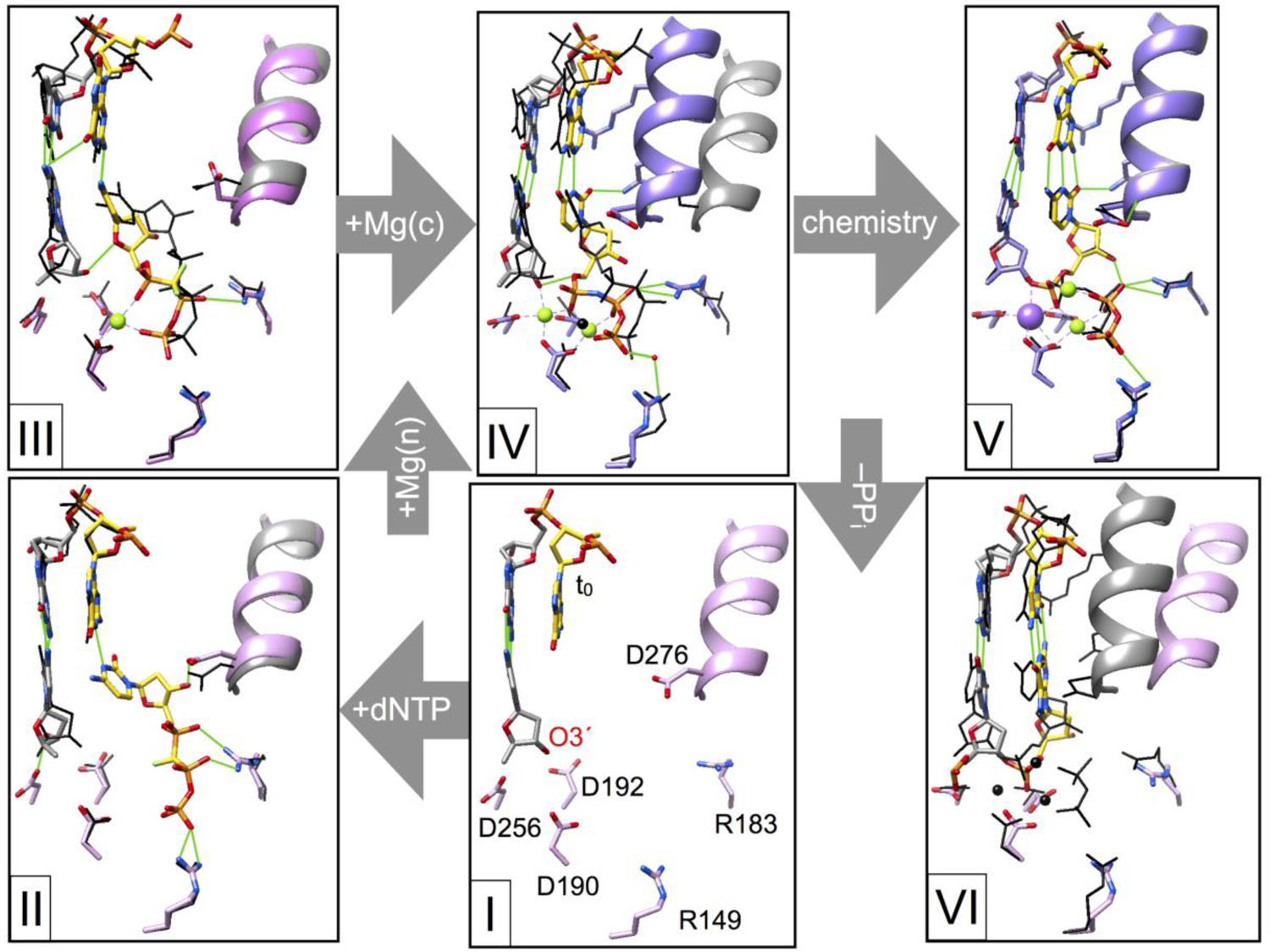
Structural attributes of intermediate complexes during correct nucleotide insertion. Crystallography has identified several protein and substrate conformational adjustments that must occur to facilitate efficient nucleotide insertion. The structure shown in complex I focuses on the active site of dpol β bound to a one-nucleotide gapped DNA substrate (i.e., binary DNA complex; PDB ID 3ISB) [29]. Key active site residues (stick representation, pink carbons) are identified as well as the templating base in the DNA gap (t0; yellow carbons) and primer terminus (O3´). The DNA primer terminal base pair is illustrated in a stick representation (gray carbons). The position of α-helix N (pink ribbon) indicates that it is in an open conformation. The correct nucleotide (dCTP, yellow carbons) binds to the open conformation (complex II; PDB ID 4F5N) [30] and can form a hydrogen bond (green line) with the templating base. In the absence of metals, the extended conformation of the triphosphate coordinates active site basic residues and the sugar moiety forms a hydrogen bond with Asp276 (D276). The position of equivalent residues observed in complex I are shown as narrow black lines and α-helix N is gray. Subsequent intermediate complex structures will be similarly compared to the previous complex illustrated with narrow black lines (gray α-helix N). Binding of the nucleotide Mg2+ (green sphere) alters the hydrogen bonding pattern of the sugar and triphosphate moieties (complex III; PDB ID 4F5O) [30]. The metal coordinates active site aspartates and non-bridging phosphate oxygens (dCTP). This one-metal structure was captured utilizing a mutant enzyme (R283K) that does not readily form a closed complex [12]. The catalytic metal is believed to bind to the closed complex IV (α-helix N and protein side chain carbons are purple; PDB ID 2FMS) [17]. Nucleotidyl transfer results in the ternary product complex V (PDB ID 4KLI) [21] described in detail in Fig. 3. Dissociation of PPi results in an open binary nicked DNA complex VI (PDB ID 1BPZ) [13].
The structure of the pre-catalytic ternary substrate complex with a correct nucleotide provides insight into the molecular attributes that optimize catalytic efficiency [i.e., hasten nucleotide binding and insertion; Fig. 3, top left image, (a)–(d)]. These key attributes are: (a) α-helix-N of the N-subdomain is in a closed position adjacent to the nascent base pair with Watson-Crick hydrogen bonding (see Fig. 4 for comparison of the open, –dNTP, and closed position of α-helix N). Additionally, α-helix N in the closed position provides key hydrogen bonds that interact in the DNA minor groove and van der Waals interactions (Fig. 3, upper images; capped dotted lines). (b) At least two active site positively charged arginine residues interact with the negatively charged phosphates of the incoming nucleotide. A water molecule coordinates one of the non-bridging oxygens of Pα (dNTP). A lysine residue is observed to coordinate this phosphate oxygen in replicative dpols suggesting that it will make Pα more reactive thereby increasing the insertion rate [19]. (c) Two closely spaced Mg2+ ions (metal A and B) are coordinating three active site aspartates (Asp190, Asp192, Asp256). These neutralize the negative charge of the phosphate oxygens facilitating nucleotide binding. The catalytic Mg2+ (metal A) coordinates Asp256 and O3´ of the primer terminus. As noted above, it lowers the pKa of the primer terminus to generate the oxyanion that will attack Pα of the incoming nucleotide [10]. Asp256 transiently acts as the proton acceptor [20] and influences the proton affinity of the primer terminus O3´ [10]. Metal A dissociates after insertion as it loses a coordinating ligand and is replaced with Na+. Another Mg2+ (metal C) is observed to transiently coordinate phosphate oxygens of the products during the forward reaction [21]. The role of this product-associated metal is believed to deter the reverse reaction [22]. (d) The sugar pucker of the incoming nucleotide and primer terminus is typically observed to be 3´-endo, a conformation typically found in RNA. This conformation encourages an intramolecular hydrogen bond between O3´ and a non-bridging oxygen on Pβ (dNTP). In the case of the primer terminus nucleotide, catalytic metal binding alters the sugar pucker sugar: 2´-endo (–Mg2+) and 3´-endo (+Mg2+) [17]. Critically, this subtle change in conformation aligns O3´ for an inline nucleophilic attach on Pα (dNTP), as well as positioning these reactive atoms more closely.
Comparing structures of the open binary DNA complex with those after binding a dNTP, thereby generating the ternary substrate complex, indicates that the N-subdomain has repositioned itself to close around the nascent base pair. This is shown by comparing the position of α-helix N of the N-subdomain in Figs. 4 an 5. This also results in subtle repositioning of the lyase domain so that interactions between these domains are enhanced, thereby stabilizing the closed conformation [1]. This transition from the binary DNA complex to the ternary substrate complex induces changes in the local hydrogen bonding networks, active site residue conformation, and substrate conformation and dynamics. For example, formation of the closed ternary substrate complex stabilizes the templating nucleotide [23] and rigidifies the N-subdomain. In contrast, side chain dynamics become localized in the C-subdomain that may modulate catalysis [24].
Fig. 5.

Structural intermediates of incorrect nucleotide insertion identify molecular strategies that deter misincorporation. Structural complexes during incorrect insertion (key residues are shown in a stick representation; α-helix N shown as a pink ribbon, protein side chain carbons are pink, DNA primer terminus base pair carbons are gray, and hydrogen bonds are green lines) are compared to similar complexes during correct insertion shown in Fig. 4 (residues shown in black wire representations; α-helix N shown as a gray ribbon and metals as black spheres). The composition and state of the liganded complex is indicated (green, active and red, inactive). Binding of an incorrect dAMPCPP/Mg2+ (templating G) results in complex III (PDB ID 4F5P) [30]. The carbons of the nascent mismatch are yellow and Mg2+ is a green sphere. The structure suggests weak initial binding since the incoming nucleotide does not interact with the templating base, but forms van der Waals interaction with α-helix N in an open conformation. Additionally, the triphosphate does not interact with basic active site side chains. Addition of Mn2+ (purple sphere), a mutagenic metal that supports catalysis, induces closure of the N-subdomain generating complex IV (PDB ID 3C2M) [31]. Importantly, this induces the template strand to move upstream so that the templating coding base moves out of the active site creating an apparent abasic site. This misaligns O3´ of the primer terminus thereby deterring misinsertion. Misincorporation results in a ternary product complex V (PDB ID 4KLT) [21]. The nascent mismatch distorts the active site and this strain induces the N-subdomain to begin to re-open (it is in an intermediate position; i.e., not fully open). Dissociation of PPi/Mn2+ results in the binary nicked DNA complex VI (PDB ID 4KLU) [21]. This product is a poor substrate for subsequent ligation and must be proofread to deter a base substitution error.
Fig. 5 compares similar structural complexes determined for right and wrong nucleotides. The structures reveal differences in active site hydrogen bonding networks and van der Waals interactions that alter substrate alignment and the stability of the closed complex. The rapid re-opening of the ternary mismatch product complex suggests that a mismatch weakens the closed post-chemistry complex. This is also pertinent to the pre-catalytic ternary substrate complex where the reverse of the conformational change (k−3) is increased [6].
Bridging the gap
The crystallographic structures described here represent snapshots of some of the intermediate steps along the reaction path. Solution studies, such as kinetic and spectroscopic NMR studies, can quantify the impact of rationale modifications of the substrates or enzyme on activity. Although conformational adjustments in response to ligand binding are difficult to experimentally quantify, computational approaches can provide models that link molecular structures and kinetic observations [25]. A key finding of these studies is that the observed open/closed transition of the N-subdomain displays an energy landscape that modulates subdomain motion rather than a single barrier. In other words, the motions of several key active site side chains do not occur in a simple concerted event. Finally, increases in computing power make it possible to perform high-level quantum and molecular mechanics calculations on the chemical reaction catalyzed by dpols [20, 26]. For example, a product-associated metal (Fig. 3, metal C) was determined to deter pyrophosphorolysis [27] without affecting the forward reaction [22] indicating a role in pulling the DNA synthesis reaction forward. There are a large number of laboratories employing novel techniques characterizing dpol β and providing biological, structural, and kinetic observations. With this foundation, a comprehensive computational analysis of the reaction cycle will provide chemical and physical context. Significantly, it will undoubtedly provide unrecognized and surprising insight into the cellular impact and biological consequences of modulating dpol activity through enzyme and substrate modifications as well as altering relevant populations (i.e., cellular concentrations).
Acknowledgments
Molecular graphics images were produced using the Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081). This research was supported by Research Project Numbers Z01-ES050158 and Z01-ES050159 to Samuel H. Wilson in the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences. The enormous progress on this topic was made possible by the strong collaborative research program assembled by Samuel H. Wilson and the many insightful discussions I have had with him over the past 30 years. Accordingly, I am indebted to the many post-doctoral fellows and collaborators who have made seminal contributions as well as Julie Horton and Rajendra Prasad who have provided needed perspectives over the years.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Beard WA, Wilson SH, Chapter One - DNA polymerase beta and other gap-filling enzymes in mammalian base excision repair, in: Zhao L, Kaguni LS (Eds.) The Enzymes, Academic Press, 2019, pp. 1–26. [DOI] [PubMed] [Google Scholar]
- [2].Beard WA, Horton JK, Prasad R, Wilson SH, Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism, Ann. Rev. Biochem, 88 (2019) 137–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Whitaker AM, Freudenthal BD, History of DNA polymerase β X-ray crystallography, DNA Repair (Amst.), (2020) in press. [DOI] [PMC free article] [PubMed]
- [4].Shock DD, Freudenthal BD, Beard WA, Wilson SH, Modulating the DNA polymerase reaction equilibrium to dissect the reverse reaction, Nature Chem. Biol, 13 (2017) 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bakhtina M, Lee S, Wang Y, Dunlap C, Lamarche B, Tsai M-D, Use of viscogens, dNTPαS, and rhodium(III) as probes in stopped-flow experiments to obtain new evidence for the mechanism of catalysis by DNA polymerase β, Biochemistry, 44 (2005) 5177–5187. [DOI] [PubMed] [Google Scholar]
- [6].Tsai Y-C, Johnson KA, A new paradigm for DNA polymerase specificity, Biochemistry, 45 (2006) 9675–9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Johnson KA, Role of induced fit in enzyme specificity: A molecular forward/reverse switch, J. Biol. Chem, 283 (2008) 26297–26301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Johnson KA, Simpson ZB, Blom T, Global kinetic explorer: A new computer program for dynamic simulation and fitting of kinetic data, Anal. Biochem, 387 (2009) 20–29. [DOI] [PubMed] [Google Scholar]
- [9].Sucato CA, Upton TG, Kashemirov BA, Osuna J, Oertell K, Beard WA, Wilson SH, Florián J, Warshel A, McKenna CE, Goodman MF, DNA polymerase β fidelity: Halomethylene-modified leaving groups in pre-steady-state kinetic analysis reveal differences at the chemical transition state, Biochemistry, 47 (2008) 870–879. [DOI] [PubMed] [Google Scholar]
- [10].Batra VK, Perera L, Lin P, Shock DD, Beard WA, Pedersen LC, Pedersen LG, Wilson SH, Amino acid substitution in the active site of DNA polymerase β explains the energy barrier of the nucleotidyl transfer reaction, J. Am. Chem. Soc, 135 (2013) 8078–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Beard WA, Batra VK, Wilson SH, DNA polymerase structure-based insight on the mutagenic properties of 8-oxoguanine, Mutat. Res, 703 (2010) 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Beard WA, Osheroff WP, Prasad R, Sawaya MR, Jaju M, Wood TG, Kraut J, Kunkel TA, Wilson SH, Enzyme-DNA interactions required for efficient nucleotide incorporation and discrimination in human DNA polymerase β, J. Biol. Chem, 271 (1996) 12141–12144. [DOI] [PubMed] [Google Scholar]
- [13].Sawaya MR, Prasad P, Wilson SH, Kraut J, Pelletier H, Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: Evidence for an induced fit mechanism, Biochemistry, 36 (1997) 11205–11215. [DOI] [PubMed] [Google Scholar]
- [14].Beard WA, Shock DD, Yang X-P, DeLauder SF, Wilson SH, Loss of DNA polymerase β stacking interactions with templating purines, but not pyrimidines, alters catalytic efficiency and fidelity, J. Biol. Chem, 277 (2002) 8235–8242. [DOI] [PubMed] [Google Scholar]
- [15].Prasad R, Shock DD, Beard WA, Wilson SH, Substrate channeling in mammalian base excision repair pathways: Passing the baton, J. Biol. Chem, 285 (2010) 40479–40488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Beese LS, Steitz TA, Structural basis for the 3’–5’ exonuclease activity of Escherichia coli DNA polymerase I: A two metal ion mechanism, EMBO J, 10 (1991) 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH, Magnesium induced assembly of a complete DNA polymerase catalytic complex, Structure, 14 (2006) 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Beard WA, Shock DD, Vande Berg BJ, Wilson SH, Efficiency of correct nucleotide insertion governs DNA polymerase fidelity, J. Biol. Chem, 277 (2002) 47393–47398. [DOI] [PubMed] [Google Scholar]
- [19].Beard WA, Wilson SH, Structural insights into the origins of DNA polymerase fidelity, Structure, 11 (2003) 489–496. [DOI] [PubMed] [Google Scholar]
- [20].Lin P, Pedersen LC, Batra VK, Beard WA, Wilson SH, Pedersen LG, Energy analysis of chemistry for correct insertion by DNA polymerase β, Proc. Natl. Acad. Sci. U.S.A, 103 (2006) 13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Freudenthal BD, Beard WA, Shock DD, Wilson SH, Observing a DNA polymerase choose right from wrong, Cell, 154 (2013) 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Perera L, Freudenthal BD, Beard WA, Pedersen LG, Wilson SH, Revealing the role of the product metal in DNA polymerase β catalysis, Nucleic Acids Res, 45 (2017) 2736–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Beard WA, Wilson SH, Structure and mechanism of DNA polymerase β, Chem. Rev, 106 (2006) 361–382. [DOI] [PubMed] [Google Scholar]
- [24].DeRose EF, Kirby TW, Mueller GA, Beard WA, Wilson SH, London RE, Transitions in DNA polymerase β μs-ms dynamics related to substrate binding and catalysis, Nucleic Acids Research, 46 (2018) 7309–7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schlick T, Arora K, Beard WA, Wilson SH, Perspective: Pre-chemistry conformational changes in DNA polymerase mechanisms, Theor. Chem. Acc, 131 (2012) 1287–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Perera L, Beard WA, Pedersen LG, Wilson SH, Chapter Four - Applications of Quantum Mechanical/Molecular Mechanical Methods to the Chemical Insertion Step of DNA and RNA Polymerization, in: Christov CZ (Ed.) Advances in Protein Chemistry and Structural Biology, Academic Press, 2014, pp. 83–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Perera L, Freudenthal BD, Beard WA, Shock DD, Pedersen LG, Wilson SH, Requirement for transient metal ions revealed through computational analysis for DNA polymerase going in reverse, Proc. Natl. Acad. Sci. U.S.A, 112 (2015) E5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Freudenthal BD, Beard WA, Wilson SH, DNA polymerase minor groove interactions modulate mutagenic bypass of a templating 8-oxoguanine lesion, Nucleic Acids Res, 41 (2013) 1848–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Beard WA, Shock DD, Batra VK, Pedersen LC, Wilson SH, DNA polymerase β substrate specificity: Side chain modulation of the “A-rule”, J. Biol. Chem, 284 (2009) 31680–31689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Freudenthal BD, Beard WA, Wilson SH, Structures of dNTP intermediate states during DNA polymerase active site assembly, Structure, 20 (2012) 1829–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH, Structures of DNA polymerase β with active site mismatches suggest a transient abasic site intermediate during misincorporation, Mol. Cell, 30 (2008) 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]