

Abstract
Polymer micelles, used extensively as vehicles in the delivery of active pharmaceutical ingredients, represent a versatile polymer architecture in drug delivery systems. We hypothesized that degree of crosslinking in the hydrophobic core of amphiphilic block copolymer micelles could be used to tune the rate of release of the biological signaling gas (gasotransmitter) hydrogen sulfide (H2S), a potential therapeutic. To test this hypothesis, we first synthesized amphiphilic block copolymers of the structure PEG-b-P(FBEA) (PEG = poly(ethylene glycol), FBEA = 2-(4-formylbenzoyloxy)ethyl acrylate). Using a modified arm-first approach, we then varied the crosslinking percentage in the core-forming block via addition of a ‘O,O’-alkanediyl bis(hydroxylamine) crosslinking agent. We followed incorporation of the crosslinker by 1H NMR spectroscopy, monitoring the appearance of the oxime signal resulting from reaction of pendant aryl aldehydes on the block copolymer with hydroxylamines on the crosslinker, which revealed crosslinking percentages of 5, 10, and 15%. We then installed H2S-releasing S-aroylthiooxime (SATO) groups on the crosslinked polymers, yielding micelles with SATO units in their hydrophobic cores after self-assembly in water. H2S release studies in water, using cysteine (Cys) as a trigger to induce H2S release from the SATO groups in the micelle core, revealed increasing half-lives of H2S release, from 117 ± 6 min to 210 ± 30 min, with increasing crosslinking density in the micelle core. This result was consistent with our hypothesis, and we speculate that core crosslinking limits the rate of Cys diffusion into the micelle core, decreasing the release rate. This method for tuning the release of covalently linked small molecules through modulation of micelle core crosslinking density may extend beyond H2S to other drug delivery systems where precise control of release rate is needed.
Graphical Abstract
1. Introduction
Macromolecular and supramolecular delivery vehicles represent a wide class of drug delivery systems with potential for controlled release rates [1–4]. Delivery vehicles that assemble into higher-order structures in solution enable modulation of physical and pharmacokinetic properties of active pharmaceutical ingredients (APIs) without drastic changes in their chemical structure, leading to effects such as tunable release rates, extended circulation times, and targeted release [5–7]. Among these classes of delivery vehicles, polymer micelles provide a particularly versatile architecture with many tunable features, such as core and corona size, the number and nature of functional groups in the core or corona, and variable core chain mobility, all of which influence donor release kinetics [8–10]. The hydrophobic nature of many APIs promotes their encapsulation or sequestration into hydrophobic micelle cores, which represents an effective strategy to overcome their low solubility in aqueous environments. As such, precise control over the diffusion of molecules into and out of micelles presents an appealing method for tuning donor release kinetics in micellar drug delivery systems.
Of particular interest to our group is delivery of endogenous biological signaling gases, known as gasotransmitters, which have recently emerged as powerful and widespread signaling molecules with vast therapeutic potential [11,12]. Gasotransmitters have short in vivo half-lives and are toxic at high concentrations, so sustained and controlled delivery is vital. Currently recognized gasotransmitters include nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S), and all are produced and regulated endogenously in mammals, are membrane permeable, and interact with each other in a medley of cellular signaling processes [13]. By exploiting these endogenous pathways, gasotransmitters may exhibit unique therapeutic advantages over conventional small molecule APIs.
H2S is the most recently recognized gasotransmitter and participates in an array of biological processes including cardioprotection [14,15], vasodilation [16–18], angiogenesis [19–21], and others [22–24]. Mammals produce H2S endogenously and systemically, with physical concentrations tightly regulated by various biosynthetic mechanisms. The presence of H2S in many biological signaling pathways has prompted researchers to develop strategies for its exogenous delivery; a plethora of small molecules and polymers designed to release H2S in response to a stimulus (termed H2S donors) have been synthesized to help evaluate its role in signaling biology and exploit its therapeutic potential [25–34]. Emerging evidence indicates that controlled delivery in a sustained manner, mimicking the body’s natural production of H2S, is critical to its success as a therapeutic [35]. As such, recent interest has focused on developing drug delivery systems with precise control over H2S release rates [36–42]. Among these systems, polymer micelles have garnered much attention recently for their versatility [34,43–46].
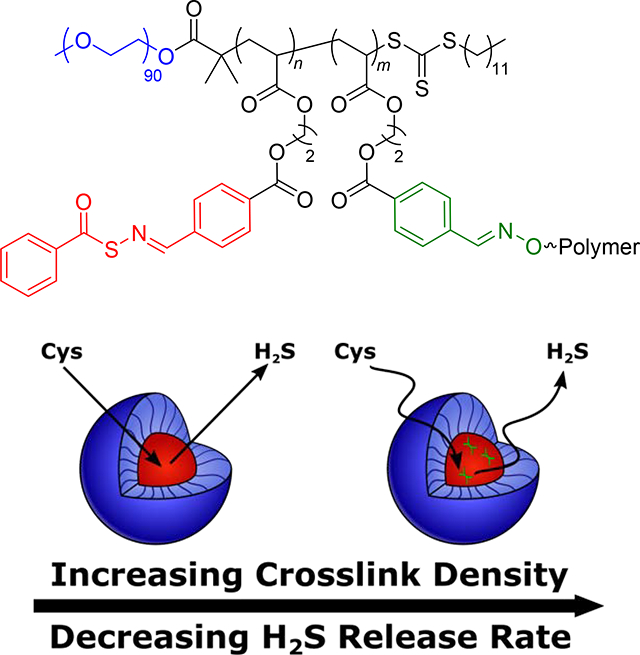
We envisioned that the wide range of structural tunability of polymer micelles would provide multiple mechanisms through which to control delivery of H2S from H2S donors tethered to the micelle core-forming block. Indeed, the idea of controlling diffusion into and out of the micelle core has been used previously to tune drug release rates [10,46,47]. For example, our group recently synthesized a series of H2S-releasing polymer micelles with tunable release kinetics through control over polymer chain mobility in the micelle core [46]. We incorporated a plasticizing comonomer into the core-forming block of an amphiphilic block copolymer with S-aroylthiooxime (SATO) H2S donors attached to the hydrophobic block to alter its glass transition temperature (Tg), with low Tg core-forming blocks resulting in highly mobile micelle cores. H2S release required diffusion of a triggering molecule (cysteine, Cys) into the micelle core (see Scheme S1 for mechanism of Cys-triggered H2S release from SATOs), so more mobile micelle cores exhibited faster Cys diffusion and consequently faster H2S release rates. While we achieved a wide range of H2S release kinetics with this method, incorporation of high levels of the plasticizing comonomer heavily diluted H2S donor concentration. To address this problem, we considered core crosslinking, which can significantly influence chain mobility in a micelle core [48–51]. Specifically, we hypothesized that small amounts of crosslinking would reduce micelle core mobility, slowing diffusion of triggering Cys molecules into the core and reducing H2S release rates (Figure 1). Thus, in this work, we aimed to utilize crosslinking in the micelle core to tune core mobility, and in turn, H2S release kinetics, without drastically lowering the H2S donor concentration.
Figure 1.

Schematic illustration showing the influence of hydrophobic block crosslinking density on H2S release rate. Higher crosslinking density results in less mobile micelle cores, slowing diffusion of triggering Cys molecules into the core and decreasing the rate of H2S release.
2. Experimental
2.1. Materials
All reagents were obtained from commercial vendors and used as received unless otherwise stated. 2,2′-Azobis(2-methylpropionitrile) (AIBN) was recrystallized from methanol prior to use. Dry solvents were purified by passage through a solvent purification system (MBraun).
2.2. Instrumentation
NMR spectra were measured on an Agilent 400 MHz spectrometer. 1H and 13C NMR chemical shifts are reported in ppm relative to internal solvent resonances. Yields refer to chromatographically and spectroscopically pure compounds unless otherwise stated. Size exclusion chromatography (SEC) was carried out in THF at 1 mL/min at 30 °C on two Agilent PLgel 10 μm MIXED-B columns connected in series with a Wyatt Dawn Heleos 2 multi-angle light scattering detector and a Wyatt Optilab Rex refractive index detector. No calibration standards were used, and dn/dc values were obtained by assuming 100% mass recovery. Dynamic light scattering (DLS) was conducted using a Malvern Zetasizer Nano operating at 25 °C. A solution of micelles was prepared at 1 mg/mL and filtered with a 0.2 μm filter prior to scanning. Calculations of the particle size distributions and distribution averages were conducted using CONTIN particle size distribution analysis routines with number, volume, and intensity averages. Measurements were made in triplicate and errors reflect standard deviations. Differential scanning calorimetry (DSC) studies were carried out on a Q-2000 DSC in aluminum pans operated with a dry nitrogen purge from −80 °C to 100 °C with a heating and cooling rate of 20 °C/min. Results are reported from the second heat cycle and figures are shown as exo down.High-resolution mass spectra were taken on an Agilent Technologies 6230 TOF LC/MS mass spectrometer.
2.3. Synthesis of 2-methyl-2-(dodecylsulfanylthiocarbonyl)sulfanyl propanoic acid (CTA)
Dodecanethiol (5.00 mL, 20.9 mmol) was dissolved in acetone (30 mL) in a round bottom flask. To the flask was added tribasic potassium phosphate (8.86 g, 41.7 mmol). The reaction mixture was stirred at rt for 10 min. CS2 (3.78 mL, 62.6 mmol) was then added dropwise, and the reaction mixture was stirred for an additional 1 h. To the flask a solution of 2-bromo-2-methyl propanoic acid (3.91 g, 23.4 mmol) in 5 mL of acetone was added dropwise. The reaction mixture was stirred overnight at rt. Reaction conversion was monitored by TLC using CH2Cl2 as the eluent and visualized via a UV lamp. The reaction mixture was diluted with ~100 mL CH2Cl2, transferred to a separatory funnel, and washed consecutively with 1N HCl and brine. The organic layer was dried over Na2SO4 and concentrated via rotary evaporation. Silica gel was added to the concentrated solution before removing the rest of the solvent via rotary evaporation. The silica gel was dry-loaded onto a silica gel column, and the product was eluted with CH2Cl2. Product containing fractions, as determined by TLC (CH2Cl2 eluent and UV lamp visualization), were combined and concentrated via rotary evaporation to yield a yellow solid. The crude product was recrystallized from hexanes to yield yellow crystals (4.88 g, 64% yield). 1H NMR (CDCl3) δ (ppm): 3.25 (2H, t, J = 7.4 Hz); 1.70 (6H, s); 1.64 (2H, m); 1.37 (2H, m); 1.25 (18H, m); 0.88 (3H, t, J = 6.9 Hz). 13C NMR (CDCl3) δ (ppm): 220.78, 178.84, 55.59, 37.12, 31.89–26.21, 23.69, 14.13. These spectra match those from published reports [52].
2.4. Synthesis of MacroCTA
A round bottom flask was charged with CTA (0.91 g, 2.50 mmol), polyethylene glycol monomethyl ether (Mn = 4,000 g/mol, 5.0 g, 1.25 mmol), 4-dimethylamino pyridine (0.15 g, 1.25 mmol), and anhydrous CH2Cl2 (5.0 mL). N,N’-Dicyclohexylcarbodiimide (DCC) (0.52 g, 2.50 mmol) was dissolved in 5.0 mL of anhydrous CH2Cl2 in a vial. The DCC solution was added dropwise to the flask containing the other reagents, and the reaction mixture was stirred at rt overnight. Reaction conversion was followed by TLC using CH2Cl2 as eluent and visualized via UV lamp. The precipitated solids were removed by filtration. The desired product was isolated via precipitation from diethyl ether and was purified by repeated precipitations (2–4) from CH2Cl2 into diethyl ether until complete removal of low molecular weight side products was confirmed by 1H NMR spectroscopy to afford the product as a yellow solid (1.50 g, 69% yield). 1H NMR (CDCl3) δ (ppm): 4.24 (2H, t, J = 5.0 Hz); 3.81 (2H, t, J = 4.8 Hz); 3.63 (~360H, m); 1.37 (2H, m); 3.37 (3H, s); 3.25 (2H, t, J = 7.4 Hz); 1.69 (6H, s); 1.64 (2H, m); 1.25 (18H, m); 0.87 (3H, t, J = 6.6 Hz). 13C NMR (CDCl3) δ (ppm): 221.38, 172.81, 71.89, 70.52, 68.75, 65.01, 58.98, 55.89, 36.86, 31.85, 29.57, 29.49, 29.38, 29.27, 29.04, 28.88, 27.81, 25.29, 22.62, 14.07. These spectra match those from published reports [53].
2.5. Synthesis of 2-(4-formylbenzoyloxy)ethyl acrylate (FBEA)
FBEA was prepared by modification of a literature procedure [54]. A round bottom flask was charged with 4-formylbenzoic acid (2.00 g, 13.3 mmol) and EDC (2.62 g, 16.0 mmol), and diluted with CH2Cl2 (20 mL). 2-Hydroxyethyl acrylate (1.61 mL, 14.0 mmol) was added in one portion, followed by 4-dimethylaminopyridine (16 mg, 0.13 mmol). Butylated hydroxytoluene (BHT) (30 mg, 0.13 mmol) was added to inhibit polymerization. The reaction mixture was stirred at rt until homogeneous (~24 h). The reaction mixture was then diluted with CH2Cl2 (30 mL) and washed with 1N HCl (3 × 30 mL). The organic layer was dried and concentrated onto silica gel. A silica column was dry-loaded with this material, and the crude reaction mixture was purified by column chromatography using 50% hexanes in CH2Cl2 as the eluent, affording the pure product after rotary evaporation of product-containing fractions, as observed by thin layer chromatography (TLC), as a clear oil (2.13 g, 64%). BHT (20 mg, 0.08 mmol) was added before storage at 0 °C. 1H NMR (CDCl3): δ 4.53 (m, 2H), 4.60 (m, 2H), 5.87 (dd, J = 1.4, 10.4 Hz, 1H), 6.15 (m, 1H), 6.45 (dd, J = 1.4, 17.3 Hz, 1H), 7.96 (d, J = 8.6 Hz, 2H), 8.20 (d, J = 8.4 Hz, 2H), 10.10 (s, 1H). 13C NMR (CDCl3): δ 191.70, 165.99, 165.42, 139.41, 134.82, 131.69, 130.42, 129.65, 128.01, 63.38, 62.16. HRMS (ESI-TOF) calcd. for C26H24NaO10 [2M+Na]+ 519.1267, found 519.1253.
2.6. Synthesis of O,O’-(decane-1,10-diyl)bis(hydroxylamine) (BHA) crosslinking agent
O,O’-(decane-1,10-diyl)bis(hydroxylamine) (BHA) was prepared using a modified literature procedure [55]. A round bottom flask was charged with N-hydroxyphthalimide (2.39 g, 14.7 mmol), triethylamine (NEt3) (2.20 mL, 15.6 mmol), potassium iodide (3 mg, 0.018 mmol) and acetonitrile (15 mL). A solution of 1,10-dibromodecane (1.47 g, 4.9 mmol) in acetonitrile (5 mL) was added to the flask dropwise while stirring. Reaction conversion was monitored by TLC using 5% ethyl acetate in hexanes as eluent and visualized by iodine staining. The reaction mixture was stirred at room temperature until complete disappearance of 1,10-dibromodecane (~24 h). The reaction mixture was diluted with CH2Cl2 (200 mL) and washed successively with alternating water and saturated sodium bicarbonate solutions until the aqueous layer was colorless. The organic layer was dried and concentrated by rotary evaporation to afford the crude 1,10-bis(N-phthalimidyloxy)decane product as a pale-yellow oil. This was used in the next step without further purification. 1,10-bis(N-phthalimidyloxy)decane (500 mg, 1.1 mmol) was dissolved in ethanol (2 mL) and distilled water (2 mL). Next, hydrazine (40% in H2O) (390 μL, 6.5 mmol) was added in one portion. Reaction conversion was monitored by TLC using CH2Cl2 as eluent and visualized by iodine staining. The reaction mixture was stirred at room temperature until complete disappearance of 1,10-bis(N-phthalimidyloxy)decane (~3 h). The reaction mixture was concentrated to dryness and diluted with 1N NaOH (50 mL). The aqueous layer was extracted with ethyl acetate (3 × 25 mL). The organic layers were combined, dried, and loaded onto silica. A silica column was dry-loaded with this material, and the crude reaction mixture was purified by column chromatography using a gradient polarity eluent starting from CH2Cl2 and adding ethyl acetate up to 50%, affording the product after rotary evaporation of product-containing fractions, as determined by TLC, as a white solid (180 mg, 20%). 1H NMR (CDCl3): δ 3.65 (t, J = 7 Hz, 4H), 1.56 (m, 4H), 1.29 (m, 6H). 13C NMR (CDCl3): δ 76.22, 29.47, 29.46, 28.38, 25.99. These spectra are consistent with previous reports [55].
2.7. Synthesis of P(FBEA)
Prior to use, FBEA monomer was passed through a plug of basic alumina to remove inhibitor (BHT). An oven-dried Schlenk tube was charged with FBEA (1.00 g, 4.0 mmol) and macroCTA (29 mg, 0.081 mmol), and diluted with DMF (2.0 mL). 100 μL of an AIBN solution (13 mg in 1.0 mL DMF) was added via syringe. The reaction mixture was deoxygenated by subjecting the contents to five freeze-pump-thaw cycles. The Schlenk tube was then backfilled with N2 and submerged in an oil bath maintained at 80 °C. Aliquots were removed periodically by N2-purged syringe to monitor molar mass evolution by SEC and conversion by 1H NMR spectroscopy. The polymerization was quenched by submerging the tube into liquid N2 and exposing the reaction mixture to air. The resulting polymer was isolated via precipitation into diethyl ether.
2.8. Synthesis of PEG-b-P(FBEA)
Prior to use, FBEA monomer was passed through a plug of basic alumina to remove inhibitor (BHT). An oven-dried Schlenk tube was charged with FBEA (3.12 g, 12.6 mmol) and macroCTA (1.09 g, 0.25 μmol), and diluted with DMF (3.0 mL). 100 μL of an AIBN solution (20 mg in 1.0 mL DMF) was added via syringe. The reaction mixture was deoxygenated by subjecting the contents to five freeze-pump-thaw cycles. The Schlenk tube was then backfilled with N2 and submerged in an oil bath maintained at 80 °C. Aliquots were removed periodically by N2-purged syringe to monitor molar mass by SEC and conversion by 1H NMR spectroscopy. The polymerization was quenched by submerging the tube into liquid N2 and exposing the reaction mixture to air. The resulting polymer was isolated via precipitation into cold diethyl ether (3.33 g, 93%).
2.9. Representative procedure for crosslinking of the core-forming block
The procedure for preparation of Polymer 1b (5% crosslinked) is as follows: A scintillation vial was charged with PEG-b-P(FBEA) (100 mg, 6.9 μmol) and CH2Cl2 (1.9 mL). A solution of BHA crosslinker (28 mg, 140 μmol) in CH2Cl2 (2.0 mL) was prepared. 100 μL of this solution was added to the reaction mixture to achieve a total volume of 2 mL. A layer of molecular sieves and trifluoroacetic acid (10 μL, 130 μmol) were added to the reaction mixture. The reaction was allowed to sit at rt overnight. The resulting crosslinked polymer was isolated via precipitation into cold diethyl ether. To prepare Polymer 1c (10% crosslinked) 200 μL of BHA solution was added to 1.8 mL CH2Cl2, and to prepare Polymer 1d (15% crosslinked) 300 μL of BHA solution was added to 1.7 mL CH2Cl2.
2.10. General procedure for preparation of H2S releasing polymers
A scintillation vial was charged with PEG-b-P(FBEA) (100 mg, 6.9 umol) and S-benzoylthiohydroxylamine (160 mg, 1.1 mmol). The solids were dissolved in CH2Cl2 (2.0 mL). A layer of molecular sieves and trifluoroacetic acid (10 μL, 130 μmol) were added to the reaction mixture. The reaction was allowed to sit at rt overnight. The resulting S-aroylthiooxime functionalized polymer was isolated via precipitation into diethyl ether.
2.11. General procedure for preparation of micelles
A scintillation vial equipped with a stir bar was charged with polymer (10 mg) and THF (3 mL). Distilled water (3.0 mL) was added to the polymer solution in one portion, and the solution was allowed to stir for 15 min afterwards. The solution was transferred to dialysis tubing (6–8 kD MWCO) and dialyzed against distilled water (1 L) for 18 h, changing the water once after 2 h and again after 4 h. The resulting aqueous solution was removed from the dialysis tubing and diluted with distilled water to a final volume of 10.0 mL (1 mg/mL polymer concentration).
2.12. Measuring H2S release kinetics by methylene blue
Reactions for kinetic studies were run in quadruplicate, with each reaction vial containing 20 μL of phosphate buffer (1 M in H2O, pH = 7), 250–288 μL micelle solution (to reach 250 μM SATO groups), 100 μL of Zn(OAc)2 solution (40 mM in H2O), 20 μL of cysteine solution (100 mM in H2O), and DI H2O to reach 2 mL total volume. Final concentrations were 250 μM SATO functional groups (0.125 – 0.144 mg/mL polymer concentrations), 2 mM Zn(OAc)2, and 1 mM cysteine. A blank vial was run for each experiment containing DI H2O instead of SATO solution. At predetermined timepoints, 100 μL aliquots were taken from each vial and diluted with 100 μL of FeCl3 solution (30 mM in 1.2 M HCl) and 100 μL of p-phenylene diamine solution (20 mM in 7.2 M HCl). Each aliquot solution remained sealed in a microcentrifuge tube for a minimum of 24 h prior to addition of 250 μL to a 96-well plate. The absorbance for each aliquot was measured at 750 nm using a plate reader.
3. Results and Discussion
3.1. Synthesis of Micelles with Crosslinks in the Hydrophobic Core
Because our previous work revealed that micelle cores with low Tg core-forming blocks released H2S faster than those with more rigid, higher Tg core-forming blocks [46], we expected core crosslinking to limit chain mobility, thus slowing H2S release. Therefore, in order to achieve a range of H2S release rates, we aimed to create H2S releasing micelles with low Tg core-forming blocks, which would exhibit fast release profiles, and incorporate varying degrees of core crosslinking to reduce the release rate. To accomplish this goal, we first required a monomer capable of yielding low Tg polymers that could subsequently be functionalized with H2S donors. Knowing that we could easily convert pendant aryl aldehydes into H2S-releasing SATO groups [56,57], and that polyacrylates exhibit lower Tg’s than their polymethacrylate counterparts [58], we selected the aryl aldehyde-functionalized acrylate 2-(4-formylbenzoyloxy)ethyl acrylate (FBEA) as the monomer for the hydrophobic, core-forming block of the polymer micelles. Based on conditions described in a previous synthesis of the methacrylate derivative of this monomer [54], we prepared FBEA by coupling 2-hydroxyethyl acrylate with 4-formylbenzoic acid using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) as the coupling agent (Scheme 1A). Compared to the methacrylate monomer we had used in previous work, we expected the more flexible polyacrylate backbone of poly(FBEA) to afford greater chain mobility and result in micelles with faster H2S release rates.
Scheme 1.

Monomer and poly(FBEA) synthesis. (A) Synthesis of FBEA monomer and (B) homopolymerization of FBEA using RAFT. Polymerization conditions: [FBEA]/[CTA]/[AIBN] = 50:1:0.1, 33% w/v FBEA in DMF, 80 °C.
Polymerization of FBEA was carried out using reversible addition-fragmentation chain-transfer (RAFT) polymerization (Scheme 1B). To our knowledge, there are no reports of a reversible-deactivation radical polymerization of FBEA. As such, we performed kinetic analysis to assess the “livingness” of its polymerization. We utilized 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DMPA) as the chain transfer agent (CTA) because it is well suited for polymerization of acrylate monomers via RAFT. Monomer conversion, determined by 1H NMR spectroscopy, progressed linearly with time, which is indicative of a polymerization with good molecular weight control (Figure 2A). We observed a short induction time of around 10 min, consistent with reports on polymerization of acrylates with this CTA [59]. A plot of polymer Mn, as measured by SEC, versus % monomer conversion, showed linear evolution of molecular weight over time and low dispersity (Đ) throughout the polymerization, both of which also indicate good molecular weight control. Furthermore, polymer Mn values agreed well with expected molecular weights based on monomer conversion. Differential scanning calorimetry (DSC) showed that the polymer had a Tg of –55 °C (Figure S10), which we expected would afford micelles with highly mobile, rubbery cores.
Figure 2.

Kinetics of RAFT polymerization of FBEA: (A) Pseudo-first-order kinetics plot of the polymerization showing monomer conversion over time; the dashed blue line shows a linear fit to the data. (B) Mn (blue circles) and dispersity (orange circles), as determined by SEC with light scattering, versus monomer conversion, as determined by 1H NMR spectroscopy; dashed line shows expected Mn based on monomer conversion. Linear evolution of molecular weight over time and good agreement between theoretical and measured molecular weight indicate a polymerization with a high degree of molecular weight control.
Next, we set out to prepare micelles with poly(FBEA) cores. We first synthesized trithiocarbonate-terminated poly(ethylene glycol) (PEG), termed MacroCTA here (Scheme 2A), according to a previously reported procedure [53]. We then utilized RAFT polymerization to grow hydrophobic poly(FBEA) from MacroCTA, resulting in an amphiphilic PEG-b-P(FBEA) block copolymer (Scheme 2B). We characterized this polymer by SEC, observing an Mn of 14 kg/mol with a dispersity of 1.13 (Figure S11). We expected the low dispersity to result in uniform micelle size distributions. Therefore, we used this batch of block copolymer for preparation of all micelles in order to have uniform micelle composition with crosslinking percentage in the hydrophobic block as the only variable.
Scheme 2.

MacroCTA and block copolymer synthesis. (A) Synthesis of Macro-CTA. (B) Preparation of PEG-b-P(FBEA) amphiphilic block copolymer. Polymerization conditions: [FBEA]/[MacroCTA]/[AIBN] = 50:1:0.1, 50% w/v FBEA in DMF, 80 °C).
Many methods for preparing crosslinked micelles have been reported [60–63]. Among these methods there are two prevalent strategies: crosslinking of pre-formed polymer assemblies, and crosslinking of unassembled polymer chains to induce formation of a polymer assembly (the arm-first approach). SATOs are reactive functional groups, so we planned to install them after crosslinking to avoid potential side reactions. Additionally, we expected that installation of SATOs into a micelle core could be hindered by the formation of crosslinks, so we avoided crosslinking pre-formed polymer assemblies. Instead, we utilized a modified version of the arm-first approach, with the goal of preparing lightly crosslinked polymer clusters, which could serve as precursor amphiphiles to H2S-releasing micelles with crosslinks in the hydrophobic core. By taking this approach we allowed for facile installation of H2S donors after the crosslinking reaction.
To achieve our goal of preparing micelles with crosslinks in the hydrophobic core, we first synthesized a difunctional crosslinker with hydroxylamine functional groups, O,O’-(1,10-decanediyl) bis(hydroxylamine) or BHA for short, according to a previously reported procedure [55]. Reaction of an aldehyde with a hydroxylamine produces an oxime linkage, which we expected to be stable throughout the functionalization of remaining unreacted aldehydes based on our previous work [46]. In this method, PEG-b-P(FBEA) was dissolved in a good solvent for both blocks (CH2Cl2), then BHA was added in different amounts (5, 10, and 15 mol% hydroxylamines with respect to aldehyde groups) to create oxime linkages between the core-forming blocks (Scheme 3). After recovering the resulting polymer clusters (Polymers 1b-d), we measured the incorporation of crosslinker by 1H NMR spectroscopy (Figure 3). By comparing the integration of the aldehyde proton signal to that of the newly formed oxime proton signal, we determined crosslinking percentages of 5, 10, and 15 mol % for Polymers 1b-d. Although reaction of BHA with the trithiocarbonate group could be possible, the integrations of the oxime signal were consistent with our reaction stoichiometry suggesting that reaction of BHA with an aldehyde group is preferential. Analysis of the polymer clusters by SEC showed increasing molar mass and dispersity with increasing crosslinking percentage (from Mn = 49.9 kg/mol and Ð = 1.43 to Mn = 61.3 kg/mol and Ð = 1.63, Figures S13–15), signifying the formation of larger polymer clusters with more incorporation of BHA crosslinker (see SI for discussion). These results demonstrated that we could tune crosslinking percentage in the core-forming block by modifying the ratio of the BHA crosslinker to pendant aryl aldehyde groups on the block copolymer backbone.
Scheme 3.

(A) Chemical structures showing the preparation of crosslinked PEG-b-P(FBEA) polymer clusters. (B) Schematic illustration of this process; blue represents the hydrophilic PEG block, orange represents the hydrophobic P(FBEA) block, and green represents the BHA crosslinker.
Figure 3.

1D 1H NMR spectra of crosslinked polymer clusters of PEG-b-P(FBEA) (Polymers 1b-d) in CDCl3, highlighting the ratio of aldehyde proton signal to oxime proton signal.
3.2. Installation of SATO H2S donors into micelle cores
After synthesizing polymer clusters with varying degrees of crosslinking in the P(FBEA) block, we sought to analyze how restricted chain mobility would impact H2S release rates after installation of H2S-releasing SATO groups and micelle formation. First, we reacted the remaining aldehydes on Polymers 1a-d with S-benzoylthiohydroxylamine (SBTHA) to produce Polymers 2a-d, which contained H2S-releasing SATO groups (Scheme 4). We monitored conversion of aldehyde to SATO by 1H NMR spectroscopy, following the disappearance of the aldehyde proton signal at 10.1 ppm and the emergence of the SATO proton signal at 8.6 ppm (Figure S12).
Scheme 4.

(A) Chemical structures showing the preparation of crosslinked, H2S-releasing amphiphilic block copolymers. H2S-releasing SATO groups are colored in red. (B) Schematic illustration of this process; blue represents the hydrophilic PEG block, orange represents the hydrophobic P(FBEA) block, red represents the hydrophobic P(FBEA-SATO) block, and green represents the BHA crosslinker.
We then prepared micelles from Polymers 2a-d via the solvent switch method (Scheme 5). Briefly, we dissolved Polymers 2a-d in a good solvent for both blocks (THF) at a concentration of 3 mg/mL. Rapid addition of water while stirring to produce a 50% THF by volume solution then induced self-assembly into micelles. Dialysis against water removed the THF, resulting in micelle solutions in water with polymer concentrations near 1.5 mg/mL. Dilution of these solutions with water to a final concentration of 1 mg/mL afforded micelle solutions of Polymers 2a-d with SATO concentrations ranging from 1.7–2.0 mM.
Scheme 5.

Preparation of H2S-releasing micelles with crosslinks in the hydrophobic core. Blue represents the hydrophilic PEG block, red represents the hydrophobic P(FBEA-SATO) block, and green represents the BHA crosslinker.
We employed dynamic light scattering (DLS) to analyze the size distributions for micelle solutions made from Polymers 2a-d (Table 1, Figures S16–18). We observed intensity-average hydrodynamic diameters (Dh) ranging from 28.0 ± 0.4 nm to 59 ± 1 nm, with micelle size increasing with increasing crosslinking percentage. We also observed an increase in micelle polydispersity index (PDI) upon crosslinking, from values near 0.03 for uncrosslinked micelles to values near 0.2 for crosslinked micelles. We attribute these results to the modified version of the arm-first crosslinking approach, in which the crosslinking step creates clusters of polymer chains that later associate with other clusters or free polymer chains to form micelles. Polymer clusters are likely more uniform in size at a lower crosslinking percentage, leading to tighter packing and overall smaller micelles. Additionally, the assembly of polymer clusters of various sizes likely leads to an increase in the range of micelle sizes. TEM images of micelles prepared from Polymers 2a-d revealed a higher frequency of larger aggregates with increasing crosslinking percentage, further supporting this idea (Figures S19–22).
Table 1.
Characterization of polymer assemblies.
| Polymer | Crosslinking Percenta | Intensity- Average Dh (nm)b | Volume-Average Dh (nm)b | Number-Average Dh (nm)b | Micelle PDIb |
|---|---|---|---|---|---|
| 2a | 0 | 28.0 ± 0.4 | 24.0 ± 0.3 | 21.1 ± 0.2 | 0.037 |
| 2b | 5 | 41 ± 2 | 32.5 ± 0.8 | 25.0 ± 0.7 | 0.212 |
| 2c | 10 | 56 ± 2 | 54 ± 4 | 29 ± 1 | 0.213 |
| 2d | 15 | 59 ± 1 | 56 ± 5 | 32 ± 1 | 0.199 |
Measured by ratio of oxime and aldehyde proton signals via 1H NMR spectroscopy (Figure 3).
Measured by dynamic light scattering (DLS) at 1 mg/mL in DI water. Each measurement was conducted in triplicate (n = 3), and error bars reflect standard deviations.
3.3. Impact of Crosslinking Percentage of the Hydrophobic Block on H2S Release Rate
To observe the effect of core crosslinking on H2S release rate, we conducted methylene blue assays on micelle samples prepared from Polymers 2a-d. The methylene blue assay is a common method for determining H2S concentrations in solution that was standardized by Siegel in 1965 [64], but dates back to Fischer’s work in the late 1800s [65]. Briefly, released H2S is scavenged by Zn(OAc)2, forming ZnS salt in the reaction vial. Aliquots are withdrawn from the reaction vial at given timepoints and diluted with FeCl3 and p-phenylene diamine, where captured sulfide reacts to form methylene blue dye. Because formation of methylene blue requires captured sulfide, the amount of H2S released at each timepoint can be determined by measuring the absorbance at 750 nm, a characteristic wavelength of absorbance for methylene blue.
In these H2S release studies, we maintained a concentration of 250 μM SATO functional groups and 1 mM cysteine across all experiments. By monitoring the production of methylene blue at varying timepoints, we evaluated H2S release kinetics for the micelles at different crosslinking percentages. As shown in Figure 4 and Figures S23–S26, the most rapid H2S release occurred from uncrosslinked micelles (Polymer 2a). In support of our hypothesis, we observed a decrease in the rate of H2S release with increasing amounts of crosslinking in the micelle core, from a half-life of 117 ± 6 min for uncrosslinked micelles to a half-life of 210 ± 30 min for 15% crosslinked micelles. These results show that crosslinking of the core-forming block decreases the rate of H2S release, which is consistent with our hypothesis. We speculate that the decreasing release rate arises from reduced core chain mobility that limits diffusion of triggering cysteine molecules into the micelle core. Alternatively, the varying release rates could result from differences in unimer percentages and/or unimer exchange rates arising from the differential molecular weights and solubilities of the polymer clusters. This is a topic that we are currently studying in depth with model systems.
Figure 4.

H2S release kinetics from micelle solutions (0.125 – 0.144 mg/mL polymer concentrations) prepared from Polymers 2a (0% crosslinked, blue circles), 2b (5% crosslinked, green stars), 2c (10% crosslinked, orange triangles), and 2d (15% crosslinked, red squares). The lines correspond to pseudo-first-order kinetics fits, from which half-life values (t1/2) were calculated. H2S release rates show a marked decrease with increasing crosslinking percentage.
We expect that the 2-fold change in release rate could be increased using different strategies. For example, a larger range of release rates might be observed when starting with a core-forming block with an even lower Tg. Additionally, a crosslinker with functionality >2 might have a more profound impact on Cys diffusion by further decreasing core mobility. As such, we are currently investigating methods to achieve H2S-releasing micelles with a more expansive range of release rates through these and other core-crosslinking approaches.
4. Conclusions
We have explored the influence of crosslinking in the micelle core on small molecule release rates by synthesizing and analyzing H2S-releasing micelles with varying amounts of crosslinking in the hydrophobic block. Using a modified arm-first approach, we followed the reaction of aryl aldehyde groups using a ‘O,O’-alkanediyl bis(hydroxylamine) crosslinker by 1H NMR spectroscopy. After micelle formation from crosslinked polymer clusters and installation of H2S-releasing SATO groups onto the remaining aryl aldehydes, we observed increasing micelle size with increasing crosslinking percentage, likely due to the assembly of larger clusters at higher amounts of crosslinking. We observed a trend of decreasing H2S release rate with increasing crosslinking percentage of the core-forming block, consistent with our hypothesis that crosslinking would lead to reduced micelle core mobility and therefore hindered diffusion of the Cys trigger necessary for H2S release. We expect that further modifications to the core-forming block or crosslinker could result in an even greater impact on the rate of H2S release. Ultimately, these findings will inform future designs for polymer micelle drug delivery systems where precise control over donor release profiles is needed.
Supplementary Material
Highlights.
Core-crosslinked micelles were prepared by assembly of polymer clusters.
Incorporation of crosslinker was monitored via 1H NMR spectroscopy.
Micelles with larger crosslinking percentages showed larger size distributions.
H2S release rates from micelles decreased with increasing crosslinking percentage.
Acknowledgements
This work was supported by the US National Science Foundation (DMR-1454754), the US National Institutes of Health (R01GM123508), and the Dreyfus Foundation. We thank Prof. Richey Davis for use of the DLS instrument, Prof. Tim Long for use of the DSC instrument, Prof. Abby Whittington for use of the plate reader, and Kearsley Dillon, Sarah Swilley, and Yumeng (Jackie) Zhu for critical readings of the manuscript. The authors also thank Dr. Mehdi Ashraf-Khorassani for help with HRMS and confirmation of molecular structure. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the funding agencies.
Footnotes
There are no conflicts of interest to declare
Data Availability
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also forms part of an ongoing study.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Associated Content
Supplementary Information
Small molecule, polymer, and polymer aggregate characterization.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Cho K, Wang X, Nie S, Chen Z,Shin DM, Therapeutic Nanoparticles for Drug Delivery in Cancer, Clin. Cancer Res. 14 (2008) 1310, 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- [2].Kataoka K, Harada A,Nagasaki Y, Block copolymer micelles for drug delivery: Design, characterization and biological significance, Adv. Drug Del. Rev. 64 (2012) 37–48, 10.1016/j.addr.2012.09.013. [DOI] [PubMed] [Google Scholar]
- [3].Petros RA,DeSimone JM, Strategies in the design of nanoparticles for therapeutic applications, Nat. Rev. Drug Discov. 9 (2010) 615–627, 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- [4].Wang Y, Cheetham AG, Angacian G, Su H, Xie L,Cui H, Peptide–drug conjugates as effective prodrug strategies for targeted delivery, Adv. Drug Del. Rev. 110–111 (2017) 112–126, 10.1016/j.addr.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ulbrich K, Hola K, Subr V, Bakandritsos A, Tucek J,Zboril R, Targeted Drug Delivery with Polymers and Magnetic Nanoparticles: Covalent and Noncovalent Approaches, Release Control, and Clinical Studies, Chem. Rev. 116 (2016) 5338–5431, 10.1021/acs.chemrev.5b00589. [DOI] [PubMed] [Google Scholar]
- [6].Tyler B, Gullotti D, Mangraviti A, Utsuki T,Brem H, Polylactic acid (PLA) controlled delivery carriers for biomedical applications, Adv. Drug Del. Rev. 107 (2016) 163–175, 10.1016/j.addr.2016.06.018. [DOI] [PubMed] [Google Scholar]
- [7].Li ZB, Ye EY, David R Lakshminarayanan XJ. Loh, Recent Advances of Using Hybrid Nanocarriers in Remotely Controlled Therapeutic Delivery, Small 12 (2016) 4782–4806, 10.1002/smll.201601129. [DOI] [PubMed] [Google Scholar]
- [8].Biswas S, Kumari P, Lakhani PM,Ghosh B, Recent advances in polymeric micelles for anti-cancer drug delivery, European Journal of Pharmaceutical Sciences 83 (2016) 184–202, 10.1016/j.ejps.2015.12.031. [DOI] [PubMed] [Google Scholar]
- [9].Cabral H, Miyata K, Osada K,Kataoka K, Block Copolymer Micelles in Nanomedicine Applications, Chem. Rev. 118 (2018) 6844–6892, 10.1021/acs.chemrev.8b00199. [DOI] [PubMed] [Google Scholar]
- [10].Chan D, Yu AC,Appel EA, Single-Chain Polymeric Nanocarriers: A Platform for Determining Structure–Function Correlations in the Delivery of Molecular Cargo, Biomacromolecules 18 (2017) 1434–1439, 10.1021/acs.biomac.7b00249. [DOI] [PubMed] [Google Scholar]
- [11].Wang R, Gasotransmitters: growing pains and joys, Trends Biochem. Sci. 39 (2014) 227–232, 10.1016/j.tibs.2014.03.003. [DOI] [PubMed] [Google Scholar]
- [12].Mustafa AK, Gadalla MM,Snyder SH, Signaling by Gasotransmitters, Sci. Signal 2 (2009) re2–re2, 10.1126/scisignal.268re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang R, Shared signaling pathways among gasotransmitters, Proc. Natl. Acad. Sci. 109 (2012) 8801, 10.1073/pnas.1206646109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Barr LA,Calvert JW, Discoveries of Hydrogen Sulfide as a Novel Cardiovascular Therapeutic, Circulation 78 (2014) 2111–2118, 10.1253/circj.CJ-14-0728. [DOI] [PubMed] [Google Scholar]
- [15].Calvert John W, Jha S, Gundewar S, Elrod John W, Ramachandran A, Pattillo Christopher B, Kevil Christopher G,Lefer David J, Hydrogen Sulfide Mediates Cardioprotection Through Nrf2 Signaling, Circ. Res. 105 (2009) 365–374, 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mustafa Asif K, Sikka G, Gazi Sadia K, Steppan J, Jung Sung M, Bhunia Anil K, Barodka Viachaslau M, Gazi Farah K, Barrow Roxanne K, Wang R, Amzel LM, Berkowitz Dan E,Snyder Solomon H, Hydrogen Sulfide as Endothelium-Derived Hyperpolarizing Factor Sulfhydrates Potassium Channels, Circ. Res. 109 (2011) 1259–1268, 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tang G, Wu L, Liang W,Wang R, Direct Stimulation of KATP Channels by Exogenous and Endogenous Hydrogen Sulfide in Vascular Smooth Muscle Cells, Mol. Pharmacol. 68 (2005) 1757, 10.1124/mol.105.017467. [DOI] [PubMed] [Google Scholar]
- [18].Zhao W, Zhang J, Lu Y,Wang R, The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener, EMBO J. 20 (2001) 6008–6016, 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Szabo C, Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: mechanisms and implications, Am. J. Physiol., Cell Physiol. 312 (2016) C3–C15, 10.1152/ajpcell.00282.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Szabo C,Papapetropoulos A, Hydrogen sulphide and angiogenesis: mechanisms and applications, Br J Pharmacol 164 (2011) 853–865, 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T,Zhu YC, The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation, Cardiovasc Res 76 (2007) 29–40, 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- [22].Bhatia M, Sidhapuriwala JN, Wei Ng S, Tamizhselvi R,Moochhala SM, Pro-inflammatory effects of hydrogen sulphide on substance P in caerulein-induced acute pancreatitis, J. Cell. Mol. Med. 12 (2008) 580–590, 10.1111/j.1582-4934.2007.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Campolo M, Esposito E, Ahmad A, Di Paola R, Wallace JL,Cuzzocrea S, A hydrogen sulfide-releasing cyclooxygenase inhibitor markedly accelerates recovery from experimental spinal cord injury, FASEB J. 27 (2013) 4489–4499, 10.1096/fj.13-234716. [DOI] [PubMed] [Google Scholar]
- [24].Giuliani D, Ottani A, Zaffe D, Galantucci M, Strinati F, Lodi R,Guarini S, Hydrogen sulfide slows down progression of experimental Alzheimer’s disease by targeting multiple pathophysiological mechanisms, Neurobiology of learning and memory 104 (2013) 82–91, 10.1016/j.nlm.2013.05.006. [DOI] [PubMed] [Google Scholar]
- [25].Zhao Y,Pluth MD, Hydrogen Sulfide Donors Activated by Reactive Oxygen Species, Angew. Chem. Int. Ed. 55 (2016) 14638–14642, 10.1002/anie.201608052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Powell CR, Dillon KM,Matson JB, A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications, Biochemical Pharmacology 149 (2018) 110–123, 10.1016/j.bcp.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kaur K, Carrazzone RJ,Matson JB, The Benefits of Macromolecular/Supramolecular Approaches in Hydrogen Sulfide Delivery: A Review of Polymeric and Self-Assembled Hydrogen Sulfide Donors, Antioxid. Redox. Sign. 32 (2019) 79–95, 10.1089/ars.2019.7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li Z, Polhemus David J,Lefer David J, Evolution of Hydrogen Sulfide Therapeutics to Treat Cardiovascular Disease, Circ. Res. 123 (2018) 590–600, 10.1161/CIRCRESAHA.118.311134. [DOI] [PubMed] [Google Scholar]
- [29].Wallace JL, Vaughan D, Dicay M, MacNaughton WK,de Nucci G, Hydrogen Sulfide-Releasing Therapeutics: Translation to the Clinic, Antioxid. Redox Signal. 28 (2017) 1533–1540, 10.1089/ars.2017.7068. [DOI] [PubMed] [Google Scholar]
- [30].Wallace J, Buret A, Nagy P, Muscara M,Nucci G.d., THU0464 phase 2 clinical trial of the GI safety of a hydrogen sulfide-releasing anti-inflammatory drug (ATB-346), Ann. Rheum. Dis. 78 (2019) 522–522, 10.1136/annrheumdis-2019-eular.568. [DOI] [Google Scholar]
- [31].Kodela R, Chattopadhyay M,Kashfi K, NOSH-Aspirin: A Novel Nitric Oxide–Hydrogen Sulfide-Releasing Hybrid: A New Class of Anti-inflammatory Pharmaceuticals, ACS Med. Chem. Lett. 3 (2012) 257–262, 10.1021/ml300002m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xiao Z, Bonnard T, Shakouri-Motlagh A, Wylie RAL, Collins J, White J, Heath DE, Hagemeyer CE,Connal LA, Triggered and Tunable Hydrogen Sulfide Release from Photogenerated Thiobenzaldehydes, Chem. Eur. J. 23 (2017) 11294–11300, 10.1002/chem.201701206. [DOI] [PubMed] [Google Scholar]
- [33].Hasegawa U,van der Vlies AJ, Polymeric micelles for hydrogen sulfide delivery, MedChemComm 6 (2015) 273–276, 10.1039/C4MD00373J. [DOI] [Google Scholar]
- [34].Foster JC, Radzinski SC, Zou X, Finkielstein CV,Matson JB, H2S-Releasing Polymer Micelles for Studying Selective Cell Toxicity, Mol. Pharm. 14 (2017) 1300–1306, 10.1021/acs.molpharmaceut.6b01117. [DOI] [PubMed] [Google Scholar]
- [35].Szabó C, Hydrogen sulphide and its therapeutic potential, Nat. Rev. Drug Discov. 6 (2007) 917, 10.1038/nrd242510.1038/nrd2425https://www.nature.com/articles/nrd2425#supplementary-informationhttps://www.nature.com/articles/nrd2425#supplementary-information . [DOI] [PubMed] [Google Scholar]
- [36].Ercole F, Mansfeld FM, Kavallaris M, Whittaker MR, Quinn JF, Halls ML,Davis TP, Macromolecular Hydrogen Sulfide Donors Trigger Spatiotemporally Confined Changes in Cell Signaling, Biomacromolecules 17 (2016) 371–383, 10.1021/acs.biomac.5b01469. [DOI] [PubMed] [Google Scholar]
- [37].Dillon KM, Carrazzone RJ, Matson JB,Kashfi K, The evolving landscape for cellular nitric oxide and hydrogen sulfide delivery systems: A new era of customized medications, Biochem. Pharmacol. (2020) 113931, 10.1016/j.bcp.2020.113931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wallace JL,Wang R, Hydrogen sulfide-based therapeutics: exploiting a unique but ubiquitous gasotransmitter, Nat. Rev. Drug Discov. 14 (2015) 329, 10.1038/nrd4433. [DOI] [PubMed] [Google Scholar]
- [39].Urquhart MC, Ercole F, Whittaker MR, Boyd BJ, Davis TP,Quinn JF, Recent advances in the delivery of hydrogen sulfide via a macromolecular approach, Polym. Chem. 9 (2018) 4431–4439, 10.1039/C8PY00938D. [DOI] [Google Scholar]
- [40].Connal LA, The benefits of macromolecular hydrogen sulfide prodrugs, J. Mater. Chem. B 6 (2018) 7122–7128, 10.1039/C8TB02352B. [DOI] [PubMed] [Google Scholar]
- [41].Ercole F, Li Y, Whittaker MR, Davis TP,Quinn JF, H2S-Donating trisulfide linkers confer unexpected biological behaviour to poly(ethylene glycol)–cholesteryl conjugates, J. Mater. Chem. B 8 (2020) 3896–3907, 10.1039/C9TB02614B. [DOI] [PubMed] [Google Scholar]
- [42].Yu SH, Esser L, Khor SY, Senyschyn D, Veldhuis NA, Whittaker MR, Ercole F, Davis TP,Quinn JF, Development of a shape-controlled H2S delivery system using epoxide-functional nanoparticles, J. Polym. Sci. A 57 (2019) 1982–1993, 10.1002/pola.29382. [DOI] [Google Scholar]
- [43].Ercole F, Whittaker MR, Halls ML, Boyd BJ, Davis TP,Quinn JF, Garlic-inspired trisulfide linkers for thiol-stimulated H2S release, Chem. Comm. 53 (2017) 8030–8033, 10.1039/C7CC03820H. [DOI] [PubMed] [Google Scholar]
- [44].Takatani-Nakase T, Katayama M, Matsui C, Hanaoka K, van der Vlies AJ, Takahashi K, Nakase I,Hasegawa U, Hydrogen sulfide donor micelles protect cardiomyocytes from ischemic cell death, Mol. BioSyst. 13 (2017) 1705–1708, 10.1039/C7MB00191F. [DOI] [PubMed] [Google Scholar]
- [45].Chen JJY, van der Vlies AJ,Hasegawa U, Hydrogen sulfide-releasing micelles for promoting angiogenesis, Polym. Chem. 11 (2020) 4454–4463, 10.1039/D0PY00495B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Foster JC, Carrazzone RJ, Spear NJ, Radzinski SC, Arrington KJ,Matson JB, Tuning H2S Release by Controlling Mobility in a Micelle Core, Macromolecules 52 (2019) 1104–1111, 10.1021/acs.macromol.8b02315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Molla MR, Rangadurai P, Antony L, Swaminathan S, de Pablo JJ,Thayumanavan S, Dynamic actuation of glassy polymersomes through isomerization of a single azobenzene unit at the block copolymer interface, Nat. Chem. 10 (2018) 659–666, 10.1038/s41557-018-0027-6. [DOI] [PubMed] [Google Scholar]
- [48].Zheng H, Yin LQ, Zhang XQ, Zhang H, Hu R, Yin YH, Qiu T, Xiong X,Wang Q, Redox Sensitive Shell and Core Crosslinked Hyaluronic Acid Nanocarriers for Tumor-Targeted Drug Delivery, J. Biomed. Nanotechnol. 12 (2016) 1641–1653, 10.1166/jbn.2016.2279. [DOI] [PubMed] [Google Scholar]
- [49].Wang TR, Ma XY, Lei Y,Luo YC, Solid lipid nanoparticles coated with cross-linked polymeric double layer for oral delivery of curcumin, Colloid Surf. B-Biointerfaces 148 (2016) 1–11, 10.1016/j.colsurfb.2016.08.047. [DOI] [PubMed] [Google Scholar]
- [50].Modarresi-Saryazdi SM, Haddadi-Asl V,Salami-Kalajahi M, N,N’-methylenebis(acrylamide)-crosslinked poly(acrylic acid) particles as doxorubicin carriers: A comparison between release behavior of physically loaded drug and conjugated drug via acid-labile hydrazone linkage, J. Biomed. Mater. Res. A 106 (2018) 342–348, 10.1002/jbm.a.36240. [DOI] [PubMed] [Google Scholar]
- [51].Talelli M, Barz M, Rijcken CJF, Kiessling F, Hennink WE,Lammers T, Core-crosslinked polymeric micelles: Principles, preparation, biomedical applications and clinical translation, Nano Today 10 (2015) 93–117, 10.1016/j.nantod.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Skey J,O’Reilly RK, Facile one pot synthesis of a range of reversible addition–fragmentation chain transfer (RAFT) agents, Chem. Commun. (2008) 4183–4185, 10.1039/B804260H. [DOI] [PubMed] [Google Scholar]
- [53].Chong D, Tan J, Zhang J, Zhou Y, Wan X,Zhang J, Dual electrical switching permeability of vesicles via redox-responsive self-assembly of amphiphilic block copolymers and polyoxometalates, Chem. Commun. 54 (2018) 7838–7841, 10.1039/C8CC03749C. [DOI] [PubMed] [Google Scholar]
- [54].Foster JC,Matson JB, Functionalization of Methacrylate Polymers with Thiooximes: A Robust Postpolymerization Modification Reaction and a Method for the Preparation of H2S-Releasing Polymers, Macromolecules 47 (2014) 5089–5095, 10.1021/ma501044b. [DOI] [Google Scholar]
- [55].Li R-Y, Jing L, Meng J-L,Li G, Synthesis and Characterization of Four New Chloro-Group Substituted Salamo-Type Bisoxime Compounds, Asian J. Chem. 26 (2014) 2733–2735, 10.14233/ajchem.2014.16295. [DOI] [Google Scholar]
- [56].Foster JC, Powell CR, Radzinski SC,Matson JB, S-aroylthiooximes: a facile route to hydrogen sulfide releasing compounds with structure-dependent release kinetics, Org. Lett. 16 (2014) 1558–1561, [DOI] [PubMed] [Google Scholar]
- [57].Lin L, Qin H, Huang J, Liang H, Quan D,Lu J, Design and synthesis of an AIE-active polymeric H2S-donor with capacity for self-tracking, Polym. Chem. 9 (2018) 2942–2950, 10.1039/C8PY00548F. [DOI] [Google Scholar]
- [58].Penzel E, Rieger J,Schneider HA, The glass transition temperature of random copolymers: 1. Experimental data and the Gordon-Taylor equation, Polymer 38 (1997) 325–337, 10.1016/S0032-3861(96)00521-6. [DOI] [Google Scholar]
- [59].Konkolewicz D, Hawkett BS, Gray-Weale A,Perrier S, RAFT polymerization kinetics: How long are the cross-terminating oligomers?, 47 (2009) 3455–3466, 10.1002/pola.23385. [DOI] [Google Scholar]
- [60].Joralemon MJ, O’Reilly RK, Hawker CJ,Wooley KL, Shell Click-crosslinked (SCC) nanoparticles: A new methodology for synthesis and orthogonal functionalization, J. Am. Chem. Soc. 127 (2005) 16892–16899, 10.1021/ja053919x. [DOI] [PubMed] [Google Scholar]
- [61].Dai J, Lin SD, Cheng D, Zou SY,Shuai XT, Interlayer-Crosslinked Micelle with Partially Hydrated Core Showing Reduction and pH Dual Sensitivity for Pinpointed Intracellular Drug Release, Angew. Chem.-Int. Edit. 50 (2011) 9404–9408, 10.1002/anie.201103806. [DOI] [PubMed] [Google Scholar]
- [62].Wu ZM, Liang H, Lu JA,Deng WL, Miktoarm Star Copolymers via Combination of RAFT Arm-First Technique and Aldehyde-Aminooxy Click Reaction, J. Polym. Sci. Pol. Chem. 48 (2010) 3323–3330, 10.1002/pola.24116. [DOI] [Google Scholar]
- [63].Wang K, Peng H, Thurecht KJ, Puttick S,Whittaker AK, Biodegradable core crosslinked star polymer nanoparticles as 19F MRI contrast agents for selective imaging, Polym. Chem. 5 (2014) 17601771, 10.1039/C3PY01311A. [DOI] [Google Scholar]
- [64].Siegel LM, A direct microdetermination for sulfide, Analytical Biochemistry 11 (1965) 126–132, 10.1016/0003-2697(65)90051-5. [DOI] [PubMed] [Google Scholar]
- [65].Fischer E, Bildung von Methylenblau als Reaktion auf Schwefelwasserstoff, Ber. Dtsch, Chem. Ges. 16 (1883) 2234–2236, 10.1002/cber.188301602138. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.