

Abstract
Major light-harvesting complex of photosystem II (LHCII) plays a dual role in light-harvesting and excited energy dissipation to protect photodamage from excess energy. The regulatory switch is induced by increased acidity, temperature or both. However, the molecular origin of the protein dynamics at the atomic level is still unknown. We carried out temperature-jump time-resolved infrared spectroscopy and molecular dynamics simulations to determine the energy quenching dynamics and conformational changes of LHCII trimers. We found that the spontaneous formation of a pair of local α-helices from the 310-helix E/loop and the C-terminal coil of the neighboring monomer, in response to the increased environmental temperature and/or acidity, induces a scissoring motion of transmembrane helices A and B, shifting the conformational equilibrium to a more open state, with an increased angle between the associated carotenoids. The dynamical allosteric conformation change leads to close contacts between the first excited state of carotenoid lutein 1 and chlorophyll pigments, facilitating the fluorescence quenching. Based on these results, we suggest a unified mechanism by which the LHCII trimer controls the dissipation of excess excited energy in response to increased temperature and acidity, as an intrinsic result of intense sun light in plant photosynthesis.
Keywords: LHCII photoprotection, conformational dynamics and allostery, protein switch, T-jump, FTIR, fluorescence quenching, excited energy transfer
1. Introduction
In higher plants, the most abundant antenna unit in photosynthesis is the major light-harvesting complex of the photosystem II (LHCII), which regulates a reversible switch from an efficient light harvesting state to a photoprotection state under intense light [1–5]. The process to dissipate the excess excitation energy is collectively known as the non-photochemical quenching (NPQ) [4,6], in which the dominant component is the pH-dependent energy quenching (qE) [1], responsible for a rapid response to natural variations in light intensity [4,7]. NPQ also exists in green algae, diatoms, and mosses [8]. For higher plants, NPQ is triggered by the transmembrane acidity gradient, leading to the LHCII aggregation and conformational changes [9], including participation of the essential protein PsbS [10] and zeaxanthin (Zea) [11]. Recently, photosynthesis efficiency with 15% increase in biomass production has been reported by accelerating the recovery process from photoprotection [12] as one of the strategies in redesigning of the photosynthesis [13], and the wavelike characteristic of the energy transfer has been proposed to explain the high efficiency of light-harvesting [14,15]. However, despite extensive studies, the underlying mechanism of protein dynamics and conformational changes that regulate light harvesting and photoprotection in the LHCII at the atomic level is still unknown [3,16,17].
The X-ray crystal structure of the LHCII (PDB ID: 1RWT) shows that each subunit consists of three transmembrane (TM)-helices (A, B, C) and one amphipathic-helix (D) along with a short 310-helix (E) at the lumenal side (Figure 1(a)) [18–22]. Two crossed TM helices A and B are interlocked by conserved salt bridges between Arg70 and Glu180, as well as Glu65 and Arg185 [18,23]. At the lumenal side, helices E and C are connected by anti-parallel β-strands together with loop segments, and these sites have been suggested to play an important role in regulating the multiple functions of the LHCII [24]. Protein conformational changes have been inferred from Raman spectroscopic studies of the carotenoids neoxanthin (Nex) and luteins (Lut) [25,26], both in crystals and in vivo in the quenched state. Nuclear magnetic resonance (NMR) studies [23] revealed rearrangements of the loops and helical segments close to the stromal and lumenal ends of LHCII aggregates. In the dissipative state, LHCII trimers are sufficient to undergo efficient energy transfer from the excited state of chlorophylls (Chls) to the dark state (S1) of Luts, supported by the two-photon studies on the electronic interaction between S1 of carotenoids and chlorophylls [26–30], and the energy, which is dissipated non-radiatively [26,31–33], is consistent with the fluorescence quenching of the LHCII embedded in gels [34] and single molecule spectroscopy experiments [35]. Furthermore, excited-state energy dissipation correlates linearly with the LHCII aggregation induced by heat both in vivo and in vitro [36]. Tang et al. investigated the aggregation of LHCII trimers in leaves of whole spinach plants subjected to heat stress (25–50 °C) in the dark for 30 min. They showed that the extent of aggregation increases with the temperature rising in a range of 35–50 °C in the thylakoid membranes isolated immediately from heat-stressed leaves by native green gels, sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting analysis. In addition, an excited energy quenching mechanism via the electron transfer from Zea to the Chla excited state resulting in the formation of Zea+ radical cations has also been proposed [37–40]. This quenching pathway has been observed only in the minor antenna complexes, but not in the isolated LHCII trimer [41]. The existence of two different quenching mechanisms acting in parallel consistently explains all available data on NPQ [42,43]. In fact, under the full sun radiation, when the ambient temperature reaches 25 °C, the steady-state temperature of a leaf can be as high as 40 °C estimated for a typical case [44]. We estimated that under a typical NPQ condition at noon, the solar flux measured is about 1.1×103 J m−2 s−1 [45], and the energy dissipation in NPQ would lead to an averaged transient local temperature jump of 14 °C per LHCII trimer within 10 μs, i.e., a transient temperature of LHCII trimers in the leaf can be as high as 54 °C at an ambient temperature of 25 °C (Details of estimation analyses are given in Supporting Information online). Then a fundamental question arises is that how the LHCII senses environmental conditions, such as pH or temperature, and transmits these signals in terms of structural changes to the remote sites, ultimately leading to an increased qE rate.
Figure 1.

Crystal structure and the fluorescence decay kinetics of LHCII trimers. (a) Schematic structures of LHCII trimers (PDB: 1RWT) along with pigments (stick model) in highlighted in one subunits. (b) Fluorescence decay curves of LHCII trimers at room temperature (RT) under different conditions as indicated, excited at 400 nm, and acquired by a streak camera with an instrument response factor (IRF) of 0.13 ns. The fluorescence decay curves for the aggregated LHCII trimer are fitted with a bi-exponential process (weakly aggregated condition: pH 5.6, β-DM: 0.003% (w/v); heavily aggregated condition: pH 5.6, β-DM: 0.0003% (w/v)) and that for non-aggregated is fitted by a mono-exponential decay (non-aggregated condition: pH 5.6, β-DM: 0.03% (w/v)). (c) Globally fitted fluorescence lifetimes for the fast and slow components at varied temperatures for the aggregated LHCII trimer (β-DM: 0.006% (w/v)) at two different pH as indicated. (d) Photos of the sucrose density gradient ultracentrifugation separation of the thermal incubated LHCII trimer for 5 min at varied temperatures as indicated and under two different aggregated conditions (aggregated condition: pH 5.6, β-DM: 0.003% (w/v); non-aggregated condition: pH 5.6, β-DM: 0.03% (w/v)). LHCII trimers corresponding to 124 μg of Chl were loaded on the sucrose gradient, and the details in fractionation were described in “Sample preparation”. (e, f) Temperature-dependent population variation of a three-state model, i.e., harvesting (H), quenching (Q), and denaturing (D) states, of non-aggregated (β-DM: 0.03% (w/v)) and aggregated LHCII trimers (β-DM: 0.006% (w/v)) at pH of 7.7 (e) and 5.6 (f) respectively (color online).
In this study, we carried out fluorescence and IR experiments along with molecular dynamics (MD) simulations to characterize the energy quenching dynamics and conformational changes of LHCII trimers. We found that the LHCII trimer undergoes local secondary structure transitions to form α-helices at the lumenal side, triggering conformational changes across the membrane, in response to ΔT. ΔpH or both, and acts as a molecular machine to pivot scissoring motions of TM helices with salt bridges as the fulcrum and hydrophobic contacts as the levers, resulting in close contacts between Luts and Chls pigments. These results suggest that the collective effects of environmental conditions and protein conformational changes lead to enhanced electronic coupling and excited energy transfer from Chls to the dark state (S1) of Lut1, providing an intrinsic mechanism for allosteric regulation of energy quenching by LHCII.
2. Materials and method
2.1. Sample preparation
Trimeric LHCII was purified from spinach leaves according to the reported protocol [21] with a few modifications. In particular, thylakoid membranes were washed with the ice-cold Hepes buffers (pH 7.5), containing 1 mM ethylenediaminetetraacetic acid (EDTA), and subsequently solubilized in 5% Triton X-100 by vortexing for 15 min. After centrifugation (Avanti J-25, Beckman Coulter, USA), the BBY particles (PSII-enriched) were homogenized in the BisTris buffer (pH 6.5), and solubilized in 1.25% (w/v) n-dodecyl β-D-maltoside (β-DM) by vortexing for 10 min. The solubilized samples were fractionated by ultra-centrifugation (CP100MX, Hitachi, Japan) on a sucrose density gradient in a SW41 rotor at 40,000 r/min for 15 h, while the gradients were prepared by freezing at −80 °C and thawing at 4 °C in the tube, containing 0.03% (w/v) β-DM, and 20 mM BisTris (pH 6.5). The trimeric LHCII band was harvested with syringes and eluted with 0.03% (w/v) β-DM solution by using 50 kD centrifugal filters (Amicon Ultra-4-Millipore). To prepare LHCII monomers, the trimers were incubated with 10 μg/mL phospholipase A2 from bee venom (Sigma, USA) for 48 h at room temperature in 20 mM CaCl2 at a Chl concentration of 500 μM and the enzymatic hydrolysate containing the monomers and the residual trimers was fractionated by ultra-centrifugation in the same sucrose density gradient as the trimer extraction [46]. The non-aggregated proteins were incubated in the dark overnight at a β-DM concentration of 0.03% (w/v), and the aggregates were achieved by lowering the β-DM concentration to 0.003% (w/v) and 0.0003% (w/v), respectively, i.e., below the critical micelle concentration (0.00817% (w/v)). Each indicated pH value was obtained by adding the additional Tris-HCl buffer giving a final concentration of Tris-HCl to be 10 mM. For infrared spectroscopy experiments, the eluted proteins were freeze-dried and preserved in a vacumn at −10 °C for 48 h, and then the lyophilized proteins were dissolved in the indicated buffer containing 99.9% D20. Unless otherwise stated, reagents were purchased from commercial suppliers and used without further purification.
2.2. Transient fluorescence spectroscopy
Time-resolved fluorescence spectra of LHCII trimers under various conditions were collected by Hamamatsu C5680 Synchroscan streak camera, with a femtosecond Ti: sapphire oscillator (Mira 900F, Coherent, USA) as the pump source [47]. The excitation wavelength was set to 400 mn at an average power density of 4.8 mW/cm2 by frequency doubling of the fundamental laser output at 800 nm. The temporal resolution of the detection system was dependent on the specific temporal detecting range, i.e., 0.13 ns for 0~5 ns and 0.30 ns for 0~10 ns. Samples in different media at a protein concentration of 0.0355 mg/mL (OD675 nm=0.12) were placed in a quartz cuvette (JGS1) of 1 mm in optical pathlength with the constant stirring during the measurement, and the ambient temperature was controlled by a water circulator. The kinetic decay traces were examined for the excitation power dependence (average power density from 2.9 to 6.4 mW/cm2). Under the current conditions, the decay traces were not affected by the selected excitation power in streak camera measurements.
2.3. Temperature-jump nanosecond time-resolved IR absorbance difference spectra
The details of the temperature-jump nanosecond time-resolved IR absorbance difference spectrometer have been described elsewhere [48], and a significant improvement of the current apparatus was to use a home-built Q-switched Cr, Tm, Ho: YAG laser delivering 1.9 μm pulses with a pulse width about 50 ns as the heating pulses to replace the previous high pressure H2 gas Raman shifting cell. The detection limit of the T-jump transient IR absorbance difference spectra has been improved to 2×10−4 ΔOD. The absorbance changes were probed by a CO mid-IR laser at a spectral spacing about 4 cm−1 (Dalian University of Technology, China) in conjunction with a MCT detector (Kolmar, New-buryport, USA) and a digital oscilloscope (Tektronix, TDS520D, Santa Clara, USA). The temporal resolution of this system was about 60 ns.
2.4. System setup of MD simulations
The trimer from the crystal structure of spinach LHCII (PDB 1RWT) [18] was used as the starting structure of all simulations. First, we removed the exogenous molecules of diglyceride, detergents and glycerol, and then manually repaired the partial phytyl chains of chlorophylls. The system was embedded into a lipid bilayer of 512 DOPC molecules via the Membrane Builder tool of CHARMM-GUI [49]. The whole structure was further solvated in a cubic solvent box with a TIP3P [50] water molecule layer extended approximately 30 Å away along the z-direction, resulting in a total of around 242k atoms. Counter ions (Na+ and Cl−) were added to ensure the electrostatic neutrality. All-atom AMBER03 [51] force fields were used for the protein and ions. Parameters for the chlorophyll cofactors CLA (chlorophyll a) and CHL (chlorophyll b) were adapted from a previous study [52] , and the parameters for various carotenoids (XAT (xanthophyll), LUT (lutein) and NEX (neoxanthin)) were generated with the antechamber program [53]. The simulation was performed using Gromacs 4.6.5 [54] with the LINCS [55] algorithm applied to constrain all covalent H-bonds. Particle-mesh Ewald [56] was used to treat long-range electrostatic interactions with a real-space cutoff of 10 Å.
The initial complex was minimized using steepest descent algorithm and set to the reference structure for starting parallel simulations at different temperatures. A set of 10 simulations were performed every 10 K ranging from 270 to 360 K. For each simulation, the complex was gradually heated to the reference temperature at a constant volume within 2 ns, using harmonic restraints with a force constant 1,000 kJ/(mol Å2) on heavy atoms of proteins and lipids. Over the following 12 ns of simulations at a constant pressure (1 atm) and temperature, the restraints were gradually released. The systems were equilibrated for an additional 20 ns without positional restraints. A Parrinello-Rahman barosta [57] was used to keep the pressure constant, while a V-rescale thermostat with a time step of 2 fs applied to the protein, lipids, and solvent groups independently. Each system was simulated for 750 ns, with snapshots recorded every 5 ps. A total of 7.5 μs trajectories were utilized for analyses.
For comparison, the simulation of the monomeric LHCII was started from the monomer A in the PDB 1RWT, and the remaining steps followed the same steps for the trimer, with a total of 7.5 μs trajectories collected over the 10 simulations from 270 to 360 K.
In order to characterize the conformational plasticity of the LHCII trimer at higher temperatures, the simulation of 350 K was extended to 1.5 μs, and the conformations were clustered into 50 microstates, and additional adaptive sampling [58] was started from the representative structure of each microstate, each lasting for 90 ns. Therefore, a total length of 6 μs trajectories was collected for the simulation at 350 K.
3. Results
3.1. Variations of fluorescence lifetimes of LHCII trimers with aggregation, acidity and temperature indicate the existence of the quenched conformer
The fluorescence lifetime was measured by a streak camera with a temporal resolution of 0.13 ns for isolated LHCII trimers at various aggregated states under acidic and neutral conditions (Figure 1(b) (acidic) and Figure S1(c), Supporting Information online) at room temperature (RT), showing the aggregation substantially enhances the fluorescence quenching while the decay kinetics can be resolved into a fast and a slow decay components by a global fitting. As shown in Table S1 Supporting Information online), under weakly aggregated states (β-DM: 0.006% or 0.003% (w/v)), the slow component has a value between 2.6–2.9 ns, while the fast component is around 0.26–0.29 ns at RT regardless of the varied pH values. Figure 1(c) further plots the two decay lifetime constants for the aggregated LHCII trimer (β-DM: 0.006% (w/v)) at acidic and neutral conditions against temperature, which reveals that the LHCII trimer undergoes a thermal-induced fluorescence quenching as temperature increases (the aggregation state at varied temperatures was examined by the dynamical light scattering, see Table S2), and the quenching efficiency is also correlated with acidity levels as reflected by the fact that a lower pH value gives rise to a lower flexion point temperature and a higher normalized pre-exponential factor for the fast component (t2) (Table S1). This flexion point temperature is coincident to the temperature for the start of monomerization of the LHCII trimer revealed by the sucrose density gradient ultra-centrifugation separation of the thermal incubated LHCII trimer at varied temperatures in Figure 1(d) (acidic) and Figure S2(d) (neutral), for the lifetime of LHCII monomers is less temperature-dependent than that of the trimer, and increases with temperature after 62 °C possibly owing to the unfolding of the monomer (Figure S3(f)). Accordingly, the two decay components would correspond to different conformers, i.e., one having a longer decay time constant is termed as the light-harvesting conformer (H) while the other belongs to a mixture of quenching (Q) and denatured (D) conformers. To rule out the complication of the observed thermal-induced fluorescence quenching resulting from the thermal denaturation of the proteins, we analyzed the reversibility of the pigment local configuration and protein secondary structures by the fluorescence and IR detection, respectively, after an annealing treatment on LHCII trimers, in which the sample was first equilibrated for 5 min at the indicated temperature, and then cooled down to RT for steady-state measurements (Figures S4(c, d) and S6(a, c, e)). The completely thermal reversible temperature is determined as about 55 °C, at which about 95% fluorescence intensity and more than 93% TM α-helices are recovered for the non-aggregated LHCII (β-DM: 0.03% (w/v), pH 7.7), in consistent with the reported result [59] and our sucrose density gradient ultra-centrifugation results as shown in Figure S2(d).
Based on above experiments, we proposed that the three different conformers in the system, i.e., H, Q and D are correlated in a way depicted in the Scheme 1, where H and Q are inter-convertible, while H and D, along with Q and D are non-interconvertible. With this model, the thermal-induced population changes of H, Q and D can be resolved from the experimental data as shown in Figure 1(e, f) (the detailed procedure is given in supporting information), which clearly reveals that the population of the quenching state Q for the aggregated LHCII trimer (β-DM: 0.006% (w/v)) reaches its maximum at a temperature of 55 °C for pH 7.7 and of 53 °C for pH 5.6, while for the non-aggregated LHCII timer, the temperature of the maximum quenching shifts to a higher temperature of 65 °C. The fact suggests that thermal-induced fluorescence quenching is also an intrinsic property of single molecules of LHCII trimers, similar to that of acidity-induced fluorescence quenching, with aggregation obviously amplifying such a quenching effect. Therefore, we can conclude that under the aggregated state, the acidity /thermal-induced fluorescence quenching efficiency is maximized in the temperature region where the protein conformations are almost completely reversible, and the protein denaturation effect can be excluded. Importantly, the fluorescence spectra for H, together with Q are quite similar as shown in Figure S2(e, f), regardless of the temperature and pH, suggesting that the fluorescence of different conformers under varied conditions involves the same fluorescence site.
Scheme 1.

Relations among three conformers for LHCII trimers.
We further analyzed the free energy difference (ΔG) between the quenching (Q) and the light-harvesting (H) conformers using their derived population at different temperatures above as the relative equilibrium concentrations [59,60]. The results (Figure S3(h)) show that for the aggregated LHCII, ΔG is about 76 kJ/mol which is less affected by the acidity, and this value is close to the enthalpy difference (ΔH) between the quenching and light-harvesting conformers about 70–85 kJ/mol reported by Santabarbara et al. [61]. For the non-aggregated LHCII, ΔG drops from 73.2 to 48.3 kJ/mol when pH changes from 7.7 to 5.6. This also accounts for the conformational change from H to Q conformers is a thermal activated process. When measured in the chloroplast, an activation energy of 64 kJ/mol for the stable organization of the LHCII in the thylakoid membrane has been reported [62]. Meanwhile, we determined the activation energy for the fluorescence quenching of Chla in ethanol solution (Figure S3(g)), which has a value of less than 5 kJ/mol, suggesting that the activation energy for the local rearrangement of pigments is small, while that for the conformational change in proteins would be large, e.g., as large as about ten folds of the former [59].
3.2. Temperature-dependence of electronic couplings from MSDFT calculations
To gain an understanding of the structural origin of the fluorescence quenching, we carried out MD simulations at 10 different temperatures, ranging from 270 to 360 K (each lasting 750 ns, except that of 6 μs at 350 K), and determined the electronic couplings for various Lut-Chl pigment pairs using multistate density functional theory (MSDFT) [63,64]. The computed average distances between selected residues, both in the same subunit and across different monomers in the LHCII, are in accordance with recent electron paramagnetic resonance (EPR) experiments (Figures S9(a–g) and S10(a–f)) [65], suggesting that the MD trajectories can provide an adequate characterization of the protein dynamic fluctuations. The possible involvement of Lut1 and Chls to modulate light-harvesting and photoprotection in the LHCII has been proposed in various context [1,26–32]. The electronic couplings between Lut1 and Chl610, and between Lut1 and Chl612 are depicted in Figure 2, which becomes progressively stronger as the temperature increases, extending to 1,600 cm−1 (~200 meV) at high temperatures. Significantly, the electronic coupling between Lut1 and Chl612 is 2–3 times stronger than that of the Lut1-Chl610 pair due to greater orbital overlap (inset of Figure 2). We conclude that Lut1-Chl612 coupling is the key energy dissipation site with strong temperature dependence, corroborating the fluorescence quenching experiments.
Figure 2.
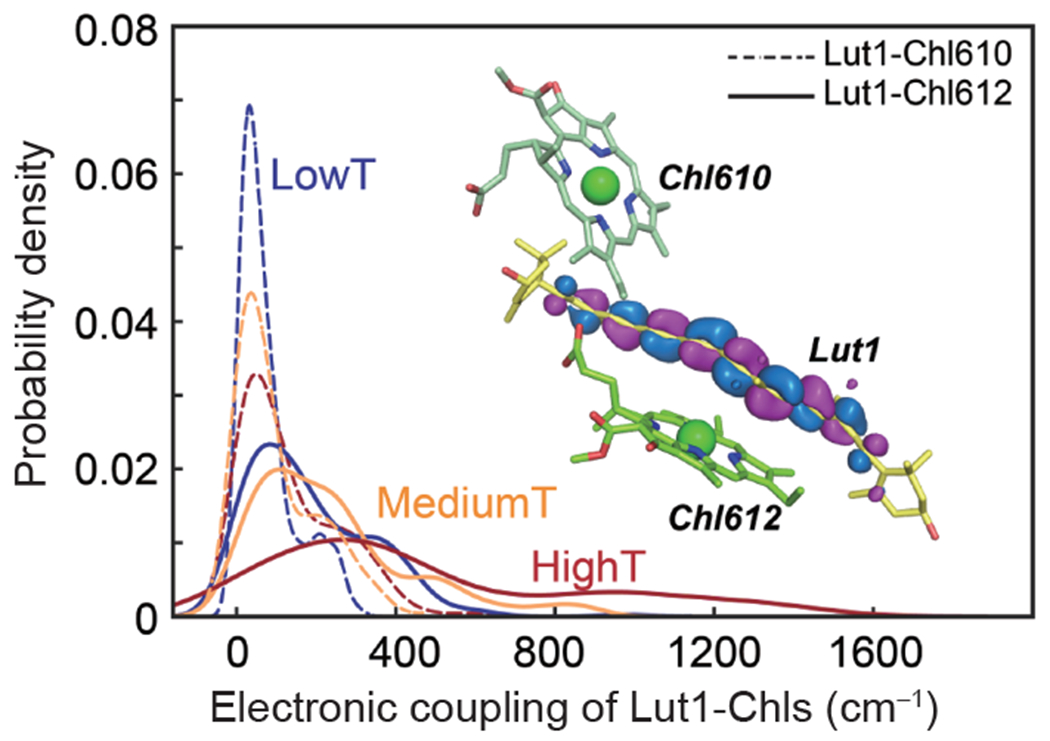
Temperature-induced electronic couplings between Lut1 and Chl and configurational twists of lutein for LHCII trimers. Distribution of computed electronic couplings between Lut1 and Chl. The computed electronic couplings are given as dashed curves for Lut1-Chl610, and as solid curves for Lut1-Ch612 at low (270–290 K) in blue, medium (300–320 K) in orange, and high (330–360 K) in maroon temperature ranges. The lowest unoccupied molecular orbital (LUMO) of Lut1 is shown along with its relative proximity to Chl610 and Chl612. Multistate density functional theory was used along with the CAM-B3LYP functional and 6– 31G(d) basis set in combined quantum-mechanical/molecular mechanical (QM/MM) calculations, in which the pigments are treated by MSDFT embedded in the rest of the protein and solvent system represented by molecular mechanics (color online).
3.3. Thermal and acidity-induced local-helical structure transitions leading to quenched conformers probed by FTIR spectroscopy
The conformational change of LHCII trimers at different temperatures, which is supposed to be responsible for the functional properties of fluorescence quenching (Figures 1(c, e, f), S2(a–c) and S4(a, b)), has been probed by Fourier transform infrared spectrometer (FTIR), which can provide reliable local structural information with the aid of crystal structures and site-directed mutations as references [66,67]. The secondary derivative FTIR spectra were employed in the present analysis (Figure 3(a)), and the full spectral assignments with corresponding references are provided in Table S3, some of which are correlated to local secondary structures at the lumenal side shown in the crystal structures (Figure 1(a)), such as 310-helix E/loops and anti-parallel β-strands. In Figure 3(a), the dominant peak at 1,651 cm−1 corresponds to the amide I′ (deuterated form) absorption of α-helices, whereas the band at 1,659 cm−1 is assigned to 310-helix E and/or loop structures since both would have the same absorption. As the temperature increases, the signal at 1,659 cm−1 decreases with the broadening of the adjacent peak at 1,651 cm−1, demonstrating that a thermal-induced bleaching of 310-helix E/loops is associated with the conformational change of α-helices (Figure S5(g)), which is clearly confirmed by the temperature-jump (T-jump) kinetics showing thermal-induced transition of 310-helix E/loops to α-helices in Figure 3(b). Furthermore, the thermal titration curves of TM-helices and 310-helix E/loops (Figure S5(a)) indicate that the latter is much less thermally stable. Significantly, when the ambient pH is lowered, the acidity-induced 310-helix E/loop transition to α-helices (Figure 3(c)) is similar to the decrease of 310-helix E/loop by the thermal effect as shown in Figure S5(g), revealing that DT and pH cause a similar local structural changes. We further investigated the S123G mutant, a key residue in close proximity of the anti-parallel β-strands and the 310-helix E at the lumenal side (the green ball in Figure 1(a)). As shown in Figure 3(d), the S123G single-site mutation induces IR spectral changes in the α-helix and 310-helix E/loop regions at RT, which is close to that of thermal-induced spectral changes in the non-aggregated LHCII trimer (Figure S5(g)). Thus, the IR spectrum of S123G mutant strongly supports that the peak at 1,659 cm−1 is from the lumenal side, which we assigned as the 310-helix E/loop.
Figure 3.

IR spectral assignments and correlation between 310-helix and α-helix in response to increased temperature and acidity for LHCII trimers. (a) Second-derivative FTIR spectra and IR assignments of LHCII trimers and monomers at RT. (b) Time-resolved spectral diffusion induced by a T-jump of ΔT=17°C. Thermal-induced transition of 310-helix/loop to α-helices is revealed by the complementary kinetics, (c) Acidity-induced spectral variation of LHCII in the amide I′ region for α-helices (1,651 cm−1) and 310-helix/loop (1,659 cm−1) at pH 5.4, 7.5 and 9.6. The isosbestic point indicates the acidity-induced interconversion between the two secondary structures, (d) Comparison of the secondary derivative spectra of wild-type (WT) LHCII trimers and S123G mutants at pH 7.4 with a β-DM concentration of 0.03% (w/v) at RT (color online).
3.4. Fast kinetics for the transition to the quenched conformer revealed by T-jump time-resolved IR spectra
T-jump transient IR spectra of LHCII trimers were obtained at two different temperatures to determine the transition kinetics of the local helices formation. Figure 4(a) shows that the transient formation of α-helices at 1,653 cm−1 accompanied by the depletion of other secondary structure elements, and Figure 4(b) and Figure S7(a–d) (together with fitted curves) display the time-resolved kinetics at several selected frequencies, based on the kinetic model described in the caption of Figure S7. A time constant of 1.04 s was obtained as the formation for the nascent α-helices and a constant of 1.24 ms was assigned to the membrane insertion of the lumenal helices (Figure S7(d)). The time-resolved bleaching kinetics for the 310-helix E/loop at 1,660 cm−1 and the anti-parallel β-strands at 1,608 cm−1 in Figure 4(b) are nearly superimposed (1.05 ms for anti-parallel β-strands in Figure S7(a) and 1.03 ms for the 310-helix E/loop in Figure S7(b)) and follow the same time constant for the formation of α-helices (Figure S7(d)), supporting a cooperative conversion of these structural segments. Furthermore, a component bleaching kinetics of the random coil at 1,645 cm−1 also follows the same time constant at 1.04 ms (Figure S7(c)), suggesting a possible cooperative transition from random coils to nascent α-helices.
Figure 4.

Experimental T-jump time-resolved IR spectra and a-helices transition of LHCII trimers from computations, (a) Differential optical absorption (ΔOD) of IR spectra of LHCII trimers at RT from T-jump values (ΔT) of 17 and 15 °C delayed by 8 μs. Positive and negative values indicate population increase and decrease of the corresponding structural elements, (b) Time-resolved spectral diffusion at selected frequencies induced by a T-jump of ΔT=17°C. The overlap in bleaching kinetics at 1,608 and 1,660 cm−1 indicates a cooperative conversion of 310-helix E/loop and antiparallel β-strands to α-helices. Details of kinetics analyses are given in Figure S7. (c, d) The distribution of helical contents of the residue 97–107 of subunit Ml vs. that of the residue 221–229 of the neighboring subunit M2 based on structures obtained from MD trajectories at 280 K (c) and 340 K (d). The helical content characterizes the number of six residue segments of the protein that are in an α-helical configuration, (e) Representative structures of the 310-helix E of Ml and C-terminal residues of M2 (blue) at 280 K. (f) A snapshot structure depicting α-helix transitions (red) at 340 K in the same regions as in (e). Note that the nascent α-helix E has been extended by two residues from Trp97-Phe105 of the initial 310-helix to Trp97-Glu107 (color online).
3.5. Secondary structural changes and hydrogen-bonding switch triggering LHCII trimers from light harvesting to the quenched conformer from MD simulations
The MD trajectories were analyzed to identity structural details revealed spectroscopically in IR experiments. Figure 4(c, d) display the α-helical contents at 280 and 340 K (other temperatures in Figure S11(a, b)) in two key structural regions, including residues 97 to 107 of the 310-helix E, and residues 221 to 229 of the C-terminal coil from a neighboring subunit at monomer contacts of LHCII trimers. In going from low to high temperature, we observed a clear, cooperative formation of nascent α-helical contents in these two structural segments (Figure 4(e, f)). The newly formed α-helix E, expanding by two residues, extrudes towards the anti-parallel β-strands, whereas the C-terminal helix, from the random coil of a neighboring subunit, is connected to helix D.
The cooperative interactions are fully consistent with the known fact that hydrophobic interactions between the helix E and the C-terminal coils of neighboring monomers are critical to the LHCII trimer stability [68], and that a single-site mutation of W222H completely abolishes the trimer formation [69]. We found that the lowest quasiharmonic mode of the lumenal interface, characterized by the first principal component (PC1), corresponding to thermal-induced α-helix E which is inserting into a hydrophobic core involving helix D (Figure 5(a–c)), reducing the distance between helices D and E as the temperature increases (Figure 5(f)). Significantly, at 20 °C, Glu(E)94 forms a salt bridge with Lys(K)99 at an average distance of 3.3 Å (Figure 5(b)), but it is replaced by a hydrogen bond between Glu94 and Gln (Q) 103 at temperatures higher than 45 °C, releasing Lys(K)99 (Figure 5(c, g)). The switch of local hydrogen-bonding interactions of Glu94 from Lys99 to Gln103 represents an initial trigger to induce a global conformation shift in favor of the fluorescence quenching. We noted that once E94 is replaced by G in a 94G mutant, the fluorescence quenching is significantly inhibited with respect to the wild type LHCII [70], while MD simulations by Yang et al. [24] reveal that such a mutation would give rise to a shorter helix E length, which is unfavorable to the fluorescence quenching.
Figure 5.

Representative structures and populations of LHCII trimers at different temperatures revealed by MD simulations. (a) Illustration of PCI modes at the lumenal interface, highlighting the intrusion motion of helix D and E towards each other. The coloring of the backbone from blue to red corresponds to average structures as temperature increases. (b) A snapshot structure from the MD trajectory at 280 K (low T), illustrating key interactions in the close proximity of TM helix A (cyan) and B (purple). One end of helix D (orange) forms a cluster of hydrophobic contacts with the helix B, involving V80, L84, and L206 in the yellow shade, denoted as Lever I. The extended loop of 310-helix E forms a second cluster of hydrophobic contacts, involving W97, FI 94 and F195 (in the yellow shade), specified as Lever II. K99 near the beginning of 310-helix E forms a salt bridge with E94, shown as the stick model, (c) A representative snapshot structure from the MD trajectory at 340 K (high T), depicting structural variations from that shown in (b). The same color scheme as that in (b) is used, superimposing to the structure of (b) in light grey. Noticeable changes are (1) the switch of the hydrogen bond of E94 from K99 in (b) to Q103 in (c), (2) the shifts of both α-helix D and E towards each other, indicated by arrows, and (3) overall tilts of TM helix A and B away from one another, (d, e) Distributions of average distances of Lever I and Lever II, respectively, from MD trajectories obtained at temperatures ranging from 270 to 360 K. Notice the relative similar distributions of each clusters of hydrophobic contacts at different temperatures. The density is given in the number of pairs per bin (0.05 Å) throughout, (f) Distribution of the distance of helix D and E at different temperatures, where the distance is represented by W97-Cα at helix E and P205-Cα at helix D. (g) Contact distance of E94-Q103 at different temperatures. The formation hydrogen bond with Q103 increases significantly as temperature increases, and the existence of hydrogen bonds is more likely attributed to the conformational change and intrusion motions of helix E. (h, i) Distributions of average hydrogen bond distances between Arg70 and Glul80 (h), Glu65 and Argl85 (i) at different temperatures, (j) Correlation of the angle between carotenoid Lut1 and Lut2 with that of TM helix A and B. (k) Distribution of the distance between Lut1 and Chl612, characterized by the minimal distance between Mg atoms of Chl and the C atoms in the conjugated π-system of lutein, at different temperatures (color online).
Analyses of the MD trajectories over the entire temperature range revealed two groups of hydrophobic contacts that are critical for transmitting the force exerted by the thermally induced local helices to remote sites of the LHCII trimer. The first group involves Leu84, Va180 (TM helix B) and Leu206 (helix D), and the second consists of Phe194 and Phe195 (TM helix A) and Trp97 (helix E) (Figure 5(b)). Figure 5(d, e) show that these hydrophobic contacts are stable, having relatively little variations on average; they act as the lever of a mechanical scissor to compress TM helices A and B (Figure 5(c)). Furthermore, the ion pair between Arg70 and Glu180, together with that between Glu65 and Arg185, has an invariant average distance of about 2.7 Å at all temperatures (Figure 5(h, i)), acting as two fulcrums between helices A and B. The persistent interactions between these two conserved residues have been observed in NMR experiments [23]. Consequently, the structural arrangement of LHCII trimers constitutes a molecular mechanical machine. The consequential variations of wedges (helices A and B) of the scissor are quantified in term of the angle between TM helices A and B in Figure 5(j), opening from about 46° to 49° on average as temperature increases. Concomitantly, the angle between Lut1 and Lut2, associated with helices A and B (Figure 5(j), also see Figure S14(e, f)), is expanded by a similar magnitude (a linear correlation slope of 0.96), together with the conformational twist of the xanthyphylls, such as Lut1 in Figure S12(d). Lut2 in Figure S12(e) and Nex in Figure S12(f), which is noticeably twisted at higher temperature, consistent with the correlation of Nex and conformational changes in the quenched state [26]. Such a pigment configuration change is along with the disruption of hydrogen bonding interactions between Lut1 and helix D (Figure S11(c, d)). Importantly, the contact between Lut1 and Chl612 markedly shifts to a shorter average distance (Figure 5(k)), while the average distance between other Lut-Chl pairs is shown in Figure S12(a–c). At elevated temperatures, thermal effect also causes the gradual breaking of hydrogen-bonding interactions, and dehydration at the interface, leading to a more hydrophobic lumenal interface (Figure S8 (b–f)). Both findings are consistent with the X-ray structure of a dehydrated LHCII crystal corresponding to a more fluorescence quenched state [71]. In addition, we have shown that MD simulations corresponding to low pH conditions also show the same structural transitions to α-helices (Figure S13(a, b)) as those of thermal effect which in turn reduces the distance between Chls and Lut1 (Figure S13(d, e)). The hydrogen bond between Q103 and E94, however, reveals an alternative pattern with the protonated E94 as the donor and the carboxyl oxygen of Q103 as the acceptor (Figure S13(f)), underlying the effects of acidification at the lumenal region.
Principal component analyses (PCA) of the MD trajectories provide further insights on the intrinsic motions of LHCII trimers, which are dictated by the three dimensional structures of the proteins. The two lowest modes of TM helices correspond to symmetric and asymmetric motions of the three subunits of the LHCII perpendicular to the membrane plane (Figure S14(a, b)). The first PCA mode is closely related to the evolution of the interface area of the three subunits on the stromal side, from uniform distribution at low temperature to bimodal structures at high temperature (Figure S14(c)), corresponding to the lateral expansion of the trimeric interfaces (Figure S14(d)). Thus, the specific scissoring motions between TM helices A and B noted above are part of the intrinsic, characteristic, and fast global dynamics of LHCII trimers (Figure S14(e, f)); the local secondary structure transition plays the role in the allosterically shifting conformational equilibrium at a slower phenomenological rate (~1 ms), in response to different environmental conditions. The present findings are in good accordance with a recent diffusion-controlled model, in which the regulatory switch of light-harvesting complexes was proposed to result from the coupling of fast protein dynamics to a specific slow coordinate [32].
Finally, our MD simulations suggest that the intrinsic protein dynamics in LHCII monomers is drastically different from LHCII trimers. As characterized by PCA in Figure S15 (a, b), the monomer has a much larger global fluctuation as temperature increases, whereas the monomeric subunit in trimers remains the similar global motions at all the temperatures. In the TM region, although the two salt bridges R70-E180 and E65-R185 in the TM helices remain stable as in the trimer (Figure S15(c, d)), the distance between Chl612 and Lut1 increases as the temperature increases (Figure S15 (e)), indicating the dissociation of the pigments from proteins, being a reverse trend to that in the trimer. In the lumenal region, the helix E reveals a random distribution of helical contents at the elevated temperatures (Figure S15(f)), in contrast to the increased helical formation found in the trimer. More importantly, helix D and E move away from each other (Figure S15(g)), as opposed to the intrusion motion in the trimer. Consequently, there is no hydrogen bond formation between E94 and Q103 in the monomer (Figure S15(h)). All these characteristic features suggest that the isolated monomer primarily expands and dissociates as the temperature increases, whereas the motion of monomeric subunits in the trimer is more restricted, with primary conformational changes taking place at the trimeric interface.
4. Discussion
The involvement of a scissoring motion between two lutein molecules, contributing to the functional switch between light-harvesting and qE has been proposed in the absence of structural informations [59]. The model was later modified to incorporate the pigment activation by conformational changes involving lumenal loops [23]. The present experimental and computational results provided structural details, demonstrating that external conditions such as increased temperature and acidity induce local structure transitions of LHCII trimers, forming two α-helices. In turn, the conformational equilibrium between TM helices A and B allosterically shifts to an open state, leading to close contacts between Lut1 and Chl612 pigments and enhanced electronic couplings. In this mechanism, the LHCII trimer acts as a molecular machine (Figure 6(a, b)): TM helices A and B constitute the wedges of a scissor that pivot around the fulcrum anchored by salt bridges between strictly conserved Arg70 and Glu180 (Figure 5(h)), as well as Glu65 and Arg185 (Figure 5(i)). The levers are connected to helices D and E on the lumenal side of the membrane through two groups of hydrophobic contacts (Figure 5(b, d, e)). Helix contents further extend to the C-terminal segment (Figure S11(a, b) for thermal triggers; Figure S13(c, d) for acid triggers).
Figure 6.

Dynamical allosteric regulation mechanism for light-harvesting and photoprotection of LHCII trimers. (a, b) LHCII trimers act as a molecular machine, in which TM helices constitute the wedges that pivot around the fulcrum anchored by the salt bridge between Arg70 and Glul80, Glu65 and Argl85 (Figure 5(h, i)), and the hydrophobic contacts connecting the helix D and E (Figure 5(b)) are the lever. The signal for photoprotection, the increase in acidity, environmental steady-state temperature together with transient temperature rising due to NPQ or both, induces a local structural transition to convert the 310-helix E and C-terminal loop into two α-helices, which triggers a switch of the hydrogen bond of Glu94 from Lys99 in the light-harvesting state (left) to Gin 103 in the photoprotection state (right) (see Figure 5(b, c)). In turn, α-helix D and E are pulled closer against TM helices A and B, shifting its conformational equilibrium to a wider intercrossing angle. The net effect of the overall conformation change of TM helices is to reduce the contact distance between Lut1 and Chl612, enhancing their electronic coupling in favor of excited energy transfer from excited Chls to the SI dark state of Lut1. ΔT: transient temperature rising; ΔpH: pH gradient (color online).
In the light-harvesting state (Figure 6(a)), the levers of the molecular machine are associated with 310-helix E and C-terminal loop structures, and the relative angle between TM helices A and B is about 46° (Figure 5(J) and Figure S14(e)), corresponding to a relaxed configuration. Under the conditions of increased external acidity and temperatures, this angle will be characteristic of the photoprotection signals. The present IR experiments and MD simulations revealed local conformational transitions of the 310-helix E and C-terminal coil into a pair of α-helices (Figures 3(b) and 4(a, e, f)). Generally, α-helix is one of the most ordered structures with stable properties and high resistance against the stress, and this structural state is the global free-energy minimum structure for the peptide at low temperatures [72]. As temperature increases, atoms become random and disordered, but the hydrophobic environment can provide additional driving forces favoring the formation of helix contents by enhanced van der Waals interactions, as an important determinant contribution to the helix stability [73,74]. In our studies, the nascent α-helix is stabilized by the burial of hydrophobic groups into the hydrophobic core from water, and this structural transition is in competition with the thermal-induced unfolding process, which finally leads to the denaturation of transmembrane helices at extreme high temperatures. The secondary structure change is accompanied by a switch of hydrogen bonds of Asp94 from Lys99 to Gln103 (Figure 5(b, c)), leading to the reduction of the average distances between helices E and D (Figure 5(f)). The latter motions play the role in a lever, compressing TM helices A and B to shift their equilibrium distribution to a more open state with an average angle of from 46° to about 49° (Figure S14(e)), along with the associated Lut1 and Lut2, to a more open, or tense state from 86° to 89° (Figure 5(j) and Figure S14(f)). The conformational change of LHCII trimers at remote sites, induced by local helix formation, greatly affects the distribution of inter-pigment contacts, notably for Lut1-Chl610, Lut2-Chl603 (Figure S12(a, c)), and Lut1-Chl612 (Figure 5(k)), as well as structural bending of the carotenoid Lut1 (Figure S12(d)). The net result is switching to a photoprotective state of LHCII, as reflected by the reduced fluorescence lifetime and intensity from experiments (Figure 1(c, e, f) and Figures S2(c), S3(e), S4(a, b)) and enhanced electronic couplings from the computation (Figure 2). We attribute the key energy dissipation site to the interactions of Chl612 to the S1 dark state of Lut1, consistent with previous proposals [26–32,75]. We noticed a recent work from Croce’s group [76] which further reported that Lut1-Chl612 was a suitable candidate for the non-photochemical quenching of CP29 in physiological conditions, suggesting that this photoprotective strategy could be generalized to other members of the LHC family. Figure 6 presents a unified mechanism for the functional switch under two key environmental variables. We propose that both the acid and thermal effect can act as a trigger for a two-state interconversion [32], whereas the subsequent heat release, as a result of non-radiative energy dissipation, plays a role in transient dynamical enhancers.
Since the temperature-dependent FTIR and fluorescence measurements of LHCII trimers are almost completely reversible when the temperature was elevated to 55 °C and then cooled down to RT with an incubation time of 5 min (Figures S6(e) and S4(c, d)), the transient heat release exerts a reversible dynamical regulation of fluorescence quenching in LHCII trimers, in agreement with CD results [59]. We note that in addition to lutein, other carotenoid pigments may also be an energy acceptor in the binding site such as Vio and Zea, which have stated well below the Qy transitions of chlorophylls in the reconstituted LHCII [75] or considered as similar as other cases [77]. The molecular machine mechanism presented in Figure 6 is consistent with existing experimental observations. The motion of helix D towards the interior of the trimer is consistent with the X-ray structure of a dehydrated LHCII crystal corresponding to the fluorescence quenching state [71]. Further, the present simulation results are also in accordance with the findings from a recent MD simulation of the LHCII monomer, in which helices D and E were found to be the primary and secondary sensitive domains to ΔpH [78]. Moreover, the coupled motions between lumenal helices D and E and TM helices A and B may be dependent on external restrictions to LHCII monomers and trimers [34], especially the effects of the protein aggregation [10]. For an unrestricted monomer or trimer, only a minimal amount of counter-forces may be transmitted to the transmembrane helix motions, leading to the least efficient fluorescence quenching. On the other hand, in restricted case such as in highly aggregated LHCII or in relatively rigid gel media [34], the forces exerted by the local conformation change can more effectively influence the equilibrium of TM helices and thus, an efficient fluorescence quenching. IR intensity thermal titration curves also show that LHCII trimers are more thermally stable in the aggregated states (Figure S5(b–d)), corresponding to a more rigid structure, and this accounts for the observed fact that the temperature-dependent decrease in the fluorescence lifetime and intensity (Figure 1(c, e, f) and Figures S2(c), S3(e) and S4(a, b)) are strongly dependent on the confinement of LHCII molecules in aggregation (Table S1). Apparently aggregation could enhance thermal-induced fluorescence quenching; however higher temperature alone can lead to fluorescence quenching once the aggregation size meets certain minimum value as shown in Figure S1(f). Experimentally, qE is known to correlate with a compressed thylakoid membrane [79], rigidity of the membrane [80], and increased stiffness of the aggregated LHCII within the membrane [81]. Single-molecule unfolding experiments revealed that the inclusion of the non-bilayer lipid MGDG can substantially increase the mechanical stability of LHCII trimers in the membrane and exert the lateral pressure on the LHCII due to the steric matching of conically formed MGDG and the hourglass shape of the trimeric LHCII [82]. Further, a recent work clearly indicates that the presence of MGDG switches the isolated LHCII trimer to a fluorescence quenching state in lipid bilayers [83]. Another consequence of the mechanical model is that the elongation of the scissoring angle between TM helices A and B would lead to a thinner membrane (Figure 6(b)). Indeed, it has been found that chloroplast membranes show as much as 13%–23% contractions in the thickness up on illumination or acid titration in dark [84].
Energy dissipation has been a subject of numerous computational studies. Duffy et al. [85] computed the change in chemical shift of Chls at different distances from Lut, providing insight on the observed 13C NMR difference between the active and qE forms. It was found that the backbone flexibility of Lut1 greatly affects the electronic coupling and excited state lifetime in the protein environment [86], consistent with the present results. Kinetically, the fluorescence quenching of Chls by luteins can be as fast as tens of picoseconds [26]. Thus, the rate-determining step to switch from light-harvesting to a quenching state involves protein motions that bring Chls and Luts in the close contact [31,32,34]. We found that the intrusion of the nascent helix E (and helix D) into the hydrophobic core has a time constant of 1.24 ms (Figure S7(d)), in good accordance with that of the experimental heat release (1.4 ms) in ΔpH-dependent quenching and the transient local temperature would relax to the steady-state temperature within 10 μs [87]. Since the irreversible denaturation of proteins involves the formation of inter-molecularly hydrogen-bonded anti-parallel β-sheet structures, leading to an irreversible re-association of unfolded peptide segments and resulting in gels or aggregates [88], the minimum time for intermolecular diffusion of proteins would be 100 ms [89], and thus a transient temperature rising, such as 14 °C, would not cause further protein denaturation than the steady-state temperature, while the fluorescence quenching efficiency is determined by the transient temperature in NPQ. The elegance of this allosteric mechanism is that the plant uses the relatively lower steady-state temperature to maintain the protein activity while using the higher transient local temperature to achieve significant qE efficiency without bringing further thermal damages to the proteins. As a result, the temperature-dependent fluorescence quenching curves in Figure 1(e, f) and Figure S2(c) can be used as the thermal-induced NPQ working curves at the combined effect of the steady-state temperature of leaves and the local transient temperature rising of the LHCII trimer. By contrast, the reverse process to restore efficient light harvesting is depending on the rate of steady-state temperature and pH recovery, and on the PsbS/zeaxanthin regulated dis-assembling of the aggregated LHCII [1,10,90]. Mutations that accelerate these processes have been demonstrated to increase photosynthesis efficiency with increased biomass production [12]. Physical means such as higher water evaporation rates to decrease the steady-state temperature would also be effective.
5. Conclusions
Green plants must cope with rapid, natural changes of light intensity in photosynthesis to prevent photodamage under full sun light, and the key component of this highly complex and cooperative process is fast energy quenching involving the major II LHCII. Based on our results, we suggest a unified mechanism by which LHCII trimers in higher plants control the dissipation of excess excited energy in response to the increased temperature and acidity under intense sun light irradiation. Explicitly, the LHCII trimer regulates its allosteric conformation as a molecular machine, with the forces arising from local structural transitions of the 310-helix E and C-terminal coil into a pair of α-helices induced by heat and acidity, pushing helix E and D into the hydrophobic core within 1.24 ms. As parts of this molecular machine, we further identified two pairs of the key conserved salt bridges (Arg70-Glu180 and Glu65-Arg185) as the fulcrum and two groups of hydrophobic contacts as helix E-B and D-A at lumenal sides, respectively, as the lever. The forces exerting on the lumenal ends of the crossed transmembrane helix pair A and B propagated to pivot scissoring motions of these two helices. Finally, such an allosteric effect leads to a decrease in the distance between the lutein-Chl pairs, especially the major Lut1-Chl612 pair, and a substantial increase in the electronic coupling between Lut1 and Chl612 pair, giving rise to an efficient quenching of the Chl612 excited state as the major pathway for the dissipation of excess excited energy. Importantly, the proposed molecular machine mechanism is consistent with existing experimental observations. In short, our study sheds light on the underlying structure-dynamics-function relationship of LHCII trimers at the atomic level.
Supplementary Material
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21433014, 11721404), the Ministry of Science and Technology (2017YFB0203400), Chinese Academy of Sciences Innovation Program (KJCX2-YW-W25) and the National Institutes of Health (GM46736, GM64742). Y.W. thanks Prof Tingyun Kuang for encouragement, Prof Xinguang Zhu for in-depth discussion, Prof Chunhong Yang for the single-site mutant S123G sample, and Miss Ju Wang and Prof Shufeng Wang from Peking University for the help in streak camera measurement.
Footnotes
Conflict of interest The authors declare no conflict of interest.
Supporting information The supporting information is available online at http://chem.scichina.com and http://link.springer.com/joumal/11426. The supporting materials are published as submitted, without typesetting or editing. The responsibility for scientific accuracy and content remains entirely with the authors.
References
- 1.Demmig-Adams B, Garab G, Adams W III, Govindjee. Non-photochemical quenching and energy dissipation in plants, algae and cyanobacteria. In: Govindjee, Sharkey TD, Eds. Advances in Photosynthesis and Respiration Including Bioenergy and Related Processes. Vol 40 The Netherlands: Springer Science & Business Media, 2014 [Google Scholar]
- 2.Rochaix JD. Annu Rev Plant Biol, 2014, 65: 287–309 [DOI] [PubMed] [Google Scholar]
- 3.Ruban AV. FEBS Lett, 2018, 592: 3030–3039 [DOI] [PubMed] [Google Scholar]
- 4.Müller P, Li XP, Niyogi KK. Plant Physiol, 2001, 125: 1558–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicol L, Nawrocki WJ, Croce R. Nat Plants, 2019, 5: 1177–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Croce R, van Amerongen H. Nat Chem Biol, 2014, 10: 492–501 [DOI] [PubMed] [Google Scholar]
- 7.Külheim C, Agren J, Jansson S. Science, 2002, 297: 91–93 [DOI] [PubMed] [Google Scholar]
- 8.Tian L, Nawrocki WJ, Liu X, Polukhina I, van Stokkum IHM, Croce R. Proc Natl Acad Sci USA, 2019, 116: 8320–8325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miloslavina Y, Wehner A, Lambrev PH, Wientjes E, Reus M, Garab G, Croce R, Holzwarth AR. FEBS Lett, 2008, 582: 3625–3631 [DOI] [PubMed] [Google Scholar]
- 10.Correa-Galvis V, Poschmann G, Melzer M, Stühler K, Jahns P. Nat Plants, 2016, 2: 15225. [DOI] [PubMed] [Google Scholar]
- 11.Holzwarth AR, Miloslavina Y, Nilkens M, Jahns P. Chem Phys Lett, 2009, 483: 262–267 [Google Scholar]
- 12.Kromdijk J, Głowacka K, Leonelli L, Gabilly ST, Iwai M, Niyogi KK, Long SP. Science, 2016, 354: 857–861 [DOI] [PubMed] [Google Scholar]
- 13.Ort DR, Merchant SS, Alric J, Barkan A, Blankenship RE, Bock R, Croce R, Hanson MR, Hibberd JM, Long SP, Moore TA, Moroney J, Niyogi KK, Parry MAJ, Peralta-Yahya PP, Prince RC, Redding KE, Spalding MH, van Wijk KJ, Vermaas WFJ, von Caemmerer S, Weber APM, Yeates TO, Yuan JS, Zhu XG. Proc Natl Acad Sci USA, 2015, 112: 8529–8536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engel GS, Calhoun TR, Read EL, Ahn TK, Mancal T, Cheng YC, Blankenship RE, Fleming GR. Nature, 2007, 446: 782–786 [DOI] [PubMed] [Google Scholar]
- 15.Arsenault EA, Yoneda Y, Iwai M, Niyogi KK, Fleming GR. Nat Commun, 2020, 11: 1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinnola A, Bassi R. Biochem Soc Trans, 2018, 46: 467–482 [DOI] [PubMed] [Google Scholar]
- 17.Ruban AV, Johnson MP, Duffy CDP. Biochim Biophysica Acta (BBA), 2012, 1817: 167–181 [DOI] [PubMed] [Google Scholar]
- 18.Liu Z, Yan H, Wang K, Kuang T, Zhang J, Gui L, An X, Chang W. Nature, 2004, 428: 287–292 [DOI] [PubMed] [Google Scholar]
- 19.Kühlbrandt W, Wang DN, Fujiyoshi Y. Nature, 1994, 367: 614–621 [DOI] [PubMed] [Google Scholar]
- 20.Standfuss J, Terwisscha van Scheltinga AC, Lamborghini M, Kühlbrandt W. EMBO J, 2005, 24: 919–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caffarri S, Kouril R, Kereïche S, Boekema EJ, Croce R. EMBO J, 2009, 28: 3052–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei X, Su X, Cao P, Liu X, Chang W, Li M, Zhang X, Liu Z. Nature, 2016, 534: 69–74 [DOI] [PubMed] [Google Scholar]
- 23.Sunku K, de Groot HJM, Pandit A. J Biol Chem, 2013, 288: 19796–19804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang C, Lambrev P, Chen Z, Jávorfi T, Kiss AZ, Paulsen H, Garab G. Biochim Biophysica Acta (BBA), 2008, 1777: 1463–1470 [DOI] [PubMed] [Google Scholar]
- 25.Pascal AA, Liu Z, Broess K, van Oort B, van Amerongen H, Wang C, Horton P, Robert B, Chang W, Ruban A. Nature, 2005, 436: 134–137 [DOI] [PubMed] [Google Scholar]
- 26.Ruban AV, Berera R, Ilioaia C, van Stokkum IHM, Kennis JTM, Pascal AA, van Amerongen H, Robert B, Horton P, van Grondelle R. Nature, 2007, 450: 575–578 [DOI] [PubMed] [Google Scholar]
- 27.Bode S, Quentmeier CC, Liao PN, Barros T, Walla PJ. Chem Phys Lett, 2008, 450: 379–385 [Google Scholar]
- 28.Liao PN, Bode S, Wilk L, Hafi N, Walla PJ. Chem Phys, 2010, 373: 50–55 [Google Scholar]
- 29.Liao PN, Pillai S, Gust D, Moore TA, Moore AL, Walla PJ. J Phys Chem A, 2011, 115: 4082–4091 [DOI] [PubMed] [Google Scholar]
- 30.Holleboom CP, Walla PJ. Photosynth Res, 2014, 119: 215–221 [DOI] [PubMed] [Google Scholar]
- 31.Bode S, Quentmeier CC, Liao PN, Hafi N, Barros T, Wilk L, Bittner F, Walla PJ. Proc Natl Acad Sci USA, 2009, 106: 12311–12316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valkunas L, Chmeliov J, Krüger TPJ, Ilioaia C, van Grondelle R. J Phys Chem Lett, 2012, 3: 2779–2784 [Google Scholar]
- 33.Daskalakis V, Maity S, Hart CL, Stergiannakos T, Duffy CDP, Kleinekathöfer U. J Phys Chem B, 2019, 123: 9609–9615 [DOI] [PubMed] [Google Scholar]
- 34.Ilioaia C, Johnson MP, Horton P, Ruban AV. J Biol Chem, 2008, 283: 29505–29512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlau-Cohen GS, Yang HY, Krüger TPJ, Xu P, Gwizdala M, van Grondelle R, Croce R, Moerner WE. J Phys Chem Lett, 2015, 6: 860–867 [DOI] [PubMed] [Google Scholar]
- 36.Tang Y, Wen X, Lu Q, Yang Z, Cheng Z, Lu C. Plant Physiol, 2007, 143: 629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holt NE, Zigmantas D, Valkunas L, Li XP, Niyogi KK, Fleming GR. Science, 2005, 307: 433–436 [DOI] [PubMed] [Google Scholar]
- 38.Ahn TK, Avenson TJ, Ballottari M, Cheng YC, Niyogi KK, Bassi R, Fleming GR. Science, 2008, 320: 794–797 [DOI] [PubMed] [Google Scholar]
- 39.Amarie S, Wilk L, Barros T, Kühlbrandt W, Dreuw A, Wachtveitl J. Biochim Biophysica Acta (BBA), 2009, 1787: 747–752 [DOI] [PubMed] [Google Scholar]
- 40.Park S, Steen CJ, Fischer AL, Fleming GR. Photosynth Res, 2019, 141: 367–376 [DOI] [PubMed] [Google Scholar]
- 41.Avenson TJ, Ahn TK, Zigmantas D, Niyogi KK, Li Z, Ballottari M, Bassi R, Fleming GR. J Biol Chem, 2008, 283: 3550–3558 [DOI] [PubMed] [Google Scholar]
- 42.Miloslavina Y, de Bianchi S, Dall’Osto L, Bassi R, Holzwarth AR. J Biol Chem, 2011, 286: 36830–36840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nilkens M, Kress E, Lambrev P, Miloslavina Y, Müller M, Holzwarth AR, Jahns P. Biochim Biophysica Acta (BBA), 2010, 1797: 466–475 [DOI] [PubMed] [Google Scholar]
- 44.Ansari AQ, Loomis WE. Am J Bot, 1959, 46: 713–717 [Google Scholar]
- 45.Pereira AB, Villa Nova NA, Galvani E. Biosyst Eng, 2003, 86: 27–34 [Google Scholar]
- 46.Ruban AV, Pesaresi P, Wacker U, Irrgang KDJ, Bassi R, Horton P. Biochemistry, 1998, 37: 11586–11591 [DOI] [PubMed] [Google Scholar]
- 47.Zhou R, Chen W, Jiang X, Wang S, Gong Q. Appl Phys Lett, 2010, 96: 133309 [Google Scholar]
- 48.Li D, Li Y, Li H, Wu X, Yu Q, Weng Y. Rev Sci Instrum, 2015, 86: 053105. [DOI] [PubMed] [Google Scholar]
- 49.Wu EL, Cheng X, Jo S, Rui H, Song KC, Dávila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, Klauda JB, Im W. J Comput Chem, 2014, 35: 1997–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys, 1983, 79: 926–935 [Google Scholar]
- 51.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. J Comput Chem, 2003, 24: 1999–2012 [DOI] [PubMed] [Google Scholar]
- 52.Zhang L, Silva DA, Yan YJ, Huang X. J Comput Chem, 2012, 33: 1969–1980 [DOI] [PubMed] [Google Scholar]
- 53.Wang JM, Wang W, Kollman PA. Abstr Am Chem Soc, 2001, 222: U403 [Google Scholar]
- 54.Hess B, Kutzner C, van der Spoel D, Lindahl E. J Chem Theor Comput, 2008, 4: 435–447 [DOI] [PubMed] [Google Scholar]
- 55.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. J Comput Chem, 1997, 18: 1463–1472 [Google Scholar]
- 56.Darden T, York D, Pedersen L. J Chem Phys, 1993, 98: 10089–10092 [Google Scholar]
- 57.Parrinello M, Rahman A. Phys Rev Lett, 1980, 45: 1196–1199 [Google Scholar]
- 58.Bowman GR, Ensign DL, Pande VS. J Chem Theory Comput, 2010, 6: 787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wentworth M, Ruban AV, Horton P. J Biol Chem, 2003, 278: 21845–21850 [DOI] [PubMed] [Google Scholar]
- 60.van Oort B, van Hoek A, Ruban AV, van Amerongen H. J Phys Chem B, 2007, 111: 7631–7637 [DOI] [PubMed] [Google Scholar]
- 61.Santabarbara S, Horton P, Ruban AV. Biophys J, 2009, 97: 1188–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krause GH. Photosynthetica, 1992, 27: 249–252 [Google Scholar]
- 63.Ren H, Provorse MR, Bao P, Qu Z, Gao J. J Phys Chem Lett, 2016, 7: 2286–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gao J, Grofe A, Ren H, Bao P. J Phys Chem Lett, 2016, 7: 5143–5149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dockter C, Müller AH, Dietz C, Volkov A, Polyhach Y, Jeschke G, Paulsen H. J Biol Chem, 2012, 287: 2915–2925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y, Li B, Xu Y, Li H, Li S, Zhang D, Mao Z, Guo S, Yang C, Weng Y, Chong K. Plant Cell, 2013, 25: 2504–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li S, Wang R, Li D, Ma J, Li H, He X, Chang Z, Weng Y. Sci Rep, 2014, 4: 4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu C, Rao Y, Zhang L, Yang C. J Biochem, 2014, 156: 203–210 [DOI] [PubMed] [Google Scholar]
- 69.Kuttkat A, Hartmann A, Hobe S, Paulsen H. Eur J Biochem, 1996, 242: 288–292 [DOI] [PubMed] [Google Scholar]
- 70.Belgio E, Duffy CDP, Ruban AV. Phys Chem Chem Phys, 2013, 15: 12253–12261 [DOI] [PubMed] [Google Scholar]
- 71.Yan H, Zhang P, Wang C, Liu Z, Chang W. Biochem Biophys Res Commun, 2007, 355: 457–463 [DOI] [PubMed] [Google Scholar]
- 72.Jas GS, Kuczera K. Biophys J, 2004, 87: 3786–3798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Seelig J, Schönfeld HJ. Quart Rev Biophys, 2016, 49: e9. [DOI] [PubMed] [Google Scholar]
- 74.Marqusee S, Robbins VH, Baldwin RL. Proc Natl Acad Sci USA, 1989, 86: 5286–5290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Polívka T, Zigmantas D, Sundström V, Formaggio E, Cinque G, Bassi R. Biochemistry, 2002, 41: 439–450 [DOI] [PubMed] [Google Scholar]
- 76.Mascoli V, Liguori N, Xu P, Roy LM, van Stokkum IHM, Croce R. Chem, 2019, 5: 2900–2912 [Google Scholar]
- 77.Wehling A, Walla PJ. Photosynth Res, 2006, 90: 101–110 [DOI] [PubMed] [Google Scholar]
- 78.Papadatos S, Charalambous AC, Daskalakis V. Sci Rep, 2017, 7: 2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnson MP, Brain APR, Ruban AV. Plant Signal Behav, 2011, 6: 1386–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tardy F, Havaux M. Biochim Biophysica Acta (BBA), 1997, 1330: 179–193 [DOI] [PubMed] [Google Scholar]
- 81.Janik E, Bednarska J, Zubik M, Puzio M, Luchowski R, Grudzinski W, Mazur R, Garstka M, Maksymiec W, Kulik A, Dietler G, Gruszecki WI. Plant Cell, 2013, 25: 2155–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Seiwert D, Witt H, Janshoff A, Paulsen H. Sci Rep, 2017, 7: 5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tietz S, Leuenberger M, Höhner R, Olson AH, Fleming GR, Kirchhoff H. J Biol Chem, 2020, 295: 1857–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Murakami S, Packer L. J Cell Biol, 1970, 47: 332–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duffy CDP, Pandit A, Ruban AV. Phys Chem Chem Phys, 2014, 16: 5571–5580 [DOI] [PubMed] [Google Scholar]
- 86.Balevičius V Jr., Fox KF, Bricker WP, Jurinovich S, Prandi IG, Mennucci B, Duffy CDP. Sci Rep, 2017, 7: 13956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mullineaux CW, Ruban AV, Horton P. Biochim Biophysica Acta (BBA), 1994, 1185: 119–123 [Google Scholar]
- 88.Clark AH, Saunderson DHP, Suggett A. Int J Peptide Protein Res, 1981, 17: 353–364 [DOI] [PubMed] [Google Scholar]
- 89.Neuweiler H, Johnson CM, Fersht AR. Proc Natl Acad Sci USA, 2009, 106: 18569–18574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dall’Osto L, Cazzaniga S, Bressan M, Paleček D, Židek K, Niyogi KK, Fleming GR, Zigmantas D, Bassi R. Nat Plants, 2017, 3: 17033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.