

Visual Abstract

Keywords: acute kidney injury and ICU nephrology, ACE inhibitors, acute renal failure, angiotensin, chronic kidney disease, diuretics, drug interactions, drug nephrotoxicity, glomerular filtration rate, hospitalization, nephrotoxicity
Abstract
Background
Nonsteroidal anti-inflammatory drugs (NSAIDS) are increasingly important alternatives to opioids for analgesia during hospitalization as health systems implement opioid-minimization initiatives. Increasing NSAID use may increase AKI rates, particularly in patients with predisposing risk factors. Inconclusive data in outpatient populations suggests that NSAID nephrotoxicity is magnified by renin-angiotensin system inhibitors (RAS-I). No studies have tested this in hospitalized patients.
Methods
Retrospective, active-comparator cohort study of patients admitted to four hospitals in Philadelphia, Pennsylvania. To minimize confounding by indication, NSAIDs were compared to oxycodone, and RAS-I were compared to amlodipine. We tested synergistic NSAID+RAS-I nephrotoxicity by comparing the difference in AKI rate between NSAID versus oxycodone in patients treated with RAS-I to the difference in AKI rate between NSAID versus oxycodone in patients treated with amlodipine. In a secondary analysis, we restricted the cohort to patients with baseline diuretic treatment. AKI rates were adjusted for 71 baseline characteristics with inverse probability of treatment-weighted Poisson regression.
Results
The analysis included 25,571 patients who received a median of 2.4 days of analgesia. The overall AKI rate was 23.6 per 1000 days. The rate difference (RD) for NSAID versus oxycodone in patients treated with amlodipine was 4.1 per 1000 days (95% CI, −2.8 to 11.1), and the rate difference for NSAID versus oxycodone in patients treated with RAS-I was 5.9 per 1000 days (95% CI, 1.9 to 10.1), resulting in a nonsignificant interaction estimate: 1.85 excess AKI events per 1000 days (95% CI, −6.23 to 9.92). Analysis in patients treated with diuretics produced a higher, albeit nonsignificant, interaction estimate: 9.89 excess AKI events per 1000 days (95% CI, −5.04 to 24.83).
Conclusions
Synergistic nephrotoxicity was not observed with short-term NSAID+RAS-I treatment in the absence of concomitant diuretics, suggesting that RAS-I treatment may not be a reason to choose opioids in lieu of NSAIDs in this population. Synergistic nephrotoxicity cannot be ruled out in patients treated with diuretics.
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are essential options for analgesia in patients who are hospitalized (1–3). They have similar efficacy compared with opioids for multiple indications (1,4–6), and are recommended as first-line treatment for mild to moderate pain (1–3). Moreover, NSAID use during hospitalization is increasing as health systems across the United States implement opioid-minimization interventions (7–10), efforts encouraged by the US Surgeon General and analgesia guideline organizations (1,2,11). A recent national survey of 81 hospitals showed that 98% had implemented such measures, with the third most common intervention being the increased use of NSAIDs and other nonopioids (7).
Increasing NSAID use in patients who are hospitalized may increase AKI rates, particularly in patients with predisposing risk factors (12–14). Minimizing NSAID-AKI risk requires avoidance of nephrotoxicity risk factors, especially modifiable factors such as drug-drug interactions. Inconclusive evidence in outpatient populations suggests that NSAID nephrotoxicity is magnified by renin-angiotensin system inhibitors (RAS-I) (13–17). NSAIDs can vasoconstrict the afferent glomerular arterioles (12), whereas RAS-I vasodilate the efferent arterioles (18). The combined effects are postulated to produce synergistic reductions in transglomerular pressure that compromise glomerular filtration (19). Some studies suggest that NSAID-AKI risk is doubled by RAS-I treatment (16,17), whereas others show no signal of synergistic nephrotoxicity (13,14,15).
Additional uncertainty stems from whether outpatient studies can be extrapolated to the inpatient setting, where patients have a higher baseline AKI risk, but also tend to receive much shorter durations of combined therapy. This represents a critical knowledge gap: if NSAID+RAS-I is truly nephrotoxic in this setting, the combination may contribute substantially to AKI and associated downstream effects including CKD (20); if not, withholding such therapy may unnecessarily expose patients to opioids, with corresponding risks of adverse drug events (21) and long-term opioid use and abuse syndromes (22,23). The potential effect on global kidney disease burden is substantial, given that approximately 15–20 million patients receive analgesia during hospitalization each year (21,24), and RAS-I are the most commonly used antihypertensives (25). We thus aimed to determine whether NSAID+RAS-I treatment has synergistic effects on AKI rates in patients who are hospitalized.
Materials and Methods
Overview of Study Design
We conducted a retrospective cohort study of patients who are hospitalized that compared AKI rate in patients treated with NSAID+RAS-I versus control patients who received comparator drugs lacking effects on kidney function (26). Oxycodone was the comparator for NSAIDs because it is a commonly used, non-nephrotoxic analgesic (21). Amlodipine was the active comparator for RAS-I because it has similar indications compared with RAS-I (27), and it does not vasodilate the efferent arterioles (28). We tested synergistic interaction by comparing the difference in AKI rate between NSAID versus oxycodone in patients treated with RAS-I to the difference in AKI rate between NSAID versus oxycodone in patients treated with amlodipine. We estimated interaction on the multiplicative (ratio of rate ratios) and additive (difference-in-differences) scales (29). The University of Pennsylvania Institutional Review Board approved the study.
Source Population
The study population was drawn from patients admitted to one of four hospitals within the University of Pennsylvania Health System (UPHS) from January 1, 2004 to June 30, 2017. Inclusion criteria were age ≥18 years and at least 24 hours of concomitant treatment with a drug pair of interest. Exclusion criteria were the presence of relative contraindications to NSAIDs (baseline platelet count <100×1011 cells/L; ESKD, RRT, or nonresolved AKI within 2 weeks before cohort entry; or baseline creatinine >2 mg/dl), relative contraindications to RAS-I treatment (pregnancy), lack of a baseline or at least one follow-up creatinine, and a history of solid organ transplant. In patients with multiple eligible episodes, only the first was included.
Drug-Drug Interaction Cohort (Cohort A)
We identified patients who received ≥24 hours of either a RAS-I or amlodipine. Eligible RAS-I were angiotensin-converting enzyme inhibitors (lisinopril, enalapril, ramipril, benazepril, quinapril, and captopril) and angiotensin II receptor blockers (losartan, irbesartan, and valsartan). Within this cohort, we identified patients who received ≥24 hours of NSAIDs or oxycodone treatment. Eligible NSAIDs were ibuprofen, ketorolac, indomethacin, naproxen, and nabumetone. Follow-up began with the first dose of concomitant exposure (i.e., the index date) and continued until a lapse in concomitant treatment >48 hours, occurrence of AKI, hospital discharge, death, or at 14 days after the index date. The specific NSAIDs and RAS-I studied were selected based on UPHS formulary availability. Detailed exposure definitions can be found in the Supplemental Methods.
Outcomes
The primary outcome was AKI at 14 days of follow-up, defined using Kidney Disease Improving Global Outcomes (KDIGO) creatinine and dialysis criteria (30). Baseline creatinine was defined as the last value before the index date. We staged AKI over 7 days after AKI onset. Secondary outcomes included KDIGO-defined AKI severity stage and AKI duration, defined as the number of days required for creatinine to return to within 25% of baseline (31). AKI duration was categorized as short (≤2 days), medium (3–6 days), and long (≥7 days) (31). Patients who required RRT were considered long duration regardless of creatinine resolution.
Data Collection
Data were obtained from Penn Data Store (PDS), Penn Medicine’s electronic health record (EHR) data warehouse. PDS includes medication administration records, laboratory values, demographics, location of care records, and diagnosis codes. PDS data on EHR procedure orders were used to define RRT episodes.
Potential confounders were selected a priori based on clinical knowledge and prior literature, with an emphasis on those associated with AKI or severity of illness (Table 1). Comorbidities were ascertained from diagnosis codes (see Supplemental Table 1). Pre-exposure AKI was defined by applying KDIGO criteria from hospital admission up to the index date (see Supplemental Methods for details). Baseline medication and laboratory variables were assessed during the 72 hours before the index date. For each laboratory measure, the value most proximate to cohort entry was collected. Baseline eGFR was calculated using the CKD Epidemiology Collaboration equation (32).
Table 1.
Baseline characteristics in cohort B (the primary analysis cohort) after inverse probability of treatment weighting
| Characteristics | RAS Cohort | Amlodipine Cohort | ||||
| NSAID, n=4034a (6249b) | Oxycodone, n=16,110a (7127b) | SMD | NSAID, n=1181a (5782b) | Oxycodone, n=4700a (6413b) | SMD | |
| Demographics | ||||||
| Age, years, mean | 62.8 | 63.1 | 0.020 | 63.8 | 63.6 | 0.012 |
| Female sex, % | 49 | 48 | 0.016 | 50 | 49 | 0.023 |
| Race, % | ||||||
| White | 54 | 54 | 0.009 | 53 | 52 | 0.019 |
| Black | 37 | 37 | 0.005 | 39 | 3 | 0.003 |
| Other/unknown | 9 | 10 | 0.007 | 8 | 9 | 0.039 |
| BMI in kg/m2, mean | 31.1 | 31.1 | 0.003 | 30.7 | 30.9 | 0.016 |
| Year, mean | 2011 | 2011 | 0.018 | 2011 | 2011 | 0.001 |
| Hospital admission characteristics | ||||||
| Center, % | ||||||
| CCH | 0.8 | 0.8 | 0.000 | 0.8 | 0.6 | 0.025 |
| HUP | 46 | 47 | 0.015 | 46 | 47 | 0.020 |
| PAH | 25 | 25 | 0.003 | 25 | 27 | 0.049 |
| PMC | 29 | 28 | 0.020 | 29 | 26 | 0.065 |
| Surgical admission, % | 61 | 62 | 0.015 | 62 | 61 | 0.016 |
| Location of initial presentation, % | ||||||
| ED | 31 | 30 | 0.018 | 32 | 30 | 0.048 |
| ICU | 8 | 7 | 0.028 | 7 | 7 | 0.005 |
| OR | 27 | 27 | 0.001 | 28 | 28 | 0.006 |
| Floor | 28 | 29 | 0.008 | 28 | 29 | 0.023 |
| Other | 6 | 7 | 0.052 | 6 | 6 | 0.034 |
| LOS before index in days, mean | 2.9 | 2.9 | 0.005 | 2.8 | 2.9 | 0.021 |
| ICU care at index date, % | 16 | 16 | 0.013 | 16 | 16 | 0.002 |
| Perioperative recency, % | ||||||
| Not in perioperative period | 74 | 73 | 0.022 | 71 | 72 | 0.008 |
| POD zero | 2 | 2 | 0.014 | 2 | 2 | 0.012 |
| POD one | 13 | 13 | 0.015 | 15 | 14 | 0.026 |
| POD two | 8 | 9 | 0.028 | 8 | 10 | 0.053 |
| POD three | 4 | 3 | 0.006 | 4 | 4 | 0.032 |
| Mechanical ventilation, % | 3 | 4 | 0.039 | 4 | 3 | 0.034 |
| Comorbidities, % | ||||||
| Heart failure | 24 | 23 | 0.011 | 22 | 21 | 0.013 |
| Myocardial infarction | 14 | 14 | 0.002 | 13 | 13 | 0.015 |
| Hypertension | 90 | 90 | 0.003 | 92 | 91 | 0.042 |
| Cardiac arrhythmias | 20 | 19 | 0.010 | 17 | 17 | 0.009 |
| Atrial fibrillation | 16 | 16 | 0.001 | 15 | 14 | 0.028 |
| Valvular disease | 16 | 15 | 0.008 | 15 | 15 | 0.013 |
| Stroke | 10 | 10 | 0.005 | 9 | 11 | 0.068 |
| Peripheral vascular disease | 14 | 15 | 0.025 | 14 | 16 | 0.061 |
| Pulmonary circulation disorder | 9 | 9 | 0.008 | 9 | 7 | 0.044 |
| Chronic pulmonary disease | 28 | 27 | 0.028 | 29 | 27 | 0.044 |
| Liver disease | 5 | 5 | 0.001 | 4 | 5 | 0.045 |
| Diabetes mellitus | ||||||
| None | 65 | 65 | 0.003 | 67 | 67 | 0.001 |
| Noncomplicated | 28 | 28 | 0.009 | 26 | 26 | 0.013 |
| Complicated | 8 | 8 | 0.021 | 7 | 8 | 0.021 |
| CKD | 10 | 11 | 0.038 | 13 | 11 | 0.058 |
| Weight loss | 6 | 6 | 0.002 | 7 | 7 | 0.005 |
| Fluid and electrolyte disorder | 26 | 26 | 0.009 | 28 | 27 | 0.040 |
| Cancer | ||||||
| None | 82 | 82 | 0.011 | 81 | 81 | 0.013 |
| Nonmetastatic | 12 | 12 | 0.003 | 13 | 13 | 0.016 |
| Metastatic | 6 | 6 | 0.015 | 6 | 6 | 0.002 |
| Obstructive sleep apnea | 15 | 14 | 0.025 | 12 | 14 | 0.075 |
| HIV/AIDS | 1 | 1 | 0.013 | 1 | 1 | 0.013 |
| Kidney function | ||||||
| GFR, ml/min per 1.73 m2, mean | 74.9 | 74.5 | 0.021 | 74.4 | 74.8 | 0.017 |
| Prior AKI, % | 10 | 10 | 0.008 | 9 | 9 | 0.002 |
| Laboratory values, mean | ||||||
| WBC, ×108 cells/L | 9.9 | 9.9 | 0.014 | 9.8 | 10.0 | 0.029 |
| Hemoglobin, g/dl | 11.2 | 11.1 | 0.037 | 11.1 | 11.1 | 0.029 |
| Platelets, ×1011 cells/L | 235.8 | 236.7 | 0.010 | 233.1 | 237.0 | 0.039 |
| Chloride, mEq/L | 103.8 | 103.7 | 0.004 | 103.9 | 103.7 | 0.039 |
| Potassium, mEq/L | 4.1 | 4.1 | 0.001 | 4.1 | 4.1 | 0.040 |
| Medications, % | ||||||
| Selective β1-blockers | 41 | 42 | 0.013 | 44 | 41 | 0.052 |
| Combined α- and β-blockers | 12 | 12 | 0.007 | 10 | 10 | 0.005 |
| Loop diuretics | 26 | 25 | 0.016 | 23 | 24 | 0.009 |
| Thiazide diuretics | 16 | 16 | 0.010 | 15 | 16 | 0.041 |
| Hydralazine | 8 | 9 | 0.037 | 10 | 10 | 0.017 |
| Other antihypertensivesc | 7 | 8 | 0.029 | 8 | 9 | 0.025 |
| Acid suppressants | ||||||
| None | 39 | 40 | 0.006 | 39 | 38 | 0.008 |
| H2RA | 25 | 25 | 0.004 | 26 | 27 | 0.012 |
| PPI | 36 | 35 | 0.009 | 35 | 35 | 0.003 |
| Broad spectrum antibioticsd | 12 | 12 | 0.008 | 10 | 13 | 0.074 |
| Narrow spectrum antibioticse | 40 | 41 | 0.026 | 44 | 42 | 0.038 |
| Vancomycin | 22 | 22 | 0.018 | 20 | 21 | 0.027 |
| Sulfamethoxazole/trimethoprim | 3 | 3 | 0.026 | 2 | 3 | 0.066 |
| Other nephrotoxic antibioticsf | 3 | 3 | 0.017 | 4 | 3 | 0.006 |
| Other nephrotoxinsg | 2 | 2 | 0.032 | 2 | 2 | 0.030 |
| Vasopressors | 4 | 4 | 0.025 | 4 | 4 | 0.002 |
RAS, renin-angiotensin system inhibitor; NSAID, nonsteroidal anti-inflammatory drug; SMD, absolute standardized mean difference; BMI, body mass index; CCH, Chester County Hospital; HUP, Hospital of the University of Pennsylvania; PAH, Pennsylvania Hospital; PMC, Presbyterian Medical Center; ED, emergency department; ICU, intensive care unit; OR, operating room; LOS, length of stay; POD, postoperative day; WBC, white blood cells; H2RA, histamine-2 receptor antagonist; PPI, proton pump inhibitor.
Actual sample size.
Effective sample size after inverse probability of treatment weighting.
Propranolol, clonidine, doxazosin, terazosin.
Carbapenems, cefepime, piperacillin-tazobactam, fluoroquinolones, aztreonam.
First- and second-generation cephalosporins, macrolides, amoxicillin, penicillin, tetracycline, nitrofurantoin, ampicillin-sulbactam.
Aminoglycosides (amikacin, gentamicin, tobramycin), colistin.
Carboplatin, cisplatin, ifosfamide, cyclosporine, tacrolimus, methotrexate, amphotericin, acyclovir.
Data Analysis
Descriptive Statistics.
We examined baseline covariate balance with standardized mean differences (SMD) (33). We examined balance across all possible contrasts between the four groups, because estimation of a causal synergistic interaction requires balanced covariates across all possible contrasts (34). Absolute SMD values >0.1 were considered meaningful imbalance (35).
Inverse Probability of Treatment Weighting Analysis.
We adjusted for covariates listed in Table 1 using inverse probability of treatment weighting (IPTW) analysis (36,37). Weights were formulated to estimate an average treatment effect (36), and were calculated from multinomial propensity scores estimated in the full cohort (cohort A). (36). Multinomial propensity scores extend standard propensity score methods to multiple treatment groups. The key difference is that there are multiple propensity scores estimated (one for each treatment group). Multinomial propensity scores were estimated using generalized boosted models (36). Generalized boosted models are machine-learning classifiers that select the propensity score model that minimizes covariate imbalance across all treatment groups (36). Covariates with absolute SMD>0.1 after IPTW were included in the outcome models (38).
Propensity Score Trimming.
The primary IPTW analysis was restricted to the subset of patients with comparable propensity scores (cohort B) (39). This restriction was accomplished by trimming the areas of nonoverlap in the propensity score distribution for each treatment category. Trimming methods are described in the Supplemental Methods, and the multinomial propensity score distributions and restriction bounds for cohort B are shown in Supplemental Figure 1.
In a sensitivity analysis, all models were repeated after restricting to the subset of patients with propensity scores between the first and 99th percentiles of the overlapping multinomial propensity score distributions (cohort C; see Supplemental Figure 1 for restriction bounds). The rationale is to remove patients who were treated contrary to prediction, because these patients may be the most likely to have unmeasured confounders (39,40).
Outcome Models.
AKI rate was estimated with IPTW Poisson regression using robust variance estimation. Interaction on the ratio scale was estimated from regression model interaction terms between NSAID and RAS-I (29). Interaction on the difference scale was determined by contrasting the predicted marginal AKI rates of each exposure group (41). AKI duration and KDIGO severity stage were modeled with IPTW multinomial logistic regression.
Sensitivity and Subgroup Analyses.
Prespecified subgroup analyses included restricting to patients who received combined exposure for ≥3 days (to examine duration response) and an analysis stratified by exposure to diuretics at baseline, because some studies suggest that synergistic NSAID–RAS-I nephrotoxicity may be limited to patients treated with diuretics (15). We also performed two additional post hoc subgroup analyses suggested during peer review: stratified analysis based on age of 65 years, and presence of diabetes mellitus II at baseline. In addition, all analyses were repeated using unweighted multivariable-adjusted Poisson regression in cohort A, because IPTW may have less power compared with multivariable regression (36,37). Multivariable modeling procedures are detailed in the Supplemental Methods. Lastly, we performed quantitative bias analysis, in which we estimated the effect of a hypothetical unmeasured confounder on our interaction estimates (see Supplemental Methods, Supplemental Table 2, and Supplemental Figures 2 and 3 for detailed methods).
Results
The selection of patients into the study is depicted in Figure 1. Our initial query identified 114,491 episodes of concomitant exposure to a drug pair of interest, of which 61,360 courses (in 43,201 patients) had a duration ≥24 hours. From this population, 27,741 patients were included in cohort A (the propensity score estimation cohort). After multinomial propensity score estimation, an additional 2170 patients were excluded due to noncomparable propensity scores, leaving 25,571 patients in cohort B (the primary analysis cohort).
Figure 1.
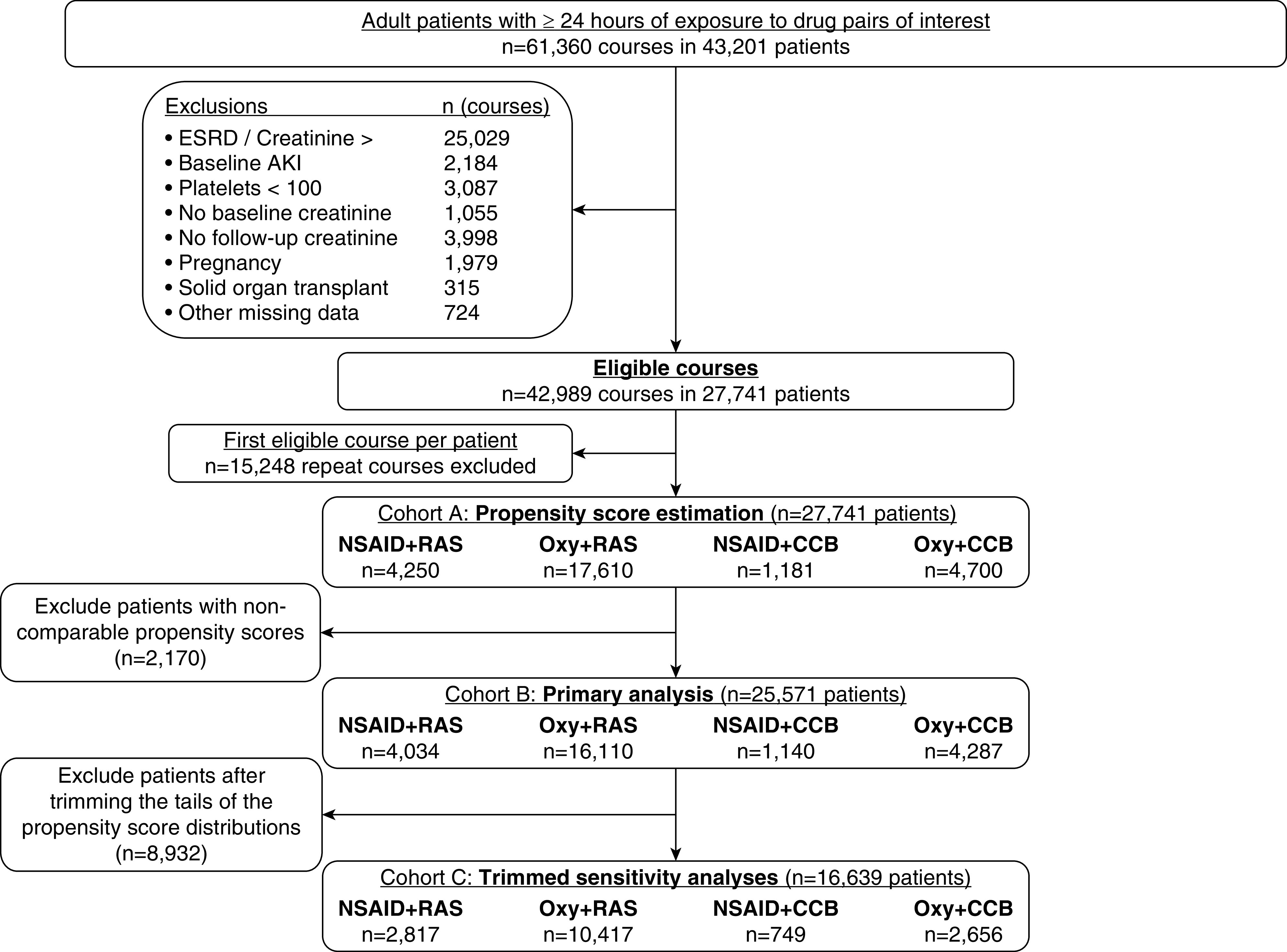
Exclusions applied to obtain the primary and sensitivity analysis cohorts. CCB, calcium channel blocker (amlodipine); NSAID, nonsteroidal anti-inflammatory drug; Oxy, oxycodone; RAS, renin-angiotensin system inhibitors.
Patient Characteristics before Weighting in Cohort A
The median duration of combined analgesic and antihypertensive treatment was approximately 2 days in all study groups, reflecting the short-term, acute pain indications that are common in the inpatient setting (Supplemental Tables 3 and 4). The daily doses of NSAID and oxycodone courses were generally low to moderate (Supplemental Table 3). In patients treated with RAS-I, patients treated with NSAIDs versus oxycodone tended to be younger, female patients admitted through the emergency department (Supplemental Table 4). They were less likely to have heart failure, atrial fibrillation, and other cardiovascular diseases. As expected, patients treated with NSAIDs had higher baseline eGFR compared with those treated with oxycodone.
In patients treated with NSAIDS, those treated with RAS-I were less likely to be in the intensive care unit or in the perioperative period at baseline (Supplemental Table 4), more often had heart failure, myocardial infarction, other cardiovascular disease, and diabetes mellitus type II. Patients treated with RAS-I also had lower baseline eGFR and were more likely to be exposed to concomitant diuretic therapy.
Patient Characteristics after Weighting in Cohort B (Primary Analysis Cohort)
The weighted population characteristics are shown in Table 1, and a summary of SMDs for all possible contrasts among the four treatment groups before and after weighting is shown in Supplemental Figure 4. IPTW successfully balanced covariates across all possible group contrasts. Obstructive sleep apnea and CKD variables had SMD values >0.1 and were thus included in the weighted outcome models.
Analysis of AKI Rate
There were 2138 AKI events observed during 90,571 person-days of follow-up. No interaction between NSAID+RAS-I was evident in the unadjusted analysis in cohort A, either on the difference (Supplemental Table 5) or the ratio (Supplemental Table 6) scales. Results of the weighted outcome analysis in cohort B are shown in Table 2 (difference scale) and Table 3 (ratio scale), which shows the effect of NSAID versus oxycodone on AKI rate to be similar in patients treated with RAS-I and those treated with amlodipine: difference in difference 1.85 (95% CI, −6.23 to 9.92) excess AKI events per 1000 days. Repeating the analysis without an interaction term between NSAID+RAS-I showed significant, independent associations between NSAID treatment, RAS-I treatment, and increased AKI rates (Table 4).
Table 2.
Weighted estimates of AKI rate and interaction analysis on the difference scale in cohort B (the primary analysis cohort)
| Antihypertensive Strata | Oxycodone Ratea | NSAID Ratea | NSAID RD within Antihypertensive Strata (95% CI)a |
| Amlodipine | 19.9 | 24.0 | 4.13 (−2.83 to 11.09) |
| RAS | 23.1 | 29.1 | 5.97 (1.88 to 10.07) |
| RAS RD within analgesic strata (95% CI)a | 3.22 (0.29 to 6.14) | 5.06 (−2.46 to 12.60) | Difference in difference: 1.85 (−6.23 to 9.92) |
The difference-in-difference estimate suggests that the effect of NSAIDs on AKI rate does not differ across antihypertensive groups. NSAID, nonsteroidal anti-inflammatory drug; RD, rate difference; RAS, renin-angiotensin system inhibitor.
AKI events/1000 person days.
Table 3.
Weighted estimates of AKI rate and interaction analysis on the ratio scale in cohort B (the primary analysis cohort)
| Antihypertensive Strata | Oxycodone Ratea | NSAID Ratea | NSAID RR within Antihypertensive Strata (95% CI) |
| Amlodipine | 19.9 | 24.0 | 1.21 (0.89 to 1.63) |
| RAS | 23.1 | 29.1 | 1.26 (1.09 to 1.45) |
| RAS RR within analgesic strata (95% CI) | 1.16 (1.00 to 1.34) | 1.21 0.89 to 1.63) | Ratio of rate ratios 1.04 (0.74 to 1.45) |
The ratio of rate ratio estimate suggests that the effect of NSAIDs on AKI rate does not differ across antihypertensive groups. NSAID, nonsteroidal anti-inflammatory drug; RR, rate ratio; RAS, renin-angiotensin system inhibitor.
AKI events/1000 person days.
Table 4.
Main effects estimates of NSAID and RAS exposure
| Exposure | IRR (95% CI) | IRD (95% CI)a |
| NSAID | ||
| Weighted analysis in cohort B | 1.24 (1.06 to 1.44) | 5.09 (1.13 to 9.05) |
| Weighted analysis in cohort C | 1.28 (1.07 to 1.53) | 5.07 (1.03 to 9.11) |
| Multivariable regression in cohort A | 1.23 (1.11 to 1.37) | 5.59 (2.51 to 8.66) |
| RAS inhibitors | ||
| Weighted analysis in cohort B | 1.18 (1.00 to 1.39) | 4.06 (0.34 to 7.78) |
| Weighted analysis in cohort C | 1.25 (1.02 to 1.54) | 4.71 (0.81 to 8.61) |
| Multivariable regression in cohort A | 1.09 (0.98 to 1.22) | 2.27 (−0.23 to 4.77) |
NSAID, nonsteroidal anti-inflammatory drug; RAS, renin-angiotensin system; IRR, incident rate ratio; IRD, incidence rate difference.
AKI events/1000 person days.
AKI Severity Stage and Duration
The majority of AKI events were stage 1 (1707/2138; 80%), with stage 2 and stage 3 events representing 14% and 6% of events, respectively. RRT was uncommon (21 events; 0.1%). There was no statistically significant evidence for synergistic effects of NSAID+RAS-I treatment on AKI severity or duration (Supplemental Figures 5 and 6).
Subgroup and Sensitivity Analyses
Figure 2 summarizes interaction estimates from the prespecified subgroup and sensitivity analyses, with the corresponding interaction tables reported in Supplemental Tables 7 and 8. Interaction estimates were more consistent with synergistic NSAID+RAS-I effects in patients treated for ≥3 days and in patients receiving diuretics at baseline. However, the confidence intervals around these estimates included the null value. Repeating all primary and subgroup analyses in cohort B (the trimmed cohort) produced similar results, as did repeating all analyses using multivariable Poisson regression (Figure 2, Supplemental Tables 7 and 8). Post hoc subgroup analysis by age and diabetes did not show strong evidence of altered NSAID–RAS-I effects (Supplemental Tables 7 and 8). Quantitative bias analysis (Supplemental Figures 2 and 3, Supplemental Table 2) showed that our primary interaction estimate is robust to unmeasured confounding under a wide range of plausible scenarios. Unmeasured confounding could change the primary conclusions only under extreme circumstances: an unmeasured confounder would need to have a very strong effect on AKI (e.g., increases AKI rate by 15–20 events/1000 days) and would need to show a degree of imbalance several fold more extreme than that observed for any of the measured covariates. Taken together, the results suggest that unmeasured confounding is unlikely to change our conclusions.
Figure 2.
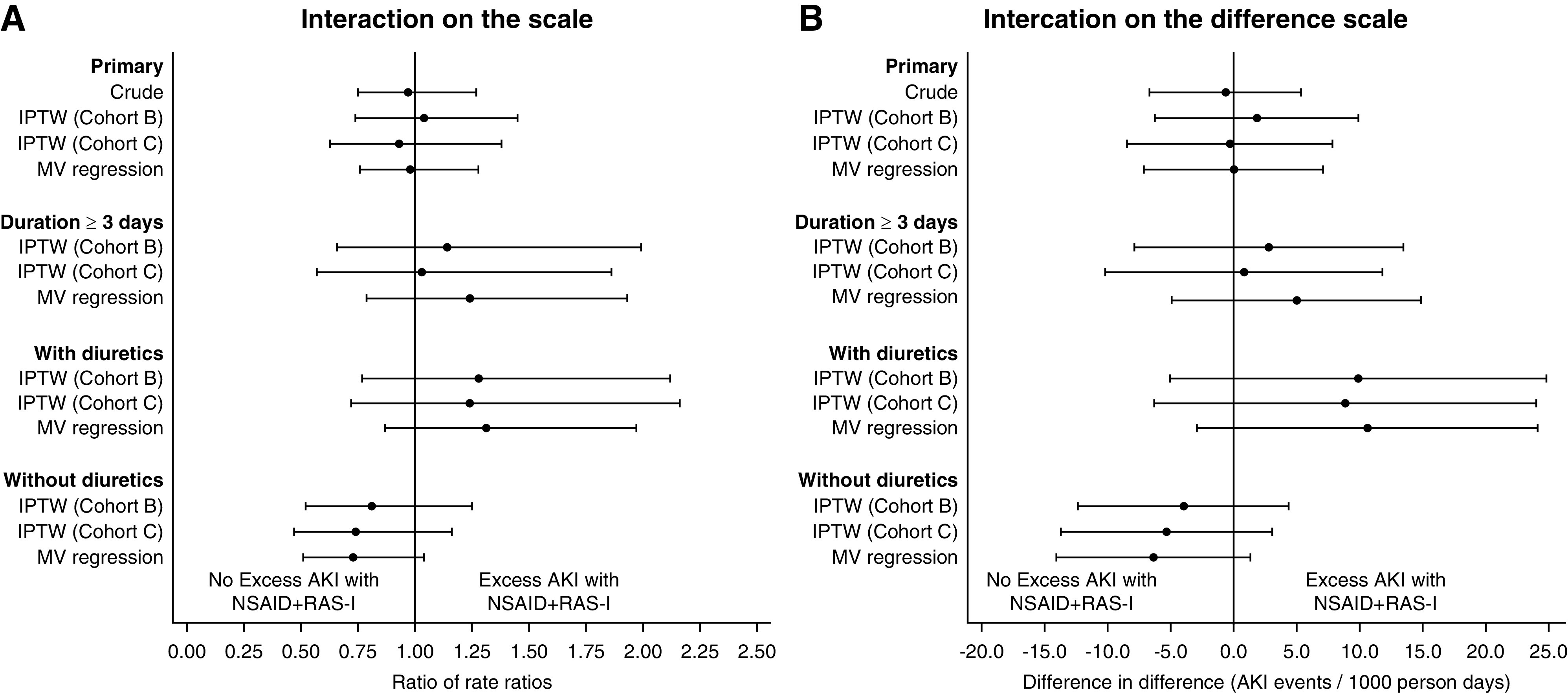
Interaction estimates from the primary analysism, sensitivity analyeses, and prespecified subgroups. (A) Interaction measures on the ratio scale (ratio of rate ratios [RRR]). Each point represents an RRR estimate, with 95% CI represented by the capped bars. Values above one favor excess AKI with NSAID+RAS-I. (B) Interaction measures on the difference scale (difference in differences [DID]). Each point represents a DID estimate, with 95% CI represented by the capped bars. Values above zero favor excess AKI with NSAID+RAS-I. IPTW, inverse probability of treatment weighting; MV, multivariable.
Discussion
We conducted a large-scale analysis of the NSAID+RAS-I interaction using a rigorous study design and analysis procedures to control confounding. To our knowledge, this is the first study to examine this question in patients who are hospitalized. We found that, despite a clear association of short-term NSAID use with AKI, NSAID nephrotoxicity was not meaningfully increased by concomitant RAS-I treatment. We could not, however, rule out the possibility of synergistic nephrotoxicity with NSAID+RAS-I among patients also taking diuretics.
Interpretation of our primary finding (lack of NSAID+RAS-I interaction) must carefully consider the confidence intervals around the interaction parameter. The upper bound of the confidence interval on the additive scale was 9.9 AKI events/1000 days. This translates into two extra AKI cases in a population of 100 patients treated with 2 days of concomitant NSAID+RAS-I (the median duration in our cohort). Given the low severity of most observed AKI events, and the risks associated with opioids (21–23,42), many clinicians might judge the upper bound of our interaction estimate to represent an acceptable risk compared with opioid analgesia.
Although there is a dearth of prior evidence in patients who are hospitalized, prior outpatient studies have examined the NSAID+RAS-I interaction with conflicting results (13–17). Dreischulte et al. (16) conducted a case-control study that compared NSAIDs versus no NSAIDs in a cohort of patients treated with RAS-I, showing a 60% increase in the odds of AKI associated with NSAID treatment. The lack of an active-comparator cohort in this study, such as patients exposed to a non-RAS antihypertensive, makes it impossible to determine whether these results reflect a true drug-drug interaction (i.e. synergistic toxicity between NSAID and RAS-I) or simply the known nephrotoxic effects of NSAIDs independent of RAS-I treatment (29). In a second case-control study, Lapi et al. (15) also compared NSAIDs versus no NSAIDs in a RAS-I-treated cohort, finding no evidence of higher AKI risk with combined NSAID+RAS-I treatment. Notably, this analysis found that NSAIDs conferred a higher AKI risk only in patients treated with both RAS-I and diuretics.
Our analyses differs from these prior outpatient studies in that we specifically tested for synergistic effects by comparing the difference in AKI rate between NSAID versus oxycodone in patients treated with RAS-I to the difference in AKI rate between NSAID versus oxycodone in patients treated with amlodipine (29). The use of active comparators for both the NSAID and RAS-I groups serves to minimize confounding by indication (26). Moreover, our approach to testing interaction directly addresses the relevant clinical question: is NSAID toxicity altered by RAS-I treatment? The presence of a synergistic interaction between two treatments implies that there are persons for whom AKI would occur if both treatments were present but not if only one or the other were present (29,43). Our results suggest that, at least for short-term (2–3 days) treatment in the absence of diuretics, RAS-I treatment does not alter AKI risk during NSAID analgesia. Thus, in patients who would otherwise be deemed candidates for NSAID therapy, RAS-I treatment may not be a reason to choose opioids over NSAIDs.
In secondary analyses, we observed stronger interaction signals in the subset of patients treated with NSAID+RAS-I for ≥3 days, and in patients who were receiving baseline diuretics. A higher risk with longer treatment duration is consistent with prior evidence describing duration-dependent nephrotoxicity with ketorolac (44). Synergistic nephrotoxicity with diuretics is consistent with the results from Lapi et al. (15), and other case reports and case series in outpatient populations (45). This three-way interaction is postulated to result from diuretic-mediated decreases of inflow to the glomerulus, combined with disrupted renal blood flow autoregulation induced by NSAID+RAS-I treatment (15). In our analysis, the upper bound of the interaction estimate in the diuretic cohort was 24.8 excess AKI events per 1000 days, an increase that many would judge as clinically meaningful. Given the uncertainty of the confidence intervals around this estimate, larger studies of individuals exposed to diuretics are warranted. However, given the similar findings in prior outpatient studies, it may be reasonable to avoid the NSAID+RAS-I combination in patients treated with diuretics.
Our study has limitations. First, the observational design is susceptible to residual confounding. We minimized confounding with the active-comparator study design (26), and by collecting and controlling for all available potential confounders in IPTW analyses. In addition, we conducted quantitative bias analysis which suggested that our results are robust to unmeasured confounding under a wide range of plausible scenarios. Second, although the use of active comparators may help to reduce confounding, this approach may limit generalizability. Strictly speaking, our results suggest that combined NSAID+RAS-I treatment may not synergistically worsen AKI risk in comparison with our control drugs (oxycodone and amlodipine). It may be the case that our inability to detect a meaningful interaction was driven by similar synergistic effects across the studied groups, rather than the absence of synergistic NSAID+RAS-I toxicity. Although possible, this seems unlikely given the proposed effects of amlodipine on renal vascular tone (28) and lack of known oxycodone nephrotoxicity (21). Third, although our primary analysis did not detect a meaningful NSAID+RAS-I interaction, this might not hold in other populations with higher AKI risk. Fourth, we were unable to examine the effect of NSAID daily dose due to the multiple different NSAIDs and RAS-I included, limiting the numbers of patients that received any specific drug and dose combination. Fifth, our findings may not apply to longer NSAID+RAS-I treatment durations. Notably, such long durations were relatively uncommon in our cohort: only 20% of the 114,491 episodes of concomitant therapy were of ≥3 days. Lastly, there may be numerous other factors that alter the effect of the NSAID–RAS-I interaction (e.g., severity of illness factors such as admission to the intensive care unit and comorbid illnesses such as heart failure). Sample size limited our ability to identify such factors because the examination of three-way interactions require severalfold greater sample sizes compared with two-way interactions.
Synergistic nephrotoxicity was not observed with short-term NSAIDs+RAS-I treatment in the absence of concomitant diuretics, suggesting that RAS-I treatment may not be a reason to choose opioids in lieu of NSAIDs in this population. Synergistic nephrotoxicity cannot be ruled out in patients treated with diuretics.
Disclosures
J. Brown has received consulting fees from Bracco Scientific. S. Hennessy has received consulting fees from the following companies: Braeburn Pharmaceuticals Inc, Esteve Pharmaceuticals LLC, Greenwich Biosciences Inc, Hoffman La Roche, Indivior Inc, Inspiron Delivery Sciences LLC, Janssen Research & Development LLC, Laboratoire HRA PHARMA, Lexicon Pharmaceuticals Inc, Lilly USA LLC, Mallincrodt Pharmaceuticals, Medulary Thyroid Cancer Consortium (AstraZeneca Pharmaceuticals LP, Eli Lilly and Company, Novo Nordisk Inc, GlaxoSmithKline LLC), Merck Research Laboratories, Nektar Therapeutics Inc, NovoNordisk, Pacira Pharmaceuticals Inc, PTC Therapeutics Inc, Purdue Pharma LP, Sage Therapeutics, Sanofi US Services Inc, Shire Human Genetic Therapies Inc, and Transdermal Immediate Release Fentanyl REMS (BioDelivery Sciences International Inc, Insys Therapeutics Inc, Mylan Inc, Par Pharmaceutical Inc, Sentynl Therapeutics Inc, SpecGX LLC [a wholly owned subsidiary of Mallinckrodt Inc], Teva Pharmaceuticals USA Inc, West Therapeutic Development LLC). In addition, S. Hennessy leads the Center for Pharmacoepidemiology Research and Training, which has received support for pharmacoepidemiology training programs from Pfizer Inc. A. Zuppa has served on an advisory committee for Pfizer. All remaining authors have nothing to disclose.
Funding
This work was funded by National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant R01DK111638 to M. Shashaty; NIH NIDDK grant R01DK113201 to J. Brown; and NIH National Institute on Aging grants R01AG025152 and R01AG060975 and NIH National Institute on Drug Abuse grant R01DA048001 to S. Hennessy. This work was also supported by the University of Pennsylvania University Research Foundation.
Supplemental Material
This article contains supplemental material online at http://kidney360.asnjournals.org/lookup/suppl/doi:10.34067/KID.0001432020/-/DCSupplemental.
Download Supplemental Methods, PDF file, 922 KB (921.6KB, pdf)
Diagnosis code algorithms. Download Supplemental Table 1, PDF file, 922 KB (921.6KB, pdf)
Overlap of the multinomial propensity score distributions. Download Supplemental Figure 1, PDF file, 922 KB (921.6KB, pdf)
Bias analysis parameters for a set of measured covariates hypothetically assumed to be unmeasured. Download Supplemental Table 2, PDF file, 922 KB (921.6KB, pdf)
Corrected interaction estimates and confidence intervals for a set of measured covariates hypothetically assumed to be unmeasured. Download Supplemental Figure 2, PDF file, 922 KB (921.6KB, pdf)
Bias as a function of the difference-in-difference of the prevalence of an unmeasured confounder and the unmeasured confounder’s association with outcome. Download Supplemental Figure 3, PDF file, 922 KB (921.6KB, pdf)
Drug dosing. Download Supplemental Table 3, PDF file, 922 KB (921.6KB, pdf)
Baseline characteristics in the unweighted population (cohort A). Download Supplemental Table 4, PDF file, 922 KB (921.6KB, pdf)
Unadjusted acute kidney injury rates and interaction analysis on the difference scale in cohort A. Download Supplemental Table 5, PDF file, 922 KB (921.6KB, pdf)
Unadjusted acute kidney injury rates and interaction analysis on the ratio scale in cohort A. Download Supplemental Table 6, PDF file, 922 KB (921.6KB, pdf)
Interaction analyses of acute kidney injury rate per 1000 days on the difference scale. Download Supplemental Table 7, PDF file, 922 KB (921.6KB, pdf)
Interaction analyses of acute kidney injury rate per 1000 days on the ratio scale. Download Supplemental Table 8, PDF file, 922 KB (921.6KB, pdf)
Absolute standardized mean differences. Download Supplemental Figure 4, PDF file, 922 KB (921.6KB, pdf)
Predicted acute kidney injury stage stratified by treatment group. Download Supplemental Figure 5, PDF file, 922 KB (921.6KB, pdf)
Predicted acute kidney injury duration stratified by treatment group. Download Supplemental Figure 6, PDF file, 922 KB (921.6KB, pdf)
Acknowledgments
We would like to thank the PDS analysts for assistance with EHR data query and cleaning. We would also like to thank Madeline D. Miano and Jordana Cohen for reviewing the manuscript and providing critical feedback.
Author Contributions
J. Brown, S. Hennessy, M. Shashaty, W. Yang, and A. Zuppa provided supervision; S. Hennessy was responsible for resources; S. Hennessy and T. Miano conceptualized the study; T. Miano wrote the original draft and was responsible for data curation, project administration, and visualization; M. Shashaty, T. Miano, and W. Yang were responsible for formal analysis; all authors were responsible for methodology and reviewed and edited the manuscript; and all authors agree to be accountable for all aspects of the work.
Footnotes
See related editorial, “Renal Safety of Nonsteroidal Anti-Inflammatory Drugs and Opioids in Hospitalized Patients on Renin-Angiotensin System Inhibitors” on pages 586–587.
References
- 1.Herzig SJ, Mosher HJ, Calcaterra SL, Jena AB, Nuckols TK: Improving the safety of opioid use for acute noncancer pain in hospitalized adults: A consensus statement from the society of hospital medicine. J Hosp Med 13: 263–271, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu CL, King AB, Geiger TM, Grant MC, Grocott MPW, Gupta R, Hah JM, Miller TE, Shaw AD, Gan TJ, Thacker JKM, Mythen MG, McEvoy MD; Fourth Perioperative Quality Initiative Workgroup : American society for enhanced recovery and perioperative quality initiative joint consensus statement on perioperative opioid minimization in opioid-naïve patients. Anesth Analg 129: 567–577, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chou R, Gordon DB, de Leon-Casasola OA, Rosenberg JM, Bickler S, Brennan T, Carter T, Cassidy CL, Chittenden EH, Degenhardt E, Griffith S, Manworren R, McCarberg B, Montgomery R, Murphy J, Perkal MF, Suresh S, Sluka K, Strassels S, Thirlby R, Viscusi E, Walco GA, Warner L, Weisman SJ, Wu CL: Management of postoperative pain: A clinical practice guideline from the American pain society, the American society of regional anesthesia and pain medicine, and the American society of anesthesiologists’ committee on regional anesthesia, executive committee, and administrative council [published correction appears in J Pain 17: 508–510, 2016]. J Pain 17: 131–157, 2016 [DOI] [PubMed] [Google Scholar]
- 4.Chang AK, Bijur PE, Esses D, Barnaby DP, Baer J: Effect of a single dose of oral opioid and nonopioid analgesics on acute extremity pain in the emergency department: a randomized clinical trial. JAMA 318: 1661–1667, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore RA, Derry S, Aldington D, Wiffen PJ: Single dose oral analgesics for acute postoperative pain in adults - an overview of Cochrane reviews. Cochrane Database Syst Rev 28: CD008659, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones P, Dalziel SR, Lamdin R, Miles-Chan JL, Frampton C: Oral non-steroidal anti-inflammatory drugs versus other oral analgesic agents for acute soft tissue injury. Cochrane Database Syst Rev [7]: CD007789, 2015 [DOI] [PubMed] [Google Scholar]
- 7.Gallegos A: Hospitals changing practices to curb opioid epidemic. ACP Hospitalist, 2019. Available at: https://acphospitalist.org/archives/2019/03/hospitals-changing-practices-to-curb-opioid-epidemic.htm. Accessed February 4, 2020 [Google Scholar]
- 8.Vizient:The opioid epidemic—A view from the front lines. Vizient, 2018. Available at: https://newsroom.vizientinc.com/sites/vha.newshq.businesswire.com/files/doc_library/file/The_Opioid_Epidemic_Survey_by_Vizient.pdf. Accessed January 29, 2020 [Google Scholar]
- 9.Mark J, Argentieri DM, Gutierrez CA, Morrell K, Eng K, Hutson AD, Mayor P, Szender JB, Starbuck K, Lynam S, Blum B, Akers S, Lele S, Paragh G, Odunsi K, de Leon-Casasola O, Frederick PJ, Zsiros E: Ultrarestrictive opioid prescription protocol for pain management after gynecologic and abdominal surgery. JAMA Netw Open 1: e185452, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meisenberg BR, Grover J, Campbell C, Korpon D: Assessment of opioid prescribing practices before and after implementation of a health system intervention to reduce opioid overprescribing. JAMA Netw Open 1: e182908, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murthy VH: Ending the opioid epidemic — a call to action. N Engl J Med 375: 2413–2415, 2016 [DOI] [PubMed] [Google Scholar]
- 12.Whelton A: Nephrotoxicity of nonsteroidal anti-inflammatory drugs: Physiologic foundations and clinical implications. Am J Med 106: 13S–24S, 1999 [DOI] [PubMed] [Google Scholar]
- 13.Lafrance JP, Miller DR: Selective and non-selective non-steroidal anti-inflammatory drugs and the risk of acute kidney injury. Pharmacoepidemiol Drug Saf 18: 923–931, 2009 [DOI] [PubMed] [Google Scholar]
- 14.Huerta C, Castellsague J, Varas-Lorenzo C, García Rodríguez LA: Nonsteroidal anti-inflammatory drugs and risk of ARF in the general population. Am J Kidney Dis 45: 531–539, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Lapi F, Azoulay L, Yin H, Nessim SJ, Suissa S: Concurrent use of diuretics, angiotensin converting enzyme inhibitors, and angiotensin receptor blockers with non-steroidal anti-inflammatory drugs and risk of acute kidney injury: Nested case-control study. BMJ 346: e8525, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dreischulte T, Morales DR, Bell S, Guthrie B: Combined use of nonsteroidal anti-inflammatory drugs with diuretics and/or renin-angiotensin system inhibitors in the community increases the risk of acute kidney injury. Kidney Int 88: 396–403, 2015 [DOI] [PubMed] [Google Scholar]
- 17.Bouvy ML, Heerdink ER, Hoes AW, Leufkens HG: Effects of NSAIDs on the incidence of hospitalisations for renal dysfunction in users of ACE inhibitors. Drug Saf 26: 983–989, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Bakris GL, Weir MR: Angiotensin-converting enzyme inhibitor-associated elevations in serum creatinine: Is this a cause for concern? Arch Intern Med 160: 685–693, 2000 [DOI] [PubMed] [Google Scholar]
- 19.Perazella MA, Luciano RL: Review of select causes of drug-induced AKI. Expert Rev Clin Pharmacol 8: 367–371, 2015 [DOI] [PubMed] [Google Scholar]
- 20.Coca SG, Singanamala S, Parikh CR: Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int 81: 442–448, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herzig SJ, Rothberg MB, Cheung M, Ngo LH, Marcantonio ER: Opioid utilization and opioid-related adverse events in nonsurgical patients in US hospitals. J Hosp Med 9: 73–81, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calcaterra SL, Yamashita TE, Min SJ, Keniston A, Frank JW, Binswanger IA: Opioid prescribing at hospital discharge contributes to chronic opioid use. J Gen Intern Med 31: 478–485, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herzig SJ: Opening the black box of inpatient opioid prescribing. J Hosp Med 11: 595–596, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.National Center for Health Statistics : Health, United States, 2016: With Chartbook on Long-term Trends in Health, Washington, D.C, National Center for Health Statistics, 2017 [PubMed] [Google Scholar]
- 25.Zhou M, Daubresse M, Stafford RS, Alexander GC: National trends in the ambulatory treatment of hypertension in the United States, 1997-2012. PLoS One 10: e0119292, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hennessy S, Leonard CE, Gagne JJ, Flory JH, Han X, Brensinger CM, Bilker WB: Pharmacoepidemiologic methods for studying the health effects of drug-drug interactions. Clin Pharmacol Ther 99: 92–100, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, Lackland DT, LeFevre ML, MacKenzie TD, Ogedegbe O, Smith SC Jr., Svetkey LP, Taler SJ, Townsend RR, Wright JT Jr., Narva AS, Ortiz E: 2014 evidence-based guideline for the management of high blood pressure in adults: Report from the panel members appointed to the eighth joint national committee (JNC 8). JAMA 311: 507–520, 2014 [DOI] [PubMed] [Google Scholar]
- 28.Hart P, Bakris GL: Calcium antagonists: Do they equally protect against kidney injury? Kidney Int 73: 795–796, 2008 [DOI] [PubMed] [Google Scholar]
- 29.VanderWeele TJ, Knol MJ: A tutorial on interaction. Epidemiol Method 3: 33–72, 2014 [Google Scholar]
- 30.Kidney Disease; Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group : KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl 2: 1–138, 2012 [Google Scholar]
- 31.Coca SG, King JT Jr., Rosenthal RA, Perkal MF, Parikh CR: The duration of postoperative acute kidney injury is an additional parameter predicting long-term survival in diabetic veterans. Kidney Int 78: 926–933, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration): A new equation to estimate glomerular filtration rate [published correction appears in Ann Intern Med 155: 408, 2011]. Ann Intern Med 150: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Austin PC: Balance diagnostics for comparing the distribution of baseline covariates between treatment groups in propensity-score matched samples. Stat Med 28: 3083–3107, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.VanderWeele TJ: On the distinction between interaction and effect modification. Epidemiology 20: 863–871, 2009 [DOI] [PubMed] [Google Scholar]
- 35.Austin PC: Using the standardized difference to compare the prevalence of a binary variable between two groups in observational research. Commun Stat Simul Comput 38: 1228–1234, 2009 [Google Scholar]
- 36.McCaffrey DF, Griffin BA, Almirall D, Slaughter ME, Ramchand R, Burgette LF: A tutorial on propensity score estimation for multiple treatments using generalized boosted models. Stat Med 32: 3388–3414, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robins JM, Hernán MA, Brumback B: Marginal structural models and causal inference in epidemiology. Epidemiology 11: 550–560, 2000 [DOI] [PubMed] [Google Scholar]
- 38.Nguyen TL, Collins GS, Spence J, Daurès JP, Devereaux PJ, Landais P, Le Manach Y: Double-adjustment in propensity score matching analysis: Choosing a threshold for considering residual imbalance. BMC Med Res Methodol 17: 78, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stürmer T, Rothman KJ, Avorn J, Glynn RJ: Treatment effects in the presence of unmeasured confounding: Dealing with observations in the tails of the propensity score distribution--a simulation study. Am J Epidemiol 172: 843–854, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshida K, Solomon DH, Haneuse S, Kim SC, Patorno E, Tedeschi SK, Lyu H, Franklin JM, Stürmer T, Hernández-Díaz S, Glynn RJ: Multinomial extension of propensity score trimming methods: A simulation study. Am J Epidemiol 188: 609–616, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Norton EC, Dowd BE, Maciejewski ML: Marginal effects-quantifying the effect of changes in risk factors in logistic regression models. JAMA 321: 1304–1305, 2019 [DOI] [PubMed] [Google Scholar]
- 42.Novick TK, Surapaneni A, Shin JI, Alexander GC, Inker LA, Wright EA, Chang AR, Grams ME: Associations of opioid prescriptions with death and hospitalization across the spectrum of estimated GFR. Clin J Am Soc Nephrol 14: 1581–1589, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.VanderWeele TJ, Robins JM: The identification of synergism in the sufficient-component-cause framework. Epidemiology 18: 329–339, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Feldman HI, Kinman JL, Berlin JA, Hennessy S, Kimmel SE, Farrar J, Carson JL, Strom BL: Parenteral ketorolac: The risk for acute renal failure. Ann Intern Med 126: 193–199, 1997 [DOI] [PubMed] [Google Scholar]
- 45.Adhiyaman V, Asghar M, Oke A, White AD, Shah IU: Nephrotoxicity in the elderly due to co-prescription of angiotensin converting enzyme inhibitors and nonsteroidal anti-inflammatory drugs. J R Soc Med 94: 512–514, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Download Supplemental Methods, PDF file, 922 KB (921.6KB, pdf)
Diagnosis code algorithms. Download Supplemental Table 1, PDF file, 922 KB (921.6KB, pdf)
Overlap of the multinomial propensity score distributions. Download Supplemental Figure 1, PDF file, 922 KB (921.6KB, pdf)
Bias analysis parameters for a set of measured covariates hypothetically assumed to be unmeasured. Download Supplemental Table 2, PDF file, 922 KB (921.6KB, pdf)
Corrected interaction estimates and confidence intervals for a set of measured covariates hypothetically assumed to be unmeasured. Download Supplemental Figure 2, PDF file, 922 KB (921.6KB, pdf)
Bias as a function of the difference-in-difference of the prevalence of an unmeasured confounder and the unmeasured confounder’s association with outcome. Download Supplemental Figure 3, PDF file, 922 KB (921.6KB, pdf)
Drug dosing. Download Supplemental Table 3, PDF file, 922 KB (921.6KB, pdf)
Baseline characteristics in the unweighted population (cohort A). Download Supplemental Table 4, PDF file, 922 KB (921.6KB, pdf)
Unadjusted acute kidney injury rates and interaction analysis on the difference scale in cohort A. Download Supplemental Table 5, PDF file, 922 KB (921.6KB, pdf)
Unadjusted acute kidney injury rates and interaction analysis on the ratio scale in cohort A. Download Supplemental Table 6, PDF file, 922 KB (921.6KB, pdf)
Interaction analyses of acute kidney injury rate per 1000 days on the difference scale. Download Supplemental Table 7, PDF file, 922 KB (921.6KB, pdf)
Interaction analyses of acute kidney injury rate per 1000 days on the ratio scale. Download Supplemental Table 8, PDF file, 922 KB (921.6KB, pdf)
Absolute standardized mean differences. Download Supplemental Figure 4, PDF file, 922 KB (921.6KB, pdf)
Predicted acute kidney injury stage stratified by treatment group. Download Supplemental Figure 5, PDF file, 922 KB (921.6KB, pdf)
Predicted acute kidney injury duration stratified by treatment group. Download Supplemental Figure 6, PDF file, 922 KB (921.6KB, pdf)