

Abstract
Protein engineering is a growing field with a variety of experimental techniques available for altering protein function. However, creating an enzyme de novo is still in its infancy, so far yielding enzymes of modest catalytic efficiency. In this study, a system of artificial retro-aldolase enzymes found to have chemistry coupled to protein dynamics was examined. The original design was created computationally, and this protein was then subjected to directed evolution to improve the initial low catalytic efficiency. We found that this re-engineering of the enzyme resulted in rapid density fluctuations throughout the enzyme being reshaped via alterations in the hydrogen bonding network. This work also led to the discovery of a second important motion which aids in the release of an intermediate product. These results provide compelling evidence that to engineer efficient protein catalysts, fast protein dynamics need to be considered in the design.
Keywords: directed evolution, protein dynamics, QMMM simulation, hydrogen bonding network, transition path sampling, Markov chain Monte Carlo
Graphical Abstract
INTRODUCTION
Protein engineering has grown into a massive area of research with a wide variety of techniques and applications. Some of the approaches being used to reduce our profound ignorance in how protein sequence dictates function include: evaluating natural evolution of functionally diverse superfamilies,1 the use of phage display,2,3 and directed evolution.4–7 One of the limitations of these techniques is the requirement for existing proteins to serve as a starting point, which is an issue that computational design has tried to address by creating novel enzymes de novo.8–10 While there has been success in creating artificial enzymes, the examples generated so far have low catalytic efficiency.11 It has been noted that these methods suffer from not including long-range electrostatics and not using an explicit water model;4 however, our group and others argue that protein dynamics must also be considered for efficient protein design.12–16 Currently, computational design is centered around a static transition state structure, and a protein scaffold is built around this transition state.
It has been shown that such computational efforts increase the probability of finding an initial variant,17 and this can provide a starting point for directed evolution in situations where no natural analogue is available. This is the case for the retro-aldol reaction of 4-hydroxy-4-(6- methoxy-2-naphthyl)-2-butanone in a series of artificial enzymes starting from the RA95.0 variant, as reported by Hilvert et al.11 This system started from a computationally designed enzyme with a catalytic lysine at position 210 which, through directed evolution, was gradually improved. The catalytic lysine was shifted to position 83, relocating the active site deeper into the scaffold. The feature chosen for optimization by directed evolution was the catalytic rate, reported as kcat/kM, which was improved from 0.17 M−1 s−1 in the initial design to 34 000 M−1 s−1 in the latest reported variant.18
Through analysis of the directed evolution proteins, we gained insight into the mechanisms of how random mutations yield improvement in the catalytic efficiency. In the most evolved variant, we found a motion on the femtosecond time scale that pushes the catalytic base toward the substrate to promote chemistry. This motion is not present in any other variant.16 The phenomenon of femtosecond motions enhancing rates in enzymes is one our group and others have been investigating in a variety of enzymes.19–23 This study focuses on how the 13 mutations between the penultimate (RA95.5–8, variant IV) and most evolved variant (RA95.5–8f, variant V) resulted in the coupling of protein dynamics to chemistry. Our goal is to develop an understanding of this process so that such capability may be designed into artificial protein catalysts from the start.
We utilize ensembles of unbiased reactive trajectories created via transition path sampling to analyze dynamic changes in evolution from variant IV to variant V. The coherent dynamical structure factor was used to probe changes in density fluctuations in the proteins, and these changes are shown to be directly connected to the change in coupling of protein motion to chemistry.
Changes in hydrogen bonding networks were examined to explain how the mutations introduced by directed evolution changed protein dynamics which in turn influence fluctuations in density. Femtosecond motions in key residues previously identified to be part of a rate promoting vibration were further examined and found to correlate with the density fluctuations. The effect of the mutations on active site volume was studied and found to have decreased. This analysis has also led to the discovery of a second motion correlated with the density fluctuations, which is involved with expediting the release of an intermediate product. This step in the mechanism was previously examined, and it was found that the off rate of 6- methoxy-2-naphthaldehyde increased going from variant IV to V.11 This evidence strongly supports the claim that the rate increase from variant IV to variant V (1600 to 34 000 M−1 s−1, respectively) is at least in part due to protein dynamics becoming coupled to chemistry, and that dynamics must be taken into account for more effective protein engineering.
RESULTS
Mechanism.
The reaction associated with 4-hydroxy-4-(6-methoxy-2-naphthyl)-2-butanone for the variants as shown in Figure 1 has multiple steps and involves the formation of a Schiff base intermediate. Our transition path sampling (TPS) study examines steps 2 and 3, the C–C bond cleavage, known to be rate limiting for chemistry in the original design. Through TPS calculations, we observe a mechanism (Figure 1A) which has an intramolecular hydrogen transfer taking the place of a catalytic base, which was observed in variant IV. The mechanism proposed by Hilvert et al. (Figure 1B) has a catalytic base transfer a hydrogen and then abstract another hydrogen, which was observed in variant V. In variant IV, residue 180 was a phenylalanine, which cannot act as a catalytic base, and as a result, our generated reactive trajectories for variant IV do not show the mechanism in Figure 1B. Directed evolution introduced 13 mutations to variant IV to produce variant V.
Figure 1.
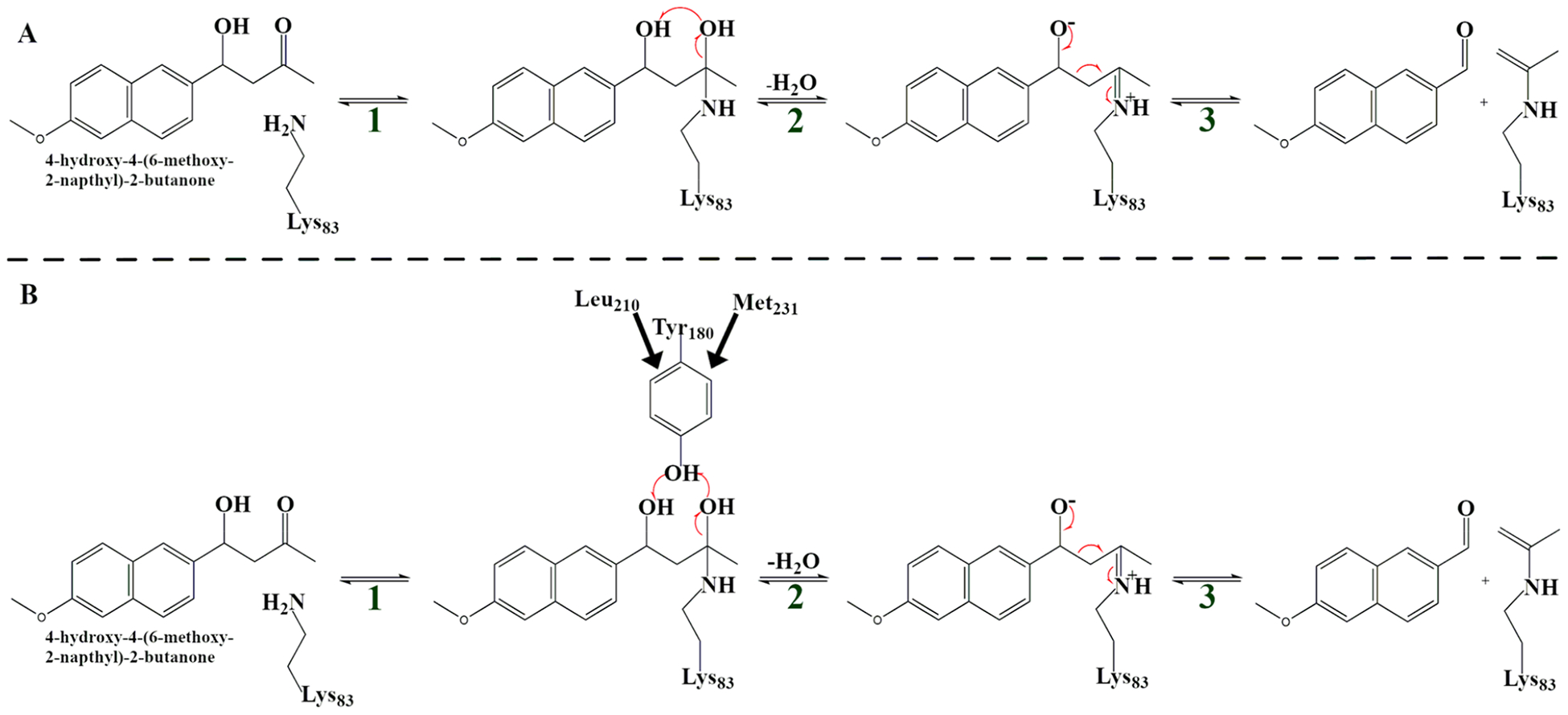
Reaction scheme detailing the major steps of the amine-catalyzed retro-aldol reaction in this study: the intramolecular mechanism (variant IV) proceeds through (A) and the mechanism that involves a catalytic tyrosine (variant V) proceeds through (B).
We generated 120 reactive trajectories of length 500 fs for variant IV and 120 reactive trajectories of length 500 fs for variant V following the reaction mechanism in Figure 1A and B, respectively. The rate promoting vibration which was previously identified16 is shown in Figure 1B. Leu210 and Met231 push downward, forcing Tyr180 to move closer to the substrate and catalyze the reaction. The C–O bond at the Schiff base breaks, and this event results in the formation of a water leaving group and subsequent C–C bond cleavage.
Volume.
We first chose to examine how the mutations directly affected the active site by comparing reactant state volumes. The volumes calculated with POVME show that the 13 mutations have significantly altered the size of the active site. The weighted average for variant IV’s active site volume is 450 Å3, while the weighted average for variant V’s active site volume is 312 Å3, Figure 2A. This shows that the active site became more tightly packed around the substrate due to the 13 mutations. We were also interested in how the volume fluctuates on a fast time scale; to calculate this, a moving average was subtracted from the calculated volumes to remove motions slower than 100 fs. The fluctuations that remain are the fastest motions associated with volume change. One may consider these as stochastic volume fluctuations, which have an approximately normal distribution (Figure 2B). Fitting a Gaussian to these distributions to calculate the standard deviation revealed a reduction of 16% going from variant IV to variant V. This shows that the active site has become more tightly packed around the substrate overall and fluctuates less, allowing chemistry to occur in a more stable environment. Because variant V develops the promoting vibration, we see that not only does the random fluctuation of active site volume go down, but what motion remains is more “functionally” directed.
Figure 2.
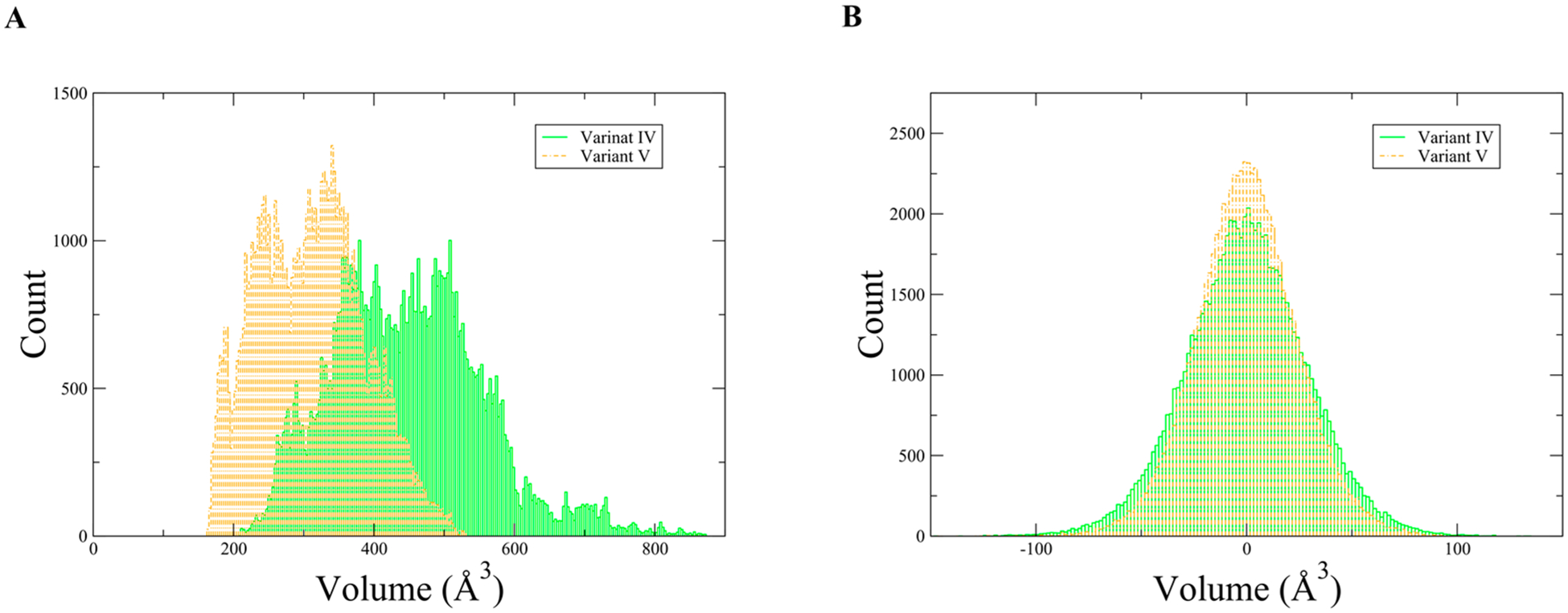
(A) Histogram of reactant state volumes associated with variant IV and variant V. (B) Histogram of fast volume fluctuations, variant IV σ = 27.18 Å3 and variant V σ = 22.81 Å3.
Hydrogen Bonding.
To understand the underlying physical changes that led to the drastic change in the mechanism between the variants, we analyzed shifts in hydrogen bonding between variant IV and V. This information is presented as a percentage of all trajectory frames that contain a hydrogen bond between two residues. The percentages for all hydrogen bonding interactions were calculated for variant IV and V, and then this information was used to calculate the change in percentage between variant IV and variant V. This way of presenting the hydrogen bonding network illustrates how the 13 mutations affected the entire hydrogen bonding network, which in turn affects protein dynamics.
Alterations in the hydrogen bonding network change the way residues can move within a protein and are likely responsible for the coupling of protein dynamics to chemistry. In analyzing the changes in hydrogen bonding networks from variant IV to V, it is clear that the largest decreases in hydrogen bonding are approximately aligned in a plane (Figure 3A) with the residues involved in the rate promoting vibration. The largest increases in hydrogen bonding flank the active site on both sides and are not aligned with the new soft axis (Figure 3B). To illustrate how these regions are spatially arranged, a least-squares analysis was done to find the plane of best fit for the soft axis (orange residues in Figure 3A) and two planes for the rigid axes flanking the active site (purple and pink residues in Figure 3B). The angle between the soft plane and a vector representing the rate promoting vibration was 3.25°. The purple and pink residues’ planes are at 48.29° and 57.22° with respect to the soft axis; this shows that the rigid regions flanking the active site are not aligned with the soft axis or the rate promoting vibration. Taking both the volume changes and hydrogen bonding changes into account suggests that the mutations have introduced a novel axis for directed motion (in the direction of the rate promoting vibration) while simultaneously strengthening the protein along other axes.
Figure 3.
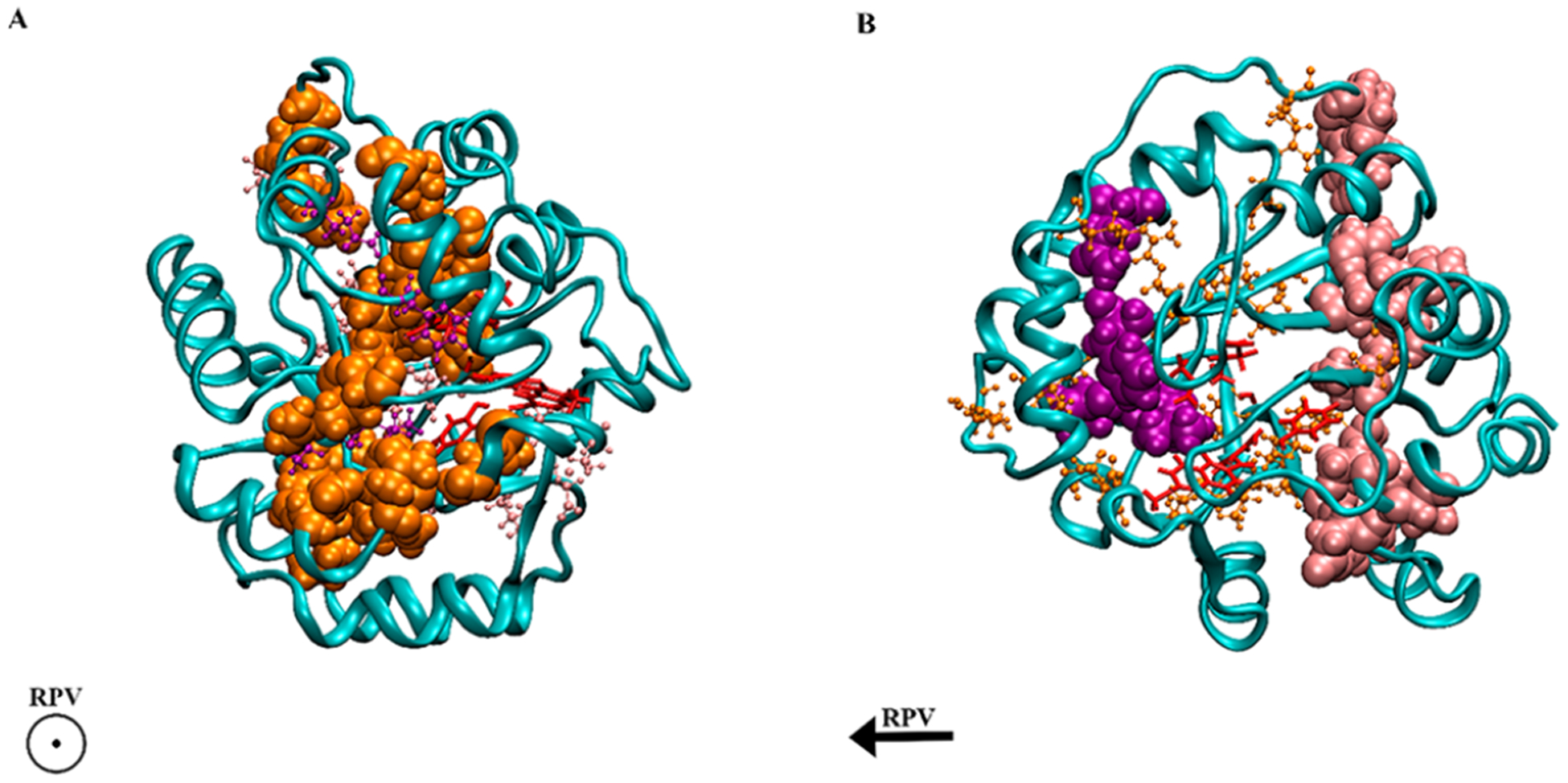
Variant V shown in both panels A and B with key interactions highlighted. Panel A has internal residues associated with decreased hydrogen bonding compared to variant IV highlighted in orange; the direction of the rate promoting vibration is out of the page. Panel B has internal residues associated with increased hydrogen bonding compared to variant IV highlighted in purple and pink; the direction of the rate promoting vibration is going from right to left. Substrate and QM residues (the active site) are highlighted in red.
Coherent Dynamical Structure Factor.
The directionality of the changes in the hydrogen bonding network motivated further analysis into the dynamics of the protein. To probe how 13 mutations altered the dynamics and mechanism of these enzymes, we calculated the coherent dynamical structure factor, S(k,ω). The coherent dynamical structure factor is defined in linear response theory and can be used to measure fluctuations in density within a system. This method is able to shed light on whether a collective motion exists in the protein in a particular direction or if residue motion is uncorrelated. In measuring S(k,ω), a direction and magnitude is chosen for k. In this work, three length scales are shown, representing an inter-residue distance (Figure 4), one-third the total length of the protein, and the length of the entire protein for variant IV and variant V, Figures SI 5 and 6. These length scales correspond to k values of 1.038, 0.346, and 0.130 for variant IV and 1.096, 0.365, and 0.132 for variant V. (Note: in each case, the magnitudes are different because the proteins have different dimensions and different average inter-residue distances). The directions that were measured are parallel to the direction of the rate promoting vibration and perpendicular to this direction. The directions chosen for variant IV are analogous to those previously described, though it is important to note that there is no rate promoting vibration in variant IV. Anisotropy in the structure factor shows that the system has a preferred direction in which a density fluctuation takes place, and this translates to a collective motion in the protein.
Figure 4.
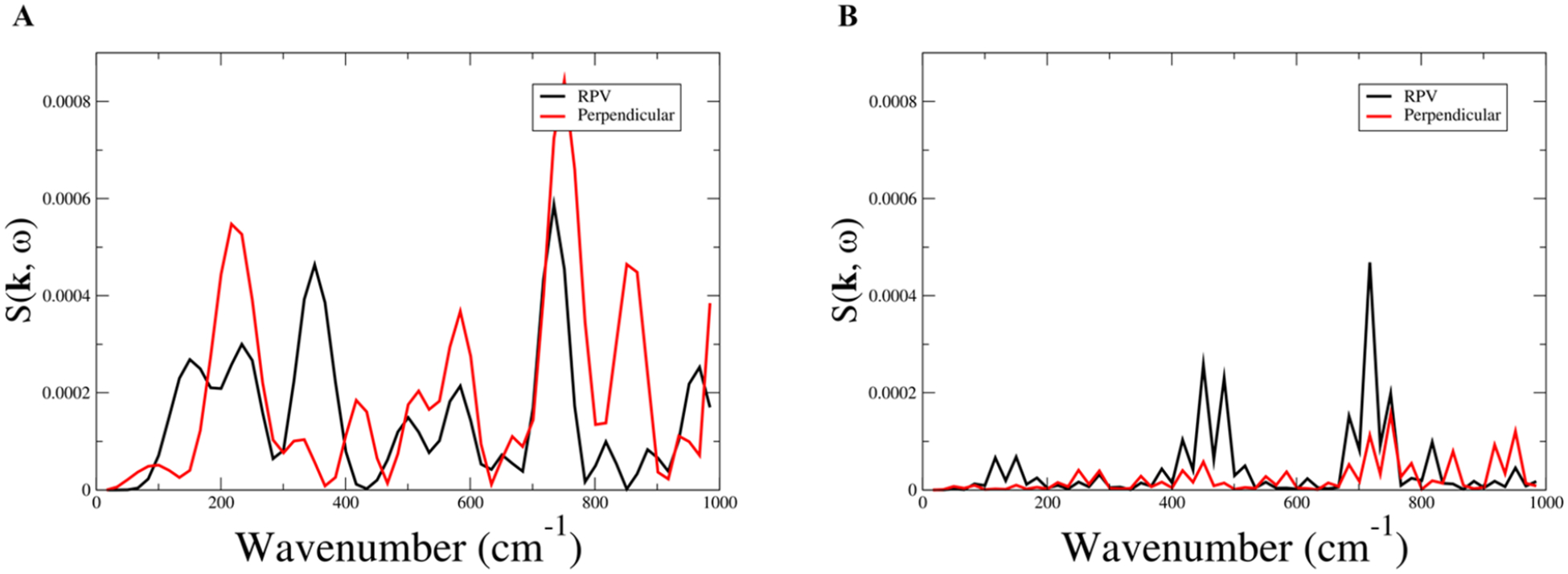
Coherent dynamical structure factor of variant IV (A) and variant V (B). Plotted are frequencies associated with an inter-residue distance, |k| = 1.038 and |k| = 1.096 for variant IV and V, respectively, in the direction of the rate promoting vibration (black) and perpendicular to the rate promoting vibration (red).
Figure 6.
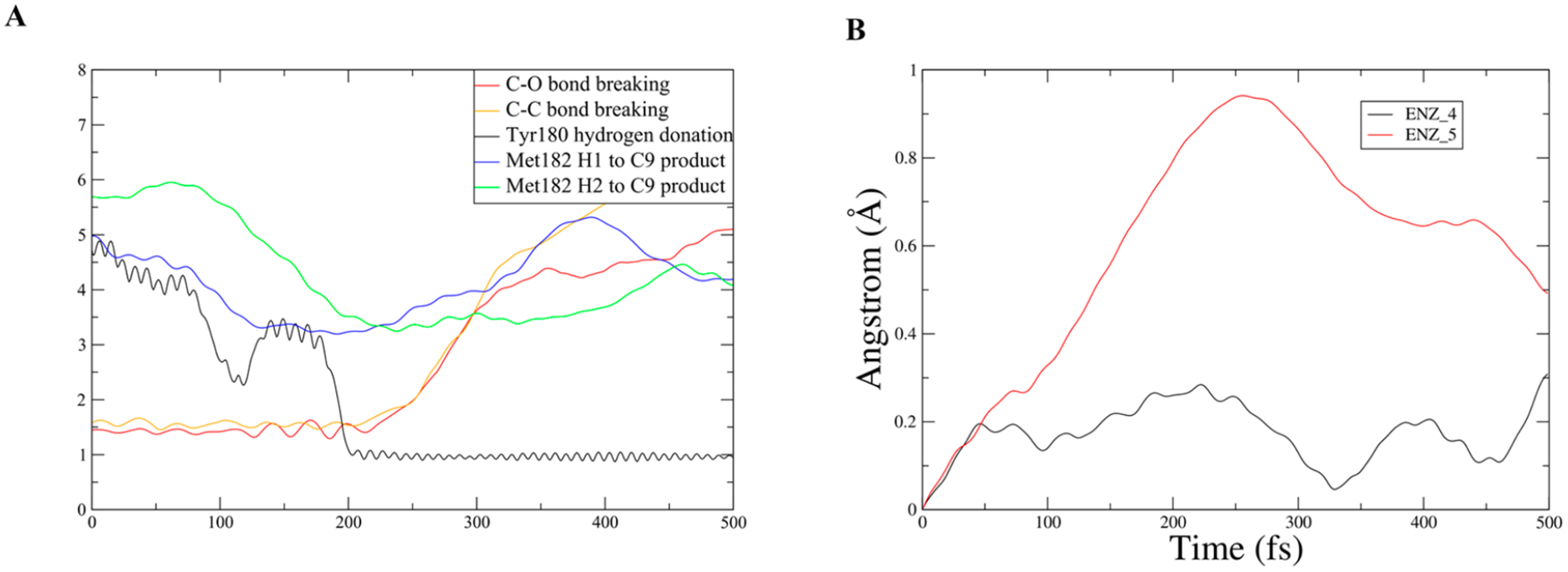
New motion identified in Met182 by analysis of changes in the hydrogen bonding network in the direction of the new softened axis. Panel A is an example unrestrained TPS trajectory showing both chemistry and the motion of Met182. Panel B is the RMSD of the terminal carbon in Met182 for both a restrained trajectory and an unrestrained trajectory.
To calculate the coherent dynamical structure factor, unbiased reactive trajectories from the TPS ensemble were taken as input, and incoherent averaging was used to improve signal-to-noise. In variant IV, the structure factor shows anisotropy at the largest possible scale (entire protein, Figure SI 4A); however, as the scale decreases, more features become apparent, and at the smallest scale (inter-residue, Figure 4A), the structure factor becomes isotropic with prominent peaks both parallel and perpendicular to the rate promoting vibration. This implies that, while a preferred direction of motion exists at the largest scale, variant IV does not have the structure to support this type of motion at the inter-residue scale. In variant V, the structure factor shows anisotropy at all scales measured (Figure SI 5), including the inter-residue scale Figure 4B, with the largest peaks being in the direction of the rate promoting vibration. Comparing the largest measured scale (Figure SI 4A and SI 5A), it is clear that the peak has shifted to a higher wavenumber, implying that the overall enzyme has become sturdier, which is reflected in both the hydrogen bonding data and in the volume fluctuations. The critical aspect of variant V’s coherent dynamical structure factor is that at all scales, the prominent peaks are parallel to the rate promoting vibration even at the inter-residue scale, which shows that variant V’s structure is capable of supporting directed local motion. The softening of the axis along the rate promoting vibration within the protein and the two newly rigid regions flanking the active site explain the anisotropy in the structure factor of variant V. This shows that directed evolution has significantly changed the density fluctuations going from variant IV to variant V, providing strong evidence for a collective motion as the genesis of the coupling of the rate promoting vibration to chemistry.
Rate Promoting Vibration.
We investigated whether the rate promoting vibration was directly involved in the density fluctuations identified in the coherent dynamical structure factor. To correlate the density fluctuations with protein dynamics, the spatial Fourier transform of femtosecond motions associated with Met231, Leu210, and Tyr180 were taken (Figure 5A–C) from the reactant state. Tyr180 is the catalytic base in the reaction, the distance measured is between the hydrogen being donated and the oxygen of the water leaving group (the oxygen down and to the right from Tyr180 in Figure 1B). Met231 and Leu210 were found to push Tyr180 toward the substrate, the distance measured is between the closest atom of Tyr180 and the closest atom of Met231 and Leu210, respectively. These are the same distance measurements involved in chemistry and the rate promoting vibration reported in our earlier work.16 In that work, we used methodology we had previously developed to estimate the rate promoting vibrations contribution to crossing the free energy barrier as approximately 1.4 kcal/mol for Tyr180 hydrogen donation, 7 kcal/mol for the C–O bond, and −0.6 kcal/mol for the C–C bond graphed in Figure 6.
Figure 5.

Spatial Fourier transform of key residues associated with the rate promoting vibration. Panel A is measured between Met231 and Tyr180; Panel B is measured between Leu210 and Tyr180, and Panel C is measured between Tyr180 and the oxygen of the water leaving group to which it will donate a hydrogen.
It is clear that the frequencies of the three residues femtosecond motion line up with the dominant inter-residue fluctuations in the coherent dynamical structure factor. Met231 has a peak around 775 cm−1 which lines up well with variant V’s largest density fluctuation which is the peak present at all measured scales of the coherent dynamical structure factor. Tyr180’s dominant peak at approximately 425 cm−1 lines up with the second largest peak in variant V’s coherent dynamical structure factor at the inter-residue scale. Leu210’s largest peak lines up with the smallest density fluctuation at 140 cm−1 in variant V’s coherent dynamical structure factor at the inter-residue scale. The fact that these residues have peaks associated with all of the significant peaks in variant V’s coherent dynamical structure factor implies that while the rate promoting vibration is seen to be a local phenomenon, the restructuring of delocalized motion is also important. It is also worth noting that the main contributors to the coherent structure factor shown in Figure 4B are well above kBT (roughly 200 cm−1), showing that the protein has been crafted via these mutations to channel thermal energy. This connects the newly reshaped density fluctuations to the motions our group has identified as being important for chemistry, implying that the new dynamics associated with variant V are the impetus of the rate promoting vibration.
Methionine 182.
In analyzing the changes to the hydrogen bonding network along the direction of the newly softened axis (direction of the rate promoting vibration), a motion was found which was overlooked in our previous work. Following a series of residues with reduced hydrogen bonding (Asp165–Asn161 and Asn161–Met182) to the active site revealed that Met182 had a significant motion which brings the terminal hydrogens in contact with the intermediate leaving group 6-methoxy-2-naphthaldehyde, Figure 6A. In the active site, Met182 is adjacent to Leu210, which is directly involved in the rate promoting vibration, and Tyr180, which is the catalytic base. While this motion is not directly coupled to chemistry like the one previously discussed, it does directly influence the motion of an intermediate leaving group. Met182 swings in and causes 6-methoxy-2-naphthaldehyde to move away from the active site, which is necessary because the active site is now much tighter and has a lower level of fluctuation. This prepares the enzyme for the next step in the reaction, which is hydrolysis. The Fourier transform of this motion was calculated and has a notable peak at approximately 425 cm−1 (Figure SI 7), which it shares with Tyr180 and again lines up well with the inter-residue dynamical structure factor of variant V. This peak is associated with the local rapid density fluctuations which characterize the rate promoting vibration. Engineering directed motion into an enzyme to not only improve a chemical transformation but also mechanically move an intermediate product in a way that is beneficial to the overall mechanism is a compelling idea. This shows that rapid protein dynamics can be operative in far subtler ways than, for example, simply the compression of a donor–acceptor distance.
To illustrate the magnitude of the effect Met182 has on 6-methoxy-2-naphthaldehyde, a restrained transition path ensemble was created where Met182 movements were restricted to create a comparison for the unrestrained ensemble. The RMSD of the terminal carbon of Met182 is plotted in Figure 6B to show the magnitude of the restraint. The RMSD of the center of mass of the ring closest to the C–C cleavage in 6-methoxy-2-naphthaldehyde was plotted for an example trajectory from the unrestrained and the restrained ensemble, and the slope of these graphs was also plotted (Figure 7 and Figure 8). In the unrestrained RMSD, it is clear that the majority of the distance traveled is in the first 150 fs after the C–C cleavage, and that while Met182 is in direct contact with the intermediate leaving group, the velocity rapidly climbs to six times its value after 150 fs (Figure 7B). In the restrained trajectory, the RMSD shows that the intermediate leaving group travels less than half the distance than in the unrestrained trajectory, and it is worth noting that the velocity becomes negative at times, which is indicative of random walk/diffusive behavior rather than a forced motion away from the active site (Figure 8B). The comparison between the motion in the two ensembles indicates that Met182’s fast dynamics accelerates the release of the intermediate product and that these dynamics are related to the restructuring of the hydrogen bonding network. The loss of motion seen in the restrained ensemble’s intermediate product shows that directed evolution has not only coupled protein motion to chemistry but also to an intermediate product release, which is vital for water to access the tighter active site and allow hydrolysis. This provides compelling data that low frequency modes in proteins are functionally significant and may be influenced by evolutionary pressure.
Figure 7.
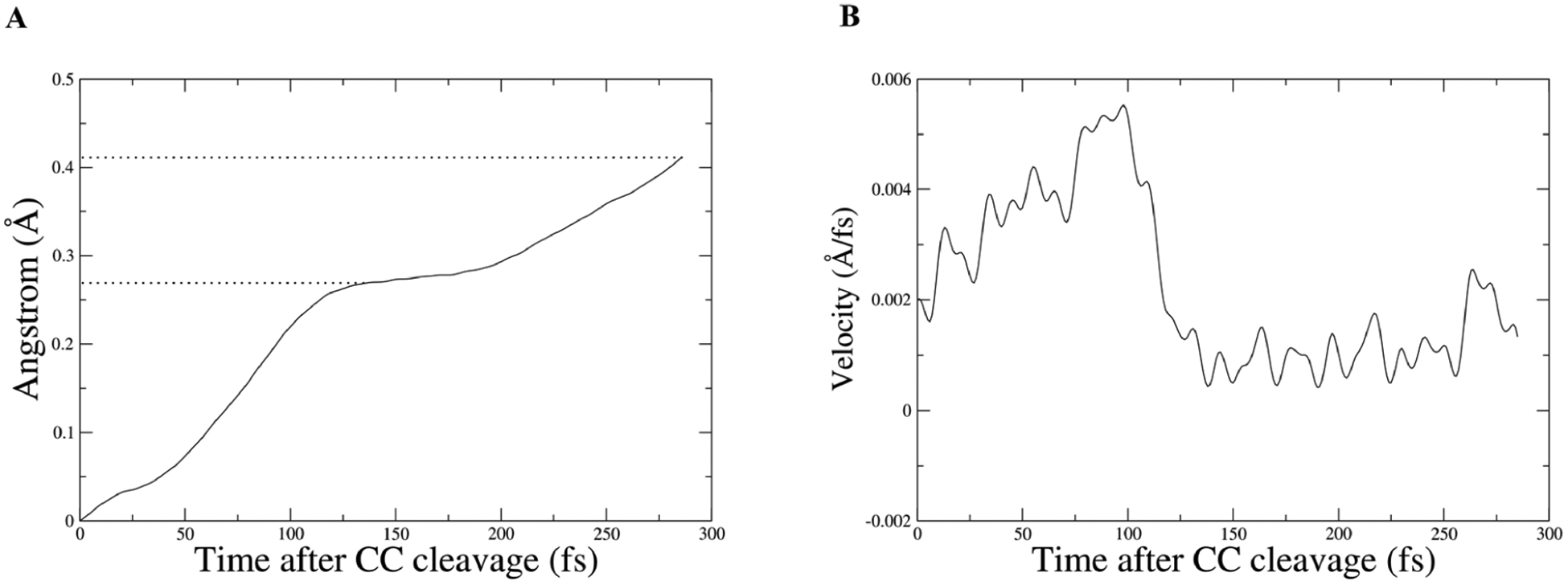
Panel A is the center of mass of ring carbons from 6-methoxy-2-naphthaldehyde, which were used to plot RMSD for the intermediate product from an unrestrained trajectory; dotted lines are to draw the eye to the time slice where the velocity drops and to the final distance. Panel B shows the corresponding velocity of the center of mass.
Figure 8.
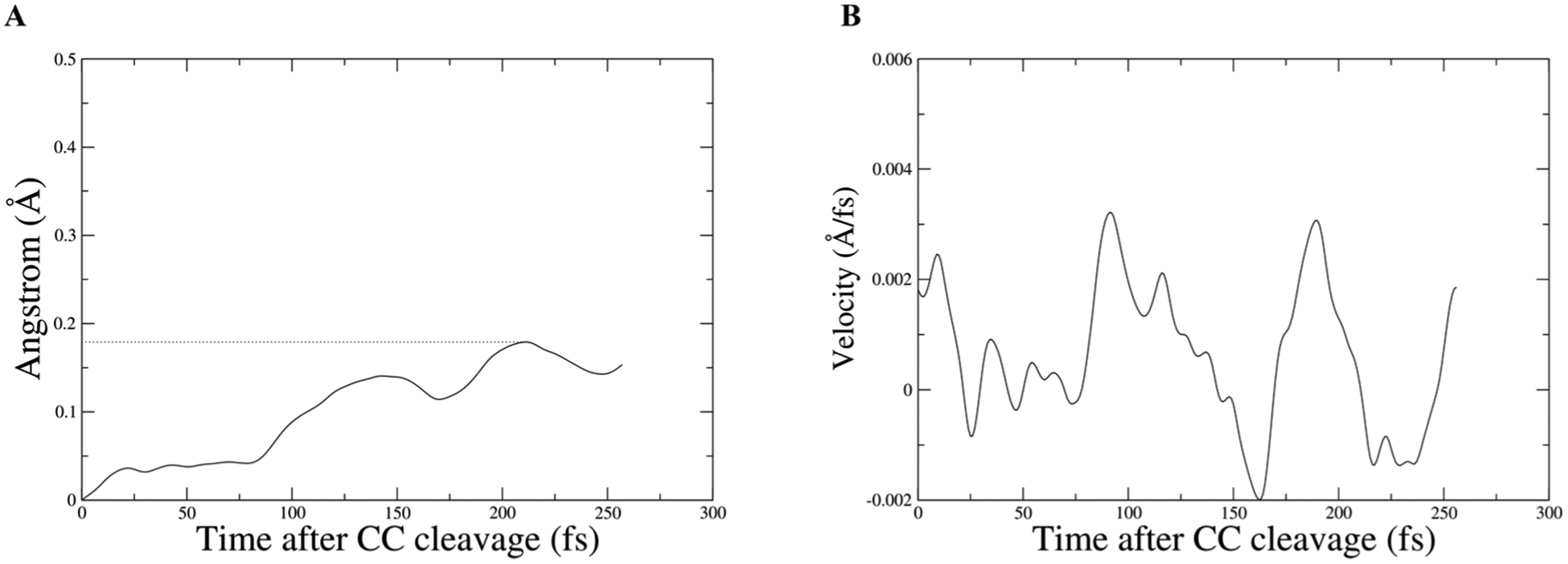
Panel A is the center of mass of ring carbons from 6-methoxy-2-naphthaldehyde, which were used to plot RMSD for the intermediate product from a restrained trajectory; dotted lines are to draw the eye to the farthest distance. Panel B shows the corresponding velocity of the center of mass.
DISCUSSION
The coupling of protein dynamics to chemistry we observed in this system inspired us to investigate how directed evolution engineered such a complex feature into the mechanism with 13 mutations scattered around the protein, changing not just the active site’s properties but affecting properties of the entire protein. The dynamic changes these mutations caused are critical to understanding why the fifth variant is a more efficient catalyst.
In analyzing the unbiased catalytic trajectories we created via transition path sampling, we have found that the 13 mutations fundamentally changed the dynamic properties of the entire protein. The density fluctuations of variant IV compared to variant V are dramatically different at the inter-residue scale, and looking at all scales measured, it is clear that the mutations altered the protein’s dynamics at all scales. This shows that the movement of atoms throughout the protein have been repurposed and coupled to chemistry because of the mutations. Correlating the density fluctuations to the motion of residues involved in the rate promoting vibration illustrates how the changes in density fluctuations are responsible for the creation of the rate promoting vibration. Dynamic changes in the hydrogen bonding network showed significant increases in hydrogen bonding flanking the active site on two sides, while a new softened axis runs approximately perpendicular to them and is in the same direction as the rate promoting vibration. The dynamic changes in the hydrogen bonding network are how the 13 mutations reorganized the way density fluctuates in variant V and also explain the anisotropy seen in the coherent dynamical structure factor.
One question that presents itself is how one could measure these effects in the laboratory. In fact, it is challenging, and this is a case where the coupling of computation to ensemble measurements like kinetics has much to offer. Experiments that might be added are inelastic X-ray scatter as an analogue to the coherent structure factor,24 and even X-ray crystallography with a careful examination of B-factors in the regions we suggest. There have also been recent advances in studying anisotropic vibrational energy transfer by incorporating noncanonical amino acids into a protein’s sequence, which can undergo ultrafast internal conversion which allows for site specific heating of the protein and subsequently probing the vibrational energy transfer.25
We also found that these dynamic changes did not sacrifice the efficiency of the active site design. The active site residues are now optimized to reduce the active site volume and the fluctuations of the active site volume, giving variant V an advantage in excluding water molecules in early steps in the reaction, which could interfere with chemistry. This tighter fit does not impede the next step in the mechanism (hydrolysis) because the active site also contains Met182, which has a newly developed motion which lines up with both the density fluctuations and the softened hydrogen bonding axis and is shown to help force the intermediate product out of the active site. This demonstrates that the reorganization of protein dynamics can be done in a way that is beneficial to more than one step of a mechanism. This information shows that directed evolution has provided significant evidence that, for certain reactions, rapid protein dynamics are critical to efficient enzyme design and further points the way to accomplish such design initially. One might speculate how to accomplish this purely ab initio. This of course would be very hard, but the lesson learned in this study is that the removal of H-bonds along a specific axis along with the addition of H-bonds in a perpendicular axis can induce flexibility in a desired fashion. Of course, this can also cause significant distortion of active sites that would render the putative changes useless. One advantage the approach presents is such changes can easily be studied computationally simply by equilibration and minimization with no reactive trajectories. The addition of such “objective functions” to the design portfolio adds complexity but may begin to allow more reliable design of protein based catalysts.
METHODS
QM/MM Simulations.
The starting point for all simulations was the crystal structure 5AN7 from the Protein Data Bank, which correspond to enzyme RA95.5–8F, variant V. Enzyme RA95.5–8, variant IV, was generated from 5AN7 using SWISSMODEL26 software to create a homology model as no crystal structure is available. To confirm that this process generated a stable structure, we ran classical MD on the system for 15 ns and saw no changes in active site residue RMSD or radius of gyration. We modified LLK((2E)-1-(6-methoxynaphthalen-2-yl)but-2-en1-one) in the crystal structure into 4-hydroxy-4-(6-methoxy2-naphthyl)-2-butanone, which is the substrate of the catalyst. We obtained parameters for 4-hydroxy-4-(6-methoxy2-naphthyl)-2-butanone according to the CHARMM force field using PARAMCHEM.27 We then deprotonated Lys83 in variants IV and V to form the Schiff base of the mechanism.
All molecular dynamics simulations were performed using the CHARMM molecular dynamics package.28,29 Each system was then partitioned into quantum and molecular mechanics regions. In enzymes IV and V, the quantum regions contain the substrate and residues 51, 83, 110, and 180. We note that residue 180 is mutated from phenylalanine in enzyme IV to tyrosine in enzyme V. The molecular mechanics region for all enzymes includes the rest of the protein, ions, and solvent.
The Generalized Hybrid Orbital method30 was used to couple the two regions through the Cβ of residues included in the quantum region. The quantum region was modeled using the PM3 semiempirical method, and the molecular region was modeled using the CHARMM36 force field. The simulations for all the enzymes were performed following the same protocol described below.
Each protein was solvated within a 60 Å sphere using TIP3 waters and neutralized using potassium ions. The system was then minimized for 100 steps using the steepest descent method followed by 2000 steps of the Adopted Basis Newton–Raphson method. The system was then heated slowly from 0 to 300 K for 150 ps and equilibrated at 300 K for 100 ps. During the previously described steps, the quantum region was switched on.
Transition Path Sampling.
We use transition path sampling31 to generate the ensemble of reactive trajectories. First, we define the order parameter to be the distance between the C atoms involved in the cleavage event. We define the reactant state to be a distance smaller than 1.70 Å and the product state to be a distance large than 1.70 Å. To begin the path ensemble, we create a biased trajectory by applying a harmonic force and allow the system to propagate for 250 fs. To generate new trajectories, we perturbed the momenta of a randomly chosen slice of a reactive trajectory, and using the new momenta, we allowed the system to propagate forward and backward in time for 250 fs. Several iterations are needed to ensure decorrelation from the initial biased trajectory.
Coherent Dynamical Structure Factor.
The unbiased 500 fs trajectories generated from transition path sampling were used to calculate the coherent dynamical structure factor. The coherent dynamical structure factor S(k,ω) was calculated for variant IV and variant V and is derived in linear response theory as the spatial Fourier transform of a density–density time correlation function.32,33
where G(r,t) is the density–density time correlation function and F(k,t) is an intermediate function. The power spectrum of F(k,t) is then taken and defined as S(k,ω),
The coherent dynamical structure factor is a measure of fluctuations in density within the system and is inversely dependent on length (k) and dependent on wavenumber (ω). Density fluctuations give insight into collective motions32 within a system, and this approach has the advantage of giving directional information associated with the motion. Three length scales were chosen for variant IV |k| = 0.130, 0.346, and 1.038 and three length scales were chosen for variant V |k| = 0.132, 0.365, and 1.096. The length scales correspond to the size of an entire protein, one-third the size of the entire protein, and an inter-residue scale. The values of |k| are different between variant IV and V because the two enzymes have slightly different sizes. To improve signal-to-noise, incoherent averaging34 was used on the structure factor from 30 unbiased reactive trajectories from the transition path ensemble. This technique improves signal-to-noise without needing to know the phase of the signal within the sample window.
Analysis of Reactant State.
To analyze differences in the reactant state of each variant, trajectories from late in the ensemble were extended using CHARMM molecular dynamics package. The initial position and negative momenta were taken and used to extend trajectories. Three 25 ps extended trajectories were created for variant IV and V to gather information on the femtosecond motions associated with key residues. These residues motions were measured, and the spatial Fourier transform was taken to compare to the dynamical structure factor. Tyr180 was measured from the hydrogen it will donate to the oxygen of the water leaving group; motion of Leu210 and Met231 was measured based on their closest atom to Tyr180, and Met182 was measured from the terminal carbon to the closest carbon of the naphthyl moiety of the intermediate product.
Seven 1.5 ns trajectories were created for variant IV and variant V with coordinates being saved every 100 steps to analyze volume fluctuations of the active site and the hydrogen bonding network. Volume calculations were done using the POVME35 software; the extended trajectories were prepared, and only ligand atoms closest to chemistry were chosen to define the origin of the pocket. This excludes ligand atoms close to the surface of the protein (far from chemistry) that will show more fluctuation as they are partially exposed to solvent. This was done to ensure the volumes calculated reflect only the interior of the active site and not space outside the active site. These trajectories were also analyzed using the VMD hydrogen bonds module, which defines a hydrogen bond as existing between donor and acceptor atoms within 3.0 Å. Hydrogen bonding results are reported as a percentage of frames where the hydrogen bond exists (Tables SI 1 and 2).
Supplementary Material
ACKNOWLEDGMENTS
All computer simulations were performed at the University of Arizona High Performance Computing Center on a Lenovo NeXtScale nx360 M5 supercomputer. This research was supported through the NIH program project grant P01GM127594 and NIH grant R01GM127594.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c01618.
Figures: (S1) POVME example; (S2) variant V with all residues with decreased hydrogen bonding highlighted and all mutations highlighted; (S3) variant V with all residues with increased hydrogen bonding highlighted and all mutations highlighted; (S4) coherent dynamical structure factor of variant IV; (S5) coherent dynamical structure factor of variant V; (S6) Fourier transform of motion associated with Met182 in variant V; Tables: (S1) list of residues with decreased hydrogen bonding; (S2) list of residues with increased hydrogen bonding (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acscatal.0c01618
REFERENCES
- (1).Akiva E; Copp JN; Tokuriki N; Babbitt PC Evolutionary and Molecular Foundations of Multiple Contemporary Functions of the Nitroreductase Superfamily. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 9549–9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Smith GP Phage Display: Simple Evolution in a Petri Dish. Angew. Chem., Int. Ed 2019, 58, 14428–14437. [DOI] [PubMed] [Google Scholar]
- (3).Esvelt KM; Carlson JC; Liu DR A System for the Continuous Directed Evolution of Biomolecules. Nature 2011, 472, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Giger L; Caner S; Obexer R; Kast P; Baker D; Ban N; Hilvert D Evolution of a Designed Retro-Aldolase Leads to Complete Active Site Remodeling. Nat. Chem. Biol 2013, 9, 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dougherty MJ; Arnold FH Directed Evolution: New Parts and Optimized Function. Curr. Opin. Biotechnol 2009, 20, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Packer MS; Liu DR Methods for the Directed Evolution of Proteins. Nat. Rev. Genet 2015, 16, 379. [DOI] [PubMed] [Google Scholar]
- (7).Romero P; Arnold F Exploring Protein Fitness Landscapes by Directed Evolution. Nat. Rev. Mol. Cell Biol 2009, 10, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).de los Santos ELC; Meyerowitz JT; Mayo SL; Murray RM Engineering Transcriptional Regulator Effector Specificity Using Computational Design and In Vitro Rapid Prototyping: Developing a Vanillin Sensor. ACS Synth. Biol 2016, 5, 287. [DOI] [PubMed] [Google Scholar]
- (9).Siegel JB; Zanghellini A; Lovick HM; Kiss G; Lambert AR; St. Clair JL; Gallaher JL; Hilvert D; Gelb MH; Stoddard BL; Houk KN; Michael FE; Baker D Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science (Washington, DC, U. S.) 2010, 329, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Jiang L; Althoff EA; Clemente FR; Doyle L; Rothlisberger D; Zanghellini A; Gallaher JL; Betker JL; Tanaka F; Barbas CF; Hilvert D; Houk KN; Stoddard BL; Baker D De Novo Computational Design of Retro-Aldol Enzymes. Science (Washington, DC, U. S.) 2008, 319, 1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zeymer C; Zschoche R; Hilvert D Optimization of Enzyme Mechanism along the Evolutionary Trajectory of a Computationally Designed (Retro-)Aldolase. J. Am. Chem. Soc 2017, 139, 12541–12549. [DOI] [PubMed] [Google Scholar]
- (12).Reetz MT The Importance of Additive and Non-Additive Mutational Effects in Protein Engineering. Angew. Chem., Int. Ed 2013, 52, 2658. [DOI] [PubMed] [Google Scholar]
- (13).Schwartz SD; Schramm VL Enzymatic Transition States and Dynamic Motion in Barrier Crossing. Nat. Chem. Biol 2009, 5, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pudney CR; Guerriero A; Baxter NJ; Johannissen LO; Waltho JP; Hay S; Scrutton NS Fast Protein Motions Are Coupled to Enzyme H-Transfer Reactions. J. Am. Chem. Soc 2013, 135, 2512–2517. [DOI] [PubMed] [Google Scholar]
- (15).Benkovic SJ; Hammes-Schiffer S Biochemistry - Enzyme Motions inside and Out. Science (Washington, DC, U. S.) 2006, 312, 208–209. [DOI] [PubMed] [Google Scholar]
- (16).Schafer JW; Zoi I; Antoniou D; Schwartz SD Optimization of the Turnover in Artificial Enzymes via Directed Evolution Results in the Coupling of Protein Dynamics to Chemistry. J. Am. Chem. Soc 2019, 141, 10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kries H; Blomberg R; Hilvert D De Novo Enzymes by Computational Design. Curr. Opin. Chem. Biol 2013, 17, 221–228. [DOI] [PubMed] [Google Scholar]
- (18).Obexer R; Godina A; Garrabou X; Mittl PRE; Baker D; Griffiths AD; Hilvert D Emergence of a Catalytic Tetrad during Evolution of a Highly Active Artificial Aldolase. Nat. Chem 2017, 9, 50–56. [DOI] [PubMed] [Google Scholar]
- (19).Masterson JE; Schwartz SD Changes in Protein Architecture and Subpicosecond Protein Dynamics Impact the Reaction Catalyzed by Lactate Dehydrogenase. J. Phys. Chem. A 2013, 117, 7107–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chen X; Schwartz SD Examining the Origin of Catalytic Power of Catechol O-Methyltransferase. ACS Catal. 2019, 9, 9870–9879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Johannissen LO; Hay S; Scrutton NS; Sutcliffe MJ Proton Tunneling in Aromatic Amine Dehydrogenase Is Driven by a Short-Range Sub-Picosecond Promoting Vibration: Consistency of Simulation and Theory with Experiment. J. Phys. Chem. B 2007, 10, 2631–2638. [DOI] [PubMed] [Google Scholar]
- (22).Kamerlin SC; Warshel A At the Dawn of the 21st Century: Is Dynamics the Missing Link for Understanding Enzyme Catalysis? Proteins: Struct., Funct., Genet 2010, 78, 1339–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zoi I; Antoniou D; Schwartz SD; Liu H; Begley T Linking Protein Dynamics to Enzyme Catalysis. Comprehensive Natural Products III 2020, 4002. [Google Scholar]
- (24).Cheatum CM Low-Frequency Protein Motions Coupled to Catalytic Sites. Annu. Rev. Phys. Chem 2020, 71 (1), 267–288. [DOI] [PubMed] [Google Scholar]
- (25).Baumann T; Hauf M; Schildhauer F; Eberl KB; Durkin PM; Deniz E; Löffler JG; Acevedo-Rocha CG; Jaric J; Martins BM; Dobbek H; Bredenbeck J; Budisa N Site-Resolved Observation of Vibrational Energy Transfer Using a Genetically Encoded Ultrafast Heater. Angew. Chem., Int. Ed 2019, 58 (9), 2899–2903. [DOI] [PubMed] [Google Scholar]
- (26).Waterhouse A; Bertoni M; Bienert S; Studer G; Tauriello G; Gumienny R; Heer FT; de Beer TAP; Rempfer C; Bordoli L; Lepore R; Schwede T SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Vanommeslaeghe K; MacKerell AD Jr. Automation of the {CHARMM} General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model 2012, 52 (12), 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Brooks BR; Brooks CL; Mackerell AD; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner AR; Feig M; Fischer S; Gao J; Hodoscek M; Im W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor RW; Post CB; PU JZ; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York DM; Karplus M CHARMM: The Biomolecular Simulation Program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M CHARMM: A Program for Macro-molecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem 1983, 4, 187–217. [Google Scholar]
- (30).Gao J; Amara P; Alhambra C; Field M A Generalized Hybrid Orbital (GHO) Approach for the Treatment of Link-Atoms Using Combined QM/MM Potentials. J. Phys. Chem. A 1998, 102, 4714–4721. [Google Scholar]
- (31).Bolhuis PG; Dellago C Practical and Conceptual Path Sampling Issues. Eur. Phys. J.: Spec. Top 2015, 224, 2409–2427. [Google Scholar]
- (32).Antoniou D; Schwartz SD Low-Frequency Collective Motions in Proteins. J. Theor. Comput. Chem 2003, 02 (02), 163–169. [Google Scholar]
- (33).Hansen JP; McDonald IR Theory of Simple Liquids, 2nd ed.; Academic Press: London, 2006. [Google Scholar]
- (34).Lyons RG Understanding Digital Signal Processing; Bernard Goodwin: Upper Saddle River, NJ, 2004. [Google Scholar]
- (35).Wagner JR; Sørensen J; Hensley N; Wong C; Zhu C; Perison T; Amaro RE POVME 3.0: Software for Mapping Binding Pocket Flexibility. J. Chem. Theory Comput 2017, 13 (9), 4584–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.