

Abstract
We assessed the virulence and anti-trypanosomal drug sensitivity patterns of Trypanosoma brucei rhodesiense (Tbr) isolates in the Kenya Agricultural and Livestock Research Organization-Biotechnology Research Institute (KALRO-BioRI) cryobank. Specifically, the study focused on Tbr clones originally isolated from the western Kenya/eastern Uganda focus of human African Trypanosomiasis (HAT). Twelve (12) Tbr clones were assessed for virulence using groups(n = 10) of Swiss White Mice monitored for 60 days post infection (dpi). Based on survival time, four classes of virulence were identified: (a) very-acute: 0–15, (b) acute: 16–30, (c) sub-acute: 31–45 and (d) chronic: 46–60 dpi. Other virulence biomarkers identified included: pre-patent period (pp), parasitaemia progression, packed cell volume (PCV) and body weight changes. The test Tbr clones together with KALRO-BioRi reference drug-resistant and drug sensitive isolates were then tested for sensitivity to melarsoprol (mel B), pentamidine, diminazene aceturate and suramin, using mice groups (n = 5) treated with single doses of each drug at 24 hours post infection. Our results showed that the clones were distributed among four classes of virulence as follows: 3/12 (very-acute), 3/12 (acute), 2/12 (sub-acute) and 4/12 (chronic) isolates. Differences in survivorship, parasitaemia progression and PCV were significant (P<0.001) and correlated. The isolate considered to be drug resistant at KALRO-BioRI, KETRI 2538, was confirmed to be resistant to melarsoprol, pentamidine and diminazene aceturate but it was not resistant to suramin. A cure rate of at least 80% was achieved for all test isolates with melarsoprol (1mg/Kg and 20 mg/kg), pentamidine (5 and 20 mg/kg), diminazene aceturate (5 mg/kg) and suramin (5 mg/kg) indicating that the isolates were not resistant to any of the drugs despite the differences in virulence. This study provides evidence of variations in virulence of Tbr clones from a single HAT focus and confirms that this variations is not a significant determinant of isolate sensitivity to anti-trypanosomal drugs.
Introduction
Human African trypanosomiasis (HAT), also known as sleeping sickness, is a vector-borne parasitic disease. It is caused by infection of humans with protozoan parasites belonging to the genus Trypanosoma. HAT is caused by two subspecies of trypanosomes, namely Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense [1]. They are transmitted to humans by tsetse flies (Glossina genus) [2]. Trypanosoma brucei gambiense is found in countries in West and Central Africa and causes a chronic infection [3]. A person can be infected for months or even years without major signs or symptoms of the disease [4]. When more evident symptoms emerge, the patient is often already in an advanced disease stage where the central nervous system is affected. Trypanosoma brucei rhodesiense is found in countries in eastern and southern Africa and causes an acute infection. Symptoms manifest within 2–4 weeks of an infective tsetse fly bite [3]. HAT develops in two stages namely, the early (hemolymphatic) and the late (meningo-encephalitic) stage. In the early stage of the disease, parasites proliferate in the blood and lymphatic system while in the late stage, parasites penetrate the blood brain barrier (BBB) and persist and proliferate in the central nervous system (CNS), causing an encephalitic reaction that leads to death if untreated or inadequately treated [5]. For first stage infections, there are no specific clinical signs and symptoms in both forms of the disease; fever, headache and loss of appetite are common [1] as well as anaemia in the monkey model [6]. With T.b. rhodesiense infections, first signs and symptoms are observed a few weeks after infection [1]. However, a mild form of chronic T. b. rhodesiense infections with incubation times of several months has been reported in Zambia [7]. The acute and the chronic HAT infections caused by T. b. rhodesiense in different foci differ in both their inflammatory response and pathology. According to Maclean et al (2008), the pathology encountered in the acute HAT infections is characterized by elevated Tumor necrosis factor alpha (TNF-α) while that encountered in the chronic HAT infections is characterized by elevated transforming growth factor (TGF-β) [8].
Treatment of Trypanosoma brucei rhodesiense infections involves the use of early stage drugs such as pentamidine and suramin [9] and late stage drugs such as melarsoprol; melarsoprol is the only drug recommended by WHO for treatment of late-stage T b rhodesiense infection, but can be lethal to 5% of patients owing to post-treatment reactive encephalopathy [10]. HAT therapy is further complicated by reports of drug resistance in different foci, including against suramin and melarsoprol in Tanzania [11] and against melarsoprol in south Sudan [12]. In their study, Pyana and colleagues [13] suggested that investigations into treatment failure in HAT and use of alternative drugs or treatment regimens should not only focus on differential genotypes of the parasites but also on differential virulence and tissue tropism as possible causes. The present study was therefore designed to investigate the occurrence of differential virulence of isolates recovered from Western Kenya/ Eastern Uganda HAT focus and the potential role of this variations on isolate sensitivity to anti-trypanosomal drugs using the mouse model. Studies on disease pathogenesis, parasite virulence, drug sensitivity and identification of new potential drug targets and staging biomarkers are commonly carried out in the mouse model based on its cost effectiveness, genetic similarity to humans estimated to be 85%, and ethical limitations of carrying out such studies in higher animal models or humans [14–16]. The study will also avail well characterized T.b.rhodesiense isolates for future studies.
Materials and methods
Ethics
All experimental protocols and procedures used in this study involving laboratory animals were reviewed and approved by Institutional Animal Care and Use Committee (IACUC) of Kenya Agricultural and Livestock Research Institute–Biotechnology Research Institute (KALRO-BioRI) Ref: C/Biori/4/325/II/53)
Experimental animals
The study used 6to 8 weeks old male Swiss White mice, each weighing 25–30 g live body weight. The animals were obtained from the Animal Breeding Unit at KALRO-BioRI, Muguga. The mice were housed in standard mouse cages and maintained on a diet consisting of commercial pellets (Unga® Kenya Ltd). All experiments were performed according to the guidelines set by the Institutional Animal Care and Use Committee (IACUC) of KALRO-BioRI. Briefly, water was provided ad libitum. All mice were acclimatized for two weeks, during which time they were screened and treated for ecto and endoparasites using ivermectin (Ivermectin®, Anupco, Suffolk, England). During the two-week acclimation period, pre-infection data were collected on body weights and packed cell volume once a week prior to parasite inoculation.
Trypanosomes and cloning
Twelve T.b. rhodesiense trypanosome stabilates (KETRI 2482, 2487, 3304, 3305, 3380, 3664, 3798, 3800, 3801, 3803, 3926, 3928) were selected from the KALRO-BioRI specimen bank. Cloning was carried out as described by [17]. Briefly, the trypanosome stabilates were inoculated into mice immunosuppressed using cyclophosphamide at 100 mg/kg for three consecutive days (total dose 300mg/kg) body weight (bwt) as previously described [18]. Animals were monitored daily for parasitaemia. When the mice attained a parasitaemia score of 3.2.x107 trypanosomes/mL [19], they were bled from the tail vein and the blood sample appropriately diluted using a mixture of PSG pH 8.0 and guinea pig serum in the ratio of 1:1. Using the hanging drop method [20], a single trypanosome was then picked using a syringe with a 25 gauge needle suspended in at least 0.2mls PSG pH 8.0 and injected intraperitoneally (ip) into a single immunosuppressed mouse. This was replicated ten times to increase the chances of success. Infected mice were then monitored for parasitaemia daily [19]. Any of the ten mice which became parasitaemic was euthanized using concentrated carbon dioxide, bled from the heart and the harvested trypanosomes cryopreserved in PSG pH 8.0 in 20% glycerol as a clone stabilate.
Virulence studies
Design of virulence study
Male Swiss White mice were housed in groups of 10 in standard mouse cages containing wood shavings as bedding material. The cryopreserved cloned parasites were thawed, and injected ip into immunosuppressed donor Swiss White mice for multiplication. The mice were euthanized using carbon dioxide [21] at peak parasitaemia and blood collected from the heart in EDTA for quantification as previously described [22]. The ten mice in each cage were infected with one Tbr clone, with each mouse receiving 1x104 trypanosomes injected intraperitoneally. The infected mice were monitored for pre-patent period, parasitaemia progression, PCV, body weight and survival time as virulence biomarkers. A control group of 10 non-infected mice was included in the study and monitored for changes in PCV, body weight and survival times.
Pre-patent period and parasitaemia progression
Blood for estimation of parasitaemia levels was collected daily for the first 14 days and thereafter three times in a week from each mouse using the tail tip amputation method [23]. The PP and parasitaemia progression were determined using the rapid matching method of [19, 24]. The infected mice were monitored for 60 days post infection.
Packed cell volume (PCV) and body weight changes
PCV was determined as outlined by [25]. Body weight (bwt) was measured once a week using a (Mettler Tolendo PB 302 ®, Switzerland) digital balance [22].
Survival times and virulence classification
The classification of trypanosome virulence was based on the survival of the infected mice as previously described [26]. The twelve T.b. rhodesiense clones were placed into four classes of virulence based on the survival of 60% or more of the infected mice as follows: very-acute (0–15 days), acute (16–30), sub-acute (31–45) and chronic classes (46–60). Each mouse’s survival time was determined on the basis of a ≥ 25% decline in PCV and consistently high parasitaemia levels of 1x109/ml for at least two consecutive days as previously described [27]. The mice were immediately euthanized by CO2 asphyxiation following the Institutional Animal Care and Use Committee (IUCAC) guidelines as earlier described by [28] and recorded as dead animal. Mice surviving at 60 dpi were equally euthanized, survival time recorded as 60 days and categorized as censored data.
Drug sensitivity study
Initially, sensitivity patterns for KALRO-BiORI laboratory reference isolates considered drug resistant (KETRI 2538) or drug-sensitive (KETRI 3738) were determined for Melarsoprol (Arsobal®, Aventis), Diminazene aceturate [(Veriben®, Ceva, France), Pentamidine (Pentacarinat®-Sanofi, UK) and Suramin (Germanin® Bayer), using dose rates ranging from 1–40 mg/kg body weight (Table 2) in order to identify cut-off points for characterizing isolates as drug resistant. Thereafter, the T. b. rhodesiense test clones were evaluated for sensitivity to the same drugs (Table 3). An isolate was considered drug-resistant if 2/5 (40%) of the infected and treated mice relapsed [11] after having been treated at 20mg/kg bwt.
Table 2. Results of drug sensitivity evaluation of reference KALRO-BioRI sensitive and resistant T b rhodesiense isolates.
| Sensitive Isolate KETRI 2537 | Resistant isolate KETRI 2538 | |||||
|---|---|---|---|---|---|---|
| Drug | Drug dose (mg/Kg) | Mice cured/5 | Status | Drug dose (mg/Kg) | Mice cured/5 | Status |
| MelB | 40 | 5 | (s) | 40 | 5 | (S) |
| 20 | 4 | (s) | 20 | 0 | (R) | |
| 10 | 5 | (s) | 10 | 0 | (R) | |
| 5 | 5 | (s) | 5 | 0 | (R) | |
| 2.5 | 5 | (s) | 2.5 | 0 | (R) | |
| 1 | 0/5 | (R | 1 | 0 | (R) | |
| diminazene aceturate | 40 | 5 | (s) | 40 | 5 | (S) |
| 20 | 5 | (s) | 20 | 0 | (R) | |
| 10 | 4 | (s) | 10 | 0 | (R) | |
| 5 | 5 | (s) | 5 | 0 | (R) | |
| 2.5 | 5 | (s) | 2.5 | 0 | (R) | |
| 1 | 2 | (R) | 1 | 0 | (R) | |
| Pentamindine | 40 | 5 | (s) | 40 | 5 | (S) |
| 20 | 5 | (s) | 20 | 1 | (R) | |
| 10 | 5 | (s) | 10 | 1 | (R) | |
| 5 | 4 | (s) | 5 | 0 | (R) | |
| 2.5 | 2 | (R) | 2.5 | 0 | (R) | |
| 1 | 0 | (R) | 1 | 0 | (R) | |
| Suramin Control | 40 | 5 | (S) | 40 | 5 | (S) |
| 20 | 5 | (S) | 20 | 5 | (S) | |
| 10 | 5 | (s) | 10 | 5 | (S) | |
| 5 | 5 | (s) | 5 | 5 | (S) | |
| 2.5 | 4 | (s) | 2.5 | 2 | (R) | |
| 1 - |
1 10 |
(R) | 1 - |
0 10 |
(R) | |
Key:- Not treated; The mice groups (n = 5) were treated 24hours post inoculation with the isolates and monitored for 60 days post treatment. An isolate is coded as sensitive (S) when at least 4/5 mice survived for at least 60 days without trypanosome relapse. All other results are coded as resistant (R).
Table 3. Results of drug sensitivity evaluation of T b rhodesiense clones in the mouse model.
| Stab. No KETRI | Virulence Class | Pentamidine Mice cured/ total treated | Melarsoprol Mice cured/ total treated | diminazene aceturate Mice cured/ total treated | Suramin Mice cured/ total treated | |||
|---|---|---|---|---|---|---|---|---|
| Very-acute | 5mg/kg | 20mg/kg | 1mg/kg | 20mg/kg | 2.5mg/kg | 20mg/kg | 2.5mg/kg | |
| 2482 | 5/5 | 5/5 | 5/5 | 5/5 | 4/5 (1) | 5/5 | 2/2 (3)b | |
| 3304 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | |
| 3803 | 4/4 (1)b | 5/5 | 5/5 | 4/4(1)b | 5/5 | 5/5 | 2/2(3)b | |
| 2487 | Acute | 4/5 (1)a | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 |
| 3801 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | |
| 3798 | Sub- acute | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 |
| 3926 | 4/5(1)a | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 3/3(2)b | |
| 3380 | Chronic | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 |
| 3928 | 4/4 (1)b | 4/4 (1)b | 4/4 (1)b | 5/5 | 1/1(4)b | 5/5 | 5/5 | |
Key: The mice were treated with single doses of various anti-trypanosomal drugs at 24 hours post infection and monitored for 60 days post treatment; a, number of mice which relapsed in each group during the 60 days of post-treatment monitoring; b, number of mice which died without parasitaemia relapse. All the isolates recorded at least 80% cure rates to all drug dose regimens and were therefore classified as sensitive.
Suramin and Pentamidine drugs (100% w/w) for the highest dosage of 40mg/kg bw were prepared by dissolving 40mg of these drugs in 10mls distilled water to give a concentration of 4 mg/ml. Diminazene aceturate (44.44% w/w active ingredient) for the highest dosage of 40mg/kg bw was prepared by dissolving 90mg of the drug powder in 10 mLs distilled water to give a concentration of 4mg/ml, whereas Melarsoprol (5 Ml vials of 180 mg) was first prepared by mixing (vortex) 1 Ml of the stock solution to 4 Ml of 50% propylene glycol to give a concentration of 7.2mg/ml (72mg/kg). This was further diluted to 40mg/kg by mixing(vortex) 5.6 ml of the 7.2mg/ml with 4.4 Mls of 50% propylene glycol to give a concentration of 4mg/ml (40mg/kg) The drug solutions for the 40mg/kg dose of each drug were then diluted serially using distilled water to give dosages for the 20, 10, 5, 2.5, 2, 1mg/kg.
Statistical analysis
Analysis was carried out to test if there are significant differences between the four classes of virulence using PP, parasitaemia progression, PCV, body weight changes and survival as the response variables. The data obtained from the study were summarized using descriptive statistics. The general linear model in SAS was used to test significance at p<0.05 level, between the means of the four virulence classes. Survival data analysis was carried out employing the Kaplan–Meier method on StatView (SAS Institute, Version 5.0.1) statistical package for determination of survival distribution function. Rank tests of homogeneity were used to determine the effect of virulence on host survival time [29].
Results
Survival time and classification
All control mice survived to the end of the experimental period of 60 days and their data (mean survival period) were therefore categorized as censored. The 12 T. b. rhodesiense clones exhibited variation in survival time and were classified into four classes of virulence based on these survival time data as shown (Table 1). A total of 3/12 clones were very acute, 3/12 were acute, 2/12 sub-acute and 4/12 chronic (Table 1).
Table 1. Changes in virulence biomarkers in mice infected with twelve clones of Trypanosoma brucei rhodesiense.
| Class | Clone ID | Locality of isolation | Iso. yr | PP passage No | Peak Para. | DPP | MST |
|---|---|---|---|---|---|---|---|
| very-acute | KETRI 2482 | Lumino, Uganda | 1969 | 5±0 7 | 1x106 | 8±0.2 | 9±0.4 |
| KETRI 3304 | Lugala, Uganda | 1971 | 5±0 64 | 7.6x108 | 7.8±0.4 | 9±0.4 | |
| KETRI 3803 | Busia, Kenya | 1961 | 4±0 2 | 9.3x108 | 6.4±0.2 | 8.2±0.3 | |
| Group mean ±SE | 4.7±0.9 | 8.9x108 | 7.4±0.5 | 8.8±0.2 | |||
| Acute | KETRI 2487 | Busoga, Uganda | 1972 | 4±0 5 | 6.2x108 | 7±0 | 18.1±0.7 |
| KETRI 3800 | Busia, Kenya | 2000 | 5.2±0.1 2 | 1.2x108 | 6±0 | 26.0±2.0 | |
| KETRI 3801 | Busia, Kenya | 1989 | 6±0 1 | 6.8x107 | 7.3±0.3 | 20.4±0.8 | |
| Group mean ±SE | 5.03±0.16 | 1.7x108 | 6.7±1.3 | 21.6±1.0 | |||
| Sub-acute | KETRI 3798 | Busia, Kenya | 1989 | 5.3±0.16 2 | 1.6x108 | 9.3±1.7 | 28.2±2.0 |
| KETRI 3926 | Busoga, Uganda | 1972 | 5±0 8 | 1.7x108 | 6.6±0.2 | 38.9±1.6 | |
| Group mean ±SE | 5.2±0.8 | 1.5x108 | 7.9±0.9 | 33.6±1.8 | |||
| Chronic | KETRI 3928 | Tororo, Uganda | 1992 | 6.3±0.3 2 | 7.9x107 | 7.0±0 | 51.8±4.1 |
| KETRI 3664 | Busia, Kenya | 1997 | 6.0±0 2 | 4.8x107 | 7.2±0.3 | 45.6±1.6 | |
| KETRI 3380 | Busoga, Uganda | 2000 | 5.9±0.5 3 | 1.3x108 | 7.3±0.2 | 55.5±2.3 | |
| KETRI 3305 | Lugala, Uganda | 1971 | 6.7±0.2 5 | 3.2x108 | 8.7±0.3 | 46.7±5.1 | |
| Group mean ±SE | 6.3±0.2 | 1.1x108 | 7.5±0.2 | 49.9±1.8 |
Control Non-infected - - - - >60
Key: PP-pre-patent period, Iso Yr–year of isolation, Par-parasitaemia, DPP -days to peak parasitaemia, MST-mean survival times,—No data
The mean survival time (MST) of isolates categorized as very acute ranged from a mean ± SEM of 8.7±0.2 days (KETRI 2482) to 9.0 ± 0.4 for KETRI 3304 (Table 1, S1 Fig (i)). These survival time data were not statistically different from each other (p > 0.05). Mice infected with the acute clones had MST ranging from a mean±SEM of 18.1±0.7 (KETRI 2487) to 26 ±2.0 for T.b rhodesiense clone KETRI 3800 (S1 Fig (ii)); these data were significantly (p < 0.0001) different from each other (S1 Fig (ii)). In mice infected with T. b. rhodesiense clones characterized as sub-acute, the survival time ranged from a mean ± SEM of 28.2±2.0 for KETRI 3798 to mean±SEM of 38.9 ± 1.6 for KETRI 3926 (S1 Fig (iii)) and were significantly (p<0.0001) different from each other. Mice infected with the T. b. rhodesiense clones characterized as chronic had MST values ranging from a mean±SEM of 45.6±1.6 for KETRI 3664 to a mean ± SEM of 55.5 ± 2.3 days for KETRI 3380 (S1 Fig (iv)) and were significantly (p<0.001) different from each other.
The survival time data for all the isolates in each virulence class were grouped together for the purpose of comparison among the groups. The overall MST was 8.7±0.2 for the very acute clones was, 21.6±1.0 for the acute clones 33.6±1.4 for the subacute clones and 50.0±1.8 for the chronic clones (Table 1, Fig 1). The Wilcoxon and Logrank tests had a p-value of 0.001, each showing that the MST of isolates in the different classes were significantly different.
Fig 1. The survival times for mice (n = 10) infected with twelve T. b. rhodesiense clones.

The clones were classified as (i) very-acute (0–15 dpi), (ii) acute (16–30 dpi), (iii) sub-acute (31–45 dpi), (iv) and chronic Tbr (46–60 dpi) all classes of virulence grouped together.
Pre-patent period and parasitaemia progression
The mean ±SEM pre-patent period in mice infected with very acute isolates ranged between 4±0.0 (KETRI 3803) to 5±0.0 (KETRI 2482 and 3304) and 4.7±0.09 when isolates are considered as a group (Table 1). The mean pre-patent period in mice infected with the acute class of the isolates ranged between 4±0.0 (KETRI 2487) and 6.0±0.0 (KETRI 3801) and 5.03±0.2 when they are considered as a class. With mice infected with sub-acute clones, the mean pre-patent period ranged between 5.0±0.0 (KETRI 3926) and 5.3±0.2 (KETRI 3798) and 5.2±0.08 when considered as a class. The pre-patent period for the mice infected with the chronic isolates was in the range of 5.9±0.5 (KETRI 3380) to 6.7±0.2 (KETRI 3305) and 6.3±0.2 dpi when considered as a class (Table 1). When the pp data were grouped together for isolates in each virulence class, the mean ±SEM were 4.7±0.09 (very acute class), 5.03±0.2 (acute class), 5.2±0.08 (sub-acute class and 6.3±0.2 (chronic class). Despite the apparently increasing trend of these data, these differences were not statistically significant (p>0.05).
The parasitaemia patterns, results of the mean peak parasitaemia (±SE), and time (days) to peak parasitaemia (DPP) of the T. b. rhodesiense clones as shown in (Table 1, Fig 2). In mice infected with the three (3) very-acute clones, the mean parasitaemia of each clone was characterized by a single wave as compared to two waves for mice infected with the acute, subacute and chronic clones (S2 Fig (i); S2 Fig (ii), S2 Fig (iii) and S2 Fig (iv)).
Fig 2. Parasitaemia progression in mice (n = 10) infected with the four classes of T. b. rhodesiense clones.

(i) Very-acute, (ii) acute, (iii) sub-acute and (iv) chronic clones all classes of virulence grouped together.
Trypanosoma b. rhodesiense clone specific variation was observed in the mean peak parasitaemia and time (days) to peak parasitaemia (Table 1). In the very acute virulence class, clone KETRI 3803 attained the highest mean peak parasitamia of 9.3 x 108 trypanosmes/mL of blood as compared to 1 x 106 trypanosomes/mL of blood for clone KETRI 2482 (Table 1), a 100-fold difference; these differences were statistically significant (p < 0.001). The KETRI 3803 clone attained peak parasitaemia faster (6 days) as compared to 8 days for the KETRI 2482 (Table 1). The third clone in this class, KETRI3304 was intermediate with respect to both parameters. Out of the three clones classified as acute, KETRI 3800 and 3801 had significantly (p 0.001) lower mean peak parasitamia parasitaemia when compared to KETRI 2487 (S2 Fig (ii)). A comparable pattern was observed for the sub-acute clones, with KETRI 3926 showing a significantly (p<0.001) lower parasitaemia than clone KETRI 3798 (S2 Fig (iii)). Similarly, clone KETRI 3305 recorded significantly lower parasitaemia (S2 Fig (iv)) when compared to the rest of the Tbr clones classified as chronic.
When parasiataemia data for T. b. rhodesiense clones in each virulence class were grouped together and compared, parasitaemia was significantly (p<0.001) higher in mice infected with very- acute clones (Fig 2).
Packed cell volume (PCV)
The pre-infection PCV data for all infected and control mice groups (Fig 3) were not statistically different (p > 0.05). The PCV of the infected mice declined significantly (p < 0.001) following infection when compared with the PCV of the non-infected control mice which remained largely constant throughout the duration of the study (Fig 3). However, the onset and severity of the anemia, as shown by the decline in PCV, was most prominent for mice infected with the isolates classified as very acute (Fig 3). In these mice, the PCV declined significantly (p<0.001), from 49.7±0.8 at baseline (day 0) to 26.0±0.5 at 14 dpi equivalent to 47.7% decline. The lowest infection-related decline in PCV (Fig 3) was recorded in the mice infected with isolates classified as chronic clones, with the PCV declining from 49.6±0.9at baseline to 43.5±1.0at 14 dpi (12.3%).
Fig 3.

Mean ± SE PCV decline in mice (n = 10) infected with T.b. rhodesiense (i) very-acute isolates, (ii) acute isolates, (iii) sub-acute isolates and (iv) chronic isolates clones all classes of virulence grouped together.
The changes in PCV varied significantly between T. b. rhodesiense clones of the same virulence class (Fig 3). In mice infected with very-acute clones, KETRI 3304 infected mice recorded the highest decline from 48.25±1.6 at day 0 to 39± 2.4 at 7 dpi equivalent to 19.2% (S3 Fig (i)) whereas within the acute clones infected mice, KETRI 2487 infected mice recorded the highest decline from 47.7±1.8 at day 0 to 38.8±1.4 at 7 dpi equivalent to 18.7% (S3 Fig (ii)). In mice infected with the sub-acute clones, KETRI 3978 recorded the highest decline from 53.4±1.1 at day 0 to 47.9±0.6 at 7dpi equivalent to 10.3% (S3 Fig (iii)) and in mice infected with the chronic clones, KETRI 3305 infected mice registered the highest decline from 54±2 at day 0 to 47±1.4 at 7dpi equivalent to 13% (S3 Fig (iv)).
When the mean PCV data of all isolates in each virulence class were grouped together and compared, the decline in PCV was significantly (p<0.001) higher in mice infected with very-acute clones (Fig 3); the PCV of mice infected with the very acute clones declined from 49.7±0.8 to 26.0±0.5 at 14 DPI by an average of 47.7% as compared to an average decline of 12.3% for mice infected with the chronic Tbr clones.
Body weight
The mean ±SE pre-infection body weight data for infected and control groups (Fig 4) were not statistically different (p > 0.05). Between 7 and 14 days post infection, all infected mice groups exhibited a decline in mean body weight (S4 Fig (i)–S4 Fig (iv)) while the body weight of un-infected control mice did not change (Fig 4). However, mice groups infected with clones classified as acute, sub-acute and chronic exhibited recovery of their body weights starting 14 dpi. Mice group infected with isolates in the very acute virulence class did not survive beyond 14 dpi (Fig 4).
Fig 4. Mean ± SE body weight changes in mice (n = 10) infected with T.b. rhodesiense.
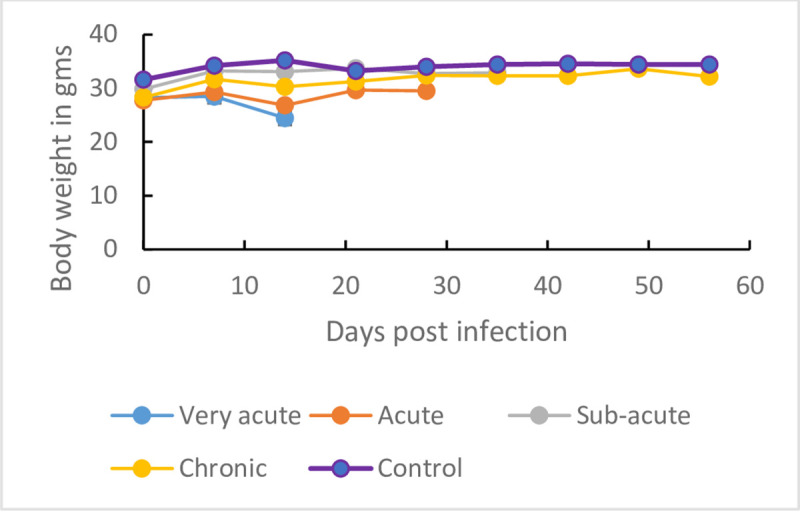
(i) very-acute isolates, (ii) acute isolates, (iii) sub-acute isolates and (iv) chronic isolates all classes of virulence grouped together.
Drug sensitivity results
The results of the drug-sensitivity testing for the reference sensitive (Tbr KETRI 3738) and drug-resistant (Tbr KETRI 2538) isolates are shown in (Table 2). The reference drug-resistant isolate was confirmed to be resistant to melarsoprol, Pentamidine and Diminazene aceturate at dose rates ranging from 1–20 mg/kg body weight (Table 2). However, infected mice were cured with all three drugs at a dose rate of 40 mg/kg body weight. With respect to suramin, the reference resistant isolate was sensitive to all doses equal to or greater than 5 mg/kg body and is therefore characterized as sensitive (Table 2). On the other hand, reference drug-sensitive isolate was confirmed to be sensitive to all doses of melarsoprol, ranging from 1–40 mg/kg bwt. It was also fully sensitive to diminazene aceturate at dose rates ranging from 2.5-40mg/kg bwt. The reference sensitive isolate was sensitive to pentamidne at all doses above 4 mg/kg bwt (Table 2). It was also sensitive to all doses of suramin equal to or greater than 2.5 mg/kg (Table 2)
The results of drug sensitivity experiments for the test Tbr clones are summarized in (Table 3). All the isolates recorded at least 80% cure rates for all drug dose regimens evaluated in this study (Table 3) and were therefore classified as sensitive. However, a few cases of relapsewere observed in 1/5 (20%) mice infected with KETRI 2482 (very-acute group) and treated with diminazene aceturate at 2.5mg/kg, and KETRI 2487 (acute) and KETRI 3926 (sub-acute) treated with pentamidine at 5mg/kg. In mice infected with KETRI 3928 (Chronic), 4/5 treated with diminazene aceturate at 2.5mg/kg and 5/5 treated at 20 mg/kg died at 47 days post treatment and without detectable parasites (Table 3), This is presumably due to pneumonia resulting from cold environment caused by a leaking water bottle. No relapses were observed in mice groups that were treated with either melarsoprol (1 and 20 mg/kg) or suramin at 2.5 mg/kg (Table 2)
Discussion
In this study, we characterized the virulence and anti-trypanosomal drug sensitivity patterns of 12 T. b. rhodesiense cloned stabilates. We used T. b rhodesiense clones because they represent a homogeneous population of genetically identical trypanosomes thus making subsequent studies based on these clones more reproducible and reliable as previously reported [30]. The results demonstrated the existence of variation in virulence of T. b. rhodesiense cloned stabilates (Table 1) which is interesting because all study isolates were originally recovered from western Kenya and eastern Uganda, regions that are considered to belong to the same Busoga focus of HAT. Our results are in agreement with a study on a number of isolates from eastern Uganda in mice which showed that distinct acute and chronic strains of T. b. rhodesiense circulated in the focus [31]. The results are also also in agreement with a previou report of variations in virulence of T. b. gambiense isolates [32].
We used mean survival time (MST) of mice post-infection as the main indicator of virulence as previously reported [26, 27, 33]; and observed that the Tbr isolates were well distributed among the four virulence classes. Infective isolates that allowed mice to have long survival times, hence chronic infections, may indicate presence of enriched population of stumpy forms which aids in prolonging host survival and enhancing the probability of parasite transmission [34]. The mean survival times for the very acute clones was 8.7 days suggesting the hosts were overwhelmed by the first parasitaemia peak before the proliferating slender forms differentiated into short stumpy forms [35]. The majority (11/12) of the Tbr clones used in this study had undergone a minimal number of (1–8) passages since isolation (Table 2). The only isolate that had undergone a significant (64) number of passages (Table 2) did exhibit notably different virulence characteristics from the other clones in the class, suggesting therefore that the observed differences in isolate virulence are an intrinsic attribute as previously reported [36].
Parasitaemia progression differed significantly among Tbr isolates assigned to different virulence classes on the basis of survival time. This finding is in agreement with previous studies in which virulence of different species of trypanosomes was characterised using parasitaemia, intensity of anaemia (PCV) and weight loss experienced by the host during the infection period [27]. In our study, parasitaemia of isolates in the very-acute virulence class was represented by a single wave whereas the acute, sub-acute and chronic virulence classes were represented by two waves. (Fig 2). This is in agreement with studies by [37] who observed that acute infections result from uncontrolled proliferation of the slender trypanosome forms without differentiation into short stumpy forms and hence kills the host before tsetse transmission takes place [37]. In contrast, chronic infection is characterized by appearance of progressive waves of parasitaemia, with each distinct wave being composed of trypanosomes with antigenically distinct coats, and with parasites easily differentiating into the transmissible short stumpy forms. This perhaps explains why highly virulent trypanosomes are not easily transmissible as was observed by [22] that tsetse flies infected with chronic T. b. brucei recorded highest mature infection as opposed to those infected with highly virulent trypanosomes. Our results are important as they reveal that the majority of T. b. rhodesiense infections are in the bracket of (acute, sub-acute and chronic) classes of virulence and can easily be transmittable.
In the present study, all infected mice recorded a decline in PCV signifying the development of T. b. rhodesiense induced anemia. Our observation was in agreement with previous studies which reported anaemia as a key feature both in humans [38] and in the monkey model [6]. As with parasitaemia and survival time parameters, the development of anemia was significantly pronounced in mice infected with very-acute clones. This finding is consistent with observations by [39] who reported that acute infection of mice with Trypanosoma cruzi was characterized by an exponential growth of parasites and high mortality accompanied by anemia. A similar observation was made by [27] in mice infected with Trypanosoma evansi. In contrast, anaemia in mice infected with clones in the other various classes of virulence (acute, sub-acute and chronic) stabilized or recovered characteristic of the chronic phase anaemia [40]. The severity of anemia is determined by parasite virulence, time lag from infection to therapeutic intervention and individual host differences [41].
Our results showed a decline in body weight in the early days of infection (7–14 dpi)
This decline was however not significant. Our observation is important as it confirms a previous observation [22] that body weight alone cannot conclusively serve as a virulence biomarker. Previous authors [42] attributed decline in body weight to reduced food intake. In our study, we did not measure the food intake. The failure by infected mice to register a significant decline in body weight calls for further investigation on the causes of body weight changes in animals infected with trypanosomes especially after previous studies have recorded an increase in body weight in T.evansi [27] and in T. b. brucei or T. congolense [22] infected mice.
Our results on drug sensitivity tests showed that all the study isolates were sensitive to melarsoprol, pentamidine, diminazene aceturate and suramin. The sensitivity of these isolates to suramin and melarsoprol is significant since currently they are the only drugs of choice recommended by WHO (2018) to treat early and late stages of Tbr HAT respectively. On the other hand the sensitivity of the Tbr isolates to diminazene aceturate, is an indicator of the utility of these drug when administered to livestock reservoirs of Tbr isolates as practiced in disease HAT control programmes in endemic countries [43] Interestingly, however, the single cases of relapses encountered in mice infected with KETRI 2482 (very- acute virulence class), KETRI 2487 (acute virulence class) and 3926 (sub-acute virulence class) were all against the two diamidines (pentamidine or dimainazene) but not against suramin or melarsoprol (Table 3) which is consistent with clinical practice of not using these specific diamidines to treat Tbr HAT (WHO, 2018). Overall, the fact that the test isolates were all sensitive (at least 80% cure rates) to the drugs suggests there was no relationship between isolates’ virulence and their sensitivity to anti-trypanosomal drugs which is in agreement with previous studies [44, 45]. In a study by Sokolova et al [46], these authors observed that resistance to nifurtimox did not compromise parasite virulence. This is despite previous studies reporting that drug resistant trypanosome have reduced virulence [47, 48].
The KALRO-BioRI reference isolate considered to be drug resistant was confirmed in this study to be resistant to melarsoprol, pentamidine and diminazene aceturate (Table 2). In general drug resistance is attributed to reduced drug uptake due the mutation or absence of a drug uptake gene [49] as well as by enhanced drug export, mediated by a multidrug resistance-associated protein [50]. The uptake of the three drugs, melarsoprol, pentamidine and diminazene is mediated by the P2 transporter [12, 51, 52] which explains why resistance to all three drugs is linked. In contrast, uptake of suramin by trypanosomes is not mediated by the P2 transporter, hence the reason why the trypanosome, KETRI 2538, retains sensitivity to suramin
In summary, this study has found that there is variation in virulence of cloned stabilates made from field isolates recovered from western Kenya/eastern Uganda HAT focus. Virulence is attributed to the production by the blood stream forms of membranous nanotubes that originate from the flagellar membrane and disassociate into free extracellular vehicles (EVs). This (EVs) contain several flagellar proteins that contribute to virulence [53]. Since our study was based on laboratory cloned T. b. rhodesiense stabilates, however, future studies should utilize the parent primary isolates. Our results are important as they have demonstrated that virulence is not a hindrance in the control of trypanosomiasis by chemotherapy.
Supporting information
(i): Mean survival times in mice infected with the very-acute clones of Trypanosoma brucei rhodesiense. (ii): Mean survival times in mice infected with the acute clones of Trypanosoma brucei rhodesiense. (iii): Mean survival times in mice infected with the sub-acute clones of Trypanosoma brucei rhodesiense. (iv): Mean survival times in mice infected with the chronic clones of Trypanosoma brucei rhodesiense.
(DOCX)
(i): Mean parasitaemia progression in mice infected with the very-acute clones of Trypanosoma brucei rhodesiense. (ii): Mean parasitaemia progression in mice infected with the acute clones of Trypanosoma brucei rhodesiense. (iii): Mean parasitaemia progression in mice infected with the sub-acute clones of Trypanosoma brucei rhodesiense. (iv): Mean parasitaemia progression in mice infected with the chronic clones of Trypanosoma brucei rhodesiense
(DOCX)
(i): Mean PCV decline in mice infected with the very acute clones of Trypanosoma brucei rhodesiense. (ii): Mean PCV decline in mice infected with the acute clones of Trypanosoma brucei rhodesiense. (iii): Mean PCV decline in mice infected with the sub-acute clones of Trypanosoma brucei rhodesiense. (iv): Mean PCV decline in mice infected with the chronic clones of Trypanosoma brucei rhodesiense.
(DOCX)
(i): Mean body weight changes in mice infected with the very acute clones of Trypanosoma brucei rhodesiense. (ii): Mean body weight changes in mice infected with the acute clones of Trypanosoma brucei rhodesiense. (iii): Mean body weight changes in mice infected with the sub-acute clones of Trypanosoma brucei rhodesiense. (iv): Mean body weight changes in mice infected with the very chronic clones of Trypanosoma brucei rhodesiense.
(DOCX)
Acknowledgments
We acknowledge the Director, KALRO for permission to publish this study. Our other acknowledgment goes to Dr. Johnson Ouma, former Center Director (Trypanosomiasis Research Center) BioRI for supervision and facilitation, technical staff of KALRO- BioRI and in particular John Ndichu, Jane Hanya for taking care of the infected mice. Gilbert Ouma and Mr. Mageto for the preparation of drugs.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
Materials were provided by the Kenya Government. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kuepfer I, Hhary E, Allan M, Edielu A, Burri C, et al. (2011) Clinical Presentation of T.b. rhodesiense Sleeping Sickness in Second Stage Patients from Tanzania and Uganda. PLoS Negl Trop Dis 5: e968 10.1371/journal.pntd.0000968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malvy D, Chappuis F (2011) Review Sleeping sickness. Clinical Microbiology and Infection 17: 986–995. 10.1111/j.1469-0691.2011.03536.x [DOI] [PubMed] [Google Scholar]
- 3.Brun R, Blum J, Chappuis F, Burri C (2010) Human African trypanosomiasis. Lancet 375: 148–159. 10.1016/S0140-6736(09)60829-1 [DOI] [PubMed] [Google Scholar]
- 4.Giroud C, Ottones F, Coustou V, Dacheux D, Biteau N, et al. (2009) Murine Models for Trypanosoma brucei gambiense disease progression—from silent to chronic infections and early brain tropism. PLoS Negl Trop Dis 3: e509 10.1371/journal.pntd.0000509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kennedy PG (2004) Human African trypanosomiasis of the CNS: current issues and challenges. Journal of clinical investigation 113: 496–504. 10.1172/JCI21052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thuita JK, Kagira JM, Mwangangi D, Matovu E, Turner CMR, et al. (2008) Trypanosoma brucei rhodesiense Transmitted by a Single Tsetse Fly Bite in Vervet Monkeys as a Model of Human African Trypanosomiasis. PLoS Negl Trop Dis 2: e238 10.1371/journal.pntd.0000238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mwanakasale Victor, Songolo Peter, Babaniyi Olusegun, Simarro P (2014) Clinical presentation of human African trypanosomiasis in Zambia is linked to the existence of strains of Trypanosoma brucei rhodesiense with varied virulence: two case reports. J Med Case Rep 8. 10.1186/1752-1947-8-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MacLean L, Chisi JE, Odiit M, Gibson WC, Ferris V, et al. (2004) Severity of Human African Trypanosomiasis in East Africa Is Associated with Geographic Location, Parasite Genotype, and Host Inflammatory Cytokine Response Profile. Infect Immun 72: 7040–7044. 10.1128/IAI.72.12.7040-7044.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Babokhov P, Sanyaolu AO, Oyibo WA, Fagbenro-Beyioku AF, Iriemenam3 NC (2013) A current analysis of chemotherapy strategies for the treatment of human African trypanosomiasis Pathog Glob Health 107: 242–252. 10.1179/2047773213Y.0000000105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kennedy P (2013) Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol 12: 186–194. 10.1016/S1474-4422(12)70296-X [DOI] [PubMed] [Google Scholar]
- 11.Kibona SN, Matemba L, Kaboya JS, Lubega GW (2006) Drug‐resistance of Trypanosoma b. rhodesiense isolates from Tanzania. Tropical Medicine and International health 11: 144–155. 10.1111/j.1365-3156.2005.01545.x [DOI] [PubMed] [Google Scholar]
- 12.Maina N, Maina K, Maser P, Brun R (2007) Genotypic and phenotypic characterization of Trypanosoma brucei gambiense isolates from Ibba, South Sudan, an area of high melarsoprol treatment failure rate. Acta Trop 104: 84–90. 10.1016/j.actatropica.2007.07.007 [DOI] [PubMed] [Google Scholar]
- 13.Pyana Pati P, Van Reet N, Mumba Ngoyi D, Ngay Lukusa I, Karhemere Bin Shamamba S, et al. (2014) Melarsoprol Sensitivity Profile of Trypanosoma brucei gambiense Isolates from Cured and Relapsed Sleeping Sickness Patients from the Democratic Republic of the Congo. PLoS Negl Trop Dis 8: e3212 10.1371/journal.pntd.0003212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kennedy PGE (2007) Animal Models of Human African Trypanosomiasis—Very Useful or Too Far Removed? Trans R Soc Trop Med Hyg 101: 1061–1062. 10.1016/j.trstmh.2007.05.001 [DOI] [PubMed] [Google Scholar]
- 15.Kennedy PGE, Rodgers J (2019) Clinical and Neuropathogenetic Aspects of Human African Trypanosomiasis. Front Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magez S, Caljon G (2011) Mouse models for pathogenic African trypanosomes: unravelling the immunology of host-parasite-vector interactions. Parasite Immunol 33: 423–429. 10.1111/j.1365-3024.2011.01293.x [DOI] [PubMed] [Google Scholar]
- 17.Otieno LH, Darji N (1985) Characterization of potentially man-infective Trypanosoma brucei from an endemic area of sleeping sickness in Kenya. Tropical Medicine and Parasitology 36: 123–126. [PubMed] [Google Scholar]
- 18.Wombou Toukam C, Solano P, Bengaly Z, Jamonneau V, Bucheton B (2011) Experimental evaluation of xenodiagnosis to detect trypanosomes at low parasitaemia levels in infected hosts. Parasite 18: 295–302. 10.1051/parasite/2011184295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herbert WJ, Lumsden WHR (1976) Trypanosoma brucei: A rapid “matching” method for estimating the host parasitaemia. Exp Parasitol 40: 423–431. 10.1016/0014-4894(76)90110-7 [DOI] [PubMed] [Google Scholar]
- 20.Chris JS, Rita FL, John MM (1982) Cloning of African Trypanosomes in Mice Immunosuppressed by Cyclophosphamide Treatment. The American Journal of Tropical Medicine and Hygiene 31: 1098–1102. 10.4269/ajtmh.1982.31.1098 [DOI] [PubMed] [Google Scholar]
- 21.Fisher S, Burgess WL, Hines KD, Mason GL, Owiny JR (2016) Interstrain Differences in CO2-Induced Pulmonary Hemorrhage in Mice. J Am Assoc Lab Anim Sci 55: 811–815. [PMC free article] [PubMed] [Google Scholar]
- 22.Gitonga PK, Ndung’u K, Murilla GA, Thande PC, Wamwiri N, et al. (2017) Differential virulence and tsetse fly transmissibility of Trypanosoma congolense and Trypanosoma brucei strains. Onderstepoort Journal of Veterinary Research 84: a1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saito M, Lourenço A, Kang H, Rodrigues C, Cabral L, et al. (2016) New approaches in tail-bleeding assay in mice: improving an important method for designing new anti-thrombotic agents. Int J Exp Pathol 97: 285–292. 10.1111/iep.12182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herbert WJ, and Lumsden WH (1976) Trypanosoma brucei: A rapid “matching” method for estimating the host parasitaemia. Experimental Parasitology 40: 427–431. 10.1016/0014-4894(76)90110-7 [DOI] [PubMed] [Google Scholar]
- 25.Naessens J, Kitani H, Nakamura Y, Yagi y, Sekikawa K, et al. (2005) TNF—α mediates the development of anaemia in murine Trypanosoma brucei brucei infection, but not the anaemia associated with murine Trypanosoma congolense infection. Clin Exp Immunol 139: 405–410. 10.1111/j.1365-2249.2004.02717.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masumu J, Marcotty T, Geysen D, Geerts S, Vercruysse J, et al. (2006) Comparison of the virulence of Trypanosoma congolense strains isolated from cattle in a trypanosomiasis endemic area of eastern Zambia. Int J Parasitol 36: 497–501. 10.1016/j.ijpara.2006.01.003 [DOI] [PubMed] [Google Scholar]
- 27.Kamidi C, Auma J, Mireji P, Ndungu K, Bateta R, et al. (2018) Differential virulence of camel Trypanosoma evansi isolates in mice. Parasitology: 1–8. 10.1017/S0031182017002359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez-Gutierrez M, Correa-Londoño L, Castellanos J, Gallego-Gómez J, Osorio J (2014) Lovastatin Delays Infection and Increases Survival Rates in AG129 Mice Infected with Dengue Virus Serotype 2. PLoS ONE 9: e87412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Everitt B, Der G, editors (1998) A Handbook of Statistical Analysis Using SAS. London. [Google Scholar]
- 30.Gomes M, Romanha A, Gonçalves A, Chiari E (1991) Stability of isoenzyme and kinetoplast DNA (k-DNA) patterns in successively cloned Trypanosoma cruzi populations. Mem Inst Oswaldo Cruz 86: 379–385. 10.1590/s0074-02761991000400001 [DOI] [PubMed] [Google Scholar]
- 31.Smith D, Bailey J (1997) Human African trypanosomiasis in south-eastern Uganda: clinical diversity and isoenzyme profiles. Ann Trop Med Parasitol 91: 851–856. 10.1080/00034989760617 [DOI] [PubMed] [Google Scholar]
- 32.Holzmuller P, Biron D, Courtois P, Koffi M, Bras-Gonçalves R, et al. (2008) Virulence and pathogenicity patterns of Trypanosoma brucei gambiense field isolates in experimentally infected mouse: differences in host immune response modulation by secretome and proteomics. Microbes Infect 10: 79–86. 10.1016/j.micinf.2007.10.008 [DOI] [PubMed] [Google Scholar]
- 33.Motloang MY, Masumu J, Mans BJ, Latif AA (2014) Virulence of Trypanosoma congolense strains isolated from cattle and African buffaloes (Syncerus caffer) in KwaZulu-Natal, South Africa. Onderstepoort Journal of Veterinary Research 81: 679,677. 10.4102/ojvr.v81i1.679 [DOI] [PubMed] [Google Scholar]
- 34.MacGregor P, Szöoõr B, Savill NJ, Matthews KR (2012) Trypanosomal immune evasion, chronicity and transmission: an elegant balancing act Nat Rev Microbiol 10. 10.1038/nrmicro2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Magez S, Schwegmann A, Atkinson R, Claes F, Drennan M, et al. (2008) The Role of B-cells and IgM Antibodies in Parasitemia, Anemia, and VSG Switching in Trypanosoma brucei-Infected Mice. PLoS Pathog 4: e1000122 10.1371/journal.ppat.1000122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sacks DL, Selkirk M, Ogilvie BM, Askonas BA (1980) Intrinsic immunosuppressive activity of different trypanosome strains varies with parasite virulence. Nature 283: 476–478. 10.1038/283476a0 [DOI] [PubMed] [Google Scholar]
- 37.Matthews KR, McCulloch R, Morrison. LJ (2015) The within-host dynamics of African trypanosome infections. Philos Trans R Soc Lond B Biol Sci 370: 20140288 10.1098/rstb.2014.0288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chisi J, Misiri H, Yuriy Z, Nkhoma A, Sternberg JM (2004) Anaemia in human African trypanosomiasis caused by Trypanosoma brucei rhodesiense. East African medical journal 81: 505–508. 10.4314/eamj.v81i10.9232 [DOI] [PubMed] [Google Scholar]
- 39.Marcondes M, Borelli P, Yoshida N, Russo M (2000) Acute Trypanosoma cruzi infection is associated with anemia, thrombocytopenia, leukopenia, and bone marrow hypoplasia: reversal by nifurtimox treatment. Microbes Infect 2: 347–352. 10.1016/s1286-4579(00)00333-6 [DOI] [PubMed] [Google Scholar]
- 40.Amole BO, Clarkson AB Jr, Shear HL (1982) Pathogenesis of anemia in Trypanosoma brucei-infected mice. Infect Immun 36: 1060–1068. 10.1128/IAI.36.3.1060-1068.1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thuita JK, Wang MZ, Kagira JM, Denton CL, Paine MF, et al. (2012) Pharmacology of DB844, an Orally Active aza Analogue of Pafuramidine, in a Monkey Model of Second Stage Human African Trypanosomiasis. PLoS Negl Trop Dis 6: e1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sandra T, Filipa R-F, Tania C, Daniel P-N, Fabien G, et al. (2016) Trypanosoma brucei Parasites Occupy and Functionally Adapt to the Adipose Tissue in Mice. Cell Host Microbe 19: 837–848. 10.1016/j.chom.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamill L, Picozzi K, Fyfe J, Wissmann Bv, Wastling S, et al. (2017) Evaluating the impact of targeting livestock for the prevention of human and animal trypanosomiasis, at village level, in districts newly affected with T b rhodesiense in Uganda Infect Dis Poverty 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Korir SC, Mburu J, Shivairo R, Serem E, Arasa G, et al. (2013) Comparison of drug Sensitivity and Pathogenicity of Trypanosoma brucei rhodesiense isolates with their respective clones originating from Busia and Busoga. Journal of Natural Sciences Research 3. [Google Scholar]
- 45.Antoaneta YS, Wyllie S, Patterson S, Oza SL, Read KD, et al. (2010) Cross-Resistance to Nitro Drugs and Implications for Treatment of Human African Trypanosomiasis. Antimicrob Agents Chemother 54: 2893–2900. 10.1128/AAC.00332-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sokolova AY, Wyllie S, Patterson S, Oza SL, Read KD, et al. (2010) Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrobial Agents and Chemotherapy 54: 2893–2900. 10.1128/AAC.00332-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kagira JMaM N. (2007) Occurrence of multiple drug resistance in Trypanosoma brucei rhodesiense isolated from sleeping sickness patients. Onderstepoort Journal of Veterinary Research 74: 17–22. 10.4102/ojvr.v74i1.135 [DOI] [PubMed] [Google Scholar]
- 48.Egbe-Nwiyi T, Igbokwe I, Onyeyili P (2005) Diminazene Aceturate Resistance on the Virulence of Trypanosoma brucei for Rats. Journal of Comparative Pathology 133: 286–288s. 10.1016/j.jcpa.2005.05.002 [DOI] [PubMed] [Google Scholar]
- 49.Baker N, Koning HPd, Mäser P, Horn. D (2013) Drug resistance in African trypanosomiasis: the melarsoprol and pentamidine story. Trends Parasitol 29. 10.1016/j.pt.2012.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gehrig S, Efferth T (2008) Development of drug resistance in Trypanosoma brucei rhodesiense and Trypanosoma brucei gambiense. Treatment of human African trypanosomiasis with natural products Int J Mol Med 22: 411–419. [PubMed] [Google Scholar]
- 51.Fairlamb AH, Horn D (2018) Melarsoprol Resistance in African Trypanosomiasis. Trends in Parasitology 34. 10.1016/j.pt.2018.04.002 [DOI] [PubMed] [Google Scholar]
- 52.Mäser P, Lüscher A, Kaminsky R (2003) Drug transport and drug resistance in African trypanosomes. Drug Resist Updat 6: 281–290. 10.1016/j.drup.2003.09.001 [DOI] [PubMed] [Google Scholar]
- 53.Szempruch AJ, Sykes SE, Kieft R, Denison L, Becker AC, et al. (2016) Extracellular vesicles from Trypanosoma brucei mediate virulence factor transfer and cause host anemia. Cell Host Microbe 164: 246–257. 10.1016/j.cell.2015.11.051 [DOI] [PMC free article] [PubMed] [Google Scholar]