

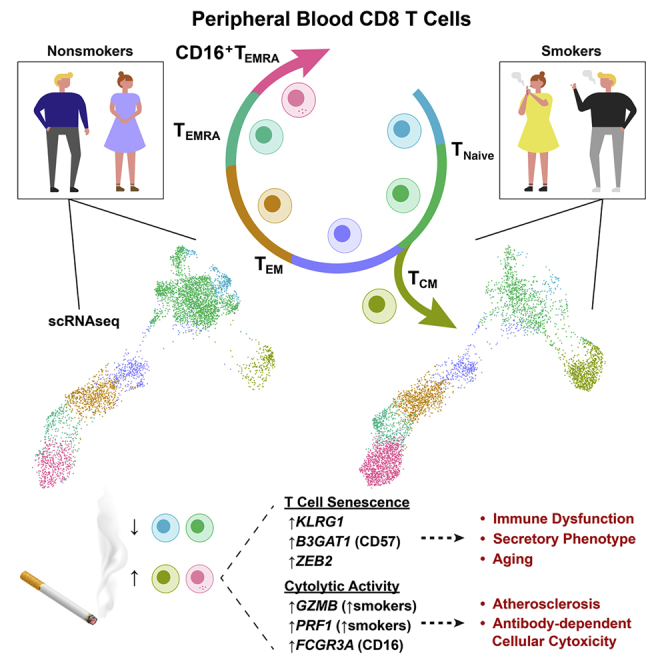
Summary
Tobacco smoke exposure contributes to the global burden of communicable and chronic diseases. To identify the immune cells affected by smoking, we use single-cell RNA sequencing on peripheral blood from smokers and nonsmokers. Transcriptomes reveal a subpopulation of FCGR3A (CD16)-expressing natural killer (NK)-like CD8 T lymphocytes that increase in smokers. Mass cytometry confirms elevated CD16+ CD8 T cells in smokers. Inferred as highly differentiated by pseudotime analysis, NK-like CD8 T cells express markers that are characteristic of effector memory re-expressing CD45RA T (TEMRA) cells. Indicative of immune aging, smokers’ CD8 T cells are biased toward differentiated cells, and smokers have fewer naive cells than nonsmokers. DNA methylation-based models show that smoking dose is associated with accelerated aging and decreased telomere length, a biomarker of T cell senescence. Immune aging accompanies T cell senescence, which can ultimately lead to impaired immune function. This suggests a role for smoking-induced, senescence-associated immune dysregulation in smoking-mediated pathologies.
Graphical Abstract
Highlights
Smoking shifts the composition of CD8 T cells from naive to differentiated states
NK-like CD16+ CD8 TEMRA cells are elevated in smokers and express GZMB and PRF1
DNA methylation links smoking dose with age acceleration and shortened telomeres
CD8 T, CD4 T, NKT, NK, and monocytes express senescence-linked genes in smokers
Smoking increases the risk of inflammatory diseases and decreases immunity. Martos et al. characterize immune cells and find that smoking reduces naive and increases late-stage CD8 T cells. They show that smoking dose is associated with age acceleration and shortened telomeres. These changes are consistent with immune aging and senescence.
Introduction
As a risk factor for human diseases, the global disease burden attributed to tobacco smoke exposure is substantial. The World Health Organization (WHO) estimates ∼6 million deaths per year from tobacco smoke exposure, resulting from both chronic and communicable diseases.1,2 In smokers, a decline in immunity and an increased risk of inflammatory diseases, such as atherosclerosis, make the argument that smoking-associated diseases are mediated by immune dysfunction. The development and progression of atherosclerotic lesions serves as an example of a complex immune-mediated pathology because T cells, monocytes, macrophages, dendritic cells (DCs), and B cells have been reported to be involved.3,4 Refining smoking-associated changes within immune populations will enhance our understanding of how dysfunctional immune subsets arise from exposure to tobacco smoke. This will facilitate the prevention of diseases by identifying immune cells to target for clinical intervention.
Smoking alters the epigenome and transcriptome of human blood leukocytes, in addition to DNA damage.5, 6, 7 In the article by Su et al.,5 we demonstrated that changes identified in isolated cell fractions, which correspond to major immune populations, were distinct from one another and whole blood. For example, ITGAL, expressed in T cells and involved in inflammation,8 had decreased methylation in smokers’ T cells but not in whole blood or isolated cell fractions. It follows that bulk data from isolated fractions, comprising multiple subtypes, would similarly mask meaningful changes, especially when differences arise in low-frequency subsets. As such, the interpretation of bulk genomic approaches is limited because changes could indicate an altered distribution of cell (sub)populations or changes in expression within (sub)populations. The recent development of single-cell methods provides the technology to resolve smoking-associated (sub)population composition changes, examine gene expression differences, and identify rare subtypes obscured by bulk fraction data. In addition, multiparameter data allow us to concordantly study multiple cell types from the same individuals.
To identify smoking-affected cell (sub)populations and connect observed immune cell changes with smoking-associated diseases, we characterized gene expression profiles and cell surface marker phenotypes from primary peripheral blood mononuclear cells (PBMCs) from four nonsmokers and four smokers by single-cell RNA sequencing (scRNA-seq) and mass cytometry. The combination of transcriptome profiling and immunophenotyping provides higher confidence in the validity of our findings than one single-cell method alone. Major population frequencies showed a strong correlation between scRNA-seq and mass cytometry. We used single-cell transcriptome profiling to further separate cell populations into multiple subsets according to differentiation, activation, or functional states. We found a population of CD16+ CD8 T cells that was increased in smokers and exhibited natural killer (NK)-like transcriptional programs. Pseudotime analysis and examination of canonical markers revealed that these NK-like CD8 T cells likely represent a terminally differentiated state. DNA methylation models demonstrated that the smoking dose was associated with accelerated immune aging and decreased telomere length (TL; a biomarker of T cell senescence) in CD8 T cells. Not limited to CD8 T cells, smokers’ other immune populations displayed senescent characteristics.
By detecting an altered abundance of a rare population, we revealed an immune target that can be isolated and explored for connections between smoking and chronic diseases. Combined with increased (pre-)senescent CD8 T cells, elevated regulatory T cells (Tregs) and induction of senescence-linked genes in multiple cell types provide evidence that smokers show signs of premature immune system aging. The potential immune function defects and inflammatory subsets demonstrated here mirror the characteristics of pathologies commonly found in smokers. Further studies of smoking-associated dysregulation of immune transcriptional programs and candidate dysfunctional T cells linked to accelerated immune system aging will lead to mechanistic insights to advance disease prevention strategies for smoking-mediated pathologies.
Results
scRNA-Seq and Mass Cytometry Profiling of Human Peripheral Blood Immune Cells in Smokers and Nonsmokers
We set out to characterize the effects of cigarette smoke on immune cells in peripheral blood using single-cell approaches to determine whether the smoking-associated gene expression changes observed within major immune cell populations resulted from an altered abundance of specific, identifiable cell subsets. We performed scRNA-seq and mass cytometry in parallel on cryopreserved peripheral blood samples from eight donors with no history of atherosclerosis, chronic obstructive pulmonary disease (COPD), or lung cancer (Figure 1A). Serum cotinine, a metabolite of nicotine and an established biomarker of recent cigarette smoke exposure,9 confirmed the smoking status of donors. Smokers (n = 4) used for single-cell analyses had serum cotinine levels ranging from 240 to 511 ng/mL; all nonsmokers (n = 4) had serum cotinine levels <2 ng/mL. Donors were matched based on gender and race; ages ranged from 31 to 56 years and were not significantly different between smokers and nonsmokers (p = 0.23). Demographic and smoking information is listed in Table S1. We obtained single-cell mRNA data from 45,049 cells and surface protein expression data from 990,748 cells.
Figure 1.

scRNA-Seq and Mass Cytometry Profiling of Human PBMCs from Smokers (n = 4) and Nonsmokers (n = 4)
(A) Experimental design: cryopreserved PBMCs from smokers and nonsmokers were thawed for scRNA-seq and mass cytometry.
(B) Uniform Manifold Approximation and Projection (UMAP) scRNA-seq plot: ∼45,000 PBMCs colored by major cell type.
(C) Force directed layout (FDL) of mass cytometry: ∼1 million cells colored by major cell type.
(D) Canonical gene expression markers for major cell types: CD8T cells (CD3D, CD8A), CD4T cells (CD3D, CD4) natural killer T (NKT) cells (CD3D, NCR3), natural killer (NK) cells (NCR3), monocytes (CD14, FCGR3A [CD16]), dendritic cells (DCs; FCER1A), and B cells (MS4A1).
(E) Cell surface protein expression for major cell types: CD8T cells (CD3, CD8a), CD4T cells (CD3, CD4), NKT cells (CD3, CD56), NK cells (CD56), monocytes (CD14, CD16), DCs (CD123) and B cells (CD19).
(F and G) Major cell type frequency distributions by individual donor (nonsmokers, NS; smokers, SM) colored by cell type for scRNA-seq (F) and mass cytometry (G).
(H) Major cell type frequencies showed strong correlation between scRNA-seq and mass cytometry (Pearson r = 0.99, r2 = 0.98, p < 0.0001). Shapes represent matched individuals, NS (unfilled) and SM (filled), are colored by cell type.
For each single-cell approach, we assigned cells to common immune populations based on mRNA (scRNA-seq) or surface protein (mass cytometry) expression (Figures 1B–1E; Table S2). For scRNA-seq, we used Seurat10,11 to integrate data and implement shared nearest neighbors (SNN) clustering (see STAR Methods). We then used Model-based Analysis of Single-cell Transcriptomics (MAST12) to identify positive and negative marker genes for each cluster and combined cells into major immune populations (Table S2). Cells in clusters expressing CD3D as a positive marker were designated as T cells (Figure 1D). T cells were further classified into CD4 T cells, CD8 T cells, or NKT cells based on the expression of CD4, CD8A, or NCR3 (Figure 1D). NK cells were identified based on CD3D as a negative marker combined with the expression of NKG7, GNLY, GZMB, PRF1, and NCR3 as positive markers (Figure 1D; Table S2). Monocytes were positive for LYZ and either CD14 or FCGR3A (encodes CD16), characteristic of classical or nonclassical monocytes (Figure 1D). DCs were similar to monocytes but could be distinguished by the expression of FCER1A (Figure 1D). B cells were defined by MS4A1 (encodes CD20; Figure 1D).
In parallel, PBMCs from each donor were assessed by mass cytometry (see STAR Methods). Viable, single-cell events were manually gated using Cytobank13 (Figure S1A). We used VorteX14 to cluster and create a force-directed layout (FDL) graph using the X-shift algorithm (see STAR Methods). A total of 122 PBMC cell clusters were determined from the 8 donors representing 983,848 cells (Figure S1B) and shown by smoking status (Figure S1C). Cell surface protein expression profiles were used to classify the cell populations (Figures 1C and 1E). T cells displayed CD3 and were classified by CD4 and CD8 as double-negative (DNT), double-positive (DPT), CD4 T, or CD8 T cells (Figures 1C and 1E). NKT cells were identified by CD3 and CD56 with CD4 or CD8 protein expression markers. Monocytes expressed CD14 and/or CD16 and DCs had CD123 above background levels (Figure 1E). B cells were positive for CD19 (Figure 1E). NK cells were positive for CD56 but negative for CD3 (Figure 1E).
To determine how well the scRNA-seq and mass cytometry corresponded with each other, we examined the individual donor proportion for each cell type. Cells colored by individual donors are shown for scRNA-seq (Figure S1D) and mass cytometry (Figure S1E). Cell-type frequencies were calculated and plotted to compare frequency distributions among individuals (Figures 1F and 1G). For both methods, all of the major populations—CD4T, CD8T, NKT, B, monocyte, and DC—were identified in all of the donors. We then compared the frequency of major populations in PBMCs by smoking status for scRNA-seq and mass cytometry using a Mann-Whitney U test. We observed no differences in the overall frequency of major cell types between smokers and nonsmokers by either scRNA-seq or mass cytometry (Figures S1F and S1G). Comparison of the major immune population percentages among individuals for scRNA-seq and mass cytometry showed significant strong correlation (Pearson r = 0.99, r2 = 0.98, p < 0.0001) between methods (Figure 1H).
scRNA-seq Reveals Increased Tregs and Altered Composition of the CD8 T Cell Population between Smokers and Nonsmokers
Single-cell transcriptome profiling can be used to separate cell populations into multiple subsets according to differentiation, activation, or functional states. Based on gene expression patterns, we clustered peripheral blood cells into 31 immune cell clusters and 1 erythroid contaminant cluster, labeled based on abundance from 0 (most abundant) through 31 (Figure 2A; Table S2). We identified 12 CD4 T cell (0, 1, 3, 4, 6, 10, 13, 14, 17, 18, 20, and 27), 7 CD8 T cell (2, 8, 11, 15, 19, 21, and 24), 3 NK cell (7, 22, and 25), 4 monocyte (5, 23, 26, and 30), and 2 B cell (9 and 12) clusters. NKTs (16), DCs (28), and MKs (31) were each contained by a single cluster. Here, clusters are referred to by major immune cell type, followed by original cluster identification (ID) (e.g., CD4T-0). To determine whether smoking altered the subtype distributions within the major cell populations, we compared the abundance of cells among clusters for each major cell type that separated into more than one cluster. Cells colored by smoking status are shown in Figure 2B. We did not observe any subset frequency shifts in B cells, monocytes, or NK cells (Figures S2A–S2F).
Figure 2.

Increased CD4 Tregs and Altered CD8 T Cell Composition Observed between Smokers (SM, n = 4) and Nonsmokers (NS, n = 4)
(A) scRNA-seq UMAP colored by cluster ID.
(B) scRNA-seq UMAP colored by smoking status. Clusters CD8T-24 and CD8T-2 had more cells from nonsmokers and CD4T-17, CD8T-15, and CD8T-8 had more cells from smokers.
(C and D) Subset frequencies within CD4 T cells. Smokers (filled) had a significant increase in cluster CD4T-17 compared to nonsmokers (unfilled). Bar = median, ∗p < 0.05 by Mann-Whitney U test (C). Individual donor distribution of CD4 T cell subsets (D).
(E) FOXP3 gene expression within CD4 T subsets.
(F and G) Subset frequencies within CD8 T cells. Individual donor distribution of CD8 T cell subsets (F). Smokers (filled) had significant decreases in clusters CD8T-24 and CD8T-2 and increases in clusters CD8T-8 and CD8T-15 compared to nonsmokers (unfilled). Bar = median, ∗p < 0.05 by Mann-Whitney U test (G).
For 11 of 12 CD4 T cell subsets, frequencies were not significantly altered by smoking (Figures 2B and 2C). Although donors exhibited interindividual variation, smoking status did not appear to have a considerable impact on the distribution of CD4 T cells (Figure 2D). Only one cluster, CD4T-17, was higher in smokers than in nonsmokers (p < 0.05; Figure 2C). This cluster was relatively low in abundance among CD4 T cell subsets: a median 3.5% in nonsmokers and 5.4% in smokers. We characterized CD4T-17 cells as Tregs based on elevated FOXP3 and IL2RA (encodes CD25; Figure 2E; Table S2). No other CD4T clusters showed FOXP3 or IL2RA as strong positive markers. DNA methylation level of the AHRR gene can be used as a dose- and duration-dependent biomarker of tobacco smoke exposure; AHRR methylation is strongly negatively correlated with smoking dose and duration.6,15,16 The elevated frequency of Tregs was associated with reduced AHRR DNA methylation in whole blood (p = 0.002, r2 = 0.81; Figure S2G).
In contrast to most CD4 T cell subsets, the variation among donors in CD8 T cells depended on smoking status, as illustrated by the differences in the dominant population(s) in CD8 T cells from smokers and nonsmokers (Figure 2F). Smokers had lower proportions of 2 CD8 T cell clusters (CD8T-24 and CD8T-2) and higher proportions of 2 CD8 T cell clusters (CD8T-8 and CD8T-15) compared to nonsmokers (p < 0.05; Figure 2G). We did not observe significant differences in the proportions of the remaining 3 CD8 T cell clusters (CD8T-19, CD8T-11, and CD8T-21).
CD8 T Cell Distribution Shifts from Naive to Differentiated States in Smokers
Smoking had substantial effects on the composition of CD8 T cells. We further analyzed each of the seven CD8 T clusters to identify distinct gene expression patterns. MAST identified positive and negative markers for each CD8 T cluster relative to other CD8 T cells (see STAR Methods). We found 71, 223, 46, 144, 71, 739, and 105 positive and 86, 978, 10, 68, 59, 214, and 52 negative markers (Bonferroni adjusted p < 0.05 in both smokers’ and nonsmokers’ cells) for CD8T-24, CD8T-2, CD8T-19, CD8T-11, CD8T-21, CD8T-8, and CD8T-15 clusters, respectively (Table S3). We examined marker lists for genes associated with T cell differentiation and function to distinguish between CD8 T subsets. Genes frequently used to classify CD8 T subsets varied among CD8 T cell clusters (Figures 3A and 3B). Elevated CCR7, SELL, and IL7R combined with low CCL5 indicated that clusters CD8T-24 and CD8T-2 represented naive cells (TN). High levels of IL7R, SELL, and FOS (activated T cell proliferation18,19) suggested that cluster CD8T-15 exhibited characteristics of long-lived memory cells, such as central memory T cells (TCM). Reduced levels of CCR7, SELL, and IL7R and elevated CCL5 and KLRG1 in CD8T-11, CD8T-21, and CD8T-8 are indicative of later differentiation stages (e.g., TEM). The lack of CD27 expression and decrease in FOS in CD8T-21 and CD8T-8 indicated highly differentiated TEM cells (i.e., TEMRA). ZEB2, associated with terminal differentiation states,20,21 was detected in approximately one-third of CD8T-8 cells (35.1% in smokers and 28.2% in nonsmokers).
Figure 3.

CD8 T Cell Subsets Shift from Naive to Differentiated CD8 T Cell States
(A) scRNA-seq UMAP (n = 8) of the 7 CD8 T cell subsets colored by cluster ID.
(B) Expression of genes to characterize CD8 T subsets. Color indicates cluster ID (n = 8).
(C) Trajectory inference (pseudotime analysis) ordered CD8 T cell clusters into 2 lineages, which originate from cluster CD8T-24 and terminate at either cluster CD8T-8 (lineage 1) or CD8T-15 (lineage 2). The black line shows lineages and the gray line shows simultaneous principle curves (n = 8).
(D) Pseudotime for CD8 T cells by individual nonsmokers (NS, n = 4) and smokers (SM, n = 4) in lineage 1 (left) and lineage 2 (right).
(E) Gene expression data17 from flow-sorted human peripheral CD8 T cells (n = 6): naive (TN; CD27+CD45RA+), central memory (TCM; CD27+CD45RA−) effector memory (TEM; CD27−CD45RA−), and EMRA (TEMRA; CD27−CD45RA+). FCGR3A (false discovery rate [FDR] = 1.38 × 10−8), PRF1 (FDR = 2.69 × 10−6), and ITGAL (FDR = 5.43 × 10−3), are positive markers for CD8T-8 cells; B3GAT1 (CD57; FDR = 1.06 × 10−5) and ZEB2 (FDR = 2.16 × 10−5) are positive markers for CD8T-8 cells and indicators of senescence; PRSS23 (FDR = 8.62 × 10−4), TNFSF10 (FDR = 6.46 × 10−3), and CTSW (FDR = 2.52 × 10−3) are secreted factors associated with SASP; and CX3CR1 (FDR = 8.95 × 10−6) is a chemokine receptor.
(F and G) Frequency of FCGR3A CD8 TEMRA (F) and CD8 TN (G) cells are correlated with smoking dose as measured by AHRR smoking biomarker (n = 8).
Several clusters exhibited intermediate expression of differentiation state genes. To organize CD8 T cell clusters by their likely differentiation trajectories, we used the Slingshot algorithm22 to perform pseudotemporal analysis. Lineage inference ordered CD8 T cells into 2 lineages, which originate from cluster CD8T-24 and terminate at either cluster CD8T-8 or CD8T-15 (Figure 3C). Lineage 1 mostly comprised cells from CD8T-24, CD8T-2, CD8T-19, CD8T-8, CD8T-21, and CD8T-8, with minimal cells from CD8T-15. Lineage 2 mostly comprised cells from CD8T-24, CD8T-2, and CD8T-15, with minimal cells from CD8T-19. Based on the altered composition of CD8 T cell subsets—lower proportions of CD8T-2 and CD8T-24 and higher proportions of CD8T-8 and CD8T-15 cells (Figures 2B, 2F, and 2G)—we propose that tobacco smoke exposure alters CD8 T cell composition by shifting CD8 T cells toward differentiated states. Smokers’ cells were biased toward later pseudotimes in both lineages (Figure 3D), demonstrating that smokers’ CD8 T cells are skewed toward differentiated states and nonsmokers’ CD8 T cells are skewed toward naive states.
We next identified temporally associated genes for each lineage. Only 10 genes within the top 100 temporally expressed genes were shared, and most shared genes exhibited the same direction of change over pseudotime in both lineages (Figures S3A and S3B). In general, temporally expressed genes for CD8 T cell lineages were consistent with effector memory (lineage 1) and central memory (lineage 2) differentiation. For example, in lineage 1, CCL5 and NKG7 increased, while SELL and IL7R decreased over the differentiation trajectory (Figures S3A and S3C). CMC1 demonstrated a nonlinear association in lineage 1, as it peaked in CD8T-21 cells and then decreased through CD8T-8 cells (Figures S3A and S3C).
The terminal cluster in the effector memory trajectory, CD8T-8, shared many features with the penultimate cluster, CD8T-21; however, CD8T-8 increased in smokers, but CD8T-21 did not. Since the low expression of CD27 and CCR7 and elevated expression of KLRG1 in both clusters would classify these cells as highly differentiated CD8 T cells (i.e., TEMRA-like), we sought to find markers within CD8T-8 that did not occur in CD8T-21. Examination of exhaustion markers—TOX, PDCD1 (encodes programmed cell death protein-1 [PD-1]), CTLA4, and HAVCR2 (encodes TIM3)—did not distinguish the clusters. Whereas smokers’ CD8T-8 cells had elevated expression of TOX (log2FC [fold change] = 0.31, p = 1.44 × 10−9), nonsmokers’ CD8T-8 cells did not, and no other exhaustion markers were significantly elevated (Table S3). We next examined senescence-associated genes KLRG1 and B3GAT1 (encodes CD57). While KRLG1 was a positive marker for CD8T-21 (smokers [SM]: log2FC = 0.65, p = 8.11 × 10−04; nonsmokers [NS]: log2FC = 1.33, p = 2.23 × 10−22) and CD8T-8 cells (SM: log2FC = 0.94, p = 6.29 × 10−47; NS: log2FC = 1.28, p = 1.94 × 10−40), B3GAT1 was unique to CD8T-8 cells (SM: log2FC = 0.19, p = 3.83 × 10−14; NS: log2FC = 0.10, p = 0.022). We also found that 2 genes reported as having smoking-associated methylation changes, GFI1 and PRSS23,15 showed elevated expression in CD8T-8 cells (GFI1 SM: log2FC = 0.20, p = 5.58 × 10−7; GFI1 NS: log2FC = 0.22, p = 0.046; PRSS23 SM: log2FC = 1.07, p = 1.5 × 10−143; PRSS23 NS: log2FC = 0.91, p = 7.7 × 10−60). FCGR3A, commonly found on NK cells and nonclassical monocytes, was identified as a strong positive marker of CD8T-8 cells in smokers (log2FC = 1.63, p = 1.17 × 10−171) and nonsmokers (log2FC = 1.72, p = 5.17 × 10−119; Figure 1D; Table S3).
We reanalyzed the gene expression data from flow-sorted human peripheral naive (CD27+CD45RA+), central memory (CD27+CD45RA−), effector memory (CD27−CD45RA−), and EMRA (CD27−CD45RA+) CD8 T cells from healthy adults.17 We found that the TEMRA subset had an increased expression of FCGR3A (Figure 3E). Combined with our finding of high FCGR3A in our most differentiated CD8 T cluster (CD8T-8), data from Callender et al.17 supports elevated FCGR3A as a characteristic of CD8 TEMRA cells. In addition, CD8 TEMRA cells showed an elevated expression of senescence-associated secretory phenotype (SASP) genes (Figure 3E).
Given that human cytomegalovirus (HCMV) infection can alter T cell composition,23,24 we tested donors for HCMV antibodies and DNA in serum (see STAR Methods). Compared to seronegative donors (immunoglobulin G [IgG]−/IgM−), seropositive donors (IgG+/IgM−) had elevated levels of CD8 TEMRA cells (CD8T-21), but not FCGR3A-expressing CD8 TEMRA cells (CD8T-8); neither naive CD8 T clusters (CD8T-24 and CD8T-2) were significantly reduced in HCMV seropositive donors (Figure S3D; Table S1).
To further support the idea that smoking elevated FCGR3A-expressing CD8 TEMRA cells (CD8T-8) and reduced naive CD8 T cells (CD8T-24 and CD8T-2), we compared the frequencies of these subpopulations against whole-blood DNA methylation of AHRR (Figures 3F and 3G; Table S1). A higher frequency of FCGR3A-expressing CD8 TEMRA cells was associated with reduced AHRR methylation (p = 0.008, r2 = 0.72), and a lower frequency of naive CD8 T cells was associated with reduced AHRR methylation (p = 0.002, r2 = 0.82). In addition, the increase in FCGR3A-expressing CD8 TEMRA cells was highly correlated with the increase in CD4 Tregs (p = 0.019, r2 = 0.63; Figure S3E).
Mass Cytometry Confirms Elevated CD16+ CD8 T Cells in Smokers
Relatively rare in nonsmokers (median: 1.8%), the FCGR3A-expressing CD8 T cell cluster (CD8T-8) comprised 7.3% of PBMCs in smokers. Reported as a low-frequency subset (∼2% of PBMCs in healthy adults), CD16+ CD8 T cells have been described.25, 26, 27 Based on these reports, we sought to ascertain whether smokers had increased levels of CD16+ CD8 T cells that expressed surface proteins for CD3, but not CD56; i.e., confirm an increase in CD16 expression within CD8 T cells that are not NKT cells. To show that smokers had increased surface protein expression of CD16 within their CD8 T cells, we ran X-shift on all CD8 T and NKT cells and visualized the FDL colored by cluster IDs (Figure 4A). We did not see any differences between smokers’ and nonsmokers’ CD8 T cells for CD56 (Figure 4B), but smokers showed an increase in the proportion of CD8 T cells expressing CD16 compared to nonsmokers (Figure 4C). We determined the frequency of CD16+ CD8 T cells in smokers and nonsmokers by manual gating (Figure S4A). CD3+CD56− negative cells were then gated to obtain single positive CD8 T cells (CD3+CD56−CD8+CD4−; Figure S4A), which were then used to determine the frequency of CD16+ CD8 T cells (CD3+CD56−CD8+CD4−CD16+; Figure S4A). Compared to nonsmokers, smokers had a significantly increased frequency of CD16+ CD8 T cells (p = 0.03; Figure 4D), confirming that smokers had elevated proportions of CD16+ CD8 T cells. In an independent group of donors, CD16+ CD8 T cells were also elevated in smokers compared to nonsmokers (p < 0.01; Figure S4B; Table S1).
Figure 4.

Characterization of CD16+ CD8 T Cells
(A–D) Mass cytometry confirms elevated proportion of CD16+ CD8 T cells in smokers.
(A) FDL (n = 8) of CD8 T and NKT cells colored by cluster ID.
(B and C) Cell surface marker intensity for NK marker CD56 (B) and CD16 (C) in nonsmokers (top, n = 4) and smokers (bottom, n = 4).
(D) Frequency of CD16+ cells increases within CD8 T cells in smokers (filled, n = 4) compared to nonsmokers (unfilled, n = 4). Bar = median, ∗p < 0.05 by 1-tailed Mann-Whitney U test.
(E and F) NK-associated transcriptome characteristics (n = 8) revealed in CD8T-8 cells by gene set enrichment analysis (GSEA). CD8T-8 cells were positively enriched (FWER < 0.05) for gene signatures of T cells induced to have NK-like phenotypes; split dot plot shows 25 genes within the induced T to NK gene set (E). CD8T-8 cells were negatively enriched (FWER < 0.05) for genes higher in naive CD8 T cells relative to NK cells; split dot plot shows 25 genes within the naive T versus NK gene set (F). Color intensity represents average per-cell expression (scale shows standard deviation), and circle size indicates the percentage of cells expressing each gene.
(G and H) CD8T-8 cells show increased GZMB expression in smokers. (G) UMAP comparison of GZMB between nonsmokers’ (top, n = 4) and smokers’ (bottom, n = 4) cells. Cluster CD8T-8 is indicated by dotted circle. (H) Individual donor GZMB levels for CD8T-8 cells for nonsmokers (NS, n = 4) and smokers (SM, n = 4).
(I and J) CD8T-8 cells have increased PRF1 expression in smokers. (I) UMAP comparison of PRF1 between nonsmokers’ (top, n = 4) and smokers’ (bottom, n = 4) cells. Cluster CD8T-8 is indicated by dotted circle. (J) Individual donor PRF1 levels for CD8T-8 cells for nonsmokers (NS, n = 4) and smokers (SM, n = 4).
To phenotype the CD16+ CD8 T cell subset, we gated CD3+ T cells by a CD45RA/CD45 biaxial plot to establish an accurate CD45RA+ gate that was then applied to the CD16+ CD8 T cells (Figure S4C). Consistent with a TEMRA phenotype, the majority of CD16+ CD8 T cells were positive for CD45RA in both smokers and nonsmokers (Figure S4D).
FCGR3A-Expressing CD8 T Cells Exhibit NK-like Transcriptome Signatures
After confirming an increase in CD16+ CD8 T cells in smokers, we further examined how the transcriptomes of FCGR3A-expressing CD8 T cells differed from other CD8 T cells. In addition to FCGR3A, CD8T-8 cells exhibited elevated expression NKG7, GNLY, FGFBP2, GZMB, and PRF1 (Tables S2 and S3). While these genes are considered cytotoxic T or NK cell-expression signatures, the presence of CD16 led us to suspect that this subset may express genes indicative of NK-like attributes. To gain insight into the functional relevance of gene expression profiles for CD8T-8 cells, we performed gene set enrichment analysis (GSEA). Consistent with an NK-like transcriptional program, “LI INDUCED T TO NATURAL KILLER UP” had a positive normalized enrichment score (NES = 1.88, FWER < 0.05) and “GSE22886 NAIVE TCELL VS NKCELL UP” had a negative NES (−2.13, FWER < 0.05). The “LI INDUCED T TO NATURAL KILLER UP” gene set encompasses expression patterns for T cells reprogrammed to have NK-like phenotypes—“induced T to NK” (iTNK) cells.28 Here, we found 67 and 71 genes from the iTNK gene signature as positive markers (p < 0.05) of FCGR3A-expressing CD8 T cells in smokers and nonsmokers, respectively. Figure 4E shows the 25 highest ranked iTNK genes, based on p value in CD8T-8 cells. CDT8-8 genes were negatively enriched (FWER < 0.05) for genes that are higher in naive CD8 T cells relative to NK cells (“GSE22886 NAIVE TCELL VS NKCELL UP”). Smokers’ and nonsmokers’ CD8T-8 cells had significantly reduced expression (i.e., negative markers) of 55 and 45 naive CD8 T versus NK genes. Figure 4F shows the 25 highest ranked naive CD8 T versus NK genes, based on p value for CD8T-8 cells.
To examine whether smokers’ “NK-like” CD8 T cells differed from those of nonsmokers, we compared the average expression of genes from smokers’ CD8T-8 cells to nonsmokers’ CD8T-8 cells (see STAR Methods). We found that 63 genes had an increased and 74 genes had a decreased average per cell expression in smokers compared to nonsmokers (p < 0.05; Table S4). Figures S4E and S4F show 25 genes with increased and decreased per cell expression, ordered by the difference in the percentage of cells expressing each gene between smokers and nonsmokers. Although cellular mRNA levels for effector molecules granzyme B (encoded by GZMB) and perforin (encoded by PRF1) exhibited interindividual variation, both increased in frequency of expression and average per-cell expression in smokers compared to nonsmokers (Figures 4G–4J, S4E, S4G, and S4H).
CD8 T Bulk Transcriptomes Reflect Differentiation State Shifts Observed at the Single-Cell Level
To assess the overall impact of smoking on CD8 T cells, we identified differentially expressed genes (DEGs) between smokers and nonsmokers by comparing the average per-cell expression for all of the cells in the 7 CD8 T clusters combined. Of 2,163 genes evaluated in the pseudobulk analysis, we found that 1,817 genes had higher expression and 344 genes had lower expression in smokers versus nonsmokers (q < 0.05; Table S5). To examine the interindividual variability in response to smoking, we performed hierarchical clustering using smoking scRNA-DEGs, which separated individual donors by smoking status (Figures 5A and S5A). To validate altered CD8 T gene expression profiles in smokers, we used RNA-seq and microarray on isolated CD8 T cells to examine the differences in bulk RNA expression. Isolated CD8 T cells for bulk RNA-seq included the 8 donors used in scRNA-seq (bulk RNA was from a previous visit) and 7 additional donors (Table S1). We identified 1,268 genes as differentially expressed—692 increased and 576 decreased (q < 0.05; Figure 5B). With the exception of F061, principal-component analysis (PCA) of bulk RNA-seq data separated smokers from nonsmokers (Figure 5C). We evaluated microarray data from isolated CD8 T cells from 19 donors (9 smokers and 10 nonsmokers). Isolated CD8T cells included 4 donors used in scRNA-seq, 4 donors used in bulk RNA-seq, and 11 additional donors (Table S1). We identified 51 DEGs (see STAR Methods)—46 increased and 5 decreased (Figure 5D).
Figure 5.

CD8 T Bulk Transcriptomes Reflect Differentiation State Shifts at Single-Cell Level and DNA Methylomes Demonstrate Accelerated Aging and Shortened Telomeres
(A) Top 350 upregulated and downregulated differentially expressed genes (DEGs) between smokers (SM, n = 4) and nonsmokers (NS, n = 4) from CD8 T cells (scRNA-seq). Donors were separated by smoking status using smoking scRNA-seq-DEGs for hierarchical clustering. Labeled genes were also significantly upregulated in the CD8 T cell microarray results.
(B) Comparison of bulk RNA-seq expression of isolated CD8 T cells between smokers (n = 7) and nonsmokers (n = 8). DEGs with higher expression in smokers are labeled in red and DEGs with lower expression in smokers are denoted in blue. The gene names in black were altered in bulk RNA-seq and scRNA-seq. The gene names in gray were altered in bulk RNA-seq and microarray. The gene names in boldface were altered in microarray, bulk RNA-seq, and scRNA-seq.
(C) Principal-component analysis (PCA) of bulk RNA-seq. Donors used in both scRNA-seq and bulk RNA-seq are labeled with “F” and “V” patient codes. See Table S1 for donor information.
(D) Comparison of microarray expression of isolated CD8 T cells between smokers (n = 9) and nonsmokers (n = 10). The genes with higher expression in smokers are red and the genes with lower expression in smokers are blue. The gene names in black were altered in microarray and scRNA-seq. The gene names in gray were altered in microarray and bulk RNA-seq. The gene names in boldface were altered in microarray, bulk RNA-seq, and scRNA-seq.
(E) Immunological signatures and chemical and genetic perturbations that were significantly enriched (FWER < 0.05) by GSEA in scRNA-seq, bulk RNA-seq, and microarray.
(F–I) In CD8 T cells, age acceleration (F, H) and shortened telomeres (G, I) are correlated with the smoking dose (AHRR DNA methylation) in scRNA-seq donors (F–G) and a larger group (H–I) of nonsmokers (n = 71, open blue circles) and smokers (n = 60, closed red circles).
We compared results from the 3 RNA analysis platforms used to identify smoking-associated DEGs in CD8 T cells. Bulk RNA-seq confirmed 241 smoking DEGs from the scRNA-seq, 162 with increased and 79 with decreased expression in smokers compared to nonsmokers (Figures 5B; Table S5). Microarray analysis confirmed 24 smoking DEGs that were identified by scRNA-seq as increased in smokers compared to nonsmokers (Figures 5A and 5D; Table S5). Two genes, FAM129A and CD58, increased in smokers’ CD8 T cells in all 3 platforms (Figures 5A, 5B, 5D, S5A, and S5B; Table S5). Genes found to be altered by at least 2 methods include LGALS1, ADAM8, and CLDND1, which were increased in bulk RNA-seq and scRNA-seq data; GPR15, which was increased in the bulk RNA-seq and microarray data; and NDFIP1, which was decreased in the bulk RNA-seq and scRNA-seq data (Figures 5A, 5B, 5D, and S5A–S5F). ITGAL, a smoking methylation biomarker,5 was only found to be significantly increased by scRNA-seq (log2FC = 0.36, p = 1.70 × 10−26), and was also elevated in the NK-like subset (SM: log2FC = 0.64, p = 6.53 × 10−29; NS: log2FC = 0.78, p = 1.50 × 10−20).
Although each platform identified smoking DEGs not found by other platforms, we expect the overall changes observed to represent a similar shift in the functional states of CD8 T cells. We used GSEA to determine that CD8 T pseudobulk and bulk transcriptomes were enriched for similar functional annotations. Pseudobulk scRNA-seq, bulk RNA-seq, and bulk microarray shared 7 positively and 8 negatively enriched gene sets (FWER < 0.05; Figure 5E). CD8 T cells were positively enriched for genes with a higher expression in memory T, TCM, TEM, PD1lo (CD8 TEM), and PD1hi (CD8 TEM) cells relative to naive T cells (Figure 5E). CD8 T cells were negatively enriched for genes that have a higher expression in naive T cells relative to memory T, TCM, TEM, PD1lo (CD8 TEM), and PD1hi (CD8 TEM) cells (Figure 5E). Therefore, GSEA of CD8 T smoking DEGs identified immunological signatures indicative of an increased expression of genes associated with effector memory and central memory functions and decreased expression of genes associated with naive T cells.
CD8 T DNA Methylomes Indicate Smoking Dose-Dependent Accelerated Aging and Decreased TL
In CD8 T cells, the shift from naive to differentiated states has been associated with aging and senescence.29 To determine whether tobacco smoke exposure was associated with immune aging and senescence, we analyzed DNA methylation data from bulk CD8 T cells (see STAR Methods). We used DNAm PhenoAge30 to calculate age acceleration (see STAR Methods). In the donors used for scRNA-seq and mass cytometry, we found a strong association between reduced AHRR DNA methylation and age acceleration (p = 0.005, r2 = 0.76; Figure 5F). Since shortened telomeres (TL) are indicative of T cell senescence, we used a DNA methylation estimator of TL (DNAmTL)31 to estimate the TL of CD8 T cells (see STAR Methods). We found a strong association between lower AHRR DNA methylation and shorter DNAmTL (p = 0.0004, r2 = 0.89; Figure 5G). To confirm these effects in a larger cohort, we analyzed CD8 T cell DNA methylation in a group of 131 individuals (Table S1). Age acceleration was highly significantly correlated with smoking dose (p < 0.00009), and ∼11% of the variance in age acceleration in this population could be attributed to smoking dose (Figure 5H). Similarly, methylation-derived CD8 T cell TL was highly significantly associated with smoking dose (p < 0.0002, r2 = 0.11; Figure 5I).
Smoking-Associated Gene Expression Changes in PBMC Populations
Since we did not observe substantial changes in subset distribution for the majority of PBMC populations, we compared average per-cell gene expression for all cells within each cell type (CD4 T, NKT, NK, monocyte, DC, and B) to identify smoking DEGs. For CD4 T cells, we found 1,563 DEGs—1,278 showed increased and 285 showed decreased expression (Table S6). Hierarchical clustering of donors by CD4 T smoking DEGs clustered individuals by smoking status (Figure 6A). Bulk gene expression of isolated CD4+ cells identified 2 upregulated (LRRN3 and GPR171) and 1 downregulated (APBA2) gene in common with the CD4 T pseudobulk analysis (Figures 6A and S6A). NKT cells had 89 smoking DEGs, 45 with increased and 44 with decreased expression, and NK cells had 238 smoking DEGs, 129 with increased and 109 with decreased expression (Table S6). Hierarchical clustering of DEGs separated donors by smoking status for NKT but not for NK cells (Figures 6B and 6C). CD56+ cells (bulk), which contain NKT and NK cells, shared 3 upregulated genes (PROK2, MX1, and TRAT1) and 1 downregulated gene (KLRB1) with NKT cells and 3 upregulated (MX1, DOCK5, and CSGALNACT1) and 3 downregulated genes (CD160, XCL2, and KLRB1) with NK cells (Figures 6B, 6C, and S6B). For monocytes, we found 488 DEGs between smokers and nonsmokers by scRNA pseudobulk analysis (Figure 6D; Table S6). Of the 290 DEGs with higher expression in smokers’ monocytes, 15 increased in smokers’ isolated CD14+ cells by microarray (Figures 6D and S6C). For DCs, we identified 21 smoking DEGs—5 showed increased and 16 showed decreased expression (Figure 6E; Table S6). In B cells, we found 190 DEGs, 111 with increased and 79 with decreased expression in smokers (Table S6). Using microarray in CD19+ cells, we confirmed the decreased expression of HLA-DQA1 in B cells from smokers compared to nonsmokers (Figures 6F and S6D). Table 1 lists the biological relevance of smoking DEGs found in PBMC populations by pseudobulk scRNA-seq and confirmed by microarray.
Figure 6.

Major Cell Types Show Gene Expression Changes in Peripheral Blood of Smokers
(A) Hierarchical clustering of the top 350 increased and decreased DEGs between smokers (n = 4) and nonsmokers (n = 4) from CD4 T cells (scRNA-seq).
(B–F) Heatmaps of smoking DEGs (scRNA-seq) in NKT cells (B), NK cells (C), monocytes (D), DCs (E), and B cells (F).
Table 1.
Smoking DEGs Altered in Both scRNA-Seq and Microarray
| Gene Name | DEG | Biological Process/Function/Pathway | |
|---|---|---|---|
| CD8 T | |||
| granzyme B | ↑ | GZMB | PD-1low44 |
| natural killer cell granule protein 7 | ↑ | NKG7 | PD-1low, PD-1high44 |
| perforin 1 | ↑ | PRF1 | PD-1low, PD-1high44 |
| killer cell lectin like receptor D1 | ↑ | KLRD1 | PD-1low, PD-1high44 |
| sterile α motif domain containing 3 | ↑ | SAMD3 | CD8 T tolerance58,59 |
| G protein-coupled receptor 171 | ↑ | GPR171 | negative regulation of myeloid differentiation60 |
| calpastatin | ↑ | CAST | PD-1low, PD-1high;44 effector memory, central memory61 |
| chromosome 1 open reading frame 21 | ↑ | C1orf21 | cytotoxic T by scRNA-seq in Crohn's disease59 |
| family with sequence similarity 129, member A | ↑ | FAM129A | PD-1low, PD-1high;44 memory, effector memory, central memory61 |
| TNF superfamily member 10 | ↑ | TNFSF10 | marker for atherosclerosis plaque formation62 |
| C-X3-C motif chemokine receptor 1 | ↑ | CX3CR1 | chemokine receptor associated with terminally differentiated effector CD8 cells63 |
| pyrin and HIN domain family member 1 | ↑ | PYHIN1 | PD-1low, PD-1high,44 memory61 |
| AT-rich interaction domain 5B | ↑ | ARID5B | central memory61 |
| zinc finger E-box binding homeobox 2 | ↑ | ZEB2a | PD-1low, PD-1high44 |
| CD58 molecule | ↑ | CD58 | PD-1low, PD-1high;44 memory, effector memory, central memory61 |
| protein kinase cAMP-dependent type I regulatory subunit α | ↑ | PRKAR1A | enhanced cytoxicity of effector T cells64 |
| acyloxyacyl hydrolase | ↑ | AOAH | effector memory phenotype, increase in chronic lymphocytic leukemia CD3 T cells65 |
| proline rich 5-like | ↑ | PRR5L | PD-1low, PD-1high;44 effector memory, central memory61 |
| MYB proto-oncogene like 1 | ↑ | MYBL1 | PD-1low, PD-1high;44 memory, effector memory, central memory61 |
| ATPase plasma membrane Ca2+ transporting 4 | ↑ | ATP2B4 | PD-1low, PD-1high;44 memory, effector memory, central memory61 |
| synaptotagmin 11 | ↑ | SYT11 | PD-1low, PD-1high44 |
| killer cell Ig-like receptor, 3 Ig domains, and long cytoplasmic tail 2 | ↑ | KIR3DL2a | T cell aging66 |
| death domain containing 1 | ↑ | DTHD1 | unknown function |
| fibrinogen like 2 | ↑ | FGL2a | replicative senescence,67 procoagulant68 |
| CD4 T | |||
| leucine-rich repeat neuronal protein 3 | ↑ | LRRN3a | smoking methylation,56 T cell replicative senescence69 |
| G protein-coupled receptor 171 | ↑ | GPR171 | negative regulation of myeloid differentiation60 |
| amyloid β precursor protein binding family A member 2 | ↓ | APBA2 | smoking methylation38 |
| NKT | |||
| prokineticin 2 | ↑ | PROK2 | inflammatory response in smoking70 and diabetes71 |
| MX dynamin like GTPase 1 | ↑ | MX1a | TNF-α induced cellular senescence37 |
| T cell receptor associated transmembrane adaptor 1 | ↑ | TRAT1 | leukocyte activation72 |
| killer cell lectin like receptor B1 | ↓ | KLRB1a | downregulated in human aged CD4+ memory T cells66 |
| NK | |||
| MX dynamin like GTPase 1 | ↑ | MX1a | TNF-α induced cellular senescence37 |
| dedicator of cytokinesis 5 | ↑ | DOCK5a | upregulated in CD57+ NK73 |
| chondroitin sulfate N-acetylgalactosaminyltransferase 1 | ↑ | CSGALNACT1 | positively correlated with smoking74 |
| CD160 molecule | ↓ | CD160a | increased in exhausted T cells75 |
| X-C motif chemokine ligand 2 | ↓ | XCL2 | overexpressed in lung cancer tumors, increases with prognosis76 |
| killer cell lectin like receptor B1 | ↓ | KLRB1a | downregulated in human aged CD4+ memory T cells66 |
| Monocytes | |||
| phospholipid scramblase 1 | ↑ | PLSCR1 | expression increased with cytokine treatment77 |
| GTPase, IMAP family member 4 | ↑ | GIMAP4 | regulates INF-γ in CD4 T cell differentiation78 |
| MX dynamin like GTPase 1 | ↑ | MX1a | TNF-α-induced cellular senescence37 |
| MX dynamin like GTPase 2 | ↑ | MX2a | TNF-α-induced cellular senescence37 |
| poly(ADP-ribose) polymerase family member 9 | ↑ | PARP9 | silences pro-inflammatory genes, found in atherosclerotic plaques41 |
| cystatin C | ↑ | CST3a | atherosclerosis,38 cellular senescence39 |
| sterile α motif domain containing 9 | ↑ | SAMD9 | anti-inflammatory factor79 |
| TNF superfamily member 13b | ↑ | TNFSF13B | regulates proliferation and differentiation of atherogenic B cells40 |
| Cysteine-rich secretory protein LCCL domain containing 2 | ↑ | CRISPLD2 | regulates anti-inflammatory effects80 |
| erythrocyte membrane protein band 4.1-like 3 | ↑ | EPB41L3 | positively correlated smoking gene and associated with cancer74 |
| heat shock protein family A (Hsp70) member 1B | ↑ | HSPA1B | overexpressed in advanced atherosclerosis42 |
| thrombospondin 1 | ↑ | THBS1 | regulation of monocyte mobility, vascular inflammation81 |
| IFN-induced protein with tetratricopeptide repeats 5 | ↑ | IFIT5a | up in TNF-α-mediated cellular senescence37 |
| formyl peptide receptor 2 | ↑ | FPR2 | increased in atherosclerotic lesions and plaque stability43 |
| IFN induced with helicase C domain 1 | ↑ | IFIH1a | up in TNF-α-mediated cellular senescence37 |
| B | |||
| major histocompatibility complex, class II, DQ α 1 | ↓ | HLA-DQA1 | presents peptides from extracellular protein in T cells82 |
cAMP, cyclic AMP; CLL, XX; DEG, differentially expressed gene; GTPase, guanosine triphosphatase; INF, interferon; TNF, tumor necrosis factor.
Gene associated with cellular senescence.
Discussion
Our study reveals CD16+ CD8 T cells and other signs of immune cell dysfunction as elevated in smokers. These cells, uncovered by scRNA-seq and confirmed by mass cytometry in human PBMCs from multiple individuals, shared transcriptomic features with iTNK cells, which acquire NK surface receptors and have increased cytotoxic potency.28 Combined with CD16 and CD45RA protein expression, the transcriptome of the NK-like CD8 T cells implies an innate-like, terminally differentiated CD8 T subset. CD16, commonly associated with NK cells, binds IgG antibodies to mediate antibody-dependent cellular cytotoxicity (ADCC); exogenous or endogenous CD16 expression enables T cells to execute ADCC.26,32 Consistent with a heightened cytolytic potential, the NK-like CD8 T cells had elevated mRNA expression of cytolytic effector molecules GZMB and PRF1; these transcripts were also higher in smokers than nonsmokers within this subset. Granzyme B and perforin-expressing CD8 T cells contribute to the development of atherosclerotic plaques in mice.33,34 As such, our results highlight a potential link between smoking-induced functional changes in human CD8 T cells and atherosclerosis.
Applying scRNA-seq and mass cytometry to PBMCs from tobacco smoke-exposed individuals, we show that major immune populations can be discerned and disparate subsets can be identified among CD4 T cells, CD8 T cells, NK cells, monocytes, and B cells. Pseudobulk analysis of immune cell populations revealed smoking DEGs, several of which were confirmed in bulk cell-type fractions. The increase in smokers’ Tregs is likely masked in bulk data from isolated CD4 T cells because Tregs are a low-frequency subset. In a larger cohort of healthy women (N = 75), smoking was found to be a positive predictor of Treg levels in peripheral blood.35 Notably, Tregs have been shown to induce T cell senescence,36 highlighting a potential role for the increase in Tregs observed here. Senescence-related genes were altered in smokers in multiple cell types (Table 1). MX1, induced in tumor necrosis factor-α (TNF-α)-mediated senescence,37 increased in smokers in NKT cells, NK cells, and monocytes.
Alluding to the shared regulation of pro-senescent and pro-atherosclerotic signaling, TNF-α-induced senescence genes are enriched for atherosclerosis signaling genes.37 CST3, associated with subclinical atherosclerosis38 and cellular senescence,39 increased in smokers’ monocytes. We also identified TNFSF13B, a critical regulator of atherogenic B cell proliferation and differentiation.40 Other notable genes connected to atherosclerosis that increased in smokers’ monocytes include PARP9,41 HSPA1B,42 and FPR2.43
Two approaches, established markers and trajectory inference, demonstrate that smokers lose naive and gain TCM-like and TEMRA-like cells. Differentiation state shifts in CD8 T cells were supported by bulk analysis methods. All 3 transcriptomic platforms identified changes associated with PD-1hi CD8 T cells. With persistent antigen stimulation, the inhibitory effect of the PD-1 pathway contributes to pathologies associated with T cell dysfunction during chronic viral infection and tumor evasion of host immune response.44 Duraiswamy et al.44 demonstrated that PD-1hi CD8 T cells obtained from healthy adults had gene expression profiles similar to those of PD-1lo CD8 T cells and did not show either exhausted gene signatures or phenotypes characteristic of PD-1hi cells obtained from humans or mice with chronic infections. Smoking DEGs found in CD8 T cells in pseudobulk scRNA-seq, bulk RNA-seq, or microarray were represented by these gene signatures (Figure 5E; Table 1). Notably, bulk RNA-seq data were acquired from prior visits from donors used for scRNA-seq, indicating a chronic or recurring state of activation in smokers’ CD8 T cells. Chronic activation suggests that CD8 T cells obtained from healthy smokers here may represent a dysfunctional phenotype.
Consistent with an end-stage TEMRA phenotype, NK-like CD8 T cells had the latest pseudotimes. GFI1, a transcriptional repressor of interleukin-7 receptor α (IL-7Rα) that drives the terminal differentiation of CD8 T cells,45 was elevated in NK-like CD8 TEMRA cells. The loss of naive and accumulation of terminally differentiated T cells, observed here in smokers, mimics the altered distribution of T cell subsets reported in aging and chronic infection that is proposed to result from repeated or persistent stimulation of immune cells, ultimately leading to the loss of immune function either due to replicative senescence or functional exhaustion.46, 47, 48 Gene expression changes in low-frequency subsets may not be discernible in pseudobulk and bulk analyses. Therefore, we looked for indicators of T cell dysfunction within the smoking-associated NK-like CD8 TEMRA subset. While TOX, a transcription factor that controls fate commitment in exhausted T cells,49 was elevated in smokers’ NK-like CD8 TEMRA cells compared to other CD8 T cells, it was only detected in 8.9% of cells within this cluster. Other exhaustion markers PDCD1, CTLA4, and HAVCR2 were not increased. Whereas exhausted CD8 T cells lack cytotoxic activity, the high expression of genes encoding proteins responsible for cytolytic activity in NK-like CD8 TEMRA cells suggests that these cells more likely represent a senescent or pre-senescent state. In support, compared to other CD8 T cells, the NK-like CD8 TEMRA cells from both smokers and nonsmokers had an elevated expression of KLRG1, an inhibitory receptor correlated with extensive proliferative history,50 and B3GAT1 (CD57), a marker of limited proliferative potential and shortened telomeres.51 Senescence is induced as the result of telomere shortening or nontelomeric DNA damage,46 both of which have been reported to occur in smokers.52,53
To further examine the effects of smoking on immune aging and senescence in CD8 T cells, we used DNA methylation-based models to demonstrate that age acceleration and reduced TL correlated with smoking dose. Accelerated immune system aging accompanies T cell senescence and can manifest as impaired immunological memory,54 which could contribute to attenuated immune responses in smokers.
In addition to impaired immune function, prolonged SASP, driven by the accumulation of senescent cells, can lead to chronic inflammation.29 Callender et al.17 showed that CD8 TEMRA cells exhibit both inflammatory and senescent phenotypes. Consistent with SASP, TNFSF10 (secreted factor) and CX3CR1 (chemokine receptor) were increased in smokers’ CD8 T cells, and NK-like CD8 TEMRA cells expressed secreted factors CTSW and PRSS23. The PRSS23 methylation level is a reproducible biomarker of tobacco smoke exposure,55, 56, 57 and altered methylation persists up to at least 30 years after smoking cessation.15,16 This indicates that epigenetic modifications likely contribute to the senescent attributes of CD8 T cells in smokers. The acquisition of CD16 and NK-like characteristics implies an underappreciated role for CD16 receptor in the maintenance of cytotoxic activity in TEMRA cells in smokers.
In conclusion, we show an association between smoking and an immune-cell subtype that can be isolated to investigate how NK-like CD8 TEMRA cells contribute to proinflammatory states in smoking-mediated chronic inflammatory conditions. Our data illustrate links between smoking-induced gene expression changes and a T cell senescent phenotype, immune system aging, and, potentially, atherosclerosis. Consequently, our use of recently developed single-cell technologies to address tobacco smoke exposure has great potential to affect global health.
Limitations of Study
The single-cell approaches have low sample numbers. Reproducing our current findings in a larger group and additional characterization of CMV status will refine the roles of smoking and CMV in CD8 T cell aging. In addition, functional studies are needed to determine the importance of the immune cell changes reported here.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45 (clone HI30) 89Y | Fluidigm | Cat# 3089003; RRID: AB_2661851 |
| CD235ab (clone HIR2) 141Pr | Fluidigm | Cat# 3141001B; RRID: AB_2651154 |
| CD19 (clone HIB19) 142Nd | Fluidigm | Cat# 3142001; RRID: AB_2651155 |
| CD4 (clone RPA-T4) 145Nd | Fluidigm | Cat# 3145001; RRID: AB_2661789 |
| CD8a (clone RPA-T8) 146Nd | Fluidigm | Cat# 3146001; RRID: AB_2687641 |
| CD7 (clone CD7-6B7) 147Sm | Fluidigm | Cat# 3147006; RRID: AB_2802104 |
| CD66 (clone CD66a-B1.1) 149Sm | Fluidigm | Cat# 3149008; RRID: AB_2802105 |
| CD61 (clone VI-PL2) 150Nd | Fluidigm | Cat# 3150001; RRID: AB_2661793 |
| CD123 (clone 6H6) 151Eu | Fluidigm | Cat# 3151001; RRID: AB_2661794 |
| CD36 (clone 5-271) 152Sm | Fluidigm | Cat# 3152007; RRID: AB_2802106 |
| CD45RA (clone H100) 153Eu | Fluidigm | Cat# 3153001; RRID: AB_2802108 |
| CD163 (clone GHI/61) 154Sm | Fluidigm | Cat# 3154007; RRID: AB_2661797 |
| CD10 (clone HI10a) 156Gd | Fluidigm | Cat# 3156001; RRID: AB_2802107 |
| CD11c (clone Bu15) 159Tb | Fluidigm | Cat# 3159001; RRID: AB_2661800 |
| CD14 (clone M5E2) 160Gd | Fluidigm | Cat# 3160001; RRID: AB_2687634 |
| CD16 (clone 3G8) 165Ho | Fluidigm | Cat# 3165001; RRID: AB_2802109 |
| CD34 (clone 581) 166Er | Fluidigm | Cat# 3166012; RRID: AB_2756424 |
| CD38 (clone HIT2) 167Er | Fluidigm | Cat# 3167001; RRID: AB_2802110 |
| CD206 (clone 15-2) 168Er | Fluidigm | Cat# 3168008; RRID: AB_2661805 |
| CD33 (clone WM53) 169Tm | Fluidigm | Cat# 3169010; RRID: AB_2802111 |
| CD3 (clone UCHT1) 170Er | Fluidigm | Cat# 3170001; RRID: AB_2661807 |
| CD20 (clone 2H7) 171Yb | Fluidigm | Cat# 3171012; RRID: AB_2802112 |
| CD15 (clone W6D3) 172Yb | Fluidigm | Cat# 3172021; RRID: AB_2802113 |
| HLA-DR (clone L243) 174Yb | Fluidigm | Cat# 3174001; RRID: AB_2665397 |
| CD56 (clone NCAM 16.2) 176Yb | Fluidigm | Cat# 3176008; RRID: AB_2661813 |
| CD11b (clone ICRF44) 209Bi | Fluidigm | Cat# 3209003; RRID: AB_2687654 |
| Human Fc Receptor Binding Inhibitor | Thermo Fisher Scientific | Cat# 14-9161-73; RRID: AB_468582 |
| Biological Samples | ||
| Whole blood from healthy donors | NIEHS Clinical Research Unit (CRU) | See Table S1 for details |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Histopaque®-1077 | Sigma Millipore | Cat# 10771 |
| ACCUSPIN Tubes | Sigma Millipore | Cat# A2055 |
| Iscove’s Modified Dulbecco’s Medium (IMDM) | Thermo Fisher Scientific | Cat# 12200036 |
| Fetal Bovine Serum (FBS) | Gemini Bio-Products | Cat# 100-106 |
| autoMACS Running Buffer | Miltenyi Biotec | Cat# 130-091-221 |
| Dimethyl sulfoxide (DMSO) | Millipore Sigma | Cat# D84-18 |
| Ultrapure BSA | Ambion by Life Technologies | Cat# AM2618 |
| Benzonase Nuclease | Millipore Sigma | Cat# E8263-25KU |
| Phosphate Buffered Saline (PBS) | Thermo Fisher Scientific | Cat# 20012-027 |
| Dynabeads CD15 | Thermo Fisher Scientific | Cat# 11137D |
| Cell-ID Cisplatin-198Pt | Fluidigm | Cat# 201198 |
| Cell-ID Intercalator-Ir—500 μM | Fluidigm | Cat# 201192B |
| Maxpar® Cell Staining Buffer | Fluidigm | Cat# 201068 |
| Maxpar® Fix and Perm Buffer | Fluidigm | Cat# 201067 |
| EQ Four Element Calibration Beads | Fluidigm | Cat# 201078 |
| Macron 0754-06 Sodium Citrate, Dihydrate | Thomas Scientific | Cat# 0562N59 |
| SPRIselect Reagent | Beckman Coulter | Cat# B23318 |
| Dynabeads MyOne Silane | Thermo Fisher Scientific | Cat# 37002D |
| Dynabeads CD19 Pan B | Thermo Fisher Scientific | Cat# 11143D |
| Dynabeads CD4 | Thermo Fisher Scientific | Cat# 11145D |
| Dynabeads CD8 | Thermo Fisher Scientific | Cat# 11147D |
| Dynabeads CD14 | Thermo Fisher Scientific | Cat# 11149D |
| CD56 MicroBeads, human | Miltenyi Biotec | Cat# 130-050-401 |
| AllPrep DNA/RNA/miRNA Universal Kit | QIAGEN | Cat# 80224 |
| Flowmi™ Tip Strainers | Bel-Art | Cat# H1680-0040 |
| Nuclease-Free Water | Thermo Fisher Scientific | Cat# AM9930 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3′ Library & Gel Bead Kit v2 | 10X Genomics, Inc. | Cat# 120237 |
| Chromium i7 Multiplex Kit | 10X Genomics, Inc. | Cat# 120262 |
| Bioanalyzer High Sensitivity DNA Analysis | Agilent | Cat# 5067-4626 |
| Ovation Pico WTA System V2 | NuGEN | Cat# 3302-60 |
| Encore Biotin Module | NuGEN | Cat# 4200-60 |
| GeneChip Hybridization, Wash, and Stain Kit | Thermo Fisher Scientific | Cat# 900720 |
| GeneChip Human Transcriptome Array 2.0 | Thermo Fisher Scientific | Cat# 902233 |
| EZ-96 DNA Methylation MagPrep | Zymo Research | Cat# D5041 |
| Infinium HumanMethylation450 BeadChip Kit | Illumina | Cat# WG-314-1002 |
| Infinium MethylationEPIC BeadChip Kit | Illumina | Cat# WG-317-1003 |
| TruSeq Stranded Total RNA Gold | Illumina | Cat# 20020598 |
| Deposited Data | ||
| Single-cell RNA sequencing | This paper | GEO: GSE138867 |
| Bulk RNA sequencing | This paper | GEO: GSE138851 |
| Microarray | This paper | GEO: GSE13897 |
| CD8 T DNA Methylation Array Profiling | This paper | GEO: GSE147430 |
| Flow-sorted CD8 T Microarray | Callender et al.17 | GEO: GSE98640 |
| Software and Algorithms | ||
| GraphPad Prism 7 | GraphPad Software, Inc. | GraphPad Prism,RRID: SCR_002798 |
| Cytobank | Cytobank | https://cytobank.org, RRID: SCR_014043 |
| Transcriptome Analysis Console (TAC) | Thermo Fisher Scientific | Transcriptome Analysis Console, RRID: SCR_016519 |
| VorteX v26 | Samusik et al.14 | https://github.com/nolanlab/vortex, RRID: SCR_017047 |
| CellRanger v2.0.2 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/installation, RRID: SCR_017344 |
| Seurat v3.0.0.9000 | Stuart et al.11 | https://satijalab.org/seurat |
| Seurat v2.3.4 | Butler et al.10 | https://satijalab.org/seurat |
| R | R Core | https://www.r-project.org/ |
| Slingshot v1.2.0 | Street et al.22 | https://bioconductor.org/packages/release/bioc/html/slingshot.html, RRID: SCR_017012 |
| GSEA | Subramanian et al.83 | https://www.gsea-msigdb.org/gsea/downloads.jsp, RRID: SCR_003199 |
| Mootha et al.84 | ||
| GenePattern | Reich et al.85 | https://www.genepattern.org, RRID: SCR_003201 |
| GEO2R | Barrett et al.86 | https://www.ncbi.nlm.nih.gov/geo/geo2r/ |
| Minfi | Aryee et al.87 | bioconductor.org/packages/release/bioc/html/minfi.html |
| PreprocessNoob | Fortin et al.88 | https://rdrr.io/bioc/minfi/man/preprocessNoob.html |
| DNAm PhenoAge | Levine et al.30 | http://dnamage.genetics.ucla.edu/home |
| DNAm TL | Lu et al.31 | http://dnamage.genetics.ucla.edu/home |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Douglas A. Bell (bell1@niehs.nih.gov), Senior Investigator, Environmental Epigenomics and Disease Group, Immunity Inflammation and Disease Laboratory, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA.
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The datasets generated during this study are available at NCBI GEO, https://www.ncbi.nlm.nih.gov/geo/. Microarray data have been deposited to NCBI GEO: GSE138974. Bulk and single-cell RNA-seq data have been deposited to NCBI GEO: GSE138851 and NCBI GEO: GSE138867. Raw idat files for DNA methylation have been deposited to NCBI GEO: GSE147430. Mass cytometry data are available from the corresponding author on request.
Experimental Model and Subject Details
Human Subjects
All donors were recruited with written informed consent under approved human IRB protocol NIEHS 10-E-0063 by the NIEHS Clinical Research Unit between March 2013 to January 2018 from the Raleigh, Durham and Chapel Hill, NC area.5,6 Whole blood was obtained from healthy (without acute disease according to self-reported medical histories) from nonsmokers, not having smoked > 100 cigarettes in their lifetime, and smokers who reported their average daily cigarette consumption for the past 3 months. Serum nicotine/cotinine levels were measured by HPLC-MS (Quest, Inc.) as an indication of their smoking exposure status. Human cytomegalovirus (HCVM) status was determined by two methods at the NIH Clinical Center in Bethesda, MD. Anti-cytomegalovirus IgG and IgM antibodies were measured in serum by a chemiluminescence immunoassay, and HCMV DNA viral load was determined using CMV-specific probes by quantitative real-time PCR. Donors were recalled matching nonsmokers/smokers on age, gender, and ethnicity for whole blood collection, cotinine levels were measured. See Table S1 for additional donor information.
Method Details
PBMC Isolation for scRNA-seq and Mass Cytometry
Whole blood was diluted 1:5 v/v with QIAGEN Buffer EL and incubated at room temperature (RT) until clarified (∼10 min) before centrifugation (300 g, RT, 10 min). After supernatant removal, leukocytes were resuspended in the same volume of Buffer EL (5 min) before spinning (300 g, 8 min). Leukocytes were then washed twice in autoMACS Running Buffer (Miltenyi Biotec), counted, and cryopreserved [20% Iscove’s Modified Dulbecco’s Medium (IMDM), 70% Fetal Bovine Serum (FBS), 10% Dimethyl sulfoxide (DMSO)] at a concentration of 1x107 cells/mL. Cryopreserved cells were thawed in nonsmoker/smoker pairs following the 10X Genomic’s protocol for “Fresh Frozen Human Peripheral Blood Mononuclear Cells for Single Cell RNA Sequencing.” Briefly, cells were serially diluted dropwise in complete media (IMDM,10% FBS) adding 50U/mL Benzonase (Millipore Sigma) for the first dilution. After centrifugation (1100 rpm, 8 min, RT), cells were resuspended in complete media and incubated with CD15 Dynabeads (Thermo Fisher Scientific) according to the manufacturer’s instructions to deplete the neutrophils from the PBMCs. PBMCs were then counted for viability and aliquoted for scRNA-seq or mass cytometry in parallel.
PBMC Preparation for Purified Cell Fractions
The mononuclear layer was isolated directly from whole blood using density gradient centrifugation with Histopaque-1077 Ficoll and ACCUSPIN Tubes (Sigma Millipore). Purified CD4+, CD8+, CD14+, CD19+, and CD56+ cell fractions were collected using antibody-coated magnetic beads (Dynabeads, Thermo Fisher Scientific; CD56, Miltenyi Biotec). Antibody-purified fractions were then extracted for DNA and RNA using the AllPrep DNA/RNA/miRNA Universal Kit according to the manufacturer’s instructions (QIAGEN).
Mass Cytometry
Thawed PBMCs (∼3x106 cells) were spun (300 g, 5 min) and resuspended in calcium magnesium-free phosphate buffered saline (PBS). 1μM Cisplatin (Fluidigm) was added for viability staining for 5 minutes before quenching the reaction with MaxPar Cell Staining Buffer (CSB, Fluidigm). After centrifugation (300 g, 5 min), cells were resuspended in CSB at a concentration of 60x106 cells/mL and incubated (RT,10 min) with Fc receptor binding inhibitor before adding 26 MaxPar metal-conjugated antibodies (Fluidigm) against immunophenotypic markers for an additional 30-minute incubation at RT. Stained cells were then washed two times before resuspension in MaxPar fix and perm buffer with 125μM 191/193Ir intercalator for either an hour at RT or 4°C overnight. Cells were then washed twice with CSB and two times with Nuclease-Free water (Thermo Fisher Scientific) followed by filtering through 40μM strainers to remove aggregates. Cells were then counted and resuspended in nuclease-free water at ∼5x105 cells/mL with 1:10 volume of four-element calibration beads (Fluidigm) and analyzed on a Helios instrument (Fluidigm) for 250,000 events for each donor at the NIEHS Flow Cytometry Center. Following the manufacturer’s instructions, downstream processing involved normalization by the calibration beads and fcs files were uploaded to Cytobank.
Preparation of Sequencing Libraries
scRNA-seq, libraries were prepared with the Chromium Single Cell 3′ Library & Gel Bead Kit v2 (10X Genomics) according to the manufacturer’s guidelines. For bulk RNA-seq, RNA from isolated CD8+ purified cell fractions were prepared using the TruSeq Stranded Total RNA Library Prep Gold (Illumina).
Microarray
Isolated RNA from antibody-purified cell fractions (CD4, CD8, CD14, CD19, CD56) from 19 individuals (9 smokers, 10 nonsmokers (See Table S1)), was amplified using NuGEN WT-Ovation Pico RNA Amplification System followed by labeling with NuGEN Encore Biotin Module according to the manufacturer’s protocol (NuGEN). Amplified biotin-cDNA was fragmented and hybridized to streptavidin/phycoerythrin-stained arrays using the GeneChip™ Hybridization, Wash and Stain Kit according to the manual protocol FS450-0004 (Thermo Fisher Scientific). The Life Technologies Human Transcriptome Arrays v2.0 were then scanned by an Affymetrix Scanner 3000 and using Transcriptome Analysis Console (TAC) Software (Thermo Fisher Scientific).
DNA Methylation arrays
Isolated DNA was bisulfite converted using the EZ-96 DNA Methylation MagPrep Kit (Zymo Research). Converted DNA was applied to either an Illumina Infinium HumanMethylation450 BeadChip (450K) or an Infinium MethylationEPIC BeadChip Kit (EPIC) according to the manufacturers’ protocol to measure methylation at ∼450,000 (450K) or 850,000 (EPIC) CpG sites genome wide.
Quantification and Statistical Analysis
Mass Cytometry Gating Strategy and Analysis
Events were gated in Cytobank to identify single viable cells (Figure S1A). Cells gated from spiked-in normalization beads were subsequently gated by Iridium (191Ir) and Cisplatin (198Pt) to obtain DNA positive cells. Single cells were identified by event length and Iridium (193Ir) and viable cells by Cisplatin-198Pt and leukocyte marker CD45. Viable cells were exported as fcs files and imported into VorteX14 using all events for each donor totaling 990,748 cells. Using the default parameter recommendations,89 all data were transformed using hyperbolic arcsin (f = 5). Applying a noise threshold of 1.0, clustering analysis was performed using a Euclidean length profile of 1.0 in X-shift and the weighted K-nearest neighbor density estimation (K). An elbow point validation was performed to determine the optimal cluster K value (K = 25) which was then used to create a Force-Directed Layout (FDL) for visualization colored by major immune cell type, expression of marker genes, cluster ID, smoking status and subject ID (Figures 1C, 1E, S1B, S1C, and S1E). 136 clusters were identified from the 990,748 events, one cluster was determined to be red blood cells (RBCs; positive expression for CD235a/b) and 13 clusters had multiple lineage markers and were determined to be doublets (e.g., positive expression profiles for CD19 and CD3) which was a total of 6900 cells that were removed prior to downstream analysis (983,848 cells retained).
scRNA-seq Processing and Analysis
scRNA-seq data was aligned to the hg19 genome and processed with CellRanger version 2.0.2. Uniquely aligned reads sharing equal barcode × unique molecular identifier (UMI) tags but annotated to multiple protein-coding transcripts (i.e., ambiguous UMIs), within each replicate were discarded from the analysis. Cells with less than 200 or greater than 3000 genes and cells with greater than 10% of UMIs from mitochondria were removed. Dataset integration, SNN clustering, and UMAP visualization of scRNA-seq data were performed with Seurat version 3.0.0.9000.11 To integrate data across eight donor samples, we used 2,500 genes and 50 dimensions. Clusters were identified by SNN clustering with Seurat FindNeighbors and FindClusters functions using 50 dimensions and a resolution parameter of 2.5. UMAP was used to visualize cells colored by major immune cell type (Figure 1B), cluster ID (Figure 2A), and smoking status (Figure 2B). Cell cluster marker genes (padj < 0.05) and smoking DEGs (padj < 0.05) were identified using Seurat version 2.3.410 implementing the MAST algorithm12 with UMI included as a latent variable. Expression of marker genes were visualized via UMAP (Figure 1D).
Slingshot version 1.2.022 was used to perform pseudotime analysis. Principal component analysis and UMAP were run on CD8 T cells using 45 dimensions. Slingshot was then used to infer cluster lineages and assign pseudotime to CD8T cells (Figure 3C). Temporally expressed genes for each CD8 T lineage were then identified by fitting a generalized additive model to each gene using loess-smoothed pseudotime as the predictor variable (Figure S3).
Bulk RNA-seq Analysis
For antibody-purified CD8 T cell fraction bulk RNA-seq, reads were aligned to the hg19 genome with STAR. Gene read counts were obtained with featureCounts from the Subread package using release 27 of the GENCODE annotation. DEGs were determined using DESeq2 with FDR-adjusted p value < 0.05 as the cutoff for significance (Figure 5B). PCA was performed with the prcomp() function in R (Figure 5C).
Microarray Analysis
For microarray, differentially expressed genes were detected using log2-transformed expression fold-change estimates with respect to the composite average of RMA-corrected fluorescence log-intensity levels (log2FC) across matched fractions (CD14, CD19, CD4, CD56, and CD8) from multiple individual female donors, both smoking and nonsmoking (N = 53 overall, with N ≥ 5 per cell fraction × smoking status group). Probe-wise log2FC values were tested across statistical groups through a resolution-weighted ANOVA; resolution weights represented relative metrics of fluorescence discrimination in the dynamic range of detection, i.e., cumulative hazard of multivariate ANOVA significance scores (probe × cell fraction × smoking status) from probe-wise generalized linear modeling of RMA-corrected fluorescence log-intensities using an exponential distribution and inverse link function.90 DEGs were detected from the annotation of probes with significance level p < 0.05 adjusted for multiple comparisons,91 then filtered against a minimum probe-wise effect size δlog2FC > 0.3 × σlog2FC and post hoc pairwise significance (Student’s t test p < 0.05) between log2FC values of at least one matched comparison between smokers and nonsmokers on same cell fraction levels. For probe-level effect size filtering, δlog2FC = 0.3 × σSSR is 5% of the 6σ-spread log2FC regression error with respect to a probe’s grand mean [where (σSSR)2 = (SSRlog2FC)/(N-1)] compared to 5% of the 6σ-spread in measurement error about the mean log2FC of each statistical group in the probe [where (σlog2FC)2 = (SSElog2FC)/(N-1)].
GSEA
We used GSEA83,84 via GenePattern85 to perform gene set enrichment analysis for Chemical and Genetic Perturbations and Immunological Signatures92 gene sets for scRNaseq, bulk RNA-seq, and microarray using FWER < 0.05 as the cutoff for significant enrichment (Figure 5E).
DNA Methylation Processing and Analysis
The raw idat files from the 450K and EPIC methylation arrays were read into R with the Minfi package separately.87 Data were combined for common CpG sites on the two arrays with Minfi combineArrays function and then preprocessed with background and dye bias correction using the Noob method.88 Methylation β values for CpG sites were calculated and values were input into a DNA Methylation Age Calculator30 to predict epigenetic biomarkers for aging (DNAm PhenoAge) and aging acceleration, determined by the correlation between DNAm PhenoAge and chronological age (DNAm PhenoAgeAccel, Figures 5F and 5H). Telomere length was estimated with DNAmTL31 (Figures 5G and 5I). Univariable linear regression was used to determine p values for the association of AHRR methylation versus DNAm PhenoAgeAccel and DNAmTL.
Reanalysis of Microarray Data
We reanalyzed Callender et al.17 gene expression data (GSE98640) from isolated CD8 T cells subsets: TN, TCM, TEM;, and TEMRA using GEO2R with default parameters86 to identify genes with differential expression among CD8 T subsets using limma (Linear Models for Microarray Analysis) with FDR-adjusted p values.93,94
Acknowledgments
This work was funded by the Intramural Research Program of the National Institute of Environmental Health Sciences-National Institutes of Health (Z01-ES100475) and a grant from the NIH/Food and Drug Administration (FDA) Intramural Center for Tobacco Research, to D.A.B. We thank the National Institute of Environmental Health Sciences (NIEHS) Clinical Research Unit (CRU) and individuals at NIEHS: Dr. Brian Papas (Integrative Bioinformatics Support Group), Marie Iannone (Flow Cytometry Center), Laura Wharey (Molecular Genomics Core), and Nicole Reeves and Jason Malphurs (Epigenomics and DNA Sequencing Core). We also thank Leonardo Albertini-Sanchez for manuscript comments.
Author Contributions
D.A.B., M.R.C., S.N.M., M.W., and G.S.P. designed the study and were involved in donor selection. M.R.C., S.N.M., and I.J.B.T. performed the experiments. S.N.M., M.R.C., O.A.L., X.W., and B.D.B. analyzed the data. S.N.M., M.R.C., and D.A.B. interpreted the data and wrote the paper, with input from O.A.L.
Declaration of Interests
The authors declare no competing interests.
Published: July 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100054.
Supplemental Information
References
- 1.World Health Organization . 2015. WHO global report on trends in prevalence of tobacco smoking 2015.https://apps.who.int/iris/handle/10665/156262 [Google Scholar]
- 2.Stämpfli M.R., Anderson G.P. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat. Rev. Immunol. 2009;9:377–384. doi: 10.1038/nri2530. [DOI] [PubMed] [Google Scholar]
- 3.Hansson G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 4.Ilhan F., Kalkanli S.T. Atherosclerosis and the role of immune cells. World J. Clin. Cases. 2015;3:345–352. doi: 10.12998/wjcc.v3.i4.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su D., Wang X., Campbell M.R., Porter D.K., Pittman G.S., Bennett B.D., Wan M., Englert N.A., Crowl C.L., Gimple R.N. Distinct Epigenetic Effects of Tobacco Smoking in Whole Blood and among Leukocyte Subtypes. PLOS ONE. 2016;11:e0166486. doi: 10.1371/journal.pone.0166486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wan M., Bennett B.D., Pittman G.S., Campbell M.R., Reynolds L.M., Porter D.K., Crowl C.L., Wang X., Su D., Englert N.A. Identification of Smoking-Associated Differentially Methylated Regions Using Reduced Representation Bisulfite Sequencing and Cell type-Specific Enhancer Activation and Gene Expression. Environ. Health Perspect. 2018;126:047015. doi: 10.1289/EHP2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reynolds L.M., Lohman K., Pittman G.S., Barr R.G., Chi G.C., Kaufman J., Wan M., Bell D.A., Blaha M.J., Rodriguez C.J., Liu Y. Tobacco exposure-related alterations in DNA methylation and gene expression in human monocytes: the Multi-Ethnic Study of Atherosclerosis (MESA) Epigenetics. 2017;12:1092–1100. doi: 10.1080/15592294.2017.1403692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y., Shu Y., Xiao Y., Wang Q., Kanekura T., Li Y., Wang J., Zhao M., Lu Q., Xiao R. Hypomethylation and overexpression of ITGAL (CD11a) in CD4(+) T cells in systemic sclerosis. Clin. Epigenetics. 2014;6:25. doi: 10.1186/1868-7083-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Florescu A., Ferrence R., Einarson T., Selby P., Soldin O., Koren G. Methods for quantification of exposure to cigarette smoking and environmental tobacco smoke: focus on developmental toxicology. Ther. Drug Monit. 2009;31:14–30. doi: 10.1097/FTD.0b013e3181957a3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kotecha N., Krutzik P.O., Irish J.M. Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom. 2010;53 doi: 10.1002/0471142956.cy1017s53. 10.17.1–10.17.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samusik N., Good Z., Spitzer M.H., Davis K.L., Nolan G.P. Automated mapping of phenotype space with single-cell data. Nat. Methods. 2016;13:493–496. doi: 10.1038/nmeth.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joehanes R., Just A.C., Marioni R.E., Pilling L.C., Reynolds L.M., Mandaviya P.R., Guan W., Xu T., Elks C.E., Aslibekyan S. Epigenetic Signatures of Cigarette Smoking. Circ. Cardiovasc. Genet. 2016;9:436–447. doi: 10.1161/CIRCGENETICS.116.001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeilinger S., Kühnel B., Klopp N., Baurecht H., Kleinschmidt A., Gieger C., Weidinger S., Lattka E., Adamski J., Peters A. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLOS ONE. 2013;8:e63812. doi: 10.1371/journal.pone.0063812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Callender L.A., Carroll E.C., Beal R.W.J., Chambers E.S., Nourshargh S., Akbar A.N., Henson S.M. Human CD8+ EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell. 2018;17:e12675. doi: 10.1111/acel.12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martins G.A., Cimmino L., Liao J., Magnusdottir E., Calame K. Blimp-1 directly represses Il2 and the Il2 activator Fos, attenuating T cell proliferation and survival. J. Exp. Med. 2008;205:1959–1965. doi: 10.1084/jem.20080526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaulian E., Karin M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 20.Omilusik K.D., Best J.A., Yu B., Goossens S., Weidemann A., Nguyen J.V., Seuntjens E., Stryjewska A., Zweier C., Roychoudhuri R. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J. Exp. Med. 2015;212:2027–2039. doi: 10.1084/jem.20150194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott C.L., Omilusik K.D. ZEBs: Novel Players in Immune Cell Development and Function. Trends Immunol. 2019;40:431–446. doi: 10.1016/j.it.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Street K., Risso D., Fletcher R.B., Das D., Ngai J., Yosef N., Purdom E., Dudoit S. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics. 2018;19:477. doi: 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson S.E., Sedikides G.X., Okecha G., Poole E.L., Sinclair J.H., Wills M.R. Latent Cytomegalovirus (CMV) Infection Does Not Detrimentally Alter T Cell Responses in the Healthy Old, But Increased Latent CMV Carriage Is Related to Expanded CMV-Specific T Cells. Front. Immunol. 2017;8:733. doi: 10.3389/fimmu.2017.00733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson S.E., Sedikides G.X., Okecha G., Wills M.R. Generation, maintenance and tissue distribution of T cell responses to human cytomegalovirus in lytic and latent infection. Med. Microbiol. Immunol. (Berl.) 2019;208:375–389. doi: 10.1007/s00430-019-00598-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Björkström N.K., Gonzalez V.D., Malmberg K.J., Falconer K., Alaeus A., Nowak G., Jorns C., Ericzon B.G., Weiland O., Sandberg J.K., Ljunggren H.G. Elevated numbers of Fc gamma RIIIA+ (CD16+) effector CD8 T cells with NK cell-like function in chronic hepatitis C virus infection. J. Immunol. 2008;181:4219–4228. doi: 10.4049/jimmunol.181.6.4219. [DOI] [PubMed] [Google Scholar]
- 26.Clémenceau B., Vivien R., Berthomé M., Robillard N., Garand R., Gallot G., Vollant S., Vié H. Effector memory alphabeta T lymphocytes can express FcgammaRIIIa and mediate antibody-dependent cellular cytotoxicity. J. Immunol. 2008;180:5327–5334. doi: 10.4049/jimmunol.180.8.5327. [DOI] [PubMed] [Google Scholar]
- 27.Clémenceau B., Vivien R., Debeaupuis E., Esbelin J., Biron C., Levy Y., Vié H. FcγRIIIa (CD16) induction on human T lymphocytes and CD16pos T-lymphocyte amplification. J. Immunother. 2011;34:542–549. doi: 10.1097/CJI.0b013e31822801d4. [DOI] [PubMed] [Google Scholar]
- 28.Li P., Burke S., Wang J., Chen X., Ortiz M., Lee S.C., Lu D., Campos L., Goulding D., Ng B.L. Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science. 2010;329:85–89. doi: 10.1126/science.1188063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goronzy J.J., Weyand C.M. Mechanisms underlying T cell ageing. Nat. Rev. Immunol. 2019;19:573–583. doi: 10.1038/s41577-019-0180-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levine M.E., Lu A.T., Quach A., Chen B.H., Assimes T.L., Bandinelli S., Hou L., Baccarelli A.A., Stewart J.D., Li Y. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany N.Y.) 2018;10:573–591. doi: 10.18632/aging.101414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu A.T., Seeboth A., Tsai P.C., Sun D., Quach A., Reiner A.P., Kooperberg C., Ferrucci L., Hou L., Baccarelli A.A. DNA methylation-based estimator of telomere length. Aging (Albany N.Y.) 2019;11:5895–5923. doi: 10.18632/aging.102173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clémenceau B., Congy-Jolivet N., Gallot G., Vivien R., Gaschet J., Thibault G., Vié H. Antibody-dependent cellular cytotoxicity (ADCC) is mediated by genetically modified antigen-specific human T lymphocytes. Blood. 2006;107:4669–4677. doi: 10.1182/blood-2005-09-3775. [DOI] [PubMed] [Google Scholar]
- 33.Kyaw T., Winship A., Tay C., Kanellakis P., Hosseini H., Cao A., Li P., Tipping P., Bobik A., Toh B.H. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE-deficient mice. Circulation. 2013;127:1028–1039. doi: 10.1161/CIRCULATIONAHA.112.001347. [DOI] [PubMed] [Google Scholar]
- 34.Hiebert P.R., Boivin W.A., Zhao H., McManus B.M., Granville D.J. Perforin and granzyme B have separate and distinct roles during atherosclerotic plaque development in apolipoprotein E knockout mice. PLOS ONE. 2013;8:e78939. doi: 10.1371/journal.pone.0078939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hampras S.S., Nesline M., Wallace P.K., Odunsi K., Furlani N., Davis W., Moysich K.B. Predictors of immunosuppressive regulatory T lymphocytes in healthy women. J. Cancer Epidemiol. 2012;2012:191090. doi: 10.1155/2012/191090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye J., Huang X., Hsueh E.C., Zhang Q., Ma C., Zhang Y., Varvares M.A., Hoft D.F., Peng G. Human regulatory T cells induce T-lymphocyte senescence. Blood. 2012;120:2021–2031. doi: 10.1182/blood-2012-03-416040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kandhaya-Pillai R., Miro-Mur F., Alijotas-Reig J., Tchkonia T., Kirkland J.L., Schwartz S. TNFα-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging (Albany N.Y.) 2017;9:2411–2435. doi: 10.18632/aging.101328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chung Y.K., Lee Y.J., Kim K.W., Cho R.K., Chung S.M., Moon J.S., Yoon J.S., Won K.C., Lee H.W. Serum cystatin C is associated with subclinical atherosclerosis in patients with type 2 diabetes: a retrospective study. Diab. Vasc. Dis. Res. 2018;15:24–30. doi: 10.1177/1479164117738156. [DOI] [PubMed] [Google Scholar]
- 39.Basisty N., Kale A., Jeon O.H., Kuehnemann C., Payne T., Rao C., Holtz A., Shah S., Sharma V., Ferrucci L. A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLOS Biol. 2020;18:e3000599. doi: 10.1371/journal.pbio.3000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kyaw T., Cui P., Tay C., Kanellakis P., Hosseini H., Liu E., Rolink A.G., Tipping P., Bobik A., Toh B.H. BAFF receptor mAb treatment ameliorates development and progression of atherosclerosis in hyperlipidemic ApoE(-/-) mice. PLOS ONE. 2013;8:e60430. doi: 10.1371/journal.pone.0060430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iwata H., Goettsch C., Sharma A., Ricchiuto P., Goh W.W., Halu A., Yamada I., Yoshida H., Hara T., Wei M. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat. Commun. 2016;7:12849. doi: 10.1038/ncomms12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kilic A., Mandal K. Heat shock proteins: pathogenic role in atherosclerosis and potential therapeutic implications. Autoimmune Dis. 2012;2012:502813. doi: 10.1155/2012/502813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petri M.H., Laguna-Fernández A., Gonzalez-Diez M., Paulsson-Berne G., Hansson G.K., Bäck M. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc. Res. 2015;105:65–74. doi: 10.1093/cvr/cvu224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duraiswamy J., Ibegbu C.C., Masopust D., Miller J.D., Araki K., Doho G.H., Tata P., Gupta S., Zilliox M.J., Nakaya H.I. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J. Immunol. 2011;186:4200–4212. doi: 10.4049/jimmunol.1001783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ligons D.L., Tuncer C., Linowes B.A., Akcay I.M., Kurtulus S., Deniz E., Atasever Arslan B., Cevik S.I., Keller H.R., Luckey M.A. CD8 lineage-specific regulation of interleukin-7 receptor expression by the transcriptional repressor Gfi1. J. Biol. Chem. 2012;287:34386–34399. doi: 10.1074/jbc.M112.378687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akbar A.N., Henson S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011;11:289–295. doi: 10.1038/nri2959. [DOI] [PubMed] [Google Scholar]
- 47.Larbi A., Fulop T. From “truly naïve” to “exhausted senescent” T cells: when markers predict functionality. Cytometry A. 2014;85:25–35. doi: 10.1002/cyto.a.22351. [DOI] [PubMed] [Google Scholar]
- 48.Reiser J., Banerjee A. Effector, Memory, and Dysfunctional CD8(+) T Cell Fates in the Antitumor Immune Response. J. Immunol. Res. 2016;2016:8941260. doi: 10.1155/2016/8941260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan O., Giles J.R., McDonald S., Manne S., Ngiow S.F., Patel K.P., Werner M.T., Huang A.C., Alexander K.A., Wu J.E. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature. 2019;571:211–218. doi: 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Voehringer D., Blaser C., Brawand P., Raulet D.H., Hanke T., Pircher H. Viral infections induce abundant numbers of senescent CD8 T cells. J. Immunol. 2001;167:4838–4843. doi: 10.4049/jimmunol.167.9.4838. [DOI] [PubMed] [Google Scholar]
- 51.Brenchley J.M., Karandikar N.J., Betts M.R., Ambrozak D.R., Hill B.J., Crotty L.E., Casazza J.P., Kuruppu J., Migueles S.A., Connors M. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood. 2003;101:2711–2720. doi: 10.1182/blood-2002-07-2103. [DOI] [PubMed] [Google Scholar]
- 52.Song Z., von Figura G., Liu Y., Kraus J.M., Torrice C., Dillon P., Rudolph-Watabe M., Ju Z., Kestler H.A., Sanoff H., Lenhard Rudolph K. Lifestyle impacts on the aging-associated expression of biomarkers of DNA damage and telomere dysfunction in human blood. Aging Cell. 2010;9:607–615. doi: 10.1111/j.1474-9726.2010.00583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valdes A.M., Andrew T., Gardner J.P., Kimura M., Oelsner E., Cherkas L.F., Aviv A., Spector T.D. Obesity, cigarette smoking, and telomere length in women. Lancet. 2005;366:662–664. doi: 10.1016/S0140-6736(05)66630-5. [DOI] [PubMed] [Google Scholar]
- 54.Reading J.L., Gálvez-Cancino F., Swanton C., Lladser A., Peggs K.S., Quezada S.A. The function and dysfunction of memory CD8+ T cells in tumor immunity. Immunol. Rev. 2018;283:194–212. doi: 10.1111/imr.12657. [DOI] [PubMed] [Google Scholar]
- 55.Besingi W., Johansson A. Smoke-related DNA methylation changes in the etiology of human disease. Hum. Mol. Genet. 2014;23:2290–2297. doi: 10.1093/hmg/ddt621. [DOI] [PubMed] [Google Scholar]
- 56.Guida F., Sandanger T.M., Castagné R., Campanella G., Polidoro S., Palli D., Krogh V., Tumino R., Sacerdote C., Panico S. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum. Mol. Genet. 2015;24:2349–2359. doi: 10.1093/hmg/ddu751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joubert B.R., Felix J.F., Yousefi P., Bakulski K.M., Just A.C., Breton C., Reese S.E., Markunas C.A., Richmond R.C., Xu C.J. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am. J. Hum. Genet. 2016;98:680–696. doi: 10.1016/j.ajhg.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schietinger A., Delrow J.J., Basom R.S., Blattman J.N., Greenberg P.D. Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science. 2012;335:723–727. doi: 10.1126/science.1214277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uniken Venema W.T., Voskuil M.D., Vich Vila A., van der Vries G., Jansen B.H., Jabri B., Faber K.N., Dijkstra G., Xavier R.J., Wijmenga C. Single-Cell RNA Sequencing of Blood and Ileal T Cells From Patients With Crohn’s Disease Reveals Tissue-Specific Characteristics and Drug Targets. Gastroenterology. 2019;156:812–815.e22. doi: 10.1053/j.gastro.2018.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rossi L., Lemoli R.M., Goodell M.A. Gpr171, a putative P2Y-like receptor, negatively regulates myeloid differentiation in murine hematopoietic progenitors. Exp. Hematol. 2013;41:102–112. doi: 10.1016/j.exphem.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abbas A.R., Wolslegel K., Seshasayee D., Modrusan Z., Clark H.F. Deconvolution of blood microarray data identifies cellular activation patterns in systemic lupus erythematosus. PLOS ONE. 2009;4:e6098. doi: 10.1371/journal.pone.0006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arcidiacono M.V., Rimondi E., Maietti E., Melloni E., Tisato V., Gallo S., Valdivielso J.M., Fernández E., Betriu À., Voltan R. Relationship between low levels of circulating TRAIL and atheromatosis progression in patients with chronic kidney disease. PLOS ONE. 2018;13:e0203716. doi: 10.1371/journal.pone.0203716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerlach C., Moseman E.A., Loughhead S.M., Alvarez D., Zwijnenburg A.J., Waanders L., Garg R., de la Torre J.C., von Andrian U.H. The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity. 2016;45:1270–1284. doi: 10.1016/j.immuni.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Panya A., Thepmalee C., Sawasdee N., Sujjitjoon J., Phanthaphol N., Junking M., Wongkham S., Yenchitsomanus P.T. Cytotoxic activity of effector T cells against cholangiocarcinoma is enhanced by self-differentiated monocyte-derived dendritic cells. Cancer Immunol. Immunother. 2018;67:1579–1588. doi: 10.1007/s00262-018-2212-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Göthert J.R., Eisele L., Klein-Hitpass L., Weber S., Zesewitz M.L., Sellmann L., Röth A., Pircher H., Dührsen U., Dürig J. Expanded CD8+ T cells of murine and human CLL are driven into a senescent KLRG1+ effector memory phenotype. Cancer Immunol. Immunother. 2013;62:1697–1709. doi: 10.1007/s00262-013-1473-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen G., Lustig A., Weng N.P. T cell aging: a review of the transcriptional changes determined from genome-wide analysis. Front. Immunol. 2013;4:121. doi: 10.3389/fimmu.2013.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Binet R., Ythier D., Robles A.I., Collado M., Larrieu D., Fonti C., Brambilla E., Brambilla C., Serrano M., Harris C.C., Pedeux R. WNT16B is a new marker of cellular senescence that regulates p53 activity and the phosphoinositide 3-kinase/AKT pathway. Cancer Res. 2009;69:9183–9191. doi: 10.1158/0008-5472.CAN-09-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu J., Yan J., Rao G., Latha K., Overwijk W.W., Heimberger A.B., Li S. The Duality of Fgl2 - Secreted Immune Checkpoint Regulator Versus Membrane-Associated Procoagulant: Therapeutic Potential and Implications. Int. Rev. Immunol. 2016;35:325–339. doi: 10.3109/08830185.2014.956360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chou J.P., Ramirez C.M., Wu J.E., Effros R.B. Accelerated aging in HIV/AIDS: novel biomarkers of senescent human CD8+ T cells. PLOS ONE. 2013;8:e64702. doi: 10.1371/journal.pone.0064702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yun J.H., Morrow J., Owen C.A., Qiu W., Glass K., Lao T., Jiang Z., Perrella M.A., Silverman E.K., Zhou X., Hersh C.P. Transcriptomic Analysis of Lung Tissue from Cigarette Smoke-Induced Emphysema Murine Models and Human Chronic Obstructive Pulmonary Disease Show Shared and Distinct Pathways. Am. J. Respir. Cell Mol. Biol. 2017;57:47–58. doi: 10.1165/rcmb.2016-0328OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Castelli M., Amodeo G., Negri L., Lattanzi R., Maftei D., Gotti C., Pistillo F., Onnis V., Congu C., Panerai A.E. Antagonism of the Prokineticin System Prevents and Reverses Allodynia and Inflammation in a Mouse Model of Diabetes. PLOS ONE. 2016;11:e0146259. doi: 10.1371/journal.pone.0146259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Taylor J., Reynolds L., Hou L., Lohman K., Cui W., Kritchevsky S., McCall C., Liu Y. Transcriptomic profiles of aging in naïve and memory CD4+ cells from mice. Immun. Ageing. 2017;14:15. doi: 10.1186/s12979-017-0092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lopez-Vergès S., Milush J.M., Pandey S., York V.A., Arakawa-Hoyt J., Pircher H., Norris P.J., Nixon D.F., Lanier L.L. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood. 2010;116:3865–3874. doi: 10.1182/blood-2010-04-282301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Charlesworth J.C., Curran J.E., Johnson M.P., Göring H.H., Dyer T.D., Diego V.P., Kent J.W., Jr., Mahaney M.C., Almasy L., MacCluer J.W. Transcriptomic epidemiology of smoking: the effect of smoking on gene expression in lymphocytes. BMC Med. Genomics. 2010;3:29. doi: 10.1186/1755-8794-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kasakovski D., Xu L., Li Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018;11:91. doi: 10.1186/s13045-018-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou H., Brekman A., Zuo W.L., Ou X., Shaykhiev R., Agosto-Perez F.J., Wang R., Walters M.S., Salit J., Strulovici-Barel Y. POU2AF1 Functions in the Human Airway Epithelium To Regulate Expression of Host Defense Genes. J. Immunol. 2016;196:3159–3167. doi: 10.4049/jimmunol.1502400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kodigepalli K.M., Bowers K., Sharp A., Nanjundan M. Roles and regulation of phospholipid scramblases. FEBS Lett. 2015;589:3–14. doi: 10.1016/j.febslet.2014.11.036. [DOI] [PubMed] [Google Scholar]
- 78.Heinonen M.T., Kanduri K., Lähdesmäki H.J., Lahesmaa R., Henttinen T.A. Tubulin- and actin-associating GIMAP4 is required for IFN-γ secretion during Th cell differentiation. Immunol. Cell Biol. 2015;93:158–166. doi: 10.1038/icb.2014.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.He P., Wu L.F., Bing P.F., Xia W., Wang L., Xie F.F., Lu X., Lei S.F., Deng F.Y. SAMD9 is a (epi-) genetically regulated anti-inflammatory factor activated in RA patients. Mol. Cell. Biochem. 2019;456:135–144. doi: 10.1007/s11010-019-03499-7. [DOI] [PubMed] [Google Scholar]
- 80.Himes B.E., Jiang X., Wagner P., Hu R., Wang Q., Klanderman B., Whitaker R.M., Duan Q., Lasky-Su J., Nikolos C. RNA-Seq transcriptome profiling identifies CRISPLD2 as a glucocorticoid responsive gene that modulates cytokine function in airway smooth muscle cells. PLOS ONE. 2014;9:e99625. doi: 10.1371/journal.pone.0099625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu Z., Morgan S., Ren J., Wang Q., Annis D.S., Mosher D.F., Zhang J., Sorenson C.M., Sheibani N., Liu B. Thrombospondin-1 (TSP1) contributes to the development of vascular inflammation by regulating monocytic cell motility in mouse models of abdominal aortic aneurysm. Circ. Res. 2015;117:129–141. doi: 10.1161/CIRCRESAHA.117.305262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zajacova M., Kotrbova-Kozak A., Cerna M. Expression of HLA-DQA1 and HLA-DQB1 genes in B lymphocytes, monocytes and whole blood. Int. J. Immunogenet. 2018;45:128–137. doi: 10.1111/iji.12367. [DOI] [PubMed] [Google Scholar]
- 83.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 85.Reich M., Liefeld T., Gould J., Lerner J., Tamayo P., Mesirov J.P. GenePattern 2.0. Nat. Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 86.Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fortin J.P., Triche T.J., Jr., Hansen K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017;33:558–560. doi: 10.1093/bioinformatics/btw691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kimball A.K., Oko L.M., Bullock B.L., Nemenoff R.A., van Dyk L.F., Clambey E.T. A Beginner’s Guide to Analyzing and Visualizing Mass Cytometry Data. J. Immunol. 2018;200:3–22. doi: 10.4049/jimmunol.1701494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lozoya O.A., Santos J.H., Woychik R.P. A Leveraged Signal-to-Noise Ratio (LSTNR) Method to Extract Differentially Expressed Genes and Multivariate Patterns of Expression From Noisy and Low-Replication RNAseq Data. Front. Genet. 2018;9:176. doi: 10.3389/fgene.2018.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 92.Godec J., Tan Y., Liberzon A., Tamayo P., Bhattacharya S., Butte A.J., Mesirov J.P., Haining W.N. Compendium of Immune Signatures Identifies Conserved and Species-Specific Biology in Response to Inflammation. Immunity. 2016;44:194–206. doi: 10.1016/j.immuni.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smyth G.K. limma: linear models for microarray data. In: Gentleman R., Carey V.J., Huber W., Irizarry R.A., Dudoit S., editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer; 2005. pp. 397–420. [Google Scholar]
- 94.Smyth G.K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at NCBI GEO, https://www.ncbi.nlm.nih.gov/geo/. Microarray data have been deposited to NCBI GEO: GSE138974. Bulk and single-cell RNA-seq data have been deposited to NCBI GEO: GSE138851 and NCBI GEO: GSE138867. Raw idat files for DNA methylation have been deposited to NCBI GEO: GSE147430. Mass cytometry data are available from the corresponding author on request.