

ABSTRACT
Acute myeloid leukemia (AML) is an aggressive, often fatal hematopoietic malignancy. All-trans retinoic acid (atRA), one of the first molecularly targeted drugs in oncology, has greatly improved the outcome of a subtype of AML, acute promyelocytic leukemia (APL). In contrast, atRA has so far provided little therapeutic benefit in the much larger group of patients with non-APL AML. Attempts to identify genetically or molecularly defined subgroups of patients that may respond to atRA have not yielded consistent results. Since AML is a stem cell-driven disease, understanding the effectiveness of atRA may require an appreciation of its impact on AML stem cells. Recent studies reported that atRA decreased stemness of AML with an FLT3-ITD mutation, yet increased it in AML1-ETO driven or EVI1-overexpressing AML. This review summarizes the role of atRA in normal hematopoiesis and in AML, focusing on its impact on AML stem cells.
KEYWORDS: AML, atRA, hematopoietic stem cell, leukemia stem cell, MECOM, FLT3
Introduction
Acute myeloid leukemia (AML) is a genetically heterogeneous disease, in which a number of recurrent genetic and molecular alterations are predictive of response to therapy [1–4]. Until recently, the great majority of patients were treated with conventional chemotherapy, and the only targeted drug used in routine clinical practice was all-trans retinoic acid (atRA), which is highly effective in a subgroup of AML characterized by rearrangements of the retinoic acid (RA) receptor, RARA [5–7]. Even though in vitro, atRA promoted blast cell differentiation, originally considered its key anti-leukemic activity, also in AML without RARA rearrangements, clinical trials did not convincingly demonstrate therapeutic utility [8–15]. Since AML is a stem cell-driven disease, a small number of studies has recently addressed the impact of atRA on leukemic stem cells (LSCs) and found that it varied widely depending on the identity of the respective driver lesions [9,16,17]. Additionally, the primitivity of an LSC is likely to influence its response to atRA [17]. This review summarizes the role of atRA in normal and leukemic hematopoiesis, with a focus on its effects on LSCs. Specific consideration is also given to EVI1 (MECOM, PRDM3), a gene with key roles both in HSCs [18] and LSCs [17].
Acute myeloid leukemia
In order to sustain the life-long renewal of blood cells, hematopoiesis is organized in a hierarchical manner. The apex of this hierarchy is formed by hematopoietic stem cells (HSCs), a rare, mostly quiescent cell type that resides in a specialized niche in the bone marrow (BM) and is able to both self-renew and give rise to proliferatively active, progressively differentiating progenitor cells [19–21]. Mutations accumulating in hematopoietic stem and progenitor cells (HSPCs) over the lifetime of an individual can lead to malignant transformation [22]. One of the most aggressive hematopoietic malignancies is acute myeloid leukemia (AML), which has an annual incidence of 3–8/100,000 and a median age of onset of around 67 years [5,6]. AML is characterized by the accumulation of immature blasts at the expense of normal myeloid cells in BM and often also peripheral blood (PB), leading to anemia, bleeding, infections, and, if left untreated, death within months. By analogy to normal hematopoiesis, leukemic hematopoiesis emerges from leukemic stem cells, which reside in the hematopoietic niche of the BM and are mostly quiescent, but able to self-renew and give rise to proliferatively active progeny [23–27]. Moreover, LSCs are considered to be able to survive chemotherapy and give rise to relapse [23–27]. Even though the view that LSCs are resistant to conventional cytotoxic therapy has been challenged recently [26], it is supported by observations that high LSC frequencies, as well as the presence of stem cell expression signatures, correlate with inferior outcome in AML [4,23,24,28].
The transforming events giving rise to an LSC may take place either in an HSC or in a progenitor cell that consequently regains stem cell characteristics [23,24,26,29]. They include cytogenetic aberrations, point mutations, copy-number alterations, and epigenetic and transcriptional changes [1–4,30]. Leukemogenic mutations occur in a nonrandom order: alterations in genes coding for epigenetic regulators and chromatin remodeling factors appear prior to mutations in genes coding for transcription factors and signaling molecules [3,31–34]. Remarkably, early-type mutations were also found in phenotypically and functionally normal HSCs in some patients with AML [31–35], and even in a subset of healthy individuals [36–38]. This has led to the concept of pre-LSCs, i.e., stem cells bearing early leukemogenic driver mutations but not yet fully transformed [39]. Aberrations recurring in the malignant cells of different patients may act as drivers of leukemogenesis, represent prognostic markers, and serve as targets for rationally designed therapies [1–4,40,41].
Standard treatment for the majority of patients with AML consists of chemotherapy based on cytosine arabinoside (araC) and an anthracycline for induction. Consolidation comprises further chemotherapy, sometimes complemented by HSC transplantation [42–44]. However, 5-year survival ranges only between <5 and ~40%, depending on a variety of prognostic parameters, e.g., age, white blood cell count, and the presence of specific genetic and gene expression alterations [2,5,6]. Recently, several targeted therapeutics, including tyrosine kinase inhibitors, BCL2 inhibitors, IDH inhibitors, and antibody-drug conjugates, have been approved for the treatment of AML [41]. Notably, one of the first examples of a molecularly targeted anti-cancer drug, albeit discovered without knowledge about its mechanism of action, is all-trans retinoic acid (atRA). atRA has greatly improved the outcome of acute promyelocytic leukemia (APL), a subtype of AML characterized by expression of an aberrant retinoic acid receptor [45–47].
atRA and its roles in normal hematopoiesis
atRA, the major biologically active metabolite of vitamin A, plays multiple roles during development and in the adult organism [48–50]. Conversion of vitamin A (retinol) into atRA requires two sequential oxidation steps, of which the second, irreversible one is catalyzed by members of the aldehyde dehydrogenase (ALDH) family, also known as retinaldehyde dehydrogenases (RALDHs)[51]. Conversely, atRA catabolism is initiated by cytochrome p450 (CYP) enzymes, primarily of the CYP26 subfamily [51]. atRA exerts its biological effects mainly through nuclear receptor type transcription factors composed of a retinoic acid receptor (RAR) and a retinoid X receptor (RXR) subunit. Each of these subunits has three isoforms that are encoded by paralogous genes – RARA, RARB, RARG, and RXRA, RXRB, RXRG, respectively, with additional diversification through alternative splicing [48,52,53]. The RAR/RXR heterodimer binds to specific retinoic acid response elements (RAREs) in the regulatory regions of numerous target genes, repressing their transcription in the absence of ligand and activating it in its presence [48,52,53]. RAR activation is followed by its degradation via the ubiquitin/proteasome pathway [53,54].
atRA plays several well-established roles in hematopoiesis, among them the promotion of granulocytic differentiation of committed progenitor cells [49,55–57]. In contrast, its roles in HSCs were controversially described (Figure 1). Some reports suggested that HSCs are subject to negative regulation by atRA: microarray analysis of human HSC enriched CD34+ CD38− cells and progenitor enriched CD34+ CD38+ cells suggested that the RA pathway was down-regulated in HSCs [58]. In vitro treatment with a pan-RAR antagonist increased the numbers of “cobblestone area forming cells-week 8” (CAFCW8) and of cells with the ability to repopulate severe combined immunodeficiency (SCID) mice (SCID repopulating cells, SRCs), both considered as readouts of human HSC activity. Likewise, co-culture of CD34+ CD38− cells with stromal cells maintained their CAFCW8 activity and SRC numbers. These effects were partially counteracted by chemical or genetic inhibition of CYP26, suggesting that stromal cells contributed to HSC maintenance by inactivating RA [58]. In a related study, an RXR antagonist maintained human lineage marker negative (lin−) CD34+ CD38− cells in G0 during culture, and substantially increased their non-obese diabetic (NOD) SCID repopulating frequency [59]. Furthermore, genetic or pharmacological inhibition of ALDH activity, and thus, presumably, RA synthesis, increased the radioprotective cell frequency and the short term (ST) repopulating potential of immunophenotypically defined, HSC enriched human and murine cell populations [60,61]. However, ALDH inhibition had no effect on the long term (LT) repopulating ability of murine HSPCs [61], indicating that its activity did not inhibit the most primitive stem cells.
Figure 1.
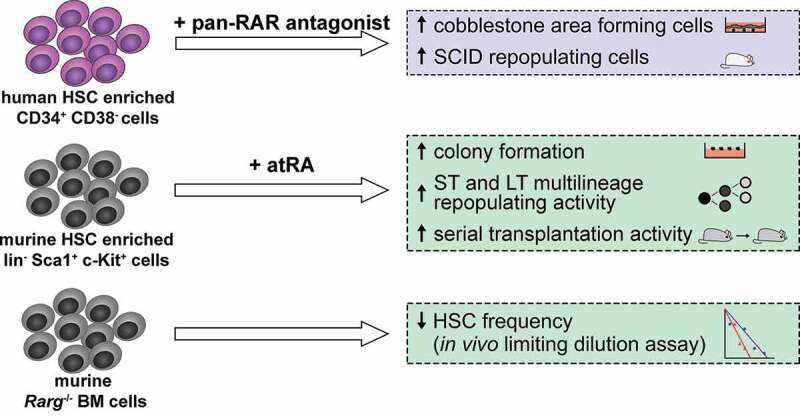
Role of all-trans retinoic acid (atRA) in hematopoietic stem cells (HSCs). Blue box summarizes key experiments leading to the conclusion that atRA negatively affects HSCs. Green boxes summarize key experiments leading to the conclusion that atRA positively affects HSCs. RAR, retinoic acid receptor; SCID, mice with severe combined immunodeficiency; ST, short term; LT, long term. Human cells are depicted in purple and murine cells in gray. The number of symbols in the serial transplantation assay is not meant to indicate the actual number of transplantations
In studies using murine HSPCs, in vitro exposure of HSC enriched lin− Sca1+ c-Kit+ (LSK) cells to the physiological agent atRA enhanced their proliferation and maintained a more immature cell surface marker profile, prolonging their ability to form immature hematopoietic colonies in semisolid media [56]. Importantly, LSK cells cultured with atRA had increased ST and LT multilineage repopulating ability in a competitive repopulation assay, while the pan-RAR antagonist AGN193109 abrogated these activities [62]. The LSK cells cultured with atRA displayed increased repopulation during serial transplantation studies, which are the gold standard test for HSC self-renewal [63]. The contrasting effects of atRA on myeloid differentiation and on HSCs were attributed to the activity of different RAR isoforms. In vitro experiments after experimental expression of RAR isoforms, as well as competitive repopulation and in vivo limited dilution assays with cells from Rara and Rarg knock-out mice suggested that RARA promoted myeloid differentiation, while RARG mediated HSC maintenance by atRA [63].
Genome-wide gene expression profiling experiments taking advantage of the refined knowledge of the immunophenotypes of murine HSPCs revealed that atRA signaling was highly enriched in dormant HSCs versus activated HSCs and early myeloid progenitor cells [64]. In vitro and in vivo treatment with atRA enhanced HSC quiescence and serial replating and serial transplantation activity, even under HSC activating stress conditions. By contrast, maintenance of mice on a vitamin A free diet for ~4 months decreased HSC quiescence and activity [64].
Possible explanations for the partially discrepant results regarding the effects of atRA on HSCs include species effects, which may reflect real differences or technical aspects (e.g., the different surface markers used to define human and murine HSPCs, and/or the need to assess human HSC activity in potentially artifact-prone xenograft assays). Also, differences in retinoid treatment – concentration, duration, and ex vs. in vivo exposure – and between the assays used may play a role [53,65]. Remarkably, in the studies claiming an inhibitory effect of atRA on HSCs, few if any experiments employed the physiological ligand itself, but rather, conclusions were mostly based on data obtained with synthetic retinoids and inhibitors. In summary, unless murine HSCs should unexpectedly behave fundamentally different from human ones, strong evidence from experiments combining agonist and antagonist treatment with knock-out models and the most stringent stem cell assays favors the interpretation that atRA promotes the abundance and activity of HSCs.
atRA in acute promyelocytic leukemia
Acute promyelocytic leukemia (APL) is a subtype of AML that is characterized by rearrangements of the RARA gene. A number of different fusion partners have been described, but about 95% of cases harbor a t(15;17)(q22;q21), which fuses the promyelocytic leukemia (PML) gene to RARA [45,46,66]. The resulting PML-RARA fusion protein acts in a dominant negative manner on both the PML and RARA pathways, but the activity of both fusion partners is at least partially restored by pharmacological doses of atRA [45,66]. Remarkably, the inclusion of atRA in the therapy of APL a few decades ago has transformed the prognosis of this disease from very poor to highly favorable [45,47,66]. Nevertheless, atRA monotherapy, even though able to enhance APL blast differentiation and effect complete morphological remissions, does not lead to long-term disease-free survival [45,66]. The outcome is ameliorated with liposomal delivery, which achieves higher intracellular atRA concentrations and definitive cures in a proportion of cases [66]. Current treatment regimens combine atRA with anthracyclines or the even more effective arsenic trioxide (ATO) and attain long-term survival in the vast majority of patients [7,47,67].
The molecular and biological mechanisms explaining the success of atRA-based therapies in APL were addressed only after the discovery of their clinical effectiveness. As mentioned above, PML-RARA hinders the functions of both of its fusion partners. It disrupts the formation of nuclear bodies (NBs), subcellular structures in whose genesis the tumor suppressor PML plays a key role, and which regulate multiple cellular functions including proliferation, apoptosis, and senescence [45,66]. It also interferes with RARA mediated transcription activation, which promotes normal granulocytic differentiation [45,66]. PML-RARA is unresponsive to physiological levels of atRA, but pharmacological atRA concentrations cause degradation of the fusion protein and restore NB formation, transcription of RARA target genes, and myeloid differentiation [45,66]. However, in contrast to initial assumptions, the induction of myeloid differentiation appears to be insufficient to cure APL. Thus, synthetic retinoids that were able to activate transcription by RARA and PML-RARA, but not their degradation, mediated granulocytic differentiation of APL blasts, but conferred a much smaller survival benefit than atRA in a mouse model of APL [54] (with the drawback that atRA and the synthetic retinoids were administered via different routes). This differential impact on survival was observed both in the originally treated mice and in secondary recipients transplanted with their BM cells, the latter being considered a readout of leukemia initiating cell (LIC) activity [54]. In contrast to the effects of synthetic retinoids, increasing doses of atRA caused increasing PML-RARA degradation in APL mice, which correlated with survival benefits both for the treated mice and for secondary recipients [68]. Together, these data indicated that restoration of RARA target gene expression and APL blast differentiation are insufficient to cure APL, but rather, PML-RARA degradation and eradication of LICs are required toward this end [45,66]. In further support of this conclusion, not only atRA, acting via the RARA-moiety, but also ATO, through the PML-moiety, caused degradation of PML-RARA. atRA and ATO cooperated both with respect to PML-RARA degradation and the survival of treated APL mice and secondary recipients [66,69]. Thus, atRA at high doses and/or in combination with ATO is able to reduce LIC activity in APL, and this reduction correlates with the clinical effectiveness of a specific therapeutic regimen.
Even though not undisputed, the presence of specific additional molecular and genetic lesions, in particular, the kinase activating FLT3 internal tandem duplication (ITD) may modulate the response of APL patients to atRA-based therapy [47,70]. In mice bearing a PML-RARA transgene, the additional presence of an FLT3-ITD knock-in allele reduced the effects of atRA on PML-RARA degradation, NB re-formation, granulocytic differentiation, in vivo blast clearance, and on the delay of disease onset in secondary recipients [71]. In contrast, but in agreement with the known clinical effectiveness of combined treatment with atRA and ATO in patients with FLT3-ITD APL, this combination promoted all of the above parameters irrespective of the presence of the FLT3 mutation [71]. Somewhat at odds with the atRA resistance conferred by the FLT3-ITD in the mouse model, an exome sequencing study on matched diagnosis-relapse samples from patients with APL that had been treated with atRA plus chemotherapy showed that FLT3 mutations present at diagnosis were consistently lost at relapse [72].
atRA in non-APL AML: clinical trials
The tremendous success of atRA in APL, together with laboratory observations that atRA promoted differentiation and chemotherapy sensitivity of non-APL AML blasts [8–14,73,74], inspired numerous trials addressing the clinical benefit of adding atRA to chemotherapy also in non-APL AML. In a phase III trial that included 242 elderly (>60 years) patients with AML, atRA, started 2 days after cytotoxic therapy, was associated with a higher response rate, and with longer event free (EFS) and overall survival (OS) as an independent parameter [75]. Later, this trial was re-analyzed for the possible predictive power of some prognostically relevant recurrent mutations in AML, namely, the NPM1, FLT3-ITD, FLT3 tyrosine kinase domain (TKD), MLL partial tandem duplication (PTD), and CEBPA mutations. This analysis suggested that the beneficial effects of atRA were restricted to the (relatively small) subgroup of patients that had a mutated NPM1 gene but no FLT3-ITD [76]. In another trial, 83 AML patients >60 years received standard chemotherapy with or without atRA, and the group of patients with below-median expression of the transcriptional co-factor MN1 experienced improved EFS and OS with atRA [8]. In contrast, in a study in which 1075 patients <60 years with non-APL AML or high-risk myelodysplastic syndrome (MDS) were randomized to receive atRA or not, atRA had no effect on response rate or survival [77]. This was true for the entire cohort, for patients with cytogenetically normal AML, and for each of the subgroups defined by FLT3-ITD, NPM1, or CEBPA mutations, or by MN1 expression [77]. Also, in a randomized study of atRA in 1100 adults <60 years with AML, atRA, started on treatment day 6, did not reveal any consistent benefit in either the entire cohort or in the subgroup of patients with NPM1 mutations [78]. A recent meta-analysis summarized eight trials comparing chemotherapy plus atRA with chemotherapy alone in a total of almost 4000 adult patients with AML, and concluded that there was no evidence for an effect of atRA on either the risk of adverse events, or on response rate, disease-free survival (DFS), or OS [15]. It should be pointed out that all discussed trials differed in numerous parameters, including patient age, the identities of the cytotoxic drugs used in conjunction with atRA, and treatment schedules, thereby precluding direct comparisons. In contrast to the so far mostly disappointing results regarding the combination of atRA with conventional chemotherapy, two recent studies suggested that atRA may be beneficial when combined with hypomethylating agents in elderly patients ineligible for induction chemotherapy [79,80]. It is assumed that hypomethylating treatment primes myeloid differentiation genes for transcription activation by RARs [81]. These recent publications, as well as the registration of at least eight currently recruiting clinical trials (clinicaltrials.gov, accessed on May 13, 2020), indicate the strong ongoing interest in exploring the activity of atRA in non-APL AML.
atRA in non-APL AML: preclinical studies
Complementing the clinical trials, numerous laboratory studies have tried to identify subgroups of AML potentially benefitting from atRA, as well as agents that may sensitize resistant AML cells to retinoids (Figure 2). MN1 encodes a transcription co-factor of the RAR/RXR complex, and its elevated expression was associated with atRA resistance in one of the clinical trials [8]. Accompanying laboratory work showed that overexpression of MN1 greatly decreased the sensitivity of a preleukemic BM cell line toward the proliferation inhibiting and differentiation promoting effects of atRA [8]. Inducible expression of MN1 in a human myeloid cell line enhanced or repressed the effects of atRA in a gene-specific manner [82].
Figure 2.
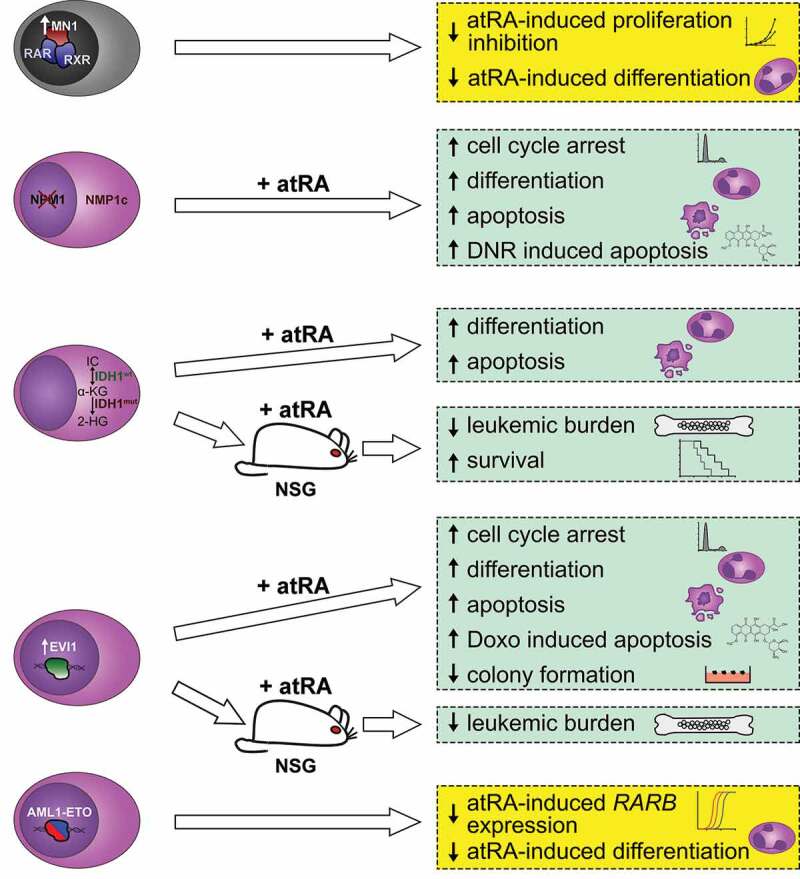
Effects of all-trans retinoic acid (atRA) on leukemic blasts of genetically or molecularly defined subgroups of AML. Yellow boxes summarize inhibition of anti-leukemic effects of atRA (note, however, that primary AML1-ETO positive blasts were found to be atRA sensitive in an independent study; see main text). Green boxes summarize anti-leukemic effects of atRA. Human cells are depicted in purple and murine cells in gray. IC, isocitrate; α-KG, α-ketoglutarate; 2-HG, 2-hydroxyglutarate; NSG, non-obese diabetic severe combined immunodeficiency IL2Rgnull mice; DNR, daunorubicin; Doxo, doxorubicin
Mutations in NPM1 represent the most frequent recurrent mutation in AML, and lead to a predominantly cytoplasmic localization of the encoded chaperone protein, which usually shuttles between nucleus, nucleolus, and cytoplasm. Building on the reported clinical association between NPM1 mutations and atRA responsiveness [76], the effects of atRA and ATO on AML cell lines and primary AML samples with and without NPM1 mutations were investigated. Both agents, and even more strongly their combination, caused proteasome-dependent down-regulation of mutated, but not wild type, NPM1, leading to re-localization of wild type NPM1 (produced from the second allele) to the nucleus [11,12]. This was accompanied by a higher propensity of NPM1-mutated AML cells to respond to atRA- and/or ATO-mediated cell cycle arrest, differentiation, and apoptosis [11,12]. Furthermore, pretreatment of an NPM1-mutated cell line with atRA and/or ATO sensitized it to daunorubicin [11]. Compassionate use of atRA and arsenic in five elderly patients with NPM1-mutated AML that were deemed unfit for chemotherapy led to transient anti-leukemic effects in three of them [12].
Pertinent clinical trials had not been analyzed for an effect of mutations in the genes encoding the tricarboxylic acid cycle enzymes IDH1 and IDH2 on atRA responsiveness, but laboratory experiments suggested a possible relation [13]. IDH1-mutated cell lines and primary samples were more sensitive to atRA induced differentiation and apoptosis than their IDH1 wild type counterparts, and this was counteracted by an inhibitor of mutated IDH1. In a mouse xenograft model, atRA reduced leukemic burden and increased survival in an IDH1-mutation-specific manner. Most provokingly, pre-treatment with a cell permeable form of 2-hydroxyglutarate, product of the neomorphic IDH1 variant and usually considered an oncometabolite, sensitized AML cell lines with wild-type IDH1 to atRA induced differentiation [13].
The ecotropic viral integration site 1 (EVI1) gene encodes a transcription factor that fulfills essential functions in HSCs, but is down-regulated during normal hematopoietic differentiation [18,83–85]. Its overexpression, observed in approximately 10% of patients with AML, is associated with a particularly poor prognosis [86–88]. Perhaps counter-intuitively, EVI1 expression was up-regulated by atRA in cell lines and in primary AML cells [14,89–91], due to both mRNA stabilization and transcriptional up-regulation through a canonical RARE [90,91]. EVI1 counteracted its own induction by atRA but enhanced that of the RARB gene in luciferase reporter assays [91]. Genome-wide gene expression analysis of human myeloid cell lines with or without experimental EVI1 expression showed that EVI1 enhanced the transcriptional responses to atRA of a number of genes [10]. Accordingly, EVI1 also augmented atRA induced cell cycle arrest, differentiation, and apoptosis in these cell lines [10]. In primary AML blasts, atRA enhanced differentiation and apoptosis, and decreased clonogenic activity and engraftment in immunodeficient mice predominantly of EVI1high, but not EVI1low, samples [14]. Preincubation with atRA also increased the doxorubicin sensitivity of two EVI1high AML samples [14]. This led to the suggestion that patients with EVI1high AML may specifically benefit from atRA containing therapy, but this assumption was not tested in the relevant clinical trials, and not affirmed by studies on LSCs (see below). However, as another interesting parallel to the situation with APL and with NPM1-mutated AML, ATO targeted the EVI1 protein for degradation via the ubiquitin-proteasome pathway [92]. Moreover, EVI1 overexpression appeared to confer sensitivity to ATO in murine myeloid cells and in a clinical trial including 28 patients with MDS [93].
In contrast to these studies, which identified lesions or gene expression states potentially sensitizing AML cells to atRA, the AML associated transcription factor fusion protein AML1-ETO was reported to confer atRA resistance [94]. In human myeloid cell lines, AML1-ETO recruited transcription corepressors to the RARE of the RARB gene, mediated increased DNA methylation and decreased histone acetylation, and prevented induction of RARB by atRA. An shRNA against AML1-ETO or treatment with 5-azacytidine reduced methylation of the RARB regulatory region, and restored the induction of RARB and differentiation in response to atRA. RARB methylation was also found in the majority of primary samples from patients with AML M2 or M4, morphological subtypes of AML often associated with the expression of AML1-ETO or functionally related fusion proteins [94]. In an independent study, however, AML1-ETO expressing primary AML blasts were found to be atRA sensitive [95].
Some authors raised the question whether the RAR pathway is functional in AML at all, since a number of genes that are mutated or misexpressed in this disease have a negative impact on it [96]. Along these lines, some reports intended to identify agents that could sensitize AML cells to atRA. As already mentioned, a cell permeable form of 2-hydroxyglutarate as well as 5-azacytidine augmented the effects of atRA on the differentiation of certain AML cell lines [13,94]. Experiments with cell lines, xenografts, and primary AML samples indicated that inhibition of the SUMO pathway may sensitize AML cells to atRA [97]. Similarly, the endoplasmatic reticulum-stress inducing drug tunicamycin was reported to cooperate with atRA and ATO to inhibit the clonogenic capacity and to promote death of human AML cell lines and primary AML cells, particularly those with a FLT3-ITD [98]. Another set of in vitro experiments led to the suggestion that inhibition of the histone acetyltransferase GCN5 and/or the lysine demethylase LSD1 augmented the anti-leukemic activities of atRA [99].
In contrast to the above described efforts, some authors have questioned the availability of atRA in the BM niche. In one study, hematopoietic cells were proposed to reside in a retinoid deplete environment in the BM based on the limited activity of a synthetic reporter gene [100]. In vitro experiments with AML cells suggested a role of BM stroma in degrading atRA: human AML cell lines with different driver lesions (PML-RARA, AML1-ETO, NPM1 mutations), as well as primary samples expressing AML1-ETO or related fusion genes, responded to atRA by differentiation and/or loss of clonogenic activity. The activity of atRA was abrogated by co-culture with BM stromal cells, and restored by incubation with a CYP26 inhibitor [95]. Accordingly, the half-life of atRA was reduced ~3-fold by stromal co-culture, but restored by CYP26 inhibition [95]. To overcome the inhibitory effects of the BM niche, the use of synthetic, CYP26-resistant retinoids was proposed. Since atRA mediated differentiation of leukemic cells via RARA, but caused feedback-induction of stromal CYP26B1 mostly via RARG, RARA-selectivity was considered an additional advantage [101]. IRX195183 fulfilled both requirements, and was able to effect differentiation of AML cell lines even in stromal co-culture. Moreover, it controlled disease burden more effectively than atRA in an NPM1-mutated AML xenograft model [101]. The RARA-selective, CYP26-resistant retinoid tamibarotene is currently being tested in a phase 2 trial including patients with AML and MDS (NCT02807558) [101].
The above-described studies are mostly based on the concept of “differentiation therapy”, i.e., the assumption that promoting the differentiation of leukemic blasts will effect, or at least support, cure of the disease. However, AML is a stem cell-driven disease and therefore can be cured only by eradication of the (small) LSC population [25]. Consequently, understanding the effects of atRA in AML requires an appreciation of its impact on AML stem cells.
Effects of atRA on AML stem cells
Both normal and cancer stem cells from a variety of tissues are characterized by high ALDH activity [102]. The human ALDH gene family contains 19 members, whose functions include the synthesis of retinoids and the metabolism of reactive aldehydes from both endogenous and exogenous sources [102,103]. ALDHs contribute to cellular resistance against a variety of anti-cancer drugs, including daunorubicin, and protect normal and cancer cells from reactive oxygen species (ROS) produced in the context of chemo- and radiotherapy [102,103]. Cellular atRA down-regulates ALDH activity as part of a negative feedback mechanism, and suppression of ALDH by exogenous atRA or synthetic retinoids sensitizes cancer cells to chemotherapeutic drugs [102]. In AML, ALDH activity displayed a complex pattern: approximately 23% of patients had a higher proportion of ALDH+ cells (identified by Aldefluor staining) than normal controls (median, 1.9%; ALDH-numerous AML), while the rest had substantially lower proportions of ALDH+ cells (ALDH-rare AML) [104]. The distribution of AML-specific aberrations between ALDH+ and ALDH− cells, gene expression profiling, and xenotransplantation experiments suggested that ALDH-numerous AML contained a higher number of LSCs and these were present among ALDH+ cells [104,105]. By contrast, in ALDH-rare AML, ALDH+ cells were enriched for normal HSCs [104]. In ALDH-numerous AML, ALDH+ cells were more resistant to araC than ALDH− cells [104]. Consistent with independent data showing that high ALDH levels were associated with poorer outcome in AML [103], patients with ALDH-numerous AML had worse DFS and OS [104].
While these studies can be interpreted as indirect evidence for a role of atRA in the LSCs of a subset of AML, a small number of studies has directly addressed the impact of atRA on AML stem cells, with heterogeneous results (Figure 3). Ma et al. investigated the effects of atRA and its interaction with the tyrosine kinase inhibitor sorafenib in AML with FLT3-ITD mutations. atRA enhanced the anti-leukemic effects of sorafenib in AML cell lines, primary samples, and xenografts with an FLT3-ITD [16]. In a congenic AML mouse model based on co-expression of an FLT3-ITD allele (which alone is insufficient to cause AML) with a Nup98-Hoxd13 fusion gene, in vivo treatment with atRA delayed disease onset, and enhanced corresponding effects of sorafenib, in secondary recipients. An in vivo limiting dilution assay with cells from the treated mice revealed LSC frequencies of 1/80, 1/1,700, 1/38,000, and <1/106 for vehicle, atRA, sorafenib, and atRA + sorafenib treated mice, respectively [16]. In summary, atRA not only enhanced the anti-leukemic effects of sorafenib on AML blasts with an FLT3-ITD mutation, but also reduced the frequency of LSCs both by itself and together with sorafenib. The ability of atRA to reduce LSC activity in Flt3-ITD driven AML was confirmed in an independent mouse model (Nguyen et al., submitted).
Figure 3.
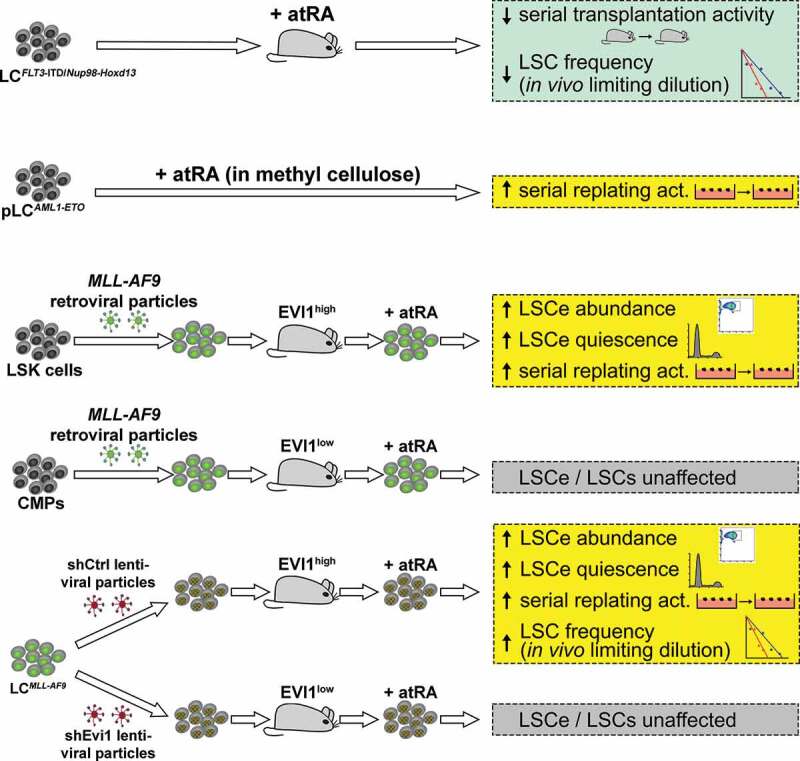
Effects of all-trans retinoic acid (atRA) on AML stem cells. Green box summarizes anti-leukemic effects of atRA; yellow boxes summarize pro-leukemic effects of atRA; gray boxes summarize absence of an effect of atRA. LC, leukemic cells; pLC, preleukemic cells; LSK cells, lin− Sca1+ c-Kit+ cells (HSC enriched); CMPs, common myeloid progenitor cells; LSCs, leukemic stem cells; LSCe, LSC enriched cells; act., activity. Numbers of symbols in serial replating or transplantation assays are not meant to indicate the actual numbers of repetitions
While atRA inhibited LSC activity in FLT3-ITD bearing non-APL AML, the opposite was reported for AML expressing the AML1-ETO fusion or overexpressing EVI1. atRA strongly increased the serial replating ability of AML1-ETO expressing murine BM cells, and resulted in larger, more immature colonies [9]. Interestingly, the effect of a RARA agonist was opposite to that of atRA, while a RARG agonist was ineffective on its own but together with the RARA agonist mimicked the effects of atRA [9]. In a congenic mouse model of AML1-ETO-driven AML, the RARA agonist did not prolong survival, but effected a transient decrease in leukemic burden and a persistent increase in myeloid differentiation [9].
Building on our own previous observations of a functional cooperation between EVI1 and atRA in malignant myeloid cells [10], we asked whether similar interactions would also exist in LSCs. To this end, a mouse model of MLL-AF9 driven AML was used. MLL-rearrangements are associated with EVI1 overexpression in human AML [88,106,107]. In mice, MLL fusion proteins were able to transform both HSC enriched LSK cells and progenitor cells, and enhanced Evi1 expression by direct promoter binding – remarkably only in LSK-, but not progenitor-, derived AML [17,106,108–111]. This suggested that the presence or absence of EVI1 overexpression, each observed in about half of the patients, reflects the cell type in which the transforming event occurred also in human MLL rearranged AML [106–109,111]. Irrespective of the cell of origin, leukemic cells with the immunophenotype of granulocyte macrophage progenitors (GMPs) are strongly enriched for LSCs in MLL-AF9 driven murine AML [109]. They were originally termed leukemic GMPs [109], but are referred to as LSC enriched cells (LSCe) by us [17].
Ex vivo treatment of leukemic cells (LCs) from BM of mice with LSK derived, MLL-AF9 driven AML (LCLSK_MLL-AF9; EVI1high) with atRA augmented the abundance and quiescence of LSCe, and the activity of LSCs as determined by serial replating and in vivo limiting dilution assays [17]. In contrast, no such response was observed with EVI1low LCCMP_MLL-AF9, i.e., LCs from mice that developed AML after transplantation with MLL-AF9 transduced common myeloid progenitors (CMPs) [17]. To investigate a possible role of Evi1 in the differential atRA responsiveness of LSCs from LSK- and CMP-derived AML, LCLSK_MLL-AF9 were transduced with shRNAs against Evi1 or with a control shRNA, and transplanted into congenic recipient mice. Experiments with LCs from these mice showed that knock-down of Evi1 per se reduced LSCe/LSC abundance, quiescence, and activity (the first demonstration of a key role of Evi1 in AML LSCs), and additionally abolished the stemness promoting effects of atRA [17]. Evi1 also strongly augmented transcriptional responses of LSCe to atRA: its knock-down reduced the number of atRA-regulated genes to less than one-half the number found in control cells. Pharmacological and genetic inhibition experiments established Notch4, one of the joint targets of EVI1 and atRA, as a relevant mediator of their effects [17]. Ex vivo exposure of BM LCs to a pan-RAR antagonist affected LSCe abundance and quiescence in a manner opposite to that of atRA. Notably, in vivo antagonist treatment significantly prolonged survival of initially treated and secondary recipient mice, and decreased the abundance, quiescence, and activity of LSCe/LSCs. Finally, atRA increased the quiescence of human AML cell lines retaining some stem cell characteristics in an EVI1-dependent manner, and enhanced clonogenicity and LSCe quiescence of primary EVI1 overexpressing, but not EVI1-negative, AML samples [17].
Together, these data show that atRA augmented leukemic stemness in AML resulting from HSC, but not progenitor cell, transformation. These differences could be largely explained by differential expression of the stem cell gene Evi1, which per se enhanced leukemic stemness, and additionally facilitated the stemness promoting activity of atRA, in AML.
Summary and conclusions
The effects of atRA in the context of AML are multiple and complex. One of its earliest known and best described consequences is the promotion of myeloid differentiation of leukemic blasts, which forms the basis of most studies aiming to identify atRA-susceptible subgroups of patients, and/or substances able to sensitize AML cells to atRA [8,10–14,97-99]. atRA also augmented the susceptibility of AML cells to chemotherapeutic drugs [11,14,73,74], possibly due to its inhibitory effect on ALDHs, an enzyme family with key roles both in retinoid metabolism and drug resistance [51,102,103]. Recent studies reported that atRA regulated the abundance, properties, and activity of AML stem cells [9,16,17]. Interestingly, the nature of these effects varied widely in a manner related to the identity of the genetic driver lesions, and/or the transformed cell type. In mouse models in which AML was driven by an FLT3-ITD in combination with a second non-APL AML typical aberration, atRA negatively affected LSC activity [16] (and Nguyen et al., submitted). However, on the background of the APL-typical PML-RARA fusion, the FLT3-ITD counteracted the inhibitory effect of atRA on LIC activity [71]. In an AML1-ETO-driven mouse model, atRA even promoted LSC properties [9]. atRA also augmented LSC abundance, quiescence, and activity in an MLL-AF9 driven murine AML model in a manner dependent on transformed cell type (LSK cells vs. CMPs) and Evi1 expression [17]. Together, these studies highlight substantial molecularly and genetically determined heterogeneity of the effects of atRA on AML LSCs. At least partially related to this, the cell of origin also may play a role in the atRA response of LSCs: in AML arising from transformed HSCs – a cell type whose self-renewal is promoted by atRA [56,62,63] – leukemic stemness may be likewise promoted by atRA. In contrast, in AML originating from transformed myeloid progenitors, which respond to atRA by growth arrest and differentiation [49,55–57], atRA may be inert or even inhibit LSC/LIC activity.
Perhaps less surprisingly on the background of such complexity, attempts to identify atRA-responsive subgroups of patients with non-APL AML have so far not yielded conclusive results [8,15,76–78]. And even though the rationale that the effects of atRA on LSCs may explain its clinical effectiveness appears compelling, translation of corresponding laboratory data does not appear straightforward: the impact of EVI1 overexpression on patients’ responses to atRA has not been investigated so far, and the inhibitory effect of atRA alone or in combination with sorafenib on LSCs from FLT3-ITD driven murine AML [16] does not correspond to clinical activity of atRA in FLT3-ITD positive patients [76,77]. Whether this can be explained by the fact that atRA was combined with chemotherapy rather than sorafenib in the clinical trials remains to be determined.
To be able to address the multifactorial effects of leukemia-associated molecular and genetic lesions on patients’ responses to atRA that are suggested by numerous laboratory studies, identification of atRA sensitive AML subpopulations will require large clinical trials accompanied by extensive molecular characterization. The resulting data need to be evaluated not only for the impact on atRA responsiveness of previously known prognostic parameters, but of all recurrent events and their interactions using advanced statistical tools. Such studies have the potential to reveal which patient subgroups respond to atRA or other RAR agonists, and which subgroups may even suffer a disadvantage from atRA but might benefit from RAR antagonists. The latter, certainly unorthodox possibility was suggested by our own recent work [17], and an earlier study pointed in a similar direction[9]. Given the multiple functions of atRA also in the adult organism [48–50], interfering with its pathway could be associated with substantial side effects. Nevertheless, RAR antagonists are being explored as treatments for diverse ailments, including malignancies and diseases of hematopoietic cells [17,112–119]. However, in view of the role of atRA in normal HSCs [56,62–64], the extent of a possible therapeutic window in the context of hematological malignancies requires specific consideration.
In addition to the decision between agonists and antagonists, degradation (CYP26) resistance and receptor isoform specificity are potentially relevant to the choice of retinoids with optimal anti-leukemic activity [9,101]. RARA specific agonists may be advantageous with respect to the induction of blast differentiation [9,101]. On the other hand, induction of stromal CYP26B1 and promotion of HSC maintenance and activity by atRA was mostly ascribed to the action of RARG [63,101]. Which RAR isoforms mediate the activity of atRA toward LSCs has been investigated only in the context of AML1-ETO, with complex results: only the combination of a RARA and a RARG agonist mimicked the LSC-promoting effects of atRA [9]. Thus, the possible superiority of receptor isoform-specific retinoids in the context of the eradication of AML LSCs requires additional thorough assessment.
Beyond the choice of the retinoids themselves, the identity of the agents used in combination with them is likely to influence their effects. All clinical studies that investigated the effects of atRA in non-APL AML to date used different protocols[15], which may contribute to the inconsistency of their results. Possibly the most promising effects were obtained when atRA was combined with hypomethylating agents [15,79,80]. The relative timing, as well as the duration of the administration of the different drugs are also likely to be important: addition of atRA prior to or together with chemotherapy may optimize the sensitization to cytotoxic drugs, while maximal effects of retinoids on LSC elimination may be achieved when using them as maintenance therapy.
The interest in retinoids for the treatment of non-APL AML is ongoing, as evidenced by numerous recent publications and several currently recruiting clinical trials (clinicaltrials.gov). Even though the effects of atRA in AML are obviously complex, this research has important potential to lead to improved therapies for additional subgroups of patients with AML.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Funding Statement
This work was supported by the Austrian Science Fund (FWF) under Grants no P28013 and P28256 to RW.
Disclosure of interest
The authors declare no conflict of interest.
References
- [1].Network CGAR . Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Grimwade D, Ivey A, Huntly B.. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ng S, Mitchell A, Kennedy J, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540:433–437. [DOI] [PubMed] [Google Scholar]
- [5].Almeida A, Ramos F. Acute myeloid leukemia in the older adults. Leuk Res Rep. 2016;6:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sanford D, Ravandi F. Management of newly diagnosed acute myeloid leukemia in the elderly: current strategies and future directions. Drugs Aging. 2015;32:983–997. [DOI] [PubMed] [Google Scholar]
- [7].Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–121. [DOI] [PubMed] [Google Scholar]
- [8].Heuser M, Argiropoulos B, Kuchenbauer F, et al. MN1 overexpression induces acute myeloid leukemia in mice and predicts ATRA resistance in patients with AML. Blood. 2007;110:1639–1647. [DOI] [PubMed] [Google Scholar]
- [9].Chee L, Hendy J, Purton L, et al. ATRA and the specific RARalpha agonist, NRX195183, have opposing effects on the clonogenicity of pre-leukemic murine AML1-ETO bone marrow cells. Leukemia. 2013;27:1369–1380. [DOI] [PubMed] [Google Scholar]
- [10].Steinmetz B, Hackl H, Slabáková E, et al. The oncogene EVI1 enhances transcriptional and biological responses of human myeloid cells to all-trans retinoic acid. Cell Cycle. 2014;13:2931–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Martelli M, Gionfriddo I, Mezzasoma F, et al. Arsenic trioxide and all-trans retinoic acid target NPM1 mutant oncoprotein levels and induce apoptosis in NPM1-mutated AML cells. Blood. 2015;125:3455–3465. [DOI] [PubMed] [Google Scholar]
- [12].El Hajj H, Dassouki Z, Berthier C, et al. Retinoic acid and arsenic trioxide trigger degradation of mutated NPM1, resulting in apoptosis of AML cells. Blood. 2015;125:3447–3454. [DOI] [PubMed] [Google Scholar]
- [13].Boutzen H, Saland E, Larrue C, et al. Isocitrate dehydrogenase 1 mutations prime the all-trans retinoic acid myeloid differentiation pathway in acute myeloid leukemia. J Exp Med. 2016;213:483–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Verhagen H, Smit M, Rutten A, et al. Primary acute myeloid leukemia cells with overexpression of EVI-1 are sensitive to all-trans retinoic acid. Blood. 2016;127:458–463. [DOI] [PubMed] [Google Scholar]
- [15].Kuley-Bagheri Y, Kreuzer K, Monsef I, et al. Effects of all-trans retinoic acid (ATRA) in addition to chemotherapy for adults with acute myeloid leukaemia (AML) (non-acute promyelocytic leukaemia (non-APL)). Cochrane Database Syst Rev. 2018;8:CD011960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ma H, Greenblatt S, Shirley C, et al. All-trans retinoic acid synergizes with FLT3 inhibition to eliminate FLT3/ITD+ leukemia stem cells in vitro and in vivo. Blood. 2016;127:2867–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nguyen C, Bauer K, Hackl H, et al. All-trans retinoic acid enhances, and a pan-RAR antagonist counteracts, the stem cell promoting activity of EVI1 in acute myeloid leukemia. Cell Death Dis. 2019;10:944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Goyama S, Yamamoto G, Shimabe M, et al. Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell. 2008;3:207–220. [DOI] [PubMed] [Google Scholar]
- [19].Haas S, Trumpp A, Milsom M. Causes and consequences of hematopoietic stem cell heterogeneity. Cell Stem Cell. 2018;22:627–638. [DOI] [PubMed] [Google Scholar]
- [20].Pinho S, Frenette P. Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol. 2019;20:303–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Laurenti E, Gottgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 2018;553:418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bowman R, Busque L, Levine R. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell. 2018;22:157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wiseman D, Greystoke B, Somervaille T. The variety of leukemic stem cells in myeloid malignancy. Oncogene. 2013;33:3091–3098. [DOI] [PubMed] [Google Scholar]
- [24].Zagozdzon R, Golab J. Cancer stem cells in haematological malignancies. Contemp Oncol (Pozn). 2015;19:A1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Stahl M, Kim T, Zeidan A. Update on acute myeloid leukemia stem cells: new discoveries and therapeutic opportunities. World J Stem Cells. 2016;8:316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vetrie D, Helgason G, Copland M. The leukaemia stem cell: similarities, differences and clinical prospects in CML and AML. Nat Rev Cancer. 2020;20:158–173. [DOI] [PubMed] [Google Scholar]
- [27].Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. 2017;129:1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Eppert K, Takenaka K, Lechman E, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–1093. [DOI] [PubMed] [Google Scholar]
- [29].Fisher J, Kalleda N, Stavropoulou V, et al. The impact of the cellular origin in acute myeloid leukemia: learning from mouse models. Hemasphere. 2019;3:e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hackl H, Astanina K, Wieser R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J Hematol Oncol. 2017;10:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jan M, Snyder T, Corces-Zimmerman M, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Corces-Zimmerman M, Hong W, Weissman I, et al. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A. 2014;111:2548–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shlush L, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hirsch P, Zhang Y, Tang R, et al. Genetic hierarchy and temporal variegation in the clonal history of acute myeloid leukaemia. Nat Commun. 2016;7:12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Garg M, Nagata Y, Kanojia D, et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood. 2015;126:2491–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Genovese G, Kahler A, Handsaker R, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Corces-Zimmerman M, Majeti R. Pre-leukemic evolution of hematopoietic stem cells: the importance of early mutations in leukemogenesis. Leukemia. 2014;28:2276–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lal R, Lind K, Heitzer E, et al. Somatic TP53 mutations characterize preleukemic stem cells in acute myeloid leukemia. Blood. 2017;129:2587–2591. [DOI] [PubMed] [Google Scholar]
- [41].Bohl S, Bullinger L, Rucker F. New targeted agents in acute myeloid leukemia: new hope on the rise. Int J Mol Sci. 2019;20:1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Koreth J, Schlenk R, Kopecky K, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA. 2009;301:2349–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dohner H, Weisdorf D, Bloomfield C. Acute Myeloid Leukemia. N Engl J Med. 2015;373:1136–1152. [DOI] [PubMed] [Google Scholar]
- [44].Magina K, Pregartner G, Zebisch A, et al. Cytarabine dose in the consolidation treatment of AML: a systematic review and meta-analysis. Blood. 2017;130:946–948. [DOI] [PubMed] [Google Scholar]
- [45].Testa U, Lo-Coco F. Targeting of leukemia-initiating cells in acute promyelocytic leukemia. Stem Cell Investig. 2015;2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Baba S, Pandith A, Shah Z, et al. Pathogenetic implication of fusion genes in acute promyelocytic leukemia and their diagnostic utility. Clin Genet. 2019;95:41–52. [DOI] [PubMed] [Google Scholar]
- [47].Sanz M, Fenaux P, Tallman M, et al. Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood. 2019;133:1630–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cunningham T, Duester G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat Rev Mol Cell Biol. 2015;16:110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Canete A, Cano E, Munoz-Chapuli R, et al. Role of vitamin A/retinoic acid in regulation of embryonic and adult hematopoiesis. Nutrients. 2017;9:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shannon S, Moise A, Trainor P. New insights and changing paradigms in the regulation of vitamin A metabolism in development. Wiley Interdiscip Rev Dev Biol. 2017;6:e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kedishvili N. Retinoic Acid Synthesis and Degradation. Subcell Biochem. 2016;81:127–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rochette-Egly C, Germain P. Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs). Nucl Recept Signal. 2009;7:e005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Grace C, Mikkola H, Dou D, et al. Protagonist or antagonist? The complex roles of retinoids in the regulation of hematopoietic stem cells and their specification from pluripotent stem cells. Exp Hematol. 2018;65:1–16. [DOI] [PubMed] [Google Scholar]
- [54].Ablain J, Leiva M, Peres L, et al. Uncoupling RARA transcriptional activation and degradation clarifies the bases for APL response to therapies. J Exp Med. 2013;210:647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Douer D, Koeffler H. Retinoic acid enhances colony-stimulating factor-induced clonal growth of normal human myeloid progenitor cells in vitro. Exp Cell Res. 1982;138:193–198. [DOI] [PubMed] [Google Scholar]
- [56].Purton L, Bernstein I, Collins S. All-trans retinoic acid delays the differentiation of primitive hematopoietic precursors (lin(-)c-kit(+)Sca-1(+)) while enhancing the terminal maturation of committed granulocyte monocyte progenitors. Blood. 1999;94(2):483–495. [PubMed] [Google Scholar]
- [57].Collins S. The role of retinoids and retinoic acid receptors in normal hematopoiesis. Leukemia. 2002;16:1896–1905. [DOI] [PubMed] [Google Scholar]
- [58].Ghiaur G, Yegnasubramanian S, Perkins B, et al. Regulation of human hematopoietic stem cell self-renewal by the microenvironment’s control of retinoic acid signaling. Proc Natl Acad Sci U S A. 2013;110:16121–16126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Safi R, Muramoto G, Salter A, et al. Pharmacological manipulation of the RAR/RXR signaling pathway maintains the repopulating capacity of hematopoietic stem cells in culture. Mol Endocrinol. 2009;23:188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chute J, Muramoto G, Whitesides J, et al. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc Natl Acad Sci U S A. 2006;103:11707–11712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Muramoto G, Russell J, Safi R, et al. Inhibition of aldehyde dehydrogenase expands hematopoietic stem cells with radioprotective capacity. Stem Cells. 2010;28:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Purton L, Bernstein I, Collins S. All-trans retinoic acid enhances the long-term repopulating activity of cultured hematopoietic stem cells. Blood. 2000;95(2):470–477. [PubMed] [Google Scholar]
- [63].Purton L, Dworkin S, Olsen G, et al. RAR gamma is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation. J Exp Med. 2006;203:1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cabezas-Wallscheid N, Buettner F, Sommerkamp P, et al. Vitamin A-retinoic acid signaling regulates hematopoietic stem cell dormancy. Cell. 2017;169:807–23 e19. [DOI] [PubMed] [Google Scholar]
- [65].Alonso S, Jones R, Ghiaur G. Retinoic acid, CYP26, and drug resistance in the stem cell niche. Exp Hematol. 2017;54:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ablain J, de The H. Retinoic acid signaling in cancer: the parable of acute promyelocytic leukemia. Int J Cancer. 2014;135:2262–2272. [DOI] [PubMed] [Google Scholar]
- [67].Zhu H, Hu J, Lo-Coco F, et al. The simpler, the better: oral arsenic for acute promyelocytic leukemia. Blood. 2019;134:597–605. [DOI] [PubMed] [Google Scholar]
- [68].Ablain J, Rice K, Soilihi H, et al. Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat Med. 2014;20:167–174. [DOI] [PubMed] [Google Scholar]
- [69].Nasr R, Guillemin M, Ferhi O, et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat Med. 2008;14:1333–1342. [DOI] [PubMed] [Google Scholar]
- [70].Lucena-Araujo A, Coelho-Silva J, Pereira-Martins D, et al. Combining gene mutation with gene expression analysis improves outcome prediction in acute promyelocytic leukemia. Blood. 2019;134:951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Esnault C, Rahme R, Rice K, et al. FLT3-ITD impedes retinoic acid, but not arsenic, responses in murine acute promyelocytic leukemias. Blood. 2019;133:1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lehmann-Che J, Bally C, Letouze E, et al. Dual origin of relapses in retinoic-acid resistant acute promyelocytic leukemia. Nat Commun. 2018;9:2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lishner M, Curtis J, Minkin S, et al. Interaction between retinoic acid and cytosine arabinoside affecting the blast cells of acute myeloblastic leukemia. Leukemia. 1989;3:784–788. [PubMed] [Google Scholar]
- [74].Yang G, Minden M, McCulloch E. Regulation by retinoic acid and hydrocortisone of the anthracycline sensitivity of blast cells of acute myeloblastic leukemia. Leukemia. 1994;8:2065–2075. [PubMed] [Google Scholar]
- [75].Schlenk R, Frohling S, Hartmann F, et al. Phase III study of all-trans retinoic acid in previously untreated patients 61 years or older with acute myeloid leukemia. Leukemia. 2004;18:1798–1803. [DOI] [PubMed] [Google Scholar]
- [76].Schlenk R, Dohner K, Kneba M, et al. Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia. Results from the AMLSG Trial AML HD98B. Haematologica. 2009;94:54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Burnett A, Hills R, Green C, et al. The impact on outcome of the addition of all-trans retinoic acid to intensive chemotherapy in younger patients with nonacute promyelocytic acute myeloid leukemia: overall results and results in genotypic subgroups defined by mutations in NPM1, FLT3, and CEBPA. Blood. 2010;115:948–956. [DOI] [PubMed] [Google Scholar]
- [78].Schlenk R, Lubbert M, Benner A, et al. All-trans retinoic acid as adjunct to intensive treatment in younger adult patients with acute myeloid leukemia: results of the randomized AMLSG 07-04 study. Ann Hematol. 2016;95:1931–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Cao Y, Liu Y, Shang L, et al. Decitabine and all-trans retinoic acid synergistically exhibit cytotoxicity against elderly AML patients via miR-34a/MYCN axis. Biomed Pharmacother. 2020;125:109878. [DOI] [PubMed] [Google Scholar]
- [80].Lubbert M, Grishina O, Schmoor C, et al. Valproate and retinoic acid in combination with decitabine in elderly nonfit patients with acute myeloid leukemia: results of a multicenter, randomized, 2 x 2, phase II trial. J Clin Oncol. 2020;38:257–270. [DOI] [PubMed] [Google Scholar]
- [81].McKeown M, Johannessen L, Lee E, et al. Antitumor synergy with SY-1425, a selective RARalpha agonist, and hypomethylating agents in retinoic acid receptor pathway activated models of acute myeloid leukemia. Haematologica. 2019;104:e138–e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Meester-Smoor M, Janssen M, Grosveld G, et al. MN1 affects expression of genes involved in hematopoiesis and can enhance as well as inhibit RAR/RXR-induced gene expression. Carcinogenesis. 2008;29:2025–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Wieser R. The oncogene and developmental regulator EVI1: expression, biochemical properties, and biological functions. Gene. 2007;396:346–357. [DOI] [PubMed] [Google Scholar]
- [84].Steinleitner K, Rampetsreiter P, Koffel R, et al. EVI1 and MDS1/EVI1 expression during primary human hematopoietic progenitor cell differentiation into various myeloid lineages. Anticancer Res. 2012;32:4883–4889. [PMC free article] [PubMed] [Google Scholar]
- [85].Kataoka K, Sato T, Yoshimi A, et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J Exp Med. 2011;208:2403–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Haas K, Kundi M, Sperr W, et al. Expression and prognostic significance of different mRNA 5ʹ-end variants of the oncogene EVI1 in 266 patients with de novo AML: EVI1 and MDS1/EVI1 overexpression both predict short remission duration. Genes Chromosomes Cancer. 2008;47:288–298. [DOI] [PubMed] [Google Scholar]
- [87].Lugthart S, van Drunen E, van Norden Y, et al. High EVI1 levels predict adverse outcome in acute myeloid leukemia: prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood. 2008;111:4329–4337. [DOI] [PubMed] [Google Scholar]
- [88].Groschel S, Lugthart S, Schlenk R, et al. High EVI1 expression predicts outcome in younger adult patients with acute myeloid leukemia and is associated with distinct cytogenetic abnormalities. J Clin Oncol. 2010;28:2101–2107. [DOI] [PubMed] [Google Scholar]
- [89].Xi Z, Russell M, Woodward S, et al. Expression of the Zn finger gene, EVI-1, in acute promyelocytic leukemia. Leukemia. 1997;11:212–220. [DOI] [PubMed] [Google Scholar]
- [90].Aytekin M, Vinatzer U, Musteanu M, et al. Regulation of the expression of the oncogene EVI1 through the use of alternative mRNA 5ʹ-ends. Gene. 2005;356:160–168. [DOI] [PubMed] [Google Scholar]
- [91].Bingemann S, Konrad T, Wieser R. Zinc finger transcription factor ecotropic viral integration site 1 is induced by all-trans retinoic acid (ATRA) and acts as a dual modulator of the ATRA response. Febs J. 2009;276:6810–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Shackelford D, Kenific C, Blusztajn A, et al. Targeted degradation of the AML1/MDS1/EVI1 oncoprotein by arsenic trioxide. Cancer Res. 2006;66:11360–11369. [DOI] [PubMed] [Google Scholar]
- [93].Raza A, Buonamici S, Lisak L, et al. Arsenic trioxide and thalidomide combination produces multi-lineage hematological responses in myelodysplastic syndromes patients, particularly in those with high pre-therapy EVI1 expression. Leuk Res. 2004;28:791–803. [DOI] [PubMed] [Google Scholar]
- [94].Fazi F, Zardo G, Gelmetti V, et al. Heterochromatic gene repression of the retinoic acid pathway in acute myeloid leukemia. Blood. 2007;109:4432–4440. [DOI] [PubMed] [Google Scholar]
- [95].Su M, Alonso S, Jones J, et al. All-trans retinoic acid activity in acute myeloid leukemia: role of cytochrome P450 enzyme expression by the microenvironment. PLoS One. 2015;10:e0127790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Johnson D, Redner R. An ATRActive future for differentiation therapy in AML. Blood Rev. 2015;29:263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Baik H, Boulanger M, Hosseini M, et al. Targeting the SUMO pathway primes all-trans retinoic acid-induced differentiation of nonpromyelocytic acute myeloid leukemias. Cancer Res. 2018;78:2601–2613. [DOI] [PubMed] [Google Scholar]
- [98].Masciarelli S, Capuano E, Ottone T, et al. Retinoic acid synergizes with the unfolded protein response and oxidative stress to induce cell death in FLT3-ITD+ AML. Blood Adv. 2019;3:4155–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Kahl M, Brioli A, Bens M, et al. The acetyltransferase GCN5 maintains ATRA-resistance in non-APL AML. Leukemia. 2019;33:2628–2639. [DOI] [PubMed] [Google Scholar]
- [100].Niu H, Chacko J, Hadwiger G, et al. Absence of natural intracellular retinoids in mouse bone marrow cells and implications for PML-RARA transformation. Blood Cancer J. 2015;5:e284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Hernandez D, Palau L, Norsworthy K, et al. Overcoming microenvironment-mediated protection from ATRA using CYP26-resistant retinoids. Leukemia. 2020. doi: 10.1038/s41375-020-0790-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Moreb J, Ucar-Bilyeu D, Khan A. Use of retinoic acid/aldehyde dehydrogenase pathway as potential targeted therapy against cancer stem cells. Cancer Chemother Pharmacol. 2017;79:295–301. [DOI] [PubMed] [Google Scholar]
- [103].Gasparetto M, Smith C. ALDHs in normal and malignant hematopoietic cells: potential new avenues for treatment of AML and other blood cancers. Chem Biol Interact. 2017;276:46–51. [DOI] [PubMed] [Google Scholar]
- [104].Hoang V, Buss E, Wang W, et al. The rarity of ALDH(+) cells is the key to separation of normal versus leukemia stem cells by ALDH activity in AML patients. Int J Cancer. 2015;137:525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Blume R, Rempel E, Manta L, et al. The molecular signature of AML with increased ALDH activity suggests a stem cell origin. Leuk Lymphoma. 2018;59:2201–2210. [DOI] [PubMed] [Google Scholar]
- [106].Bindels E, Havermans M, Lugthart S, et al. EVI1 is critical for the pathogenesis of a subset of MLL-AF9-rearranged AMLs. Blood. 2012;119:5838–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Groschel S, Schlenk R, Engelmann J, et al. Deregulated expression of EVI1 defines a poor prognostic subset of MLL-rearranged acute myeloid leukemias: a study of the German-Austrian Acute Myeloid Leukemia Study Group and the Dutch-Belgian-Swiss HOVON/SAKK Cooperative Group. J Clin Oncol. 2013;31:95–103. [DOI] [PubMed] [Google Scholar]
- [108].Arai S, Yoshimi A, Shimabe M, et al. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood. 2011;117:6304–6314. [DOI] [PubMed] [Google Scholar]
- [109].Krivtsov A, Figueroa M, Sinha A, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27:852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].George J, Uyar A, Young K, et al. Leukaemia cell of origin identified by chromatin landscape of bulk tumour cells. Nat Commun. 2016;7:12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Stavropoulou V, Kaspar S, Brault L, et al. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell. 2016;30:43–58. [DOI] [PubMed] [Google Scholar]
- [112].Hammond L, Van Krinks C, Durham J, et al. Antagonists of retinoic acid receptors (RARs) are potent growth inhibitors of prostate carcinoma cells. Br J Cancer. 2001;85:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Weiss F, Marques I, Woltering J, et al. Retinoic acid receptor antagonists inhibit miR-10a expression and block metastatic behavior of pancreatic cancer. Gastroenterology. 2009;137(2136–45):e1–7. [DOI] [PubMed] [Google Scholar]
- [114].Marchwicka A, Cunningham A, Marcinkowska E, et al. Therapeutic use of selective synthetic ligands for retinoic acid receptors: a patent review. Expert Opin Ther Pat. 2016;26:957–971. [DOI] [PubMed] [Google Scholar]
- [115].Toma S, Emionite L, Scaramuccia A, et al. Retinoids and human breast cancer: in vivo effects of an antagonist for RAR-alpha. Cancer Lett. 2005;219:27–31. [DOI] [PubMed] [Google Scholar]
- [116].Hong S, Dvorak-Ewell M, Stevens H, et al. Rescue of a primary myelofibrosis model by retinoid-antagonist therapy. Proc Natl Acad Sci U S A. 2013;110:18820–18825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Farinello D, Wozinska M, Lenti E, et al. A retinoic acid-dependent stroma-leukemia crosstalk promotes chronic lymphocytic leukemia progression. Nat Commun. 2018;9:1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Devalaraja S, To T, Folkert I, et al. Tumor-derived retinoic acid regulates intratumoral monocyte differentiation to promote immune suppression. Cell. 2020;180:1098–114 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Chung S, Wang X, Wolgemuth D. Prolonged oral administration of a pan-retinoic acid receptor antagonist inhibits spermatogenesis in mice with a rapid recovery and changes in the expression of influx and efflux transporters. Endocrinology. 2016;157:1601–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]