

ABSTRACT
Zika is an arboviral illness caused by infection with the Zika flavivirus. Transmission most commonly occurs during a feeding event involving an infected Aedes mosquito or vertical transmission between an infected mother to her fetus. Infection outcomes range from asymptomatic to devastating neurologic injuries in children infected in utero. The recognition of Congenital Zika Syndrome prompted the declaration of an international health emergency and a call to rapidly develop medical countermeasures such as vaccines and therapeutics. A flurry of research and development activity in industry, government, non-governmental organizations, and academia during the most recent Zika epidemic (2015) stimulated the development of a number of vaccine candidate prototypes, generation of pre-clinical data, and the conduct of early phase human trials. The safety and immunogenicity of different vaccine platforms were demonstrated and mouse and non-human primate passive transfer studies hinted at the potential for clinical benefit in humans and defining an immune correlate of protection. A rapid decline in regional transmission, however, prevented the conduct a clinical endpoint efficacy trial. The pathway to licensure of a Zika vaccine remains unclear.
KEYWORDS: Zika, vaccine, animal, human
Introduction
Zika virus (ZIKV) is a flavivirus that is most often transmitted by Aedes mosquito species vectors but may also be transmitted through sexual contact or vertically from mother to fetus. As of July 2, 2019, a total of 87 countries and territories have had evidence of autochthonous mosquito-borne ZIKV transmission across four World Health Organization (WHO) Regions (African Region, Region of the Americas, South-East Asia Region, and the Western Pacific Region) (https://www.who.int/emergencies/diseases/zika/countries-with-zika-and-vectors-table.pdf, accessed January 19, 2020). Between 2015 and 2019 there were more than a quarter million confirmed ZIKV infections in the Americas with the majority (>150,000) occurring in the Southern Cone sub-region (Figure 1).
Figure 1.
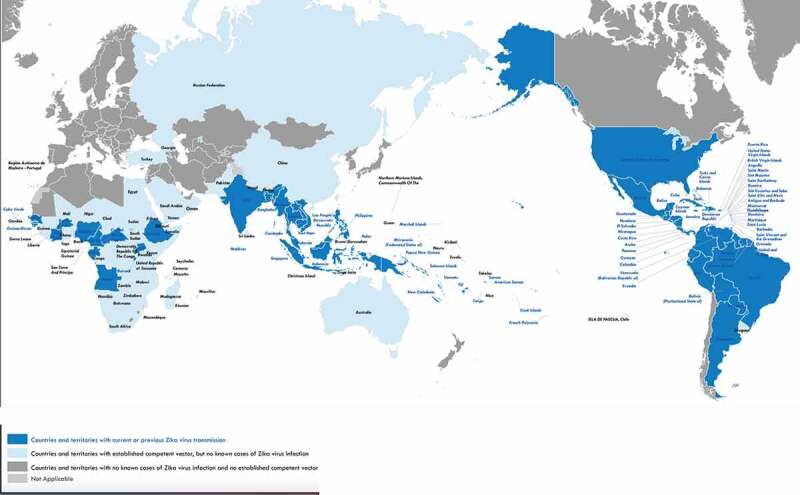
Countries and territories with current or previous Zika virus transmission
*World Health Organization (June 5, 2019)* https://www.who.int/ith/Zika_map.pdf?ua=1; accessed 21 JAN 2020
As with most flavivirus infections, the majority of ZIKV-infected people do not experience clinically apparent disease but in approximately 20% of those infected there may be a mild to moderate febrile illness with conjunctivitis, rash, joint pain, and headache.1 More serious complications, such as Guillain–Barré syndrome (GBS), encephalitis/meningoencephalitis, myelitis, and inflammatory demyelinating polyneuropathy are also possible.2–4 In utero, ZIKV infection is associated with numerous severe neurologic issues such as central nervous system calcifications, microcephaly, hydrocephalus, ocular findings (macular lesions, retinal pigment mottling, optic nerve abnormalities), and musculoskeletal abnormalities (i.e. arthrogryposis).5 Currently, there is no specific anti-ZIKV therapeutic or prophylactic antiviral drug available for use but numerous efforts are being made to re-purpose existing compounds or develop new ones.6
Efforts to develop Zika vaccine candidates and explorations of an optimal vaccine target product profile (TPP) were initiated soon after the 2015 Latin America epidemic erupted (https://www.who.int/immunization/research/development/Zika_Vaccine_Development_Technology_Roadmap_after_consultation_April_2019.pdf?ua=1; https://www.who.int/immunization/research/development/WHO_UNICEF_Zikavac_TPP_Feb2017.pdf?ua=1, accessed 30 JUL 2019).7 The WHO coordinated numerous meetings and discussions of vaccine developers and have continued to track candidate vaccine development. Nuances of ZIKV transmission and disease that differentiate Zika vaccine development from other flavivirus vaccine efforts include: 1) the ability of ZIKV to be sexually transmitted; 2) persistence of ZIKV in immunologically protected anatomic locations; 3) vertical transmission of ZIKV from mother to fetus; and 4) the transient nature of epidemics with high rates of infection, and consequently clinically apparent disease.
Numerous Zika vaccine candidates entered into pre-clinical development at the start of the outbreak but only a few advanced to clinical development.8 Candidates which did enter into clinical testing were faced with a declining epidemic and questions about how a clinical efficacy endpoint study may be conducted to support licensure. Similar to Ebola, chikungunya, and MERS-CoV, regulatory agencies were required to think how developers would demonstrate safety and clinical benefit outside of a traditional efficacy trial.9–12 Ultimately, only a single candidate entered into advanced clinical testing (phase 2, Latin America) but it would fall short of meeting the desired objectives and endpoints due to lack of Zika clinical disease.13,14
Below we provide an up to date review of the various Zika vaccine candidates in pre-clinical and clinical development. Unique aspects of the candidates are highlighted. Finally, we will provide our perspective on the future of Zika vaccine development in the absence of an epidemic and high clinical attack rates.
Zika vaccine candidates in preclinical development
Nearly every vaccine platform technology has been applied to the development of ZIKV vaccine candidates (Table 1). An ideal ZIKV vaccine candidate would be safe, quickly induce enduring protective immunity after a single dose, would not be contraindicated in pregnancy, and could prevent or significantly attenuate the vertical transmission of a ZIKV infection from mother to fetus. These characteristics would support prophylactic immunization in the prime target population (i.e. women of childbearing potential) and have the potential to support outbreak interruption. Unfortunately, the ideal Zika vaccine will be difficult to obtain as both “non-replicating” and “replicating” vaccines each have advantages and disadvantages impacting safety and/or their potential for a clinical benefit at the individual and population levels (Table 2).
Table 1.
Zika vaccine candidate platforms and components
| Platform technology | Type |
|---|---|
| Live attenuated |
|
| Inactivated |
|
| Recombinant protein |
|
| Virus-like particles (VLPs) |
|
| Live vectored |
|
| None-live vectored |
|
| DNA |
|
| RNA |
|
*prM = pre-membrane; E = envelope; NS1 = nonstructural protein 1
Table 2.
Characteristics of candidate Zika vaccines
| NON-REPLICATING | LIVE REPLICATING |
|---|---|
| ● No risk of reversion to virulence | ● Risk of reversion to virulence |
| ● No risk of mosquito-competence | ● Risk of mosquito-competence |
| ● Variable induction of arms of the immune response | ● Induces all arms of immune response |
| ● Usually, multiple doses needed to give protective immunity | ● One dose likely gives protective immunity |
| ● Booster doses needed to maintain immunity | ● Long-lasting immunity only induced by live vaccine |
| ● Long-lasting immunity is important if low-level Zika activity intersperses Zika epidemics |
Licensed flavivirus vaccines
Because of the lack of knowledge on ZIKV at the start of the epidemic, candidate Zika vaccines were benchmarked based on what is known about currently licensed flavivirus vaccines, namely live attenuated yellow fever (YF) and Japanese encephalitis (JE), and formalin-inactivated JE and tick-borne encephalitis (TBE). For all but live attenuated YF, a 50% plaque reduction neutralization test (PRNT50) value of 1 in 10 is found to be a serologic surrogate of protection while for YF the exact PRNT50 is not known but is thought to be somewhere between 1 in 40 and 1 in 50. The animal models used for preclinical evaluation for licensure are immunocompetent mice as a marker of neurotropic disease except for YF where rhesus or cynomolgus macaques are used to evaluate viscerotropic (liver) and neurotropic disease; mice are not approved for preclinical evaluation of YF vaccines due to this model not recapitulating human disease, i.e., mice exhibit neurotropic disease and not a viscerotropic disease. Comparing candidate Zika vaccines with licensed flavivirus vaccines was a very logical approach, however, the clinical disease caused by ZIKV involves multiple tissue tropisms and was found to be very different to that of other flaviviruses where the tissue tropism involved either brain (JE and TBE) or liver (YF) making extrapolation of both immunologic biomarkers and definition of protection from licensed flavivirus vaccines to Zika difficult.
Zika clinical disease
Great strides have been made in understanding ZIKV infection and clinical disease since the transmission was first recognized in South America in 2015. The result has been the identification of a relatively large range of clinical syndromes associated with ZIKV infection compared to other flaviviruses, including JE, TBE, and YF viruses. ZIKV infection has been shown to cause not only neurotropic disease and GBS but also infection of the reproductive tissues and eyes, and the ability to cross the placenta to infect the fetus.15 This complex clinical picture has made the pre-clinical evaluation of candidate Zika vaccines very difficult. Unfortunately, there is no animal model for GBS, but there are a number of cell culture and animal models which have been developed to study other Zika disease syndromes and their use in evaluating vaccine candidates are described below. Nonetheless, an important point to note is that the most of the studies were conducted under ‘research conditions’ and were not conducted under Good Laboratory Practices (GLP) using the quality systems required to generate data appropriate to support regulatory submissions. In particular, standardized reagents are often not used for the conduct of neutralization or reverse transcriptase-polymerase chain reactions (RT-PCR) assays. Of note, the WHO makes reference reagents available (National Institute of Biological Standards and Control, UK and Paul Ehrlich Institute, Germany, respectively).16
Early preclinical studies
When Zika vaccine research started in earnest in 2015 there was a paucity of animal models as little was known about the clinical disease in humans or animals. As such, preclinical evaluation of candidate vaccines was based on the induction of neutralizing antibodies in animal models (mice and/or non-human primates) and no detectable viremia in immunized animals following wild-type ZIKV challenge.17 Many candidates were able to meet the relatively low bar of preventing viremia (measured by infectivity or RT-PCR) following challenge of immunized animals and there was no correlate of protection with which to evaluate vaccine efficacy. A significant number of candidates entered clinical evaluation and these efforts are described below.
Mouse models
Early studies showed that immunocompetent mice were not susceptible to clinical disease following infection with natural ZIKV strains, although strains isolated from the prototype MR766 in 1947 through the next 25 years were isolated by a passage in mouse brain and correspondingly more virulent in mice than more recently isolated non-mouse passaged strains; nonetheless, no ZIKV strain was uniformly lethal in immunocompetent mice. Thus, we believe that mice have great value as a research tool to undertake discovery research on Zika vaccine candidates, however, the mouse models are not suitable for use in the preclinical regulatory evaluation of candidate Zika vaccines as the mouse model does not recapitulate the disease seen in humans. In comparison, the lethal virus challenge mouse models for JE and TBE vaccine evaluation uses immunocompetent mice as the disease recapitulates that seen in humans, i.e., neurotropism. Nonetheless, it should be stressed immunocompetent that immunocompetent mice have proved to be a good model for vaccine-ZIKV challenge studies where viremia is the endpoint for evaluation in the vaccine discovery phase.18 Positive results in the mouse model have been used to advance a vaccine candidate to studies in non-human primates (see below).
Many RNA viruses are sensitive to interferon and so studies turned to utilizing mouse strains that had disruption of the interferon signaling pathway, including signal transducer and activator of transcription 2 (STAT-2) gene, interferon α/β receptor, or interferon α/β/γ receptor-deficient mice.19,20 Not surprisingly, these immunocompromised mouse strains were susceptible to ZIKV infections. ZIKV infection of interferon α/β deficient (A129 or IFNAR−/-) mice tend to show an age-related resistance to lethal infection in mice older than 6 weeks while interferon α/β/γ receptor-deficient (AG129) mice tend to show no age-related resistance and succumb to lethal infections in adult animals. In addition, humanized mice have also been used to evaluate a Zika DNA vaccine.21 While the immunocompromised mouse strains offer the opportunity to immunize mice with vaccine candidates and then challenge with wild-type ZIKV to look for protective immunity, there are major concerns about such mice as they do not have an intact immune system and have the potential to not induce the appropriate vaccine-induced immune response following immunization. As such, these mouse models have important value in vaccine discovery research but their value for preclinical evaluation is unclear. This conundrum has not been resolved and is unlikely to be in the near future as different vaccine candidates induce different immune responses as exemplified when different platform technologies are compared, e.g. RNA vs DNA vs live attenuated vs inactivated vs non-replicating vector vs single round replicating particles. Thus, overall there is no ideal mouse model that recapitulates human disease for evaluating Zika vaccine candidates and their value in preclinical evaluation of vaccine candidates to select those to proceed to human testing is unclear. Nonetheless, mouse models have been the “workhorse” of initial in vivo evaluation of Zika vaccine candidates.
Following the identification of congenital Zika syndrome, much research has been undertaken in establishing mouse models of Zika infections during pregnancy.22,23 These models have generated a lot of information on Congenital Zika Syndrome. As stated above, these models are based on immunocompromised mice and, to date, an immunocompetent mouse model of CZS has not been developed. Thus, the mouse CZS models are excellent research tools but the differences between the physiology of the mouse and human reproductive system make extrapolation of data from mice to humans difficult and at present preclude the mouse CZS models being used to select vaccine candidates that will proceed to clinical trials.
The immunocompromised mouse models (mostly IFNAR−/- mice) have been used to investigate the ability of candidate vaccines to protect from infection of the placenta, the fetus, and testes. There is no doubt that candidate vaccines reduce the viral load in different organs and the placenta, and prevent infection of the fetus, and in some cases, there is no detectable ZIKV, even when measuring for viral RNA. For example, it has been shown that a single-dose of chimeric JE SA14-14-2/ZIKV prM+E live attenuated vaccine fully prevented infection of pregnant mice and maternal-to-fetal transmission as evidenced by lack of detectable viral RNA in the fetus plus lack of placental or fetal damage.24 Similar results have been obtained by with a YF 17D/ZIKV prM+E chimera.25 Studies with other vaccine platforms (mRNA, recombinant envelope (E) protein, and with a live ZIKV vaccine) have shown that these candidate vaccines will also protect pregnant mice and offspring plus testes from ZIKV infection.26–28 Overall, these studies suggest that candidate ZIKV vaccines have potential as vaccines against CZS. However, while the above results provide optimism, this assumes that results in mice can be translated to humans. This may, or may not, be straightforward as the reproductive anatomy and physiology of mice are different to that of humans
Hamster models
The hamster models are similar to mouse models in that immunocompetent animals do not succumb to ZIKV infection. However, a STAT2 gene knock-out hamster model has been developed and this model succumbs to ZIKV infection, specifically animals show clinical signs of disease and mortality, as well as infection of the uterus, placenta, brain, spinal cord, and testicles can be demonstrated.29 This model has been used to evaluate protection mediated by ZIKV-specific human polyclonal antibodies (termed SAB-155) that have been produced in transchromosomal cows. The antibodies provide protection when given either prophylactically or therapeutically, and also prevented testicular lesions in this hamster model.30 No vaccines have been evaluated in this model.
Guinea pig models
The guinea pig has been an attractive model for studying transplacental infections, particularly herpes viruses, listeria, and syphilis, as unlike mice and hamsters, the guinea pig has a reproductive system similar to humans. Specifically, both have hemomonochorial placentas, similar in utero neural development, and like humans are born precocial. As such, it is an attractive model to be considered for evaluating Zika disease. Miller et al. used strain 13 guinea pigs and found no clinical signs of disease nor induction of neutralizing antibodies following intraperitoneal inoculation, whereas Kumar et al. used Dunkin-Hartley guinea pigs and found animals had clinical signs of infection including fever, lethargy, hunched back, ruffled fur, and decrease in mobility following subcutaneous inoculation. Virus was detected in blood, serum, spleen and brain, with the highest titers in the brain, indicating neurotropic disease.31,32 Interestingly, Deng et al. found that subcutaneous inoculation of Hartley guinea pigs resulted in no clinical signs of disease but viremia and infection of the spleen, intestine, and testes were detected.33 To date, only one study has investigated the infection of pregnant animals demonstrating that subcutaneous inoculation of non-pregnant Hartley guinea pigs resulted in no clinical signs of infection but viremia was detectable as was NS1 protein indicating some level of virus replication had taken place.34 Pregnant guinea pigs were challenged early in the pregnancy at between 18 and 21 days gestational age, and neither viremia nor NS1 protein was detected in maternal or pup blood, plasma, or tissues but anti-ZIKV antibodies were detected in both the pups and dams indicative of a sub-clinical infection.
In summary, interpretation of data from these limited studies must be made with care as the route of infection, inoculum, virus strain, and age and strain of animals all varied. However, the guinea pig model probably merits further investigation for the study of CZS.
Non-human primate models
A limited number of studies have been published on the evaluation of Zika vaccine candidates in non-human primates (NHPs) and are summarized in Table 3. All of the studies, irrelevant of the platform, show that the candidate vaccines induce neutralizing antibody titers >1 in 100 resulting in protection from wild-type ZIKV challenge defined as no detectable viremia post-challenge. In a number of ways, these NHP studies are equivalent to the studies undertaken in mice. Some studies not only demonstrated undetectable virus/viral RNA in the peripheral bloodstream but also in select tissues. Unfortunately, NHPs show few clinical signs of ZIKV infection and so interpretation of lack of viremia post-challenge needs to be undertaken with care. For example, Abbink et al. have shown that neutralizing antibody titers are sustained in non-human primates when using RhAd52 or purified formalin inactivated vaccines but not by a DNA vaccine.41,42 Whether or not the DNA vaccine used in this study is representative of all DNA vaccines remains to be seen. Overall, the ability of vaccine candidates to induce a significant immune response in NHPs is promising as demonstrated by no detectable viremia post-challenge and some vaccine candidates able to maintain reasonable levels of neutralizing antibodies over time. In particular, the HuAd26 vaccine candidate may have induced sterilizing immunity as evidenced by undetectable viral RNA loads measured by qRT-PCR in Balb/c serum samples post-wild-type ZIKV challenge, in addition, there was undetectable viral RNA in plasma, CSF, urine, and saliva in rhesus macaques post-wild-type ZIKV challenge plus the macaques had an apparent lack of increase in neutralizing antibody titers post wild-type ZIKV challenge, although anti-NS1antibody titers were low to undetectable.36 Exploring vaccination in a NHP pregnancy model demonstrated that a DNA vaccine constructs (VRC5283), when compared to unvaccinated controls, reduced peak magnitude, and duration of viremia in the mother, reduced early fetal loss and infection, and reduced pathologic findings in the placenta and fetal brain. Also of interest, was the observation that the neutralizing antibody titer present at the time of first ZIKV exposure inversely correlated with maternal viremia and was associated with better fetal outcomes.43
Table 3.
Candidate Zika vaccines evaluated in non-human primates
| Candidate vaccine | Non-human primate species | Dosage | Route of immunization | Neutralization titer | Challenge Virus/Dose Route |
Viremia post challenge | Reference |
|---|---|---|---|---|---|---|---|
| N-terminal 80% envelope protein | Cynomolgus macaque | Two doses of 50 µg (days 0,21) + alhydrogel | intramuscular | 3500 (by PRNT50) | 10,000 TCID50 PRVABC550 subcutaneous |
4/4 animals had no detectable viremia | 35 |
| HuAd26 | Rhesus macaques | One dose of 1 × 1011 virus particles | Intramuscular | 500 (by FRNT50) | 1000 pfu ZIKV-BR subcutaneous |
5/5 animals had no detectable viremia, nor detection of virus in CSF, saliva or urine | 36 |
| DNA prM/E |
Rhesus macaques | Two doses | Electoporation Intradermal | Not known | Strain and dose unknown | No detectable, or significantly reduced, viremia | 37 |
| mRNA | Rhesus macaques | One dose (50 µg) | Lipid-nanoparticle encapsidated Intradermal |
400 (by PRNT50) | 10,000 TCID50 PRVABC550 subcutaneous |
4/5 animals had no detectable viremia | 38 |
| RhAd52 | Rhesus Macaques |
One dose 1 × 1011 virus particles | Approx. 200 (by MN50) | 1000 pfu ZIKV-BR subcutaneous |
4/4 animals had no detectable viremia | 39 | |
| Formalin Inactivated | Rhesus macaques |
Two doses (5 µg) (days 0,28) | subcutaneous | 5000 (by MN50) | 1000 pfu ZIKV-BR subcutaneous |
8/8 animals had no detectable viremia, nor virus in CSF, urine, colorectal or cervicovaginal secretions | 39 |
| DNA prM/E |
Rhesus Macaques |
Two doses of 5 mg (days 0,28) | Approx. 200 (by MN50) | 1000 pfu ZIKV-BR subcutaneous |
4/4 animals had no detectable viremia | 39 | |
| DNA prM/E |
Rhesus macaques | Two doses of 1 mg or 4 mg (days 0,28) | Needle-free injector intramuscular | 322 (by EC50) | 1000 TCID50 PRVABC550 subcutaneous |
17/18 animals had no detectable viremia | 40 |
Neutralizing antibodies and protection
There is very good evidence from multiple independent studies that neutralizing antibodies are correlated with protection based on studies of candidate vaccines in both mice and NHPs.18,39 Similarly, adaptive transfer of antibodies from either mouse or NHPs, or humans into mice and/or NHPs also mediate protection from ZIKV challenge, including human monoclonal antibodies.44 The exact neutralizing antibody titer that is a correlate of protection has yet to be determined. Nevertheless, studies by Dowd et al. show that a protective threshold of vaccine-induced neutralizing antibodies is significantly higher than that for YF, JE, or TBE vaccines.40 Values of 1 in 80 or 1 in 100 have been proposed, but differing neutralization assays and lack of standards have made an accurate figure difficult to establish.40–42
Virus strains
As stated above, vaccine studies conducted under ‘research conditions’ have made great progress in the last 3 years utilizing numerous virus strains, doses, and routes of delivery that complicate interpretation of data and comparison between studies. The fact that ZIKV is a single serotype, genetic variation of wild-type challenge strains is also a potential issue. Standardized reagents and animal models would greatly aid vaccine development, especially given the many candidates that have been undergoing preclinical evaluation. Such a process took place for filovirus drug and vaccine research in the United States where the Filovirus Animal Non-Clinical Group (FANG) was created; FANG is a US interdepartmental/interagency group established to support and facilitate the advanced development of filovirus drugs and vaccines.45 In terms of Zika vaccine research, to date, only one paper has made such suggestions.46 While standardized reagents are critical, it will also be important to test the ability of sera following vaccine immunization to neutralize the virus from at least examples of the two major genetic lineages of ZIKV.
Zika vaccine candidates in clinical development
To date, the only information generated in animal models useful in progressing a vaccine candidate into clinical development has been the ability of a vaccine candidate to induce an immune response in the animal resistant (i.e. prevent viremia) to wild-type ZIKV challenge. These studies have been undertaken in either mice and/or NHPs.
According to clinicaltrials.gov (accessed 8 AUG 2019), ZIKV vaccines are being, or have been, tested at 17 sites conducting phase 1 studies and a single phase 2 trial was initiated at numerous sites (Table 4.). A diverse range of vaccine platforms is being explored to include purified inactivated virus (PIV), Zika, and flavivirus chimerics (dengue, Yellow fever, Japanese encephalitis viruses), measles vectored (MV), messenger RNA (mRNA), DNA, and chimpanzee Adenovirus vectored (ChAd) candidates. The age range of volunteers spans 15 to 65 years and includes both flavivirus naïve and flavivirus primed individuals. The phase 1 studies are (were) being primarily conducted in ZIKV non-endemic settings in North America and Europe with a few candidates being tested in the setting of previous or active ZIKV transmission (i.e. Puerto Rico). The phase 2 trial testing the U.S. National Institute of Health’s DNA vaccine candidate was planned for initiation in 17 total sites including the U.S. (i.e. Texas and Florida) and numerous sites in Latin America with known ZIKV transmission (Ecuador, Costa Rica, Mexico, Brazil, etc.).
Table 4.
ZIKV vaccine candidates in clinical testing as of January 2020 (clinicaltrials.gov)
| Title | Status | Interventions | Sponsor/Collaborators | Age Range (yrs) | Phase | Sample Size | Locations |
|---|---|---|---|---|---|---|---|
| Zika Virus Purified Inactivated Vaccine (ZPIV) Accelerated Vaccination Schedule Study | Completed | Biological: Zika Virus Purified Inactivated Vaccine|Other: Placebo | Kathryn Stephenson|Walter Reed Army Institute of Research (WRAIR)|National Institute of Allergy and Infectious Diseases (NIAID)|Beth Israel Deaconess Medical Center | 18 − 50 | 1 | 36 | US |
| ZIKA Vaccine in Naive Subjects | Completed | Drug: Saline|Biological: Zika Virus Purified Inactivated Vaccine (ZPIV) | National Institute of Allergy and Infectious Diseases (NIAID) | 18 – 49 | 1 | 91 | US |
| Safety and Immunogenicity of a Novel Vaccine Formulation MV-ZIKA-RSP | Recruiting | Biological: Two MV-ZIKA-RSP vaccinations (high dose)|Biological: Two MV-ZIKA-RSP vaccination (low dose)|Biological: One MV-ZIKA-RSP vaccination (high dose) and one placebo|Other: Two placebo injection | Themis Bioscience GmbH | 18 – 55 | 1 | 48 | Austria |
| Safety and Immunogenicity of a Zika Virus DNA Vaccine, VRC-ZKADNA085-00-VP, in Healthy Adults | Completed | Biological: VRC-ZKADNA085-00-VP | National Institute of Allergy and Infectious Diseases (NIAID)|National Institutes of Health Clinical Center (CC) | 18 – 35 | 1 | 80 | US |
| Phase I, Randomized, Double-blinded, Placebo-Controlled Dose De-escalation Study to Evaluate Safety and Immunogenicity of Alum Adjuvanted Zika Virus Purified Inactivated Vaccine (ZPIV) in Adults in a Flavivirus Endemic Area | Active, not recruiting | Other: Placebo|Biological: Zika Virus Purified Inactivated Vaccine (ZPIV) | National Institute of Allergy and Infectious Diseases (NIAID) | 21 – 49 | 1 | 91 | Puerto Rico |
| Evaluation of the Safety and Immunogenicity of the Live Attenuated Zika Vaccine rZIKV/D4Δ30-713 in Flavivirus-naïve Adults | Completed | Biological: rZIKV/D4Δ30-713|Biological: Placebo | National Institute of Allergy and Infectious Diseases (NIAID) | 18 – 50 | 1 | 28 | US |
| Safety, Tolerability, and Immunogenicity of Zika Vaccine mRNA-1893 in Healthy Flavivirus Seropositive and Seronegative Adults | Recruiting | Biological: mRNA-1893|Other: Placebo | ModernaTX, Inc.|Biomedical Advanced Research and Development Authority | 18 – 49 | 1 | 120 | US Puerto Rico |
| VRC 320: A Phase I, Randomized Clinical Trial to Evaluate the Safety and Immunogenicity of a Zika Virus DNA Vaccine, VRC-ZKADNA090-00-VP, Administered Via Needle and Syringe or Needle-free Injector, PharmaJet, inHealthy Adults | Completed | Biological: VRC-ZKADNA090-00-VP | National Institute of Allergy and Infectious Diseases (NIAID)|National Institutes of Health Clinical Center (CC) | 18 – 50 | 1 | 45 | US |
| Randomized, Placebo-controlled, Observer-blinded Phase 1 Safety and Immunogenicity Study of Inactivated Zika Virus Vaccine Candidate in Healthy Adults | Completed | Biological: VLA1601|Biological: Placebo | Valneva Austria GmbH|Emergent BioSolutions | 18 – 49 | 1 | 67 | US |
| Safety and Immunogenicity of a Candidate ZIKV Vaccine (ZIKA001) | Recruiting | Biological: ChAdOx1 Zika|Biological: ChAdOx1 Zika, ChAdOx1 Chik | University of Oxford | 18 – 50 | 1 | 57 | UK |
| A Phase 1, First-in-human, Double-blinded, Randomized, Placebo-controlled Trial of a Zika Virus Purified Inactivated Vaccine (ZPIV) With Alum Adjuvant in Healthy Flavivirus-naive and Flavivirus-Primed Subjects. | Completed | Biological: IXIARO|Other: Placebo|Biological: YF Vax 17D Strain|Biological: Zika Virus Purified Inactivated Vaccine (ZPIV) | National Institute of Allergy and Infectious Diseases (NIAID) | 18 – 49 | 1 | 75 | US |
| Zika-Vaccine Dose Finding Study Regarding Safety, Immunogenicity and Tolerability | Completed | Biological: MV-ZIKA|Other: Placebo | Themis Bioscience GmbH | 18 – 55 | 1 | 48 | Austria |
| Safety, Immunogenicity and Dose Ranging Study of Inactivated Zika Virus Vaccine in Healthy Adult Participants | Active, not recruiting | Drug: Placebo|Biological: PIZV | Takeda | 18 – 49 | 1 | 271 | US Puerto Rico |
| VRC 705: A Zika Virus DNA Vaccine in Healthy Adults and Adolescents | Completed | Biological: VRC-ZKADNA090-00-VP|Other: VRC-PBSPLA043-00-VP | National Institute of Allergy and Infectious Diseases (NIAID)|The Emmes Company, LLC|Leidos Biomedical Research, Inc.|FHI Clinical, Inc.|PPD | 15 – 35 | 2 | 2338 | US Brazil Colombia Costa Rica Ecuador Mexico Panama Peru Puerto Rico |
| Safety, Tolerability, and Immunogenicity of mRNA-1325 in Healthy Adult Subjects | Completed | Biological: mRNA-1325|Other: Placebo | ModernaTX, Inc.|Biomedical Advanced Research and Development Authority | 18 – 49 | 1 | 90 | US |
| Study of GLS-5700 in Dengue Virus Seropositive Adults | Completed | Biological: GLS-5700|Biological: Placebo | GeneOne Life Science, Inc.|Inovio Pharmaceuticals | 18 – 65 | 1 | 160 | US Puerto Rico |
| Study of GLS-5700 in Healthy Volunteers | Completed | Biological: GLS-5700 | GeneOne Life Science, Inc.|Inovio Pharmaceuticals | 18 – 65 | 1 | 40 | US Canada |
To date, there have been three publications in the literature describing the results of clinical trials. Two manuscripts describe DNA vaccine candidates from the NIH and Inovio Pharmaceuticals and GeneOne Life Sciences and a single manuscript describes the inactivated vaccine (ZPIV) developed by the U.S. Army.
NIH – DNA Vaccine
The NIH tested two different ZIKV vaccine candidates in a randomized open-label study. Two-phase 1 studies tested vaccine candidates using a ZIKV-Japanese encephalitis (JE) plasmid (VRC319 trial) and a stand-alone ZIKV plasmid (VRC320 trial). The vaccines used mammalian expression control elements coding for ZIKV pre-Membrane (prM) and E protein sequences from a French Polynesian isolate (strain H/PF/2013). On the ZIKV-JE vaccine, JE virus sequences from the stem and transmembrane regions of E were used to modify the ZIKV coding. In both vaccines the prM signal sequence in the ZIKV coding was exchanged with an analogous JEV region.47
VRC319 enrolled 80 volunteers 18–35 years of age while VRC320 enrolled 45 volunteers 18–50 years of age, all of who received 4 mg of an intramuscular injection. The different dosing schedules for VRC319 included: 1) 0 and 8 weeks, 2) 0 and 12 weeks, 3) 0, 4 and 8 weeks, and 4) 0, 4, and 20 weeks. The VRC320 trial dosed at 0, 4, and 8 weeks using: 1) a single-dose needle and syringe in one deltoid or 2) a split-dose needle and syringe, or 3) a needle-free injection in each deltoid. Safety was assessed out to 24 months while the immunogenicity endpoint was measured 4 weeks after the last vaccination. A reporter virus particle assay was used to measure neutralizing antibody responses while T cell responses were measured using intracellular cytokine staining techniques.40,48
Both vaccines were well tolerated and had an acceptable safety profile. In VRC319, 46% of the overall cohort (i.e. groups 1–4) experienced mild pain at the injection site, 1% experienced mild swelling, and 6% experienced mild redness. In VRC320, 73% and 7% of the total cohort (i.e. groups 1–3) experienced mild and moderate pain, respectively, 7% experienced mild swelling, and 2% experienced mild redness. In terms of systemic symptoms, there were no recorded fevers in VRC319 nor VRC320. Mild malaise (30%), myalgia (25%), headache (25%), chills (15%), nausea (20%), and joint pain (15%) occurred in VRC319 along with moderate myalgia (5%) and headache (5%). VRC320 had similar rates of systemic symptoms with moderate malaise (4%), myalgia (7%), headache (4%), chills (2%), and joint pain (2%) occurring with low frequency. There were no deaths or vaccine-related serious adverse events (SAEs).
Mean CD4 and CD8 responses compared to baseline were measured across all groups in both vaccine constructs using E, M, pr, and pooled peptides. In VRC319, CD4 changes from baseline were significant for groups 1, 2, and 4 (dosing at 0, 4, and 20 weeks) using E peptides and group 4 using pooled peptides (p = .0108). Significant changes were seen in CD8 counts in groups 1, 3, and 4 and in groups 3 (p = .0304) and 4 (p = .0039) using pooled peptides. Similar measurements in VRC320 demonstrated group 3 (needle-free injection) had significant increases in CD4 counts using E (p = .0001) and pooled peptides (p = .0004) and group 2 with a significant increase using pooled peptides (p = .0353). Significant CD8 responses were only observed in group 3 to E (p = .0004) and pooled (p = = 0.0166) peptides.
Neutralizing antibody endpoints were measured using a virus reporter particle assay at 4 weeks after the last vaccination in each cohort. Two to four separate assays were conducted for each sample to generate the standard deviation around the geometric mean titers (GMTs). In VRC319, antibody responses were measured in 60%, 75%, 80%, and 89% of the volunteers in groups 1–4, respectively. GMTs for each of the four groups were 67, 55, 81, and 120, respectively. Antibody responses in VRC320 were measured in groups 1–3 in 77%, 93%, and 100% of volunteers with GMTs of 48, 150, 304, respectively. As with the CD4 and CD8 responses, groups 4 (VRC319) and 3 (VRC320) had superior neutralizing antibody responses compared to the other cohorts.
Based on these data both vaccine constructs and administration strategies had acceptable safety profiles. Group 3 (ZIKV only construct; dosing 0, 4, 8 weeks; needle-free split dose) had superior CD4, CD8, and neutralizing antibody responses compared to the other groups in both studies. The observed reactogenicity was higher in group 3 but likely not significant. Small sample sizes across all groups limit the ability to make conclusive data interpretations but were sufficient to advance this candidate to phase 2 testing.
GeneOne/Inovio – DNA vaccine
The DNA ZIKV vaccine candidate, GLS-5700, contains a plasmid that encodes ZIKV prM and E proteins using a consensus of pre-2016 human ZIKV strain sequences available in GenBank. The sequences were cloned into a modified pVax1 expression vector.49 The open-label, phase 1 study enrolled two groups of 20 volunteers per group. Volunteers would receive either a 1 mg or 2 mg dose of the candidate vaccine in a 0.1 ml intradermal injection in the deltoid region followed by electroporation. One or two injections were provided at 0, 4, and 12 weeks.50
Antibody measurements to a vaccine-matched recombinant envelop protein were made using an enzyme-linked immunosorbent assay (ELISA). Neutralizing antibodies were measured using a microneutralization platform (Vero cell, ZIKV-PRVABC59) and an immunofluorescence-based neutralization assay using human glioblastoma cells (U87 MG) as a model for ZIKV
Infection of neural progenitor cells.51 T cell responses were explored by measuring the number of cells (collected time 0, 4, 6, and 14 weeks) secreting interferon-γ in response to stimulation with ZIKV prM and E protein peptides using an enzyme-linked immunospot (ELISPOT) assay. Finally, passive transfer experiments were performed using IFNAR−/- mice. The ability of volunteer serum collected after three doses of vaccine to protect from mice from ZIKV challenge was explored.
No SAEs were reported during the trial. There were no grade 2 local reactions with the 1 mg dose vaccine and grade 2 injection site erythema and pain with the 2 mg dose. Grade 2 fatigue was recorded with the 1 mg dose and grade 2 nightmares, headache, and musculoskeletal pain were reported with low frequency (<20% of the recipients) in the 2 mg dose group. The sponsor reports more than half of the adverse events recorded were determined to not be due to the investigational product.
Binding antibodies were measured after each vaccine dose with the final measurement 2 weeks after dose 3. Volunteer response to 1 mg and 2 mg doses was 25% and 58% at week 4, 70% and 95% at week 12, and 100% in both groups at week 14. Inter-group differences were only significant at week 6 (65% versus 84%). Neutralizing antibody responses were measured in Vero cells. These were less robust with a 1 mg dose generating responses at 6, 12, and 14 weeks of 5%, 15%, and 60%, respectively. The 2 mg candidate generated responses in 37%, 31%, and 63%; there was no significant difference between groups. GMTs for the 1 mg dose at 4, 6, and 14 weeks were 3.086, 21.28, and 1642, respectively. Neutralizing antibody measured at the same timepoints following three 2 mg doses was 14.23, 125, and 2871. The authors also used an alternative neutralization assay using U87 MG glioblastoma cells. Here, a 1:25 dilution of volunteer serum collected after dose 3 demonstrated 50% inhibition of infection (i.e. cell fluorescence) in >90% (both 1 mg and 2 mg dose) and 90% inhibition in >70%.
Media and interquartile ranges (IQR) were determined for each vaccine group based on ELISPOT measurements. Compared to a baseline (day 0) measurement of 7.085, the largest T cell response was measured in the 1 mg dose group after dose 3 (week 14) with a median value of 35 and IQR of 14.17–63.75. The 2 mg dose group experienced the greatest increase from baseline (value 0) after dose 2 with a value of 58.33, IQR 25–95; the median value declined after dose 3 to 28.33. The post-dose 2 responses (measured week 6) in the 2 mg group were significantly higher than those in the 1-mg dose group (P = .006 by the Mann–Whitney test) but this difference disappeared after dose 3. Of importance, the authors reported issues with cell viability making interpretation of the data difficult.
IFNAR−/- mice were administered serum taken from volunteers either at day 0 (before vaccination) or 2 weeks after a third vaccine dose. One hour later the animals were challenged with a ZIKV-PR209 isolate at a concentration of 1 × 105 plaque forming units. Mice which received serum from day 0 uniformly succumbed to infection (0% survival 14 days after challenge) whereas in the week 14 serum group, 92% of the mice (103/112) survived a challenge at 14 days. Interestingly, five of the volunteers who donated sera for this experiment did not have neutralizing antibodies measured at week 14 and the associated survival rates among mice for the five was 80%, 80%, 80%, 100%, 100% suggesting that non-neutralizing antibodies contribute to protection. Kaplan–Meier curves clearly demonstrate divergence of the curves (day 0 serum, control (saline), week 14 serum) approximately 6 days after infection.
In summary, the GLS-5700 vaccine candidate was well tolerated at both 1 mg and 2 mg doses. Binding antibody profiles were superior to neutralizing antibody profiles and there was no significant difference between the doses. There was inhibition of infectivity in an assay using U87 MG glioblastoma cells but the significance of this in vitro representation of an in vivo human infection experience is unknown. T cell responses were measurable but assay quality due to cell viability may require a caveat. Finally, in vivo, passive protection studies in IFNAR−/- mice clearly differentiated the protective effects of serum from vaccine recipients from control or serum collected on day 0. Again, the generalizability of data from an immunocompromised mouse to the human experience remains to be determined.
Walter reed army institute of research (WRAIR) – purified inactivated virus vaccine
The U.S. Army developed an inactivated vaccine candidate using a 2015 Puerto Rican ZIKV strain (PRVABC59). Three single-center studies (WRAIR, St. Louis University [SLU], and Beth Israel Deaconess Medical Center [BIDMC]) were performed to assess safety and immunogenicity in flavivirus naïve volunteers and those previously vaccinated with yellow fever or Japanese encephalitis vaccine (WRAIR). One site (SLU) assessed three different doses (5.0ug, 2.5ug, and 10.0ug) while another (BIDMC) assessed a single dose, two doses separated by 2 weeks, or two doses separated by 4 weeks. Importantly, the BIDMC site did not pre-screen volunteers for previous flavivirus exposure but did so retrospectively to support immunogenicity analyses. A total of six volunteers were found by microneutralization assay to have been already flavivirus primed on the day of their first vaccination. The data below are taken from the first published manuscript describing the evaluation of two doses of a 5ug adjuvanted vaccine with aluminum hydroxide separated by 4 weeks.52
Neutralizing antibody measurements were completed for all sites using a microneutralization assay conducted at WRAIR.18 Passive transfer experiments were performed in Balb/c mice (i.e., an immunocompetent mouse strain where viremia is used as a surrogate for clinical signs of infection) using plasma from 10 vaccine recipients compared to two control recipients. Polyclonal IgG was purified from plasma collected 2 weeks following dose two and intravenously infused into mice at varying concentrations. Mice were then challenged with 1 × 102 plaque-forming units of a Brazilian ZIKV strain (SPH2015). Peripheral RNAemia was measured following challenge in placebo versus IgG recipients.
There was no vaccine-related serious adverse events. The frequency of local symptoms was similar after dose 1 and 2. Of 67 volunteers, 64% experienced a mild local symptom at the injection site while 3% experienced a moderate symptom. Pain and tenderness at the injection site occurred in 60% and 47% of volunteers, respectively. Systemic symptoms occurred in 49% (mild), 15% (moderate), and 2% (severe) of volunteers; the severe event was not attributed to vaccination. The most common systemic complaints included fatigue (43%), headache (39%), and malaise (22%) with fatigue having the highest percentage of moderate severity (11.9%). The authors report clinical abnormalities were mostly mild and infrequent.
Neutralizing antibody measurements occurred at different times based on the study site. Common measurements among all sites occurred on day 1 (day of vaccination), day 29, and day 57. Day 29 data from all sites indicate 10.9% (95%CI 4.1–22.2%) of volunteers who received vaccine seroconverted measured using a titer threshold of ≥1:10. When using a titer threshold ≥1:100 the seroconversion rate was 5.5% (95%CI 1.1–15.1%). One month following two doses (day 57) the ≥1:10 and ≥1:100 seroconversion rates were 92.2% (95%CI 81.1–97.8%) and 68.6% (95%CI 54.1–80.9%), respectively. GMTs on day 29 (after a single dose of vaccine) for all groups was 7.0 (95%CI 5.2–9.5) while day 57 GMTs (after two doses of vaccine) for all groups was 173.1 (95%CI 104.6–286.5). There was variance in GMTs among the sites with the WRAIR, SLU, and BIDMC demonstrating day 57 GMTs of 100.8, 142.9, and 820.6, respectively. The peak titers measured after dose 2 for all groups were 100.8 (WRAIR), 354.6 (SLU), and 1061.7 (BIDMC). The GMTs according to baseline flavivirus priming status was evaluable for N = 50 naïve and N = 5 primed volunteers. The day 29 GMT for the naïve group was 6.5 (95%CI 5.1–8.3) compared to 15.1 (95%CI 0.7–326) for the primed group. At day 57 the naïve group GMT was 161.3 (95%CI 944–276) compared to 332.0 (95%CI 48–2314) for the primed group with peak post-dose 2 GMTs of 271 (95%CI 155–472) for the naïve group and 493 (95%CI 70–3462) for the primed group. These differences were not statistically significant but there was a trend toward higher titers in the primed group. Day 57 GMTs were significantly lower for WRAIR and SLU volunteers compared to those enrolled at BIDMC. One hypothesis for this difference was that the BIDMC cohort was younger than the cohort enrolled at the other two sites. In fact, neutralizing antibody titers negatively correlated with age across the entire aggregate dataset (R = −0.46 (P = .006)).
Naïve Balb/c mice infected with SPH2014 develop RNAemia for approximately 7 days following challenge; this was similar for mice that received purified IgG from non-vaccinated volunteers and placebo recipients. Mice that received IgG from vaccinated volunteers experienced complete or partial protection from RNAemia following challenge. Furthermore, the neutralizing antibody titers of the IgG preparations administered to the mice correlated with the observed percent protection (r = 0 · 744, p = 0 · 009) following the challenge.
Perspectives
Clinical development of vaccines against epidemic diseases
A major hurdle to licensing any vaccine using traditional field-based efficacy studies is confirming infection and clinical attack rates are sufficient to support properly (and practically) designed effectiveness trials. One must first determine the primary efficacy (clinical) endpoint to be measured which is, typically, determined by the clinical syndrome constituting the greatest public health burden. In the case of Zika, most would agree on the clinical outcomes of CZS account for the greatest burden and are what rallied the global call for rapid countermeasure development. The question is whether or not it is possible to design a trial to capture this outcome with sufficient numbers to allow the formulation of adequately powered conclusions about the vaccine’s clinical benefit.
Recent examples of diseases such as chikungunya, Ebola, and Zika demonstrate how epidemic and non-sustained transmission create scenarios where traditional efficacy trials may not be possible; in these cases, there are three options for moving forward. First, candidate vaccines that look promising in early clinical evaluations of safety and immunogenicity may be stockpiled for use awaiting future Zika outbreaks. Second, efforts can be made to utilize the “Animal Efficacy Rule” where licensure is based on efficacy studies in animal models (U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER)/Center for Biologics Evaluation and Research (CBER). Product Development Under the Animal Rule, Guidance for Industry. October 2015). This has possibilities but, as discussed above, it is unclear if any of the current animal models recapitulate human disease.12 Finally, Controlled Human Infection Model studies (CHIMS) could be used to evaluate candidate vaccines.53 This approach has garnered interest in the last 18 months but will require well-characterized wild-type strains and additional conversations regarding the ethics of such an approach (https://www.thehastingscenter.org/navigating-ethics-review-human-infection-trials-zika/; accessed 22 SEP 2019). In particular, the characteristics of such wild-type strains are unclear as studies in animal models may not predict their phenotype in humans.
Comparing vaccines
Vaccine developers, funders, and policymakers are eager to contrast and compare vaccine constructs. In the face of a public health emergency this is especially true as groups hope to identify, support, and advance the most promising vaccine candidates with the thought there may be an opportunity to quickly demonstrate safety and clinical benefit in the field. Head to head comparisons of arboviral vaccine candidates (i.e. dengue, chikungunya, West Nile) has traditionally been difficult and Zika is no exception. Numerous factors contribute to this difficulty including but not limited to:
the increasing knowledge about Zika virus infection and disease has modified the desired vaccine target product profile;
animal models of disease imperfectly recapitulate the human infection and disease experience obscuring what an optimal immune mechanism of action may be for a vaccine construct;
different developers explore vaccine immunogenicity using different assay platforms or variations of similar platforms;
variance in study designs do not allow for direct comparison of immune response kinetics or durability;
the absence of an immune correlate or surrogate of protection; and
the typically small sample sizes used in early phase clinical trials which limit statistically significant comparisons especially when considering subgroups.
While many vaccines look promising in pre-clinical development, most have been evaluated using different animal models based on differences in age and strain of animal, virus challenge strain, and dose, and neutralization assay employed to measure immunogenicity endpoints. These methodological variations make direct comparisons of performance difficult and very few studies have undertaken a head-to-head comparison of multiple vaccine candidates. One outlier is a study where formalin-inactivated, DNA, and rhesus adenovirus vaccine platforms were compared.41,42 The conduct of challenge or passive transfer and protection experiments in animal models can also be helpful in understanding, and characterizing, different candidates’ potential for clinical benefit. During clinical development, it is possible to compare acute safety and reactogenicity profiles between candidates using standardized objective and subjective metrics. Direct comparisons beyond these are difficult for the reasons cited above. Ultimately, a licensed Zika vaccine will have to meet the criteria described in the WHO TPP, which was outlined at the start of this paper.
What next?
As with Ebola, significant advances in both pre-clinical and clinical vaccine development are possible in the context of outbreaks. We are currently in the inter-epidemic period for Zika and have a number of vaccine candidates awaiting evaluation during the next epidemic. A major challenge is that the date and geographic location of the next epidemic is largely unknown and both pieces of information are critical to maximize the opportunity. In terms of date, vaccines normally have a shelf-life of 3 years, meaning if we do not have an outbreak by 2021 it is likely that new clinical-grade material (i.e. vaccines manufactured under Good Manufacturing Practice standards) will be needed and this is not an inexpensive or rapid endeavor. A great deal of time is also required to negotiate with host nation ethical, regulatory, and other review committees and agencies prior to being able to conduct an interventional study on its citizens. Building the needed trial infrastructure including reliable electricity, security for documents and biologic samples, and shipping networks can also take time. Finally, in resource limited settings it may be difficult to find qualified and available clinical trialists.
In terms of geographic location, ZIKV has been found in many parts of the world and, as such, preparation and infrastructure for the next outbreak is difficult when compared to Ebola where we know the next outbreak will take place on the African continent. Arbovirus epidemics take place at irregular intervals and are impossible to predict accurately due to a mosquito-primate transmission cycle and the contribution of a number of ecological factors that are poorly understood. This is demonstrated by West Nile virus where the periodicity of epidemics in the United States have proved very difficult to predict. The development of infectious disease outbreak prediction models has become popular but their accuracy often varies due to the large number of factors which can impact a zoonotic or vector-borne diseases.54
How do we proceed without efficacy trials? One possibility would be to use the “Animal Efficacy Rule,” or equivalent, but this would be potentially very difficult for regulators. As stated above, it is unclear whether or not animal models recapitulate human disease and a correlate of protection has not been established, although it is likely to be neutralizing antibodies based on studies with other flavivirus vaccines; however, since neutralization assays vary it is currently not possible to quantitate accurately what titer of neutralizing antibodies would be the correlate of protection.
Unfortunately, the above does not give confidence we will have a licensed ZIKV vaccine in the near future. However, it should be remembered the enormous strides in our understanding of Zika, including vaccine development, have been made since the start of the epidemic in 2015 and it is very likely that enormous strides will also be made during the next ZIKV epidemic. Our goal now must be not to lose momentum in ZIKV research and be prepared for the next epidemic.
Disclosure of potential conflicts of interest
All authors declare that there exist no commercial or financial relationships that could, in any way, lead to a potential conflict of interest.
Disclaimer
SJT is listed as an inventor on the U.S. Army’s Zika vaccine candidate and has assigned all rights to the U.S. government. He also consults for a number of industry entities developing Zika vaccine candidates and he is compensated for his time.
References
- 1.Anderson KB, Thomas SJ, Endy TP.. The emergence of zika virus: a narrative review. Ann Intern Med. 2016;165:175–83. doi: 10.7326/M16-0617. [DOI] [PubMed] [Google Scholar]
- 2.Mier YT-RL, Delorey MJ, Sejvar JJ, Johansson MA. Guillain-Barre syndrome risk among individuals infected with Zika virus: a multi-country assessment. BMC Med. 2018;16:67. doi: 10.1186/s12916-018-1052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carod-Artal FJ. Neurological complications of Zika virus infection. Expert Rev Anti Infect Ther. 2018;16:399–410. doi: 10.1080/14787210.2018.1466702. [DOI] [PubMed] [Google Scholar]
- 4.da Silva IRF, Frontera JA, Bispo de Filippis AM, Nascimento O, Group R-G-ZR . Neurologic complications associated with the Zika virus in Brazilian adults. JAMA Neurol. 2017. doi: 10.1001/jamaneurol.2017.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marques VM, Santos CS, Santiago IG, Marques SM, Nunes Brasil MDG, Lima TT, Costa PS. Neurological complications of congenital Zika virus infection. Pediatr Neurol. 2019;91:3–10. doi: 10.1016/j.pediatrneurol.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Zou J, Shi PY. Strategies for Zika drug discovery. Curr Opin Virol. 2019;35:19–26. doi: 10.1016/j.coviro.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Maurice J. WHO reveals its shopping list for weapons against Zika. Lancet. 2016;387:733. doi: 10.1016/S0140-6736(16)00390-1. [DOI] [PubMed] [Google Scholar]
- 8.Thomas SJ, Phimister EG. Zika Virus Vaccines - A Full Field and Looking for the Closers. N Engl J Med. 2017;376:1883–86. doi: 10.1056/NEJMcibr1701402. [DOI] [PubMed] [Google Scholar]
- 9.Vannice KS, Giersing BK, Kaslow DC, Griffiths E, Meyer H, Barrett A, Durbin AP, Wood D, Hombach J. Meeting Report: WHO consultation on considerations for regulatory expectations of Zika virus vaccines for use during an emergency. Vaccine. 2019;37:7443–50. [DOI] [PubMed] [Google Scholar]
- 10.Vannice KS, Cassetti MC, Eisinger RW, Hombach J, Knezevic I, Marston HD, Wilder-Smith A, Cavaleri M, Krause PR. Demonstrating vaccine effectiveness during a waning epidemic: A WHO/NIH meeting report on approaches to development and licensure of Zika vaccine candidates. Vaccine. 2019;37:863–68. doi: 10.1016/j.vaccine.2018.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gruber MF, Krause PR. Regulating vaccines at the FDA: development and licensure of Zika vaccines. Expert Rev Vaccines. 2017;16:525–27. doi: 10.1080/14760584.2017.1324304. [DOI] [PubMed] [Google Scholar]
- 12.Gruber MF, Farizo KM, Pratt RD, Fink DL, Finn TM, Krause PR, Borio LL, Marks PW. Clinical development strategies and considerations for zika vaccine licensure. J Infect Dis. 2017;216:S964–S70. doi: 10.1093/infdis/jix433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen J. Steep drop in Zika cases undermines vaccine trial. Science. 2018;361:1055–56. doi: 10.1126/science.361.6407.1055. [DOI] [PubMed] [Google Scholar]
- 14.Cohen J. Where has all the Zika gone? Science. 2017;357:631–32. doi: 10.1126/science.357.6352.631. [DOI] [PubMed] [Google Scholar]
- 15.Miner JJ, Diamond MS. Zika virus pathogenesis and tissue tropism. Cell Host Microbe. 2017;21:134–42. doi: 10.1016/j.chom.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mattiuzzo G, Knezevic I, Hassall M, Ashall J, Myhill S, Faulkner V, Hockley J, Rigsby P, Wilkinson DE, Page M, collaborative study participants . Harmonization of Zika neutralization assays by using the WHO International Standard for anti-Zika virus antibody. NPJ Vaccines. 2019. October;4:42. eCollection 2019. PMID: 31632743. doi: 10.1038/s41541-019-0135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrett ADT. Current status of Zika vaccine development: zika vaccines advance into clinical evaluation. NPJ Vaccines. 2018;3:24. doi: 10.1038/s41541-018-0061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larocca RA, Abbink P, Peron JP, Zanotto PM, Iampietro MJ, Badamchi-Zadeh A, Boyd M, Ng’ang’a D, Kirilova M, Nityanandam R, et al. Vaccine protection against Zika virus from Brazil. Nature. 2016;536:474–78. doi: 10.1038/nature18952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bowen JR, Zimmerman MG, Suthar MS. Taking the defensive: immune control of Zika virus infection. Virus Res. 2018;254:21–26. doi: 10.1016/j.virusres.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrison TE, Diamond MS. Animal models of Zika virus infection, pathogenesis, and immunity. J Virol. 2017;91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yi G, Xu X, Abraham S, Petersen S, Guo H, Ortega N, Shankar P, Manjunath N. A DNA vaccine protects human immune cells against Zika virus infection in humanized mice. EBioMedicine. 2017;25:87–94. doi: 10.1016/j.ebiom.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao B, Diamond MS, Mysorekar IU. Maternal-Fetal Transmission of Zika Virus: routes and Signals for Infection. J Interferon Cytokine Res. 2017;37:287–94. doi: 10.1089/jir.2017.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caine EA, Jagger BW, Diamond MS. Animal models of Zika virus infection during pregnancy. Viruses. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li XF, Dong HL, Wang HJ, Huang XY, Qiu YF, Ji X, Ye Q, Li C, Liu Y, Deng Y-Q, et al. Development of a chimeric Zika vaccine using a licensed live-attenuated flavivirus vaccine as backbone. Nat Commun. 2018;9:673. doi: 10.1038/s41467-018-02975-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kum DB, Mishra N, Boudewijns R, Gladwyn-Ng I, Alfano C, Ma J, Schmid MA, Marques RE, Schols D, Kaptein S, et al. A yellow fever-Zika chimeric virus vaccine candidate protects against Zika infection and congenital malformations in mice. NPJ Vaccines. 2018;3:56. doi: 10.1038/s41541-018-0092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, et al. Modified mRNA vaccines protect against zika virus infection. Cell. 2017;168:1114–25 e10. doi: 10.1016/j.cell.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu X, Li C, Afridi SK, Zu S, Xu JW, Quanquin N, Yang H, Cheng G, Xu Z. E90 subunit vaccine protects mice from Zika virus infection and microcephaly. Acta Neuropathol Commun. 2018;6:77. doi: 10.1186/s40478-018-0572-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shan C, Muruato AE, Jagger BW, Richner J, Nunes BTD, Medeiros DBA, Xie X, Nunes JGC, Morabito KM, Kong W-P, et al. A single-dose live-attenuated vaccine prevents Zika virus pregnancy transmission and testis damage. Nat Commun. 2017;8:676. doi: 10.1038/s41467-017-00737-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siddharthan V, Van Wettere AJ, Li R, Miao J, Wang Z, Morrey JD, Julander JG. Zika virus infection of adult and fetal STAT2 knock-out hamsters. Virology. 2017;507:89–95. doi: 10.1016/j.virol.2017.04.013. [DOI] [PubMed] [Google Scholar]
- 30.Siddharthan V, Miao J, Van Wettere AJ, Li R, Wu H, Sullivan E, Jiao J, Hooper JW, Safronetz D, Morrey JD. Human polyclonal antibodies produced from transchromosomal bovine provides prophylactic and therapeutic protections against Zika virus infection in STAT2 KO Syrian Hamsters. Viruses. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller LJ, Nasar F, Schellhase CW, Norris SL, Kimmel AE, Valdez SM, Wollen-Roberts SE, Shamblin JD, Sprague TR, Lugo-Roman LA, et al. Zika virus infection in Syrian golden hamsters and strain 13 guinea pigs. Am J Trop Med Hyg. 2018;98:864–67. doi: 10.4269/ajtmh.17-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar M, Krause KK, Azouz F, Nakano E, Nerurkar VR. A guinea pig model of Zika virus infection. Virol J. 2017;14:75. doi: 10.1186/s12985-017-0750-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng YQ, Zhang NN, Li XF, Wang YQ, Tian M, Qiu YF, Fan J-W, Hao J-N, Huang X-Y, Dong H-L, et al. Intranasal infection and contact transmission of Zika virus in guinea pigs. Nat Commun. 2017;8:1648. doi: 10.1038/s41467-017-01923-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bierle CJ, Fernandez-Alarcon C, Hernandez-Alvarado N, Zabeli JC, Janus BC, Putri DS, Schleiss MR. Assessing Zika virus replication and the development of Zika-specific antibodies after a mid-gestation viral challenge in guinea pigs. PLoS One. 2017;12:e0187720. doi: 10.1371/journal.pone.0187720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Medina LO, To A, Lieberman MM, Wong TAS, Namekar M, Nakano E, Andersen H, Yalley-Ogunro J, Greenhouse J, Higgs S, et al. a recombinant subunit based zika virus vaccine is efficacious in non-human primates. Front Immunol. 2018;9:2464. doi: 10.3389/fimmu.2018.02464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cox F, van der Fits L, Abbink P, Larocca RA, van Huizen E, Saeland E, Verhagen J, Peterson R, Tolboom J, Kaufmann B, et al. Adenoviral vector type 26 encoding Zika virus (ZIKV) M-Env antigen induces humoral and cellular immune responses and protects mice and nonhuman primates against ZIKV challenge. PLoS One. 2018;13:e0202820. doi: 10.1371/journal.pone.0202820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kudchodkar SB, Choi H, Reuschel EL, Esquivel R, Jin-Ah Kwon J, Jeong M, Maslow JN, Reed CC, White S, Kim JJ, et al. Rapid response to an emerging infectious disease - Lessons learned from development of a synthetic DNA vaccine targeting Zika virus. Microbes Infect. 2018;20:676–84. doi: 10.1016/j.micinf.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pardi N, Hogan MJ, Pelc RS, Muramatsu H, Andersen H, DeMaso CR, Dowd KA, Sutherland LL, Scearce RM, Parks R, et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature. 2017;543:248–51. doi: 10.1038/nature21428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abbink P, Larocca RA, De La Barrera RA, Bricault CA, Moseley ET, Boyd M, Kirilova M, Li Z, Nganga D, Nanayakkara O, et al. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science. 2016;353:1129–32. doi: 10.1126/science.aah6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dowd KA, Ko SY, Morabito KM, Yang ES, Pelc RS, DeMaso CR, Castilho LR, Abbink P, Boyd M, Nityanandam R, et al. Rapid development of a DNA vaccine for Zika virus. Science. 2016;354:237–40. doi: 10.1126/science.aai9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Erratum for the Research Article: “Durability and correlates of vaccine protection against Zika virus in rhesus monkeys” by P. Abbink, R. A. Larocca, K. Visitsunthorn, M. Boyd, R. A. De La Barrera, G. D. Gromowski, M. Kirilova, R. Peterson, Z. Li, O. Nanayakkara, R. Nityanandam, N. B. Mercado, E. N. Borducchi, A. Chandrashekar, D. Jetton, S. Mojta, P. Gandhi, J. LeSuer, S. Khatiwada, M. G. Lewis, K. Modjarrad, R. G. Jarman, K. H. Eckels, S. J. Thomas, N. L. Michael, D. H. Barouch. Sci Transl Med. 2018;10(450):eaau6861. doi: 10.1126/scitranslmed.aau6861. [DOI] [PubMed] [Google Scholar]
- 42.Abbink P, Larocca RA, Visitsunthorn K, Boyd M, De La Barrera RA, Gromowski GD, Kirilova M, Peterson R, Li Z, Nanayakkara O, et al. Durability and correlates of vaccine protection against Zika virus in rhesus monkeys. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Rompay KKA, Keesler RI, Ardeshir A, Watanabe J, Usachenko J, Singapuri A, Cruzen C, Bliss-Moreau E, Murphy AM, Yee JL , et al. DNA vaccination before conception protects Zika virus-exposed pregnant macaques against prolonged viremia and improves fetal outcomes. Sci Transl Med. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Magnani DM, Rogers TF, Beutler N, Ricciardi MJ, Bailey VK, Gonzalez-Nieto L, Briney B, Sok D, Le K, Strubel A, et al. Neutralizing human monoclonal antibodies prevent Zika virus infection in macaques. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kilgore N, Nuzum EO. An interagency collaboration to facilitate development of filovirus medical countermeasures. Viruses. 2012;4:2312–16. doi: 10.3390/v4102312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coelho SVA, Neris RLS, Papa MP, Schnellrath LC, Meuren LM, Tschoeke DA, Leomil L, Verçoza BRF, Miranda M, Thompson FL, et al. Development of standard methods for Zika virus propagation, titration, and purification. J Virol Methods. 2017;246:65–74. doi: 10.1016/j.jviromet.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 47.Gaudinski MR, Houser KV, Morabito KM, Hu Z, Yamshchikov G, Rothwell RS, Berkowitz N, Mendoza F, Saunders JG, Novik L, et al. Safety, tolerability, and immunogenicity of two Zika virus DNA vaccine candidates in healthy adults: randomised, open-label, phase 1 clinical trials. Lancet. 2018;391:552–62. doi: 10.1016/S0140-6736(17)33105-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarwar UN, Costner P, Enama ME, Berkowitz N, Hu Z, Hendel CS, Sitar S, Plummer S, Mulangu S, Bailer RT, et al. Safety and immunogenicity of DNA vaccines encoding Ebolavirus and Marburgvirus wild-type glycoproteins in a phase I clinical trial. J Infect Dis. 2015;211:549–57. doi: 10.1093/infdis/jiu511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muthumani K, Block P, Flingai S, Muruganantham N, Chaaithanya IK, Tingey C, Wise M, Reuschel EL, Chung C, Muthumani A, et al. Rapid and long-term immunity elicited by dna-encoded antibody prophylaxis and DNA vaccination against chikungunya virus. J Infect Dis. 2016;214:369–78. doi: 10.1093/infdis/jiw111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tebas P, Roberts CC, Muthumani K, Reuschel EL, Kudchodkar SB, Zaidi FI, White S, Khan AS, Racine T, Choi H, et al. Safety and Immunogenicity of an Anti-Zika Virus DNA Vaccine - Preliminary Report. N Engl J Med. 2017. doi: 10.1056/NEJMoa1708120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anfasa F, Siegers JY, van der Kroeg M, Mumtaz N, Stalin Raj V, de Vrij FMS, Widagdo W, Gabriel G, Salinas S, Simonin Y, et al. Phenotypic differences between Asian and African Lineage Zika viruses in human neural progenitor cells. mSphere. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Modjarrad K, Lin L, George SL, Stephenson KE, Eckels KH, De La Barrera RA, Jarman RG, Sondergaard E, Tennant J, Ansel JL, et al. Preliminary aggregate safety and immunogenicity results from three trials of a purified inactivated Zika virus vaccine candidate: phase 1, randomised, double-blind, placebo-controlled clinical trials. Lancet. 2018;391:563–71. doi: 10.1016/S0140-6736(17)33106-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramanathan R, Stibitz S, Pratt D, Roberts J. Use of controlled human infection models (CHIMs) to support vaccine development: US regulatory considerations. Vaccine. 2019;37:4256–61. doi: 10.1016/j.vaccine.2019.06.009. [DOI] [PubMed] [Google Scholar]
- 54.Pedro SA, Abelman S, Tonnang HE. Predicting rift valley fever inter-epidemic activities and outbreak patterns: insights from a stochastic host-vector model. PLoS Negl Trop Dis. 2016;10:e0005167. doi: 10.1371/journal.pntd.0005167. [DOI] [PMC free article] [PubMed] [Google Scholar]