

Abstract
Ferroptosis is a regulated form of necrotic cell death that is caused by the accumulation of oxidized phospholipids, leading to membrane damage and cell lysis1, 2. While other types of necrotic death such as pyroptosis and necroptosis are mediated by active mechanisms of execution3–6, ferroptosis is thought to result from the accumulation of unrepaired cell damage1. Previous studies suggested that ferroptosis has the ability to spread through cell populations in a wave-like manner, resulting in a distinct spatiotemporal pattern of cell death7, 8. Here we investigate the mechanism of ferroptosis execution and discover that ferroptotic cell rupture is mediated by plasma membrane pores, similarly to cell lysis in pyroptosis and necroptosis3, 4. We further find that intercellular propagation of death occurs following treatment with some ferroptosis-inducing agents, including erastin2, 9 and C’ dot nanoparticles8, but not upon direct inhibition of the ferroptosis-inhibiting enzyme Glutathione Peroxidase 4 (GPX4)10. Propagation of a ferroptosis-inducing signal occurs upstream of cell rupture, and involves the spreading of a cell swelling effect through cell populations in a lipid peroxide- and iron-dependent manner.
The proper regulation of cell death is important for normal organismal development and the maintenance of tissue homeostasis in adulthood. It was once thought that programmed cell death occurred exclusively through apoptosis, whereas necrotic death resulted only from acute cell stress or injury. However, numerous new cell death modalities have recently been discovered, including programmed forms of necrosis that are regulated by specific and distinct cellular machineries11. One form of regulated necrosis called ferroptosis involves the iron-dependent accumulation of lipid peroxide species in cell membranes1, 2. Under physiological conditions, ferroptosis is prevented by antioxidant enzymes that limit the buildup of oxidized lipids, including GPX4, which uses glutathione as a cofactor to detoxify peroxidation products12. Cell death can be triggered by GPX4 inactivation, either through direct inhibition or depletion of cellular glutathione, thereby allowing the accumulation of phospholipid peroxides and cell damage. Recent work has uncovered an additional ferroptosis-preventing mechanism controlled by Ferroptosis Suppressor Protein 1 (FSP1), which catalyzes the reduction of the lipophilic antioxidant coenzyme Q10 (CoQ) 13, 14.
Ferroptosis was previously shown to spread through cell populations, resulting in spatiotemporal patterns of cell death with a wave-like appearance not previously observed in other forms of cell death7, 8. It is unknown what mechanism underlies this phenomenon and whether death propagation between neighboring cells is a consistent feature of ferroptosis or occurs only under certain conditions. Given emerging links between ferroptosis and degenerative diseases that often involve large, continuous areas of tissue damage, the propagative nature of ferroptosis is important to understand15. Furthermore, while factors that affect the accumulation of lipid peroxides and thereby modulate ferroptosis have been elucidated1, 16, little is known about how lipid peroxidation leads to plasma membrane permeabilization. Whether cell lysis is involved in the intercellular propagation of ferroptosis is also unknown17. Here we investigated the wave-like nature of ferroptosis, the mechanism of ferroptotic cell rupture, and the link between the two processes.
We previously observed wave-like spreading of ferroptosis when cells were treated with ferroptosis-inducing nanoparticles called C’ dots (Fig. 1a,b and Supplementary Video 1)8, and a similar phenomenon was reported in mouse renal tubules treated with the ferroptosis-inducing agent erastin7. However, the spatiotemporal patterns of ferroptosis have not been systematically investigated15. To quantitatively study propagation, we performed live cell imaging of several cell lines (MCF10A mammary epithelium, MCF7 breast cancer, U937 promonocytic leukemia, HAP1 chronic myelogenous leukemia, and B16F10 melanoma) in the presence of the cell death indicator SYTOX Green and different ferroptosis-inducing agents (C’ dots8, erastin2, the GPX4 inhibitor ML16210, 18, or a combination of ferric ammonium citrate (FAC) and buthionine sulfoxamine (BSO), Extended Data Fig. 1a,b). We then used a bootstrapping approach to quantify potential non-random patterns of cell death. For each movie, we calculated the mean time difference between neighboring cell deaths, μexpΔt, and compared this experimental value to a distribution of means derived from computationally generated permutations representing random orders of death (Fig. 1c,d). Consistent with wave-like propagation, ferroptosis occurred with non-random spatiotemporal patterns when it was induced by erastin, C’ dots, or FAC and BSO, as determined by comparing μexpΔt to the 95th percentile of the random distribution, μperm95Δt (Fig. 1b, d, g; Extended Data Fig. 1c). Interestingly, when ferroptosis was induced by inhibition of GPX4 through treatment with ML162, μexpΔt was more similar to the 95th percentile of the random permutations (Fig. 1g and Extended Data Fig. 1d, e).
Figure 1.
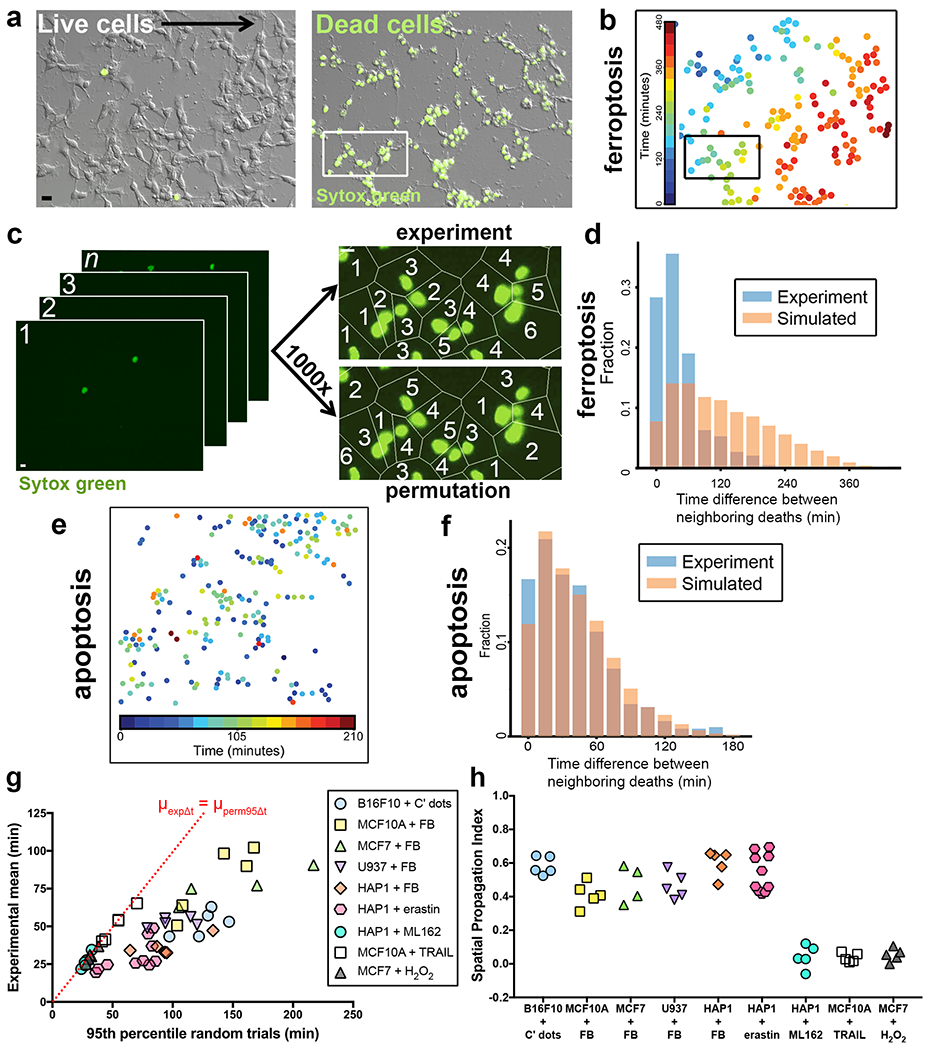
Ferroptosis exhibits non-random spatiotemporal patterns. (a) B16F10 cells treated with C’ dot nanoparticles in amino acid-free (-AA) media to induce ferroptosis. Images show DIC and SYTOX Green; SYTOX-positive cells are dead. Scale bar = 20μm. Images are representative of five movies from one experiment. (b) Nuclei of ferroptotic cells in panel a, pseudocolored to indicate relative timing of cell death, as determined by time-lapse microscopy. See Supplementary Video 1. (c) Schematic summarizing our method to quantify cell death patterns. Images from time-lapse microscopy (left) are processed to determine relative timing of neighboring cell deaths (top right image, “experiment”) versus permuted trials (bottom right image, “permutation”) to detect potential non-random patterns. Images match insets in panels a and b. Scale bar = 10μm (d) Distribution of time differences between neighboring deaths (Δt) from experiment in panels a-c shown in blue, versus averaged distribution of the set of random permutations shown in orange. Graph shows fraction of total deaths with given time differences.. (e) Spatiotemporal distribution of apoptosis in MCF10A cells treated with TRAIL. Each dot represents a cell; colors indicate relative times of cell death as determined by cell morphology. Data are representative of five fields of view from one experiment. (f) Distribution of experimental time differences between neighboring deaths (Δt) in blue and averaged distribution of Δts from the corresponding permuted data in orange. Data belong to the experiment shown in panel e. (g) Ferroptosis, apoptosis, and H2O2-induced necrosis show non-random spatiotemporal patterns. Graph shows μexpΔt vs. μperm95Δt of different cell lines undergoing ferroptosis induced by the indicated treatment (FB = FAC+BSO), apoptosis induced with TRAIL, or necrosis induced with H2O2. Dashed line indicates μexpΔt = μperm95Δt. Each data point represents one movie. Data are from two independent experiments for MCF7+H2O2 and one experiment for all other conditions. (h) Spatial Propagation Index generated from data in panel e.
To measure intercellular death propagation in different forms of cell death, we induced necrosis by treatment with hydrogen peroxide (H2O2), and apoptosis using TNF-related apoptosis-inducing ligand (TRAIL). While H2O2-induced necrosis and TRAIL-induced apoptosis displayed no visually obvious wave-like spreading of death (Fig. 1e, f, Supplementary Video 2), they did result in death patterns with non-random spatiotemporal features (Fig. 1g). In order to better compare the propagative features of these different forms of cell death, we devised a measure termed the spatial propagation index (SPI). When propagation is not the major determinant of the spatiotemporal distribution of cell death across a population, we expect μexpΔt to have similar or larger values than μperm95Δt, as death occurs independently of neighboring cell deaths in the vicinity. However, when propagation does play a major role, i.e. cells are affected by the death of their neighbors, we expect μexpΔt to be much smaller than μperm95Δt due to the non-random spatial order of death. Thus we defined the , the spatial contribution to the observed death patterns as a fraction of the neighboring death times expected if death order were spatially random. The SPI indicates that cell death propagation plays a dominant role when ferroptosis is induced by erastin, C’ dots, or FAC and BSO, but not when induced by ML162 (Fig. 1h). Similarly, H2O2-induced necrosis and TRAIL-induced apoptosis also do not exhibit propagative features (Fig. 1h). These results demonstrate that the ability to spread in wave-like patterns by propagating between neighboring cells is a feature of particular forms of ferroptosis.
To examine the mechanism of ferroptotic propagation, we first asked whether iron and lipid peroxidation, two known drivers of ferroptosis, are required for propagation. The addition of the lipid peroxidation inhibitor liproxstatin-1 or the iron chelator deferoxamine (DFO) to cell cultures after the initiation of ferroptosis stopped death from spreading (Fig. 2a–d, Supplementary Video 3), demonstrating that iron and lipid peroxidation are both required for continuous ferroptosis propagation. Because iron and lipid peroxidation are also necessary for ferroptosis to occur in individual cells, these results suggested that the full execution of ferroptosis, including cell lysis, could be required for the spreading of death between cells.
Figure 2.
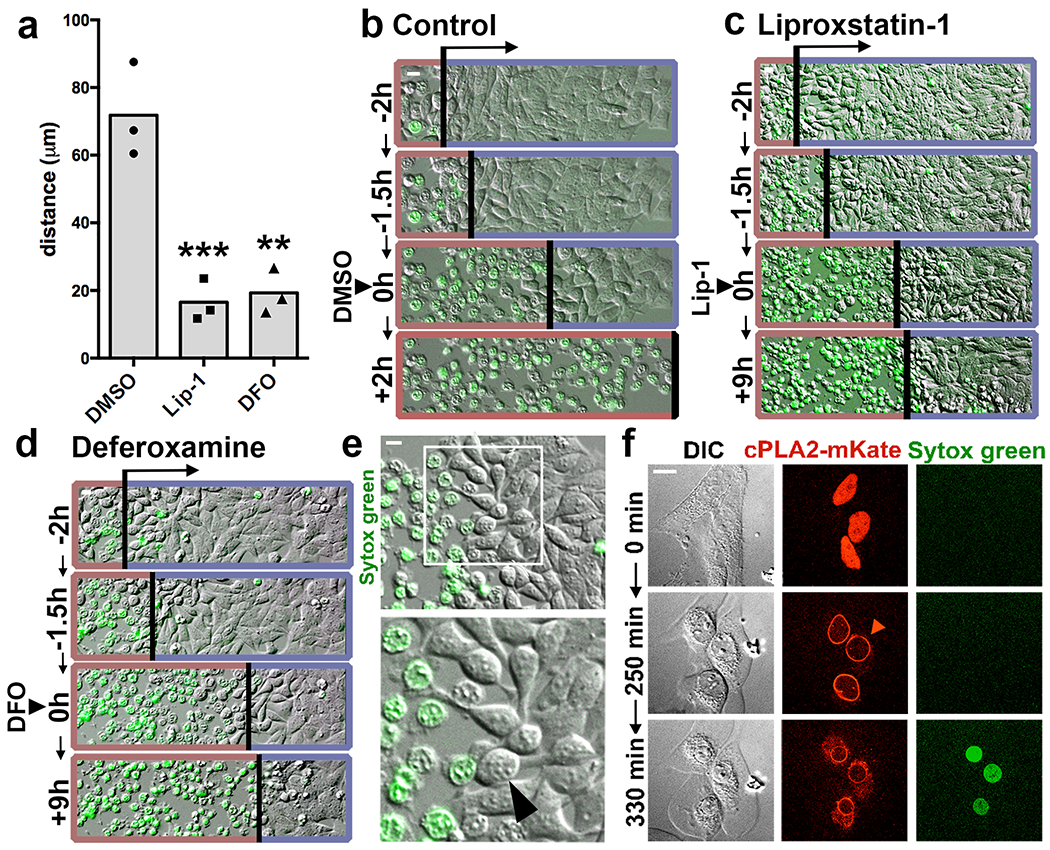
Ferroptosis spreading requires lipid peroxidation and iron and involves cell swelling. (a) Distance of ferroptosis spreading in HAP1 cells incubated with FAC and BSO, and treated with Liproxstatin-1 (Lip-1), Deferoxamine (DFO), or DMSO control after wave initiation. Distance was quantified 2h after drug addition. N = three independent experiments, averaged across three or four microscopic fields of view per replicate. Dunnett’s test; ***p=0.0008, **p=0.001. (b-d) Representative images from experiments quantified in panel a. Timing of treatment with DMSO (b), Lip-1 (c), or DFO (d) is indicated as 0h. Images show DIC and SYTOX Green fluorescence. Death waves are indicated by an arrow and a red border, live cells are indicated with a blue border on each image. Note Lip-1 and DFO-treated cells are shown 9 hours after treatment (+9h), versus 2 hours after treatment for DMSO (+2h). See Supplementary Video 3. (e) HAP1 cells treated with FAC and BSO round prior to ferroptotic cell rupture (arrowhead). Images are representative of four independent experiments. (f) The cell swelling marker cPLA2-mKate translocates to the nuclear envelope (arrowhead) prior to SYTOX Green labeling in HeLa cells treated with FAC and BSO. See Supplementary Video 4. Images are representative of two independent experiments. All scale bars = 10μm. Statistical source data can be found at Source data figure 2.
How ferroptosis is executed downstream of lipid peroxidation is not clearly defined. We noted from time-lapse imaging that ferroptotic cells appeared to round and swell prior to cell death (Fig. 2e). Like cell death, swelling also appeared to spread through cell populations in a manner that was blocked by treatment with liproxstatin-1 or DFO (Supplementary Video 3). Expression of an mKate-tagged version of the zebrafish cPLA2 enzyme, which localizes to the nuclear envelope upon osmotic swelling in HeLa cells19, confirmed that ferroptotic cells indeed swell prior to undergoing rupture (Fig. 2f, Supplementary Video 4). Cell swelling is also known to occur during pyroptosis and necroptosis, both of which involve the formation of pores in the plasma membrane, leading to the influx of extracellular ions and water molecules3, 4. Pore-mediated cell rupture can be inhibited by incubating cells with large carbohydrates known as osmoprotectants4. Osmoprotectants with a diameter larger than the pores protect cells from lysis by osmotically balancing large intracellular molecules that cannot diffuse freely across the perforated membrane, while smaller osmoprotectants do not. Thus, while osmoprotectants of sufficient size do not block plasma membrane permeabilization, pore-mediated ion exchange, or cell death, they prevent osmotic cell lysis caused by pore formation. Indeed, cell rupture resulting from FAC and BSO-induced ferroptosis, as measured by the release of lactate dehydrogenase (LDH), was inhibited by the addition of polyethylene glycols (PEGs) with molecular weights of 1450 and 3350 Da, but not by the smaller osmoprotectants sucrose and raffinose (Fig. 3a). The translocation of cPLA2-mKate to the nuclear envelope was also reduced by PEG1450 and PEG3350 (Fig. 3b), suggesting that ferroptotic swelling and rupture may be caused by the opening of nano-scale pores in the plasma membrane. Induction of ferroptosis with erastin or the GPX4 inhibitors RSL3 and ML162 likewise resulted in cell rupture that was inhibited by treatment with PEG 1450 or 3350 (Fig. 3d,e). LDH release caused by H2O2-induced death, on the other hand, was not affected by osmoprotectants (Fig. 3c).
Figure 3.
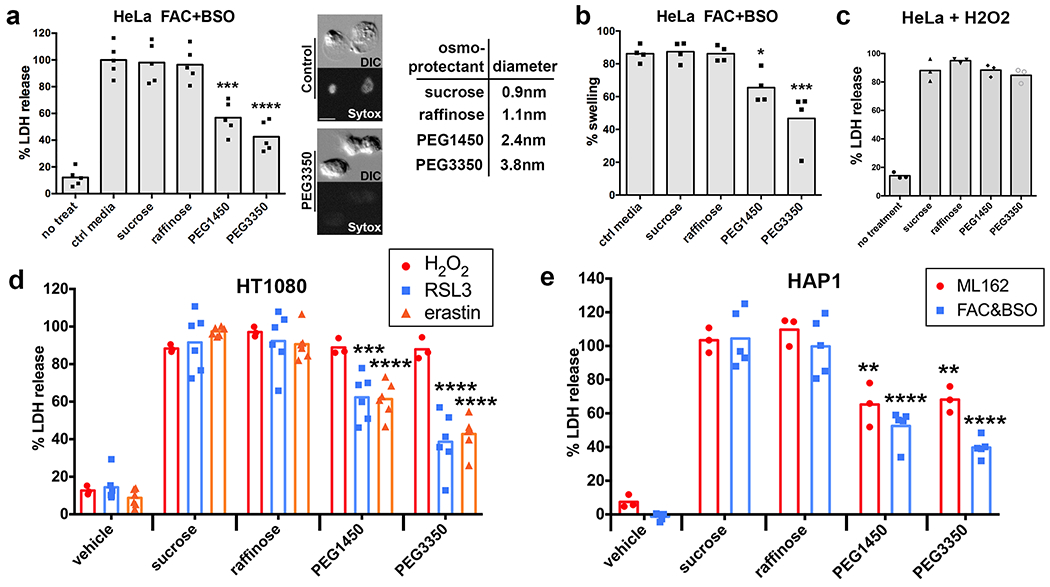
Ferroptotic cell rupture is inhibited by osmoprotectants. (a) Percent lactate dehydrogenase (LDH) released from ferroptotic HeLa cells treated with FAC and BSO and the indicated osmoprotectants: sucrose, raffinose, PEG1450, and PEG3350. Images show DIC and Sytox Green fluorescence for Hela cells treated with FAC + BSO in the presence or absence of PEG3350. Scale bar = 10μm. Diameters of osmoprotectants are shown in the table. N=5 biologically independent experiments. Dunnett’s test; ***p=0.0001, ****p=0.0001. (b) Swelling of ferroptotic HeLa cells treated with FAC and BSO as measured by recruitment of cPLA2-mKate to the nuclear envelope, determined by time-lapse microscopy. N=4 biologically independent experiments. Dunnett’s test; *p=0.0318, ***p=0.0002. (c) LDH release by HeLa cells treated with H2O2 and the indicated osmoprotectants, relative to HeLa cells treated with H2O2 only. N=3 biologically independent experiments. Dunnett’s test; all comparisons not significant. Raffinose: p=0.307, PEG1450: p=0.9999, PEG3350: p=0.7764. (d, e) LDH release in HT1080 cells treated with H2O2, RSL3, or erastin and HAP1 cells treated with ML162 or FAC and BSO and the indicated osmoprotectants, relative to the treatment alone. N=6 (RSL3 and erastin), 3 (H2O2 and ML162), or 5 (FAC + BSO) biologically independent experiments. Dunnett’s test; RSL3+PEG1450: p=0.0067, erastin+PEG1450: p=0.0001, RSL3+PEG3350: p=0.0001, erastin+PEG3350: p=0.0001, ML162+PEG1450: p=0.003, FAC&BSO+PEG1450: p=0.0001, ML162+PEG3350: p=0.0048, FAC&BSO+PEG3350: p=0.0001. Statistical source data can be found at Source data figure 3.
As ferroptotic cell rupture could be inhibited using osmoprotectants, we sought to examine whether cell lysis is required for ferroptosis propagation. When HAP1 cells were treated with FAC and BSO in the presence of the osmoprotectant PEG1450, we observed waves of cell rounding that spread through cell colonies and appeared similar to waves of cell death (Supplementary Video 5). However, SYTOX uptake was reduced, consistent with the inhibition of cell rupture. To quantify these waves, we expressed a fluorescent sensor of nuclear calcium (GCaMP6-NLS) in HAP1 cells, reasoning that pore formation might lead to a spike in intracellular calcium levels that could be used as a readout of cell permeabilization. Live imaging of ferroptotic cells demonstrated that GCaMP fluorescence indeed increased prior to the uptake of SYTOX, and that GCaMP signals spread through cell populations in a similar manner to SYTOX and cell rounding (Fig. 4a, Supplementary Video 6). We compared the relative timing of GCaMP and SYTOX fluorescence for individual cells and found a high degree of correlation, indicating that GCaMP signals could be used instead of SYTOX uptake to assess propagation (Fig. 4b). When cells were treated with PEG1450 to inhibit rupture, wave-like spreading of GCaMP fluorescence still occurred (Fig. 4c, Supplementary Video 7). We quantitatively examined the spatiotemporal GCaMP patterns, and found that their non-random nature was similar to SYTOX death waves in both the presence and absence of osmoprotectants (Fig. 4d,e), demonstrating that propagation occurs in the absence of cell rupture.
Figure 4.
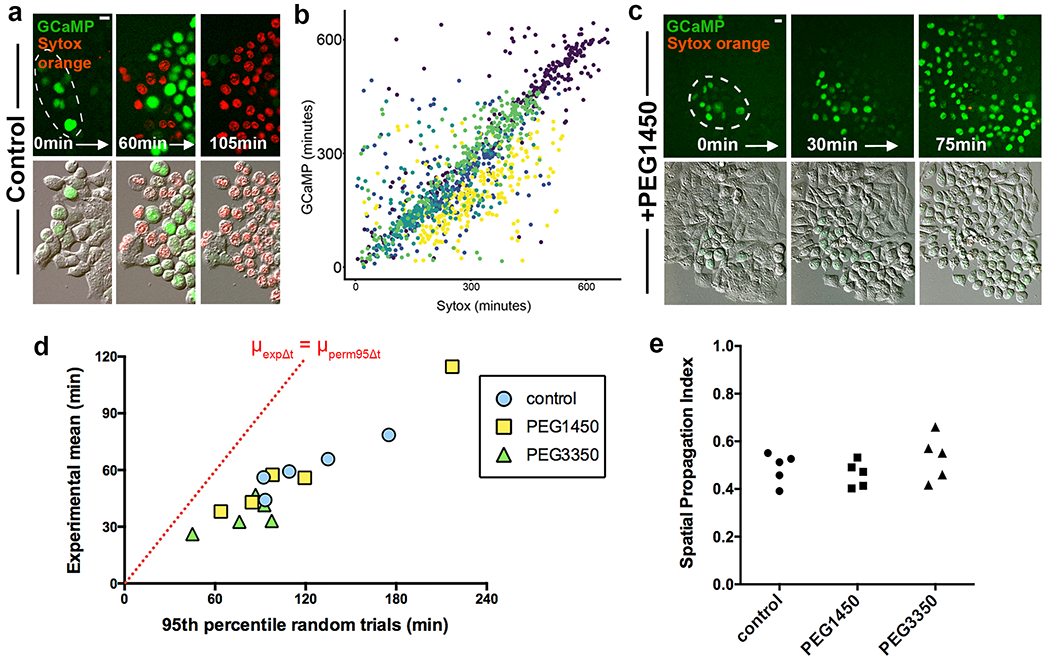
Ferroptosis spreading involves calcium flux and does not require cell rupture. (a) Images show spreading of GCaMP fluorescence (green) prior to cell rupture marked by SYTOX Orange (red) in HAP1 cells treated with FAC and BSO. Dashed circles show origin of death spreading. Note that cells lose GCaMP fluorescence upon cell rupture, likely due to GCaMP efflux. See Supplementary Video 6. Images are representative of three independent experiments. (b) Correlation between relative timing of GCaMP fluorescence and SYTOX labeling in HAP1 cells treated with FAC and BSO. Each dot represents a cell and each color represents a different field of view. Data from one experiment.. (c) Images show spreading of GCaMP fluorescence (green) and SYTOX Orange (red) in HAP1 cells treated with FAC and BSO and PEG1450. Dashed circles show origin of death spreading. Note that PEG1450-treated cells maintain GCaMP fluorescence and do not label with SYTOX Orange, unlike control cells in panel a. See Supplementary Video 7. Images are representative of three independent experiments. (d) Graph showing μexpΔt vs. μperm95Δt of movies of HAP1 cells treated with FAC and BSO and the indicated osmoprotectants, analyzed using GCaMP fluorescence. Dashed line indicates μexpΔt = μperm95Δt. Each data point represents one movie. Data are from one experiment. (e) Spatial Propagation Index calculated for experiments shown in panel d. All scale bars = 10μm. Statistical source data can be found at Source data figure 4.
While treatment with osmoprotectants did not prevent propagation, we wondered if it might affect wave speed. To test this we used U937 cells, which exhibit long-lived, unidirectional waves of ferroptosis that can be imaged by differential interference contrast (DIC) microscopy even in the absence of SYTOX staining (Fig. 5a). Treatment of U937 cells with PEG3350 inhibited cell lysis (Fig. 5b) yet had no effect on the induction of cell death waves (Supplementary Video 8), consistent with the HAP1 data. However, when we measured the speed of these ferroptosis waves, we found them to be slightly but significantly slower in the presence of PEG3350 (1.66 vs. 1.37 μm/min, see Fig. 5c), demonstrating that ferroptosis propagation is faster when cells are able to fully lyse.
Figure 5.
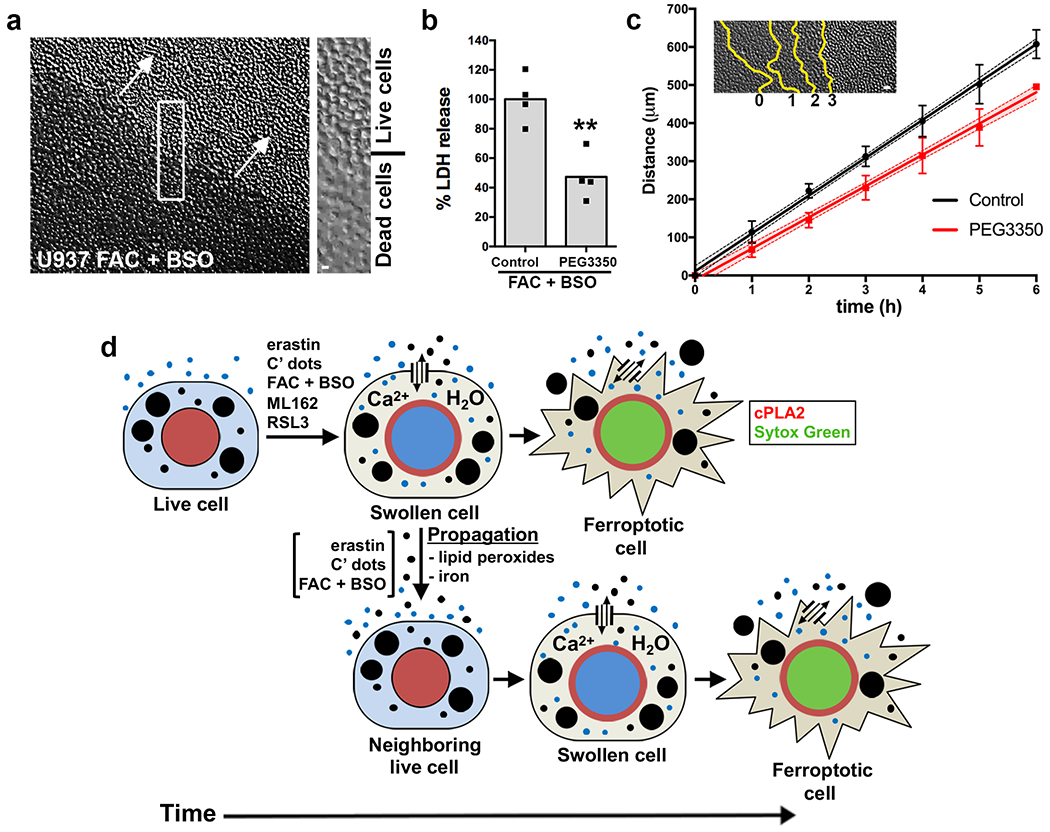
PEG3350 slows ferroptosis propagation (a) Wave-like spreading of ferroptosis in U937 cells treated with FAC and BSO, imaged by DIC microscopy. Arrows indicate direction of wave spreading; inset shows boundary between live and dead cells. See Supplementary Video 8. Image is representative of four independent experiments. Scale bar = 10μm (b) Percent LDH release in U937 cells treated with FAC and BSO in control and PEG3350-treated conditions. Data are from four biological replicates. **p=0.004 and was obtained using a two-sided t-test. (c) Wave-like spreading of ferroptosis is slower in the presence of PEG3350. Inset shows a representative example of death progression at each time point indicated by yellow lines. Graph shows distance over time of wave spreading in U937 cells treated with FAC and BSO. Data points indicate means from five independent waves per condition; error bars represent SD; line shows linear regression and its 95% confidence interval (shaded regions). Scale bar = 25μm. (d) Model for osmotic regulation of ferroptosis and cell death propagation. Ferroptosis induction involves the opening of plasma membrane pores that allow for solute exchange with the external environment, leading to cell swelling that occurs priors to cell death and is marked by cPLA2 translocation to the nuclear membrane (red). After swelling, ferroptotic cells undergo rupture and death marked by the rapid influx of death-indicating dyes such as Sytox Green. When ferroptosis is induced by treatment with erastin, C’ dots, or FAC and BSO, but not by treatment with the GPX4 inhibitor ML162, death propagates to neighboring cells in an iron and lipid peroxide-dependent manner, through a signal that is sent independently of cell rupture. Statistical source data can be found at Source data figure 5.
Together these data indicate that wave-like spreading is a feature of specific forms of ferroptosis that requires the continuous presence of iron and lipid peroxidation, and involves a signal that propagates upstream of cell rupture. While ferroptosis propagation has been previously observed7, 8, here we quantitatively establish the existence of non-random spatiotemporal patterns of ferroptosis in multiple contexts. Our method allowed us to distinguish two types of ferroptosis: cell-autonomous or “single-cell ferroptosis” observed in response to GPX4 inhibition, and propagative or “multicellular ferroptosis” that is induced by treatments that inhibit the generation of glutathione (erastin, BSO) and/or increase cellular iron concentrations (FAC, C’ dots). Why direct GPX4 inhibition does not induce propagative ferroptosis is important to examine in future studies, and may relate to activities of iron or functions of glutathione that do not directly involve GPX420.
Non-autonomous cell death effects have been described elsewhere, most notably in the radiation-induced bystander effect (RIBE), where damage and death rates are increased in cells adjacent to those exposed to radiation21. While RIBE may increase death frequencies, such phenotypes appear distinct from the wave-like death observed during ferroptosis that, in many cases, leads to the near-complete elimination of a cell population7, 8, 15. Further discovery of the underlying molecular mechanisms is required to determine whether death propagation in these different systems involves similar signaling mechanisms. Numerous factors are proposed to mediate RIBE, including gap junctions, TGFβ22, p53, and cyclooxygenase-2 (COX-2) signaling21. While our U937 data suggest that gap junctions are not involved in ferroptosis propagation, since these cells do not form cell junctions (Supplementary Video 8), whether other RIBE signals could play a role in ferroptosis spreading is not yet known. Our finding that the presence of an osmoprotectant slows propagation could suggest that the release of a spreadable factor is enhanced by cell rupture, although further experiments are needed to test this.
Our data also indicate that ferroptosis is an osmotic process, as it involves cell swelling (Fig. 5d), and can be blocked by the addition of large osmoprotectants. The ability of osmoprotectants to block lysis following induction of necroptosis or pyroptosis both in culture and in vivo, and the observed size-dependence of the protective effects of different osmoprotectants (Fig. 3), have been interpreted previously as evidence for the existence of pore-like structures that trigger lysis in these forms of necrosis3, 4. This is indeed known to occur during pyroptosis, in which the caspase-dependent cleavage of Gasdermin D (GSDMD) triggers its oligomerization in the plasma membrane23, 24. Similarly, necroptosis may involve plasma membrane permeabilization mediated by the pseudokinase MLKL25, 26. Our data thus suggest that ferroptotic rupture is mediated by the formation of plasma membrane pores of a few nanometers in diameter and that cell permeabilization during ferroptosis could be a regulated process. Intriguingly, lipid peroxidation has been proposed to lead to conformational changes in lipid domains and plasma membrane regions27, 28, raising the possibility that pore formation could occur through a lipid-based mechanism rather than by activation of a pore-forming protein. Ferroptotic pore formation could regulate not only cell death execution but also the potential release of pro-inflammatory cytokines or DAMPs, which is known to occur during pyroptosis29. We previously showed that ferroptosis induction in mouse xenografts leads to tumor regression and a concomitant immune response, implying that ferroptosis-inducing agents may be promising cancer therapies8, 15. Ferroptosis is also implicated in cell death resulting from ischemia reperfusion injury during stroke or myocardial infarction, as well as in acute kidney injury, all of which result in the formation of large zones of necrotic tissue, possibly indicating a role for ferroptosis propagation in these diseases1, 15. Intriguingly, the paper by Katikaneni et al. published in this issue shows large waves of cellular deformation occurring in intact zebrafish larvae following microperfusion of arachidonic acid (AA). As AA is a known driver of ferroptosis, this finding suggests that wave-like propagation of ferroptosis may also occur in vivo, causing widespread tissue damage30. Uncovering the molecular mechanisms that regulate ferroptosis execution and propagation through cell populations will ultimately further our understanding of how modulators of ferroptosis may be leveraged for therapeutic benefit.
Methods
Cell culture
HT1080 cells (ATCC), HeLa cells (ATCC), HAP1 chronic myelogenous leukemia cells (the kind gift of Dr. Jan Carette, Stanford School of Medicine), and MCF7 breast cancer cells (Lombardi Cancer Center, Georgetown University) were cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM) (MSKCC Media Preparation Facility) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (F2442; Sigma-Aldrich) and penicillin/streptomycin (30-002-CI; Mediatech). B16F10 melanoma cells (ATCC) and U937 promonocytic leukemia cells (ATCC) were grown in RPMI-1640 (11875-093; ThermoFisher) containing 10% heat-inactivated FBS and penicillin/streptomycin, and MCF10A mammary epithelium cells (ATCC) were cultured in DMEM/F12 (11320-033; ThermoFisher) supplemented with 5% horse serum (HS) (S12150; Atlanta Biologicals), 20ng/mL epidermal growth factor (EGF) (AF-100-15; Peprotech), 10μg/mL insulin (I9278; Sigma-Aldrich), 0.5μg/mL hydrocortisone (H-0888; Sigma-Aldrich), 100ng/mL cholera toxin (C-8052; Sigma-Aldrich), and penicillin/streptomycin. HeLa cells expressing Danio rerio cytosolic phospholipase A2 (cPLA2)-mKate have been described previously12. HAP1 cells expressing GCaMP6-NLS were generated using the Sleeping Beauty transposase system. HAP1 cells were transfected with pSB-CMV-MCS-Puro GCaMP6-NLS and pSB-cag-100x-Transposase, using Lipofectamine 3000 (L3000-015; ThermoFisher) as recommended by the manufacturer. Amino acid-free culture medium was prepared by dialyzing FBS or HS in PBS (MSKCC Media Preparation Facility) using MWCO 3500 dialysis tubing (21-152-9; Fisherbrand) and adding it to amino acid-free base media (MSKCC Media Preparation Facility). These media were used in combination with FAC and BSO to induce ferroptosis in MCF10A and MCF7 cells, and with αMSH-tagged C’ dots to induce ferroptosis in B16F10 cells.
Reagents
Ferric ammonium citrate (FAC) (F5879; Sigma-Aldrich), L-buthionine sulfoximine (BSO) (B2515; Sigma-Aldrich), and deferoxamine (DFO) (D9533; Sigma-Aldrich) were dissolved in water and stock solutions were stored at -20°C. FAC and BSO were used at 400μM and DFO at 200μM. SuperKillerTRAIL (ALX-201-115-C010; Enzo) stock solution was diluted to 100μg/mL in KillerTRAIL dilution buffer (20mM HEPES, 300mM NaCl, 0.01% Tween-20, 1% sucrose, 1mM DTT), aliquotted, and stored at -80°C. It was used at a final concentration of 50ng/mL. Hydrogen peroxide (216763; Sigma-Aldrich) was first diluted in water and then added to cell culture media at a final concentration of 1mM. C11-BODIPY (D3861; Molecular Probes) was dissolved in DMSO, stored at -20°C, and diluted to a final concentration of 5μM prior to use. Sucrose (S7903; Sigma-Aldrich), raffinose (R0514; Sigma-Aldrich), polyethylene glycol (PEG) 1450 (P7181; Sigma-Aldrich), and PEG3350 (P3640; Sigma-Aldrich) were dissolved directly into cell culture media at a concentration of 20mM. All other compounds were prepared as stock solutions in DMSO and stored at -20°C. Erastin (E7781, used at 7.5μM with HAP1s and 2μM with HT1080s), ferrostatin-1 (SML0583, used at 4μM), and Trolox (238813, used at 100μM) were from Sigma-Aldrich; RSL3 (S8155, used at 1μM) and liproxstatin-1 (S7699, used at 200nM) were from SelleckChem. ML162 was a kind gift from Dr. Scott Dixon and was used at 4μM. αMSH-tagged C’ dots were synthesized as described previously 8, 31 in water, stored at 4°C, and used at a concentration of 15μM. SYTOX Green (S7020; Molecular Probes) was used at a concentration of 10nM and SYTOX Orange (S11368; Molecular Probes) at a concentration of 50nM for all experiments.
Analysis
Single cell time of death annotation: we used a custom MATLAB (R2016A) script to record the xy position and time of death (for Sytox) or time of calcium influx (for GCaMP) of each cell. All other spatiotemporal analysis (measuring propagation and statistical testing, see below) was performed in Python 3.
Measuring propagation: we used Voronoi tessellation32, a computational geometry method to divide a plane into regions given a set of loci where each locus corresponds to a cell nucleus’s xy location. Each point on the plane belongs to a single region and each region consists of all the closest points to the associated nuclear locus. Neighbors were defined as regions sharing a border, which allowed us to identify all pairs of neighboring cells. Note that neighboring cells in the tessellation do not necessarily share cell-cell junctions in the experiment. For each field of view we calculated the mean difference in time of death between all neighboring cell pairs as a measure for propagation speed, marked with μexpΔt.
Statistical test: we devised a statistical test to determine the statistical significance of the hypothesis that there is a relationship between a cell’s and its neighbors’ times of death. This was achieved by using a non-parametric permutation test to reject the null hypothesis that the cells’ time of death is independent of their neighbors’ time of death. For every field of view, the following procedure was repeated for 1000 iterations. The recorded time of death was randomly permuted between the cells (i.e., each cell was assigned a random time of death, with the same number of deaths at each time point as in the experimental data) and the mean difference in time of death between all neighboring cell pairs was recorded. The p-value was calculated as the fraction of iterations in which the mean time difference between neighbors using the permuted (“random”) cell death was faster than the experimental measurement. We considered a p-value of 0.05 as statistically significant.
We devised the spatial propagation index to quantify the contribution of the spatial component to the observed experimental cell-cell propagation. This measure was defined as the deviation of the experimental mean propagation (μexpΔt) from the 95th percentile of the mean randomly permuted death times (μperm95Δt) normalized to μperm95Δt: . This measure can be interpreted as the fraction of the μperm95Δt needed to reconstruct back the spatial information in the experimental data.
Quantifying ferroptosis propagation from DIC: to quantify the distance covered by the ferroptosis wave in each field of view, lines delineating live and dead cell regions were drawn manually in NIS elements AR (Nikon, 3.22.15) at one-hour intervals, starting from a timepoint at which smaller initiation points, if present, had converged into larger waves. For each interval, the area between two lines was measured and divided by their average length to obtain the mean distance travelled during a given interval in a given field of view. Information from different fields of view in the same condition was then combined to plot the mean distance covered in each condition, and a linear regression performed to calculate the speed of the corresponding ferroptosis wave.
Live cell imaging
Cells were seeded on glass-bottom plates (P24G-1.5-13-F; Mattek) and treated in fresh culture media the next day. For amino acid-free conditions, cells were washed with PBS twice prior to treatment. Imaging was performed in live-cell incubation chambers maintained at 37°C and 5% CO2. Images were acquired every 5 to 30 min for 24-48 hours using a Nikon Ti-E inverted microscope attached to a CoolSNAP charge-coupled device camera (Photometrics) and NIS Elements AR software (Nikon, 3.22.15). For experiments with ML162, time-lapse imaging was performed on plastic tissue culture plates (3527; Corning) on a Zeiss microscope using ZEN software (Zeiss). Images were quantified manually in NIS Elements AR (Nikon) or ZEN (Zeiss) and processed using ImageJ (2.0.0) and Adobe Photoshop (CS6 13.0.5).
LDH assays
LDH assays were performed using the Pierce LDH Cytotoxicity Assay Kit (88954; ThermoFisher) following the manufacturer’s instructions. Briefly, cells were seeded on 96 well plates and treated in triplicate the next day. At the indicated time, 50μL media was transferred from each well to a well containing 50μL assay buffer, and plates were incubated at RT for 30 mins. At this point 50μL stop solution was added to each well, bubbles were removed using a syringe, and the absorbance was read on a BioTek Synergy H1 Hybrid Reader at 490nm and 680nm wavelengths. The 490nm absorbance was subtracted from the 680nm absorbance, and background from cell culture medium was subtracted from all values. Data were averaged across technical replicates and normalized to the indicated treatment without osmoprotectants to calculate percent LDH release. For suspension cells, cells were treated in 24 well tissue culture plates containing 1mL media per well. At the indicated time, 200μL media was taken from each well, spun down to remove dead cells, and supernatant was used to perform the assay as described.
Crystal Violet assays
Cells were seeded on 24-well tissue culture plates (3527; Corning) and treated in triplicate the next day. After 24 hours, when most control cells had died, wells were washed twice with PBS, then fixed in 4% paraformaldehyde (15710-S; Electron Microscopy Sciences) in PBS for 15 minutes. After washing with water, cells were stained with 0.1% crystal violet (61135; Sigma-Aldrich) in 10% ethanol for 20 minutes, then washed again with water until clear and allowed to air dry. After drying, crystal violet stain was dissolved in 10% acetic acid by shaking plate at room temperature for 30 minutes. This solution was diluted 1:4 with water and absorbance measured at 590nm on a BioTek Synergy H1 Hybrid Reader. Background absorbance from wells containing only tissue culture medium was subtracted from all readings, and values were averaged across technical replicates and normalized to the untreated controls to obtain percent viability.
Confocal imaging
Cells were plated on glass-bottom dishes and treated the next day. For C11-BODIPY581/591 imaging, cells were washed twice with Hank’s Balanced Salt Solution (HBSS) (14025-092; ThermoFisher) 24 hours after treatment, stained in 5μM C11-BODIPY581/591 in HBSS for 10 minutes at 37°C and 5% CO2, and again washed twice in HBSS. Cells were imaged at 37°C and 5% CO2 using the Ultraview Vox spinning-disk confocal system (PerkinElmer) equipped with 488nm and 568nm lasers and an electron-multiplying charge-coupled device camera (Hamamatsu C9100-13), and attached to a Nikon Ti-E microscope. For cPLA2 imaging shown in Figure 2f, a single confocal plane is shown from the indicated time points. For C11-BODIPY581/591 imaging in Extended Data Figure 1, maximum projections are shown. Images were acquired and processed using Volocity software (Perkin Elmer, version 5.2.0).
Statistics and Reproducibility
Data were analyzed in Microsoft Excel (Office 2011) and GraphPad Prism 7 and are represented as mean with individual data points. P values were obtained using two-sided Dunnett’s multiple comparisons test unless otherwise indicated. * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001. The following analyses were done using data from one experiment: spatiotemporal analyses in Figures 1d–h, 4b, d, and e, and Extended Data Figure 1c and e, except data for MCF7+H2O2 in Figure 1g and h which was from two independent experiments. The images in Figure 1a are representative of one experiment and images in Figure 2f are representative of two independent experiments. All other data were derived from three or more biologically independent experiments; exact n are reported in the figure legends. For LDH measurements in Figures 3a–d and Figure 5b, and Crystal Violet assay in Extended Data Figure 1a, each experiment consisted of three technical replicates that were averaged for each condition.
Code Availability
Our source code is available via GitHub at https://github.com/AssafZaritskyLab/PropagationOfCellDeath. This repository includes all code used to measure the mean time difference between neighboring deaths and to run the random simulations, as well as a demo dataset.
Data Availability
The statistical source data that support the findings of this study have been provided as part of this publication. All other data are available from the corresponding authors upon request.
Extended Data
Extended Data Fig. 1. Treatment of cells with FAC and BSO induces ferroptosis.
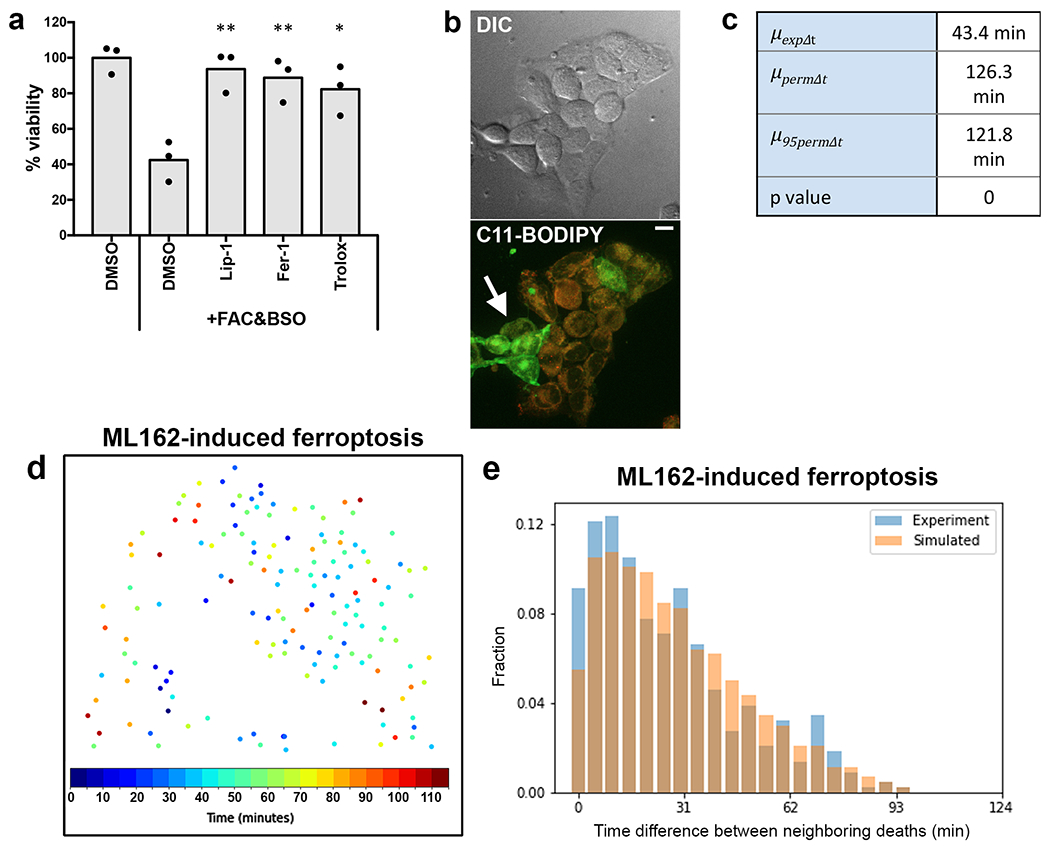
(a) Viability of HAP1 cells after treatment with FAC and BSO and either DMSO or ferroptosis inhibitors as measured by crystal violet staining. N = three independent experiments. Dunnett’s test; **p=0.0024 for Lip-1; **p=0.0045 for Fer-1; *p=0.0107 for Trolox. (b) Confocal images of HAP1 cells treated with FAC and BSO and stained with C11-BODIPY581/591. Non-oxidized probe is shown in red, oxidized probe is shown in green (arrow). Scale bar = 10μm. Images are representative of three independent experiments. (c) Values from the analysis of the experiment shown in panels 1c and d. Note that the experimental mean time difference between neighbors (μexpΔt) is much smaller than the mean (μpermΔt) and 95th percentile (μpermΔt) obtained from the randomly permuted data. (d) Spatiotemporal distribution of cell death in HAP1 cells treated with ML162 to induce ferroptosis. Each dot represents a cell from a single movie representative of five fields of view from one experiment. Colors indicate relative times of cell death as determined by SYTOX Green staining. (e) Distribution of experimental time differences between neighboring deaths in blue and averaged distribution of the corresponding permuted data in orange. Data belong to the experiment shown in panel d and are representative of five fields of view from one experiment. Statistical source data can be found at Source data Extended Data Figure 1.
Supplementary Material
Acknowledgements
This research was supported by 1R01GM122923 from the NIH to S.J.D. and CA154649 to M.O. from NCI. A.Z. was supported by the Data Science Research Center, Ben-Gurion University of the Negev, Israel. The authors thank members of the Overholtzer lab for helpful discussions.
Footnotes
Competing Interests
Memorial Sloan-Kettering Cancer Center and three investigators involved in this study (M.S.B., U.W., and M.O.) have financial interests in Elucida Oncology, Inc. Research involving C’ dots may involve one or more U.S. or international patent applications.
References
- 1.Stockwell BR et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixon SJ et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ros U et al. Necroptosis Execution Is Mediated by Plasma Membrane Nanopores Independent of Calcium. Cell Rep 19, 175–187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fink SL & Cookson BT Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 8, 1812–1825 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Degterev A et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1, 112–119 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Cookson BT & Brennan MA Pro-inflammatory programmed cell death. Trends Microbiol 9, 113–114 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Linkermann A et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A 111, 16836–16841 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim SE et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol 11, 977–985 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dolma S, Lessnick SL, Hahn WC & Stockwell BR Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Yang WS et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galluzzi L et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seibt TM, Proneth B & Conrad M Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med 133, 144–152 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Bersuker K et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doll S et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Riegman M, Bradbury MS & Overholtzer M Population Dynamics in Cell Death: Mechanisms of Propagation. Trends Cancer 5, 558–568 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang WS & Stockwell BR Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol 26, 165–176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng H & Stockwell BR Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol 16, e2006203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bittker JA et al. Screen for RAS-Selective Lethal Compounds and VDAC Ligands - Probe 1, in Probe Reports from the NIH Molecular Libraries Program (Bethesda (MD: ); 2010). [Google Scholar]
- 19.Enyedi B, Jelcic M & Niethammer P The Cell Nucleus Serves as a Mechanotransducer of Tissue Damage-Induced Inflammation. Cell 165, 1160–1170 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao JY et al. A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell Rep 26, 1544–1556 e1548 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou H et al. Mechanism of radiation-induced bystander effect: role of the cyclooxygenase-2 signaling pathway. Proc Natl Acad Sci U S A 102, 14641–14646 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iyer R, Lehnert BE & Svensson R Factors underlying the cell growth-related bystander responses to alpha particles. Cancer Res 60, 1290–1298 (2000). [PubMed] [Google Scholar]
- 23.Chen X et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res 26, 1007–1020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sborgi L et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35, 1766–1778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54, 133–146 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Chen X, Gueydan C & Han J Plasma membrane changes during programmed cell deaths. Cell Res 28, 9–21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agmon E, Solon J, Bassereau P & Stockwell BR Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep 8, 5155 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Runas KA, Acharya SJ, Schmidt JJ & Malmstadt N Addition of Cleaved Tail Fragments during Lipid Oxidation Stabilizes Membrane Permeability Behavior. Langmuir 32, 779–786 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Evavold CL et al. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35-44 e36 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katikaneni A, J.M., Gerlach G, Ma Y, Overholtzer M, Niethammer P Lipid peroxidation instructs long-range wound detection through 5-lipoxygenase in zebrafish. Nature Cell Biology (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma K et al. Control of Ultrasmall Sub-10 nm Ligand-Functionalized Fluorescent Core–Shell Silica Nanoparticle Growth in Water. Chemistry of Materials 27, 4119–4133 (2015). [Google Scholar]
- 32.Du Q & Gunzburger M Grid generation and optimization based on centroidal Voronoi tessellations. Applied Mathematics and Computation 133, 591–607 (2002). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The statistical source data that support the findings of this study have been provided as part of this publication. All other data are available from the corresponding authors upon request.