

Abstract
Purpose
Emerging evidence has shown that pinocembrin protects the myocardium from ischemic injury in animals. However, it is unknown whether it has cardioprotection when given at the onset of reperfusion. Also, mechanisms mediating the cardioprotective actions of pinocembrin were largely unknown. Thus, this study is aimed at investigating the effects of pinocembrin postconditioning on ischemia-reperfusion (I/R) injury and the underlying mechanisms.
Methods
The in vivo mouse model of myocardial I/R injury, ex vivo isolated rat heart with global I/R, and in vitro hypoxia/reoxygenation (H/R) injury model for primary cardiomyocytes were used.
Results
We found that pinocembrin postconditioning significantly reduced the infarct size and improved cardiac contractile function after acute myocardial I/R. Mechanically, in primary cardiomyocytes, we found that pinocembrin may confer protection in part via direct stimulation of cardiac glycolysis via promoting the expression of the glycolytic enzyme, PFKFB3. Besides, PFKFB3 inhibition abolished pinocembrin-induced glycolysis and protection in cardiomyocytes. More importantly, PFKFB3 knockdown via cardiotropic adeno-associated virus (AAV) abrogated cardioprotective effects of pinocembrin. Moreover, we demonstrated that HIF1α is a key transcription factor driving pinocembrin-induced PFKFB3 expression in cardiomyocytes.
Conclusions
In conclusion, these results established that the acute cardioprotective benefits of pinocembrin are mediated in part via enhancing PFKFB3-mediated glycolysis via HIF1α, which may provide a new therapeutic target to impede the progression of myocardial I/R injury.
1. Introduction
Cardiovascular disease remains the main cause of death worldwide [1]. Currently, there are few effective drugs to protect the heart after ischemia/reperfusion (I/R) injury. Therefore, it has become a hot research field to find the molecular mechanism of coronary artery disease progression and development that can protect the myocardium from I/R injury. With this in mind, more and more attention is focused on pharmacological interventions because it can induce cardioprotection and easy to implement [2]. However, for patients with ischemic heart disease, there are still many restrictive drugs available clinically.
Pinocembrin (5,7-dihydroxyflavone) is an abundant flavonoid isolated from propolis and some plants. It has various biological functions such as anti-inflammatory, antioxidation, and antibacterial [3]. Previous studies have confirmed that pinocembrin confers neuroprotective effects during cerebral ischemic I/R [4–6]. Recent research has also tried to determine whether pinocembrin is beneficial for cardiac injury. For example, several studies suggest that pinocembrin can improve the cardiac function of myocardial I/R rats, reduce ventricular arrhythmia, and reduce the area of myocardial infarction [7, 8]. However, it is still unclear whether pinocembrin given at the onset of reperfusion has cardioprotective effects, which is a more clinically effective method. In addition, the underlying mechanism by which pinocembrin can provide cardioprotection is largely unknown.
Recent findings report that changes in energy metabolism relate to many human diseases, and targeted energy metabolism intervention may have beneficial effects on these diseases [9]. Due to the mechanical function of the heart, it is an organ with high energy requirements. In general, most of the energy (about 70%) of a healthy heart comes from the β-oxidation of fatty acids, and the rest of the energy comes from glucose oxidation [10]. Nevertheless, in a pathological environment, substrate utilization may change [11]. In patients with diabetes, circulating blood glucose levels at admission are related to the clinical outcome after acute myocardial infarction (AMI), suggesting that it may be related to myocardial metabolism [12]. A metabolic shift from β-oxidation to glycolysis metabolism will reduce the cell's need for oxygen by 11-13%, and NAD+ precursors have been shown to activate cellular glycolysis to protect the heart from ischemic injury [13]. It is worth noting that redirect energy metabolism to glycolysis can reduce oxidative damage and inhibit apoptosis [14–16]. However, few agents that target energy metabolism are clinically safe and useful for patients. At the same time, it is unclear whether and how pinocembrin regulates acute myocardial I/R glycolysis.
In this study, we (i) examine the role of pinocembrin in rat and mouse cardiac I/R injury ex vivo and in vivo, respectively, (ii) clarify the effects of pinocembrin on the glycolytic metabolism during I/R, and (iii) investigate the underlying molecular basis that contributes to the pinocembrin-induced cardioprotection. Our results uncover a novel mechanism for pinocembrin-induced cardioprotection and suggest a potential application value of pinocembrin in the protection of hearts against I/R injury.
2. Materials and Methods
2.1. Chemicals and Reagents
3-(3-Pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) was purchased from EMD Millipore (MA, USA). Pinocembrin and other reagents were supplied by Sigma-Aldrich (St. Louis, MO, USA).
2.2. Animals
Male Sprague-Dawley rats (aged 6-8 weeks) and C57BL/6 mice (aged 8-10 weeks) were from Shanghai Slac Laboratory Animal Co. Ltd. All animals were housed in a temperature-controlled (24°C) and humidity-controlled (40-70%) barrier system with a 12-hour light/dark cycle. All animal experiments were conducted to the guidelines for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (revised in 2011), and all procedures were approved by the Shanghai University of Health and Medicine. Rats and mice were selected for treatment and observed in a randomized, double-blind trial.
2.3. Ex Vivo Global I/R Injury Model
As mentioned before, the heart was rapidly excised at 37°C and perfused at a constant pressure of 80 mmHg with Krebs-Henseleit buffer (KHB) using the Langendorff technique (Xie et al., 2005). The PowerLab system (AD instrument, Australia) was used to monitor the left ventricular imaging pressure (LVDP), left ventricular imaging pressure (LVEDP), maximum rate of pressure changes over time (+dP/dtmax), and pressure decay over time (-dP/dtmax). Pinocembrin's final concentration of 10, 30, and 100 μM perfusate was added with 5 minutes of reperfusion. In order to assess the infarct size, the isolated rat heart was reperfused for 2 hours after 30 minutes of ischemia. The slices were incubated in 1% w/v triphenyltetrazolium chloride (TTC, pH 7.4) for 15 min and then fixed in 10% formaldehyde. The Image-Pro Plus software (Media Cybernetics) was used to calculate the infarct area. The infarct area was expressed as a percentage of the LV area at risk.
2.4. In Vivo Myocardial I/R Model
Surgical ligation of the left coronary artery (LCA) was performed as described previously [17]. Mice were anesthetized with pentobarbital sodium (50 mg/kg) and ketamine (50 mg/kg) by intraperitoneal injection, followed by orally intubating and ventilating. The core body temperature is always maintained at 37°C. An internal sternotomy was then performed with electrocautery, and then, the proximal LCA was displayed and ligated. We investigated the magnitude of protection afforded by two different doses of pinocembrin (5 and 10 mg/kg i.v.). Accordingly, the mice received at random an intravenous injection of either saline, 5 mg/kg of pinocembrin, and 10 mg/kg of pinocembrin. These intravenous injections were performed during the last 5 min of ischemia, prior to reperfusion. After 30 minutes of coronary artery occlusion, the suture was cut and the blood vessels were allowed to reperfuse. After 24 hours of reperfusion, the mice were anesthetized with isoflurane. Transthoracic echocardiography was used to determine the left ventricular ejection fraction. After reperfusion, blood samples were taken followed by centrifugation at 3000 rpm for cardiac troponin T (cTnT) and lactate dehydrogenase (LDH) measurements. Measurement of an area at risk and infarct size was performed as reported previously.
2.5. Isolation and Culture of Primary Cardiomyocytes
Continuous enzymatic digestion and isolation were used to obtain neonatal rat and mouse cardiomyocytes [18, 19]. Neonatal rats were decapitated, and hearts were immediately placed in HBSS. The ventricle was taken and digested with trypsin and collagenase several times for 10 min at 37°C. Cardiomyocytes were suspended in sterile DMEM. The cells were preplated twice (37°C for 45 minutes) to reduce fibroblast contamination. Hypoxia/reoxygenation (H/R) was conducted as previously described in cardiomyocytes to simulate MI/R in vivo [20].
2.6. Measurement of LDH Release and cTnT Release
Necrotic cell death was assessed by the activity of the supernatant LDH, just like in previous studies [21]. The plasma cTnT level was used as an indicator of myocardial cell damage and was measured using a mouse cTnT ELISA kit (Wuhan Elabo Biotech Co., Ltd., China) according to the instructions.
2.7. Quantitative Real-Time PCR
Total RNA was extracted from the heart tissue using Trizol Reagent (Invitrogen, Carlsbad, CA, USA). Relative quantitation by real-time PCR was performed with SYBR Premix Ex Taq kits (TaKaRa) with the ABI PRISM 7900 System (Applied Biosystems). The primers targeting Gck, Galm, Gp1, Hk2, PFKFB3, Pdk1, Pdk2, Pdk3, Glut10, Glut4, Glut2, Glut1, Pdk4, Eno2, Bpgm, Aldoa, Aldob, Aldoc, and Pfk1 genes were list in Supplemental Table 1. GAPDH was used as internal normalization. The reactions were done at 95°C for 5 min followed by 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s.
2.8. Western Blot Analysis
Western blot was performed as described previously [22]. Left ventricles were homogenized, and the cells were directly lysed in ice-cold RIPA buffer. The samples were analyzed by SDS-PAGE. Transfer the protein to a polyvinylidene fluoride microporous membrane (Bio-Rad) with primary antibodies PFKFB3 (Abcam, USA; 1: 1000), HIF1α (CST, USA; 1: 1000), and anti-GAPDH (internal control; Kangcheng Co., Ltd., China; 1 : 8000). The bands were detected by horseradish peroxidase-coupled secondary antibodies (Cell Signaling Technology, 1 : 6000) and were visualized using the ECL detection kit (Amersham Pharmacia Biotech) and quantified with a video documentation system (Gel Doc 2000, Bio-Rad).
2.9. Generation and Administration of Adeno-Associated Virus (AAV)
Serotype 9 AAV vectors (AAV9) encoding shNC or shPFKFB3 (AAV9-shNC and AAV9–shPFKFB3) were prepared as previously described [23]. 3 × 1011 vg of AAV9-shNC or AAV9–shPFKFB3 was injected intravenously into tail veins as previously described [24] of 4-5-week-old male C57 mice. Sham or myocardial ischemia surgeries were conducted 4 weeks after AAV9 injection.
2.10. Seahorse Extracellular Flux Analyzer Assays
Cellular bioenergetics was measured using a Seahorse XFe24 extracellular flux analyzer in intact cardiomyocytes. We conducted glycolysis stress testing following the manufacturer's instructions as previously reported [25]. Glycolysis stress test: cells were incubated in glucose-free Seahorse assay media supplemented with 1 mM pyruvate at 37°C in an incubator without CO2 for 1 h prior to the assay. Injectors were loaded to add 20 mM glucose, 1 μM oligomycin, and 100 mM 2 deoxy-glucose (2-DG), and glycolysis, glycolytic capacity, and glycolytic reserves were calculated as an extracellular acidification rate (ECAR).
2.11. Generation of Plasmids and Transfection
p-GL4-basic luciferase expression vectors containing various lengths of 5′-flanking regions from the human PFKFB3 gene promoter and an HIF1-luciferase reporter gene were prepared by gene synthesis by General Biol (Anhui, China). siRNAs for mouse HIF1α were obtained from RiboBio (Guangzhou, China). Transfection was performed with Lipofectamine 3000 (Thermo, MA, USA) following the manufacturer's protocol. Luciferase activities were measured by using a Dual-Luciferase® Reporter Assay System (Promega, USA).
2.12. Statistical Analysis
Statistical analysis was undertaken only for studies where each group size was at least n = 5 using software GraphPad Prism (v7, GraphPad Software, USA). Data were presented as means ± SEM. Comparisons between 2 groups were performed using unpaired, two-tailed t test, and ANOVA with post hoc Dunnett's test was used for among multiple groups. A P value < 0.05 was deemed statistically significant.
3. Results
3.1. Compound Pinocembrin Significantly Improved the Cardiac Function and Reduced Infarct Size after I/R Ex Vivo
We perfused isolated rat hearts to explore the cardioprotective effects of pinocembrin against I/R injury. Ten to 100 μM pinocembrin were delivered during the first 5 min of reperfusion (Figure 1(a)). During 45 min of reperfusion following 30 min ischemia, the contractile function of the left ventricle (LV) was significantly suppressed (Figures 2(a)–2(d)). Pinocembrin itself does not affect the heart rate during reperfusion (Supplementary Figure 1), while it dose-dependently improves the postischemic myocardial performance from 10 to 100 μM (Figures 2(a)–2(d)).
Figure 1.
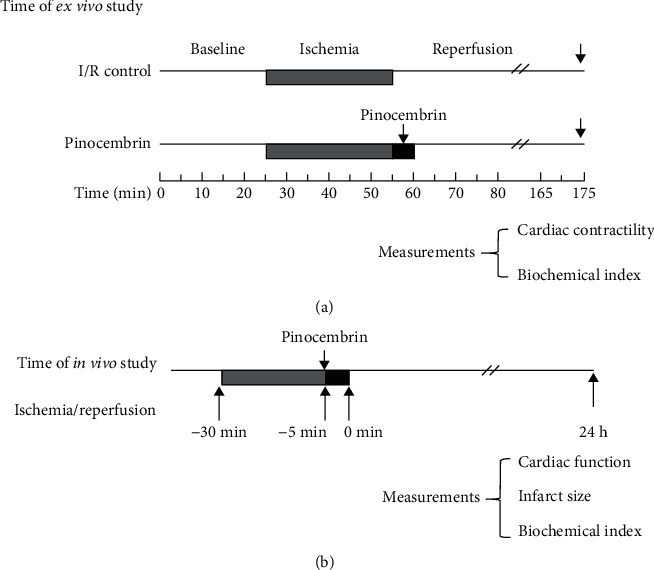
Experimental protocols to investigate the effects of pinocembrin on MI/R: (a) protocol of I/R ex vivo study (1); (b) protocol of MI/R in vivo study.
Figure 2.
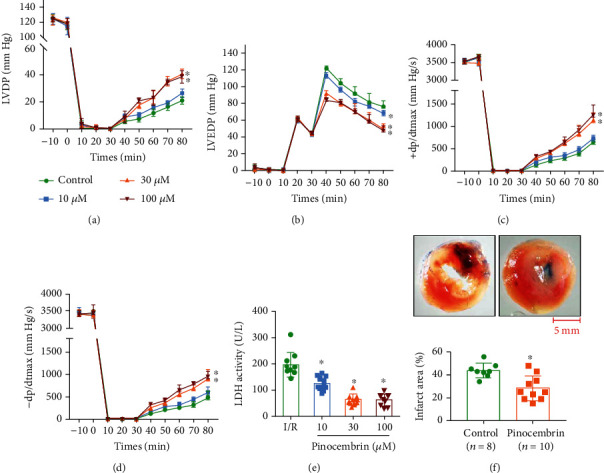
Compound pinocembrin significantly improves cardiac contractile recovery and reduces infarct size and LDH leakage after ischemia-reperfusion (I/R) ex vivo. (a–d) Time course of LVDP (a), LVEDP (b), and +dP/dtmax (c) and -dP/dtmax (d); n = 10 each. (e) Effect of pinocembrin on the LDH activity in the coronary effluent; n = 10 each. (f) Representative images and analysis of the infarct size in isolated I/R (30 min/2 h) hearts; n = 7 in the control group; n = 10 in the pinocembrin group. Data represent the mean ± SEM. ∗P < 0.05 versus I/R control group.
Next, we explore whether pinocembrin improves cell survival during I/R via examining lactate dehydrogenase (LDH) release, an indicator of myocardial injury. Little LDH release was detected in the coronary efflux before ischemia, while LDH release was obviously induced at the end of reperfusion, while pinocembrin significantly inhibited the release of LDH from 10 to 100 μM (Figure 2(e)). Consistently, pinocembrin significantly reduced the I/R-induced infarction after 2 h of reperfusion at the concentration of 30 μM (Figure 2(f)). These results demonstrated that pinocembrin exhibits protective effects on cardiac I/R injury ex vivo.
3.2. Compound Pinocembrin Protects Hearts from Myocardial I/R Injury In Vivo
To explore the cardioprotective effects of pinocembrin on myocardial I/R injury in vivo, pinocembrin was intravenously injected into wild-type (WT) mice 5 minutes before the end of sustained ischemia (i.v., 5 mg/kg and 10 mg/kg), followed by reperfusion for 24 hours (Figure 1(b)). Evans-blue/TTC dye method was used to determine the infarct size. After reperfusion for 24 hours, no difference of area at risk (AAR) was observed between each group (Figures 3(a) and 3(b)). Nevertheless, compared with the I/R group, pinocembrin i.v. treatment significantly decreased the infarct size by 20% (Figures 3(a) and 3(c)). Besides, plasma levels of cTnI and LDH activity were markedly elevated during myocardial I/R, which were both suppressed with pinocembrin i.v. treatment (Figures 3(d) and 3(e)). Furthermore, the echocardiographic results showed that pinocembrin can significantly improve I/R-suppressed ejection fraction (EF) and fractional shortening (FS) (Figure 3(f)).
Figure 3.
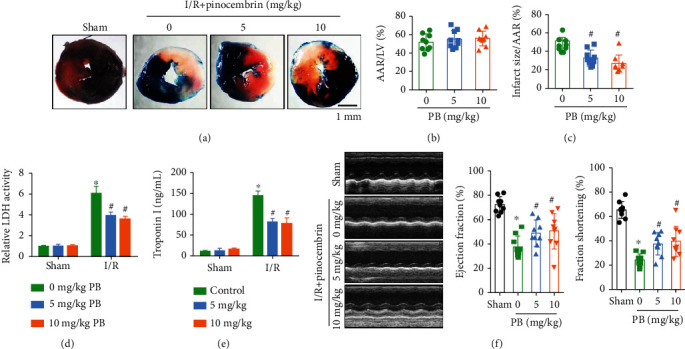
Pinocembrin protects from myocardial I/R (MI/R) injury in vivo in C57/BL6 mice. Wild-type (WT) mice were subjected to 30 minutes of the left anterior descending coronary artery (LAD) ligation, followed by 24 h of reperfusion, vehicle or pinocembrin intravenously (5 and 10 mg/kg) 5 min before reperfusion. (a) Representative TTC-stained LV transverse slices (scale bar, 1 mm). Areas stained dark blue, white, and red represented nonrisk, infarcted, and ischemic but noninfarcted zones, respectively. (b) Analysis of AAR (% total LV). (c) Analysis of myocardial infarct size (%AAR). (d, e) Plasma LDH activity and cTnI levels. (f) Representative images and analysis of left ventricular EF and FS, n = 9 each. Data represent the mean ± SEM. ∗P < 0.05 versus sham group. #P < 0.05 versus I/R control group.
3.3. Protective Effects of Pinocembrin against H/R Induced Cardiomyocyte Injury
To determine whether pinocembrin confers cardioprotective effects through its direct action on the cardiomyocytes, isolated neonatal rat and mouse cardiomyocytes were subjected to H/R and applied 30 μM pinocembrin during the onset of reperfusion. In accordance with the effects of pinocembrin on the myocardial I/R injury, simulated I/R-reduced cell viability was significantly improved by pinocembrin treatment (data not shown). Moreover, our data demonstrated that pinocembrin rescued cardiac troponin I (cTnI) release and LDH release postsimulated I/R in vitro, in both rat and mouse cardiomyocytes (Figures 4(a)–4(d)).
Figure 4.
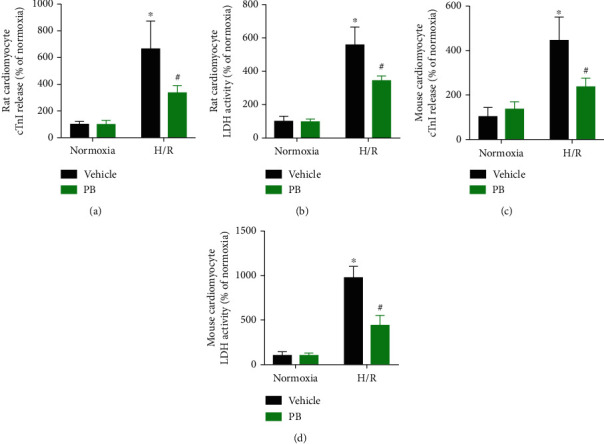
Protective actions of pinocembrin against cardiomyocyte injury responses in vitro. (a) Effect of pinocembrin (30 μM, added during reperfusion) on rat cardiomyocyte cTnI release subsequent to H/R. (b) Effect of pinocembrin on rat cardiomyocyte LDH release subsequent to H/R. (c) Effect of pinocembrin on mouse cardiomyocyte cTnI release subsequent to H/R. (d) Effect of pinocembrin on mouse cardiomyocyte LDH release subsequent to H/R. n = 5 each. Data represent the mean ± SEM. ∗P < 0.05 versus normoxia with the vehicle group. #P < 0.05 versus H/R with the vehicle group.
3.4. Pinocembrin Increases Glycolysis in Cardiomyocyte
During myocardial ischemia, enhanced glycolytic metabolism is essential for maintaining homeostasis of cardiomyocytes. In addition, previous studies reported that pinocembrin was involved in regulating glucose uptake in cancer cells [26]. Subsequently, we explored the effects of pinocembrin on cellular bioenergetics with the Seahorse extracellular flux analyzer and performed glycolysis stress tests to measure glycolysis and glycolytic capacity both in intact rat and mouse cardiomyocytes. Our results indicate that delivery with pinocembrin versus the control increased glycolysis by 21.4% (extracellular acidification rate (ECAR)) after H/R. Pinocembrin also increased glycolytic capacity in cardiomyocyte by 23.7% (Figures 5(a) and 5(b)).
Figure 5.
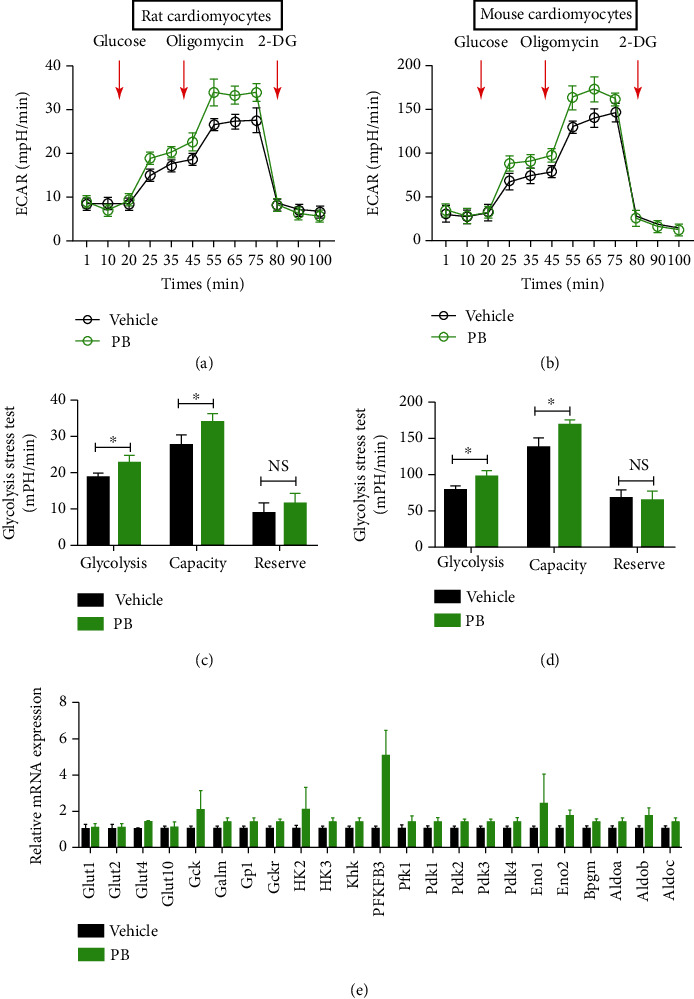
Pinocembrin increases glycolysis in primary cardiomyocytes. (a) Glycolysis stress test in rat cardiomyocytes measured as the ECAR by sequential injection of 20 mM glucose, 1 μM oligomycin followed by 100 mM 2-deoxy-d-glucose (2-DG). (b) Quantitative analysis of glycolysis, glycolytic capacity, and glycolytic reserves in rat cardiomyocytes. (c) Glycolysis stress test in mouse cardiomyocytes measured as the ECAR by sequential injection of 20 mM glucose, 1 μM oligomycin, and 100 mM 2-DG. (d) Quantitative analysis of glycolysis, glycolytic capacity, and glycolytic reserves in mouse cardiomyocytes pretreated with pinocembrin or control. (e) Mouse cardiomyocytes were treated with pinocembrin for 24 h, and then, the glycolysis-related genes were detected by qRT-PCR. n = 3/treatment in all Seahorse assays, each in triplicate. Data represent the mean ± SEM. ∗P < 0.05 indicates a significant difference. NS indicates no significant difference
To figure out the underlying mechanism of pinocembrin promoting myocardial glycolysis, mRNA expressions of glycolysis-related genes in cardiomyocytes after H/R were determined with qRT-PCR. As shown in Figure 5(c), pinocembrin significantly increased the expression of glycolysis-related genes, especially the PFKFB3 gene.
3.5. PFKFB3 Inhibition Alters Glycolysis and Abolished Pinocembrin-Induced Cardioprotection in Cardiomyocytes
Next, we explored whether blockade of PFKFB3 inhibits cardiomyocyte glycolysis and abolishes pinocembrin-afforded cardioprotective effects. As shown in Figures 6(a)–6(d), exposure of cardiomyocyte to 10 μM PFKFB3 inhibitor, 3PO remarkably reversed pinocembrin-enhanced glycolysis. What is more, inhibition of PFKFB3 resulted in a significant increase of cTnI release and LDH release post-H/R in vitro (Figures 6(e)–6(h)), suggesting that disruption of glucose metabolism leads to impaired cardioprotection of pinocembrin during H/R.
Figure 6.
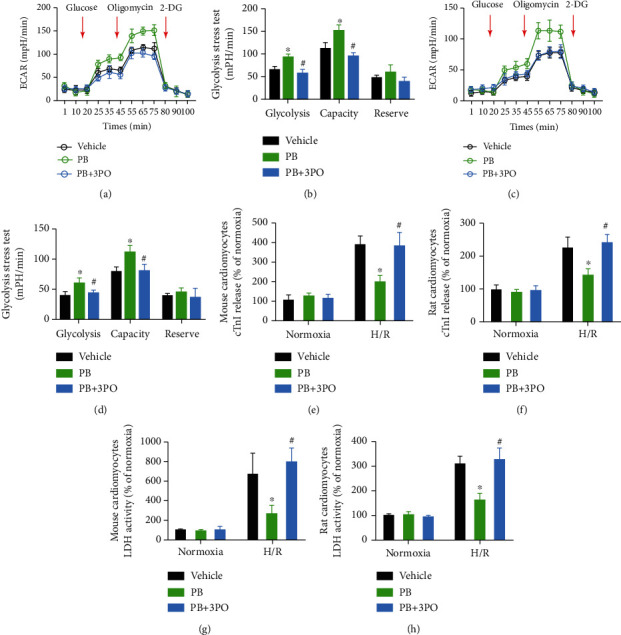
Pinocembrin protects cardiomyocytes from simulated I/R injury via enhancing glycolysis. (a–d) Treatment with PFKFB3 inhibitor, 3PO, results in a decline of glycolysis and glycolytic capacity in mouse and rat cardiomyocytes. n = 3/treatment in all Seahorse assays, each in triplicate. (e, f) Effect of PFKFB3 inhibitor, 3PO, on mouse and rat cardiomyocyte cTnI release subsequent to H/R. n = 5 each. (g, h) Effect of PFKFB3 inhibitor, 3PO, on mouse and rat cardiomyocyte LDH release subsequent to H/R. n = 5 each. Data represent the mean ± SEM. ∗P < 0.05 versus H/R with the vehicle group. #P < 0.05 versus H/R with the PB group.
3.6. PFKFB3 Deficiency in Normal Mice Using AAV9 Abolished Pinocembrin-Afforded Cardioprotective Effects after MI/R
Since pinocembrin alleviated H/R-induced cardiomyocyte death by upregulating glycolysis via PFKFB3, we therefore evaluated the roles of PFKFB3 on myocardial I/R injury in vivo. To determine whether PFKFB3 regulation is directly involved in pinocembrin-related MI/R injury improvement, the WT mice were injected with AAV9 encoding PFKFB3 shRNA to knockdown endogenous PFKFB3. Intravenous injection of AAV9-shPFKFB3 on mice has successfully reduced the protein level of PFKFB3 in the heart (Supplementary Figure 2). Subsequently, mice were insulted with in situ myocardial ischemia-reperfusion to assess the functional role of myocardial-specific PFKFB3 in pinocembrin-afforded cardioprotection. Myocardial injury was measured by infarct size area, serum cTnT levels, and LDH activity. Exposure of mice infected with AAV9-shPFKFB3 to MI/R presented larger infarct sizes and enhanced release of troponin I and LDH (Figures 7(a)–7(c)). More importantly, the deletion of myocardial PFKFB3 abolished pinocembrin-conferred protective effects on those indexes. Furthermore, the pinocembrin-improved EF and FS were also abolished by PFKFB3 deficiency with AAV9-shPFKFB3 (Figure 7(d)). Collectively, these data confirm that pinocembrin confers cardioprotective effects by enhancing cardiomyocyte glycolysis through activation of PFKFB3.
Figure 7.
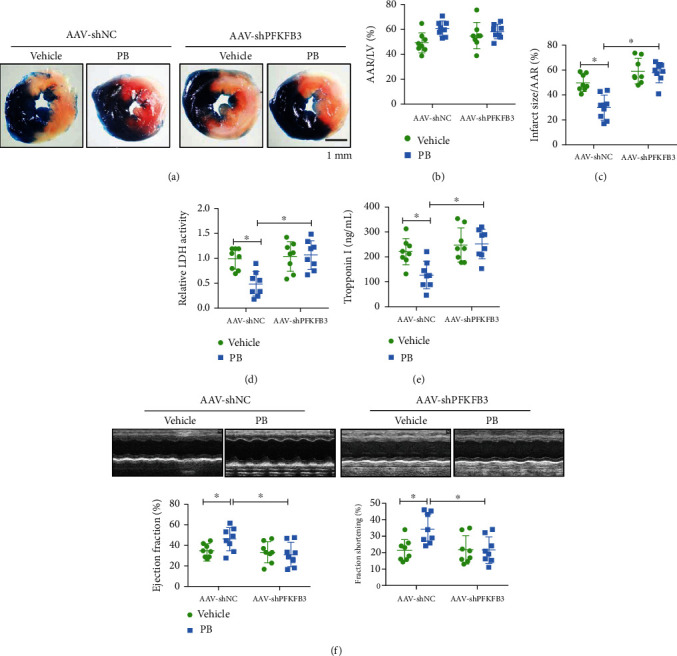
Knockdown of PFKFB3 in the myocardium using AAV9 impairs pinocembrin-induced cardioprotection after MI/R. (a) Representative infarct staining from AAV-shNC or AAV-shPFKFB3 mice treated with vehicle or pinocembrin. (b, c) AAR and infarct sizes from AAV-shNC or AAV-shPFKFB3 mice treated with vehicle or pinocembrin. (d, e) Plasma LDH activity and cTnI levels. (f) Representative echocardiographic images, assessment of LV EF and FS by echocardiography from AAV-shNC or AAV-shPFKFB3 mice treated with vehicle or pinocembrin. n = 8 each. Data represent the mean ± SEM. ∗P < 0.05, significantly different as indicated.
3.7. HIF1α Is a Key Transcription Factor Driving Pinocembrin-Induced PFKFB3 Expression
Pinocembrin notably promoted PFKFB3 gene expression in cardiomyocytes exposed to H/R (Figure 8(a)). To explore the underlying mechanisms responsible for the increased expression of PFKFB3 in cardiomyocytes induced by pinocembrin, we examined the activity of the mouse PFKFB3 promoter both in the absence or in the presence of pinocembrin. We transfected cardiomyocytes with a construct in which luciferase expression was driven by a 3,308 bp fragment from the mouse PFKFB3 promoter. As shown in Figure 8(b), pinocembrin by itself exerted little effect on PFKFB3 promoter activity, but it cooperated with H/R to increase luciferase activity (Figure 8(b)). Furthermore, we studied the DNA regions required for pinocembrin to increase PFKFB3 promoter activity by using a series of promoter constructs containing successive deletions from the 5′ end. As shown in Figure 8(c), 5′ deletion mutants revealed that sequences between -50 and -200 and -1108 and -2108 from the transcription start site are essential for the induction of the PFKFB3 gene promoter by pinocembrin.
Figure 8.
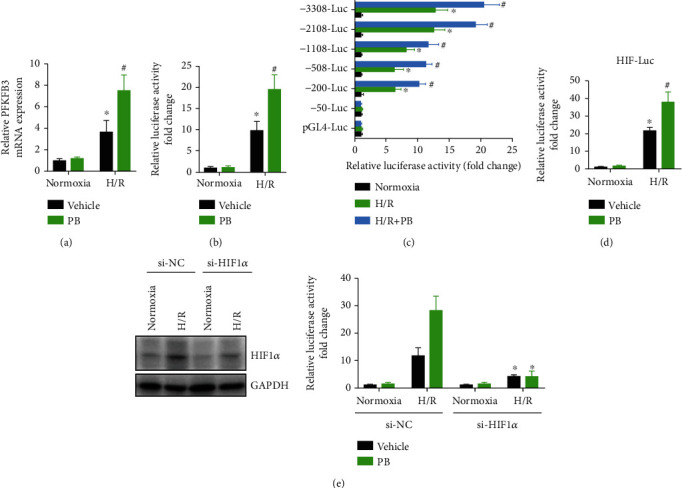
Pinocembrin promotes PFKFB3 expression through activating transcription factor HIF1α. (a) Cardiomyocytes were treated with pinocembrin and subsequent to H/R, and then, PFKFB3 expression was detected by qRT-PCR. ∗P < 0.05 versus normoxia with the vehicle group. #P < 0.05 versus H/R with the vehicle group. (b) Cardiomyocytes were transiently cotransfected with a pGL4-basic luciferase expression vector, containing 3,308 bp of the mouse PFKFB3 gene promoter. Cells were exposed to H/R with or without pinocembrin. Transfections were performed in triplicate. ∗P < 0.05 versus normoxia with the vehicle group. #P < 0.05 versus H/R with the vehicle group. (c) Cardiomyocytes were transiently transfected with pGL4-basic luciferase expression vectors containing various lengths of the PFKFB3 gene promoter 5′-flanking region, and cells were treated with pinocembrin subsequent to H/R. ∗P < 0.05 versus normoxia with the vehicle group. #P < 0.05 versus H/R with the vehicle group. (d) Transcriptional activity of reporter constructs carrying binding sites for HIF1α transfected in cardiomyocytes insulted with H/R. ∗P < 0.05 versus normoxia with the vehicle group. #P < 0.05 versus H/R with the vehicle group. (e) Analysis of PFKFB3 promoter activity in cardiomyocytes transfected with control, HIF1α-specific siRNA. n = 5 each. Data represent the mean ± SEM. ∗P < 0.05 versus corresponding si-NC group.
HIF1α is considered to be an important regulator of glycolysis, and its target genes include PFKFB3 and glucose transporter-1 (GLUT1) [27, 28]. Also, pinocembrin increased the expression of HIF1α, which are key transcription factors for PFKFB3 expression (data not shown). To identify transcription factor HIF1α that could modulate PFKFB3 induction by pinocembrin, reporter constructs carrying binding sites for HIF1 were transfected into cardiomyocytes, which were stimulated afterward with pinocembrin. The results showed that the activity of HIF1 reporter constructs increased upon activation with H/R, and this activity was enhanced by the pinocembrin (Figure 8(d)). Moreover, we observed that reduced expression of HIF1α with specific siRNA greatly decreased PFKFB3 induction by pinocembrin during H/R, corroborating its key role in pinocembrin-induced PFKFB3 expression (Figures 8(e) and 8(f)). Together, our data demonstrates that HIF1α is a key transcription factor driving pinocembrin-induced PFKFB3 expression during H/R.
4. Discussion
In the present study, we observed that glycolysis functionally mediated the cardioprotective responses of pinocembrin. We demonstrated that (i) pinocembrin delivered at the onset of reperfusion (postconditioning) significantly improved postischemic myocardial function and reduced infarct size after I/R ex vivo; (ii) pinocembrin significantly protected mouse hearts from acute myocardial I/R injury in vivo; (iii) these protections are partially related to enhanced glycolysis by pinocembrin in the I/R cardiomyocytes; and (iv) the cardioprotective effects of pinocembrin are mediated by the activation of PFKFB3. These results extend previous findings indicating the cardioprotection of pinocembrin against I/R injury and reveal the new mechanisms of pinocembrin in the cardioprotection.
It would be an attractive treatment principle to reduce the infarct size through pharmaceutical intervention to assist classic reperfusion intervention [29]. Pinocembrin is a potential cardiovascular drug with potential neuroprotective effects on transient and long-term ischemic stroke in rats [30]. Also, a previous study indicated that the administration of pinocembrin before myocardial ischemia improved LV function [7]. Pharmacological postconditioning is easier to implement and has therapeutically potential in both clinical and experimental settings, which avoids the potential injury induced by ischemic conditioning, and therefore has good clinical application prospects [31]. Therefore, our study aims to explore the cardioprotective effects of pinocembrin postconditioning. The ex vivo results showed that pinocembrin from 10 to 100 μM delivered at the first 5 minutes of reperfusion remarkably improved postischemic myocardial function and attenuates cell death in a concentration-dependent manner. Furthermore, the in vivo mouse myocardial I/R injury model was prepared and pinocembrin postconditioning was fulfilled by an intraperitoneal injection of pinocembrin (5 mg/kg and 10 mg/kg body weight) 5 min before reperfusion. Cardiac function, serum LDH activity and cTnT content, and infarct size were significantly improved with pinocembrin treatment. The cardioprotective effects of pinocembrin are consistent with other reports. This suggests that pinocembrin could be a candidate compound that may be used in the clinical treatment of myocardial infarction.
Various metabolic abnormalities occur during myocardial I/R, such as increased fatty acid oxidation and decreased glucose oxidation [32]. This phenomenon is related to the uncoupling of mitochondrial respiration, increased proton leakage, ROS formation, and, more importantly, increased myocardial oxygen consumption [33]. During myocardial I/R, the metabolic shift aimed at increasing glucose oxidation has proved to be beneficial. Although some therapeutic strategies have tried to reverse this metabolic imbalance, there is still no approved treatment regimen so far [34–36]. In addition, strategies aimed at increasing clinical glucose consumption have different results and have not yet reached routine clinical practice. Looking for drugs that can safely induce the transfer of cellular energy metabolism and a better understanding of the protective mechanisms of increased glucose oxidation may facilitate transfer to the clinic. Our results show that increasing glycolysis is responsible for pinocembrin-induced protective effects. This is consistent with previous studies that metabolic shift towards increased glycolysis protects the heart from I/R injury [13]. Moreover, PFKFB3 expression is notably upregulated during I/R, which directs cellular glucose metabolism from PPP to aerobic glycolysis [37]. Our data have shown that PFKFB3 was significantly upregulated with pinocembrin treatment and specific inhibitor of PFKFB3 abolished pinocembrin-afforded protective effects in cardiomyocytes. Most importantly, knockdown of PFKFB3 in the myocardium using AAV9 reversed the cardioprotection of pinocembrin postconditioning. Previous studies have shown that the main reason responsible for the protection offered by the metabolic shift is the fact that glycolysis is able to produce two molecules of ATP without the need for oxygen, so that uncoupling between mitochondrial full glucose oxidation and glycolysis leads to increased cardiac efficiency [38]. However, potential protective mechanisms of the metabolic shift are largely unknown. Further studies are needed to investigate how pinocembrin regulate the glycolysis and search for its putative downstream targets.
5. Conclusions
In summary, our findings demonstrate that compound pinocembrin exhibits significant protective effects on cardiac I/R injury when delivered at the beginning of or before reperfusion in ex vivo rat and in vivo mouse models through enhancing glycolysis. Pinocembrin may be considered as an effective lead compound for large animal experiments and is expected to be used in clinical research of acute myocardial infarction.
Acknowledgments
This work was supported by the Project of National Natural Science Foundation of China (grant numbers 81700354 and 81970229), Construction Project of Shanghai Key Laboratory of Molecular Imaging (18DZ2260400), Shanghai Municipal Education Commission (Class II Plateau Disciplinary Construction Program of Medical Technology of SUMHS, 2018-2020), the Outstanding Clinical Discipline Project of Shanghai Pudong Health Commission (No. PW2016D-13 and No. PWYgy2018-03), and the Shanghai Medical Key Specialty of Shanghai Health Commission (No. ZK2019B25).
Contributor Information
Xuefeng Gu, Email: guxf@sumhs.edu.cn.
Jingrong Lin, Email: jingrong.lin@163.com.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare no conflict of interest.
Authors' Contributions
ZYJ and GXF performed and analyzed the experiments. ZYJ wrote the first draft of the manuscript. WGQ and YB participated in the analysis of the experiments. LJR revised the manuscript. ZYJ and LJR contributed to the experimental design and revised the manuscript.
Supplementary Materials
Supplemental Figure 1: effects of pinocembrin on the heart rate and coronary flow (CF) in isolated rat hearts subjected to 30 min no-flow global ischemia followed by 45 min of reperfusion. Supplemental Figure 2: (A) schematic time scale and experimental strategy to evaluate the effect of PFKFB3 expression on pinocembrin-afforded cardioprotection. Adeno-associated virus of serotype 9 (AAV9) encoding nontarget (NC) or shRNA for PFKFB3 was injected intravenously into tail veins of mice (3∗1011 vg) at week 0. Four weeks later, sham or myocardial ischemia surgeries were conducted. (B) Protein levels of PFKFB3 in myocardium treated with control shRNA (AAV-shNC) or FetB shRNA (AAV-shPFKFB3). Data represent the mean ± SEM. ∗P < 0.05 versus indicated group. Supplemental Table 1: primers used in this study.
References
- 1.Bice J. S., Baxter G. F. Postconditioning signalling in the heart: mechanisms and translatability. British Journal of Pharmacology. 2015;172(8):1933–1946. doi: 10.1111/bph.12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng Y., Gu S., Li X., et al. Berbamine postconditioning protects the heart from ischemia/reperfusion injury through modulation of autophagy. Cell Death & Disease. 2017;8(2, article e2577) doi: 10.1038/cddis.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanieh H., Hairul Islam V. I., Saravanan S., et al. Pinocembrin, a novel histidine decarboxylase inhibitor with anti-allergic potential in in vitro. European Journal of Pharmacology. 2017;814:178–186. doi: 10.1016/j.ejphar.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Saad M. A., Abdel Salam R. M., Kenawy S. A., Attia A. S. Pinocembrin attenuates hippocampal inflammation, oxidative perturbations and apoptosis in a rat model of global cerebral ischemia reperfusion. Pharmacological Reports. 2015;67(1):115–122. doi: 10.1016/j.pharep.2014.08.014. [DOI] [PubMed] [Google Scholar]
- 5.Shi L. L., Chen B. N., Gao M., et al. The characteristics of therapeutic effect of pinocembrin in transient global brain ischemia/reperfusion rats. Life Sciences. 2011;88(11-12):521–528. doi: 10.1016/j.lfs.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 6.Tao J., Shen C., Sun Y., Chen W., Yan G. Neuroprotective effects of pinocembrin on ischemia/reperfusion-induced brain injury by inhibiting autophagy. Biomedicine & Pharmacotherapy. 2018;106:1003–1010. doi: 10.1016/j.biopha.2018.07.026. [DOI] [PubMed] [Google Scholar]
- 7.Lungkaphin A., Pongchaidecha A., Palee S., Arjinajarn P., Pompimon W., Chattipakorn N. Pinocembrin reduces cardiac arrhythmia and infarct size in rats subjected to acute myocardial ischemia/reperfusion. Applied Physiology, Nutrition, and Metabolism. 2015;40(10):1031–1037. doi: 10.1139/apnm-2015-0108. [DOI] [PubMed] [Google Scholar]
- 8.Zhang P., Xu J., Hu W., Yu D., Bai X. Effects of pinocembrin pretreatment on connexin 43 (Cx43) protein expression after rat myocardial ischemia-reperfusion and cardiac arrhythmia. Medical Science Monitor. 2018;24:5008–5014. doi: 10.12659/MSM.909162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gohil V. M., Sheth S. A., Nilsson R., et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nature Biotechnology. 2010;28(3):249–255. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopaschuk G. D., Ussher J. R., Folmes C. D. L., Jaswal J. S., Stanley W. C. Myocardial fatty acid metabolism in health and disease. Physiological Reviews. 2010;90(1):207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 11.Lionetti V., Stanley W. C., Recchia F. A. Modulating fatty acid oxidation in heart failure. Cardiovascular Research. 2011;90(2):202–209. doi: 10.1093/cvr/cvr038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malmberg K., Norhammar A., Wedel H., Ryden L. Glycometabolic state at admission: important risk marker of mortality in conventionally treated patients with diabetes mellitus and acute myocardial infarction: long-term results from the Diabetes and Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI) study. Circulation. 1999;99(20):2626–2632. doi: 10.1161/01.cir.99.20.2626. [DOI] [PubMed] [Google Scholar]
- 13.Nadtochiy S. M., Wang Y. T., Nehrke K., Munger J., Brookes P. S. Cardioprotection by nicotinamide mononucleotide (NMN): involvement of glycolysis and acidic pH. Journal of Molecular and Cellular Cardiology. 2018;121:155–162. doi: 10.1016/j.yjmcc.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeong D. W., Kim T. S., Cho I. T., Kim I. Y. Modification of glycolysis affects cell sensitivity to apoptosis induced by oxidative stress and mediated by mitochondria. Biochemical and Biophysical Research Communications. 2004;313(4):984–991. doi: 10.1016/j.bbrc.2003.12.033. [DOI] [PubMed] [Google Scholar]
- 15.Hunter A. J., Hendrikse A. S., Renan M. J. Can radiation-induced apoptosis be modulated by inhibitors of energy metabolism? International Journal of Radiation Biology. 2009;83(2):105–114. doi: 10.1080/09553000601121157. [DOI] [PubMed] [Google Scholar]
- 16.Vaughn A. E., Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nature Cell Biology. 2008;10(12):1477–1483. doi: 10.1038/ncb1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin C. X., May L. T., Li R., et al. Small-molecule-biased formyl peptide receptor agonist compound 17b protects against myocardial ischaemia-reperfusion injury in mice. Nature Communications. 2017;8(1) doi: 10.1038/ncomms14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irvine J. C., Cao N., Gossain S., et al. HNO/cGMP-dependent antihypertrophic actions of isopropylamine-NONOate in neonatal rat cardiomyocytes: potential therapeutic advantages of HNO over NO. American Journal of Physiology. Heart and Circulatory Physiology. 2013;305(3):H365–H377. doi: 10.1152/ajpheart.00495.2012. [DOI] [PubMed] [Google Scholar]
- 19.Irvine J. C., Ganthavee V., Love J. E., et al. The soluble guanylyl cyclase activator bay 58-2667 selectively limits cardiomyocyte hypertrophy. PLoS One. 2012;7(11, article e44481) doi: 10.1371/journal.pone.0044481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou Z., Qin X., Hu Y., et al. Longterm exercise-derived exosomal miR-342-5p: a novel exerkine for cardioprotection. Circulation Research. 2019;124(9):1386–1400. doi: 10.1161/CIRCRESAHA.118.314635. [DOI] [PubMed] [Google Scholar]
- 21.Kishi S., Campanholle G., Gohil V. M., et al. Meclizine preconditioning protects the kidney against ischemia-reperfusion injury. eBioMedicine. 2015;2(9):1090–1101. doi: 10.1016/j.ebiom.2015.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang L. L., Zhang D. M., Jiao R. Q., et al. Pterostilbene attenuates fructose-induced myocardial fibrosis by inhibiting ROS-driven Pitx2c/miR-15b pathway. Oxidative Medicine and Cellular Longevity. 2019;2019:25. doi: 10.1155/2019/1243215.1243215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie C., Zhang Y. P., Song L., et al. Genome editing with CRISPR/Cas9 in postnatal mice corrects PRKAG2 cardiac syndrome. Cell Research. 2016;26(10):1099–1111. doi: 10.1038/cr.2016.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Somanathan S., Jacobs F., Wang Q., Hanlon A. L., Wilson J. M., Rader D. J. AAV vectors expressing LDLR gain-of-function variants demonstrate increased efficacy in mouse models of familial hypercholesterolemia. Circulation Research. 2014;115(6):591–599. doi: 10.1161/CIRCRESAHA.115.304008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He X., Zeng H., Chen S. T., et al. Endothelial specific SIRT3 deletion impairs glycolysis and angiogenesis and causes diastolic dysfunction. Journal of Molecular and Cellular Cardiology. 2017;112:104–113. doi: 10.1016/j.yjmcc.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y., Liang X., Zhang G., Kong L., Peng W., Zhang H. Galangin and pinocembrin from propolis ameliorate insulin resistance in HepG2 cells via regulating Akt/mTOR signaling. Evidence-based Complementary and Alternative Medicine. 2018;2018:10. doi: 10.1155/2018/7971842.7971842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merighi S., Borea P. A., Stefanelli A., et al. A2a and a2b adenosine receptors affect HIF-1α signaling in activated primary microglial cells. Glia. 2015;63(11):1933–1952. doi: 10.1002/glia.22861. [DOI] [PubMed] [Google Scholar]
- 28.Obach M., Navarro-Sabate A., Caro J., et al. 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. The Journal of Biological Chemistry. 2004;279(51):53562–53570. doi: 10.1074/jbc.M406096200. [DOI] [PubMed] [Google Scholar]
- 29.Morel O., Perret T., Delarche N., et al. Pharmacological approaches to reperfusion therapy. Cardiovascular Research. 2012;94(2):246–252. doi: 10.1093/cvr/cvs114. [DOI] [PubMed] [Google Scholar]
- 30.Wu C. X., Liu R., Gao M., et al. Pinocembrin protects brain against ischemia/reperfusion injury by attenuating endoplasmic reticulum stress induced apoptosis. Neuroscience Letters. 2013;546:57–62. doi: 10.1016/j.neulet.2013.04.060. [DOI] [PubMed] [Google Scholar]
- 31.Heusch G. Treatment of myocardial ischemia/reperfusion injury by ischemic and pharmacological postconditioning. Comprehensive Physiology. 2015;5(3):1123–1145. doi: 10.1002/cphy.c140075. [DOI] [PubMed] [Google Scholar]
- 32.Fillmore N., Mori J., Lopaschuk G. D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. British Journal of Pharmacology. 2014;171(8):2080–2090. doi: 10.1111/bph.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boudina S., Abel E. D. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology. 2006;21(4):250–258. doi: 10.1152/physiol.00008.2006. [DOI] [PubMed] [Google Scholar]
- 34.Jaswal J. S., Keung W., Wang W., Ussher J. R., Lopaschuk G. D. Targeting fatty acid and carbohydrate oxidation--a novel therapeutic intervention in the ischemic and failing heart. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2011;1813(7):1333–1350. doi: 10.1016/j.bbamcr.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 35.Kantor P. F., Lucien A., Kozak R., Lopaschuk G. D. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circulation Research. 2000;86(5):580–588. doi: 10.1161/01.RES.86.5.580. [DOI] [PubMed] [Google Scholar]
- 36.Taniguchi M., Wilson C., Hunter C. A., Pehowich D. J., Clanachan A. S., Lopaschuk G. D. Dichloroacetate improves cardiac efficiency after ischemia independent of changes in mitochondrial proton leak. American Journal of Physiology-Heart and Circulatory Physiology. 2001;280(4):H1762–H1769. doi: 10.1152/ajpheart.2001.280.4.H1762. [DOI] [PubMed] [Google Scholar]
- 37.Li Z., Zhang B., Yao W., Zhang C., Wan L., Zhang Y. APC-Cdh1 regulates neuronal apoptosis through modulating glycolysis and pentose-phosphate pathway after oxygen-glucose deprivation and reperfusion. Cellular and Molecular Neurobiology. 2019;39(1):123–135. doi: 10.1007/s10571-018-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lopaschuk G. D. Metabolic modulators in heart disease: past, present, and future. The Canadian Journal of Cardiology. 2017;33(7):838–849. doi: 10.1016/j.cjca.2016.12.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: effects of pinocembrin on the heart rate and coronary flow (CF) in isolated rat hearts subjected to 30 min no-flow global ischemia followed by 45 min of reperfusion. Supplemental Figure 2: (A) schematic time scale and experimental strategy to evaluate the effect of PFKFB3 expression on pinocembrin-afforded cardioprotection. Adeno-associated virus of serotype 9 (AAV9) encoding nontarget (NC) or shRNA for PFKFB3 was injected intravenously into tail veins of mice (3∗1011 vg) at week 0. Four weeks later, sham or myocardial ischemia surgeries were conducted. (B) Protein levels of PFKFB3 in myocardium treated with control shRNA (AAV-shNC) or FetB shRNA (AAV-shPFKFB3). Data represent the mean ± SEM. ∗P < 0.05 versus indicated group. Supplemental Table 1: primers used in this study.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.