

Abstract
Objectives:
Colistin is a ‘last-line’ antibiotic used to treat multidrug-resistant Gram-negative bacteria, but colistin resistance has emerged. Colistin normally binds to the lipid A moiety on the bacterial outer membrane, where it then destroys the bacterial membrane. Mobilize colistin resistance (MCR, encoded by mcr-1 and others) is a phosphoethanolamine transferase that modifies lipid A, preventing colistin binding. We hypothesized that combining pore-forming AMPs and colistin will circumvent this mechanism and reduce the minimum inhibitory concentration (MIC) of colistin for both colistin- and multidrug-resistant Gram-negative bacteria.
Methods:
In vitro cultures were incubated for 18 h after combining bacteria (Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii and Pseudomonas aeruginosa) with serially diluted colistin and a fixed concentration of peptide MSI-78 or OTD-244.
Results:
When combined with either peptide, the colistin MIC decreased more than 4-fold for 88% of all tested isolates (n = 17; range, 4–64-fold reduction) and for 75% of colistin-resistant isolates (n = 8; range, 4–64-fold reduction). The concentrations used had no effect on red blood cells based on a conventional haemolysis assay.
Conclusions:
These findings are consistent with two membrane-damaging compounds having an additive effect on bacterial killing. Combining antimicrobial peptides with colistin is a promising strategy for bypassing MCR-mediated colistin resistance, but also for improving the susceptibility of other Gram-negative bacteria while potentially reducing the therapeutic concentration of colistin needed to treat infections.
Keywords: Antimicrobial peptides, Colistin, Multidrug-resistance
1. Introduction
Untreatable multidrug-resistant (MDR) infections have become increasingly common around the world [1]. In light of these challenges, physicians have begun using older antibiotics (e.g. colistin) to treat recalcitrant infections [2]. Colistin was first discovered in the 1940s and was used clinically in the 1950s for the treatment of Gram-negative infections. Use was largely discontinued, however, owing to reports of neuro- and nephrotoxicity and with increasing availability of less toxic classes of antibiotics, such as aminoglycosides [2]. With the emergence of MDR bacteria, such as Acinetobacter baumannii, Pseudomonas aeruginosa, Klebsiella pneumoniae [2] and Escherichia coli [3], colistin has been used increasingly as a ‘last-line’ antibiotic, despite the risk of toxic effects [3].
For bacteria that are susceptible to colistin, positively charged colistin binds the outer membrane via electrostatic interactions with the negatively charged lipid A phosphate group. After colistin binds, it damages the integrity of the proximal outer membrane, allowing colistin to diffuse into the periplasm, where it can then disrupt the inner membrane. This interaction disrupts the osmotic equilibrium of the cell and causes leakage of the bacterial cell components [4] and eventual formation of membrane pores that lead to cell lysis and death [5].
There are multiple mechanisms that convey resistance to colistin [6–8], but the most alarming is a plasmid-mediated, mobilizable colistin resistance (MCR) [6]. The mcr gene encodes a phosphoethanolamine transferase that alters 4′-phosphoethanolamine so that the lipid A moiety that normally binds colistin is changed from a negative to a neutral charge [4]. This change prevents colistin from binding, effectively preventing colistin from interacting with the inner membrane.
The unique mechanism of colistin activity—binding, diffusion and damage to the cytoplasmic membrane—suggests that if alternative pathways are available for colistin to reach the periplasm, then MCR-mediated resistance could become irrelevant. That is, if the outer membrane can be disrupted by an alternative mechanism, this may compensate for MCR-mediated changes to the cell membrane and allow colistin to reach the periplasm. One possible strategy is to permeabilize the outer membrane with antimicrobial peptides (AMPs). For example, MSI-78 (also called pexiganan) is an AMP modified from magainin-2 [9,10]. It is a potent antibacterial peptide that forms pores in the outer membrane, but its use has been limited by toxicity to host cells when used at high concentrations. Similarly, human β-defensin-2 is a membrane-disrupting peptide that was originally identified as part of the human innate immune system [11]. Human β-defensin-2 acts by binding the negatively charged phospholipids, causing hyperpolarization and inducing efflux of intracellular components, eventually leading to cell death [12,13].
For the current study, we combined either MSI-78 or a modified human β-defensin-2 (OTD-244) with colistin to test the hypothesis that these combinations can circumvent MCR-mediated resistance. Human β-defensin-2 was modified by adding a glycine and a serine to the N-terminal glycine. We found that relatively low concentrations of either peptide, when combined with colistin, lowered the MIC of colistin against subsets of colistin-resistant and colistin-sensitive strains of bacteria. Consequently, not only was our hypothesis supported, but the findings suggest that combining AMPs with colistin may reduce the therapeutic concentration of both colistin and the peptides that would otherwise be needed to fight Gram-negative bacterial infections. Such a reduction might permit effective control of infections at concentrations of colistin and peptides that are less toxic to people.
2. Methods
2.1. Bacterial strains
Seventeen Gram-negative isolates from the US Food and Drug Administration (FDA) and Centers for Disease Control and Prevention (CDC) Antimicrobial Resistance Isolate Bank were used [14]. These included six isolates that harbour mcr-1 and one mcr-2-positive isolate (Table 1). All selected isolates came from clinical cases.
Table 1.
Isolates used in this study with genotypes and minimum inhibitory concentrations (MICs) for MSI-78, OTD-244 and colistin.
| Isolate | Antibiotic resistance genesa | MIC (μg/mL)c |
||
|---|---|---|---|---|
| MSI-78 | OTD-244 | Colistind | ||
| Escherichia coli AR bank no. 0346b | mcr-1, blaCMY-2, blaCTX-M-55, aad1_5, rmtB, strA, strB, fosA, mph(A), catA1, sul1, sul2, dfrA17 | 2.5 | >500 | 1.25 |
| Escherichia coli AR bank no. 0349b | mcr-1, blaCTX-M-14, bla CTX-M-55, blaTEM-B, aph(4)-Ia, strA, strB, fosA, mph(A), tet(A), dfrA14 | 5 | >500 | 1.25 |
| Escherichia coli AR bank no. 0350b | mcr-1 | 5 | >500 | 1.25 |
| Escherichia coli AR bank no. 0493b | mcr-1 | 2.5 | >500 | 1.25 |
| Escherichia coli AR bank no. 0494b | mcr-1 | 2.5 | >500 | 2.5 |
| Escherichia coli AR bank no. 0495b | mcr-1 | 5 | >500 | 1.25 |
| Escherichia coli AR bank no. 538b | mcr-2 | 5 | >500 | 1.25 |
| Escherichia coli AR bank no. 0061 | blaKPC-3, bla OXA-9, blaTEM-1, strB, aadA2, aadA1, strA, aac(6′)-Ib, sul1, sul3, sul2, tet(A), dfrA12, dfrA14 | 2.5 | >500 | 0.0625 |
| Escherichia coli AR bank no. 0089 | blaCMY-2, strB, strA, sul2, tet(B) | 2.5 | >500 | 0.0625 |
| Klebsiella pneumoniae AR bank no. 0497b | mcr-1 | 10 | >500 | 5 |
| Klebsiella pneumoniae AR bank no. 0145 | blaNDM-1, blaOXA-9, blaTEM-1A, blaCTX-M-15, blaSHV-11, blaOXA-1 OmpK35, aadA1, oqxA,QnrS1, catB3, sul1 | >10 | >500 | 2.5 |
| Acinetobacter baumannii AR bank no. 0035 | blaTEM-1D, blaADC-25, blaOXA-66, blaOXA-72, strB, aph(3′)-Ic, strA, mph(E), msr(E), sul2 | 2.5 | >500 | <0.016 |
| Acinetobacter baumannii AR bank no. 0078 | blaADC-25, blaSHV-5, blaOXA-71, aac(3)-IIa, aadA11, mph(E), msr(E), sul1, dfrB1 | 2.5 | >500 | <0.016 |
| Acinetobacter baumannii AR bank no. 0083 | blaNDM-1, blaPER-7, blaOXA-23, blaOXA-69, aph(3′)-Ic, armA, strA, mph(E), msr(E), cmLA1, ARR-3, sul1, sul2, tet(B), dfrA1 | 2.5 | >500 | <0.016 |
| Pseudomonas aeruginosa AR bank no. 0054 | blaVIM-4, blaOXA-50, blaPAO, strB, aadB, strA, catB7, tet(A) | 10 | >500 | 0.125 |
| Pseudomonas aeruginosa AR bank no. 0353 | blaGES-1, blaPDC-19A | 5 | >500 | 0.125 |
| Pseudomonas aeruginosa AR bank no. 0356 | blaKPC-2, blaPDC-42 | 5 | >500 | 0.0625 |
Genotypes from Centers for Disease Control Antimicrobial Resistance Isolate Database (https://wwwn.cdc.gov/arisolatebank/).
Isolates harbouring either the mcr-1 or mcr-2 resistance gene.
MIC is defined by the concentration of antibiotic or peptide at which bacteria culture growth after 18 h incubation (37 °C) remains <0.1 below the background optical density (600 nm).
Colistin MIC values can differ based on the type of plastic microplate used, treatment method and manufacturer [17,18]. Note that when this panel of strains was tested using a different plate type, the MIC values for 15/17 isolates were within one serial dilution of values reported in the CDC Antimicrobial Resistance Isolate Bank (Supplementary Table S1).
2.2. Antibiotics and antimicrobial peptides
We used colistin sulfate from Research Products International (Mount Prospect, IL; lot no. 32466-38298). The MSI-78 was a synthesized peptide prepared by Mimotopes and has the sequence H-GIGKFLKKAKKFGKAFVKILKK-OH. The modified human β-defensin-2, labelled here as Optide 244 (OTD-244), was made by the Fred Hutchison Cancer Research Center described by Crook et al. [15]. The amino acid sequence of OTD-244-2 is GSGIGDPVTCLKSGAICHPVFCPRRYKQIGTCGLPGTKCCKKP.
2.3. Minimum inhibitory concentration assay
A microdilution assay was used to find the minimum inhibitory concentration (MIC) of each antibiotic or AMP against each isolate. Here, we define the MIC as the concentration of drug that inhibited growth, as measured by an OD600, to be equivalent to the sterility control (<0.1 after 18 h). The results of the MIC assay for each organism were determined by making 2-fold serial dilutions of each drug at varying concentrations (in Luria Broth [LB]).
We recovered frozen stock by streaking isolates onto LB agar plates and incubating overnight at 37 °C. We then plated one isolated colony onto antibiotic-containing plates. For isolates harbouring mcr, we used colistin plates at 4 μg/mL and, for all other isolates, we used carbenicillin plates at 8 μg/mL. From the antibiotic selection plates, we used one well-isolated colony to make an overnight culture, shaking at 37 °C in 3 mL of LB. We diluted overnight cultures 1:4 into new media and grew for 1 h shaking at 37 °C. We diluted these bacterial subcultures from ~0.8 OD600 to 1:10,000. The diluted cultures were then added to the media with treatment (colistin, peptide or both) in 100-well honeycomb plates. The plates were incubated in a Labsystems Bioscreen C plate reader (Elsichrom AB, Knivsta, Sweden) for 18 h, taking the OD600 measurement every hour at 37 °C (shaking for 5 min before each reading). Leftover cultures were diluted, and 100 μL was plated onto LB agar and grown at 37 °C overnight to ensure that the OD measurements reflected a consistent number of colony-forming units (CFU) across isolates. After 18 h of incubation, we transferred culture (5 μL) from wells onto LB agar plates with no antibiotics. After overnight incubation (37 °C), if no bacteria grew below the estimated MIC concentrations (or higher), the treatment was considered bactericidal. Every experiment included bacterial culture only (growth control), and LB only (sterility/background control). At least two independent experiments were completed for each isolate, with each treatment having at least two technical replicates.
Because the type of plastic used can influence MIC estimates for colistin [16,17], we verified that mcr-1–positive strains used in this study had similar MIC values, as indicated by the CDC. Assays were described as above, except 96-well, U-bottom microplates were used (Greiner, Millipore Sigma, Burlington, MA, USA), and the dilution series ranged from 16 to 0 μg/mL colistin. Plates were incubated at 37 °C, not shaking, for ~18 h. Aliquots (5 μL) were taken from each well, plated onto LB agar plates and grown at 37 °C overnight. Plates were examined for growth, and the lowest treatment concentration with no growth was considered the MIC. Three independent assays were completed for each isolate; each included two technical replicates.
2.4. Combinatorial assays
Combinatorial assays were performed with a fixed concentration of MSI-78 or OTD-244 combined with 2-fold serial dilutions of colistin. Initial concentrations for MSI-78 or OTD-244 were set at half the MIC value, as measured for each strain used in this study. In addition to half the MIC, we used a checkerboard assay to select additional concentrations above and below half the MIC that reflected the greatest combinatorial effect (data not shown). Controls included MSI-78 or OTD-244 only, bacterial culture only (growth control) and LB only (sterility/background control). The plates were incubated in a Labsystems Bioscreen C plate reader for 18 h, taking the OD600 every hour at 37 °C and shaking for 5 min before each reading.
Statistical analysis was done using PRISM version 8 (GraphPad Systems, San Diego, CA). Significant fold reduction was found by first normalizing OD600 values to remove the background absorbance (OD600 at hour 0 - OD600 at hour 18). Next, we determined the MIC as OD600 ≥ 0.1 μg/mL. We then determined if there was a significant difference in OD600 values at any given concentration below the MIC for colistin with either MSI-78 or OTD-244 versus colistin alone by using multiple Student t tests with a two-stage, step-up method of the Benjamini, Krieger and Yekutieli false discovery method (Q = 5%) [18]. Once we identified a significant difference, we then calculated the fold reduction by identifying the number of concentrations for which there were significant differences between the OD600 values for colistin with either MSI-78 or OTD-244 and colistin alone. Because we used 2-fold dilutions, we used the formula 2n, where n is the number of concentrations tested with significant differences. We considered the fold reduction to be biologically significant if 2n ≥ 4.
2.5. Haemolysis assay
The haemolysis assay was adapted from Monteiro et al. [19]. Defibrinated sheep blood (100 mL; <2 weeks old; Hardy Diagnostics, Santa Maria, CA) was centrifuged (500 × g at 4 °C) and resuspended in 10 mL of phosphate buffered saline (PBS). This process was repeated three times before red blood cells (RBCs) were enumerated by using a haemocytometer and diluted to 1 × 109 RBC/mL in PBS. We aliquoted 100 μL of the diluted RBCs into each well of a 96-well white polystyrene plate (Thermo Fisher Scientific, Waltham, MA). The plate was centrifuged at 500 g at 4 °C, and the supernatant was aspirated from every well. We then added 100 μL of each treatment to the RBC pellet. We used 100 μL of PBS as a negative control and 100 μL of 0.1% Triton X-100 as a positive control. The cells were then incubated at 37 °C for 1 h. Supernatant was then transferred into a flat-bottomed, clear, 96-well plate. We then read the OD450 in a plate reader (Tecan Spark, Tecan Group, Mannedorf, Switzerland). The readings were normalized using the equation ([treatment – negative control]/[positive control – negative control] × 100) to get the percentage of haemolysis in each well. Two independent experiments were performed with duplicates.
3. Results
3.1. Bacterial isolates
We tested 17 Gram-negative isolates that originated from the FDA-CDC Antimicrobial Resistance Isolate Bank [14]. All 17 isolates (4 species) have been sequenced at least partially, and antibiotic resistance genes were previously identified (Table 1). Seven isolates harboured mcr-1, and one isolate harboured mcr-2. The remaining nine isolates were resistant to other classes of antibiotics (Table 1).
3.2. Minimum inhibitory concentration
The MIC was defined as the concentration of inhibiting compound that limited the optical density (600 nm) of inoculated culture media to <0.1 after an 18-h incubation. The colistin MIC in 100-well honeycomb plates for strains lacking mcr-1 and mcr-2 ranged from <0.016 to 2.5 μg/mL, whereas strains harbouring these resistance genes had MIC values from 1.25 to 5 μg/mL (Table 1). The MIC results in 96-well Greiner plates ranged from 1 to 8 μg/mL (Supplementary Table S1). The MIC for MSI-78 ranged from 2.5 μg/mL(least resistant) to >10 μg/mL (most resistant). The MIC for OTD-244 for all organisms was >500 μg/mL, consistent with limited antimicrobial activity against the tested organisms. No bacteria were recoverable after 18 h of treatment at the recorded MIC concentration for colistin or MSI-78.
3.3. Colistin MIC reduced when combined with antimicrobial peptides
For these experiments, the concentration of MSI-78 or OTD-244 that we used ranged from 1.25 to 4.25 μg/mL or 25 to 50 μg/mL, respectively (see Section 2). It had been shown previously that colistin MIC values can vary based on the type of plastic used in the assay. Because of this, we chose to compare fold change rather than specific MIC values (Table 1, Supplementary Table S1). When colistin was used in combination with MSI-78 (1.25 μg/mL), there was a profound 64-fold reduction in the colistin MIC for mcr-1-positive E. coli isolate 346 (Fig. 1A), but the reduction for other strains varied from none to 32-fold (Table 2). Representative examples for different species included MDR K. pneumoniae isolate 145, for which combining colistin with 3.5 μg/mL MSI-78 resulted in a 4-fold reduction in colistin MIC compared with colistin alone (Fig. 1B). A 4-fold reduction was also evident for MDR P. aeruginosa isolate 356 when 3.25 μg/mL of MSI-78 was combined with colistin (Fig. 1C). E. coli isolate 349, harbouring mcr-1, showed only a 2-fold MIC reduction when colistin was combined with 1.25 μg/mL of MSI-78 (Fig. 1D). When used in combination with OTD-244 at 50 μg/mL, there was a 64-fold reduction in the colistin MIC for A. baumannii isolate 83 (Fig. 2A), whereas A. baumannii isolate 35 showed a 16-fold reduction (Fig. 2B). The colistin MIC for E. coli isolate 495, positive for mcr-1, did not improve when OTD-244-2 was combined with colistin (Fig. 2C).
Fig. 1.
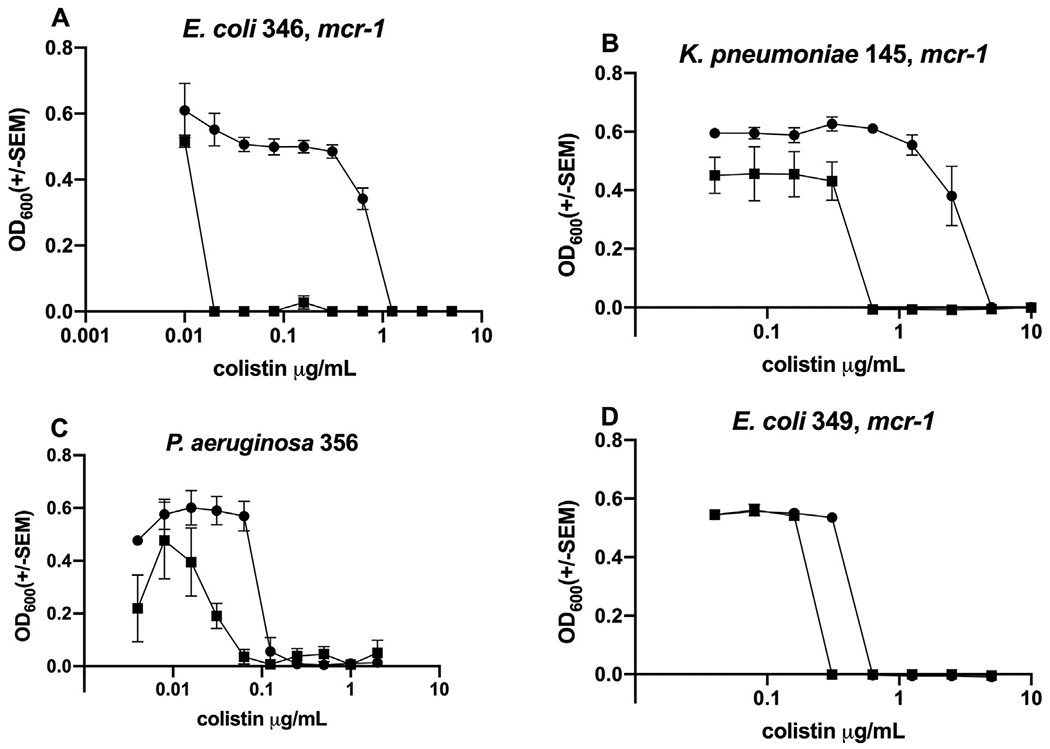
Representative endpoint concentration curves comparing the effects of colistin alone (solid circle) versus colistin combined with MSI-78 (solid squares; see also Table 2). Tested strains show how the addition of MSI-78 reduces the MIC for colistin as follows: (A) 64-fold; (B) 4-fold; (C) 4-fold; (D) 2-fold. SEM, Standard error of the mean.
Table 2.
Fold reduction in the MIC when comparing colistin alone vs. colistin with MSI-78 or colistin with OTD-244.
| Species and CDC ID no.a | MSI-78 [μg/mL] | MIC colistin with MSI-78 | Fold reductionc | OTD-244 [μg/mL]b | MIC colistin with OTD-244 | Fold reductionc |
|---|---|---|---|---|---|---|
| Escherichia coli AR bank no. 0346 | 1.25 | 0.01 | 64 | 25 | 0.08 | 4 |
| Escherichia coli AR bank no. 0349d | 1.25 | 0.16 | 2 | 50 | 0.08 | 4 |
| Escherichia coli AR bank no. 0350d | 2 | ≤0.04 | 32 | 25 | 0.16 | 2 |
| Escherichia coli AR bank no. 0493d | 1.75 | ≤0.04 | 32 | 50 | 0.16 | 2 |
| Escherichia coli AR bank no. 0494d | 2 | 1.25 | 2 | 50 | 2.5 | 1 |
| Escherichia coli AR bank no. 0495d | 2 | 0.08 | 4 | 50 | 0.08 | 2 |
| Escherichia coli AR bank no. 538d | 1.75 | 0.04 | 16 | 50 | 1.25 | 1 |
| Escherichia coli AR bank no. 0061 | 2 | 0.004 | 16 | 50 | 0.004 | 16 |
| Escherichia coli AR bank no. 0089 | 2.5 | 0.03 | 4 | 50 | 0.01 | 8 |
| Klebsiella pneumoniae AR bank no. 0497d | 3.5 | 2.5 | 1 | 50 | 2.5 | 1 |
| Klebsiella pneumoniae AR bank no. 0145 | 3.5 | 0.31 | 4 | 50 | 0.04 | 8 |
| Acinetobacter baumannii AR bank no. 0035 | 1.25 | 0.016 | 1 | 50 | 0.001 | 16 |
| Acinetobacter baumannii AR bank no. 0078 | 1.25 | 0.016 | 1 | 50 | 0.0005 | 16 |
| Acinetobacter baumannii AR bank no. 0083 | 1.25 | 0.016 | 1 | 50 | 0.0005 | 64 |
| Pseudomonas aeruginosa AR bank no. 0054 | 4.25 | 0.008 | 16 | 50 | 0.125 | 1 |
| Pseudomonas aeruginosa AR bank no. 0353 | 3.0 | 0.031 | 4 | 50 | 0.125 | 1 |
| Pseudomonas aeruginosa AR bank no. 0356 | 3.25 | 0.016 | 4 | 50 | 0.0625 | 1 |
See Table 1 for isolate information; CDC, Centers for Disease Control and Prevention.
Fixed concentration used for MSI-78 or OTD-244 was found by starting combination treatments with half the MIC (see Table 1).
Values in bold are considered a significant fold reduction. See Section 2.
Isolates positive for either mcr-1 or mcr-2.
Fig. 2.
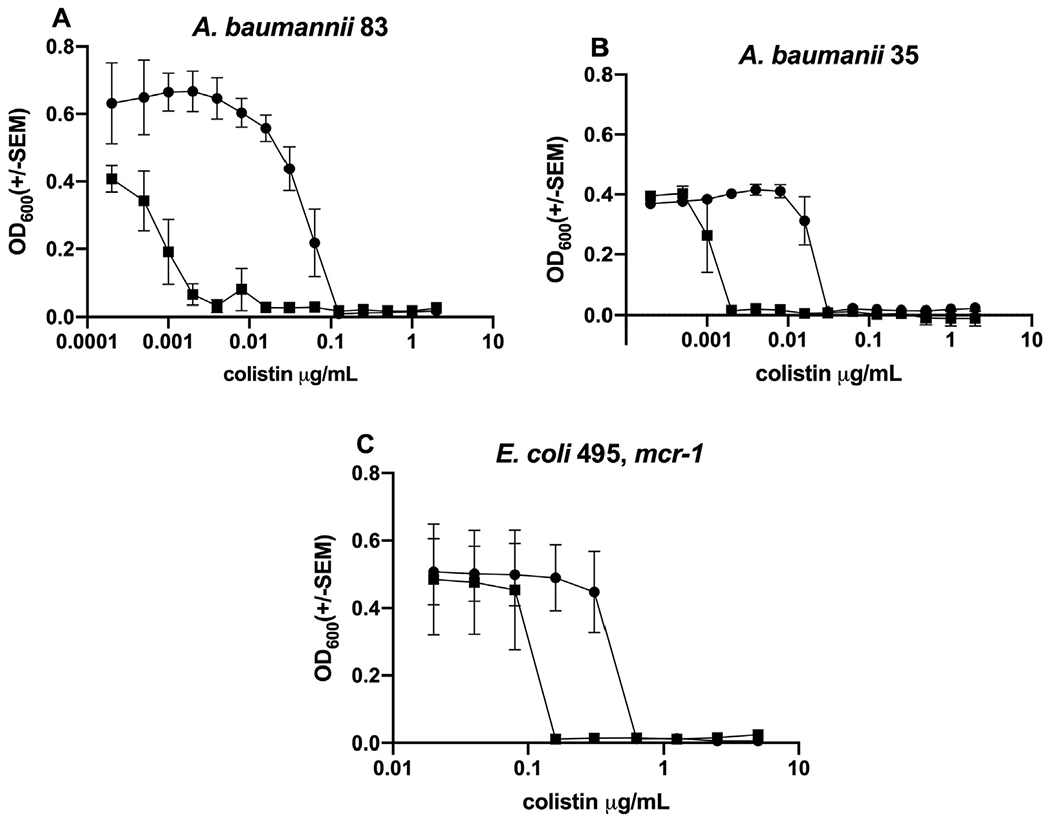
Representative endpoint concentration curves comparing the effects of colistin alone (solid circle) versus colistin combined with OTD-244 (solid squares; see also Table 2). Tested strains show how the addition of OTD-244 reduces the MIC for colistin as follows: (A) 64-fold; (B) 16-fold; (C) 2-fold.
Overall, 15/17 (88.2%) isolates showed significant (≥4-fold) reductions in MIC when colistin and an AMP were used jointly (Table 2). A reduction was observed for 11/17 (64.7%) isolates when MSI-78 and colistin were combined and for 8/17 (47.1%) isolates when OTD-244 and colistin were combined. The MIC for colistin-resistant isolates was reduced significantly in five of eight (62.5%) cases, and for OTD-244 and colistin there was a significant reduction in two of eight (25%) cases (Table 2, Fig. 3). Overall, the MIC for colistin alone included eight strains (47.1%) that had an MIC <1 μg/mL (Table 1), which is clinically applicable for most patients [20]. When combined with an AMP tested here, 94.1% of the tested isolates met this clinical threshold for colistin efficacy.
Fig. 3.
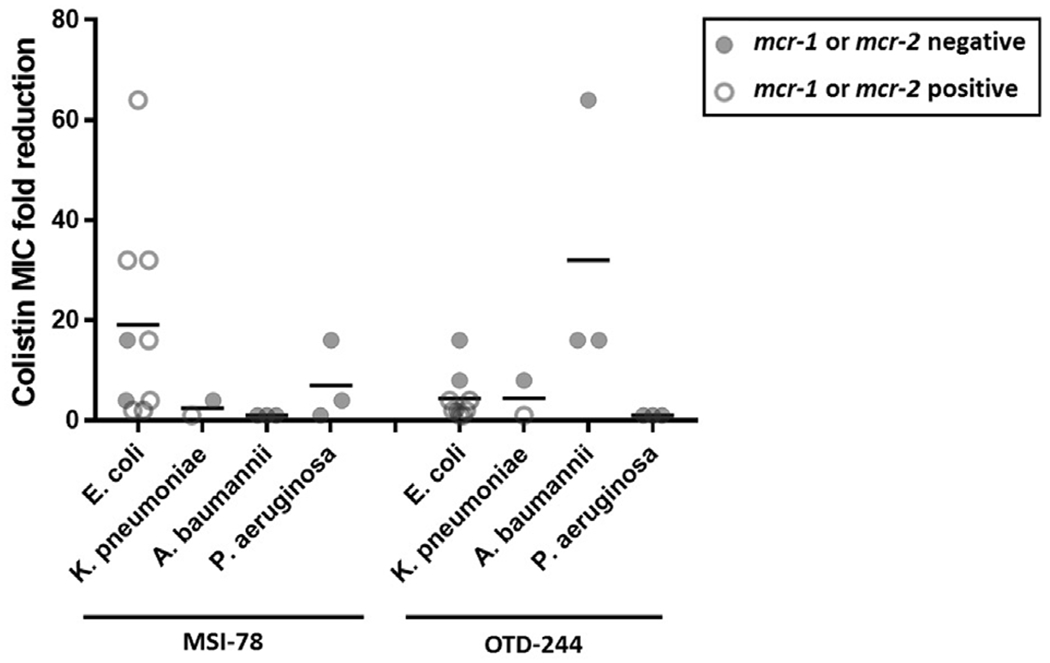
Summary of MIC-fold reduction results (Table 2) for mcr-1- or mcr-2-positive strains (open circles) and mcr-1- and mcr-2-negative strains (closed circles).
3.4. Lower concentrations translate into reduced toxicity by haemolysis assay
Haemolysis of RBCs is commonly used as a first assessment of potential toxicity of membrane-damaging compounds; colistin causes haemolysis at concentrations well above 1 μg/mL with this assay (Fig. 4A). The OTD-244 peptide exhibited no haemolysis, even when RBCs were exposed to 512 μg/mL (Fig. 4B). MSI-78 appears far more toxic (>50% haemolysis with 200 μg/mL; Fig. 4C), but when used at concentrations tested here that have additive effects with colistin (1.25–10 μg/mL), there was no evidence for haemolytic effects against RBCs (Fig. 4D).
Fig. 4.
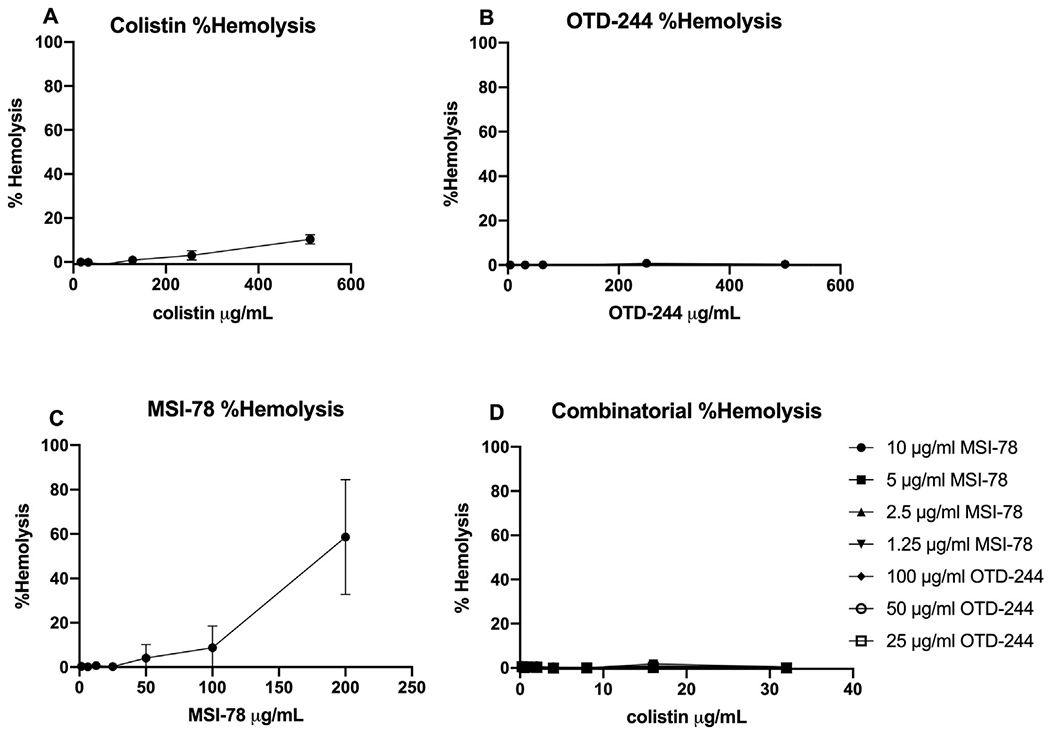
Percentage haemolysis of sheep red blood cells of colistin, OTD-244, MSI-78 and combination after 1 h incubation at 37 °C. Shown is the percentage haemolysis of each compound ± the standard error of the mean. (A) Colistin; LD50 of red blood cells not found. (B) OTD-244; LD50 of red blood cells not found. (C) MSI-78; LD50 of red blood cells, 122.52. (D) Combination of colistin and antimicrobial peptides at representative concentrations used in this study; LD50 of red blood cells not found.
4. Discussion
With the emergence of colistin-resistant organisms in combination with multidrug resistance, monotherapy may in many cases no longer be a sustainable option [21]. By using new antimicrobial compounds, particularly in the absence of genetically encoded resistance, it may be possible to treat otherwise recalcitrant colistin-resistant and other MDR infections successfully. Furthermore, combining new antimicrobials with last-line antibiotics might thwart the rapid increase in antibiotic resistance, given that there is no evidence of cross-resistance between AMPs and medically important antibiotics [22]. For the current study, combining colistin with an AMP produced significant reductions in the MIC (>4-fold) for most strains tested. The combinatorial effect worked with concentrations of MSI-78 and OTD-244 that produced no haemolysis of red blood cells while allowing the colistin concentration to be reduced by more than 100-fold in some cases.
Used alone, MSI-78 is more potent against every organism tested compared with OTD-244. This difference is probably as a result of the manner in which each peptide interacts with the cell membrane [22]. MSI-78 has a higher isoelectric point compared with OTD-244 (10.9 and 9.3, respectively [24]). Such a difference might lead to greater avidity between MSI-78 and bacterial phospholipids, thus creating more pores. MSI-78 combined with colistin appears to kill both mcr-positive bacteria and MDR bacteria equally, whereas OTD-244 combined with colistin was more effective against MDR bacteria compared with mcr-positive bacteria. When combined with colistin, OTD-244 was particularly effective against MDR A. baumannii strains that can be particularly problematic nosocomial infectious agents [25].
Additional studies may address why some organisms respond to an MSI-78 and colistin combination but not to OTD-244 and colistin, or vice versa. Some reasons might include the size of the molecules and the quaternary structures that they form. MSI-78 may bind more effectively than OTD-244 to bacterial membranes due to its smaller size and structure [9,26–28]. MSI-78 is approximately 2.5 kDa in size, whereas OTD-244 is much larger, 4.5 kDa [24]. In addition, the shape of the peptide might alter how well the peptides can bind and create pores leading to cell death [23]. MSI-78 forms an α-helical structure when it comes into contact with the membrane and forms dimers when creating pores [9,26–28]. OTD-244 creates pores by making octamers, of which each monomer contains three antiparallel β sheets and one α helix [26,28]. The traits that enhance the antibacterial activity of MSI-78 may also explain the greater potential toxicity of this peptide.
The advantage of using AMPs that interact with negatively charged phospholipids is that compared with conventional antibiotics, their mechanism of action makes the development of complete resistance difficult [29–31]. Guo et al. [32] suggested that the modification of lipid A could produce resistance to a wide range of AMPs but, in the case of MCR-mediated changes, this is not a universal resistance mechanism. For example, we found that when used alone, MSI-78 susceptibility was unchanged for mcr-positive strains that presumably undergo lipid A modification. We believe that this is because MSI-78 is highly cationic and can bind to many negatively charged phospholipids on the bacterial membrane, rather than being restricted to a specific binding interaction, as is the case for colistin [19]. Human defensins, from which OTD-244 was derived, behave in a similar manner, being highly cationic and relying on electrostatic interactions to form pores in the bacterial membranes. Nevertheless, mcr-positive strains were less vulnerable to colistin combined with OTD-244 as compared with when colistin is combined with MSI-78. This finding suggests that modification to the lipid A moiety might provide some protection against some AMPs. The specific mechanism of binding to the bacterial membrane likely varies for different peptides; thus, changes to lipid A and other membrane moieties are unlikely to provide universal resistance to all AMPs. Notably, when used in conjunction with colistin, the overall concentrations of AMP and colistin can be reduced, which likely reduces potential toxicological complications while also reducing the overall selection pressure for resistant infectious organisms. Based on data from the antimicrobial peptide database, there are over 3,200 verified AMPs and an estimated 1.3 million natural AMPs in existence [33,34]. The research shown here supports the need for broader screening of natural and modified AMPs for potential combinations with antibiotics currently in use and for other potentially therapeutic AMP cocktails.
Although AMPs occur naturally in nature, it is possible for them to be synthesized on an industrial scale, as would be needed for them to be potential drug candidates. There are several possible ways to make AMPs in large quantities, such as using recombinant production or solid-phase synthesis [35,36]. Solid-phase synthesis first came into use in 1963 and is a well-established method for producing peptides; it is currently used for the synthesis of several therapeutic peptides. Some significant advantages to using this method is that it is fast and easily scalable, and the products can be purified very easily by techniques such as high-performance liquid chromatography (HPLC). However, this method can be costly. In contrast with the already established solid-phase method, the recombinant method still needs more development in terms of scaling up production. The advantage to using the recombinant method is that it is cost-effective and a sustainable way of producing AMPs. However, this method has its limitations in that the scalability needs to be refined [35]. Nevertheless, it is important to note that new technologies are being studied to consider AMPs as an alternative therapeutic market.
Supplementary Material
Acknowledgments
Funding
This publication was supported in part by Project Violet at the Fred Hutchinson Cancer Research Center, United States of America, the National Institutes of Health/National Institute of General Medical Sciences through institutional training grant award T32-GM008336, United States of America, and the College of Veterinary Medicine, Washington State University, United States of America.
Footnotes
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethical approval
This was not required.
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.jgar.2020.05.013.
References
- [1].Laxminarayan R, Duse A, Wattal C, Zaidi AKM, Wertheim HFL, Sumpradit N, et al. Antibiotic resistance—the need for global solutions. Lancet Infect Dis 2013;13:1057–98. [DOI] [PubMed] [Google Scholar]
- [2].Lim LM, Ly N, Anderson D, Yang JC, Macander L,Jarkowski A 3rd, et al. Resurgence of colistin: a review of resistance, toxicity, pharmacodynamics, and dosing. Pharmacotherapy 2010;30:1279–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Brennan-Krohn T, Pironti A, Kirby E. Synergistic activity of colistin-containing combinations against colistin-resistant Enterobacteriaceae. Antimicrob Agents Chemother 2018;62: e00873–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Vidaillac C, Benichou L, Duval RE. Acinetobacter baumanniiIn vitro synergy of colistin combinations against colistin-resistant, Pseudomonas aeruginosa, and Klebsiella pneumoniae isolates. Antimicrob Agents Chemother 2012;56:4856–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gao R, Hu Y, Li Z, Sun J, Wang Q, Lin J, et al. Dissemination and mechanism for the MCR-1 colistin resistance. PLoS Pathog 2016;12:1–19, doi: 10.1371/journal.ppat.1005957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang X, Wang Y, Zhou Y, Li J, Yin W, Wang S, et al. Emergence of a novel mobile colistin resistance gene, mcr-8, in NDM-producing Klebsiella pneumoniae. Emerg Microbes Infect 2018;7:1–9, doi: 10.1038/s41426-018-0124-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Adams MD, Nickel GC, Bajaksouzian S, Lavender H, Murthy AR, Jacobs MR, et al. Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrob Agents Chemother 2009;53:3628–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cannatelli A, D’Andrea MM, Giani T, Di Pilato V, Arena F, Ambretti S, et al. In vivo emergence of colistin resistance in Klebsiella pneumoniae producing KPC-type carbapenemases mediated by insertional inactivation of the PhoQ/PhoP mgrB regulator. Antimicrob Agents Chemother 2013;57:5521–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gottler LM, Ramamoorthy A. Structure, membrane orientation, mechanism, and function of pexiganan—a highly potent antimicrobial peptide designed from magainin. Biochim Biophys Acta 2009;1788:1680–6, doi: 10.1016/j.bbamem.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Matsuzaki K, Sugishita K, Harada M, Fujii N, Miyajima K. Interactions of an antimicrobial peptide, magainin 2, with outer and inner membranes of Gram-negative bacteria. Biochim Biophys Acta 1997;1327:119–30. [DOI] [PubMed] [Google Scholar]
- [11].Joly S, Maze C, McCray PB, Guthmiller JM. Human β-defensins 2 and 3 demonstrate strain-selective activity against oral microorganisms. J Clin Microbiol 2004;42:1024–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Amerikova M, El-tibi IP, Maslarska V, Tachkov K. Antimicrobial activity, mechanism of action, and methods for stabilisation of defensins as new therapeutic agents. Biotechnol Equip 2019;33:671–82. [Google Scholar]
- [13].Yount NY, Kupferwasser D, Spisni A, Dutz SM, Ramjan ZH, Sharma S, et al. Selective reciprocity in antimicrobial activity versus cytotoxicity of hBD-2 and crotamine. Proc Natl Acad Sci U S A 2009;106:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Centers for Disease Control and Prevention, Food and Drug Administration. Antibiotic resistance isolate bank. 2018. [Accessed 30 April 2018] https://wwwn.cdc.gov/arisolatebank.
- [15].Crook ZR, Sevilla GP, Friend D, Brusniak MY, Bandaranayake AD, Clarke M, et al. Mammalian display screening of diverse cystine-dense peptides for difficult to drug targets. Nat Commun 2017;8:2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sharafi T, Ardebili A. Plastic binding feature of polymyxins: the effect on MIC susceptibility measurements. Infect Drug Resist 2019;12:2649–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Karvanen M, Malmberg C, Friberg LE, Cars O. Colistin is extensively lost during standard in vitro experimental conditions. Antimicrob Agents Chemother 2017;61:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Benjamini Y, Krieger AM, Yekutieli D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 2006;93:491–50 . [Google Scholar]
- [19].Monteiro C, Pinheiro M, Fernandes M, Maia S, Seabra CL, Ferreira-Da-Silva F, et al. A 17-mer membrane-active MSI-78 derivative with improved selectivity toward bacterial cells. Mol Pharm 2015;12:2904–11. [DOI] [PubMed] [Google Scholar]
- [20].Nation RL, Garonzik SM, Tham Likitkul V, Giamarellos-Bourboulis EJ, Forrest A, Paterson DL, et al. Dosing guidance for intravenous colistin in critically ill patients. Clin Infect Dis 2017;64:565–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Worthington RJ, Melander C. Combination approaches to combat multi-drug resistant bacteria. Nat Commun 2017;31:177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zasloff M Antimicrobial peptides of multicellular organisms. Nature 2002;415:389–95. [DOI] [PubMed] [Google Scholar]
- [23].Shai Y Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta 1999;1462:55–70. [DOI] [PubMed] [Google Scholar]
- [24].Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein identification and analysis tools on the ExPASy server In: Walker JM, editor. The proteomics protocols handbook. Totowa, NJ: Humana Press; 2005. p. 571–608. [Google Scholar]
- [25].Munier A, Biard L, Legrand M, Rousseau C, Lafaurie M, Donay J, et al. Incidence, risk factors, and outcome of multi-drug resistant Acinetobacter baumannii nosocomial infections during an outbreak in a burn unit. Int J Infect Dis 2019;79:179–84. [DOI] [PubMed] [Google Scholar]
- [26].Hoover DM, Rajashankar KR, Blumenthal R, Puri A, Oppenheim JJ, Chertov O, et al. The structure of human β-defensin-2 shows evidence of higher order oligomerization. J Biol Chem 2000;275:32911–8. [DOI] [PubMed] [Google Scholar]
- [27].Hallock KJ, Lee DK, Ramamoorthy A. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys J 2003;84:3052–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sawai MV, Jia HP, Liu L, Aseyev V, Wiencek JM, McCray PB, et al. The NMR structure of human β-defensin-2 reveals a novel α-helical segment. Biochemistry 2001;40:3810–6. [DOI] [PubMed] [Google Scholar]
- [29].Flamm RK, Rhomberg PR, Simpson KM, Farrell DJ, Sader HS, Jones RN. In vitro spectrum of pexiganan activity when tested against pathogens from diabetic foot infections and with selected resistance mechanisms. Antimicrob Agents Chemother 2015;59:1751–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schro J, Harder É. Molecules in focus: human beta-defensin-2. Int J Biochem Cell Biol 1999;31:645–51. [DOI] [PubMed] [Google Scholar]
- [31].Bahar AA, Ren D. Antimicrobial peptides. Pharmaceuticals (Basel) 2013;6:1543–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, Hackett M, et al. Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 1998;95:189–98. [DOI] [PubMed] [Google Scholar]
- [33].Wang G, Li X, Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res 2016;44: D1087–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].http://aps.unmc.edu/AP/; 2020. [Accessed 21 July 2020].
- [35].Wibowo D, Zhao CX. Recent achievements and perspectives for large-scale recombinant production of antimicrobial peptides. Appl Microbiol Biotechnol 2019;103:659–71. [DOI] [PubMed] [Google Scholar]
- [36].Andersson L, Blomberg L, Flegel M, Lepsa L, Nilsson B, Verlander M. Large-scale synthesis of peptides. Biopolymers 2000;55:227–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.