

Abstract
COVID-19 has intensified into a global pandemic with over a million deaths worldwide. Experimental research analyses have been implemented and executed with the sole rationale to counteract SARS-CoV-2, which has initiated potent therapeutic strategy development in coherence with computational biology validation focusing on the characterized viral drug targets signified by proteomic and genomic data. Spike glycoprotein is one of such potential drug target that promotes viral attachment to the host cellular membrane by binding to its receptor ACE-2 via its Receptor-Binding Domain (RBD). Multiple Sequence alignment and relative phylogenetic analysis revealed significant sequential disparities of SARS-CoV-2 as compared to previously encountered SARS-CoV and MERS-CoV strains. We implemented a drug re-purposing approach wherein the inhibitory efficacy of a cluster of thirty known drug candidates comprising of antivirals, antibiotics and phytochemicals (selection contingent on their present developmental status in underway clinical trials) was elucidated by subjecting them to molecular docking analyses against the spike protein RBD model (developed using homology modelling and validated using SAVES server 5.0) and the composite trimeric structures of spike glycoprotein of SARS-CoV-2. Our results indicated that Camostat, Favipiravir, Tenofovir, Raltegravir and Stavudine showed significant interactions with spike RBD of SARS-CoV-2. Proficient bioavailability coupled with no predicted in silico toxicity rendered them as prospective alternatives for designing and development of novel combinatorial therapy formulations for improving existing treatment regimes to combat COVID-19.
Keywords: Coronavirus, COVID-19, Drug repurposing, Molecular docking, SARS-CoV, SARS-CoV-2
Graphical abstract

Highlights
-
•
COVID-19 has notoriously accentuated as a global pandemic.
-
•
The Spike Glycoprotein RBD represents a promising therapeutic drug target.
-
•
10 drugs displayed significant interactions with Spike glycoprotein.
-
•
Camostat, Favipiravir, Tenofovir, Raltegravir & Stavudine showed future prospects.
1. Introduction
SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus-2), a member of the beta-coronavirus genus, which includes SARS-CoV (Severe Acute Respiratory Syndrome Coronavirus), MERS-CoV (Middle East Respiratory Syndrome Coronavirus) and other coronaviruses, is a novel discovered virus that causes COVID-19 (A Chronicle on the SARS Epidemic, 2003). The occurrence of SARS-CoV-2 was first epidemiologically linked in December 2019 to patients suffering from pneumonia of an unknown cause who were linked to a seafood market in Wuhan, Hubei province, China. On January 30, 2020, WHO (World Health Organisation) declared SARS-CoV-2 as a “Public Health Emergency of International Concern” and officially named it as Corona Virus Disease 2019(COVID-19/2019-nCoV) (Wu et al., 2020; Li et al., 2020). As of July 27, 2020, Global statistics reported by WHO regarding this intensifying pandemic indicate 16,114,449 confirmed COVID-19 cases, including 6,46,641 deaths collectively in the 216 affected countries (https://www.who.int/emergencies/diseases/novel-coronavirus-2019).
Pathological diagnoses of SARS-CoV-2 infections designate severe acute respiratory distress syndrome along with secondary ailments like sore throat, high fever, muscle weakness, encephalitis and diarrhoea in infected patients (Zhu et al., 2020). It primarily targets the lower respiratory system by invading the pulmonary epithelial cells, releasing the nucleocapsid component and arrogating the host's cellular machinery for cytoplasmic replication (Shah et al., 2020).
Comprehensive structural genomics analyses exemplify that the SARS- CoV-2 genome codes for a variety of structural and non-structural proteins which constitute potential drug targets for scientific experimental investigations to impede the COVID-19 epidemic (Prajapat et al., 2020). The spike glycoprotein (S protein) is a prospective drug target as it initiates the first crucial step in SARS-CoV-2 attachment to the host cell. It is a homotrimeric protein (each chain containing 1273 amino acids) which recognizes and binds to its receptor Angiotensin Converting Enzyme-2 (ACE-2) with a greater affinity attributed to the mutations at L455, F486, Q493, S494, N501 and Y505 residues (Yan et al., 2020). Microbiological digital resources data insinuate a comprehensive schematic of the spike glycoprotein (Fig. 1, Fig. 2).
Fig. 1.

ACE-2 receptor-mediated SARS-CoV-2 internalization occurs via three steps. The first step involves the recognition and attachment of the spike glycoprotein (SP, olive green) to its receptor ACE-2 (Red). This binding instigates the activation process wherein the spike protein is cleaved into its subunits, S1 (receptor-binding domain, light blue) and S2 (fusion peptide, yellow) via the adjacent transmembrane protease TMPRSS2 (light green). This fusion peptide integrates with the host cell membrane, resulting in subsequent release and internalization of the SARS-CoV-2 single-stranded RNA (ssRNA, blue) and other components. Once internalized, the virus utilizes the host cell machinery for replication and extrusion of novel viral particles (Colour, Double-column fitting image).
Fig. 2.

Spike glycoprotein gene schematics. The spike glycoprotein gene comprises of eight distinct segments. Residues 1–12 act as the Signal peptide (SP, Red). Residues 13–685 and 686–1273 indicate the S1 and S2 subunits of the spike glycoprotein respectively. The S1 subunit comprises of the N-Terminal Domain (NTD, residues 13–303, Olive green) and the Receptor-Binding Domain (RBD, residues 319–541, yellow). The RBD domain is further sub-divided into the C-Terminal Domain (CTD, residues 334–427, Pink) and the Receptor-Binding Motif (RBM, 437–508, Light blue). The cleavage site 1 (685–686) releases the S2 subunit which comprises of the Fusion Peptide region (FP: 788–806, Brown) followed by a second cleavage site (815–816) which generates the S2′ Domain (816–1273). The S2 subunit comprises of two heptad repeats HR1 (920–927) and HR2 (1163–1202) (Green) and two Coiled-Coil Regions CCR1 (949–993) and CCR2 (1176–1203) (Cyan) respectively. A transmembrane Helical Domain exists (1214–1234, Bright yellow) followed by a Topological Cytoplasmic Domain (TPC, 1235–1273, Magenta) which encompasses a Short Sequence Motif (SSF, 1269–1273, light red). The Extracellular Topological domain spans from residues 13–1213. (Colour, Double-column fitting image).
1.1. Alternative strategies targeting SARS-CoV-2 spike glycoprotein
Several potential therapeutic strategies are being explored to combat SARS-CoV-2 targeting the spike glycoprotein as depicted in Fig. 3 (Wu et al., 2020). Of these strategies, ‘Drugs Re-purposing’ has emerged as a strong pharmaceutical rationale due to its reduced perils of drug development costs and shortening of the time gap between identification of prospective drug candidates and designing of treatment regimes for patients. This is due to the availability of extensive data regarding its pharmacokinetics, pharmacodynamics, toxicity, clinical trial results, etc (Zhou et al., 2020; Ciliberto, 2020).
Fig. 3.

Alternative therapeutic strategies to counteract SARS-CoV-2 targeting the spike Glycoprotein. (Black and White, Single-column fitting image).
Drug-spike protein molecular docking studies were executed to elucidate their inhibitory competence to counteract SARS-CoV-2 infections targeting spike glycoprotein receptor-binding domain. Computational pharmacokinetic analyses coupled with in silico toxicity prediction studies were used to evaluate the therapeutic proficiency of the selected drug candidates for futuristic COVID-19 treatment regimes.
2. Materials and methods
2.1. Workstation
The entire study was executed on a Dell Laptop having an i5-6200U Intel core (2.30 GHz) processor, 8 GB RAM, 1 TB hard disk and a 64-bit operating system. All softwares utilized for the present study were open-source tools and freely downloadable.
2.2. Sequence retrieval
The protein sequences, structures and other relevant data regarding the surface spike glycoprotein from SARS-CoV-2 and the whole genome sequence of SARS-CoV-2 Wuhan-Hu-1 isolate were downloaded from digital resources such as NCBI (National Centre for Biotechnology Information) (https://www.ncbi.nlm.nih.gov/), Universal Protein Knowledgebase (UniProtKB) and RCSB-PDB (Research Collaboratory for Structural Bioinformatics-Protein Data Bank) databases (Sayers et al., 2018; Apweiler et al., 2004; Berman et al., 2000). For comparative analysis, SARS-CoV (first identified in Foshan, Guangdong, China in 2002 from bats) data was also collected and analyzed (Chinese Law and Government, 2003; Li et al., 2005) (Table 1 ).
Table 1.
Spike Glycoprotein sequence information.
| Sr. No. | Digital resource | IDa | Description |
|---|---|---|---|
| 1 | UniProtKB | P0DTC2 | SARS-CoV-2b spike glycoprotein |
| 2 | NCBI | YP_009724390.1 | SARS-CoV-2 surface glycoprotein |
| 3 | NCBI | MN908947.3 | Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome |
| 4 | RCSB-PDB | 6VYB | SARS-CoV-2 spike ectodomain structure (open state) |
| 5 | RCSB-PDB | 6VSB | Prefusion 2019-nCoV spike glycoprotein with a single receptor-binding domain up |
| 6 | RCSB-PDB | 6VW1 | Structure of SARS-CoV-2 chimeric receptor-binding domain complexed with its receptor human ACE2c |
| 7 | RCSB-PDB | 6LZG | Structure of novel coronavirus spike receptor-binding domain complexed with its receptor ACE2 |
| 8 | RCSB-PDB | 6YLA | Crystal structure of the SARS-CoV-2 receptor binding domain in complex with CR3022 Fabd |
| 9 | RCSB-PDB | 2AJF | Structure of SARS coronavirus spike receptor-binding domain complexed with its receptor |
ID = Identification.
Severe Acute Respiratory Syndrome – Coronavirus-2.
Angiotensin Converting Enzyme-2.
antigen-binding Fragment.
2.3. Crystallizability and protein- disorderness
The crystallization propensity of the SARS-CoV-2 spike glycoprotein sequence (UniProtKB ID: P0DTC2) was predicted by the PPCpred, CRYSTALP2 and XTalPred servers respectively (Mizianty and Kurgan, 2011; Kurgan et al., 2009; Slabinski et al., 2007). XTalPred also disclosed diverse physicochemical information such as Molecular Weight (MW), hypothetical isoelectric point (pI), amino-acid length, instability index and Grand Average of Hydropathicity (GRAVY) index, etc. The prediction of the conformational diversity in the ordered globular domains and intrinsically disordered regions of the spike glycoprotein sequence was derived using IUPred2A, MobiDB web tool, Meta disorder web server and PONDR servers (Meszaros et al., 2018; Piovesan et al., 2018; Kozlowski and Bujnicki, 2012; Romero et al., 1997). The unstructured regions of proteins are stipulated to play a crucial role in diverse cellular activities.
2.4. Physicochemical characterization and post-Translational modifications (PTMs)
The computation of the physicochemical properties and amino acid scale representation to determine the significant amino acid enrichment and/or depletion patterns were resolved by ProtParam and ProtScale tools of ExPasy (a SIB Bioinformatics resource portal) and Protein Calculator v 3.4 servers. (Gasteiger et al., 2005; Anthis and Clore, 2013).
SignalP 4.1 server was used to identify the secretory signal peptides and its cleavage sites (Nielson, 2017). N-Terminal acetylation sites were predicted by NetAcet 1.0 server (Kiemer et al., 2005). High specificity Coronavirus 3C-like proteinase cleavage sites were predicted by the NetCorona-1.0 server (Kiemer et al., 2004).
N-linked and O-linked glycosylation sites were predicted using the NetNGlyc-1.0 and NetOGlyc-4.0 servers respectively (Gupta and Brunak, 2002; Steentoft et al., 2013). NetPhos 3.1 server was used to predict the serine, threonine or tyrosine phosphorylation sites (Blom et al., 1999). DiANNA 1.1 webserver was used to predict the disulfide bond connectivity (Ferre and Clote, 2005). The surface accessibility, secondary structure, disorder, and phi/psi dihedral angles of amino acids were predicted by NetSurfP-2.0 server (Klausen et al., 2019). DeepLoc 1.0 server was used to predict the sub-cellular localization of the spike glycoprotein by deep neural network algorithms (Armenteros et al., 2017).
2.5. Secondary structure prediction, tertiary structure prediction, refinement and model evaluation
PSIPRED 4.0 and JPred v.4 servers were utilized to elucidate the secondary structure characteristics of the spike glycoprotein (McGuffin et al., 2000; Drozdetskiy et al., 2015). The UniProtKB spike glycoprotein sequence (P0DTC2) was submitted in FASTA format to Modeller v9.24 which generated its 3D structure employing a restraint-based approach using a model-single module (Webb and Sali, 2017; Sali and Blundell, 1993). The best model was selected based on low discrete optimized protein energy (DOPE) score. It was then submitted to RAMPAGE server to determine the geometrical structural validity (Lovell et al., 2003). Loop refinement of the outlier residues was carried out by ModLoop server (Fiser et al., 2000).
The above steps were repeated until the outlier residues were within the designated values. The final model geometry was subjected to energy minimization by Chiron server wherein the atomic steric clashes between the side-chain and backbone were resolved (Ramachandran et al., 2011). The overall quality assessment of the final model in terms of packing density (total void volume), unsatisfied hydrogen bonds, steric clashes and the accessible surface area scaling was exemplified by Gaia server (Kota et al., 2011). The stereochemical quality evaluation of the pre- and post-refinement 3D models of spike glycoprotein was done via PROCHECK, ERRAT (v 2.0) and Verify3D environment profile analysis methods by submitting to SAVES v5.0 (Structural Analysis and Verification Server) (Pradeepkiran et al., 2015).
The Root Mean Square Deviation (RMSD) statistics between the target model and template were calculated by structural superimposition in SuperPose v1.0 program which utilizes a modified quaternion eigenvalue approach under the default parameters (Maiti et al., 2004). This final model was selected as the receptor for further studies. Composition profiler was utilized to investigate the amino acid composition bias in the modeled spike glycoprotein and the template protein (Vacic et al., 2007).
2.6. Multiple Sequence Alignment (MSA) and comparative phylogeny
MSA of the sequence of the generated 3-D model with the existing PDB structures was carried out using Jalview v2.11.1.0 (Waterhouse et al., 2009). Evolutionary analyses of the selected sequences was carried out by phylogenetic tree construction using MEGA X v10.1.8 maintaining the default values of the selected parameters (Kumar et al., 2018).
2.7. Binding sites prediction
Binding site prediction to determine the active site residues of the 3-D spike glycoprotein model was carried out by COACH server (Yang et al., 2013; Yang et al., 2013). These were also determined by the receptor cavity method using Biovia Discovery studio v17.2.0.16349 (Dassault Systemes, 2017). The functional domain prediction was carried out by MOTIF search, a subserver of GenomeNet, using the Pfam and PROSITE data (Al-Khayyat and Al-Dabbagh, 2016). The cut-off score (E-value) was set at 1.0.
2.8. Ligand selection and molecular docking
The computational molecular docking of ACE-2 (PDB ID: 1R42) (receptor) with the spike glycoprotein model (ligand) was performed using the HawkDock server. It is an integrated web server that utilizes ATTRACT for protein-protein docking, HawkRank scoring function for re-ranking docked complexes based on desolvation potentials and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) free energy decomposition methodology for the identification of key residues in Protein-Protein Interactions [PPIs] (Weng et al., 2019; Sarkar et al., 2020).
The drug candidates for virtual screening against the spike glycoprotein model were selected from an extensive literature survey of potential therapeutic agents undergoing trials against SARS-CoV-2 and/or those which were previously developed as antiviral/anti-malarial therapeutic inhibitors as shown in Table 1. The. sdf files of all molecules were retrieved from the PubChem database and were converted to. pdb files and saved for further docking protocols (Kim et al., 2015).
Molecular docking of the selected ligands with the spike glycoprotein 3-D model (receptor) was carried out by iGEMDOCK v2.1 (Hsu et al., 2011; Kashyap, 2019). It is an integrated virtual screening graphical automatic drug design system dealing with preparations through post-screening analysis depicting crucial pharmacological interactions based on protein-compound interaction profiles. It employs a generic evolutionary method algorithm. The docking accuracy settings (GA parameters) for all docking operations were set as follows: Population size: 300; Generations: 80; Number of solutions: 10. The mode of docking was stable (slow docking).
The scoring function was set to GEMDOCK and the hydrophobic and electrostatic preferences for the ligands were set to 1.00. The molecular interactions of the ligand-receptor docked complexes were analyzed using Discovery studio v20.1.0.19295, PyMol v1.7.4.5 and UCSF Chimera v1.11.2 respectively (Seeliger and de Groot, 2010; Sadati et al., 2019; Pettersen et al., 2004). The best docked complex of each ligand-receptor combination was selected based on the least binding energy values for further analysis.
2.9. Galaxy refine complex
Refinement of protein-protein complex structure was done by GalxyWEB. This tool allows us to understand the flexibility, at the protein interface (modeled RBD with RBD-ligand complex) and gives us an idea regarding overall conformational changes that occurs upon binding (Ko et al., 2012). GalaxyRefine refines the docked structure by a hybrid technique that involves the triaxial loop closure (TLC) algorithm during global optimization by conformational space annealing (CSA) technique (Park and Seok, 2012). The molecular interactions of the ligand-receptor docked complexes were analyzed using PyMol v1.7.4 (Seeliger and de Groot, 2010). Further, Favipiravir, Hydroxychloroquine and Nafamostat were taken to study the possible structural changes occurring to spike protein when drug binds.
2.10. Argus Lab analysis
Molecular docking interactions of the selected ligands with the SARS-CoV-2 whole trimeric spike glycoprotein (PDB ID's 6VSB and 6VYB respectively) were analyzed and visualized using Argus Lab 4.0.1 (Thompson, 2004). Argus Lab platform has distinct advantages over other softwares because it performs meticulous docking analyses of huge peptides/proteins with a precise chain or domain selection within minutes. It follows the Lamarckian genetic algorithm along with precise grid resolution calculation in coherence with the ligand binding sites.
It was performed under the following parametric considerations: scoring function selected as ‘AScore’, docking algorithm as Argus dock (exhaustive search), a grid resolution of 0.4 A0 and the binding site box dimensions measuring 60 A0 x 60 A0 x 60 A0 respectively. The docking precision was set to “Regular precision” and “Flexible” ligand docking mode was utilized for all docking runs. The individual docked pose stability was evaluated by ArgusLab energy calculations and the number of hydrogen bonds formed (Hafeez et al., 2013).
The best docking model was selected according to the lowest AScore computed and the most pertinent binding conformation was selected on the basis of ligand-receptor hydrogen bond interactions in the vicinity of the substrate binding site. The lowest energy poses implied the highest binding affinity because high energy values represent unstable conformations.
2.11. In silico toxicity prediction
The analysis of the pharmacokinetic properties of the selected ligands such as absorption, distribution metabolism and excretion (ADME), drug-likeness and medicinal chemistry features was carried out by SWISS ADME (Daina et al., 2014, 2017; Daina and Zoete, 2016). OSIRIS Property Explorer was used to evaluate the risks of side effects, such as mutagenicity, tumorigenicity, irritant and reproductive consequences, as well as drug-relevant properties including cLogP, LogS (solubility), molecular weight, Topological Polar Surface Area (TPSA), drug-likeness and overall drug-score (Ayati et al., 2012).
3. Results
3.1. Crystallization propensity and protein- disorderness
PPCPred and CRYSTALP2 servers both predicted the sequence to be non-crystallizable. PPCPred predicted the crystallization propensity to be 0.084 which was significantly lower than the probability threshold value of 0.43. The individual step values of the crystallization process were as follows: probability that production of protein material fails: 0.831, probability that purification fails: 0.806, probability that crystallization fails: 0.01 and probability that target will yield diffraction-quality crystals: 0.355. CRYSTALP2 predicted the sequence as non-crystallizable with a confidence of 0.28. Xtalpred results depicted that the protein was non-crystallizable (EP crystallization class 5: very difficult; RF protein crystallization class 4). It also enumerated an assortment of molecular characterization parameters (Table 2 ). Overall, the spike glycoprotein RBD model was deemed non-crystallizable in its native form.
Table 2.
Molecular characterization of P0DTC2 sequence predicted by XTalPred server.
| Sr.No. | Server | Molecular feature (s) | Prediction |
|---|---|---|---|
| 1 | XtalPred | Molecular weight | 141.196 KDa |
| Amino acid length | 1273 | ||
| Theoretical pI | 6.24 | ||
| Instability index | 33.02 | ||
| GRAVY | −0.08 | ||
| Molecular surface feature (EP and RF crystallization): | |||
| a) Surface entropy | −1.06 | ||
| b) Surface hydrophobicity | −0.09 | ||
| c) Surface ruggedness | 4.04 | ||
| 2 | COILS | Coiled-coils regions | 28 |
| 3 | DISOPRED 2 | Longest disorder region | 16 (1%) |
| 4 | TMHMM | Transmembrane helices | 1 |
| 5 | SignalP | Signal peptides | No |
| 6 | SEG | Longest low-complexity region | 23 |
| 7 | PSIPRED | Secondary structure prediction: | |
| Coil residues% | 38 | ||
| Helix residues % | 22 | ||
| Strand residues % | 40 | ||
| 8 | – | Residue content | |
| Cysteine residues | 40 | ||
| Methionine residues | 14 | ||
| Tryptophan residues | 12 | ||
| Tyrosine residues | 54 | ||
| Phenylalanine residues | 77 |
pI: Isoelectric point; GRAVY: Grand Average of Hydropathicity; EP: Expert Pool; RF: Random Forest.
The MobiDB web tool indicated 0% disorderness in the spike glycoprotein sequence (UniProtKB: P0DTC2) while IUPred2A and Metadisorder web server (global disorder tendency = 0.3474) indicated disordered residues. PONDR server depicted 11 disordered regions, comprising a total of 98 disordered amino acid residues (7.70%) with the longest disorder region comprising of 38 amino residues (Average prediction score = 0.1374). DEPP (Disorder Enhanced Phosphorylation Predictor) in the PONDR server predicted the phosphorylation potentiality of serine (6 out of 99; 6.0606%), threonine (2 out of 97; 2.06%) and tyrosine (3 out of 54; 5.56%) residues of the spike glycoprotein. (Table S1; Fig. S1 in supplementary data).
3.2. Physicochemical characterization and post-translational modifications (PTMs)
SARS-CoV-2 encompasses a variety of structural proteins such as the spike glycoprotein, small envelope protein (E), Matrix protein (M) and the nucleocapsid protein (N) while the ORF1a/b region (initial ~20 kb) is translated into two polyproteins (PP1A and PPIB) which encode the majority of the Non-Structural Proteins (NSPs) (Khodadadi et al., 2020, Wu et al., 2020).ProtParam and ProtScale servers explicated several physicochemical parameters of SARS-CoV-2 spike glycoprotein viz. Molecular weight: 141.178Kda; theoretical pI: 6.24; Instability index: 33.01 (stable); aliphatic index: 84.67; GRAVY: −0.079. The estimated half-life (t1/2) was predicted to be 30 h in mammalian reticulocytes (in vitro); >20 h in yeast (in vivo) and >10 h in E. coli (in vivo) (Kar et al., 2020). ProtScale also depicted an anthology of hydrophobicity plots of the spike glycoprotein. Protein calculator v3.4 further corroborated the above results. SignalP 4.1 server predicted residues 1–13 as the signal peptide (cleavage site between the 13th and 14th residues) with a score (D value) of 0.837 which was higher than the threshold cut-off value (D = 0.450).
NetAcet 1.0 server predicted no N-terminal acetylation as alanine, glycine, serine or threonine residues were not present at positions 1–3. One specific coronavirus 3C-like proteinase cleavage site was found by NetCorona 1.0 server at position 1002 with a score of 0.685 (TGRLQ^SLQTY). N-linked glycosylation sites predicted by NetNGlyc 1.0 server were in accordance with the data retrieved from UniProtKB for the spike glycoprotein while O-linked glycosylation sites were predicted at positions 673, 678 and 686 by NetOGlyc server 4.0 which was in accordance with the previous research analysis (Andersen et al., 2020).
NetPhos 3.1 server predicted phosphorylation of serine, tyrosine and threonine residues at 136 positions in the spike glycoprotein. DiANNA 1.1 web server revealed 20 disulfide bond connectivity predictions. NetSurfP 2.0 results validated the disorder predictions and also depicted surface accessibility and phi/psi angles for each amino acid. DeepLoc 1.0 server predicted that the spike glycoprotein was a soluble (score = 0.0577), membrane type protein (score = 0.9423).
3.3. Secondary structure prediction, tertiary structure prediction, refinement and model evaluation
PSIPRED 4.0 and JPred v.4 servers deduced the secondary structure of the spike glycoprotein which comprised of α-helices, β-sheets and coils. It also predicted a putative domain boundary at position 164 (Asparagine) (Fig. S2 in supplementary data).
The spike glycoprotein sequence from UniProtKB was submitted to MODELLER v9.24. It generated two models whose characteristics are listed in Table 3 . GA341 (range: 0–1, value near 1 being preferred), z-DOPE (lowest score preferred), MPQS (ModPipe Quality Score; highest value preferred) and sequence identity (highest value preferred) scores were employed for model quality assessment.
Table 3.
Model characteristics generated by MODELLER v.9.24.
| Sr.No. | Features | Model 1 | Model 2 |
|---|---|---|---|
| 1 | Target region | 14–302 | 330–529 |
| 2 | Protein length | 1273 | 1273 |
| 3 | PDB template code | 5 × 4S A | 6YLA E |
| 4 | Template region | 18–289 | 330–529 |
| 5 | Sequence identity | 54% | 100% |
| 6 | E-value | 0 | 0 |
| 7 | GA341 | 1 | 1 |
| 8 | MPQS | 0.786723 | 1.37941 |
| 9 | z-DOPE | −0.3 | −1.32 |
| 10 | TSVMod method | MSALL | MSALL |
| 11 | TSVMod RMSD | 3.711 | 0.464 |
| 12 | TSVMod NO35 | 0.923 | 0.972 |
E-value: no: of expected hits of similar quality; MPQS: ModPipe Quality Score; z-DOPE: Discrete Optimized Protein Energy Score of Salilab Model evaluation server; TSVMod RMSD: Root Mean Square Deviations; TSVModNO35: Native Overlap prediction of fraction of Cα atoms within 3.5 Å of the native structure.
The results indicated that model 2 was the most reliable one as compared to model 1. The MPQS score of model 1 was also predicted to be unreliable as compared to that of model 2. Model 2 represented a segment of the S1 subunit of the SARS-CoV-2 spike glycoprotein as deduced from the UniProtKB data while model 1 corresponded to the N-Terminal Domain (NTD) region. It depicted 2 N-Acetyl glycosylation sites at asparagine residues (positions 331 and 343 respectively). The model (residues 1–200) formed the majority portion of the Receptor Binding Domain (RBD: residues 319–541) containing the Receptor Binding Motif (RBM; residues 437–508).
The selected model (Fig. 4 a ) was analyzed by Ramachandran plot generation by submission to the RAMPAGE server which elucidated 186 favored residues (93.9%), 10 allowed residues (5.1%) and 2 outlier residues (TRP353 and PHE497; 1%) respectively. The model was subjected to further structural refinement by ModLoop server which then depicted 186 favored residues (93.9%), 12 allowed residues (6.1%) and zero outlier residues respectively (Fig. 4b). The refined model was then subjected to energy minimization by Chiron server with side chain constraints.
Fig. 4.

The refined spike RBD model 3D structure comprising of helix (red), sheet (yellow) and loop (green) regions. b) The Ramachandran plot of the spike RBD model exhibiting zero outlier residues. (Colour, Double-column fitting image).
After energy minimization, the model was subjected to structural validation by Gaia server (with side chain constraints) which elucidated the following data: Clash score = 0.0381; 69 unsatisfied HBS (Hydrogen Bonds in Shell); 4 HBC (Hydrogen Bonds in Core); solvent accessible (10225.4 Å2) and solvent excluded (8623.33 Å2) surface areas and a void volume score of 0.53. All bond lengths were within the acceptable range. The side-chain integrity discrepancies were refined and the model was then submitted to the SAVES V5.0 server for structure validation.
SAVES server validated the refined model yielding an overall quality factor of 83.3333. VERIFY3D substantiated that about 80% amino acids scored≥0.2 in the 3D/1D profile. PROVE results depicted about 25 buried outlier protein atoms (3.6%). Overall results validated the refined model (spike RBD model). Discovery studio construed characteristics of the spike RBD model viz. 1947 atoms; Molecular formula: C1016 H1522 N265 O295 S8; molecular weight: 22.411Kda and a net formal charge of +5.
The spike RBD model and the template (6YLA chain E) were superimposed via Superpose 1.0 server which depicted a resulting local and global RMSD value of 0.37 over 200 Cα atoms and a value of 0.43 over 800 atoms in the protein backbone (Fig. 5 a).
Fig. 5.

a) Superimposition of spike RBD model (red) with its template 6YLA chain E (green). b) Superimposition of the spike RBD model (yellow) with 6VSB (red). c) Superimposition of the spike RBD model (yellow) with 6VYB (yellow). (Colour, Double-column fitting image).
Composition profiler revealed that no significant enrichment or depletion patterns in the amino acid composition had occurred. The relative entropy had a value of 0.00092 with a p-value of 1. The spike model was also superposed with the SARS-CoV-2 spike open-state ectodomain structure (PDB ID: 6VYB) and the Prefusion 2019-nCoV spike glycoprotein with a single receptor-binding domain up structure (PDB ID: 6VSB). The spike RBD model-6VYB superimposition illustrated a local and global RMSD value of 1.183 over the Cα atoms and atoms in the protein backbone whereas the spike RBD model-6VSB superimposition elucidated a local and global RMSD values of 1.655 and 1.648 over the Cα atoms and atoms in the protein backbone respectively (Fig. 5b and c). The MOTIF server detected diverse proteomic segments in the whole spike glycoprotein and the spike RBD model (Table 4 ).
Table 4.
Functional domain prediction by MOTIF search server.
| Whole Spike Glycoprotein | |||||
|---|---|---|---|---|---|
| Sr.No. | PfamaID | Pfam | Position | IE-valueb | Descripton |
| 1 | PF01601 | Corona_S2 | 686–1270 | 1.4e-266 | Coronavirus S2 glycoprotein |
| 2 | PF09408 | Spike_rec_bind | 330–583 | 6.6e-75 | spike receptor binding domain |
| 3 | PF16451 | Spike_NTD | 262–294 | 0.0014 | spike glycoprotein N-terminal domain |
| 4 | PF04513 | Baculo_PEP_C | 931–989 | 0.11 | Baculovirus polyhedron envelope protein, PEP, C-terminus |
| 5 | PF11172 | DUF2959 | 917–1015 | 0.057 | Unknown functional protein |
| 6 | PF17192 | MukF_M | 906–969 | 0.22 | MukF middle domain |
| 7 | PF16025 | CALM_bind | 853–871 | 0.26 | Calcium-dependent calmodulin binding |
| 8 | PF16519 | TRPM_tetra | 934–962 | 0.21 | TRPM tetramerization domain |
| 9 | PF00517 | GP41 | 1149–1241 | 0.26 | Retroviral envelope protein |
| Spike RBD model | |||||
| 10 | PF09408 | Spike_rec_bind | 1–200 | 2.7e-60 | spike receptor binding domain |
Pfam = Protein family.
IE-value = Independent E-value.
Sequential analysis of the spike RBD model and the template (6YLA chain E) revealed that our RBD model lacked the first three residues (GLU327, THR328 and GLY329) incorporated by 6YLA chain E. In addition, the residues 1, 2, 3, 4 …. in the spike RBD model corresponded to residues 330, 331, 332, 333… and so on. Hence, the numbering of residues in the spike RBD model could be correlated to the residue position in 6YLA chain E as follows:
Where R.N = Residue Number.
3.4. Multiple Sequence Alignment (MSA), comparative phylogeny and binding sites prediction
SARS-CoV-2 is a more potent virus as it depicts important genomic dissimilarities when compared to the SARS-CoV (79%) and MERS-CoV (50%) genomes (Walls et al., 2020). Previous studies elucidated that amino acid mutations in the Spike glycoprotein influenced its credited ACE-2 (receptor) binding capability in humans (Walls et al., 2020; Coutard et al., 2020).
MSA of the selected sequences listed in Table 1 including the spike RBD model was carried out using Jalview v2.11.1.0 (Fig. 6 ). It revealed that large number of significant amino acid substitutions/mutations had occurred in the SARS-CoV-2 as compared to the SARS-CoV spike protein sequences reported earlier viz. R403K, E406D, K417V, L441I, S443A, K444T, V445S, G446T, L452K and K501N respectively.
Fig. 6.

MSA (Multiple Sequence Alignment) of the refined spike model with the assorted spike glycoprotein sequences listed in Table 1. The Receptor-Binding Domain (RBD, 319–541) consisted of the Receptor-Binding Motif (RBM, 437–508). 15 RBD residues were implicated in molecular interactions with ACE-2 (red and green). The cysteine residues (dark blue) were involved in disulphide bond interactions. MSA revealed mutation of amino acid residues of SARS-CoV-2 viz. V417K, T446G, P475A, W490F, N493Q, and T501N respectively (green). The other possible mutations within the strains are reported by white within the strains. (Colour, Double-column fitting image).
Furthermore, SARS-CoV-2 spike glycoprotein asserted 80% sequence identity to the previously validated spike protein structures. These residues formed a part of loops 1e, 1 d, 2 g and 3f respectively. Loop 2 g accounted for 50% of these mutations. These mutations accentuate the augmented binding affinity of the spike protein to its receptor ACE-2 and were in good co-relation with earlier investigations (Ortega et al., 2020).
An evolutionary analysis of the Spike RBD model with the selected sequences in Table 1 was achieved by phylogenetic tree construction utilizing the Neighbour-Joining method (Saitou and Nei, 1987) (Fig. 7 ). The evolutionary distances were computed using the Poisson correction method and are in the units of the number of amino acid substitutions per site (Zuckerkandl and Pauling, 1965).
Fig. 7.

Phylogenetic tree analysis using MEGA X software. The optimal tree with the sum of branch length = 0.35413322 is shown. Data coverage is shown in percentage (%) while the nodal branch lengths are depicted in decimals. (Black and White, Single-column fitting image).
COACH and TM-SITE results predicted the binding site residues of the Spike RBD model viz. ASN354, LYS356, SER399, VAL401 and ARG509 with C-score (confidence of prediction) values of 0.10 and 0.22 respectively. Using Discovery studio visualizer, we found 6 possible receptor-binding cavities in the Spike RBD model (Table S3 in supplementary data).
3.5. ACE-2-spike RBD model molecular docking and analysis
The spike RBD model (ligand) and ACE-2 (receptor) were docked using the HawkDock server which generated 10 ligand-receptor docked complexes. The best one was selected based on the MM/GBSA score (binding free energy in Kcal/mol) having a value of −52.33 (Fig. 8 ). A total of 20 molecular interactions were observed upon analysis using the aforementioned molecular analysis software, of which 15 interactions were considered significant (bond-length of ≤4.0 Å) (Table 5 ).
Fig. 8.

Docked complex of spike RBD model (ligand, red) with ACE-2 (receptor, blue). Residues GLY446, GLY496 and ASN501 (RBD) formed a single hydrogen bond with residues ASN259, ASN136 and GLU132 of ACE-2 (bond lengths: 2.9, 2.2 and 2.7 respectively). TYR449 depicted 2 hydrogen bonds with residues ASN259 (bond length 2.8) and THR258 (bond length 2.7) respectively. The yellow-dashed lines indicate hydrogen bonds. (Colour, Double-column fitting image).
Table 5.
ACE2-spike RBD model molecular interactions analysis.
| Bond | Bond length | Bond category | Bond type | Donor | Acceptor | ||
|---|---|---|---|---|---|---|---|
| A:LYS229:NZ - B:GLU484:OE1 | 3.10837* | Electrostatic | Attractive Charge | A:LYS229:NZ | Positive | B:GLU484:OE1 | Negative |
| B:LYS444:NZ - A:ASP274:OD1 | 3.3582* | Electrostatic | Attractive Charge | B:LYS444:NZ | Positive | A:ASP274:OD1 | Negative |
| B:LYS444:NZ - A:ASP349:OD1 | 5.53446 | Electrostatic | Attractive Charge | B:LYS444:NZ | Positive | A:ASP349:OD1 | Negative |
| B:ARG509:NH2 - A:ASP277:OD1 | 4.66804 | Electrostatic | Attractive Charge | B:ARG509:NH2 | Positive | A:ASP277:OD1 | Negative |
| A: ASN136: ND2 - B: GLY496: O | 2.19044* | Hydrogen Bond | Conventional Hydrogen Bond | A:ASN136:ND2 | H-Donor | B: GLY496: O | H-Acceptor |
| A:ASN232:ND2 - B:GLU484:OE2 | 1.07015* | Hydrogen Bond | Conventional Hydrogen Bond | A:ASN232:ND2 | H-Donor | B:GLU484:OE2 | H-Acceptor |
| A: ASN259: ND2 - B: GLY446: O | 2.85426* | Hydrogen Bond | Conventional Hydrogen Bond | A:ASN259:ND2 | H-Donor | B: GLY446: O | H-Acceptor |
| A:ASN259:ND2 - B:TYR449:OH | 2.81454* | Hydrogen Bond | Conventional Hydrogen Bond | A:ASN259:ND2 | H-Donor | B:TYR449:OH | H-Acceptor |
| A: SER262: OG - B: TYR495: O | 2.58607* | Hydrogen Bond | Conventional Hydrogen Bond | A:SER262:OG | H-Donor | B: TYR495: O | H-Acceptor |
| B: TYR449: OH - A: THR258: O | 2.68899* | Hydrogen Bond | Conventional Hydrogen Bond | B:TYR449:OH | H-Donor | A: THR258: O | H-Acceptor |
| B: SER494: N - A: SER262: O | 2.68145* | Hydrogen Bond | Conventional Hydrogen Bond | B:SER494:N | H-Donor | A: SER262: O | H-Acceptor |
| B:ASN501:ND2 - A:GLU132:OE1 | 2.64768* | Hydrogen Bond | Conventional Hydrogen Bond | B:ASN501:ND2 | H-Donor | A:GLU132:OE1 | H-Acceptor |
| B:ASN501:ND2 - A:ASN136:OD1 | 1.59889* | Hydrogen Bond | Conventional Hydrogen Bond | B:ASN501:ND2 | H-Donor | A:ASN136:OD1 | H-Acceptor |
| A:LYS229:CA - B:GLU484:OE2 | 3.48252* | Hydrogen Bond | Carbon Hydrogen Bond | A:LYS229:CA | H-Donor | B:GLU484:OE2 | H-Acceptor |
| A:ASN259:CA - B:TYR449:OH | 2.46628* | Hydrogen Bond | Carbon Hydrogen Bond | A:ASN259:CA | H-Donor | B:TYR449:OH | H-Acceptor |
| B:LYS444:CE - A:ASP274:OD1 | 3.0135* | Hydrogen Bond | Carbon Hydrogen Bond | B:LYS444:CE | H-Donor | A:ASP274:OD1 | H-Acceptor |
| B:TYR449:CA - A:SER262:OG | 3.60435* | Hydrogen Bond | Carbon Hydrogen Bond | B:TYR449:CA | H-Donor | A:SER262:OG | H-Acceptor |
| A:LYS229:NZ - B:PHE490 | 4.31429 | Electrostatic | Pi-Cation | A:LYS229:NZ | Positive | B:PHE490 | Pi-Orbitals |
| A:GLU132:OE1 - B:TYR505 | 4.89665 | Electrostatic | Pi-Anion | A:GLU132:OE1 | Negative | B:TYR505 | Pi-Orbitals |
| B:TYR489 - A:PRO235 | 5.23117 | Hydrophobic | Pi-Alkyl | B:TYR489 | Pi-Orbitals | A:PRO235 | Alkyl |
* = Significant; H-Donor = Hydrogen Donor; H-Acceptor = Hydrogen Acceptor.
Earlier studies expounded that the spike RBD comprised of significant loops (a-f) which were then re-categorized into loop 1, 2 and 3 sub-segments. It was observed that the spike protein interactions with ACE-2 receptor comprised of loops 3a, 3 b and 3f respectively (Robson, 2020). It also contained nine cysteine residues of which eight are involved in disulfide bond formation viz. CYS336-CYS361, CYS379-CYS432, CYS391-CYS525 and CYS480-CYS488 respectively. Of these, the first three disulphide bonds facilitate structural stabilization while the last one acts as a loop connector in the distal RBM region (Lan et al., 2020). From the results obtained, it was observed that the loop regions of the spike RBD model (2 g, 3 b and 3f respectively) displayed maximum significant interactions.
A total of thirty ligands were selected from an extensive literature survey, WHO (www.who.int/covid-19/information) and FDA (www.fda.gov/drugs/emergency-preparedness-drugs/) organizations’ list of approved drugs designated for the treatment of SARS-CoV-2 to evaluate their therapeutic inhibitory potential against the spike Glycoprotein. These ligands were docked against Spike RBD model (receptor) utilizing iGEMDOCK v2.1 and their molecular interactions were analyzed (Table 6 ).
Table 6.
Molecular docking analysis of he selected ligands with the spike glycoprotein RBD model.
| Sr. No | Selected | PubChema | Drug Class | Drug Type | Ligand-spike RBD model Docked complex analysis |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Energy | VDWb | AverConPair | TIc | SId | LIe | |||||
| 1. | Azithromycin | 447043 | Antibiotic | Macrolide | −90.34 | −90.34 | 13.78 | 9 | 5 | No |
| 2. | Hydroxychloroquine | 3652 | Anti-malarial | Chloroquine derivative | −69.19 | −69.19 | 20.17 | 3 | 2 | No |
| 3. | Remdesivir | 121304016 | Anti-viral | ATP analogue | −83.36 | −83.36 | 14.59 | 9 | 8 | No |
| 4. | Peramivir | 154234 | Anti-viral | Neuraminidase inhibitor | −63.94 | −63.94 | 18.78 | 1 | 1 | No |
| 5. | Abacavir | 441300 | Anti-viral | Nucleoside RT*inhibitor Guanosine analogue | −73.03 | −73.03 | 25.52 | 4 | 4 | No |
| 6. | Didanosine | 135398739 | Anti-viral | Nucleoside RT inhibitor Purine analogue | −68.11 | −68.11 | 27.23 | 8 | 4 | No |
| 7. | Tenofovir | 464205 | Anti-viral | Acyclic Adenosine nucleotide analogue | −64.91 | −64.91 | 20.79 | 9 | 5 | Yes |
| 8. | Colistin | 5311054 | Antibiotic | Cyclic polypeptide | −85.92 | −85.92 | 13.33 | 9 | 5 | No |
| 9. | Eugenol | 3314 | Allylbenzene | Phenylpropanoid, Guaiacol derivative | −57.03 | −57.03 | 29.33 | 8 | 4 | Yes |
| 10. | Liquiritin | 503737 | Flavanone glycoside | Liquiritigenin derivative | −91.47 | −91.47 | 21.13 | 10 | 5 | No |
| 11. | Emblicanin A | 119058016 | Polyphenol | Antioxidant | −146.89 | −121.98 | 17.39 | 9 | 7 | No |
| 12. | 3-Carene | 26049 | Bicyclic monoterpene | Carane hydride derivative | −48.05 | −48.05 | 29.8 | 4 | 0 | No |
| 13. | Allicin | 65036 | Sulfenic acid thioester | Allylthiosulfonate | −51.36 | −51.36 | 30.22 | 6 | 4 | Yes |
| 14. | Glycyrrhizic acid | 14982 | Triterpene glycoside | Hepatoprotective drug | −99.17 | −99.17 | 13.19 | 12 | 7 | No |
| 15. | Nafamostat | 4413 | Anti-viral, Anti-cancer | Synthetic Serine protease inhibitor | −92.44 | −92.44 | 22.5 | 9 | 6 | No |
| 16. | Oseltamivir | 65028 | Anti-viral | Synthetic derivative prodrug of ethyl ester | −81.45 | −81.45 | 25.59 | 4 | 2 | No |
| 17. | Telbivudine | 159269 | Anti-viral | Synthetic Thymidine nucleoside analogue | −67.42 | −67.42 | 28.35 | 8 | 7 | No |
| 18. | Zanamavir | 60855 | Anti-viral | Sialic acid-analogue neuraminidase inhibitor | −67.26 | −67.26 | 20.26 | 4 | 3 | No |
| 19. | Stavudine | 18283 | Anti-viral | Nucleoside RT inhibitor Thymidine analogue | −68.16 | −68.16 | 25.93 | 7 | 5 | Yes |
| 20. | Raltegravir | 54671008 | Anti-viral | Integrase inhibitor | −93.62 | −93.62 | 20.34 | 12 | 7 | Yes |
| 21. | Zalcitabine | 24066 | Anti-viral | Synthetic dideoxynucleotide, RT inhibitor | −66.87 | −66.87 | 27.66 | 5 | 1 | Yes |
| 22. | Favipiravir | 492405 | Anti-viral | Pyrazine analogue, RNA-dependent RNA polymerase inhibitor | −53.70 | −53.70 | 27.90 | 5 | 3 | Yes |
| 23. | Ribavirin | 37542 | Anti-viral | Synthetic Ribofuranose nucleoside analogue | −66.63 | −66.63 | 26.94 | 6 | 4 | No |
| 24. | Galidesivir | 10445549 | Anti-viral | Adenosine analogue | −67.80 | −67.80 | 22.79 | 6 | 2 | No |
| 25. | Lopinavir | 92727 | Anti-viral | Anti-retroviral protease inhibitor | −103.75 | −103.75 | 14.52 | 8 | 3 | No |
| 26. | Ritonavir | 392622 | Anti-viral | Anti-retroviral protease inhibitor | −100.1 | −100.1 | 13.34 | 8 | 7 | No |
| 27. | Azadirachtin | 5281303 | Limonoids | Hepatoprotective drug | −85.92 | −85.92 | 13.37 | 9 | 5 | No |
| 28. | Camostat | 2536 | Anti-viral | Carbonyl compound | −88.78 | −88.78 | 21.24 | 7 | 2 | Yes |
| 29. | Doxycycline | 54671203 | Antibiotic | Synthetic tetracycline derived protein synthesis inhibitor | −77.46 | −77.46 | 15.31 | 7 | 4 | Yes |
| 30. | Ivermectin | 6321424 | Antiparasitic | Macrocyclic lactone | −102.63 | −102.63 | 11.98 | 10 | 5 | Yes |
PubChem CID = PubChem Chemical Identifier; b VDW = Van der Waal's interactions; c TI = Total Interactions; d SI = Significant Interactions (Bold); e LI = Loop Interactions; * RT = Reverse Transcriptase, Red colour indicates interaction with spike glycoprotein RBD-ACE-2 receptor residues.
The criteria for selection of the potential drug candidates as potential inhibitors of SARS-CoV-2 spike glycoprotein RBD model hinged on various factors viz. number and strength of the hydrogen bonds, number and strength of other interactions (electrostatic/hydrophobic/van der Waals), number of loop interactions (2 g, 3 b and 3f respectively), number of mutated residues involved, and number of non-significant interactions involving loop residues, etc. Based on the above factors, we selected ten drugs out of the thirty studied for further analysis whose detailed significant molecular interactions are entailed (Table 7 ). All selected drug candidates are illustrated in Fig. 9 .
Table 7.
Detailed analysis of selected drugs with spike glycoprotein RBD model involving significant interactions.
| Sr.No. | Drug | Significant spike RBD model interacting residues | Bond Type | Bond Length (in Å) | Interacting residues of Actual spike Glycoprotein |
Precise Loop Interactions |
Contact residues of the SARS-CoV-2 RBD–ACE2 | Cysteine residue interactions | |
|---|---|---|---|---|---|---|---|---|---|
| RBD (319–541) | RBM (437–508) | ||||||||
| 1 | Tenofovir | W436 | Conventional Hydrogen bond | 2.62 | Yes | No | No | No | No |
| F342 | Carbon Hydrogen bond | 3.30 | Yes | No | No | No | No | ||
| W436 | Electrostatic (pi-anion) | 3.48 | Yes | No | No | No | No | ||
| L441* | Hydrophobic (pi-sigma) | 3.58 | Yes | Yes | 2 g | No | No | ||
| W436 | Hydrophobic (pi-sigma) | 3.80 | Yes | No | No | No | No | ||
| 2 | Eugenol | E471 | Carbon Hydrogen bond | 3.30 | Yes | Yes | 3a | No | No |
| E471 | Carbon Hydrogen bond | 3.29 | Yes | Yes | 3a | No | No | ||
| D467 | Electrostatic (pi-anion) | 3.74 | Yes | Yes | No | No | No | ||
| K458 | Hydrophobic (pi-sigma) | 3.81 | Yes | Yes | 2 g | No | No | ||
| 3 | Allicin | E471 | Electrostatic interaction | 3.78 | Yes | Yes | 3a | No | No |
| K458 | Hydrogen bond | 2.70 | Yes | Yes | 2 g | No | No | ||
| S469 | Hydrogen bond | 3.08 | Yes | Yes | No | No | No | ||
| P491 | Hydrophobic interaction | 3.98 | Yes | Yes | 3 b | No | No | ||
| 4 | Stavudine | Y453 | Conventional Hydrogen bond | 2.35 | Yes | Yes | 2 g | Yes | No |
| N501* | Conventional Hydrogen bond | 3.05 | Yes | Yes | 3f | Yes | No | ||
| R403* | Carbon Hydrogen bond | 3.30 | Yes | Yes | 1e | No | No | ||
| Y495 | Carbon Hydrogen bond | 3.23 | Yes | Yes | 3f | No | No | ||
| S494 | Carbon Hydrogen bond | 3.30 | Yes | Yes | 3 b | No | No | ||
| 5 | Raltegravir | S349 | Conventional Hydrogen bond | 2.59 | Yes | No | No | No | No |
| S399 | Conventional Hydrogen bond | 2.98 | Yes | No | No | No | No | ||
| S349 | Conventional Hydrogen bond | 2.96 | Yes | No | No | No | No | ||
| F347 | Conventional Hydrogen bond | 2.90 | Yes | No | No | No | No | ||
| L441* | Halogen (Fluorine) | 3.65 | Yes | Yes | 2 g | Yes | No | ||
| N354 | Pi-Donor Hydrogen bond | 2.44 | Yes | No | No | No | No | ||
| A348 | Hydrophobic (alkyl) | 3.54 | Yes | No | No | No | No | ||
| 6 | Zalcitabine | R403* | Carbon Hydrogen bond | 3.30 | Yes | No | 1e | No | No |
| 7 | Favipiravir | R403* | Conventional Hydrogen bond | 3.05 | Yes | No | 1e | No | No |
| Y505 | Conventional Hydrogen bond | 3.30 | Yes | Yes | 3f | Yes | No | ||
| R403* | Carbon Hydrogen bond | 3.40 | Yes | No | 1e | No | No | ||
| 8 | Camostat | L441* | Conventional Hydrogen bond | 2.91 | Yes | Yes | 2 g | No | No |
| N354 | Carbon Hydrogen bond | 3.30 | Yes | No | No | No | No | ||
| 9 | Doxycycline | Y449 | Conventional Hydrogen bond | 1.87 | Yes | Yes | 2 g | Yes | No |
| N501* | Conventional Hydrogen bond | 2.35 | Yes | Yes | 3f | Yes | No | ||
| S494 | Conventional Hydrogen bond | 3.22 | Yes | Yes | 3 b | No | No | ||
| Y505 | Hydrophobic (pi-sigma) | 3.63 | Yes | Yes | 3f | Yes | No | ||
| 10 | Ivermectin | G381 | Conventional Hydrogen bond | 2.32 | Yes | No | No | No | No |
| F377 | Carbon Hydrogen bond | 2.99 | Yes | No | No | No | No | ||
| Y380 | Carbon Hydrogen bond | 3.74 | Yes | No | No | No | No | ||
| C379 | Sulfur-X | 2.99 | Yes | No | No | No | Yes | ||
| Y369 | Pi-Donor Hydrogen bond | 3.28 | Yes | No | No | No | No | ||
RBD = Receptor Binding Domain; RBM = Receptor Binding Motif; ACE-2 = Angiotensin Converting Enzyme type II; * = Possible mutated amino acids in SARS-CoV-2.
Fig. 9.

Structures of all ten selected drugs colored by CPK. Hydrogen atoms (white), Oxygen atoms (red), Chlorine atoms (green), Nitrogen atoms (blue) and Phosphorous atoms (orange). (Colour, Double-column fitting image).
Most significant interactions were observed in case of Stavudine, Doxycycline and Favipiravir (Fig. 10 a, b and 10c). Our results also indicated that Tenofovir, Eugenol, Allicin, Raltegravir, Zalcitabine, Camostat and Ivermectin depicted significant molecular interactions with the ACE-2 residues involved in spike glycoprotein recognition elucidating their inhibitory potentiality. Moreover, Raltegravir also interacted with loop 1e (LYS441) but via a halogen (fluorine) interaction. (Fig. S3a/b - S9a/b in supplementary data).
Fig. 10.

The docked complexes of Stavudine (10a), Doxycycline (10 b) and Favipiravir (10c) with the spike RBD model are depicted in Fig. i), iii) and v) respectively while ii), iv) and vi) represent the 2D interaction diagrams of Stavudine-, Doxycycline- and Favipiravir–spike RBD model docked complexes comprising of van der Waals (medium green), Conventional Hydrogen bonds (green), Carbon–Hydrogen bonds (light green), Pi-Pi T-Shaped (dark pink), Pi-Alkyl (light pink), Pi-Sigma (purple), Amide-Pi stacked (magenta) and Unfavourable Donor-Donor (red) interactions. (Colour, Double-column fitting image).
Remdesivir, Liquiritin, Nafamostat, Oseltamivir, Zanamavir, Lopinavir, EmblicaninA and Ivermectin depicted strong interactions with CYS336 and CYS379 residues of the Spike RBD model, thus hypothesizing their role in structural destabilization of the Spike protein (Table S2; Fig. 6 supplementary data).
In Fig. 11 we can see the residues at the interface of RBD of the spike protein and the ACE-2 receptor as listed earlier (Table 5). To understand whether the binding of inhibitor causes any structural changes in the RBD-ACE2 complex, we superimposed the structure of the RBD with and without the inhibitors. Favipiravir was found to interact directly with residues Y505, R403 and L441 of the RBD, where R403 is considered to be a possible mutation (Fig. 12 a, Fig. 6, Table 7). Surprisingly, upon superimposition of RBD and the RBD-Favipiravir complex we found deviation in side chain conformation of R403 (Fig. 12a). Apart from that few changes were seen in V401 and R509. This may envisage that Favipiravir alters the spike binding efficiency with ACE-2 receptor. Further, we also did a similar study with Hydroxycholoroquine and Nafamostat, both of which did not directly bind near the interface region of the Spike and ACE2 proteins. As it can be seen from Fig. 12 b, amino acids L517, T430, F429, F464, E516 and D428 were directly interacting with the hydroxychloroquine. The large number of non-polar residues promotes primarily hydrophobic interactions with the ligand. After superimposition we could observe the switch in the polar side chain of D428 of the spike protein. Although, we could not see much difference at the interface region of Spike-ACE2 complex, we could again find a prominent side chain shift of R403 towards RBD, in the presence of inhibitor, similar to favipiravir. Nafamostat showed interactions with S371, S373 A363, C336, F368, G339, S373, S371, V367, L335 and D 364 residues and not much difference was seen in the absence of the ligand (Fig. 12 c). C336 forms disulphide linkage with C361 and plays a crucial role in structureal stabilization of RBD. However, R403 shift remained consistent in all the three complexes. This indicates that inhibitor binding impacts the binding energetics of the RBD to the ACE2 receptor.
Fig. 11.

The protein-protein complex of RBD of Spike protein (magenta) and the ACE-2 receptor (yellow) are shown as cartoon. A large number of polar residues from the Spike protein interact with the ACE2 receptor by hydrogen bonding and hydrophobic interactions (Table 5). The primary residues from the Spike protein which interact with the ACE2 are shown as sticks and colored by CPK. (Colour, Double-column fitting image).
Fig. 12a.
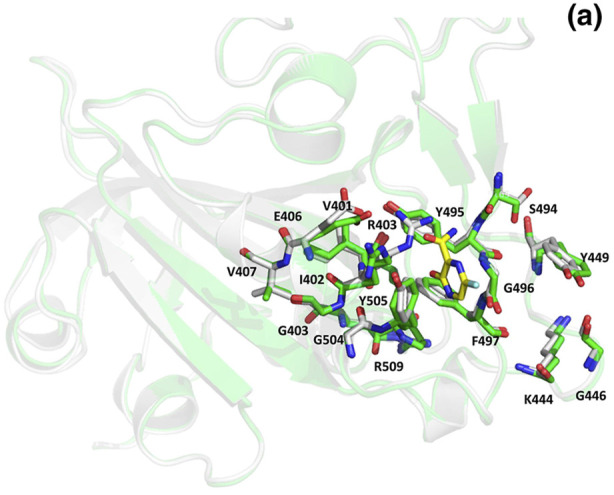
Superposed structures of Spike protein with (green) and without Favipiravir (white). The residues and the inhibitor are shown as sticks and colored by CPK. The carbon atoms of Favipiravir are shown in yellow. (Colour, Double-column fitting image).
Fig. 12b.
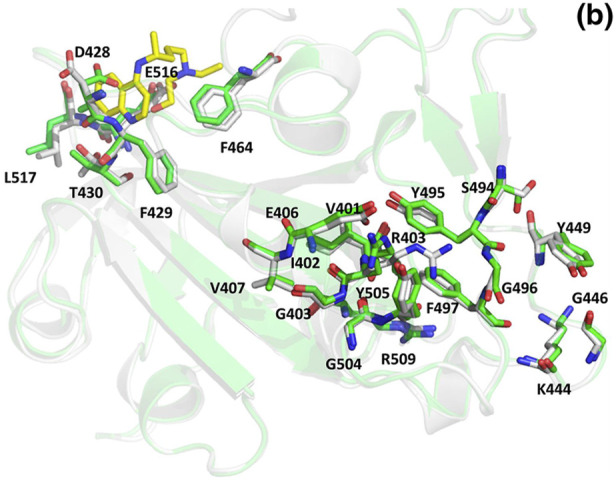
Superposed structures of Spike protein with (green) and without Hydroxychloroquine (white). The residues and the inhibitor are shown as sticks and colored by CPK. The carbon atoms of Hydroxychloroquine are shown in yellow. (Colour, Double-column fitting image).
Fig. 12c.
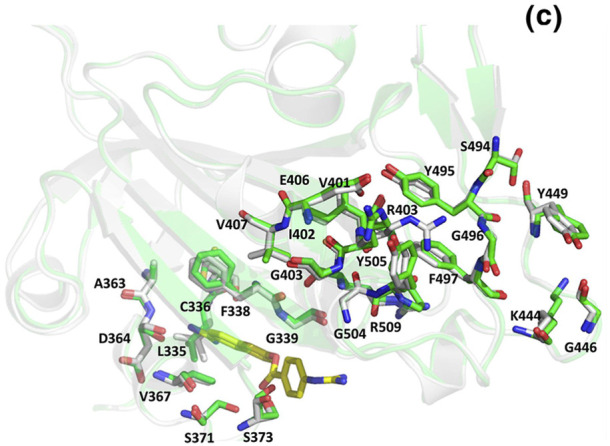
Superposed structures of Spike protein with (green) and without Nafamostat (white). The residues and the inhibitor are shown as sticks and colored by CPK. The carbon atoms of Camostat are shown in yellow. (Colour, Double-column fitting image).
Validation of the drug-Spike molecular interactions were achieved by docking studies of the selected drugs with the Spike glycoprotein trimeric structures available (PDB ID's 6VSB and 6VYB respectively) (Tables S4–S5 in supplementary data). We observed that Eugenol, Allicin, Doxycycline and Ivermectin did not depict any significant interactions with 6VSB. Furthermore, Tenofovir, Zalcitabine and Favipiravir depicted four significant interactions each with the Extracellular Topological Domain (ETD) of the S1 subunit while Camostat exhibited five significant interactions with the S2 subunit ETD. Likewise, Allicin, Stavudine, Raltegravir and Ivermectin elucidated no interactions with 6VYB. However, Camostat and Zalcitabine exhibited 1 and 4 interactions respectively with the Coiled-Coil Region 1 (CCR-1) of the S2 subunit. Eugenol and Doxycycline exhibited 1 interaction each with the ETD of S2′ domain and S1 subunit respectively (Figs. S10–S11 in supplementary data).
3.6. In silico toxicity prediction
SWISS-ADME determined the in silico physico-chemistry and pharmacokinetic analysis of the selected drugs. It was perceived that Ivermectin had the highest Po/w coefficient (iLOG Po/w = 5.86) while Favipiravir had the lowest value (iLOG Po/w = 0.39) signifying its higher solubility. Furthermore, Allicin exhibited the lowest skin permeation rate (log Kp = −9.89 cm/s) whereas Eugenol depicted the highest (log Kp = −5.69 cm/s). All drugs, except Ivermectin, complied with the Lipinski rules accompanied by minor violations. The GI absorption rate was high for all drugs except Tenofovir, Raltegravir, Doxycycline and Ivermectin. Out of all drugs, Eugenol and Allicin were predicted as Blood-Brain Barrier (BBB) permeants. Ultimately, all drugs had an acceptable bioavailability score, except for Doxycycline and Ivermectin (Tables S6–S7 in supplementary data).
OSIRIS Property Explorer was utilized to compute the systemic toxicity of the selected drugs. Our analysis showed that Raltegravir displayed the highest drug-likeness capacity (score = 0.999) while Tenofovir had the least possible value (0.0). Moreover, Eugenol and exemplified a high risk of mutagenicity, tumorigenicity and irritant capacity (scores of 0.6 in each) while Zalcitabine and Doxycycline exhibited a high risk of reproductive toxicity (score of 0.6). Favipiravir had the best drug score (0.933) amongst all potential drug candidates (Tables S8–S9 in supplementary data).
4. Discussion
Coronaviruses (CoV) are positive single-stranded RNA viruses classified as pathogenic agents reported since the 1960's. These viruses are responsible for causing acute and chronic respiratory diseases as well as enteric, hepatic and neurologic infections as it has a broad host range (avian, murine, porcine, bovine and other domestic mammalian species including humans) (Weiss and Martin, 2005; Di Gennaro et al., 2020).
SARS-CoV-2 has materialized as a potent etiological agent with a current infection rate of ~20,000 individuals/day. Till date, no confirmed drug/vaccine candidates have been reported to counteract COVID-19 epidemic rendering healthcare facilities to face comprehensive obstacles to restore the healthiness of COVID-19 affected individuals. This is predominantly accredited to the genomic discrepancy of SARS-CoV-2 as compared to previously established SARS-CoV and MERS-CoV strains, making SARS-CoV-2 more virulent than its predecessors. These anomalies have commanded stringent amendments in conventional treatment regimes in terms of efficacy for eradication of SARS-CoV-2 (Fig. 6).
Diverse experimental research analyses have yielded abundant information regarding SARS-CoV-2 life cycle, epidemiology, etc (Wu et al., 2020; Li et al., 2020). Molecular experimentation in coherence with computational biology has resulted in the fabrication of credible therapeutic strategies for combating COVID-19 viz. identification of proteomic and genomic drug targets, traditional epidemiological drug re-purposing, peptidomimetics studies for antibody/vaccine development against structural and non-structural proteins of SARS-CoV-2 targeting host attachment and viral replication processes, etc. (Alanagreh et al., 2020) (Fig. 1, Fig. 2).
The present study focused on the drug re-purposing based inhibition of SARS-CoV-2 attachment wherein the spike glycoprotein of SARS-CoV-2 undergoes conformational reorganization to distinguish its receptor ACE-2 and initiates a cascade of molecular progressions resulting in the integration with the cell membrane and liberation of its genome inside the host cell for replication.
We developed our validated model of the spike glycoprotein RBD based on the UniProtKB sequence P0DTC2 utilizing 6YLA chain E as the template (Fig. 5a). Multiple sequence alignment studies of the constructed spike RBD model with previously constructed 3-D structures demonstrated that SARS-CoV-2 exhibited significant mutations as compared to SARS-CoV and MERS-CoV strains resulting in an amplified affinity for ACE-2 (Fig. 6). The protein was predicted to be non-crystallizable and depicted minute disorderness. The spike glycoprotein RBD harboured many such mutations and exhibited additional sequential diversity which designates it as a latent therapeutic drug target (Table 2, Table 3, Table 4; Fig. 10a/10b/10c).
We implemented a computational biology–based approach for drug re-purposing to screen drug candidates as potential therapeutic inhibitors via molecular docking analyses impacting the spike glycoprotein mechanism of action (Tables 6–7).
Molecular docking analysis involving the spike glycoprotein RBD model (ligand) with ACE-2 (receptor) depicted twenty interactions of which fifteen were deemed significant and implicated loop 2 g, 3 b and 3f residues. Considering the above results in conjunction with the number and type of significant bonds formed, loop interactions, mutated amino acid residues concerned, etc., we observed that ten molecules out of the assorted set of thirty drug candidates exposited molecular interactions with the loop residues of the spike glycoprotein (Table 6, Table 7). Additionally, eight drugs exhibited significant interactions with the cysteine residues of the spike RBD model constituted in disulfide bond formation for structural stabilization (Table S2 in supplementary data).
Eugenol, an allyl-chain substituted guaiacol, is derived from essential oils such as clove oil, etc. It retains diverse pharmacological roles, including antiviral potentiality. It has been reported to have a detrimental effect on the viral envelope of newly formed virions as observed in studies with Herpes Simplex viruses HSV-1 and HSV-2 (Pramod et al., 2010). Similarly, Allicin (allyl 2-propenethiosulfinate) is a bioactive compound present in garlic extract and is reported to display antiviral activity against Influenza viruses A and B, HIV, herpes simplex viruses, rotavirus, etc. (Bayan et al., 2014). Eugenol exhibited interactions with the loop residues of spike glycoprotein but was deemed to be a BBB permeant, along with high risks of mutagenicity and tumorigenicity, questioning its administration for treatment of COVID-19 (Fig. S4; Table S6).
Stavudine, a thymidine nucleoside analogue, is a reverse transcriptase (RT) inhibitor and has been selectively utilized against HIV strains. Stavudine is intracellularly phosphorylated by cellular kinases to Stavudine triphosphate which competes with deoxythymidine triphosphate, the natural substrate of RT. It also inhibits DNA polymerases Beta and Gamma (Hurwitz and Schinazi, 2012). Favipiravir, a purine nucleoside analogue (6-fluoro-3-hydroxy-2-pyrazinecarboxamide), is a potent RNA-dependent RNA polymerase inhibitor where its phosphorylated form (T-705-RTP) exhibits broad-spectrum antiviral potency against retroviruses viz. arenavirus, bunyavirus, flavivirus, etc. (Du and Chen, 2020). Both depicted significant interactions with the ACE-2 binding regions of spike glycoprotein with no inherent risk of predicted systemic toxicity (Tables S6- S7).
Zalcitabine, another thymidine analogue, acts in the same way as Stavudine and is more potent (Leandro et al., 2013). It depicted two interactions with ARG403 (loop 1e) of the spike protein with a high risk of hazardous reproductive consequences. Tenofovir, an acyclic adenosine nucleotide analogue, is used in combinatorial therapy with other anti-retroviral drugs to treat HIV and singularly to treat Hepatitis B infections, acts as a reverse transcriptase inhibitor and depicted no predicted systemic toxicity upon administration (Delaney et al., 2006) (Fig. S7; Table S7).
Raltegravir is an anti-retroviral drug belonging to the class of integrase inhibitors, ultimately affecting viral DNA insertion and its subsequent integration into the host cell's DNA. It is primarily used to treat HIV-1 infections (Boesecke and Gelgor, 2009). It exhibited seven significant interactions of which only one targeted the loop residue of spike protein (LEU441) (Fig. S7). Similarly, Camostat (a carbonyl compound) is a synthetic serine protease inhibitor targeting the host cell TMPRSS2 (viral entry mediator) thereby inhibiting viral infection and replication (Uno, 2020). It also exhibits anti-inflammatory and anti-fibriotic capacity. Like Raltegravir, it interacted with LEU441 but overall had only two significant bonds. Both drugs lacked predicted toxicity upon administration (Fig. S8).
Doxycycline (a semi-synthetic derivative of oxytetracycline) is a protein synthesis inhibitor and exhibits broad spectrum anti-bacterial potency (Holmes and Charles, 2009). It also exhibits antiviral competence by inhibition of matrix metalloproteases in case of Dengue and other retroviruses or transcriptional up-regulation of intracellular zinc finger antiviral protein (ZAP) (Malek et al., 2020). Although it illustrated three significant spike RBD loop interactions, it was predicted to have a low bioavailability and posed a high risk of reproductive toxicity (Fig. 10b; Table S9). Ivermectin (an avermectin derivative), a broad-spectrum anti-parasitic agent is a potent endectocide and has been reported to inhibit the HIV-1 integrase and host cell Importin (responsible for nuclear import of viral proteins) subsequently hindering viral replication and also exhibits antiviral potency against other retroviruses (Caly et al., 2020). Ivermectin did not interact with any ACE-2 binding regions of Spike protein. However, it did depict a significant interaction with CYS379 which is involved in disulfide bond formation (CYS379-CYS432) and depicted no systemic toxicity whatsoever. Docking of the selected drug candidates with the spike glycoprotein trimeric structures elucidated distinctive subsidiary spike protein target regions in addition to the RBD, contriving the segmented drug targets within the spike protein for future drug development experiments (Fig. S9).
Several studies corroborate the usage of the drugs selected in the present study to counteract COVID-19. Remdesivir has reached the phase III level of clinical trials for the treatment of mild and moderate SARS-CoV-2 infections (Senanayake, 2020). Favipiravir, an antiviral/anti-influenza drug has been shortlisted in ten hospitals across India for a phase III trial with mild and moderately infected COVID-19 patients while an another combination of Doxycycline and Ivermectin has been recommended to treat acute symptoms of COVID-19 in Bangladesh which has reached to the experimental stage in India (Hafeez et al., 2020).
Another example of combinatorial therapy combines Emtricitabine/Tenofovir-Alafenamide and Lopinavir/Ritonavir to treat COVID-19 patients (Duan et al., 2020). Camostat and Nefamostat and their derivatives are certified protease inhibitors which are currently being investigated in Japan as therapeutic alternatives to combat SARS-Co-V 2 (Hoffman et al., 2020). No significant interaction was observed between RBD and hydroxychloroquine thus concluding that the mode of action of hydroxychloroquine is different than that of direct interaction with spike protein (Table 6;Fig. 12b). Chloroquine inhibits the viral attachment to its receptor along with the subsequent molecular processes involved in the final viral particle extrusion (Narkhede et al., 2020; Devaux et al., 2020). Nafamostat, primarily used for anticoagulant therapy, exhibited spike-mediated membrane fusion inhibitory potentiality (Li and Clercq, 2020). These experimental studies advocate drug-repurposing as a viable and resourceful technique to eradicate the infectivity of SARS-CoV-2 because of its cost-effectiveness and reduction of administration complexity in treatment regimes with high compatibility efficacy.
5. Conclusion
The present work comprised of selection and molecular docking analysis of a collection of 30 plausible drug candidates having diverse in silico mechanisms with the spike glycoprotein of SARS-CoV-2 aimed at identifying novel inhibitory competency of the same. Our results suggested that ten drugs out of the thirty selected could be utilized as promising drugs as they interacted with the experimentally validated ACE-2 binding residues and also depicted additional interactions with the ETD regions. Camostat, Favipiravir, Tenofovir, Raltegravir and Stavudine illustrated maximum interactions with the spike glycoprotein RBD model and the inherent trimeric structure displaying optimal bioavailability (score = 0.55) with absence of predicted systemic toxicity. As these drugs can be a good candidate for further in vitro or in vivo studies so dosage appropriate amalgamation of these drugs/drug derivatives in conjunction with refined experimental validation can serve as the platform for combinatorial drug therapy design and development to counter COVID-19 for futuristic applications.
Support
The authors have credited the sources of support received in the Acknowledgement page.
Permissions
The authors have duly credited all the sources which were utilized during the manuscript preparation.
Sources of funding
The present work has received no specific grant from any funding agency in the public, commercial or not-for-profit sectors in India or elsewhere.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
CRediT authorship contribution statement
Himanshu G. Toor: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing - original draft, Writing - review & editing, Final manuscript approval. Devjani I. Banerjee: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing - original draft, Writing - review & editing, Final manuscript approval. Soumya Lipsa Rath: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing - review & editing, Final manuscript approval. Siddhi A. Darji: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing - original draft, Writing - review & editing, Final manuscript approval.
Declaration of competing interest
The authors declare that they have no conflict(s) of interest.
Acknowledgements
The authors would like to express their sincere gratitude to the Dr. Vikram Sarabhai Institute of Cell and Molecular Biology, Faculty of Science, The Maharaja Sayajirao University of Baroda, Gujarat, India for providing the platform to conduct the present study. We are also thankful to Dr. Rina Soni (DST-Woman Scientist), Department of Chemistry, The Maharaja Sayajirao University of Baroda for her valuable suggestions. We are also grateful to Ashok and Rita Patel Institute of Integrated Study and Research in Biotechnology and Allied Sciences (ARIBAS), Gujarat; Charutar Vidya Mandal (CVM) and Sardar Patel University Gujarat, India for providing unconditional support for the duration of the present study.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejphar.2020.173720.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- Al-Khayyat M.Z.S., Al-Dabbagh A.G.A. In silico prediction and docking of tertiary structure of LuxI, an inducer synthase of Vibrio fischeri. Rep. Biochem. Mol. Biol. 2016;4(2):66–75. [PMC free article] [PubMed] [Google Scholar]
- A Chronicle on the SARS Epidemic Chin. Law Govern. 2003;36(4):12–15. doi: 10.2753/CLG0009-4609360412. [DOI] [Google Scholar]
- Alanagreh L., Alzoughool F., Atoum M. The human coronavirus disease COVID-19: its origin, characteristics, and insights into potential drugs and its mechanisms. Pathogens. 2020;9(331):1–11. doi: 10.3390/pathogens9050331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020;26:450–455. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthis N.J., Clore G.M. Sequence-specific determination of protein and peptide concentrations by absorbance at 205 nm. Proteome Sci. 2013;22:851–858. doi: 10.1002/pro.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R., Bairoch A., Wu C.H., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Martin M.J., Natale D.A., O'Donovan C., Redaschi N., Yeh L.L. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32:115–119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenteros J.J.A., Sonderby C.K., Sonderby S.K., Nielsen H., Winther O. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics. 2017;33(21):3387–3395. doi: 10.1093/bioinformatics/btx431. [DOI] [PubMed] [Google Scholar]
- Ayati A., Falahati M., Irannejad H., Emami S. Synthesis, in vitro antifungal evaluation and in silico study of 3-azolyl-4-chromanone phenylhydrazones. DARU J. Pharm. Sci. 2012;20(46):1–7. doi: 10.1186/2008-2231-20-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayan L., Koulivand P.H., Gorji A. Garlic: a review of potential therapeutic effects. Avicenna J. Phytomed. 2014;4(1):1–14. [PMC free article] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom N., Gammeltoft S., Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999;294(5):1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- Boesecke C., Gelgor L. A review of Raltegravir and its use in HIV-1 infection. Clin. Med. Therapeut. 2009;1:1159–1171. doi: 10.4137/CMT.S1985. [DOI] [Google Scholar]
- Caly L., Druce J.D., Catton M.G., Jans D.A., Wagstaff K.M. The FDA-approved drug Ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020;178:1–4. doi: 10.1016/j.antiviral.2020.104787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciliberto G. Boosting the arsenal against COVID-19 through computational drug repurposing. Drug Discov. Today. 2020;1–3 doi: 10.1016/j.drudis.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutard B., Valle C., de Lamballerie X., Canard B., Seidah N.G., Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020;176:1–5. doi: 10.1016/j.antiviral.2020.104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A., Zoete V. A BOILED-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11:1117–1121. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A., Michielin O., Zoete V. iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 2014;54(12):3284–3301. doi: 10.1021/ci500467k. [DOI] [PubMed] [Google Scholar]
- Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:1–13. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassault Systèmes Biovia . Dassault Systèmes; San Diego: 2017. Discovery Studio, v20.1.0.19295. [Google Scholar]
- Delaney IV W.E., Ray A.S., Yang H., Qi X., Xiong S., Zhu Y., Miller M.D. Intracellular metabolism and in vitro activity of Tenofovir against Hepatitis B virus. Antimicrob. Agents Chemother. 2006;50(7):2471–2477. doi: 10.1128/AAC.00138-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux C.A., Rolain J.M., Colson P., Raoult D. New insights on the antiviral effects of chloroquine against coronavirus: what to expect for COVID-19? Int. J. Antimicrob. Agents. 2020;55:1–6. doi: 10.1016/j.ijantimicag.2020.105938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Gennaro F., Pizzol D., Marotta C., Antunes M., Racalbuto V., Veronese N., Smith L. Coronavirus Diseases (COVID-19) current status and future perspectives: a narrative review. Int. J. Environ. Res. Publ. Health. 2020;17(2690):1–11. doi: 10.3390/ijerph17082690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdetskiy A., Cole C., Proctor J., Barton G.J. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015;43:389–394. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y., Chen X. Favipiravir: pharmacokinetics and concerns about clinical trials for 2019-nCoV Infection. Clin. Pharmacol. Therapeut. 2020;108(2):1–6. doi: 10.1002/cpt.1844. [DOI] [PubMed] [Google Scholar]
- Duan Y., Zhu H., Zhou C. Advance of promising targets and agents against COVID-19 in China. Drug Discov. Today. 2020;25(5):810–812. doi: 10.1016/j.drudis.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre F., Clote P. DiANNA: a web server for disulfide connectivity Prediction. Nucleic Acids Res. 2005;33:230–232. doi: 10.1093/nar/gki412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A., Do R.K.G., Sali A. Modeling of loops in protein structures. Proteome Sci. 2000;9(9):1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker J.M., editor. The Proteomics Protocols Handbook. Humana Press Inc.; Totowa, New Jersey: 2005. pp. 571–607. [Google Scholar]
- Gupta R., Brunak S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2002;7:310–322. [PubMed] [Google Scholar]
- Hafeez A., Saify Z.S., Naz A., Yasmin F., Akhtar N. Molecular docking study on the interaction of riboflavin (vitamin B2) and cyanocobalamin (vitamin B12) coenzymes. J. Comput. Med. 2013;1–5 doi: 10.1155/2013/312183. [DOI] [Google Scholar]
- Hafeez A., Ahmad S., Siddiqui S.A., Ahmad M., Mishra S. A review of COVID-19 (coronavirus disease-2019) diagnosis, treatments and prevention. EJMO. 2020;4(2):116–125. doi: 10.14744/ejmo.2020.90853. [DOI] [Google Scholar]
- Holmes N.E., Charles P.G.P. Safety and efficacy review of Doxycycline. Clin. Med. Therapeut. 2009;1:471–482. doi: 10.4137/CMT.S2035. [DOI] [Google Scholar]
- Hsu K., Chen Y., Lin S., Yang J. iGEMDOCK: a graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinf. 2011;12(1):1–11. doi: 10.1186/1471-2105-12-S1-S33. http://www.biomedcentral.com/1471-2105/12/S1/S33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz S.J., Schinazi R.F. Practical considerations for developing nucleoside reverse transcriptase inhibitors. Drug Discov. 2012;9(3):e183–e193. doi: 10.1016/j.ddtec.2012.09.003. Today Technol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar T., Nardaria U., Basak S., Deb D., Castiglione F., Mueller D.M., Srivastava A.P. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 2020;10:10895. doi: 10.1038/s41598-020-67749-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap S. Comparative in Silico studies on phytochemicals of ocimum as natural inhibitors of Ebola VP-35 protein. Indo American J. Pharm. Res. 2019;498–511 doi: 10.5281/zenodo.3524053. [DOI] [Google Scholar]
- Khodadadi E., Maroufi P., Khodadadi E., Esposito I., Ganbarov K., Esposito S., Yousefi M., Zeinalzadeh E., Kafil H.S. Study of combining virtual screening and antiviral treatments of the Sars-CoV-2 (Covid-19) Microb. Pathog. 2020;146:1–9. doi: 10.1016/j.micpath.2020.104241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiemer L., Lund O., Brunak S., Blom N. Coronavirus 3CLpro proteinase cleavage sites: possible relevance to SARS virus pathology. Bioinformatics. 2004;5(72):1–9. doi: 10.1186/1471-2105-5-72. http://www.biomedcentral.com/1471-2105/5/72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiemer L., Bendsten J.D., Blom N. NetAcet: prediction of N-terminal acetylation sites. Bioinformatics. 2005;21(7):1289. doi: 10.1093/bioinformatics/bti130. 1270. [DOI] [PubMed] [Google Scholar]
- Kim S., Thiessen P.A., Bolton E.E., Chen J., Fu G., Gindulyte A., Han L., He J., He S., Shoemaker B.A., Wang J., Yu B., Zhang J., Bryant S.H. PubChem substance and compound databases. Nucleic Acids Res. 2015;44:1202–1213. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausen M.S., Jespersen M.C., Nielsen H., Jensen K.K., Jurtz V.I., Sonderby C.K., Sommer M.O.A., Winther O., Nielsen M., Petersen B., Marcatili P. NetSurfP-2.0: improved prediction of protein structural features by integrated deep learning. Proteins. 2019;87(6):520–527. doi: 10.1002/prot.25674. [DOI] [PubMed] [Google Scholar]
- Ko J., Park H., Heo L., Seok C. GalxyWEBserver for protein structure prediction and refinement. Nucleic Acids Res. 2012;40:W294–W297. doi: 10.1093/nar/gks493. https://doi:10.1093/nar/gks493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota P., Ding F., Ramachandran S., Dokholyan N.V. Gaia: automated quality assessment of protein structure models. Bioinformatics. 2011;27(16):2209–2215. doi: 10.1093/bioinformatics/btr374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski L.P., Bujnicki J.M. MetaDisorder: a meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinf. 2012;13(111):1–11. doi: 10.1186/1471-2105-13-111. http://www.biomedcentral.com/1471-2105/13/111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Li M., Knyaz C., Tamura K. Mega X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurgan L., Razib A.A., Aghakhani S., Dick S., Mizianty M., Jahandideh S. CRYSTALP2: sequence-based protein crystallization propensity prediction. BMC Struct. Biol. 2009;9(50):1–14. doi: 10.1186/1472-6807-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Xhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;1581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Leandro K.C., Moreira J.C., Farias P.A.M. Determination of Zalcitabine in medicaments by differential pulse voltammetry. J. Pharm. (Lahore) 2013;1–6 doi: 10.1155/2013/495814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- Li X., Wang W., Zhao X., Zai J., Zhao Q., Li Y., Chaillon A. Transmission dynamics and evolutionary history of 2019‐nCoV. J. Med. Virol. 2020;92:501–511. doi: 10.1002/jmv.25701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell S.C., Davis I.W., Arendall W.B., III, de Bakker P.I.W., Word J.M., Prisant M.G., Richardson J.S., Richardson D.C. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Protein Struct. Funct. Genet. 2003;50(3):437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Maiti R., Van Domselaar G.H., Zhang H., Wishart D.S. SuperPose: a simple server for sophisticated structural superposition. Nucleic Acids Res. 2004;32:590–594. doi: 10.1093/nar/gkh477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek A.E., Granwehr B.P., Kontoyiannis D.P. Doxycycline as a potential partner of COVID-19 therapies. IDCases. 2020;21:1–3. doi: 10.1016/j.idcr.2020.e00864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin L.J., Bryson K., Jones D.T. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16(4):404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Meszaros B., Erdos G., Dosztanyi Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018;46:329–337. doi: 10.1093/nar/gky384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizianty M.J., Kurgan L. Sequence-based prediction of protein crystallization, purification and production propensity. Bioinformatics. 2011;27:24–33. doi: 10.1093/bioinformatics/btr229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkhede R.R., Cheke R.S., Ambhore J.P., Shinde S.D. The molecular docking study of potential drug candidates showing Anti-COVID-19 activity by exploring of therapeutic targets of SARS-CoV-2. Eurasian J. Med. Oncol. 2020;4(3):185–195. doi: 10.14744/ejmo.2020.31503. [DOI] [Google Scholar]
- Nielsen H. Predicting secretory proteins with SignalP. Methods Mol. Biol. 2017;1611:59–73. doi: 10.1007/978-1-4939-7015-5_6. [DOI] [PubMed] [Google Scholar]
- Ortega J.T., Serrano M.L., Pujol F.H., Rangel H.R. Role of Changes in SARS-COV-2 Spike protein in the interaction with the human ace2 receptor: an in silico analysis. Exp. Clin. Sci. 2020;19:410–417. doi: 10.17179/excli2020-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H., Seok C. Refinement of unreliable local regions in template-based protein models. Proteins. 2012;80:1974–1986. doi: 10.1002/prot.24086. [DOI] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Piovesan D., Tabaro F., Paladin L., Necci M., Micetic I., Camilloni C., Davey N., Dosztanyi Z., Meszaros B., Monzon A.M., Parisi G., Schad E., Sormanni P., Tompa P., Vendruscolo M., Vranken W.F., Tossato S.C.E. MobiDB 3.0: more annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res. 2018;46:471–476. doi: 10.1093/nar/gkx1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeepkiran J.A., Kumar K.K., Kumar Y.N., Bhaskar M. Modeling, molecular dynamics, and docking assessment of transcription factor rho: a potential drug target in Brucella melitensis 16M. Drug Des. Dev. Ther. 2015;9:1897–1912. doi: 10.2147/DDDT.S77020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prajapat M., Sarma P., Shekhar N., Avti P., Sinha S., Kaur H., Kumar S., Bhattacharyya A., Kumar H., Bansal S., Medji B. Drug targets for corona virus: a systematic review. Indian J. Pharmacol. 2020;52(1):56–65. doi: 10.4103/ijp.IJP_115_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramod K., Ansari S.H., Ali J. Eugenol: a natural compound with versatile pharmacological actions. Nat. Prod. Commun. 2010;5(12):1999–2006. doi: 10.1177/F1934578X1000501236. [DOI] [PubMed] [Google Scholar]
- Ramachandran S., Kota P., Ding F., Dokholyan N.V. Automated minimization of steric clashes in protein structures. Proteins. 2011;79(1):261–270. doi: 10.1002/prot.22879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson B. COVID-19 Coronavirus spike protein analysis for synthetic vaccines, a peptidomimetic antagonist, and therapeutic drugs, and analysis of a proposed achilles' heel conserved region to minimize probability of escape mutations and drug resistance. Comput. Biol. Med. 2020:1–67. doi: 10.1016/j.compbiomed.2020.103749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P., Obradovic Z., Kissinger C., Villafranca J.E., Dunker A.K. Identifying disordered regions in proteins from amino acid sequence. Neural Network. 1997:1–6. [Google Scholar]
- Sadati S.M., Gheibi N., Ranjbar S., Hashemzadeh M.S. Docking study of flavonoid derivatives as potent inhibitors of influenza H1N1 virus neuraminidase. Biomed. Rep. 2019;10:33–38. doi: 10.3892/br.2018.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Sarkar B., Ullah M.A., Araf Y. A systematic and reverse vaccinology approach to design novel subunit vaccines against Dengue virus type-1 (DENV-1) and human Papillomavirus-16 (HPV-16) Inform. Med. Unlocked. 2020;19:1–32. doi: 10.1016/j.imu.2020.100343. [DOI] [Google Scholar]
- Sayers E.W., Agarwala R., Bolton E.E., Brister J.R., Canese K., Clark K., Connor R., Fiorini N., Funk K., Hefferon T., Holmes J.B., Kim S., Kimchi A., Kitts P.A., Lathrop S., Lu Z., Madden T.L., Marchler-Bauer A., Phan L., Schneider V.A., Schoch C.L., Pruitt K.D., Ostell J. Database resources of the national center for Biotechnology information. Nucleic Acids Res. 2019;47:23–28. doi: 10.1093/nar/gky1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeliger D., de Groot B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010;24:417–422. doi: 10.1007/s10822-010-9352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senanayake S.L. Drug repurposing strategies for COVID-19. Future Drug. 2020:1–3. doi: 10.4155/fdd-2020-0010. Discov. [DOI] [Google Scholar]
- Shah B., Modi P., Sagar S.R. In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sci. 2020;252:1–12. doi: 10.1016/j.lfs.2020.117652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slabinski L., Jarozewski L., Rychlewski L., Wilson I.A., Lesley S.A., Godzik A. XtalPred: a web server for prediction of protein crystallizability. Bioinformatics. 2007;23(24):3403–3405. doi: 10.1093/bioinformatics/btm477. [DOI] [PubMed] [Google Scholar]
- Steentoft C., Vakhrushev S.Y., Joshi H.J., Kong Y., Vester-Christensen M.B., Scholdager K.T.-B.G., Lavrsen K., Dabelsteen S., Pedersen N.B., Marcos-Silva L., Gupta R., Bennet E.P., Mandel U., Brunak S., Wandall H.H., Levery S.B., Clausen H. EMBO J. 2013;32(10):1478–1488. doi: 10.1038/emboj.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson M. Planaria software LLC; Seattle, Wash, USA: 2004. ArgusLab 4.0.1. [Google Scholar]
- Uno Y. Camostat mesilate therapy for COVID-19. Intern. Emerg. Med. 2020:1–2. doi: 10.1007/s11739-020-02345-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacic V., Uversky V.N., Dunker A.K., Lonardi S. Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinf. 2007;8(211):1–7. doi: 10.1186/1471-2105-8-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.J., Tortoricci M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;180:281–292. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A.M., Procter J.B., Martin D.M.A., Clamp M., Barton G.J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B., Sali A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics. 2017;54(5):1–37. doi: 10.1002/cpbi.3. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S.R., Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005;69(4):635–664. doi: 10.1128/MMBR.69.4.635–664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng G., Wang E., Wang Z., Liu H., Zhu F., Li D., Hou T. HawkDock: a web server to predict and analyze the protein–protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019;47:322–330. doi: 10.1093/nar/gkz397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M., Chen L., Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020;10(5):766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D., Wu T., Liu Q., Yang Z. The SARS-CoV-2 outbreak: what we know. Int. J. Infect. Dis. 2020;94:44–48. doi: 10.1016/j.ijid.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Roy A., Zhang Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics. 2013;29(20):2588–2595. doi: 10.1093/bioinformatics/btt447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Roy A., Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 2013;41:1096–1103. doi: 10.1093/nar/gks966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6(14):1–18. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., tan W. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382(8):727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerkandl E., Pauling L. Evolving Genes and Proteins by V. Bryson and H.J. Vogel. Academic Press; New York: 1965. Evolutionary divergence and convergence, in proteins; pp. 97–166. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.