

Abstract
Alzheimer’s disease (AD) is a multifactorial disorder characterized by cognitive deficit and memory loss. The pathological feature of the disease involves β-amyloid senile plaques, reduced levels of acetylcholine neurotransmitter, oxidative stress and neurofibrillary tangles formation within the brain of AD patients. The present study aims to screen the inhibitory activity of newly synthesized and existing novel 4-methylthiocoumarin derivative against acetylcholinesterase, butyrylcholinesterase, BACE1, β-amyloid aggregation and oxidative stress involved in the AD pathogenesis. The in vitro assays used in this study were Ellman’s assay, FRET assays, Thioflavin T, transmission electron microscopy, circular dichroism, FRAP, and TEAC. Molecular docking and dynamics studies were performed to correlate the results. C3 and C7 (thiocoumarin derivatives) were found to be the most potent inhibitors of acetylcholinesterase (IC50-5.63 µM) and butyrylcholinesterase (IC50-3.40 µM) using Ellman’s assays. Enzyme kinetic studies showed that C3 and C7 compounds followed by the mixed mode of inhibition using LB plot. C3 also moderately inhibited the BACE1 using FRET assay. C3 inhibited the fibrillization of β-amyloid peptides in a concentration-dependent manner as observed by Thioflavin T, TEM studies and Circular dichroism data. Molecular modeling studies were performed to understand the probable mode of binding of C3 and C7 in the binding pocket of acetylcholinesterase, butyrylcholinesterase, BACE1 and amyloid β peptides. This indicates the important role of hydrophobic interactions between C3 and acetylcholinesterase. C3 also exhibited significant antioxidant potential by FRAP and TEAC assays. Hence, C3 might serve as a promising lead for developing novel multi target-directed ligand for the treatment of AD.
Electronic supplementary material
The online version of this article (10.1007/s13205-020-02481-1) contains supplementary material, which is available to authorized users.
Keywords: 4-Methylthiocoumarin derivative, Alzheimer’s disease, BACE1 inhibitor, Cholinesterase inhibitor, In silico studies
Introduction
Alzheimer’s disease (AD) is a complex multifactorial neurodegenerative disease of the central nervous system (CNS) characterized by cognitive impairment, behavioral abnormalities, memory loss, and a decline in language skill (Güvenalp et al. 2017). Age is one of the significant risk factors associated with AD. Various socio-economic factors like low education as well as small occupation class which increases the risk factor of AD (Hasselgren et al. 2018). This leads to poor nutrition and hence decreases medical treatment or health care facilities (Alzheimer's Association 2019). At present, 33–50% of people at the age of 85 are suffering from AD, while 20% are in the range of 75–80 years old. It is estimated that by 2050, there will be around 70 million AD patients. The disease pathology of AD involves multiple factors such as low levels of acetylcholine (ACh) in CNS, the formation of β-amyloid (Aβ) senile plaques, oxidative stress, and formation of neurofibrillary tangles. Therefore, “one molecule and one target” approach is not suitable to cure a complex disease such as AD. Because of this, scientists are emphasizing to multi-target-directed ligands (MTDL) approach, where a single compound can concomitantly interact with different receptors or enzymes involved in the pathology of AD (Czarnecka et al. 2017). Hence, the present study focusses on different targets, namely cholinesterase enzymes, β-secretase cleavage enzyme (BACE1), monoamine oxidase B (MAO B) and Aβ which can be inhibited by a single compound.
The most important known therapeutic target of AD is acetylcholinesterase (AChE; EC 3.1.1.7) and butyrylcholinesterase (BuChE; EC 3.1.1.8). These enzymes belong to the family of cholinesterase and are present in the CNS, which cleaves carboxylic ester chemical bonds from its substrate ACh (Lionetto et al. 2013). Both the enzymes share 65% sequence homology. However, they differ in kinetics and substrate specificity in addition to differential localization in the brain. In the early stages of AD, ACh levels are predominantly regulated by AChE (80%). Therefore, AChE inhibitors are efficiently preventing its hydrolysis, but in advanced AD, the ratio of BuChE/AChE slowly increases; hence, compounds inhibiting BuChE may also be important in the treatment of AD (Larik et al. 2020). Therefore, cholinesterase inhibitors are one of the potent therapeutic options recommended for the treatment of AD. The X-ray crystallographic structure of AChE reveals the substrate-binding site of AChE lies within a deep groove of 20 Å called ‘gorge’. The binding of ACh starts at the rim of ‘gorge’ called ‘peripheral anionic site’ (PAS), whereas hydrolysis takes place at the bottom of ‘gorge’ called the ‘catalytic anionic site’ (CAS). The main factor regulating the substrate specificity of AChE is aromatic amino acids lining the binding site that limits the size of the substrate, which can bind to AChE. Three aromatic residues present in PAS of AChE, whereas it is absent in BuChE; hence, AChE PAS ligands like donepezil have a weaker affinity for BuChE (Das et al. 2017).
According to the “amyloid hypothesis”, the production of Aβ peptides is the result of sequential cleavage of amyloid precursor protein (APP) by BACE 1 and γ-secretase cleavage enzyme resulting in aggregation of amyloid plaques that eventually leads to neuronal cell death (Kumar et al. 2018). BACE1 cleaves APP, which generates two different peptide lengths, i.e., Aβ40 and Aβ42, both are neurotoxic. Evidence shows that AChE binds to amyloid-β (Aβ) by hydrophobic environment close to PAS accelerates the pro-aggregation of Aβ monomers into oligomers and fibril, resulting in aggravating AChE dependent Aβ neurotoxicity. In view of this, the development of dual inhibitors was proposed that can inhibit both AChE and BACE-1, a new class of therapeutics that can prevent amyloid aggregation (Green et al. 2018).
Another hypothesis responsible for the pathogenesis of AD is ‘oxidative stress’ which suggests that with age or under stress condition, there is an increased formation of reactive oxygen species (ROS) within the mitochondria with no efficient antioxidant system and hence, the risk of developing AD increases (Nita and Grzybowski 2016). The underlying mechanism behind the induction of oxidative stress is metal accumulation, mitochondrial dysfunction, Aβ, hyperphosphorylated tau, and inflammation (Chen and Zhong 2014). Oxidative stress is responsible for lipid peroxidation, decreased polyunsaturated fatty acids, increased DNA and protein oxidation, diminished energy metabolism in the brain AD patients (Droge 2002; Sarma et al. 2010). Other pieces of evidence show that hyperphosphorylated tau protein aggregates bind to Fe3+, which leads to the production of neurofibrillary tangles, whereas Aβ chelates with transition metal ions (Zn2+, Fe3+, Cu2+) (Phaniendra et al. 2015).
Furthermore, MAO (EC 1.4.3.4) enzyme has been proposed as a therapeutic target as it is involved in elevated production of reactive oxygen species (ROS), which is responsible for neuronal cell injury and death in AD. It is a family of flavin adenine dinucleotide (FAD) containing enzymes that are found in the outer membrane of mitochondria in various cells present in the nerve terminals, liver, gut mucosa, and other tissues. There exist two isoforms of MAO, i.e., MAO-A and MAO B, which is responsible for the catalysis of the deamination of amines, like norepinephrine, dopamine, and serotonin. MAO B is abundant in the brain, is of therapeutic use in various neurological diseases like AD and PD (Kumar et al. 2017). MAO produces hydrogen peroxide (H2O2) and oxygen radicals in AD patients’ results in neuronal cell death (Borroni et al. 2017). Hence, compound inhibiting MAO-B activity and anti-oxidant property may reduce the oxidative stress and neurodegeneration, thus retarding the progression of AD.
Currently, few small molecules are present which inhibit BACE1, γ-secretase, have anti-amyloidogenic property, tau aggregation or microglial activation, and immunotherapeutic agents are under clinical trial. Still, only a few FDA approved drugs available for the treatment of AD (Zhu et al. 2019). Available drugs for AD such as donepezil, tacrine, physostigmine, galantamine, rivastigmine, neostigmine, and memantine only provide symptomatic relief to the patients. They are also associated with several side effects such as nausea, vomiting, hepatoxicity, gastrointestinal toxicity, and many more (Czarnecka et al. 2018). Because of this, there is a continuous search for small molecules which have disease-modifying abilities and comparatively lesser side effects on the multifactorial disease like AD.
Coumarin is a class of benzopyrones that interacts with several enzymes exhibiting a wide range of pharmacological properties like neuroprotective properties, antioxidant, anti-inflammatory, anticoagulant, antiviral, antihyperglycemic, anticonvulsant, antibacterial, antihypertensive, antifungal, anti-adipogenic, anti-tubercular and anti-cancer (Detsi et al. 2017). The aromatic center of coumarins can be used as a scaffold to synthesize various novel derivatives that might have improved therapeutic activity. Studies have shown thiocoumarin to have many therapeutic properties like anticoagulants, anthelmintic, antiallergic, mitochondrial swelling inhibition, choleretic activity (Borg et al. 1999), 15-lipoxygenase inhibitor (Khavari et al. 2017), anticonvulsant activity (Mokrov et al. 2019), anti-bacterial properties (De et al. 1962) and also has an apoptogenic effect (Goel et al. 2009). Several pieces of evidence show coumarin derivatives as an AChE inhibitor (AChEI) due to their ability to bind to PAS of AChE via T-stacking and steric interactions (Modh et al. 2013). Still, none has observed the same for thiocoumarins. Studies have shown that the 4th position of coumarin has found to be more favorable for inhibiting AChE by binding to the catalytic site contributing to the dual-site inhibitory-activity. Torres et al. have shown that 4-methylcoumarins/1,4-substituted 1,2,3-triazole conjugates inhibit E. electricus AChE efficiently (Torres et al. 2016). Another derivative of coumarin, AP2238, has demonstrated inhibitory activity against AChE (Tarozzi et al. 2014). Recent studies have proven coumarin derivatives to be of a therapeutic role in AD (Joubert et al. 2017; Hamulakova et al. 2018; He et al. 2018; Jalili-Baleh et al. 2018; Tehrani et al. 2019; Shi et al. 2020), but none has shown the importance of introducing the thio group in the coumarin scaffold. Therefore, the present study involves the synthesis of novel thiocoumarin derivative and comparison of 4-methylcoumarin and 4-methylthiocoumarin derivatives for their anti-cholinesterase, anti-BACE1, anti-MAO B, anti-amyloidogenic and antioxidant potential by in vitro assays and in silico studies.
Materials and methods
Chemicals/reagents
AChE (EC 3.1.1.7) from Electrophorus electricus (electric eel) and BuChE (EC 3.1.1.8) from equine serum, acetythiolcholine iodide (ATChI), butyrylcholine iodide (BuChI), 5:5-dithiobis-2-nitrobenzoic acid (DTNB), 4,7-dihydroxy coumarin sodium bicarbonate, thioflavin T, and were purchased from Sigma, India. Glacial acetic acid, TPTZ, ferric chloride, ferrous sulphate, ABTS glycine, sodium hydroxide from SRL chemicals.
Test compounds
Most of the test compounds (coumarins and thiocoumarins) mentioned in Fig. 1 were synthesized in the laboratory using the Pechmann condensation process and by a slight modification of previous literature reports (Pechmann and Duisberg 1883; Kumar et al. 2005; Tyagi et al. 2008; Raj et al.1998). To do the acetylation, propylation, and butylation, reference was taken from the previous publication (Tyagi et al. 2010). The structures of synthesized compounds were confirmed by their melting points and spectral data reported in the literature (Kumar et al. 2005; Tyagi et al. 2008).
Fig. 1.

Structures of the 4-methylcoumarin, thiocoumarin derivatives and reference compounds (other coumarin derivatives available against Alzheimer’s disease)
General procedure for the preparation of novel 7, 8-dipropoxy-4-methylthiocoumarin
The compound 2 (1.00 g, 3.12 mmol) was added to the solution of Phosphorus pentasulfide (2.08 g, 9.37 mmol) in 1,4-dioxane (20 mL), and the reaction mixture was refluxed at 110 °C for overnight (Scheme 1). The reaction was monitored on TLC. After completion of reaction, the solvent was evaporated on vacuum and the crude compound was purified by the column chromatography using ethylacetate: hexane (2:3 v/v) to yield a green amorphous compound 3 with 52% yield, mp 225–230 °C; compound was characterized by FTIR, UV–Vis, 1H, 13C NMR spectral data. UV (CHCl3λ(nm)): 400, 272; FTIR (KBr): 2771, 2319, 1771 (C=O), 1612, 1562, 1437, 1367, 1306, 1252, 1100 cm−1; 1H NMR spectrum (400 MHz, DMSO-d6, ppm): δ 1.32 (t, 3H, J = 5.6 Hz, CH3), δ 1.92 (t, 3H, J = 5.6 Hz, CH3), δ 2.66–2.72 (4H, m, 2×OCOCH2CH3), δ 7.26 (3H, s, CH3), δ 7.40 (1H, d, J = 7.2 Hz, C-5H), δ 7.79 (1H, d, J = 7.2 Hz, C-6H); 13C NMR spectrum (100.5 MHz, DMSO-d6): δ = 18.042 (CH3), 26.93 and 27.12 (2×OCOCH2CH3), 39.30 and 39.80 (2×OCOCH2CH3), 116.25 (C-10), 120.45 (C-6) 120.854 (C-5) 128.41 (C-3), 145.65 and 146.27 (C-7 and C-8) 147.79 (C-4), 150.46 (C-9), 171.34 (2×C=O) and 195.73 (C=S); LC–MS for C16H16O5S [M + Na]+found 343.943.
Scheme 1.

Synthesis of 7,8-dipropoxy-4-methylthiocoumarin (3)
Cholinesterase assays
An evaluation of cholinesterase inhibition was carried out in 96-well flat-bottom microtitre plates. Using the colorimetric method of Okello (Okello et al. 2004). A typical run consisted of 0.1 M phosphate buffer pH 8 (200µL); AChE/BuChE enzyme solution (5 µL), at final assay concentrations of 0.06 U/mL and 0.03 U/mL, respectively; test compound (5 µL) and finally DTNB (5 µL) prepared in 0.1 M phosphate buffer pH 7 at a final concentration of 0.3 mM with 0.12 M of sodium bicarbonate. The reactants were then pre-incubated at 30 °C for 15 min. By adding substrate ATChI/BuChI (5 µL) at final concentrations of 0.5 mM, the reaction was initiated. The buffer solution was added instead of the test compounds in the control and assay was done in triplicates. To evaluate any non-enzymatic hydrolysis in the reaction, two blanks for each run were prepared in triplicates. One blank consisting of enzyme instead of buffer and other contains the substrate. Absorbance was measured at 412 nm on 96-well microplate reader SpectraMax M2, for 6 min at 30 °C. Using linear regression analysis, IC50 of the test compounds that inhibited the substrate ATChI/BuChI was calculated by plotting the percentage inhibition curve vs. concentration graph. The concentration required to inhibit half of the maximum biological activity is the IC50 value of the test compound.
Kinetic study of the enzyme AChE and BuChE was performed with three different concentrations of test compounds (Kumar et al. 2011). The data was analyzed using Lineweaver–Burk methods where at a varying concentration of substrate (0.5 mM to 0.0625 mM), 1/velocity against 1/substrate was plotted.
BACE1 inhibition assay
BACE 1 inhibition assay was performed by the kit purchased from Sigma, India (Sharma et al. 2019). The substrate was prepared in the assay buffer at a concentration of 50 µM, and the enzyme was made in the assay buffer at the final concentration of 0.3 units/µL. Stock solutions of test compounds (C1–C15) were prepared in DMSO and then diluted in the assay buffer to 20 µM (final concentration). BACE1 enzyme, substrate, standard, test compounds, and buffer were then added to the 96 well microlitre black plate for a total volume of 100 µl, with the BACE1 enzyme being added last. Baseline fluorescence was recorded immediately after the addition of the BACE1 enzyme at excitation 320 nm, emission 405 nm. Black plates were then covered and incubated at 37 °C for 3 h, following which fluorescence was recorded again.
Modelling studies
Modelling studies were performed to understand the potential binding mode and interactions of the most active compounds C3 with AChE and BACE1, compound C7 with BuChE. Crystal structure co-ordinates of AChE, BuChE and BACE1 were used as their respective three-dimensional (3D) receptor models. In contrast, 3D co-ordinates of compound C3 and C7 were calculated by the molecular modelling approach. These compounds were modelled in their respective binding pocket followed by refinement of docked conformation using Molecular Dynamics (MD) simulations in the presence of water solvent models.
Protein preparation
Each of the protein structures, AChE(PDB ID: 4EY6) (Cheung et al. 2012), BuChE (PDB ID: 1P0M) (Nicolet et al. 2003), and BACE 1 (PDB ID: 4D8C) (Rueeger et al. 2012) (supplementary data, Fig. 2) was prepared as a receptor model for docking studies with the help of ‘Protein Preparation Wizard’ workflow in Schrodinger Suite of molecular modelling programs. During preparation, all the non-protein atoms, including water, were removed, proper bond order was assigned, the protonation state of side chains was assigned, hydrogens were added, and force field parameters of OPLS_2005 and partial charges (Kaminski et al. 2001) was assigned to the receptor atoms. Then protein structure was energy minimized to remove the steric clash and maximize the intramolecular interactions. Energy minimization was performed using the conjugate gradient algorithm with the convergent threshold of 0.05 kcal/mol/Å energy gradient. The binding site for docking of the compound was defined around the active/binding site of the protein, and the energy grid was generated around the binding site with the help of the “Receptor Grid Generation” workflow in Schrödinger suite.
Fig. 2.

Kinetic study of different concentrations of lead compound with cholinesterase enzyme using LB plot; a AChE of C7; b BuChEof C7; c AChE of C3; d BuChEof C3; filled square 80 µM; filled triangle 40 µM; cross mark 20 µM; filled diamond control
Ligand preparation
2D chemical structure of each compound (C3 and C7) was sketched in Maestro, ionization state of the compound at neutral pH was assigned using Epik (Shelley et al. 2007), and their 3D coordinates were generated using ConfGen program (Watts et al. 2010) in the “Ligand Preparation” workflow of Schrodinger suite. Generated 3D structure of the compound was energy minimized by conjugate gradient energy minimization algorithm with a convergence threshold of 0.05 kcal/mol/Å with the help of OPLS force field parameters similar to protein structure. These prepared ligands were used for docking studies with AChE, BuChE, and BACE 1.
Molecular docking studies
Glide docking protocol was used for docking studies of compounds C3 and C7 with AChE and BuChE, respectively. BACE1 was docked with compound C3. Glide uses incremental construction algorithm for the conformational sampling of the ligand to generate the compatible docked conformation in the binding pocket of receptor and then score the generated docked poses (positional conformer) using an empirical scoring function, Glide score, which is an approximate measure of free energy of binding (ΔG). However, before docking of compounds C3 and C7, docking protocol was validated by re-docking the co-crystallized ligand in the binding pocket of prepared protein structures (PDB ID: 4EY6, 1P0M and 4D8C) to assess the predictive power of docking protocol to generate the native conformation of the ligand. Based on the validation results, prepared compounds C3 and C7 were docked in the defined binding site using the same protocol used for validation. Ligands and selected side chains in the binding site were treated flexibly during the docking process while keeping the rest of the protein restrained.
Molecular dynamics simulations
Docked complex of compound C3 and C7 was energy minimized to remove van der Waal clash and further refined to relax the complex in the solvated state by molecular dynamics (MD) simulation. MD simulation of the minimized docked complex was performed for 10 ns using the MD simulation program Desmond (Bowers et al. 2006) with the help of OPLS_2005 force field parameters. The docked complex was solvated by TIP3P (Jorgensen et al. 1983) water model in an orthorhombic simulation box having a boundary extension of 10 Å from protein atoms in each direction. Simulations were performed under NPT (isothermal–isobaric) condition at 300 K and 1-atmosphere pressure and periodic boundary condition using reversible reference system propagator algorithm (RESPA) (Tuckerman et al. 1992) dynamics integrator with the time step of 2 fs in the presence of SHAKE (Ryckaert et al. 1977) constraint. Nose–Hoover chain thermostat model(Martyna et al. 1992) with a relaxation time of 1.0 ps was used to regulate the temperature. In comparison, the Martyna–Tobias–Klein barostat method (Martyna et al. 1994) with a relaxation time of 2.0 ps was used to control the pressure. Short-range electrostatic interactions and van der Waals interactions were calculated using the cut-off method at a radius of 9.0 Å. In contrast, long-range electrostatic interactions were calculated by the ‘smooth particle mesh Ewald’ method. Trajectories after equilibration point were considered for evaluation. Similar approaches and run parameters were used in previous studies (Kumar et al. 2015; Pandey et al. 2019; Tripathi et al. 2020). The conformation at the end of simulation was energy minimized and used as the final model complex to analyze the potential binding mode of ligand and interactions between ligand and receptor.
Aβ fibril formation
All Aβ samples (44 µM) with and without different test compounds (C3 and C7; 44 µM) were prepared immediately before each experiment. To generate amyloid fibrils, Aβ alone, and Aβ with test compound solutions were incubated at 37 °C for 48hours and being stirred at 200 rpm (pH-7) (Kumar et al. 2010). Control reactions were carried out in the presence of 1% DMSO vehicle. Compound showing the highest anti-amyloidogenic property was analyzed at three concentrations, i.e., 11 µM, 22 µM, and 44 µM.
Inhibition of Aβ fibrillization by ThT assay
20 μl of Aβ sample with and without test compound (C3 and C7) was added to 80 μl of 10 μM thioflavin T (ThT) solution, in the glycine–NaOH buffer (50 mM; pH-9), and mixed gently. Readings were taken at 440, 490 nm. Fluorescence emission spectra (400–650 nm) were taken using an excitation wavelength of 440 nm (Necula et al. 2007). Tannic acid was used as a positive control in the assay (Ono et al. 2004).
Inhibition of Aβ fibrillization by TEM
The Aβ sample (44 µM) was incubated in the presence and absence of test compound (C3, 44 µM) at 37 °C and being stirred at 200 rpm. After incubation for 48 h, the samples (40 µL) were placed on a carbon-coated copper grid. The grid was negatively stained with 1% uranyl acetate for 5 min at room temperature. The specimen was transferred in TEM (JEOL JEM-2100F) for imaging, after draining off the excess staining solution (Meena et al. 2016).
Inhibition of Aβ fibrillization by CD
Sample containing Aβ fibrils with or without C3 at a concentration of 0.1 mg/mL in a solution for used for CD analysis. CD spectra were recorded at room temperature in a 0.1 cm path length cell on Applied PhotoPhysics spectropolarimeter over the range of 200–250 nm (Chenet al. 2006). Results were depicted as CD (mdeg) vs wavelength. CD data were analyzed by K2D2 software (Perez-Iratxeta and Andrade-Navarro 2008).
Inhibition of Aβ fibrillization by in silico study
To study the potential interaction mechanism of Aβ peptide with test compounds (C3 and C7), molecular docking studies were performed using the Schrodinger suite. For Aβ peptide, initial coordinates were taken from the NMR structure (PDB ID: 1IYT) (Crescenzi et al. 2002). Before performing molecular docking, the peptide was pre-processed using the ‘Protein Preparation Wizard’ workflow. Hydrogen atoms were added, and bond order was assigned. Energy minimization was done using the OPLS_2005 force field. To get an insight of the amyloidogenic region, FoldAmyloid software was used (Garbuzynskiy et al. 2009). Grid was generated around the same region using the ‘Receptor grid generation’ workflow to keep all the functional residues in the grid (Sastry et al. 2013). With this model, docking studies were performed using Glide extra precision docking module (Friesner et al. 2006). Homotaurine was used as a positive control for studying in silico anti-amyloidogenic potential (Kocis et al. 2017).
MAO B inhibition assay
This assay was performed according to the protocol given by the kit purchased from Biovision, India (Liu et al. 2018). Assay buffer and the test compounds (20 µM) were added to 96 well black plates followed by diluted MAO-B enzyme and were incubated for 10 min at 37 °C. Then, the substrate solution was added and fluorescence (Ex/Em = 535/587 nm) was recorded.
Antioxidant assay
ABTS assay
The stock solution contained 9:1 volumes of 7 mM ABTS salt and 2.45 mM potassium persulfate, respectively, which was kept in the dark at room temperature for 16 h. Before experimentation, the stock ABTS*+ solution was diluted with a phosphate buffer solution (PBS) until an absorbance of 0.700 was obtained at 734 nm. Varying concentrations of the test compound (20 µM) were allowed to react with ABTS*+ solution and the absorbance was taken at 734 nm. Trolox was used as a standard in the experiment. The percentage scavenging capacity of the test compound (C1–C15) was calculated as:
where Acontrol is the absorbance of ABTS radical + ethanol and Asample is the absorbance of ABTS radical + test compound/trolox. A graph was plotted between percentage of scavenging versus compound concentration (Pisoschi and Negulescu 2011). All reactions were carried out in triplicates.
Ferric reducing antioxidant power (FRAP) assay
Another method used to measure the antioxidant capacity of 4-methylcoumarin and 4-methylthiocoumarin derivatives is Ferric reducing antioxidant power (FRAP), uses the method of Benzie and Strain (1996). This is a spectrophotometric assay which involves the reduction of Fe3+–TPTZ complex (colorless complex) to Fe2+–tripyridyltriazine (blue colored complex) formed at low pH in the presence of electron-donating antioxidants at 593 nm. The FRAP reagent was prepared by mixing 300 mM acetate buffer, 10 ml TPTZ in 40 mM HCl and 20 mM FeCl3·6H2O in the proportion of 10:1:1 at 37°. In 96-well micro titre plate, 180 µL of frap reagent was added, followed by 20 µL of concentrations of test compound (20 µM) or FeSO4 (standard used) was incubated for 10 min at 37 °C. All reactions were carried out in triplicates. The FRAP values of test compounds were determined by comparing the change in absorbance w.r.t to that of the standard used and expressed as µM Fe equivalent of the test compounds (C1–C15).
Pharmacokinetic parameters
ADME (absorption, distribution, metabolism, and excretion) studies of these 4-methylcoumarin and 4-methylthiocoumarin derivatives were predicted using Qikprop software. Various parameters like molecular weight, QPlogPo/w, no of hydrogen bond acceptors, number of hydrogen bond donors, QPlog BB, and Percent Human Oral Absorption were calculated to analyze for violation of the Lipinski’s rule of five, the rule of three, its human oral absorption and permeability through Blood–brain barrier (BBB).
Results
Synthesis of 7, 8-dipropoxy-4-methylthiocoumarin
As illustrated in Scheme 1, the novel target compound 7,8-dipropoxy-4-methylthiocoumarin (C3) was prepared based on a well-known method. Briefly, the reaction of pyrogallic acid with ethyl acetoacetate in the presence of H2SO4 gave 7,8-dihydroxy-4-methylcoumarin C1. The 7,8-dipropoxy-4-methylcoumarin C2 was prepared from the reaction of 7, 8-dihydroxy-4-methylcoumarin with propionic anhydride in the presence of pyridine. Both compound C1 and C2 were confirmed by the spectral data reported in the literature (Tyagi et al. 2010). The final novel product 7,8-dipropoxy-4-methylthiocoumarin was synthesized by thionation of compound 2 with the help of phosphorous pentasulfide in the presence of dioxane.
Cholinesterase inhibitory activity
The inhibitory potential of test compounds (C1–C15) was measured spectrophotometrically against AChE (electric eel) and BuChE (equine serum) by Ellman’s method. The structure and inhibitory activity of test compounds are summarised in Table 1. Ten and nine out of the fifteen derivatives exhibited concentration-dependent inhibitory activity against AChE and BuChE respectively. Four compounds showed dual cholinesterase inhibition, with all of them having higher selectivity for AChE except for C7.
Table 1.
Cholinesterase inhibition, anti-BACE1, anti-MAO B and antioxidant capacity of 4-methylcoumarin and thiocoumarin derivatives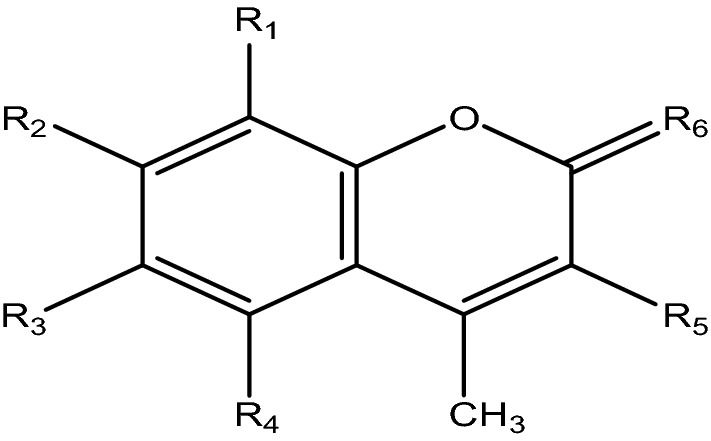
| Compounds | Structures | IC50 (µM) or % inhibition at 80 µM | Selectivity | Percentage inhibition for BACE 1 (*) | Percentage inhibition for MAO B (*) | Fe equivalent (µM ± SEM) (*) | Trolox equivalent (µM ± SEM) (*) | |
|---|---|---|---|---|---|---|---|---|
| AChE | BuChE | |||||||
| C1 | R1 = R2 = OH, R3 = R4 = R5 = H,R6 = O | n.a | 55.44 ± 5.30 | – | 19.35 ± 1.23% | n.d | 831.93 ± 17.81 | 393.35 ± 10.25 |
| C2 | R1 = R2 = OPr, R3 = R4 = R5 = H, R6 = O | 40.59 ± 0.95% | 35.78 ± 0.13% | – | n.a. | n.a. | – | – |
| C3 | R1 = R2 = OPr, R3 = R4 = R5 = H, R6 = S | 5.63 ± 1.68 | 17.02 ± 3.67 | 3.03 | 57.23 ± 1.76% | 31.99 ± 1.56 | 799.83 ± 9.13 | 357.36 ± 5.63 |
| C4 | R1 = R2 = OAc, R3 = R4 = R5 = H, R6 = O | 17.87 ± 2.37 | 38.93 ± 3.57 | 2.17 | 11.92 ± 0.78% | 25.62 ± 2.62 | 733.22 ± 15.98 | 364.92 ± 15.32 |
| C5 | R2 = OAc, R1 = R3 = R4 = R5 = H, R6 = O | 32.46 ± 1.35 | 10.23 ± 1.52% | – | n.a. | n.d. | – | – |
| C6 | R2 = R4 = OAc, R1 = R3 = R5 = H, R6 = O | 43.56 ± 2.32 | 92.85 ± 1.20 | 2.13 | 9.74 ± 0.89% | n.d. | – | – |
| C7 | R1 = R2 = OAc, R3 = R4 = R5 = H, R6 = S | 14.91 ± 2.29 | 3.40 ± 0.20 | 0.22 | 43.62 ± 0.95% | 17.06 ± 1.02 | 626.27 ± 5.42 | 260.52 ± 4.25 |
| C8 | R2 = R4 = OH, R1 = R3 = H, R5 = EtOAc, R6 = O | 41.99 ± 6.93 | > 100 | – | n.a. | n.a. | 249.31 ± 3.17 | 329.48 ± 7.35 |
| C9 | R1 = R2 = OBu, R3 = R4 = R5 = H, R6 = O | n.a | n.a | – | 20.06 ± 1.51% | n.d. | 126.33 ± 3.14 | 196.38 ± 1.23 |
| C10 | R2 = R3 = OAc, R1 = R4 = R5 = H, R6 = O | 29.49 ± 2.89 | > 100 | – | n.a. | n.d. | 518.28 ± 2.07 | 346.42 ± 2.36 |
| C11 | R1 = NHAc, R2 = OAc, R3 = R4 = R5 = H, R6 = O | 61.96 ± 5.62 | 98.54 ± 4.91 | – | 41.66 ± 1.36% | 12.05 ± 2.93 | – | – |
| C12 | R1 = R2 = OH, R4 = COOH, R3 = R5 = H, R6 = O | n.a | > 100 | – | 15.27 ± 0.76% | n.d. | 998.09 ± 12.66 | 421.59 ± 11.89 |
| C13 | R2 = OH, R3 = COOH, R1 = R4 = R5 = H, R6 = O | > 100 | 28.60 ± 1.25% | – | 30.65 ± 0.58% | 4.98 ± 0.89 | – | – |
| C14 | R2 = OAc, R3 = COOH, R1 = R4 = R5 = H, R6 = O | 79.43 ± 3.94 | n.a. | – | n.a. | n.d. | – | – |
| C15 | R2 = R5 = OH, R1 = R3 = R4 = H, R6 = O | n.a. | n.a. | – | 31.09 ± 1.83% | 12.91 ± 1.27 | 790.81 ± 36.41 | 311.46 ± 13.85 |
| Donepezil | 0.19 ± 0.02 | 5.75 ± 0.36 | – | – | – | |||
| Selegiline | – | – | – | 81.36 ± 0.38 | ||||
n.a. no activity was observed
n.d. activity was not determined
*All compounds were screened at 20 µM
aIC50, inhibitor concentration (mean ± SEM) resulting in 50% inhibition of the enzyme AChE
bIC50, inhibitor concentration (mean ± SEM) resulting in 50% inhibition of the enzyme BuChE
cSelectivity for AChE: (IC50 of BuChE)/(IC50 of AChE
Among them, C3 is most potent as it exhibits highest inhibitory potential (IC50 = 5.60 μM) for AChE (p < 0.001) followed by C7 and C4, whereas, IC50 value for BuChE demonstrated that C7 was more potent as its significant inhibitory potential (IC50 = 3.40 μM) (p < 0.001) followed by C3 and C4. IC50 values of donepezil, which is used as s standard drug against AChE and BuChE was 0.19 µM and 5.75 μM, respectively (Table 1).
Furthermore, the kinetic study of two lead compounds, C3 and C7, was performed using Line weaver Burk plot. Analysis of kinetic study showed that both the compounds have higher Km and decreased Vmax at higher concentrations of test compounds (Table 2). This pattern revealed a mixed type of inhibition for C3 and C7 for both AChE as well as BuChE (Fig. 2).
Table 2.
Vmax and Km of C7 and C3 for AChE and BuChE
| Concentration (µM) | Vmax (mM/min) | Km (mM) | ||||||
|---|---|---|---|---|---|---|---|---|
| AChE | BuChE | AChE | BuChE | |||||
| C7 | C3 | C7 | C3 | C7 | C3 | C7 | C3 | |
| 80 | 0.26 | 0.446 | 0.864 | 0.791 | 0.865 | − 1.300 | − 0.499 | − 3.090 |
| 40 | − 7.75 | 0.668 | 0.249 | 0.676 | − 0.080 | − 1.358 | − 5.409 | − 2.081 |
| 20 | − 0.94 | 0.757 | 0.244 | 0.629 | − 1.463 | − 1.992 | − 9.191 | − 3.127 |
BACE 1 inhibition assay
BACE 1 enzyme cleaves APP into Aβ peptide. Therefore, it is considered a disease-modifying therapeutic target of AD (Vassar and Cole 2007). The present study demonstrates the BACE 1 inhibition of all the test compounds by the FRET-based BACE-1 fluorescence assay kit (Table 1). The test compounds were screened for anti-BACE activity. Out of fifteen compounds screened, only one compound (C3) exhibited more than 50% inhibition.
Modelling studies
Modelling studies for binding mode of inhibitor C3 in the binding pocket of AChE indicates that this compound occupies the hydrophobic pocket surrounded by mainly side chains of aromatic amino acids (Fig. 3a). It appears pertinent as C3 itself has an aromatic scaffold. This scaffold of C3 is mainly stabilized by the aromatic side chain of Trp86 of AChE through π–π stacking interactions which is a specific and strong non-covalent interaction. Binding of C3 is further stabilized by hydrogen-bonded interactions with the side-chain hydroxyl of Ser125 and Tyr337. Besides, van der Waals interactions between ethyl moiety of C3 and side chain of Phe338 also helped in stabilizing the binding. Majority of these interactions were observed throughout MD simulations and this indicates the importance of these three interactions in the binding of C3. The RMSD plot of its MD trajectory clearly indicates the stable binding of C3 derivative in the binding pocket with fluctuation of around 1.5 Å which is within the thermal fluctuation range at 300 K (Fig. 4a). The RMSF data shows that residues involved in interactions with C3 have fluctuation within the thermal limit while residues in the loop and terminal region has higher fluctuation (Fig. 4b).
Fig. 3.

Probable binding mode of identified potent inhibitors, C3 (ball and stick, pink) and C7 (ball and stick, orange), in the binding pocket of a AChE [PDB: 4EY6]; b BuChE [PDB: 1P0M] and c binding site residues of AChE [PDB: 4EY6] (cartoon, green) superimposed on BuChE [PDB: 1P0M] (cartoon, cyan); d BACE1 [PDB: 4D8C], respectively. Side chains of selected interacting residues are shown as ball and stick in respective colour and hydrogen bonded interactions are shown as black dotted lines
Fig. 4.

a RMSD and b RMSF plot of 10.2 ns MD trajectory of C3–AChE complex; c RMSD and d RMSF plot of 10.2 ns MD trajectory of C7–BuChE complex
Though C7 was also a 4-methylthiocoumarin derivative similar to C3, its binding mode was slightly different in BuChE (Fig. 3b) as compared to C3 in the binding pocket of AChE. The binding site of BuChE is also hydrophobic similar to AChE. Still, there are three notable substitutions (Gln119 in place of Tyr24, Val288 in place of Phe297 and Ala328 in place of Tyr337) (Fig. 3c) that influenced the binding mode of C7 in its binding pocket. The aromatic side chain of Trp82 stabilizes the aromatic ring of C7 through π–π stacking interaction similar to corresponding residue Trp86 with C3. However, no hydrogen bonded interaction was observed between C7 and BuChE, unlike C3 with AChE. Lack of hydrogen bond led to a change in the binding orientation of C7 during MD simulation (Fig. 4c). This resulted in r.m.s deviation of more than 5 Å with respect to the original docked position. However, a significantly higher number of van der Waals interaction was observed between C7 and non-polar side chains of Ala328, Phe329, Trp430, and Met437 of BuChE in comparison to C3 and AChE. The RMSF pattern of protein Cα is similar to the C3 complex (Fig. 4d). Comparative RMSD data of C3 and C7 compounds from their trajectory also indicates that C3 has higher binding stability with AChE in comparison to C7 with BuChE protein. Hence these modelling data correlates with the higher inhibitory activity of C3 against AChE compared to C7 against BuChE observed in the enzyme inhibition assays.
Modelling studies indicate that C3 also binds in the predominantly hydrophobic pocket of BACE1 and interacts with residues Leu30, Asp54, Tyr71, Thr72, Phe108, Ile118, Trp215, Thr220 and Val321 (Fig. 3d). Aromatic scaffold of C3 interacts with π–π stacking interaction with the side chain of Tyr71 and rest of the non-polar moiety interacts with side chains of other hydrophobic residues through van der Waal interaction. Binding of C3 is further stabilized by three hydrogen-bonded interactions with side-chain hydroxyl of Thr72, Thr220 and side chain carboxylate of Asp54. This shows a similar pattern of hydrophobic and hydrogen bond interactions of C3 with AChE.
Inhibition of Aβ fibrillization by ThT fluorescence spectroscopic measurements
The intensity of ThT emission is assumed to be correlated with the formation of amyloid fibrils, as it binds specifically to the amyloid region (Soto and Castaño 1996). Compounds C3 and C7 were analyzed for their inhibitory potential against Aβ fibrillization. It was observed that both the compounds C3 and C7 showed significant anti-amyloidogenic properties as compared to control (p < 0.001) (Fig. 5a). It was found that the inhibitory potential of C3 was increased from 45.42 to 75.75%, and C7 was increased from 63.25 to 70.69%, whereas that of the positive control, tannic acid, there was no significant change from 24 to 48 h. As C3 exhibits the highest anti-amyloidogenic property after 48hours, so concentration-dependent ThT assay was also analyzed (Fig. 5b, c). It was observed that at three different concentrations used (11, 22, 44 µM), the inhibitory percentage increases from 29.37 to 76.69%.
Fig. 5.

Inhibition of Aβ fibrillization by lead compounds and known inhibitor using ThT fluorescence spectroscopic measurements. a Anti-amyloidogenic property of lead compounds C3, C7 and known inhibitor tannic acid (TA) by ThT assay at 44 µM concentration at 24 h and 48 h (p < 0.001); b concentration dependent inhibition of Aβ fibrillization with C3 (p < 0.001); c ThT fluorescence emission spectra (400–650 nm) of different concentration of C3 (11, 22, 44 µM) with Aβ and Aβ alone were taken using an excitation wavelength of 440 nm after 48hours of incubation
Inhibition of Aβ fibrillization by in silico study
The interaction of Aβ was analyzed with anti-aggregatory molecules using computational methods. The amyloidogenic region was determined by FoldAmyloid software based on the average value of the parameter being greater to the threshold (Nair et al. 2011). Docking study was performed by making a grid around the above mentioned amyloidogenic region.
In the present study, FoldAmyloid software predicted two amyloidogenic regions in Aβ42 predicted 16–21 (KLVFFA) and 32–36 (IGLMV). Docking studies revealed that C3 interacts via hydrophobic interaction with Leu17, Phe20, Ala21, Val24, Ile31, Leu34, Met35, and it forms polar interaction with Lys28 (Fig. 6Aa). C7 forms hydrogen bonding with Lys28 and hydrophobic interactions with Phe20, Ala21, Val24, Leu34, and Met35 (Fig. 6Ab). Homotaurine used as a known anti-aggregatory molecule was also analyzed for its interaction with Aβ (PDB ID: 1IYT). Homotaurine forms two hydrogen bonds with Phe20, Asp23, and hydrophobic interaction with Val24, Ile31, and Leu34 (Fig. 6Ac).
Fig. 6.

Inhibitory potential of C3 against Aβ fibril formation by in silico, TEM and CD spectrophotometric studies A: Molecular docking studies of a: C3; b: C7; c: homotaurine with Aβ (1IYT); B: TEM image analysis of the sample, a: Aβ (44 μM) alone, b: Aβ + C3 (44μM); C: Circular dichroism studies of Aβ (red line) and Aβ + C3 (blue line) (both at 44 μM)
Inhibition of Aβ fibrillization by transmission electron microscopy (TEM)
TEM analysis demonstrated inhibition of Aβ fibril formation by C3 as compared to control (Aβ only) (Fig. 6B). Aβ control sample showed the formation of large aggregates with characteristic fibrillar structure. In contrast, shorter and fewer aggregates were seen in the sample that was incubated with the C3 compound. TEM results were correlated with ThT data that confirmed that compound C3 effectively inhibited Aβ fibril formation.
Inhibition of Aβ fibrillization by circular dichroism (CD)
The difference in the conformation of Aβ fibril formation of different variants is due to the presence of specific amino acid residues (Li et al. 2016). The formation of β sheets in Aβin the presence of C3 is significantly reduced as compared to control (Aβ only). This was confirmed by comparison of CD spectra between Aβalone and a sample containing Aβincubated with C3 compound (Fig. 6C). β-sheets observed in the case of Aβ alone is 50.01% as compared to 35.17% in C3 compounds. This implies that there is a relatively less transition of α helix to β sheet formation in C3 as compared to Aβ only sample.
MAO B inhibition assay
Test compounds (C1–C15) were screened for their MAO-B inhibitory activity at 20 µM concentration. The majority of compounds do not any significant inhibitory potential against MAO B. C3 showed the highest inhibition which was 31.09% followed by C7, i.e., 17.06%. The positive control used in this case was selegiline whose percentage inhibition was 81.36% (Table 1).
Antioxidant assay
ABTS assay
ABTS [2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid)] is a widely used method in producing ABTS + radical cation, which gives a green coloured solution and can be measured spectrophotometric method at 734 nm. The antioxidants hinder the green color of ABTS + radical cation as it suppresses the reaction by electron(s) donation. Hence, the formation of ABTS radicals is inversely proportioned to the amount of antioxidants present in the test compound (Mareček et al. 2017). All the fifteen compounds were tested for their antioxidant potential by FRAP and ABTS assay. Nine compounds showed significant antioxidant capacity at 20 μM from all the compounds tested. The two lead compounds (C3 and C7) showed significant scavenging activity of 74.71 ± 0.77% and 81.85 ± 0.50% for respectively at 20 μM (highest concentration) in the case of ABTS assay.
FRAP assay
Fe2+–tripyridyltriazine is formed by the reduction of Fe3+ TPTZ complex, which is an intense blue-colored complex and can be measured at 593 nm. The reducing power of compounds is directly correlated with the amount of complex formed (Kumar et al. 2013). The lead compounds show significant reducing power. Fe equivalent of C3 and C7 was 799.83 ± 9.13 µM and 626.27 ± 5.42 µM respectively at 20 μM (highest concentration) (Table 1).
Pharmacokinetic parameters
An important hurdle in the drug discovery process, especially in the later stages involves clearance of ADME (absorption, distribution, metabolism and excretion) profile of the lead compounds. During the drug development process, any discrepancy with the ADME profile is one of the significant drawbacks of the failure of the candidate molecule. To avoid this failure in later stages, in silico ADME screening was done using the QikProp module of Schrodinger software. Approximately forty-five important descriptors and pharmacologically relevant parameters were studied. In the current study, these nine descriptors were given special significance, which is molecular weight, CNS activity, hydrogen bond donor (donorHB), hydrogen bond acceptor (accptHB), QPlog Po/w, QPlog BB, PercentHumanOralAbsorption, Rule of five and rule of three. In the present study, none of the test compounds violated the Lipinski’s rule of five (MW < 500, QPlogPo/w < 5, donorHB ≤ 5, accptHB ≤ 10) but two of them (C13 and C15) violated the rule of three. PercentHumanOralAbsorption of the test compounds was above 50%, and six of them have above 80%, which is considered good enough. Predicted octanol/water partition coefficient is between the ranges except for one compound (C13). Based on the predicted value for CNS activity and QPlog BB, C3 and C7 might penetrate BBB. Polar surface area (PSA), which is another significant parameter for BBB penetration, it has been observed that CNS drugs tend to have a lower PSA generally in the range of 60–70 Å (Kumar et al. 2016a, b). The predicted values for various descriptors revealed that C3 and C7 have drug-like properties and might be the right candidate for drug development in the future for AD (Table 3).
Table 3.
In silico ADME profiling of the compounds
| Compounds | Mol. weight | CNS | donorHB | accptHB | QPlogPo/w | QPlogBB | Percent human oral absorption | Rule of five violation | Rule of three violation |
|---|---|---|---|---|---|---|---|---|---|
| C1 | 192.17 | − 1 | 2 | 4 | 0.341 | − 0.977 | 70.84 | 0 | 0 |
| C3 | 320.35 | 0 | 0 | 7 | 2.276 | − 0.291 | 96.85 | 0 | 0 |
| C4 | 276.24 | − 1 | 0 | 7.5 | 0.446 | − 0.927 | 75.22 | 0 | 0 |
| C5 | 220.22 | 0 | 0 | 4.95 | 1.268 | − 0.217 | 93.6 | 0 | 0 |
| C6 | 340.33 | − 1 | 0 | 7.5 | 1.642 | − 0.82 | 84.31 | 0 | 0 |
| C7 | 292.30 | 0 | 0 | 7 | 1.675 | − 0.305 | 91.63 | 0 | 0 |
| C8 | 292.28 | − 2 | 2 | 6 | 1.297 | − 1.871 | 70.27 | 0 | 0 |
| C9 | 332.35 | − 2 | 0 | 7.5 | 1.996 | − 1.141 | 86.42 | 0 | 0 |
| C10 | 276.24 | − 1 | 0 | 7.5 | 0.38 | − 0.969 | 74.13 | 0 | 0 |
| C11 | 275.26 | − 1 | 1 | 7.5 | 0.817 | − 0.971 | 76.92 | 0 | 0 |
| C12 | 236.11 | − 2 | 3 | 6 | − 0.127 | − 1.758 | 43.26 | 0 | 1 |
| C13 | 220.18 | − 2 | 1 | 4.25 | 0.891 | − 1.321 | 56.34 | 0 | 1 |
| C14 | 262.21 | − 2 | 1 | 7 | 0.648 | − 1.321 | 56.99 | 0 | 0 |
| C15 | 292.30 | 0 | 0 | 7 | 1.005 | − 0.62 | 82.47 | 0 | 0 |
Mol. Weight molecular weight, donorHB hydrogen bond donor, accptHB hydrogen bond acceptor, BBB blood brain barrier, PSA polar surface area
Discussion
Alzheimer’s disease is characterized by oxidative stress, synaptic dysfunction, neuroinflammation, and blood–brain barrier disruption, which can be due to abnormal extracellular accumulation Aβ peptide, loss of cholinergic neurons, oxidative stress and hyperphosphorylation of tau protein (Yadav et al. 2019). There are various targets implicated in AD are AChE, BuChE, Aβ peptide, BACE 1, MAO-B and free radicals (Kumar et al. 2016a, b). The most conventional therapeutic target of AD is AChE and BuChE. A plethora of previously published research papers has reported the newly synthesized coumarin derivatives as an inhibitor of AChE (Hamulakova et al. 2016, 2017) but none has shown the same for thiocoumarin derivatives. In view of this, the synthesis of novel 7,8-dipropoxy-4-methylthiocoumarin (C3) was performed by substituting sulphur at 2nd position of C2. Previous study has shown that electron-donating groups such as –OCH3 on coumarin scaffold contributes to the increased AChE inhibitory activity (Anand et al. 2012), therefore, derivatives which contains acetoxy (C4, C5, C6, C7, C10, C11 and C14) and propoxy group (C2, C3) exhibited better inhibition of AChE. In addition, C3 exhibits enhanced AChE inhibition of all the tested derivatives which might be due to the presence of propoxy group which is also observed in the previous study where they supported the argument of lipophilic interaction of electron-donating groups such as methyl and methoxy with the active site (Yusufzai et al. 2018). Whereas, C7 showed significant BuChE inhibition, which might be due to the presence of acetoxy group on the 7th and 8th position followed by C3 and C4, which suggest that lesser methyl group and more electronegativity is responsible for selectivity BuChE inhibition as compared to AChE. Hence, it was hypothesized that a smaller methyl group and more electronegativity are responsible for selectivity BuChE inhibition as compared to AChE. To validate these results, docking studies reveal C3 interacts with the PAS residues of AChE, whereas C7 interacts with the active site residues of BuChE.
Another pathological hallmark of AD is the Aβ plaque formation, which is mainly due to the cleavage of APP by BACE 1. The current study showed that C3 exhibits moderate inhibition of the BACE 1 enzyme, which might be due to the presence of thio group that interacts with the target protein, whereas other coumarin derivatives like C2 and C4 show negligible inhibition because of the absence of sulfur on 2nd position. Previous studies have shown that the importance of thio group where sulfonamido substitution in the carbazoles compounds has played a significant role in the BACE1 inhibition (Bertini et al. 2017). Furthermore, in vitro data was validated by in silico study, wherein both C3 and C7 forms polar interactions with the active site residue, i.e., Asp32 of BACE 1 enzyme. It is evident from previous studies that compounds which interact with PAS residues of AChE also inhibit BACE1 enzyme (Wang et al. 2016). The present study also showed the interaction of C3 with PAS residue of AChE that might be the reason for BACE1 inhibition and induce anti-amyloidogenic activity.
Previous reports have shown the role of coumarin derivatives in inhibiting Aβ fibrillization (Soto-Ortega et al. 2011; Ranade et al. 2016; Chen et al. 2017). C3 and C7 exhibited significant anti-amyloidogenic activity as compared to control as demonstrated by ThT assay. TEM studies in which we observed that samples with C3 had lesser and shorter fibrils as compared to that of control. Hence, the dense network of fibrils was observed in control (Aβ only) which was demonstrated by increased ThT fluorescence measurement in control as compared to the sample containing C3. Besides, anti-aggregation was also shown by CD spectroscopy, where smaller β sheets were observed in Aβ with C3 as compared to the control sample. Furthermore, in silico results showed that both the compounds (C3 and C7) form hydrophobic interactions with Met35 residue of Aβ which has been proved to be an essential residue that is responsible for inducing neurotoxicity and oxidative stress (Butterfield and Sultana 2011). Another residue, Lys 28, is critical for stabilizing β sheets conformation in Aβ peptide (Sinha et al. 2012) demonstrated polar interaction with C3 and forms a hydrogen bond with C7.
Another mechanism involved in the pathogenesis of AD is oxidative stress, which is primarily due to the imbalance of antioxidant defense systems and free radicals. Hence, antioxidants are used to neutralize the effect of these radicals, which contribute to the development of diseases like AD, cancer, diabetes mellitus (Ighodaro and Akinloye 2018). C3 and C7 exhibited significant activity both by scavenging and reducing the free radicals by ABTS and FRAP methods, respectively. As MAO B is also involved in the oxidative stress mechanism of AD, C3 and C7 do not show any significant inhibitory effect, so it can be concluded that C3 and C7 are showing antioxidant properties by scavenging mechanism but not through MAO B enzyme pathway.
Furthermore, in silico, ADME property has become a basis of screening compounds in the early stages of drug development and discovery process. C3 and C7 follow Lipinski’s rule of five, Rule of three has good oral absorption and can penetrate BBB. Moreover, C3 has a propoxy group on the 7th position, whereas C7 has an acetoxy group, as the increases the lipophilicity is increased in both the cases the druggability of a compound.
Conclusion
In conclusion, novel 4-methylthiocoumarin derivative was synthesized. This compound was further compared with other 4-methylthiocoumarin and 4-methylcoumarin derivatives for their potential anti-cholinesterase, anti-BACE1, anti-aggregatory potential, anti-MAO B, and antioxidant activity. Among the fifteen coumarin derivatives, new 4-methylthiocoumarin derivative C3 was a potent AChE inhibitor, whereas C7 exhibited potent BuChE inhibition. C3 compound was also able to inhibit BACE1 moderately and weakly inhibit MAO B enzymes. Moreover, both the compound C3 and C7 showed potent anti-aggregatory and antioxidant capacities. Molecular docking and dynamics studies of C3 and C7 calculated their potential binding mode with AChE, BuChE and BACE1 which correlate with their in vitro results. Together, this novel compound has shown a promising activity towards cholinesterase enzyme, BACE1, Aβ, and oxidative stress, and further chemical modification can provide a better lead that might be useful in the amelioration of AD.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Abbreviations
- AD
Alzheimer’s disease
- CNS
Central nervous system
- Aβ
Amyloid β
- MTDLs
Multi-target-directed ligands
- BACE1
β-Secretase cleavage enzyme
- MAO B
Monoamine oxidase B
- AChE
Acetylcholinesterase
- BuChE
Butyrylcholinesterase
- Ach
Acetylcholine
- APP
Amyloid precursor protein
- ROS
Reactive oxygen species
- FAD
Flavin adenine dinucleotide
- H2O2
Hydrogen peroxide
- AChEI
AChE inhibitor
- TEM
Transmission electron microscope
- CD
Circular dichroism
- donorHB
Hydrogen bond donor
- accptHB
Hydrogen bond acceptor
- BBB
Blood–brain barrier
- PSA
Polar surface area
- ATChI
Acetythiolcholine iodide
- BuChI
Butyrylcholine iodide
- DTNB
5:5-Dithiobis-2-nitrobenzoic acid
- ADME
Absorption, distribution, metabolism and excretion
Funding
This work was supported by FRGS, Guru Gobind Singh Indraparatha University, New Delhi, India.
Compliance with ethical standards
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Alzheimer’s Association Alzheimer's disease facts and figures. Alzheimers Dement. 2019;15(3):321–387. doi: 10.1016/j.jalz.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Anand P, Singh B, Singh N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg Med Chem. 2012;20(3):1175–1180. doi: 10.1016/j.bmc.2011.12.042. [DOI] [PubMed] [Google Scholar]
- Benzie IF, Strain JJ. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem. 1996;239(1):70–76. doi: 10.1006/abio.1996.0292. [DOI] [PubMed] [Google Scholar]
- Bertini S, et al. Sulfonamido-derivatives of unsubstituted carbazoles as BACE1 inhibitors. Bioorg Med Chem Lett. 2017;27(21):4812–4816. doi: 10.1016/j.bmcl.2017.09.058. [DOI] [PubMed] [Google Scholar]
- Borg S, et al. Design, synthesis, and evaluation of Phe-Gly mimetics: heterocyclic building blocks for pseudopeptides. J Med Chem. 1999;42(21):4331–4342. doi: 10.1021/jm990197+. [DOI] [PubMed] [Google Scholar]
- Borroni E, et al. Sembragiline: a novel, selective monoamine oxidase type B inhibitor for the treatment of Alzheimer’s disease. J Pharmacol Exp Ther. 2017;362(3):413–423. doi: 10.1124/jpet.117.241653. [DOI] [PubMed] [Google Scholar]
- Bowers KJ et al (2006) Scalable algorithms for molecular dynamics simulations on commodity clusters. In: Proceedings of the ACM/IEEE conference on supercomputing (SC06), Tampa, Florida, pp 17.
- Butterfield DA, Sultana R. Methionine-35 of Aβ (1–42): importance for oxidative stress in Alzheimer disease. J Amino Acids. 2011;2011:1–10. doi: 10.4061/2011/198430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhong C. Oxidative stress in Alzheimer’s disease. Neurosci Bull. 2014;30(2):271–281. doi: 10.1007/s12264-013-1423-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YR, et al. The effect of Aβ conformation on the metal affinity and aggregation mechanism studied by circular dichroism spectroscopy. J Biochem. 2006;139(4):733–740. doi: 10.1093/jb/mvj083. [DOI] [PubMed] [Google Scholar]
- Chen GF, et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin. 2017;38(9):1205. doi: 10.1038/aps.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung J, et al. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J Med Chem. 2012;55:10282–10286. doi: 10.1021/jm300871x. [DOI] [PubMed] [Google Scholar]
- Crescenzi O, et al. Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvironment: similarity with a virus fusion domain. Eur J Biochem. 2002;269(22):5642–5648. doi: 10.1046/j.1432-1033.2002.03271.x. [DOI] [PubMed] [Google Scholar]
- Czarnecka K, et al. Tetrahydroacridine derivatives with fluorobenzoic acid moiety as multifunctional agents for Alzheimer’s disease treatment. Bioorg Chem. 2017;72:315–322. doi: 10.1016/j.bioorg.2017.05.003. [DOI] [PubMed] [Google Scholar]
- Czarnecka K, et al. New cyclopentaquinoline hybrids with multifunctional capacities for the treatment of Alzheimer’s disease. J Enzyme Inhib Med Chem. 2018;33(1):158–170. doi: 10.1080/14756366.2017.1406485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, et al. Prediction of anti-Alzheimer's activity of flavonoids targeting acetylcholinesterase in silico. Phytochem Anal. 2017;28(4):324–331. doi: 10.1002/pca.2679. [DOI] [PubMed] [Google Scholar]
- De LP, Pitzurra M, Negri M. Antibacterial properties of thiocoumarin and its 7-substituted derivatives. Boll Chim Farm. 1962;101:376–379. [PubMed] [Google Scholar]
- Detsi A, Kontogiorgis C, Hadjipavlou-Litina D. Coumarin derivatives: an updated patent review (2015–2016) Expert Opin Ther Pat. 2017;27(11):1201–1226. doi: 10.1080/13543776.2017.1360284. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82(1):47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Friesner RA, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J Med Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Garbuzynskiy SO, Lobanov MY, Galzitskaya OV. Fold amyloid: a method of prediction of amyloidogenic regions from protein sequence. Bioinformatics. 2009;26(3):326–332. doi: 10.1093/bioinformatics/btp691. [DOI] [PubMed] [Google Scholar]
- Goel A, et al. Apoptogenic effect of 7, 8-diacetoxy-4-methylcoumarin and 7, 8-diacetoxy-4-methylthiocoumarin in human lung adenocarcinoma cell line: role of NF-κB, Akt, ROS and MAP kinase pathway. Chem Biol Interact. 2009;179(2–3):363–374. doi: 10.1016/j.cbi.2008.10.060. [DOI] [PubMed] [Google Scholar]
- Green K, Fosso M, Garneau-Tsodikova S. multifunctional donepezil analogues as cholinesterase and BACE1 inhibitors. Molecules. 2018;23(12):3252. doi: 10.3390/molecules23123252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güvenalp Z, et al. Cholinesterase inhibition and molecular docking studies of sesquiterpene coumarin ethers from Heptapteracilicica. Rec Nat Prod. 2017;11(5):462–467. [Google Scholar]
- Hamulakova S, et al. Targeting copper (II)-induced oxidative stress and the acetylcholinesterase system in Alzheimer's disease using multifunctional tacrine-coumarin hybrid molecules. J Inorg Biochem. 2016;161:52–62. doi: 10.1016/j.jinorgbio.2016.05.001. [DOI] [PubMed] [Google Scholar]
- Hamulakova S, et al. Synthesis, in vitro acetylcholinesterase inhibitory activity and molecular docking of new acridine-coumarin hybrids. Int J Biol Macromol. 2017;104:333–338. doi: 10.1016/j.ijbiomac.2017.06.006. [DOI] [PubMed] [Google Scholar]
- Hamulakova S, et al. Tacrine-coumarin and tacrine-7-chloroquinoline hybrids with thiourea linkers: cholinesterase inhibition properties, kinetic study, molecular docking and permeability assay for blood-brain barrier. Curr Alzheimer Res. 2018;15(12):1096–1105. doi: 10.2174/1567205015666180711110750. [DOI] [PubMed] [Google Scholar]
- Hasselgren C, et al. Socioeconomic status, gender and dementia: the influence of work environment exposures and their interactions with APOE ɛ4. SSM Popul Health. 2018;5:171–179. doi: 10.1016/j.ssmph.2018.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, et al. Coumarin-dithiocarbamate hybrids as novel multitarget AChE and MAO-B inhibitors against Alzheimer’s disease: design, synthesis and biological evaluation. Bioorg Chem. 2018;81:512–528. doi: 10.1016/j.bioorg.2018.09.010. [DOI] [PubMed] [Google Scholar]
- Ighodaro OM, Akinloye OA. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): their fundamental role in the entire antioxidant defence grid. Alexandria J Med. 2018;54(4):287–293. [Google Scholar]
- Jalili-Baleh L, et al. Design, synthesis and evaluation of novel multi-target-directed ligands for treatment of Alzheimer's disease based on coumarin and lipoic acid scaffolds. Eur J Med Chem. 2018;152:600–614. doi: 10.1016/j.ejmech.2018.04.058. [DOI] [PubMed] [Google Scholar]
- Jannat S, et al. Inhibition of β-site amyloid precursor protein cleaving enzyme 1 and cholinesterases by pterosins via a specific structure−activity relationship with a strong BBB permeability. Exp Mol Med. 2019;51(2):12. doi: 10.1038/s12276-019-0205-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- Joubert J, et al. Synthesis and evaluation of 7-substituted coumarin derivatives as multimodal monoamine oxidase-B and cholinesterase inhibitors for the treatment of Alzheimer's disease. Eur J Med Chem. 2017;125:853–864. doi: 10.1016/j.ejmech.2016.09.041. [DOI] [PubMed] [Google Scholar]
- Kaminski GA, et al. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105(28):6474–6487. [Google Scholar]
- Kocis P, et al. Hey, Elucidating the Aβ42 anti-aggregation mechanism of action of tramiprosate in Alzheimer’s disease: integrating molecular analytical methods, pharmacokinetic and clinical data. CNS Drugs. 2017;31(6):495–509. doi: 10.1007/s40263-017-0434-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, et al. Novel thiocoumarins as inhibitors of TNF-α induced ICAM-1 expression on human umbilical vein endothelial cells (HUVECs) and microsomal lipid peroxidation. Bioorg Med Chem. 2005;13(5):1605–1613. doi: 10.1016/j.bmc.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Kumar S, et al. In vitro protective effects of Withaniasomnifera (L.) dunal root extract against hydrogen peroxide and β-amyloid (1–42)-induced cytotoxicity in differentiated PC12 cells. Phytother Res. 2010;24(10):1567–1574. doi: 10.1002/ptr.3261. [DOI] [PubMed] [Google Scholar]
- Kumar S, Seal CJ, Okello EJ. Kinetics of acetylcholinesterase inhibition by an aqueous extract of Withaniasomnifera roots. Int J Pharm Sci. 2011;2(5):1188. [Google Scholar]
- Kumar CS, et al. Structural correlation of some heterocyclic chalcone analogues and evaluation of their antioxidant potential. Molecules. 2013;18(10):11996–12011. doi: 10.3390/molecules181011996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, et al. Structure based in silico analysis of quinolone resistance in clinical isolates of Salmonella Typhi from India. PLoS ONE. 2015;10(5):e0126560. doi: 10.1371/journal.pone.0126560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, et al. Current and novel therapeutic molecules and targets in Alzheimer's disease. J Formos Med Assoc. 2016;115(1):3–10. doi: 10.1016/j.jfma.2015.04.001. [DOI] [PubMed] [Google Scholar]
- Kumar J, et al. Synthesis and screening of triazolopyrimidine scaffold as multi-functional agents for Alzheimer's disease therapies. Eur J Med Chem. 2016;119:260–277. doi: 10.1016/j.ejmech.2016.04.053. [DOI] [PubMed] [Google Scholar]
- Kumar S, Chowdhury S, Kumar S. In silico repurposing of antipsychotic drugs for Alzheimer’s disease. BMC Neurosci. 2017;18(1):76–92. doi: 10.1186/s12868-017-0394-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, et al. Secretase inhibitors for the treatment of Alzheimer's disease: long road ahead. Eur J Med Chem. 2018;148:436–452. doi: 10.1016/j.ejmech.2018.02.035. [DOI] [PubMed] [Google Scholar]
- Larik FA, et al. Synthesis, inhibition studies against AChE and BChE, drug-like profiling, kinetic analysis and molecular docking studies of N-(4-phenyl-3-aroyl-2 (3H)-ylidene) substituted acetamides. J Mol Struct. 2020;1203:127459. [Google Scholar]
- Li H, Rahimi F, Bitan G. Modulation of amyloid β-protein (Aβ) assembly by homologous C-terminal fragments as a strategy for inhibiting aβ toxicity. ACS Chem Neurosci. 2016;7(7):845–856. doi: 10.1021/acschemneuro.6b00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionetto MG, et al. Acetylcholinesterase as a biomarker in environmental and occupational medicine: new insights and future perspectives. Biomed Res Int. 2013;2013:1–8. doi: 10.1155/2013/321213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CZ, et al. Rasagiline, an inhibitor of MAO-B, decreases colonic motility through elevating colonic dopamine content. J Neurogastroenterol Motil. 2018;30(11):e13390. doi: 10.1111/nmo.13390. [DOI] [PubMed] [Google Scholar]
- Mareček V, et al. ABTS and DPPH methods as a tool for studying antioxidant capacity of spring barley and malt. J Cereal Sci. 2017;73:40–45. [Google Scholar]
- Martyna GJ, Klein ML, Tuckerman M. Nosé-Hoover chains: the canonical ensemble via continuous dynamics. J Chem Phys. 1992;97(4):2635–2643. [Google Scholar]
- Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101(5):4177–4189. [Google Scholar]
- Meena P, et al. Novel insights into multitargeted potential of N′-(4-benzylpiperidin-1-yl) alkylamine derivatives in the management of Alzheimer's disease associated pathogenesis. RSC Adv. 2016;6(106):104847–104867. [Google Scholar]
- Modh RP, et al. Design, synthesis, biological evaluation, and molecular modelling of coumarin-piperazine derivatives as acetylcholinesterase inhibitors. Arch Pharm. 2013;346(11):793–804. doi: 10.1002/ardp.201300242. [DOI] [PubMed] [Google Scholar]
- Moghadam EK, Seyed SM, Gholizadeh M (2017) Design and synthesis of new derivatives of 2-Thiocoumarin 15-lipoxygenase inhibitors. In the 19th Iranian chemistry congress, Shiraz University
- Mokrov GV, et al. Synthesis and anticonvulsant activity of 4-amino-3-nitro-1-thiocoumarins and 4-amino-3-nitroquinolin-2-ones. Pharm Chem J. 2019;53(3):1–7. [Google Scholar]
- Nair SSK, Reddy NS, Hareesha KS. Exploiting heterogeneous features to improve in silico prediction of peptide status–amyloidogenic or non-amyloidogenic. BMC Bioinform. 2011;12(13):S21. doi: 10.1186/1471-2105-12-S13-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necula M, et al. Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J Biol Chem. 2007;282(14):10311–10324. doi: 10.1074/jbc.M608207200. [DOI] [PubMed] [Google Scholar]
- Nicolet Y, et al. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J Biol Chem. 2003;278:41141–41147. doi: 10.1074/jbc.M210241200. [DOI] [PubMed] [Google Scholar]
- Nita M, Grzybowski A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid Med Cell Longev. 2016;2016:1–23. doi: 10.1155/2016/3164734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okello EJ, Savelev SU, Perry EK. In vitro anti-β-secretase and dual anti-cholinesterase activities of Camellia sinensis L. (tea) relevant to treatment of dementia. Phytother Res. 2004;18(8):624–627. doi: 10.1002/ptr.1519. [DOI] [PubMed] [Google Scholar]
- Ono K, et al. Anti-amyloidogenic activity of tannic acid and its activity to destabilize Alzheimer's β-amyloid fibrils in vitro. Biochim Biophys Acta. 2004;1690(3):193–202. doi: 10.1016/j.bbadis.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Pandey G, et al. Prognostic and therapeutic relevance of cathepsin B in pediatric acute myeloid leukemia. Am J Cancer Res. 2019;9(12):2634–2649. [PMC free article] [PubMed] [Google Scholar]
- Pechmann HV, Duisberg C. Ueber die verbindungen der phenolemitacetessigäther. Berichte der deutschenchemischen Gesellschaft. 1883;16(2):2119–2128. [Google Scholar]
- Perez-Iratxeta C, Andrade-Navarro MA. K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct Biol. 2008;8(1):25. doi: 10.1186/1472-6807-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem. 2015;30(1):11–26. doi: 10.1007/s12291-014-0446-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisoschi AM, Negulescu GP. Methods for total antioxidant activity determination: a review. Biochem Anal Biochem. 2011;1(1):106. [Google Scholar]
- Raj HG, et al. Mechanism of biochemical action of substituted 4-methylbenzopyran-2-ones. Part I: dioxygenated 4-methyl coumarins as superb antioxidant and radical scavenging agents. Bioorg Med Chem. 1998;6(6):833–839. doi: 10.1016/s0968-0896(98)00043-1. [DOI] [PubMed] [Google Scholar]
- Ranade DS, et al. Thiosemicarbazone modification of 3-acetyl coumarin inhibits Aβ peptide aggregation and protect against Aβ-induced cytotoxicity. Eur J Med Chem. 2016;121:803–809. doi: 10.1016/j.ejmech.2015.07.028. [DOI] [PubMed] [Google Scholar]
- Rueeger H, et al. Discovery of cyclic sulfone hydroxyethylamines as potent and selective β-site APP-cleaving enzyme 1 (BACE1) inhibitors: structure-based design and in vivo reduction of amyloid β-peptides. J Med Chem. 2012;55:3364–3386. doi: 10.1021/jm300069y. [DOI] [PubMed] [Google Scholar]
- Ryckaert JP, Ciccotti G, Berendsen HJ. Numerical integration of the cartesian equations of motion of a system with constraints, molecular dynamics of n-alkanes. J Comput Phys. 1977;23(3):327–341. [Google Scholar]
- Sarma AD, Mallick AR, Ghosh AK. Free radicals and their role in different clinical conditions: an overview. Int J Pharm Sci Res. 2010;1(3):185–192. [Google Scholar]
- Sastry GM, et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Sharma P, et al. Design and development of multitarget-directed N-benzylpiperidine analogs as potential candidates for the treatment of Alzheimer's disease. Eur J Med Chem. 2019;167:510–524. doi: 10.1016/j.ejmech.2019.02.030. [DOI] [PubMed] [Google Scholar]
- Shelley JC, et al. Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des. 2007;21(12):681–691. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- Shi DH, et al. Liu, synthesis, characterization, crystal structure and evaluation of four carbazole-coumarin hybrids as multifunctional agents for the treatment of Alzheimer's disease. J Mol Struct. 2020;1209:127897. [Google Scholar]
- Sinha S, Lopes DH, Bitan G. A key role for lysine residues in amyloid β-protein folding, assembly, and toxicity. ACS Chem Neurosci. 2012;3(6):473–481. doi: 10.1021/cn3000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C, Castaño EM. The conformation of Alzheimer's β peptide determines the rate of amyloid formation and its resistance to proteolysis. Biochem J. 1996;314(2):701–707. doi: 10.1042/bj3140701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto-Ortega DD, et al. Inhibition of amyloid-β aggregation by coumarin analogs can be manipulated by functionalization of the aromatic center. Bioorg Med Chem. 2011;19(8):2596–2602. doi: 10.1016/j.bmc.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Tarozzi A, et al. From the dual function lead AP2238 to AP2469, a multi target directed ligand for the treatment of Alzheimer’s disease. Pharmacol Res Perspect. 2014;2(2):e00023. doi: 10.1002/prp2.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehrani MB, et al. Design, synthesis, and cholinesterase inhibition assay of coumarin-3-carboxamide-N-morpholine hybrids as new anti-Alzheimer agents. Chem Biodivers. 2019;16(7):e1900144. doi: 10.1002/cbdv.201900144. [DOI] [PubMed] [Google Scholar]
- Torres FC, et al. Combining the pharmacophore features of coumarins and 1,4-substituted 1,2,3-triazoles to design new acetylcholinesterase inhibitors: fast and easy generation of 4-methylcoumarins/1,2,3-triazoles conjugates via click chemistry. J Brazil Chem Soc. 2016;27(9):1541–1550. [Google Scholar]
- Tripathi A, et al. Efficacy of Quercetin as a potent sensitizer of β2 AR in combating the impairment of fluid clearance in lungs of rats under hypoxia. Respir Physiol Neurobiol. 2020;273:103334. doi: 10.1016/j.resp.2019.103334. [DOI] [PubMed] [Google Scholar]
- Tuckerman MBBJM, Berne BJ, Martyna GJ. Reversible multiple time scale molecular dynamics. J Chem Phys. 1992;97(3):1990–2001. [Google Scholar]
- Tyagi YK, et al. Synthesis of novel 4-methylcoumarins and comparative specificities of substituted derivatives for acetoxy drug: protein transacetylase. Sci Pharm. 2008;76(3):395–414. [Google Scholar]
- Tyagi YK, et al. In vitro antioxidant activity evaluation of 4-methyl coumarin derivatives. Asian J Chem. 2010;22(5):3622–3628. [Google Scholar]
- Vassar R, Cole SL. The basic biology of BACE1: a key therapeutic target for Alzheimer's disease. Curr Genom. 2007;8(8):509–530. doi: 10.2174/138920207783769512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang H, Chen HZ. AChE inhibition-based multi-target-directed ligands, a novel pharmacological approach for the symptomatic and disease-modifying therapy of Alzheimer’s disease. Curr Neuropharmacol. 2016;14(4):364–375. doi: 10.2174/1570159X14666160119094820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts KS, et al. ConfGen: a conformational search method for efficient generation of bioactive conformers. J Chem Inf Model. 2010;50(4):534–546. doi: 10.1021/ci100015j. [DOI] [PubMed] [Google Scholar]
- Yadav K, et al. Review on aetiology, diagnosis and treatment of Alzheimer’s disease. J Drug Deliv Ther. 2019;9(3):626–633. [Google Scholar]
- Yusufzai SK, et al. Molecular docking studies of coumarin hybrids as potential acetylcholinesterase, butyrylcholinesterase, monoamine oxidase A/B and β-amyloid inhibitors for Alzheimer’s disease. Chem Cent J. 2018;12(1):1–57. doi: 10.1186/s13065-018-0497-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, et al. Inhibiting Aβ toxicity in Alzheimer's disease by a pyridine amine derivative. Eur J Med Chem. 2019;168:330–339. doi: 10.1016/j.ejmech.2019.02.052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.