

Abstract
Worldwide, many mangrove species are experiencing significant population declines, including Rhizophora apiculata, which is one of the most widespread and economically important species in tropical Asia. In Malaysia, there has been an alarming decline in R. apiculata populations driven primarily by anthropogenic activities. However, the lack of genetic and demographic information on this species has hampered local efforts to conserve it. To address these gaps, we generated novel genetic information for R. apiculata, based on 1,120 samples collected from 39 natural populations in Peninsular Malaysia. We investigated its genetic diversity and genetic structure with 19 transcriptome and three nuclear microsatellite markers. Our analyses revealed a low genetic diversity (mean He: 0.352) with significant genetic differentiation (FST: 0.315) among populations of R. apiculata. Approximately two-third of the populations showed significant excess of homozygotes, indicating persistent inbreeding which might be due to the decrease in population size or fragmentation. From the cluster analyses, the populations investigated were divided into two distinct clusters, comprising the west and east coasts of Peninsular Malaysia. The western cluster was further divided into two sub-clusters with one of the sub-clusters showing strong admixture pattern that harbours high levels of genetic diversity, thus deserving high priority for conservation.
Subject terms: Ecology, Genetics
Introduction
Southeast Asia holds the greatest diversity of mangrove species in the world1. The mangroves, which form the habitat for hundreds of species at all levels of near-shore food webs, provide numerous ecosystems functions such as storm surge protection and carbon sequestration for climate change mitigation2,3. However, in recent decades, mangrove populations have plummeted owing mainly to various anthropogenic activities, which contribute to widespread mangrove forest degradation through land clearing, illegal logging and the over-exploitation of their highly valuable wood1,4,5. These were evident with around 16% mangrove species are in danger of extinction6.
Habitat fragmentation can affect the genetic structure of a species, primarily by increasing levels of genetic drift and inbreeding within populations, which consequently reduces the genetic diversity within the gene pool7. Given that populations with a lack of genetic diversity often exhibit a greater risk of extinction8, examining the genetic diversity patterns of the threatened mangrove populations will be the key to ensure their long-term survival. The patterns of genetic diversity in a population can be shaped by both natural (evolution) and environmental events such as human-induced climate change. Unveiling insights into the mechanisms underlying these patterns are crucial to ensure informed conservation and management of the threatened species.
Rhizophora is the most widely distributed species among all the mangroves species9. Among them, Rhizophora apiculata is a dominant commercial mangrove species that plays important ecological and economic role in Malaysia10. Locally known as Bakau Minyak, this species is preferred for its hard, strong and heavy wood, harvested mostly for wood chips, furniture, and charcoal11,12. While the population size of R. apiculata has been reduced dramatically over the past few decades13, little is known about the population genetics of this species. Previous work on the genus Rhizophora revealed low genetic diversity and high genetic differentiation, which seemed to be common traits for mangroves14–18. However, as most studies aimed to cover a large geographical area, sampling was not thorough, e.g. for Malaysia, only a few populations were chosen to represent the country (see15,18). The genetic information generated from a few populations is not sufficient to develop sound conservation plans for the species in Malaysia.
Here, we developed a new set of genic microsatellite markers (EST-SSR) in R. apiculata using next generation sequencing (NGS) approach, to assess its level of genetic diversity and population differentiation throughout Peninsular Malaysia. Microsatellite markers have been widely used for population analyses because of their co-dominant inheritance and high degree of polymorphism (for recent work see19,20). The markers developed in this study will provide a detailed genetic diversity database of R. apiculata which can facilitate the formulation of conservation guidelines in Peninsular Malaysia.
Results
Identification and development of microsatellite markers in R. apiculata
A total of 25,938,686 raw RNA-seq reads were generated by the Illumina HiSeq 4000 sequencer, with a total of 25,627,792 clean reads remained for further analysis after the ambiguous and low-quality sequences were filtered. These clean reads were then assembled, resulting in 141,915 contigs with an overall GC content of 44%. Using MISA software, these contigs were analysed and found to harbour 18,674 microsatellites, with the highest distribution in dinucleotide (15,898, 85.13%), followed by trinucleotide (2,403, 12.87%) and tetranucleotide microsatellites (373, 2.00%). Three highest frequencies of dinucleotide motifs that were detected were CT (16.80%), AG (16.68%) and TC (13.45%). Out of the 60 primer pairs tested, 46 yielded successful amplification. These primers were then used to screen 24 individuals of R. apiculata via PCR amplification and fragment analysis. Nineteen out of 46 primer pairs which showed clear genetic polymorphisms along with three other primer pairs (RM111, RM116, and RM121) previously developed for R. mucronata21 were selected (Table 1). The assembled contig sequences from which the newly developed microsatellite markers derived were blasted against the genome sequence of R. apiculata22 to check the reliability (Supplementary Table S1). These markers were subsequently used to genotype the 1,120 R. apiculata samples collected throughout Peninsular Malaysia (Fig. 1 and Table 2).
Table 1.
Information on the 22 microsatellite markers for R. apiculata and the corresponding Genbank accession number.
| Locus | Repeat motif | Forward primer sequence (5′–3′) | Reverse primer sequence (5′–3′) | Expected PCR product size | Genbank accession no |
|---|---|---|---|---|---|
| RapT01 | (AT)10 | CCACACATTTTGCACCGAGC | CTCAATCGCCTCGGAATGA | 298 | MT364422 |
| RapT02 | (CT)11 | TAAAGTGCGTGAATCCGCCA | ACTGCAACTCCACCGCTTAA | 138 | MT364423 |
| RapT06 | (CT)14 | TCCTCCTTGCTCCAAGCTTG | ACATGGAGCCTGTAGCCAAA | 349 | MT364424 |
| RapT08 | (GA)16 | TGATTCACTGCAAATGAGGCC | ACGAAACAAGCTTGCTCCCT | 102 | MT364425 |
| RapT09 | (TC)16 | AGGCTCGGGATTTTCCTTCG | CTGGGAAGCATGGGGTAGTG | 170 | MT364426 |
| RapT16 | (AG)11 | GGTGGATTGAGGGAAGTGGG | GGACACCTGGCAAACCCTTA | 296 | MT364427 |
| RapT17 | (GA)18 | GTGGGAGCCTGCAAAGTTTG | GCGCGTTGAGCTCAGTTTAG | 281 | MT364428 |
| RapT18 | (AT)11 | CACAGCAGTGTCATGGGCTA | AGGAGGAAGCTCATGCACAA | 165 | MT364429 |
| RapT20 | (CT)12 | TCAACCGCCTTTTCCCTCTC | ACCTTAAGCAATCTCCGGCA | 102 | MT364430 |
| RapT21 | (GA)15 | TATGTGGTCTGTGCAACCCC | GAAGCAGGCTGGATGGACTT | 156 | MT364431 |
| RapT23 | (AGA)11 | TGGAGTGAAACACCTGATCCA | TGGGGCCAACAAAATGAGGA | 148 | MT364432 |
| RapT25 | (ATT)11 | ATGCGACGTCACTCTGGTTT | CTCCTTCAGCTCTCCGCAAA | 267 | MT364433 |
| RapT31 | (CAT)13 | TCGCTTAAGTCCCTGCCTTG | CCTTTGTAGGAGTGGCTGCA | 217 | MT364434 |
| RapT38 | (AAT)10 | ACAGCCTTTACTCCAGCGAG | AGGTAAGGAACGCCGTGTTG | 347 | MT364435 |
| RapT43 | (CAG)11 | GCATAGACAGGGCAGGGATC | CACCACGAGACCAAGTCTGA | 153 | MT364436 |
| RapT46 | (CAG)12 | CCCAATCCGAACCCTAACCC | TGCTGCACATTCTGCCCTAA | 116 | MT364437 |
| RapT51 | (AGAC)6 | TCTGAACCCGTTTTCTCCGG | GCCAGCAGCAAAATGAGGAC | 359 | MT364438 |
| RapT53 | (TCTT)6 | CCGCAACGGTTCTTCAAAGG | GACCACAACAGTAGACAGCC | 338 | MT364439 |
| RapT60 | (AAAG)9 | TCCACTTCGGTCTTTTCCCT | GGGCTAGAAGCGTCCGATTT | 369 | MT364440 |
| RM111* | (TC)13 | AACCGTTACTCGCGTATGCT | CATTGCCTCCATTCCATT | 149 | AB721976 |
| RM116* | (TA)12 | ATAAGACCATATGTAACACCCATTT | CCTCCTCTCATTCTTCATTTCA | 152 | AB721980 |
| RM121* | (ATC)12 | TGGCCTATAGAGAAAGCGGA | CCTTCAATCCCAAACAGC | 179 | AB721982 |
*Nuclear microsatellite markers developed for R. mucronata21.
Figure 1.

An overview of sample collection in Peninsular Malaysia. See Table 2 for detailed information on sampling sites. The map was generated using the ArcGIS v10.5 (https://desktop.arcgis.com/en/, ESRI). The Malaysian administrative boundary data for mapping was downloaded from the iGISMAP (https://map.igismap.com/share-map/export-layer/Malaysia_Polygon/cedebb6e872f539bef8c3f919874e9d7).
Table 2.
Sampling location, habitat condition and genetic diversity measures (number of alleles per locus, Aa; observed heterozygosity, Ho; expected heterozygosity, He; and allelic richness, Rs) and inbreeding coefficient (FIS) of the 39 populations of R. apiculata from Peninsular Malaysia.
| No | Population (Code) | Habitat conditiona | State | N | Latitude | Longitude | Genetic diversity measures | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Aa | Ho | He | Rs | FIS | |||||||
| 1 | Kubang Badak (KBa) | Developed | Kedah | 30 | 06°24' | 99°43' | 2.7 | 0.219 | 0.261 | 2.30 | 0.161* |
| 2 | Kisap (Kis) | Undisturbed | Kedah | 30 | 06°23' | 99°51' | 4.3 | 0.321 | 0.404 | 3.19 | 0.207* |
| 3 | Dayang Bunting (DBu) | Undisturbed | Kedah | 29 | 06°13' | 99°49' | 4.0 | 0.242 | 0.436 | 3.17 | 0.450* |
| 4 | Merbok (Mer) | Developed | Kedah | 28 | 05°40' | 100°23' | 2.7 | 0.278 | 0.290 | 2.32 | 0.044* |
| 5 | Balik Pulau (BPu) | Developed | Pulau Pinang | 28 | 05°18' | 100°11' | 2.6 | 0.234 | 0.247 | 2.13 | 0.054* |
| 6 | Pulau Gula (PGu) | Undisturbed | Perak | 30 | 04°55' | 100°29' | 2.9 | 0.312 | 0.322 | 2.34 | 0.029 |
| 7 | Teluk Kertang (TKe) | Logged | Perak | 30 | 04°50' | 100°38' | 3.0 | 0.305 | 0.327 | 2.39 | 0.071 |
| 8 | Trong (Tro) | Developed | Perak | 30 | 04°42' | 100°41' | 2.8 | 0.250 | 0.308 | 2.31 | 0.191* |
| 9 | Sungai Tinggi (STi) | Undisturbed | Perak | 30 | 04°35' | 100°40' | 2.9 | 0.274 | 0.301 | 2.24 | 0.091 |
| 10 | Sungai Batang (SBa) | Developed | Perak | 28 | 03°50' | 100°46' | 3.1 | 0.194 | 0.302 | 2.43 | 0.363* |
| 11 | Banjar Utara (BUt) | Developed | Selangor | 29 | 03°21' | 101°14' | 3.0 | 0.227 | 0.281 | 2.35 | 0.193* |
| 12 | Pulau Klang (PKl) | Logged | Selangor | 26 | 03°03' | 101°19' | 3.4 | 0.366 | 0.375 | 2.86 | 0.025 |
| 13 | Pulau Ketam (PKe) | Developed | Selangor | 26 | 03°01' | 101°15' | 4.6 | 0.358 | 0.478 | 3.63 | 0.256* |
| 14 | Pulau Tengah (PTe) | Undisturbed | Selangor | 29 | 02°58' | 101°14' | 3.7 | 0.409 | 0.395 | 2.87 | -0.035 |
| 15 | Pulau Pintu Gedong (PPi) | Undisturbed | Selangor | 29 | 02°56' | 101°15' | 3.7 | 0.353 | 0.355 | 2.83 | 0.007 |
| 16 | Pulau Che Mat Zin (PCh) | Undisturbed | Selangor | 28 | 02°58' | 101°17' | 3.6 | 0.377 | 0.386 | 2.89 | 0.022 |
| 17 | Telok Gedong (TGe) | Logged | Selangor | 31 | 02°58' | 101°22' | 3.6 | 0.322 | 0.364 | 2.76 | 0.118* |
| 18 | Sepang Besar (SBe) | Developed | Selangor | 26 | 02°40' | 101°44' | 3.2 | 0.369 | 0.392 | 2.77 | 0.061* |
| 19 | Sepang Kecil (SKe) | Developed | Selangor | 28 | 02°37' | 101°40' | 3.1 | 0.339 | 0.376 | 2.68 | 0.101* |
| 20 | Jimah (PBJ) | Undisturbed | Negeri Sembilan | 30 | 02°36' | 101°43' | 3.5 | 0.405 | 0.411 | 2.94 | 0.017 |
| 21 | Sungai Linggi (SLi) | Undisturbed | Negeri Sembilan | 30 | 02°23' | 101°59' | 3.7 | 0.456 | 0.463 | 3.07 | 0.019 |
| 22 | Pulau Besar (PBe) | Developed | Melaka | 9 | 02°06' | 102°19' | 2.9 | 0.404 | 0.455 | 2.91 | 0.118* |
| 23 | Merlimau Tambahan (MTa) | Developed | Melaka | 30 | 02°07' | 102°25' | 3.9 | 0.483 | 0.473 | 3.20 | -0.023 |
| 24 | Muar (Mua) | Developed | Johor | 30 | 01°58' | 102°36' | 4.5 | 0.370 | 0.503 | 3.44 | 0.268* |
| 25 | Pulau Kukup (PKu) | Developed | Johor | 29 | 01°19' | 103°26' | 4.6 | 0.373 | 0.498 | 3.61 | 0.256* |
| 26 | Tanjung Piai (TPi) | Developed | Johor | 29 | 01°16' | 103°30' | 3.7 | 0.423 | 0.467 | 3.11 | 0.096* |
| 27 | Sungai Pulai (SPu) | Undisturbed | Johor | 30 | 01°26' | 103°35' | 3.7 | 0.404 | 0.474 | 3.07 | 0.150* |
| 28 | Pulau Juling (PJu) | Undisturbed | Johor | 30 | 01°30' | 104°00' | 2.9 | 0.328 | 0.366 | 2.49 | 0.106* |
| 29 | Endau (End) | Undisturbed | Pahang | 30 | 02°45' | 103°30' | 2.7 | 0.312 | 0.370 | 2.36 | 0.159* |
| 30 | Kuantan (PBK) | Developed | Pahang | 30 | 03°47' | 103°18' | 2.5 | 0.273 | 0.323 | 2.16 | 0.157* |
| 31 | Peramu (Per) | Developed | Pahang | 30 | 03°48' | 103°20' | 2.8 | 0.310 | 0.369 | 2.33 | 0.165* |
| 32 | Balok (Bal) | Undisturbed | Pahang | 30 | 03°56' | 103°22' | 2.5 | 0.274 | 0.312 | 2.16 | 0.123 |
| 33 | Cherating (Che) | Developed | Pahang | 30 | 04°07' | 103°23' | 2.8 | 0.302 | 0.329 | 2.25 | 0.085* |
| 34 | Kuala Kemaman (KKe) | Developed | Terengganu | 30 | 04°17' | 103°24' | 2.4 | 0.226 | 0.284 | 2.07 | 0.205* |
| 35 | Kuala Paka (KPa) | Undisturbed | Terengganu | 30 | 04°38' | 103°24' | 2.3 | 0.292 | 0.319 | 2.12 | 0.085* |
| 36 | Sungai Pimpin (SPi) | Undisturbed | Terengganu | 30 | 04°47' | 103°24' | 2.6 | 0.303 | 0.312 | 2.21 | 0.031 |
| 37 | Merchang (Mrc) | Logged | Terengganu | 28 | 05°01' | 103°18' | 3.6 | 0.256 | 0.399 | 2.87 | 0.363* |
| 38 | Kuala Terengganu (KTe) | Developed | Terengganu | 30 | 05°16' | 103°09' | 2.6 | 0.309 | 0.343 | 2.31 | 0.101* |
| 39 | Pengkalan Gelap (PGe) | Undisturbed | Terengganu | 30 | 05°40' | 102°43' | 2.7 | 0.314 | 0.327 | 2.32 | 0.042 |
| 1,120 | 3.2 | 0.299 | 0.352 | ||||||||
aLogged logged over for firewood, charcoal production and construction, Developed development for human settlement, aquaculture, ecotourism, road construction and land use for plantation, and Undisturbed undisturbed population.
*Significant at 95% confidence interval.
Genetic diversity
Descriptive statistics for the 39 populations of R. apiculata based on the 22 polymorphic microsatellite markers are listed in Table 2. These microsatellite markers had number of alleles per locus (Aa) ranging from 2.3 to 4.6 alleles for all samples, with an average of 3.2 alleles per locus. The mean values for observed heterozygosity (Ho) and expected heterozygosity (He) were 0.299 and 0.325, respectively. The expected heterozygosity (He) at the population level ranged from 0.247 (Balik Pulau) to 0.503 (Muar). The allelic richness (Rs) ranged from 2.07 to 3.63, showing marginal differences among the populations investigated. All loci deviated from Hardy–Weinberg equilibrium (HWE), with heterozygote frequencies being either lower or higher than expected (Table 2). Approximately 95.9% of the 39 tested populations had excess of homozygotes with positive inbreeding coefficient values (FIS) ranging from 0.007 to 0.450. Of that, 66.7% (26 populations) were found to be statistically significant (p < 0.05), mainly of the populations with disturbed habitat, either due to logging for firewood, charcoal production and construction (Telok Gedong and Merchang) or development for human settlement, aquaculture, ecotourism, road construction and land use for plantation (Kubang Badak, Merbok, Balik Pulau, Trong, Sungai Batang, Banjar Utara, Pulau Ketam, Sepang Besar, Sepang Kecil, Pulau Besar, Muar, Pulau Kukup, Tanjung Piai, Kuantan, Peramu, Cherating, Kuala Kemaman and Kuala Terengganu). There were only two populations with negative FIS values (Pulau Tengah, FIS = − 0.035 and Merlimau Tambahan; FIS = − 0.023) or excess of heterozygotes but were not statistically significant (p < 0.05) (Table 2).
Most of the total genetic diversity (HT = 0.532) was partitioned within population (HS = 0.370) (Table 3). The values recorded for Wright index (FST) ranged from 0.079 to 0.638, while those for the Slatkin’s divergence parameter (RST), varied from 0.038 to 0.617. The proportion of genetic variation distributed among populations (GST) was estimated at 0.305, indicating that 30.5% of genetic variability was distributed among populations (Table 3). The mean FST (0.315) estimate was slightly higher than GST and was significantly greater than zero (p < 0.05), while the mean RST (0.242) was lower than FST (Table 3).
Table 3.
Genetic diversity assessment in R. apiculata.
| Locus | HT | HS | GST | FST | RST | Genbank accession no |
|---|---|---|---|---|---|---|
| RapT01 | 0.489 | 0.403 | 0.176 | 0.180 | 0.249 | MT364422 |
| RapT02 | 0.532 | 0.283 | 0.469 | 0.487 | 0.617 | MT364423 |
| RapT06 | 0.648 | 0.430 | 0.337 | 0.350 | 0.305 | MT364424 |
| RapT08 | 0.615 | 0.564 | 0.084 | 0.089 | 0.083 | MT364425 |
| RapT09 | 0.818 | 0.645 | 0.212 | 0.221 | 0.400 | MT364426 |
| RapT16 | 0.512 | 0.198 | 0.614 | 0.638 | 0.402 | MT364427 |
| RapT17 | 0.842 | 0.683 | 0.189 | 0.195 | 0.145 | MT364428 |
| RapT18 | 0.633 | 0.395 | 0.377 | 0.392 | 0.042 | MT364429 |
| RapT20 | 0.518 | 0.456 | 0.121 | 0.126 | 0.070 | MT364430 |
| RapT21 | 0.656 | 0.396 | 0.396 | 0.412 | 0.528 | MT364431 |
| RapT23 | 0.439 | 0.166 | 0.622 | 0.616 | 0.090 | MT364432 |
| RapT25 | 0.529 | 0.217 | 0.590 | 0.609 | 0.581 | MT364433 |
| RapT31 | 0.560 | 0.432 | 0.230 | 0.229 | 0.270 | MT364434 |
| RapT38 | 0.584 | 0.460 | 0.212 | 0.222 | 0.038 | MT364435 |
| RapT43 | 0.257 | 0.237 | 0.079 | 0.079 | 0.071 | MT364436 |
| RapT46 | 0.404 | 0.183 | 0.548 | 0.564 | 0.070 | MT364437 |
| RapT51 | 0.470 | 0.275 | 0.415 | 0.431 | 0.273 | MT364438 |
| RapT53 | 0.357 | 0.188 | 0.474 | 0.480 | 0.508 | MT364439 |
| RapT60 | 0.201 | 0.180 | 0.103 | 0.106 | 0.117 | MT364440 |
| RM111* | 0.694 | 0.603 | 0.132 | 0.134 | 0.275 | AB721976 |
| RM116* | 0.690 | 0.526 | 0.237 | 0.249 | 0.094 | AB721980 |
| RM121* | 0.259 | 0.222 | 0.140 | 0.147 | 0.070 | AB721982 |
| 0.532 | 0.370 | 0.305 | 0.315** | 0.242 |
*Nuclear microsatellite markers developed for R. mucronata21.
**Significant at 95% confidence interval.
Population genetic structure
Genetic differentiation between populations increased as distance between populations increased (R2 = 0.2602) (Fig. 2a). Additionally, the results from the model-based Bayesian clustering analysis using STRUCTURE v2.3.1 indicated the presence of two main clusters within Peninsular Malaysia, one formed by the populations in western Peninsular Malaysia (Cluster 1: populations 1–27), and the other by those in eastern Peninsular Malaysia (Cluster 2: populations 28–39). The STRUCTURE algorithm showed the best clustering at K = 2, providing good biological explanation since the clusters coincided with two geographical groups23 corresponding to the Straits of Malacca (western Peninsular Malaysia) and South China Sea (eastern Peninsular Malaysia) (Fig. 2b). A gradually increasing level of admixture was observed in populations between Kedah and west part of Johor (Cluster 1: populations 1–27). When genetic variation is hierarchically organised, the algorithm underlying STRUCTURE detects only the uppermost level of population structure23. Hence, STRUCTURE analysis within cluster level further divided western cluster into two sub-clusters: Sub-cluster 1a (populations 1–19) and Sub-cluster 1b (populations 20–27) (Fig. 2c). Interestingly, the observed strong admixture pattern in Sub-cluster 1b suggests that the populations within this sub-cluster harbour higher levels of genetic diversity and could be an important genetic reservoir for R. apiculata in Peninsular Malaysia.
Figure 2.

(a) Result of Mantel test for Isolation-by-Distance using Nei’s genetic distance; (b) STRUCTURE showing the division of R. apiculata populations into two main clusters; (c) Sub-structuring of Cluster 1, forming Sub-clusters 1a and 1b; (d) Principal component analysis (PCA) based on pairwise FST of 39 R. apiculata populations, assigning the populations into two distinct clusters that coincide with western (orange color: Straits of Malacca) and eastern (blue color: South China Sea) Peninsular Malaysia and (e) PCA showing Cluster 1 further divided into two sub-clusters.
Results from the principal component analysis (PCA) corroborates with the STRUCTURE analysis, whereby the 39 populations were also divided into two distinct clusters (Fig. 2d), i.e. Cluster 1 along the Straits of Malacca (western Peninsular Malaysia) and Cluster 2 facing the South China Sea (eastern Peninsular Malaysia). Similarly, Cluster 1 was further divided into two sub-clusters (Fig. 2e). To complement the population structure analysis, a UPGMA dendrogram was constructed with MEGA v5.0 based on Nei’s DA24, producing two main branches that illustrate the two major clusters and two sub-clusters within Cluster 1 (Fig. 3).
Figure 3.
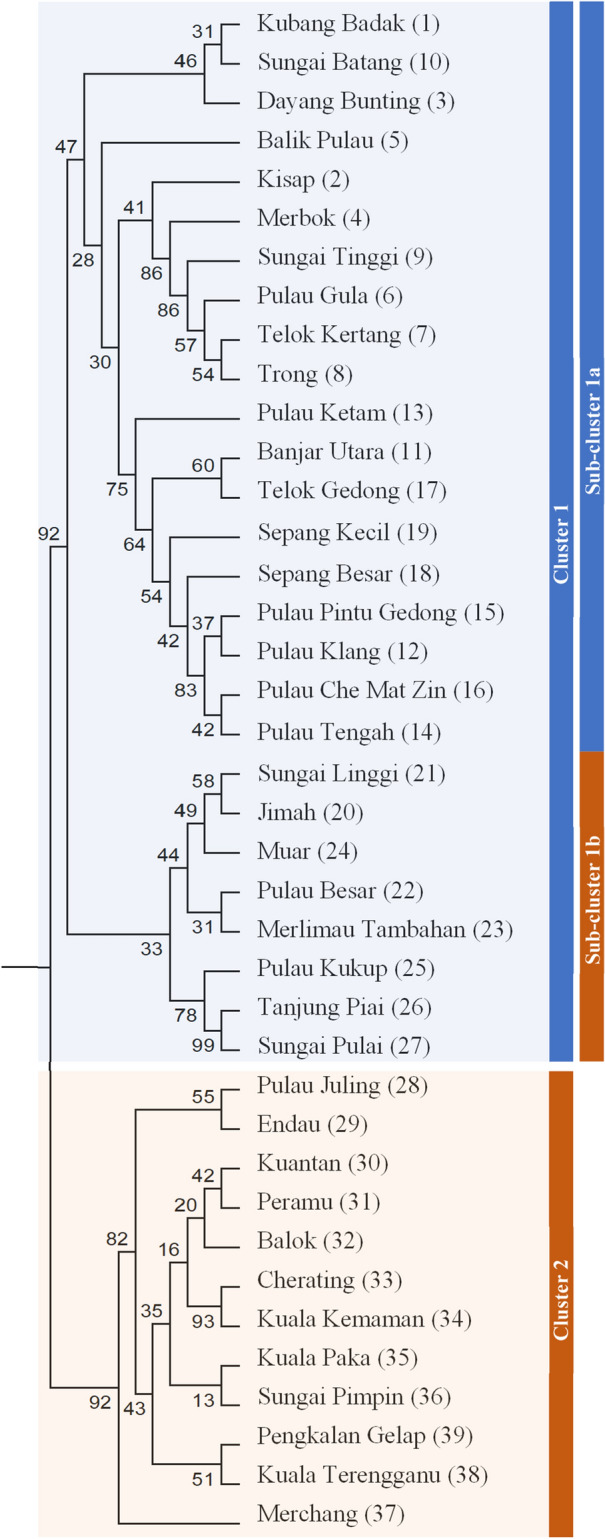
UPGMA dendrogram based on mean character differences estimated from microsatellite data of 1,120 R. apiculata individuals in 39 populations.
When the 39 populations were grouped based on the two main clusters, AMOVA revealed that 45% of the variation was apportioned between the western and eastern regions of Peninsular Malaysia, 13% among populations within regions, and 42% within populations (Table 4).
Table 4.
Analysis of molecular variance (AMOVA) performed by dividing the 39 populations into geographical regions.
| Source of variation | Degree of freedom | Sum of squares | Variance component | Percentage of variance |
|---|---|---|---|---|
| Among regions | 1 | 4950.041 | 9.966 | 45 |
| Among populations within regions | 37 | 3443.394 | 2.918 | 13 |
| Within populations | 1081 | 10,125.069 | 9.366 | 42 |
| Total | 1119 | 18,518.504 | 22.250 | 100 |
Discussion
From our transcriptome data, we identified a total of 18,674 microsatellites, with dinucleotide repeat motifs (85.13%) being the most frequent type of microsatellite. Similar observations, whereby dinucleotides was the most abundant motif in the plants’ genomes, have been reported for other tree species such as grey mangrove (Avicennia marina)25, downy oak (Quercus pubescens)26 and crape myrtle (Lagerstroemia spp.)27. A total of 19 transcriptome-based microsatellite markers (EST-SSR) were developed in this study. To increase the number of markers for genotyping, three additional nuclear microsatellite markers (gSSR) which were previously developed for R. mucronata21 have been validated and utilised in our study. Increasing the number of polymorphic loci can greatly enhance the precision of estimates of genetic distance28. The high cross-species transferability of microsatellite markers between R. apiculata and R. mucronata has previously been demonstrated9.
Generally, low genetic diversity is common in mangrove plant species particularly the Rhizophora species9,16,22. Low levels of genetic diversity (mean He = 0.352, Table 2) in R. apiculata were observed in the present study. The result is comparable to other mangrove species such as R. mucronata (He = 0.354)9, R. stylosa (He = 0.321)9, and Sonneratia alba (He = 0.280)29. The low level of genetic diversity occurring within R. apiculata populations suggests that this species may have experienced severe habitat fragmentation or population size reduction.
Our results also showed significant excess of homozygotes or positive inbreeding coefficient values (FIS) in most of the R. apiculata populations, consistent with the observed habitat destruction found in most of these populations (Table 2). Hence, the observed positive FIS values might be the result of selfing and/or mating between close relatives due to the reduction in population size caused by the anthropogenic activities. This is also a common result when drift proceeds in small population. As drift proceeds and each population becomes different from one other, the genetic variation among populations increases30,31.
R. apiculata is considered a threatened mangrove species in Southeast Asia, and its population decline is due mainly to deforestation and land conversion4,32,33. The high wood density is an attractive selling factor for R. apiculata (0.600 gcm-3), causing extensive over-exploitation of the species12,34. Owing to this, decline in effective population size and aggravate loss of genetic diversity ultimately reduced resilience of populations to anthropogenic climate change35. The genetic fragility in R. apiculata is of concern, considering that future impacts of environmental changes, whether natural or otherwise, will likely to further reduce its genetic diversity and threaten its long-term viability.
Wright’s F-statistics36 is among the most common methods used to estimate the level of heterozygosity in a population. The FST is more sensitive in detecting intraspecific differentiation as compared to its analogue, the RST37,38. The RST, however, is thought to be a better predictor for interspecific divergence because it can effectively reflect the mutation patterns of microsatellites39. Therefore, both models were applied in the present study, revealing a strong population genetic structure of R. apiculata (FST = 0.315, RST = 0.242). We found that approximately 68.5% and 31.5% of genetic variations were distributed within and among R. apiculata populations, respectively. Other mangrove studies also revealed high population differentiations, namely in Avicennia marina (0.410)25, Ceriops tagal (0.529)40, and A. germinans (0.410)41.
All the three cluster analyses performed in this study showed similar results, whereby the 39 populations were divided into two major geographical clusters (Cluster 1: populations 1–27; and Cluster 2: populations 28–39), with strong admixture pattern observed between southern part of western and eastern regions of Peninsular Malaysia (Fig. 2b), sub-structuring further divided Cluster 1 (Fig. 2c) into two sub-clusters. Due to the strong admixture of alleles, Sub-cluster 1b (populations 20–27) harbours higher genetic diversity (Table 2, Fig. 2c). The observed pattern may be best explained by ocean current movements, in which the Straits of Malacca act as a channel connecting the Andaman Sea with South China Sea, flanked by Indonesian island of Sumatra and Peninsular Malaysia land mass, linking the mangrove ecosystems between the western and eastern regions through hydrological cycle33,42. The ocean current movements along the Straits of Malacca and South China Sea are highly dependent on the monsoon winds during the north-east (December–January) and south-west (June–July) monsoons33. This enables the mangroves propagules to float in ocean water for an extended period and follow the ocean currents, transported from the source to the sink43. Due to the shallow waters at water depth ~ 30 m, which is narrow in the southern part of the Straits of Malacca before meeting the South China Sea42, the propagules of either region travelled to the sink population and formed the observed admixed populations (Sub-cluster 1b).
Studies have shown that R. apiculata are strong dispersers with high propagules survivorship in seawater with capability of long-distance dispersal44. Nevertheless, the potential of long-distance dispersal can be limited due to several factors such as ocean circulations, wind, large distances, longevity and land barriers14,44,45. Peninsular Malaysia has been reported to serve as a land barrier to gene flow15,45, thus promoting genetic differentiation between R. apiculata populations, particularly those from the eastern and western regions of Peninsular Malaysia. Previous study suggested that limited gene dispersal likely played an important role in the evolutionary history of Rhizophora species, as frequent sea level fluctuations associated with climate changes would negatively impact their effective population sizes9. In addition, human activities such as logging and developments near mangrove habitat (as recorded in Table 2) might have created anthropogenic barriers that pose serious threats to gene flow. Such barriers can effectively split a species' range into isolated fragments, and dispersal from one population to another can prove difficult.
The low genetic diversity of R. apiculata, along with environmental fragility caused primarily by anthropogenic activities, can decrease its fitness and affect its long-term survival. Prior to this study, there was a lack of genetic and demographic information on R. apiculata, hampering local efforts to develop a conservation plan for the species. The newly generated genetic information will enable the formulation of comprehensive conservation guidelines for R. apiculata in Peninsular Malaysia. Since Sub-cluster 1b of the western cluster exhibited strong admixture pattern that harbours higher levels of genetic diversity, as a reservoir rich with admixed alleles from both western and eastern clusters, this sub-cluster deserves high priority for conservation. Besides, the selection of in situ conservation areas can be considered independently from the two main clusters with priority given to habitat protection to combat the potentially detrimental effects of inbreeding in the remaining R. apiculata populations.
Methods
Plant material collection and habitat condition survey
Sampling was carried out along sheltered coasts of Peninsular Malaysia, where R. apiculata grow abundantly in water zones where saltwater meets fresh water. We collected a total of 1,120 samples from 39 natural populations of R. apiculata between 2017 and 2018, with an average of 29 samples per population (Fig. 1, Table 2). Leaf samples were collected randomly from individual trees, cleaned, and kept in liquid nitrogen prior to DNA extraction. Genomic DNA of R. apiculata was extracted with a modified cetyl trimethylammonium bromide (CTAB) method46 and purified using High Pure PCR Template Preparation Kit ver. 20 (Roche, USA).
Besides, habitat condition survey was also carried out based on systematic observation of each population by boat and/or walking on foot, providing visual coverage on the anthropogenic activities of the surrounding area that could contribute to mangrove habitat destruction. We categorised the anthropogenic activities into two categories, ‘logged’ as logging for firewood, charcoal production and construction, and ‘developed’ as development for human settlements, aquaculture, ecotourism, road construction and land use for plantation, while undisturbed populations were recorded as ‘undisturbed’. Most of the R. apiculata sampling sites have experienced some form of anthropogenic activities either due to logging or development (Table 2). Several sites were considered undisturbed mostly due to poor accessibility and far from human settlements and activities.
Microsatellite marker development and genotyping
The total RNA was extracted using Qiagen RNeasy kit (Qiagen, USA) and purified using the TURBO DNA-free kit (Ambion, USA). The quality of RNA sample was then checked using NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). Transcriptome sequencing was carried out on an Illumina HiSeq 4000 sequencer (Illumina, USA) with default parameters at Novogene Bioinformatics Technology Co. Ltd. (Beijing, China). The raw data underwent quality checking, trimming, and assembling using FastQC47, Trimmomatic v0.3248 and Trinity v2.4.049, respectively. Sequencing reads with many ambiguous (N) bases and more than 50% low-quality bases in raw reads were filtered out from the raw data set.
MicroSAtellite program (MISA, https://pgrc.ipk-gatersleben.de/misa)50 was used to identify microsatellite sequences of di-, tri, and tetranucleotide motifs. Markers were then designed with Primer3 (https://bioinfo.ut.ee/primer3-0.4.0/)51. We used the following criteria for marker selection: microsatellite motif length of ≤ 30 base pairs (bp); amplicon size of between 80 and 400 bp; and rejecting markers and amplicons with multiple mononucleotide repeat sequences. Based on these criteria, 60 primer pairs were synthesised and those that showed clear polymerase chain reaction (PCR) products on 1.5% agarose gel were fluorescently-labelled at the forward primer with HEX or 6-FAM. We performed PCR on a GeneAmp PCR System 9700 (Applied Biosystems, USA) in a final reaction volume of 10 μL. The PCR thermocycling parameters consisted of an initial denaturation step of 4 min at 94 °C followed by 40 cycles 94 °C for 1 min, 55 °C annealing temperature for 30 s, and 72 °C for 40 s, and the final extension step at 72 °C for 30 min. The PCR products were genotyped on an ABI 3130xl Genetic Analyzer (Applied Biosystems, USA) with ROX400 as the internal size standard. The alleles were scored using GeneMarker v2.6.4 (SoftGenetics, USA). The assembled contig sequences of the successfully developed microsatellite markers were assessed for reliability by blasting them to the R. apiculata genome sequence22 using BLASTN.
Data analysis
Micro-Checker software was used to detect possible genotyping errors and null alleles52. We examined deviations from Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD) using Fisher’s exact test in Genetic Data Analysis (GDA) v1.153. The sequential Bonferroni’s correction was conducted to adjust critical values for multiple comparisons, with a significance level of 5%54. Allelic frequencies for each locus in each population were obtained. The levels of genetic diversity within each population were estimated using Microsatellite Toolkit55 based on several parameters, including the average number of alleles per polymorphic locus (Aa), effective number of alleles per polymorphic locus (Ae), observed heterozygosity (Ho), and expected heterozygosity (He, Nei’s genetic diversity)28. Allelic richness (Rs) was estimated using FSTAT v2.9.356 and GDA v1.1, respectively. On the other hand, the inbreeding coefficient (FIS) was calculated using FSTAT v2.9.356, and its significance was tested with GDA v1.1. Additional statistics, such as polymorphic information content (PIC) and Nei’s genetic distance (Nei’s DA)57, were calculated using POWERMARKER v3.2558.
We also analysed the levels of genetic differentiation in R. apiculata populations using the molecular fixation index (FST)59, the divergence parameter (RST)38, and Analysis of Molecular Variance (AMOVA), which were all determined by GenAlEx v6.5 at the significance level of 5%60. A Mantel test based on 9,999 permutations was carried out to examine if there was correlation between geographical distance and genetic differentiation (FST, RST and FST/1 − FST), by analysing the Isolation-by-Distance (IBD) in GenAlEx v6.5. To infer the populations’ genetic structure, we applied a model-based Bayesian clustering method using STRUCTURE v2.3.161. Dataset was explored using admixture model, which can detect structure among populations that are potentially similar due to shared ancestry or migration, with a burn-in of 250,000 steps followed by 850,000 Markov Chain Monte Carlo (MCMC) iterations. The StructureSelector software62 was then used to select and visualise the optimal number of clusters (K) in order to identify the highest level of genetic division hierarchy. Additionally, principal component analysis (PCA) was carried out using PCAGEN v1.263 to assess the goodness-of-fit between simulated and real datasets, visualizing genetic distance and relatedness between populations in a two dimensional (2D) standard plot. To complement the analysis of the population structure, a UPGMA dendrogram was constructed based on Nei’s DA using MEGA v5.064. Bootstrap of 1,000 times was applied to acquire a reliable tree with correct branch topology.
Supplementary information
Acknowledgements
We thank the state Forest Departments of Kedah, Pulau Pinang, Perak, Selangor, Negeri Sembilan, Melaka, Johor, Pahang and Terengganu for granting permissions to access the forest reserves. We are indebted to the District Forest Officers for logistic assistance during sample collection. Also, we are particularly grateful to Shahril Mohamad, Abdullah Mat Isa, Mohd Suhairi Mohd Supian, Ramli Ponyoh, Yasri Baya, Sharifah Talib, Yahya Marhani, Ghazali Jaafar and the late Suryani Che Seman for their excellent assistance in the field and laboratory. This study was funded by the Forestry Department of Peninsular Malaysia under the RMK11 Grant (FRIM[S].600-1/19/21).
Author contributions
S.L.L., K.K.S.N., C.H.N., C.T.L., L.H.T. and N.F.Z obtained funding, conceived and designed the study; K.K.S.N., C.H.N., S.L.L., S.M., K.P., M.Z.A.K., A.A. and C.T.L. collected samples from the field; A.A., K.K.S.N., C.H.N. and C.T.L. performed experiments; A.A., K.K.S.N., C.H.N., C.T.L., L.H.T., N.F.Z and S.L.L. analysed the data; A.A., A.C., S.L.L., K.K.S.N., C.H.N., C.T.L., L.H.T. and N.F.Z. wrote the article.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Acga Cheng, Email: acgacheng@um.edu.my.
Soon-Leong Lee, Email: leesl@frim.gov.my.
Supplementary information
is available for this paper at 10.1038/s41598-020-76092-4.
References
- 1.Gandhi S, Jones TG. Identifying mangrove deforestation hotspots in South Asia, Southeast Asia and Asia-Pacific. Remote Sens. 2019;11:728. doi: 10.3390/rs11060728. [DOI] [Google Scholar]
- 2.Hamdan, O., Khali-Aziz, H., Shamsudin, I. & Raja-Barizan, R.S. Status of Mangroves in Peninsular Malaysia. 153 (Forest Research Institute Malaysia, 2012).
- 3.Taillardat P, Friess DA, Lupascu M. Mangrove blue carbon strategies for climate change mitigation are most effective at the national scale. Biol. Lett. 2018;14:20180251. doi: 10.1098/rsbl.2018.0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richards DR, Friess DA. Rates and drivers of mangrove deforestation in Southeast Asia, 2000–2012. Proc. Natl. Acad. Sci. USA. 2016;113:344–349. doi: 10.1073/pnas.1510272113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friess DA, et al. Mangroves give cause for conservation optimism, for now. Curr. Biol. 2020;30:R153–R154. doi: 10.1016/j.cub.2019.12.054. [DOI] [PubMed] [Google Scholar]
- 6.Polidoro BA, et al. The loss of species: mangrove extinction risk and geographic areas of global concern. PLoS ONE. 2010;5:e10095. doi: 10.1371/journal.pone.0010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matesanz S, Rubio-Teso ML, García-Fernández A, Escudero A. Habitat fragmentation differentially affects genetic variation, phenotypic plasticity and survival in populations of a gypsum endemic. Front. Plant Sci. 2017;8:843. doi: 10.3389/fpls.2017.00843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furches MS, Small RL, Furches A. Genetic diversity in three endangered pitcher plant species (Sarracenia; Sarraceniaceae) is lower than widespread congeners. Am. J. Bot. 2013;100:2092–2101. doi: 10.3732/ajb.1300037. [DOI] [PubMed] [Google Scholar]
- 9.Yan YB, Duke NC, Sun M. Comparative analysis of the pattern of population genetic diversity in three Indo-West Pacific Rhizophora mangrove species. Front. Plant Sci. 2016;7:1434. doi: 10.3389/fpls.2016.01434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wan-Ismail WN, Wan-Ahmad WJ, Salam MR, Latiff A. Structural and floristic pattern in a disturbed mangrove tropical swamp forest: a case study from the Langkawi UNESCO Global Geopark Forest, Peninsular Malaysia. Sains Malays. 2018;47:861–869. doi: 10.17576/jsm-2018-4705-01. [DOI] [Google Scholar]
- 11.Setyawan, A.D., Ulumuddin, Y.I. & Ragavan, P. Mangrove hybrid of Rhizophora and its parentals species in Indo-Malayan region. Nusantara Biosci. 6 (2014).
- 12.Lahjie, A.M., Nouval, B., Lahjie, A.A., Ruslim, Y. & Kristiningrum, R. Economic valuation from direct use of mangrove forest restoration in Balikpapan Bay, East Kalimantan, Indonesia. F1000Res. 8 (2019). [DOI] [PMC free article] [PubMed]
- 13.Omar, H., Misman, M.A. & Musa, S. GIS and remote sensing for mangroves mapping and monitoring. Geographic Information Systems and Science. IntechOpenhttps://www.intechopen.com/books/geographic-information-systems-and-science/gis-and-remote-sensing-for-mangroves-mapping-and-monitoring (2019).
- 14.Takayama K, Tamura M, Tateishi Y, Webb EL, Kajita T. Strong genetic structure over the American continents and transoceanic dispersal in the mangrove genus Rhizophora (Rhizophoraceae) revealed by broad-scale nuclear and chloroplast DNA analysis. Am. J. Bot. 2013;100:1191–1201. doi: 10.3732/ajb.1200567. [DOI] [PubMed] [Google Scholar]
- 15.Ng WL, et al. Closely related and sympatric but not all the same: genetic variation of Indo-West Pacific Rhizophora mangroves across the Malay Peninsula. Conserv. Genet. 2015;16:137–150. doi: 10.1007/s10592-014-0647-3. [DOI] [Google Scholar]
- 16.Yahya AF, et al. Genetic variation and population genetic structure of Rhizophora apiculata (Rhizophoraceae) in the greater Sunda Islands, Indonesia using microsatellite markers. J. Plant Res. 2014;127:287–297. doi: 10.1007/s10265-013-0613-z. [DOI] [PubMed] [Google Scholar]
- 17.Chen, Y. et al. Applications of multiple nuclear genes to the molecular phylogeny, population genetics and hybrid identification in the mangrove genus Rhizophora. PLoS ONE. 10 (2015). [DOI] [PMC free article] [PubMed]
- 18.Guo Z, et al. Genetic discontinuities in a dominant mangrove Rhizophora apiculata (Rhizophoraceae) in the Indo-Malesian region. J. Biogeogr. 2016;43:1856–1868. doi: 10.1111/jbi.12770. [DOI] [Google Scholar]
- 19.Cheng A, et al. Molecular marker technology for genetic improvement of underutilised crops. In: Abdullah S, Chai-Ling H, Wagstaff C, et al., editors. Crop improvement. Cham: Springer; 2017. pp. 47–70. [Google Scholar]
- 20.Ali A, et al. Genetic diversity and population structure analysis of Saccharum and Erianthus genera using microsatellite (SSR) markers. Sci. Rep. 2019;9:1–10. doi: 10.1038/s41598-018-37186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shinmura Y, et al. Isolation and characterization of 14 microsatellite markers for Rhizophora mucronata (Rhizophoraceae) and their potential use in range-wide population studies. Conserv. Genet. Resour. 2012;4:951–954. doi: 10.1007/s12686-012-9681-y. [DOI] [Google Scholar]
- 22.Xu S, et al. The origin, diversification and adaptation of a major mangrove clade (Rhizophoraceae) revealed by whole-genome sequencing. Natl. Sci. Rev. 2017;4:721–734. doi: 10.1093/nsr/nwx065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 24.Nei M, Tajima F, Tateno Y. Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol. 1983;19:153–170. doi: 10.1007/BF02300753. [DOI] [PubMed] [Google Scholar]
- 25.Maguire, T.L., Edwards, K.J., Saenger, P. & Henry, R. Characterisation and analysis of microsatellite loci in a mangrove species, Avicennia marina (Forsk.) Vierh. (Avicenniaceae). Theor. Appl. Genet. 101, 279–285 (2000).
- 26.Torre S, et al. RNA-seq analysis of Quercus pubescens leaves: de novo transcriptome assembly, annotation and functional markers development. PLoS ONE. 2014;9:e112487. doi: 10.1371/journal.pone.0112487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye Y, et al. Characterization, validation, and cross-species transferability of newly developed EST-SSR markers and their application for genetic evaluation in crape myrtle (Lagerstroemia spp) Mol. Breed. 2019;39:26. doi: 10.1007/s11032-019-0936-0. [DOI] [Google Scholar]
- 28.Nei M. Molecular Evolutionary Genetics. London: Columbia University Press; 1987. [Google Scholar]
- 29.Wee AK, et al. Vicariance and oceanic barriers drive contemporary genetic structure of widespread mangrove species Sonneratia alba, J. Sm in the Indo-West Pacific. Forests. 2017;8:483. doi: 10.3390/f8120483. [DOI] [Google Scholar]
- 30.Ellstrand NC, Elam DR. Population genetic consequences of small population size: implications for plant conservation. Annu. Rev. Ecol. Evol. Syst. 1993;24:217–242. doi: 10.1146/annurev.es.24.110193.001245. [DOI] [Google Scholar]
- 31.Feder JL, Gejji R, Yeaman S, Nosil P. Establishment of new mutations under divergence and genome hitchhiking. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2012;367:461–474. doi: 10.1098/rstb.2011.0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Annuar AS, Latip NA. Mangrove contributions towards environmental conservation and tourism in Balik Pulau. Adv. Conserv. Sci. Technol. 2020;1:1–7. [Google Scholar]
- 33.Wee, A.K. et al. Oceanic currents, not land masses, maintain the genetic structure of the mangrove Rhizophora mucronata Lam. (Rhizophoraceae) in Southeast Asia. J. Biogeogr. 41, 954–964 (2014).
- 34.Ismail MH, Zaki PH, Hamed AA. Wood density and carbon estimates of mangrove species in Kuala Sepetang, Perak, Malaysia. Malays. For. 2015;78:115–124. [Google Scholar]
- 35.Vitorino CA, Nogueira F, Souza IL, Araripe J, Venere PC. Low genetic diversity and structuring of the Arapaima (Osteoglossiformes, Arapaimidae) population of the Araguaia-Tocantins basin. Front. Genet. 2017;8:159. doi: 10.3389/fgene.2017.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wright S. The genetical structure of populations. Ann. Eugen. 1951;15:323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 37.Slatkin M. A measure of population subdivision based on microsatellite allele frequencies. Genetics. 1995;139:457–462. doi: 10.1093/genetics/139.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodman SJ. RST calc: a collection of computer programs for calculating estimates of genetic differentiation from microsatellite data and determining their significance. Mol. Ecol. 1997;6:881–885. doi: 10.1111/j.1365-294X.1997.tb00143.x. [DOI] [Google Scholar]
- 39.Moulin NL, Wyttenbach A, Brüunner H, Goudet J, Hausser J. Study of gene flow through a hybrid zone in the common shrew (Sorex araneus) using microsatellites. Hereditas. 1996;125:159–168. doi: 10.1111/j.1601-5223.1996.00159.x. [DOI] [Google Scholar]
- 40.Ge XJ, Sun M. Population genetic structure of Ceriops tagal (Rhizophoraceae) in Thailand and China. Wetl. Ecol. Manag. 2001;9:213–219. doi: 10.1023/A:1011156707160. [DOI] [Google Scholar]
- 41.Dodd, R.S., Afzal-Rafii, Z., Kashani, N. & Budrick, J. Land barriers and open oceans: effects on gene diversity and population structure in Avicennia germinans L. (Avicenniaceae). Mol. Ecol. 11, 1327–1338 (2002). [DOI] [PubMed]
- 42.Rizal S, et al. General circulation in the Malacca strait and Andaman Sea: a numerical model study. Am. J. Environ. Sci. 2012;8:479–488. doi: 10.3844/ajessp.2012.479.488. [DOI] [Google Scholar]
- 43.Nathan R, et al. Mechanisms of long-distance seed dispersal. Trends Ecol. Evol. 2008;23:638–647. doi: 10.1016/j.tree.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 44.Drexler, J.Z. Maximum longevities of Rhizophora apiculata and R. mucronata propagules. Pac. Sci. 55, 17–22 (2001).
- 45.Li J, et al. Pronounced genetic differentiation and recent secondary contact in the mangrove tree Lumnitzera racemosa revealed by population genomic analyses. Sci. Rep. 2016;6:29486. doi: 10.1038/srep29486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucl. Acids Res. 1980;8:4321–4326. doi: 10.1093/nar/8.19.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andrews, S. FastQC: a quality control tool for high throughput sequence data. Babraham Bioinformaticshttps://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
- 48.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinform. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grabherr MG, et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011;29:644. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Varshney RK, Thiel T, Stein N, Langridge P, Graner A. In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species. Cell Mol. Biol. Lett. 2002;7:537–546. [PubMed] [Google Scholar]
- 51.Rozen, S. & Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers in Bioinformatics methods and protocols. 365–386 (Humana Press, 2000). [DOI] [PubMed]
- 52.Van-Oosterhout C, Hutchinson WF, Wills DP, Shipley P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes. 2004;4:535–538. doi: 10.1111/j.1471-8286.2004.00684.x. [DOI] [Google Scholar]
- 53.Lewis, P.O. & Zaykin, D. Genetic Data Analysis (GDA) version 1.1: a computer program for the analysis of allelic data. UConnhttps://phylogeny.uconn.edu/software/ (2002).
- 54.Rice WR. Analyzing tables of statistical tests. Evol. 1989;43:223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x. [DOI] [PubMed] [Google Scholar]
- 55.Park, S.D.E. Trypanotolerance in West African cattle and the population genetic effects of selection. Ph. D (University of Dublin, 2001).
- 56.Goudet, J. FSTAT version 2.9.3.2: a program to estimate and test gene diversities and fixation indices. Unilhttps://www2.unil.ch/popgen/softwares/fstat.htm (2002).
- 57.Nei, M., Tajima, F. & Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol.19, 153–170 (1983). [DOI] [PubMed]
- 58.Liu K, Muse SV. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinform. 2005;21:2128–2129. doi: 10.1093/bioinformatics/bti282. [DOI] [PubMed] [Google Scholar]
- 59.Nei M. F-statistics and analysis of gene diversity in subdivided populations. Ann. Hum. Genet. 1977;41:225–233. doi: 10.1111/j.1469-1809.1977.tb01918.x. [DOI] [PubMed] [Google Scholar]
- 60.Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinform. 2012;28:2537–2539. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li YL, Liu JX. StructureSelector: a web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 2018;18:176–177. doi: 10.1111/1755-0998.12719. [DOI] [PubMed] [Google Scholar]
- 63.Goudet, J. PCAGEN version 1.2: a program to perform a principal component analysis (PCA) on genetic data. Unilhttps://www2.unil.ch/popgen/softwares/pcagen.htm (1999).
- 64.Tamura K, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.