

Abstract
A previous study on hepatoblastoma revealed novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex, including the tumor suppressor speckle-type BTB/POZ (SPOP). Moreover, the SPOP gene affected cell growth, and its S119N mutation was identified as a loss-of-function mutation in hepatoblastoma. This study aimed to explore more functions and the potential mechanism of SPOP and its S119N mutation. The in vitro effects of SPOP on cell proliferation, invasion, apoptosis, and in vivo tumor growth were investigated by western blot analysis, Cell Counting Kit-8, colony formation assay, flow cytometry, and xenograft animal experiments. The substrate of SPOP was discovered by a protein quantification assay and quantitative ubiquitination modification assay. The present study further proved that SPOP functioned as an anti-oncogene through the phosphatidylinositol 3-kinase/Akt signaling pathway to affect various malignant biological behaviors of hepatoblastoma both in vitro and in vivo. Furthermore, experimental results also suggested that solute carrier family 7 member 1 (SLC7A1) might be a substrate of SPOP and influence cell phenotype by regulating arginine metabolism. In conclusion, these findings demonstrated the function of SPOP and revealed a potential substrate related to hepatoblastoma tumorigenesis, which might thus provide a novel therapeutic target for hepatoblastoma.
Keywords: amino acid metabolism, epithelial-mesenchymal transition, hepatoblastoma, mutation, ubiquitin degradation
Graphical Abstract
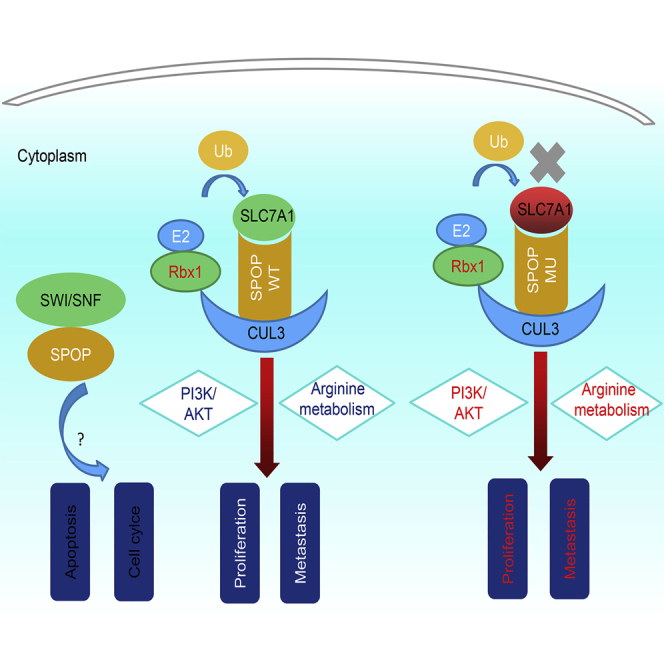
SPOP, an E3 ubiquitin ligase, inhibits hepatoblastoma progression via inactivating the PI3K/AKT pathway, and its anti-cancer function could be destroyed by S119N mutation. Moreover, SLC7A1 may be a substrate of SPOP and thus promote hepatoblastoma by affecting arginine metabolism. SPOP and SLC7A1 could be novel targets of childhood hepatoblastoma in the future.
Introduction
Hepatoblastoma (HB) is the most common primary malignant tumor of the liver in children, accounting for 80% of pediatric hepatic malignancies and 1% of all malignant tumors in children, with an increasing trend in recent years.1, 2, 3, 4, 5 HB is an embryonal tumor, and overactivation of Wnt/β-catenin, hedgehog, and phosphatidylinositol 3-kinase (PI3K)/Akt signaling has been reported to be associated with its development.5, 6, 7, 8, 9 Recent progress in research and development of comprehensive treatments has significantly improved the prognosis of HB.4,5,10, 11, 12, 13, 14 However, high-risk HB, especially in patients with extrahepatic involvement, multifocal lesions, or distant metastases, still has a poor prognosis.15, 16, 17, 18, 19 Present studies on molecular-targeted drugs are thus in progress with the aim of improving the prognosis for children with high-risk HB.
The speckle-type BTB/POZ (SPOP) protein is a component of the multi-subunit cullin 3-dependent ubiquitin ligase complex. It is mutated at high frequency in prostate and endometrial cancers.20,21 SPOP has been shown to be downregulated in HB compared with matched nontumor tissues and may thus function as a tumor suppressor gene. Moreover, SPOP S119N was found to be a loss-of-function mutation that inhibited HB cell proliferation by upregulating cyclin-dependent kinase inhibitor 2B (CDKN2B).6 This mutation may affect the substrate recognition domain and impair substrate binding. However, the mechanism of SPOP and the substrate affected by the SPOP S119N mutation remain unclear.
In this study, SPOP was further confirmed as a tumor suppressor participating in HB development through the PI3K/Akt pathway, whose anti-cancer function was impaired by S119N mutation. Moreover, solute carrier family 7 member 1 (SLC7A1) might act as a substrate of SPOP and promote HB progression via disturbing arginine metabolism.
Results
SPOP Promoted HB Cell Apoptosis and Inhibited Cell Proliferation
We first constructed stably transfected HB cells with SPOP-knockdown, SPOP-overexpressing, and S119N mutations, respectively. The efficiency of transfection was confirmed by both real-time PCR and western blot (Figures 1A–1C). Cell counting kit-8 (CCK8) and colony-formation assays indicated that SPOP downregulation facilitated cell proliferation and colony formation, while SPOP upregulation conversely suppressed cell proliferation and colony formation. SPOP S119N mutation promoted cell proliferation and colony formation (Figures 1D–1F). 5-Ethynyl-2′-deoxyuridine (EdU) assays also verified the function of SPOP in cell proliferation (Figures S1A and S1B). Flow cytometry showed that SPOP increased apoptosis in HepG2 and Huh6 cells (Figures 1G–1I). Moreover, knockdown of SPOP increased the expression of Bcl2 (anti-apoptotic) and decreased the expression of Bax (pro-apoptotic), while the opposite results were obtained from SPOP overexpression. Moreover, SPOP S119N mutation also reduced Bax but increased Bcl2 levels compared with SPOP overexpression groups (Figure 1J).
Figure 1.

SPOP Promoted Apoptosis and Inhibited Cell Proliferation
(A and B) Real-time PCR was performed to detect the expression of SPOP after downregulation, upregulation, or S119N mutation of SPOP compared with the control in hepatoblastoma HepG2 cells (A) and Huh6 cells (B). (C) Western blot analysis was performed to check the expression of SPOP after downregulation, upregulation, or S119N mutation of SPOP compared with the control in HepG2 and Huh6 cells. (D and E) CCK8 assays showed that the knockdown of SPOP increased cell proliferation, the overexpression of SPOP decreased cell proliferation, and S119N mutation also promoted proliferation. (D) HepG2 cells; (E) Huh6 cells. (F) Colony formation assays showed that the silencing of SPOP promoted cell proliferation, the upregulation of SPOP inhibited cell proliferation, and S119N mutation also increased proliferation. (G–I) Flow cytometry showed that the knockdown of SPOP decreased cell apoptosis, the overexpression of SPOP increased cell apoptosis, and S119N mutation also inhibited apoptosis. (J) Apoptosis marker proteins Bcl2 and Bax were detected by western blot analysis in HepG2 cells. Mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
SPOP Affected Cell Cycle and Tumor Metastasis
The influence of SPOP on the cell cycle was studied using flow cytometry. SPOP knockdown promoted cell cycle transition from the G1 to S phase, while its overexpression caused cell cycle arrest at the G2 phase, although this latter effect was lost in SPOP S119N-mutated cells (Figures 2A–2C). Moreover, the cell cycle proteins p21 and p27, but not other cyclin-dependent kinases, were significantly decreased in the SPOP-knockdown and S119N mutation groups compared with the relative control groups (Figures 2D–2F and S1E). In addition, transwell assays showed that SPOP downregulation increased, while SPOP upregulation decreased, both cell migration and invasion, and SPOP S119N mutation prevented its inhibition of cell migration and invasion in both HepG2 and Huh6 cells (Figures 2G–2K). Furthermore, wound-healing experiments demonstrated similar results in HepG2 cells (Figures S1C and S1D). Western blotting indicated that SPOP affected tumor metastasis, possibly via affecting the epithelial-mesenchymal transition (EMT) (Figures 2L–2O).
Figure 2.

SPOP Affected Cell Migration, Invasion, and Cell Cycle
(A–C) Representative results and statistical analysis of the cell cycle were analyzed by flow cytometry in HepG2 and Huh6 cells. (D–F) Western blot analysis was performed for cell cycle proteins in HepG2 cells. (G–K) Transwell assays showed that the silencing of SPOP promoted migration and invasion, while the upregulation of SPOP inhibited migration and invasion, and S119N mutation also increased migration and invasion. (L–O) The EMT marker was detected by western blot analysis, and relative protein grayscale was accounted. Mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
RNA-Seq and Protein Mass Spectrometry in Cells with SPOP Overexpression and S119N Mutation
We explored the function of the SPOP S119N mutation by RNA sequencing (RNA-seq) and identified numerous differentially expressed genes. The top 40 differentially expressed genes were shown (Figure 3A). These genes included various cell cycle proteins, including CDKN2B (which we previously found to be associated with SPOP), stratifin, and cyclin-dependent kinase 1. We also performed protein mass spectrometry in SPOP-overexpressing and S119N-mutated cells (Figure 3B). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis indicated that SPOP was related to the PI3K/Akt signaling pathway (Figures 3D, 3E, S1F, and S1G). Western blot also demonstrated that downregulation of SPOP or SPOP S119N mutation could activate the PI3K/Akt signaling pathway, while upregulation of SPOP inhibited the pathway (Figure 3C).
Figure 3.

RNA-Seq and Mass Spectrometry Were Performed in the SPOP Overexpression and S119N Mutation Groups
(A) Heatmap was drawn for the top 40 differential genes of RNA-seq in SPOP overexpression (OE) and SPOP S119N-mutated HepG2 cells. (B) Western blot analysis for immunoprecipitation of SPOP OE and SPOP S119N compared with negative control group immunoglobulin G (IgG). (C) Western blot analysis for the PI3K/Akt signaling pathway enriched by protein mass spectrometry showing control, shSPOP, SPOP OE, and SPOP S119N groups in HepG2 cells. (D and E) KEGG analysis for protein mass spectrometry of SPOP OE (D) and SPOP S119N (E) in HepG2 cells.
Protein Quantification and Quantitative Ubiquitination Modification in SPOP-Overexpressing and S119N-Mutated Cells
Protein quantification and quantitative ubiquitination modification in SPOP-overexpressing and S119N-mutated cells were performed to explore the substrate of SPOP. A total of 4,746 proteins were identified using label-free quantitative proteomics technology (Figures 4A–4D). Forty-three proteins were increased and 31 were decreased (significantly changed in abundance) in SPOP S119N-mutated compared with SPOP-overexpressing cells. In addition, 96 proteins were increased and 106 were decreased (consistent presence/absence expression profile) (Figure 4E). The heatmap showed several differentially expressed proteins (Figure 4F). Gene Ontology (GO) and KEGG analyses demonstrated that the SPOP S119N mutation participated in various biological processes and signaling pathways, such as mitogen-activated protein kinase (MAPK) signaling and ubiquitin-mediated proteolysis (Figures 4G and 4H). Western blot also showed that SPOP and S119N mutation could affect the MAPK signaling pathway (Figure 4I). Ubiquitinated label-free quantitative proteomics technology identified 4,327 ubiquitination sites, 4,002 ubiquitinated peptides, and 1,753 ubiquitinated proteins (Figures 5A–5E). Two proteins were increased and 225 were decreased (significantly changed in abundance) between the SPOP S119N mutation and overexpression groups. Moreover, 43 proteins were increased and 543 were decreased (consistent presence/absence expression profile) (Figure 5F). Ubiquitin protein ligase binding, the PI3K/Akt signaling pathway, and cell cycle were identified by bioinformatics analysis (Figures 5G and 5H).
Figure 4.

Label-free Quantitative Proteomics between SPOP OE and S119N Mutation
(A) Three repeated protein Venn map showing the SPOP OE sample group by label-free relative quantitative proteomics. (B) Three repeated protein Venn map showing the SPOP S119N sample group by label-free relative quantitative proteomics. (C) Protein Venn map identified between the SPOP OE and SPOP S119N groups. (D) Volcano plot using two factors showing the difference in protein expression between the two sets of samples, and the p value obtained by the t test showing the difference between the two groups of samples. (E) Differentially expressed proteins were found between the SPOP OE and SPOP S119N groups (upregulation more than two times or downregulation less than 0.5, and p value less than 0.05). (F) Heatmap for differentially expressed proteins between SPOP OE and SPOP S119N through label-free relative quantitative proteomics. (G and H) GO (G) and KEGG (H) analyses were performed for differentially expressed proteins between SPOP OE and SPOP S119N through label-free relative quantitative proteomics. (I) Western blot analysis was performed for key proteins of the MAPK signaling pathway.
Figure 5.

Ubiquitinated Label-Free Quantitative Proteomics between SPOP OE and S119N Mutation
(A) Ubiquitinated label-free quantitative proteomics identified 4,327 modified sites, 4,002 modified peptides, and 1,753 modified proteins. (B and C) Three repeated protein Venn maps showing the SPOP OE (B) and SPOP S119N (C) sample groups by ubiquitinated label-free quantitative proteomics. (D) Protein Venn map showing SPOP OE and SPOP S119N groups. (E) Volcano plot showing the differential proteins with ubiquitination of SPOP OE and SPOP S119N groups. (F) Differentially expressed ubiquitination proteins shown between SPOP OE and SPOP S119N groups (upregulation more than two times or downregulation less than 0.5, and p value less than 0.05). (G and H) GO (G) and KEGG (H) analyses were performed for differentially expressed ubiquitination proteins between SPOP OE and SPOP S119N groups.
SLC7A1 Was a Substrate of SPOP
To investigate the mechanism of SPOP in HB, we combined the upregulated proteins identified in the protein quantification experiment and downregulated proteins in the ubiquitination modification experiment. Nine proteins were identified, among which we were most interested in the arginine transporter protein SLC7A1 (Figure 6A). Western blot revealed that overexpression of SPOP decreased SLC7A1 expression, while SPOP S119N mutation increased SLC7A1 expression (Figure 6B). However, SLC7A1 mRNA levels did not differ significantly among the groups (Figure 6C). Immunoprecipitation experiments indicated that SPOP may interact with SLC7A1, and that ubiquitin could also bind with SLC7A1 (Figure 6D). Co-localization by immunofluorescence verified the interactions between SPOP and SLC7A1 (Figure 6E). SLC7A1 expression was increased in HepG2 cells treated with 50 μM MG132 (proteasome inhibitor) at 0, 0.75, 1.5, 3, and 6 h (Figure 6F). Moreover, SLC7A1 protein stability was determined in SPOP-overexpressing, S119N-mutated, and control HepG2 cells using cycloheximide, which indicated that SLC7A1 was more stable in SPOP S119N compared with SPOP-overexpressing cells (Figure 6G). SLC7A1 was knocked down and overexpressed in HepG2 cells to verify the function of SLC7A1 (Figure S3A). Both CCK8 and EdU results showed that the knockdown inhibited cell proliferation, while the overexpression of SLC7A1 promoted proliferation (Figures S3B–S3D). Moreover, the silencing of SLC7A1 decreased cell migration and invasion, and the upregulation of SLC7A1 increased migration and invasion (Figures S3E–S3G).
Figure 6.

SLC7A1 Might Be a Ubiquitination Substrate of SPOP
(A) Venn map of upregulated proteins and downregulated ubiquitin-modified protein for the SPOP S119N group compared with the SPOP OE group. (B) Expression of SLC7A1 (substrate of SPOP) in the control, shSPOP, SPOP OE, and SPOP S119N groups was detected by western blot analysis. (C) Real-time PCR was used for the detection of relative SLC7A1 mRNA level in the control, shSPOP, SPOP OE, and SPOP S119N groups. (D) A coimmunoprecipitation (coIP) assay was performed for SPOP and SLC7A1, SLC7A1, and ubiquitin. (E) A co-localization assay was performed for SPOP and SLC7A1 in HepG2 and Huh6 cells (original magnification ×63). (F) Western blot analysis showed the expression of SLC7A1 with 50 μM MG132 treatment after 0, 0.75, 1.5, 3, and 6 h. (G) Western blot analysis showed the expression of SLC7A1 with 0.1 μM cycloheximide after 0, 0.75, 1.5, 3, 6, and 12 h in the control, SPOP OE, and SPOP S119N groups, respectively.
SPOP S119N Mutation Promoted Tumor Growth and Metastasis In Vivo
We investigated the functions of SPOP and the SPOP S119N mutation in vivo by injecting stably transfected cells into nude mice. Tumor volumes differed significantly between the groups after 6 weeks (Figures 7A–7C). SPOP knockdown or S119N mutation increased tumor growth (Figure 7D). We also injected the same cells into the tail vein of nude mice to study the relationship between SPOP and HB metastasis. In vivo fluorescence imaging demonstrated that overexpression of SPOP inhibited tumor metastasis, while SPOP knockdown or S119N mutation promoted metastasis (Figures 7E and 7F). Hematoxylin and eosin staining and Ki67 immunohistochemistry of lung tissues from mice showed similar results (Figure 7G). Furthermore, immunofluorescence assays for EMT markers verified the previous western blot results (Figure 7H).
Figure 7.

SPOP Suppressed Tumor Growth and Metastasis In Vivo
(A) Images of tumors of nude mice injected with control, SPOP OE, and SPOP S119N HepG2 cells. (B) Tumor growth curve of the xenograft tumor of nude mice injected with control, SPOP OE, and SPOP S119N HepG2 cells. (C and D) Relative tumor volume (C) and tumor weight (D) of the control, SPOP OE, and SPOP S119N groups in nude mice. Mean ± SEM. ∗p < 0.05, ∗∗p < 0.01. (E and F) In vivo imaging shown after tail vein injection in the control, shSPOP, SPOP OE, and SPOP S119N groups. (G) Hematoxylin and eosin staining and Ki67 immunohistochemistry were performed for lung tissues of tail vein injection mice in the control, shSPOP, SPOP OE, and SPOP S119N groups (original magnification ×100). (H) Immunofluorescence was performed for E-cadherin, vimentin, and N-cadherin in the control, shSPOP, and SPOP OE groups, or SPOP S119N groups (original magnification ×100). Mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Downregulation of SLC7A1 in SPOP S119N Mutation Cells Partly Rescued Malignant Biological Behaviors
Given that S119N mutation could increase SLC7A1 expression by decreasing ubiquitination-mediated degradation of SPOP, we knocked down SLC7A1 and examined its effects on cell proliferation and migration. Downregulation of SLC7A1 inhibited cell proliferation indicated by CCK8 and colony-formation assays (Figures 8A–8C) and decreased cell migration and invasion indicated by transwell assays (Figures 8D–8F). However, flow cytometry showed no difference in apoptosis or cell cycle between the SPOP S119N and SLC7A1 downregulation groups (Figures S2A and S2B). The PI3K/Akt signaling pathway was suppressed by downregulation of SLC7A1 (Figure 8G). As SLC7A1 is an arginine transporter, we performed metabolomics analysis targeting this amino acid. Arginine was increased in SPOP S119N and decreased in SLC7A1-downregulated cells compared with their respective control groups (Figures 8H and S2C).
Figure 8.

Knockdown of SLC7A1 in SPOP S119N Mutation Cells Rescued Cell Proliferation, Migration, and Invasion Phenotype
(A) CCK8 assays showed that SLC7A1 knockdown in SPOP S119N cells inhibited cell proliferation. (B and C) Colony formation assays showed that downregulation of SLC7A1 in SPOP S119N cells inhibited colony formation. (D–F) Transwell assays showed that knockdown of SLC7A1 decreased migration and invasion in SPOP S119N cells. (G) The PI3K/AKT signaling pathway was inhibited by SLC7A1 knockdown in SPOP S119N cells. (H) Relative arginine content was shown in SPOP OE, SPOP S119N, and SLC7A1 knockdown in SPOP S119N cells, respectively. Mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
We previously identified 24 somatic nonsynonymous mutations in 21 genes in HB by whole-exome sequencing. SPOP mRNA and protein expression levels were detected by real-time PCR and immunohistochemistry, respectively, in HB and adjacent non-tumor tissues, and we found that SPOP expression was significantly downregulated in the tumors. This indicated that SPOP might be a critical candidate tumor suppressor in HB carcinogenesis. The SPOP S119N mutation was also previously shown to decrease the tumor suppressor ability of the SPOP gene in HB cells, possibly by regulating the expression of CDKN2B.6 However, the functions and substrates of SPOP remain to be explored.
Wang et al.22 revealed that SPOP-mediated NANOG degradation, controlled by the AMPK-BRAF signaling axis via phosphorylation of NANOG, played a crucial role in prostate cancer (PCa).23 Furthermore, the novel dual BET and CBP/p300 inhibitor NEO2734 was active in SPOP Q165P-mutated PCa cells. The results of the present study also showed that SPOP and the S119N mutation affected CBP/p300 of the SWI/SNF complex (Figure S2C).24 Yu et al.25 demonstrated that TRIM28 interacted with TRIM24 to prevent its ubiquitination and degradation by SPOP, thus providing a novel mechanism for TRIM24 protein stabilization and establishing TRIM28 as a promising therapeutic target. SPOP was also found as an important regulator of luminal epithelial cell proliferation, and c-Myc as a novel SPOP substrate, and helped to explain the frequent inactivation of SPOP in human PCa.26 A meta-analysis of nine studies including 928 patients with various cancers suggested that SPOP protein may be a useful prognostic biomarker in cancer patients.27
We previously found that SPOP downregulation or S119N mutation resulted in loss of function in HB. We carried out various experiments to verify SPOP as an anti-oncogene and S119N as a loss-of-function mutation. SPOP promoted HB cell apoptosis and inhibited cell proliferation, migration, and invasion in vivo and in vitro. Furthermore, S119N mutation damaged the anti-cancer function of SPOP. In addition, SPOP overexpression caused cell cycle G2 arrest, while SPOP S119N mutation lost this potential. Moreover, the knockdown of SPOP led to cell cycle transition from the G1 to S phase. The upregulation or downregulation of SPOP caused various changes in the cell cycle, indicating that the level of SPOP had many effects on the cell cycle. RNA-seq and protein mass spectrometry revealed that SPOP and the S119N mutation were associated with the cell cycle pathway and with the PI3K/Akt signaling pathway. SPOP is known to be an E3 ubiquitin ligase, and the MATH domain could bind with its substrate.28,29 The S119N mutation is located in the MATH domain and may thus affect the interaction between SPOP and its substrate. We therefore performed protein quantification and quantitative ubiquitination modification experiments to identify the substrate of SPOP. In addition, we synthesized the upregulated proteins and downregulated ubiquitination modification proteins and detected nine proteins (CD320, SNRPG, SUMO2, RPL14, HIST1H1E, GABARAP, SLC7A1, C21orf59, and ACTR10). Among these, SLC7A1 is an arginine transporter reportedly related to liver cancer.30, 31, 32 Our results demonstrated that SLC7A1 could interact with SPOP and ubiquitin. Moreover, SLC7A1 was degraded by the ubiquitin proteasome pathway, and amino acid metabolomics revealed that SLC7A1 affected arginine, but not other amino acids. SPOP may thus affect the malignant biological behaviors of HB via arginine metabolism. Rescue assays confirmed these results. SLC7A1 affected cell proliferation and metastasis, but not apoptosis and the cell cycle, leading to the question of how SPOP affects apoptosis and the cell cycle. It was assumed that this effect may be mediated through the SWI/SNF complex, given that SPOP was shown to bind to various proteins (CBP, p300, ARID1A, ARID1B, BRG1, and BRM) in this complex by an immunoprecipitation assay. However, further studies are needed to confirm this hypothesis.
In summary, SPOP is a tumor suppressor gene that participates in HB development via the PI3K/Akt signaling pathway, and its anti-cancer function can be destroyed by the S119N mutation. Moreover, SLC7A1 is a substrate of SPOP and may promote HB development by affecting arginine metabolism. SPOP and SLC7A1 may thus be joint targets for the future treatment of HB.
Materials and Methods
HB Cell Lines
HepG2 and Huh6 cells were obtained from the Cell Bank of the Chinese Academy of Science (Shanghai, China). HEK293T cells were gifted by the School of Basic Medical Sciences, Fudan University (China). All cells were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin solution in a humidified incubator with 5% CO2 at 37°C. All cell culture dishes and culture plates were purchased from Hangzhou Xinyou Biotechnology (China).
Quantitative Real-Time PCR
Total RNA was extracted with TRIzol reagent (Takara, Japan) and used to synthesize cDNA using a cDNA reverse transcription kit (Takara). The procedure was carried out at 42°C for 2 min to erase genomic DNA (gDNA), followed by 37°C for 15 min and 85°C for 5 s for reverse transcription. Quantitative real-time PCR was performed using SYBR Green PCR master mix (Takara) with a Roche machine. PCR was carried out as follows: an initial denaturation step at 95°C for 5 min, followed by 35 cycles at 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. Primer sequences were as follows: SPOP, forward, 5′-GAAATGGTGTTTGCGAGTAAACC-3′, reverse, 5′-GCCCGAACTTCACTCTTTGGA-3′; SLC7A1, forward, 5′-GCCTGTGCTATGGCGAGTTT-3′, reverse, 5′-ACGCTTGAAGTACCGATGATGTA-3′; GAPDH, forward, 5′-GGAGCGAGATCCCTCCAAAAT-3′, reverse, 5′-GGCTGTTGTCATACTTCTCATGG-3′. Relative gene expression was determined by the 2−ΔΔCt method, and data were analyzed using GraphPad Prism 5 software.
Construction of Stably Transfected Cell Lines
SPOP short hairpin RNA (shRNA) (shSPOP) plasmids were designed and constructed by GenePharma (Shanghai, China) using the LV3 vector. The shRNA target for SPOP was 5′-GCTGATGATTTGCTGGCAGCT-3′, and for SLC7A1 it was 5′-GCTGAGGATGGACTGCTATTT-3′. SPOP-overexpressing and SPOP S119N mutation plasmids were also constructed by GenePharma using the LV5 vector. 293T cells were cultured in a 10-cm culture dish to 80%–90% confluence for virus packaging using a standard procedure. The virus was collected after 48 h and transfected into tumor cells using 10 μg/mL Polybrene (Genomiditech, Shanghai, China).
Western Blot
Cell proteins were extracted using cell lysis buffer for western blot and immunoprecipitation (Beyotime Institute of Biotechnology, Nantong, China). Protein electrophoresis was carried out and the protein bands were transferred to membranes, following standard procedures. The membranes were blocked with 8% skim milk and then incubated with primary antibodies (Table S1) overnight at 4°C. After washing three times with Tris-buffered saline with Tween 20 (TBST) (New Cell & Molecular Biotech, Suzhou, China), the membranes were incubated with the relative secondary antibody for 2 h at room temperature.
Cell Proliferation Assay
Cells were cultured in a 96-well plate at a concentration of 1,000 cells in 100 μL of medium. CCK8 reagent (10 μL; Yeasen, Shanghai, China) was then added to the medium and detected 2 h later using a microplate reader. HepG2 cells were also treated with 50 μM EdU for 2 h, followed by staining according to the protocol described by the instructions (RiboBio, Guangzhou, China). For the colony formation assay, 1,000 cells were cultured in a six-well plate for 2 weeks, fixed with 4% paraformaldehyde for 10 min at room temperature, stained with crystal violet for 30 min, washed three times with phosphate-buffered saline (PBS), and then imaged under a microscope.
Cell Migration and Invasion Assays
Cell migration and invasion assays were performed in 24-well culture plates with 8-μm Micropore inserts. For the migration experiments, 1 × 105 HB cells were placed into the upper wells without FBS for 48 h. For the cell invasion assays, the upper wells were coated with Matrigel (BD Biosciences, USA), followed 30 min later by the addition of 2 × 105 HB cells into the upper wells without FBS for 48 h. The cells were washed three times with PBS, fixed with 4% paraformaldehyde, and stained with 0.2% crystal violet for 30 min. For the wound-healing assay, 1 × 104 HepG2 cells were seeded in two wells of culture inserts (Ibidi, Germany). The inserts were removed after 1 day, leaving a gap between the cells. The cells were then cultured with 2% FBS and observed and imaged under a light microscope (Thermo Fisher Scientific) at ×100 magnification after 0 and 24 h.
Apoptosis and Cell Cycle Assays
Apoptosis and cell cycle assays were performed by flow cytometry. Cells were digested and washed twice with pre-cooled PBS, followed by the addition of 5 μL of annexin V-allophycocyanin and 10 μL of propidium iodide (PI) (Yeasen Biotechnology) in the dark at room temperature for 20 min. Apoptosis rates were then detected within 1 h and analyzed using FlowJo software. For cell cycle assay, cells in 6-cm dishes were digested with trypsin and stained using PI/RNase staining buffer (BD Pharmingen, USA), and the results were analyzed using ModFit software.
Immunofluorescence Experiment
Cells were cultured in 48-well plates for the immunofluorescence assay. When the cell density reached 80%, the cells were fixed with 4% paraformaldehyde for 15 min and washed three times with PBS for 5 min each. After blocking with goat serum for 1 h, primary antibodies and DAPI were mixed and added to the cells overnight at 4°C. Fluorescent secondary antibodies were purchased from Yeason (Shanghai, China). For tissue immunofluorescence, paraffin sections of tissues were treated according to the above procedure for cellular immunofluorescence.
Integrated Analysis of Multiple Omics
An RNA-seq assay and protein mass spectrometry were performed by Lianchuan Biotechnology (Hangzhou, China).33,34 Protein quantification and quantitative ubiquitination modification were carried out by Shanghai Applied Protein Technology (Shanghai, China).35 Amino acid metabolomics analysis was performed by Novogene Bioinformatics Technology (Beijing, China).36
Tumor Xenografts
Animal housing and procedures were approved by the local Animal Care and Use Committee at the Children’s Hospital of Fudan University and complied with humane animal care standards. HepG2 cells were cultured for animal experiments. The cells were collected by trypsin digestion, washed with precooled PBS, and converted into single-cell suspensions in PBS. Then, 2 × 106 cells were subcutaneously inoculated into 5-week-old BALB/c nude mice (five mice in every group) randomly. Six weeks later, mice were sacrificed by cervical vertebrae dislocation. The tumors were quickly removed for imaging and weight. Next, 2 × 106 cells were inoculated into the tail vein of mice (two mice from every group) randomly, and 150 mg/kg d-luciferin was intraperitoneally injected 4 weeks later. In vivo imaging was performed 5 min later, and fluorescence was quantified. After the experiment, the mice were sacrificed by cervical dislocation, the lung tissue was stripped, and images were taken. Then, the lung tissue was immersed in formalin and fixed.
Statistical Analysis
The data were analyzed using GraphPad Prism 5 software (GraphPad Software) and expressed as mean ± standard error of the mean (SEM). Differences were analyzed using the Student t test, and p < 0.05 was considered significant.
Data Availability
The authors declare that all the other data supporting the findings of this study are available within the article and its additional files and from the corresponding author upon reasonable request.
Authors Contributions
W.H., J.Z., and B.L. carried out the molecular genetic studies, participated in the sequence alignment, and drafted the manuscript. X.L. and J.M. carried out the immunoassays and participated in the sequence alignment. G.L., L.X., J.H., and M.W. participated in the design of the study and performed the statistical analyses. M.Y., K.D., D.M., R.D., and K.L. conceived of the study, participated in its design and coordination, and helped to draft the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Prof. Deshui Jia and Prof. Xianghuo He for their previous work and for help with our study. This study was supported by the National Natural Science Foundation of China (81771633 and 81572324).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.09.008.
Contributor Information
Duan Ma, Email: duanma@fudan.edu.cn.
Kuiran Dong, Email: kuirand@hotmail.com.
Mujie Ye, Email: mujiey0629@163.com.
Supplemental Information
References
- 1.Kobayashi T., Kubota M., Kinoshita Y., Arai Y., Oyama T., Yokota N., Saito K., Matsuda Y., Osawa M. Epidermal growth factor receptor/heme oxygenase-1 axis is involved in chemoresistance to cisplatin and pirarubicin in HepG2 cell lines and hepatoblastoma specimens. Pediatr. Surg. Int. 2019;35:1369–1378. doi: 10.1007/s00383-019-04563-5. [DOI] [PubMed] [Google Scholar]
- 2.Ranganathan S., Lopez-Terrada D., Alaggio R. Hepatoblastoma and pediatric hepatocellular carcinoma: an update. Pediatr. Dev. Pathol. 2020;23:79–95. doi: 10.1177/1093526619875228. [DOI] [PubMed] [Google Scholar]
- 3.Luo Z., Cao P. Long noncoding RNA PVT1 promotes hepatoblastoma cell proliferation through activating STAT3. Cancer Manag. Res. 2019;11:8517–8527. doi: 10.2147/CMAR.S213707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsumoto S., Yamamichi T., Shinzawa K., Kasahara Y., Nojima S., Kodama T., Obika S., Takehara T., Morii E., Okuyama H., Kikuchi A. GREB1 induced by Wnt signaling promotes development of hepatoblastoma by suppressing TGFβ signaling. Nat. Commun. 2019;10:3882. doi: 10.1038/s41467-019-11533-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mavila N., Thundimadathil J. The emerging roles of cancer stem cells and Wnt/beta-catenin signaling in hepatoblastoma. Cancers (Basel) 2019;11:1406. doi: 10.3390/cancers11101406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia D., Dong R., Jing Y., Xu D., Wang Q., Chen L., Li Q., Huang Y., Zhang Y., Zhang Z. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology. 2014;60:1686–1696. doi: 10.1002/hep.27243. [DOI] [PubMed] [Google Scholar]
- 7.Sha Y.L., Liu S., Yan W.W., Dong B. Wnt/β-catenin signaling as a useful therapeutic target in hepatoblastoma. Biosci. Rep. 2019;39 doi: 10.1042/BSR20192466. BSR20192466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui X., Liu X., Han Q., Zhu J., Li J., Ren Z., Liu L., Luo Y., Wang Z., Zhang D. DPEP1 is a direct target of miR-193a-5p and promotes hepatoblastoma progression by PI3K/Akt/mTOR pathway. Cell Death Dis. 2019;10:701. doi: 10.1038/s41419-019-1943-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mi Y., Xiao C., Du Q., Wu W., Qi G., Liu X. Momordin Ic couples apoptosis with autophagy in human hepatoblastoma cancer cells by reactive oxygen species (ROS)-mediated PI3K/Akt and MAPK signaling pathways. Free Radic. Biol. Med. 2016;90:230–242. doi: 10.1016/j.freeradbiomed.2015.11.022. [DOI] [PubMed] [Google Scholar]
- 10.El-Gendi A., Fadel S., El-Shafei M., Shawky A. Avoiding liver transplantation in post-treatment extent of disease III and IV hepatoblastoma. Pediatr. Int. (Roma) 2018;60:862–868. doi: 10.1111/ped.13634. [DOI] [PubMed] [Google Scholar]
- 11.Fonseca A., Gupta A., Shaikh F., Ramphal R., Ng V., McGilvray I., Gerstle J.T. Extreme hepatic resections for the treatment of advanced hepatoblastoma: are planned close margins an acceptable approach? Pediatr. Blood Cancer. 2018;65:e26820. doi: 10.1002/pbc.26820. [DOI] [PubMed] [Google Scholar]
- 12.Uchida H., Sakamoto S., Sasaki K., Takeda M., Hirata Y., Fukuda A., Hishiki T., Irie R., Nakazawa A., Miyazaki O. Surgical treatment strategy for advanced hepatoblastoma: resection versus transplantation. Pediatr. Blood Cancer. 2018;65:e27383. doi: 10.1002/pbc.27383. [DOI] [PubMed] [Google Scholar]
- 13.Zsíros J., Maibach R., Shafford E., Brugieres L., Brock P., Czauderna P., Roebuck D., Childs M., Zimmermann A., Laithier V. Successful treatment of childhood high-risk hepatoblastoma with dose-intensive multiagent chemotherapy and surgery: final results of the SIOPEL-3HR study. J. Clin. Oncol. 2010;28:2584–2590. doi: 10.1200/JCO.2009.22.4857. [DOI] [PubMed] [Google Scholar]
- 14.Yada K., Ishibashi H., Mori H., Sato H., Shimada M. The role of surgical treatment in the multidisciplinary therapy for hepatoblastoma. Hepatogastroenterology. 2014;61:553–556. [PubMed] [Google Scholar]
- 15.Powers J.M., Pacheco M.M., Wickiser J.E. Addition of vincristine and irinotecan to standard therapy in a patient with refractory high-risk hepatoblastoma achieving long-term relapse-free survival. J. Pediatr. Hematol. Oncol. 2019;41:e171–e173. doi: 10.1097/MPH.0000000000001171. [DOI] [PubMed] [Google Scholar]
- 16.Le N., Rivard D.C., Rentea R.M., Manalang M., Andrews W., Kane B., Hendrickson R.J. Right trisegmentectomy after portal vein embolization in a high-risk toddler with hepatoblastoma. Pediatr. Surg. Int. 2018;34:573–578. doi: 10.1007/s00383-018-4250-4. [DOI] [PubMed] [Google Scholar]
- 17.Beck A., Eberherr C., Hagemann M., Cairo S., Häberle B., Vokuhl C., von Schweinitz D., Kappler R. Connectivity map identifies HDAC inhibition as a treatment option of high-risk hepatoblastoma. Cancer Biol. Ther. 2016;17:1168–1176. doi: 10.1080/15384047.2016.1235664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barrena S., Hernandez F., Miguel M., de la Torre C.A., Moreno A.M., Encinas J.L., Leal N., Murcia J., Martinez L., Gamez M. High-risk hepatoblastoma: results in a pediatric liver transplantation center. Eur. J. Pediatr. Surg. 2011;21:18–20. doi: 10.1055/s-0030-1262798. [DOI] [PubMed] [Google Scholar]
- 19.Katzenstein H.M., Furman W.L., Malogolowkin M.H., Krailo M.D., McCarville M.B., Towbin A.J., Tiao G.M., Finegold M.J., Ranganathan S., Dunn S.P. Upfront window vincristine/irinotecan treatment of high-risk hepatoblastoma: a report from the Children’s Oncology Group AHEP0731 study committee. Cancer. 2017;123:2360–2367. doi: 10.1002/cncr.30591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hernández-Llodrà S., Segalés L., Safont A., Juanpere N., Lorenzo M., Fumadó L., Rodríguez-Vida A., Cecchini L., Bellmunt J., Lloreta-Trull J. SPOP and FOXA1 mutations are associated with PSA recurrence in ERG wt tumors, and SPOP downregulation with ERG-rearranged prostate cancer. Prostate. 2019;79:1156–1165. doi: 10.1002/pros.23830. [DOI] [PubMed] [Google Scholar]
- 21.Cuneo M.J., Mittag T. The ubiquitin ligase adaptor SPOP in cancer. FEBS J. 2019;286:3946–3958. doi: 10.1111/febs.15056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X., Jin J., Wan F., Zhao L., Chu H., Chen C., Liao G., Liu J., Yu Y., Teng H. AMPK promotes SPOP-mediated NANOG degradation to regulate prostate cancer cell stemness. Dev. Cell. 2019;48:345–360.e7. doi: 10.1016/j.devcel.2018.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J., Chen M., Zhu Y., Dai X., Dang F., Ren J., Ren S., Shulga Y.V., Beca F., Gan W. SPOP promotes Nanog destruction to suppress stem cell traits and prostate cancer progression. Dev. Cell. 2019;48:329–344.e5. doi: 10.1016/j.devcel.2018.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan Y., Ma J., Wang D., Lin D., Pang X., Wang S., Zhao Y., Shi L., Xue H., Pan Y. The novel BET-CBP/p300 dual inhibitor NEO2734 is active in SPOP mutant and wild-type prostate cancer. EMBO Mol. Med. 2019;11:e10659. doi: 10.15252/emmm.201910659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fong K.W., Zhao J.C., Song B., Zheng B., Yu J. TRIM28 protects TRIM24 from SPOP-mediated degradation and promotes prostate cancer progression. Nat. Commun. 2018;9:5007. doi: 10.1038/s41467-018-07475-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geng C., Kaochar S., Li M., Rajapakshe K., Fiskus W., Dong J., Foley C., Dong B., Zhang L., Kwon O.J. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene. 2017;36:4767–4777. doi: 10.1038/onc.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng F., Zeng C., Zeng L., Wu C., Chen Y. The association of speckle-type POZ protein with lymph node metastasis and prognosis in cancer patients: A meta-analysis. Medicine (Baltimore) 2019;98:e17439. doi: 10.1097/MD.0000000000017439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mani R.S. The emerging role of speckle-type POZ protein (SPOP) in cancer development. Drug Discov. Today. 2014;19:1498–1502. doi: 10.1016/j.drudis.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Q., Shi Q., Chen Y., Yue T., Li S., Wang B., Jiang J. Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA. 2009;106:21191–21196. doi: 10.1073/pnas.0912008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chafai A., Fromm M.F., König J., Maas R. The prognostic biomarker L-homoarginine is a substrate of the cationic amino acid transporters CAT1, CAT2A and CAT2B. Sci. Rep. 2017;7:4767. doi: 10.1038/s41598-017-04965-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdelmagid S.A., Rickard J.A., McDonald W.J., Thomas L.N., Too C.K. CAT-1-mediated arginine uptake and regulation of nitric oxide synthases for the survival of human breast cancer cell lines. J. Cell. Biochem. 2011;112:1084–1092. doi: 10.1002/jcb.23022. [DOI] [PubMed] [Google Scholar]
- 32.Chang J., Nicolas E., Marks D., Sander C., Lerro A., Buendia M.A., Xu C., Mason W.S., Moloshok T., Bort R. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1:106–113. doi: 10.4161/rna.1.2.1066. [DOI] [PubMed] [Google Scholar]
- 33.Xia W., Hu J., Ma J., Huang J., Jing T., Deng L., Zhang J., Jiang N., Ma D., Ma Z. Mutations in TOP2B cause autosomal-dominant hereditary hearing loss via inhibition of the PI3K-Akt signalling pathway. FEBS Lett. 2019;593:2008–2018. doi: 10.1002/1873-3468.13482. [DOI] [PubMed] [Google Scholar]
- 34.Martins L., Amorim B.R., Salmon C.R., Leme A., Kantovitz K.R., Nociti F.J. Novel LRAP-binding partner revealing the plasminogen activation system as a regulator of cementoblast differentiation and mineral nodule formation in vitro. J. Cell. Physiol. 2020;235:4545–4558. doi: 10.1002/jcp.29331. [DOI] [PubMed] [Google Scholar]
- 35.Liu B., Jiang S., Li M., Xiong X., Zhu M., Li D., Zhao L., Qian L., Zhai L., Li J. Proteome-wide analysis of USP14 substrates revealed its role in hepatosteatosis via stabilization of FASN. Nat. Commun. 2018;9:4770. doi: 10.1038/s41467-018-07185-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hattori A., Tsunoda M., Konuma T., Kobayashi M., Nagy T., Glushka J., Tayyari F., McSkimming D., Kannan N., Tojo A. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature. 2017;545:500–504. doi: 10.1038/nature22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all the other data supporting the findings of this study are available within the article and its additional files and from the corresponding author upon reasonable request.