

Abstract
Bacterial secretory proteins are translocated post‐translationally by the SecA ATPase through the protein‐conducting SecY channel in the plasma membrane. During the ATP hydrolysis cycle, SecA undergoes large conformational changes of its two‐helix finger and clamp domains, but how these changes result in polypeptide movement is unclear. Here, we use a reconstituted purified system and protease protection assays to show that ATP binding to SecA results in a segment of the translocation substrate being pushed into the channel. This motion is prevented by mutation of conserved residues at the finger's tip. Mutation of SecA's clamp causes backsliding of the substrate in the ATP‐bound state. Together, these data support a power stroke model of translocation in which, upon ATP binding, the two‐helix finger pushes the substrate into the channel, where it is held by the clamp until nucleotide hydrolysis has occurred.
Keywords: AAA ATPase, E. coli, protein translocation, SecA, SecYEG
Subject Categories: Membrane & Intracellular Transport
Protease protection experiments show that the SecA ATPase uses a power‐stroke mechanism to directly push translocating polypeptides through the bacterial SecY channel.
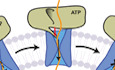
Introduction
In bacteria, most secretory proteins are translocated post‐translationally across the plasma membrane. They are synthesized as precursors (preproteins) containing N‐terminal signal sequences, recruited from the cytosol in an unfolded state, and moved through the protein‐conducting SecY channel by the ATPase SecA (for review, see Rapoport et al, 2017; Seinen & Driessen, 2019). SecA is a monomeric member of the AAA ATPase family. Other members, such as p97 (Cdc48 in yeast), the Clp's, and NSF form hexameric rings that remodel or relocate proteins (for review, see Zhao et al, 2007; Sauer & Baker, 2011; Bodnar & Rapoport, 2017). All these ATPases perform mechanical work on polypeptide substrates, but how SecA translocates polypeptides is unclear.
Previous studies led to a proposed “push‐and‐slide” model for SecA (Erlandson et al, 2008a,b; Zimmer et al, 2008; Bauer et al, 2014; Catipovic et al, 2019). Upon ATP binding to SecA, a two‐helix finger (THF), consisting of two α‐helices connected by a loop, inserts into the SecY channel, providing a power stroke that pushes the interacting polypeptide substrate into the SecY channel (Erlandson et al, 2008a; Catipovic et al, 2019). During ATP hydrolysis, a clamp positioned above the channel closes around the polypeptide substrate (Catipovic et al, 2019; Ma et al, 2019). Clamp closure then allows the THF to reset without dragging the substrate backwards with it. In the ADP‐bound state, after the dissociation of inorganic phosphate, the clamp reopens. With the clamp open and the THF de‐inserted, the polypeptide is free to slide forwards and backwards through the channel, until the next power stroke occurs (Erlandson et al, 2008b; Bauer et al, 2014). The conformational changes of SecA are supported by single‐molecule FRET experiments (Catipovic et al, 2019), and the sliding of the polypeptide chain in the ADP‐bound state by protease protection experiments (Erlandson et al, 2008b; Bauer et al, 2014).
However, a crucial point of the power stroke model remains uncertain: does the THF actually push a polypeptide into the SecY channel? Previous reports have not directly observed the forward movement of the polypeptide proposed to coincide with ATP binding and THF insertion (Bauer et al, 2014; Catipovic et al, 2019). As such, it is possible that the conformational changes of the THF seen by single‐molecule FRET are not directly linked to substrate translocation. Instead, they could be indirectly involved, as proposed in an alternative ratcheting model (Allen et al, 2016; Corey et al, 2019). According to this model, SecA in its ADP‐bound state would cause the SecY channel to be constricted, limiting the substrate's ability to slide through the channel. Large amino acids would trigger exchange of ADP for ATP, causing the SecY channel to open and allowing these bulky residues to diffuse through it. Subsequent ATP hydrolysis would then close the channel, trapping the residues on the periplasmic side. The THF conformational change would either be coupled to opening and closing the SecY channel or required for sensing the residue identities of the substrate as it passes by the tip of the finger.
Here, we use protease protection experiments to directly observe the forward movement of the translocating polypeptide. Our results show that ATP binding induces a forward step of the translocating polypeptide dependent on SecA's THF. Passive sliding in the ATP‐bound state is limited by the SecA clamp and is observed when the clamp is fixed in an open conformation.
Results and Discussion
Purified SecA lacking cysteines was mixed with a model translocation substrate and proteoliposomes containing purified SecYEG. The substrate consisted of a truncated variant of proOmpA, with two cysteines added into its C‐terminal segment (Fig 1A). It was synthesized in an in vitro translation system in the presence of 35S‐methionine, with the two cysteines remaining in a reduced state. When ATP was added, SecA moved the substrate through the SecY channel into the lumen of the proteoliposomes, where it was protected during subsequent proteinase K treatment; the protected population could be visualized by SDS–PAGE followed by autoradiography (Fig 1B lane 2). If ATP was omitted, or detergent was present during proteolysis, no substrate was protected (Fig 1B lanes 3,4). In the presence of the oxidant sodium tetrathionate (NaTT), the two cysteines formed a disulfide bond creating a loop too large to fit through the SecY channel (Fig 1A) (Schiebel et al, 1991; Erlandson et al, 2008b; Bauer et al, 2014). Addition of proteinase K cleaved off the non‐translocated substrate loop, forming an intermediate‐sized species. The presence of the reducing agent dithiothreitol (DTT) prevented disulfide formation and allowed translocation to be completed, as shown by protease protection of the entire polypeptide (Fig 1A and B lanes 5,6).
Figure 1. Forward movement of a translocating polypeptide into the SecY channel.
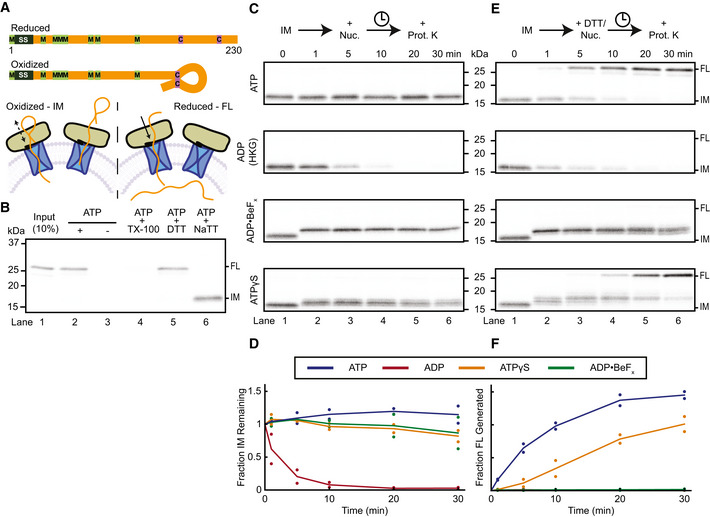
- Schematic of model substrates and translocation assay. Polypeptide substrates consist of truncated variants of proOmpA (orange), including the signal sequence (SS) (black). Methionines and cysteines are indicated in green and purple, respectively. Substrate was incubated with SecA (tan), proteoliposomes containing SecYEG (blue), and ATP. When the cysteines are oxidized, they form a disulfide, which is too large to allow complete translocation of the substrate, generating a translocation intermediate (IM) (bottom left). Depending on the added nucleotide, the polypeptide can slide back and forth in the channel or make a single step into the channel. When the cysteines are reduced, translocation of the full‐length (FL) substrate can be completed with hydrolysable nucleotides (bottom right).
- Substrate was synthesized in reticulocyte lysate in the presence of 35S‐methionine and mixed with SecA and SecYEG proteoliposomes. After incubation with ATP, the samples were treated with proteinase K to degrade any non‐translocated material. Where indicated, the reaction was performed in the absence of ATP and proteolysis in the presence of Triton X‐100 (TX‐100). Translocation reactions were performed in the presence of the reducing agent DTT or oxidizing agent NaTT, generating protease‐protected full‐length (FL) substrate or a translocation intermediate (IM), respectively. All samples were analyzed by SDS–PAGE followed by autoradiography. Lane 1 shows 10% of the input sample. The gel is representative of three replicates. Molecular weight markers are estimated from Coomassie‐stained gels run in parallel.
- Translocation intermediates (IM) generated as in (B) were further incubated in the presence of different nucleotides (Nuc.) for different time periods and then treated with proteinase K (Prot. K). One sample received hexokinase and glucose (HKG) to generate ADP, other samples ADP•BeFx or ATPγS. The gel is representative of two replicates.
- Quantification of (C). Points show IM band intensities normalized to the 0‐min time point for each replicate. Lines show the average of replicates. Colors correspond to different nucleotides as shown in the inset box.
- As in (C), but DTT was added concurrently with HKG or nucleotides. The gel is representative of two replicates.
- Quantification of (E) as in (D), except intensities, were calculated for the FL bands.
Source data are available online for this figure.
In the presence of ATP, the intermediate was stable throughout the experiment (Fig 1C and D). However, if ATP was converted to ADP by the addition of hexokinase and glucose (HKG), the substrate quickly slid back out of the SecY channel, resulting in the disappearance of the intermediate‐sized fragment (Fig 1C and D). To observe the effect of a single ATP‐binding event, we first formed the intermediate and then added an excess of either the non‐hydrolysable ATP analog ADP•BeFx or slowly hydrolysable ATPγS (Burton et al, 2003). Both analogs caused a rapid shift of the protected species to a higher molecular weight, consistent with the polypeptide being pushed further into the SecY channel (Fig 1C). The intensity of this protected species then remained almost constant (Fig 1D). The size shift was not due to SecA protecting a portion of the substrate outside the SecY channel: SecA was readily degraded by proteinase K, as shown by inclusion of fluorescently labeled SecA, while the size of the protected translocation intermediates remained unchanged (Fig EV1A and B).
Figure EV1. Protease resistance of membrane‐protected fragments.
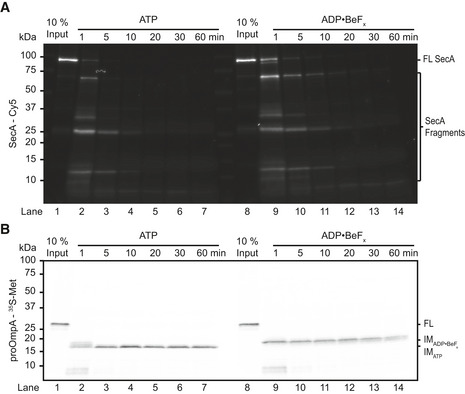
- Translocation intermediates were generated in the presence of ATP using SecA labeled at position 285 with a Cy5 dye. To some samples, ADP•BeFx was added, as indicated. Proteolysis was initiated by the addition of proteinase K at 23°C and quenched after incubation for the indicated times. All samples were analyzed by SDS–PAGE followed by fluorescence scanning. The gel is representative of two replicates.
- The same gel as in (A) imaged by autoradiography to visualize the radiolabeled proOmpA substrate.
Source data are available online for this figure.
Addition of ADP•BeFx to the translocation intermediate resulted in a single band, whereas ATPγS generated several closely spaced fragments (Fig 1C), possibly because its slow hydrolysis allows the substrate to slide in the channel during the ADP‐bound phase, blurring the observed protected band. Indeed, when the disulfide bond was reduced by DTT, ATPγS addition caused translocation to be completed, albeit slower than with ATP, indicating that the analog is hydrolyzed (Fig 1E and F). Translocation in the presence of ATPγS proceeded through the same fragments observed with the stalled complex. Larger intermediates were not visualized, likely because the SecA molecules quickly become asynchronous after the initial translocation step. The addition of ADP•BeFx to the reduced complex shifted the protected species to a higher molecular weight, but translocation was not completed because this analog is truly non‐hydrolysable. In the presence of ADP, the substrate slid backwards out of the channel, even when the stalling disulfide was reduced (Fig 1E), consistent with power strokes depending on ATP binding.
As an alternative method to keep SecA in its ATP‐bound state, we used the D209N Walker B (WB) mutant of SecA, which binds, but does not efficiently hydrolyze, ATP (Mitchell & Oliver, 1993; van der Wolk et al, 1995). This SecA mutant functions as a dominant negative inhibitor of wild‐type SecA (Mitchell & Oliver, 1993; Economou et al, 1995; Bauer et al, 2014). When added to a preformed translocation intermediate, WB SecA replaced wild‐type SecA and pushed the substrate further into the channel, producing a shifted fragment similar to that formed with ATPγS (Figs 2A and B, and EV2A); the intensity of this fragment decreased only little during the time course (Fig 2A and B). Upon the addition of DTT, some full‐length substrate was rapidly produced, likely by wild‐type SecA that was not yet displaced from the translocon (Fig EV2A and B); once the WB mutant replaced the wild‐type, the mutant's low ATPase activity allowed only slow completion of translocation, proceeding through the shifted fragment (Fig EV2A and B).
Figure 2. Substrate movements by Walker B SecA mutants.
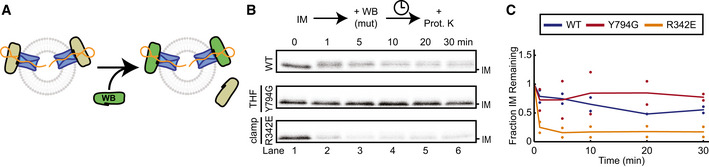
- Schematic showing replacement of wild‐type SecA in a translocation intermediate by Walker B (WB) SecA mutants (green).
- WB SecA with additional mutations (WT, Y794G, R342E) was added to preformed intermediates (IM) and incubated for the indicated times before proteolysis. The gel is representative of two replicates.
- Quantification of (B). Points show IM band intensities normalized to the 0‐min time point for each replicate. Lines show average of replicates. Colors correspond to WB SecA carrying additional mutations, as shown in the inset box.
Source data are available online for this figure.
Figure EV2. Translocation activity of SecA mutants.
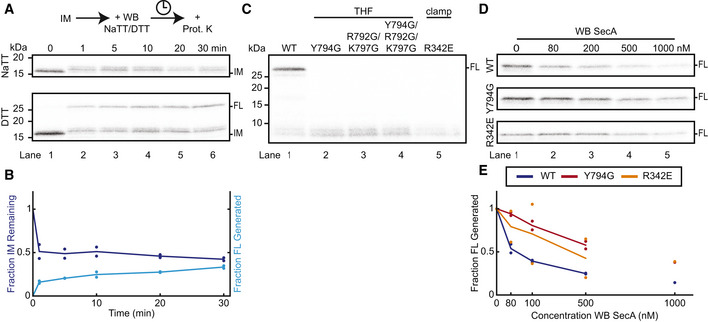
- WB SecA was added to preformed intermediates (IM) in the presence of NaTT or DTT and incubated for the indicated times before proteolysis by proteinase K (Prot. K). The gel is representative of two replicates.
- Quantification of the lower panel of (A). Points show band intensities normalized to the 0‐min time point for each replicate. Lines show average of replicates. The intensities of both the IM (dark blue) and FL (light blue) bands were measured.
- WT SecA, or the indicated mutants, was incubated with SecYEG proteoliposomes and in vitro translated substrate in the presence of ATP and DTT. All samples were treated with proteinase K. The gel is representative of three replicates.
- WB SecA carrying additional mutations (WT, Y794G, R342E) was added at increasing concentrations to translocation reactions with WT SecA, SecYEG proteoliposomes, and in vitro translated substrate in the presence of ATP and DTT. Shown is full‐length substrate protected from proteinase K treatment. The gel is representative of two replicates, except for the 1,000 nM concentration point.
- Quantification of (D). Points show FL band intensities normalized to the value with 0 nM WB for each replicate. Lines show average of replicates. Colors correspond to WB SecA carrying additional mutations, as shown in the inset box.
Source data are available online for this figure.
Because of its ability to displace wild‐type SecA in translocation intermediates, WB SecA was used to introduce additional SecA mutations into preformed wild‐type translocation complexes. Single point mutations at the tip of the THF (Y794G) or in the clamp (R342E) abolished de novo translocation activity (Fig EV2C) (Erlandson et al, 2008a; Chen et al, 2015), but together with the WB mutation, these mutants still served as dominant negative inhibitors of wild‐type SecA. When added at increasing concentrations in the presence of ATP and DTT, these mutants inhibited wild‐type SecA from initiating and completing translocation (Fig EV2D and E), suggesting they still bind to the translocon. When WB SecA carrying the additional THF mutation was added to a preformed translocation intermediate, it did not cause the forward movement seen with WB SecA (Fig 2B). This mutant could, however, largely prevent the polypeptide from sliding backwards, consistent with it having a functional clamp (Fig 2B and C). In contrast, the WB mutant carrying the additional R342E mutation, which prevents clamp closure (Chen et al, 2015), could not prevent the substrate from backsliding (Fig 2B and C).
We next assessed the effect of the THF and clamp mutations on their own, without the additional WB mutation. To this end, we first generated a translocation intermediate with SecA lacking a positively charged N‐terminal helix necessary for binding to acidic phospholipids (Bauer et al, 2014). This helix was replaced by a His6‐tag (dN20::H6 SecA), which can recruit the ATPase to liposomes containing Ni‐modified lipids (Figs 3A and EV3A) (Bauer et al, 2014). SecA targeted in this manner could form a translocation intermediate (Fig EV3B). Upon addition of imidazole, dN20::H6 SecA was released from the liposomes (Fig EV3A), allowing the stalled polypeptide substrate to slide out of the SecY channel (Fig 3B and C). Concurrent addition of wild‐type SecA replaced the dissociated dN20::H6 SecA and prevented the substrate from backsliding (Fig 3B and C).
Figure 3. The roles of the THF and clamp in substrate movement.
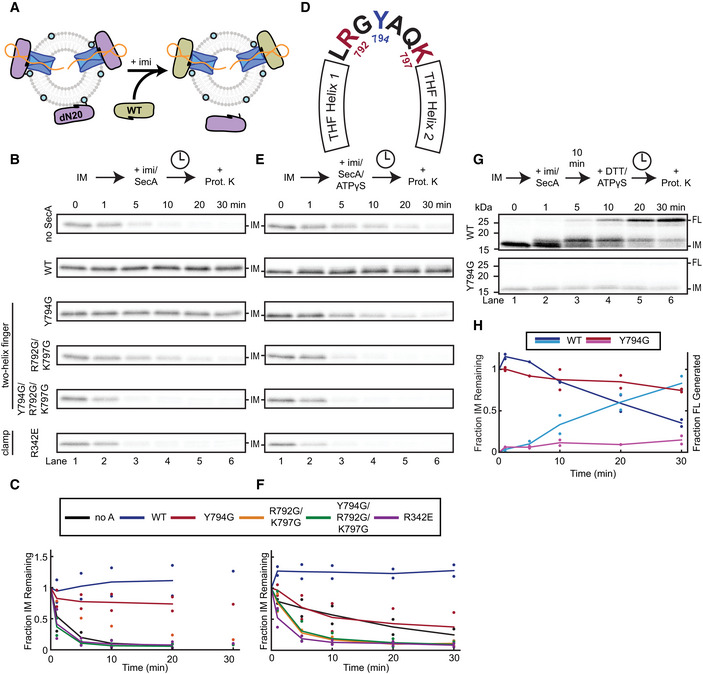
- Schematic showing the generation of a translocation intermediate with SecA dN20::H6 (purple) recruited to SecYEG liposomes containing Ni‐modified lipids (cyan circles). Addition of imidazole dissociates dN20::H6 SecA, which can then be replaced by WT or mutant SecA (tan).
- Imidazole (imi) and either WT or mutant SecA were added to translocation intermediates (IM) preformed with dN20::H6 SecA. The samples were incubated for the indicated times before proteolysis with proteinase K (Prot. K). The gel is representative of two replicates, except for the experiment with the R792G/K797G mutant and the 30‐min time points.
- Quantification of (B). Points show IM band intensities normalized to the 0‐min time point for each replicate. Lines show average of replicates. Colors correspond to different SecA mutants as shown in the inset box.
- Schematic of the residues at the tip of the SecA two‐helix finger. The conserved tyrosine Y794 is indicated in blue, and the conserved basic residues R792 and K797 are in red.
- As in (B), but ATPγS was added together with imidazole. The gel is representative of two replicates.
- Quantification of (E) as in (C).
- As in (D), but DTT was added with ATPγS 10 min after the addition of imidazole and WT or mutant SecA. The gel is representative of two replicates.
- Quantification of (G) as in (F), except that both the intensities of the IM (darker colors) and FL (lighter colors) bands were measured.
Source data are available online for this figure.
Figure EV3. dN20::H6 SecA binds to Ni‐lipid‐containing liposomes and translocates substrates.
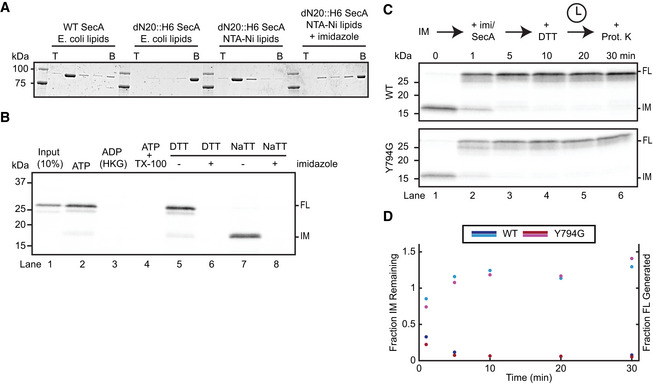
- Binding of SecA to liposomes was determined with a flotation assay. WT or dN20::H6 SecA was incubated with liposomes containing E. coli polar lipids with or without Ni‐NTA lipids. Imidazole was added, where indicated. The samples were loaded at the bottom (B) of a Nycodenz step gradient. After centrifugation, fractions were collected from the top (T) and analyzed by SDS–PAGE and Coomassie‐blue staining. The gel is representative of two replicates.
- ProOmpA was synthesized in reticulocyte lysate in the presence of 35S‐methionine and mixed with dN20::H6 SecA and SecYEG proteoliposomes containing Ni‐NTA lipids. Imidazole was added to the reactions where indicated. After incubation in the presence of ATP, the samples were treated with proteinase K to degrade any non‐translocated material. Where indicated, the reaction was performed in the absence of ATP and proteolysis in the presence of Triton X‐100 (TX‐100). Translocation reactions were performed in the presence of the reducing agent DTT or oxidizing agent NaTT, generating protease‐protected full‐length (FL) substrate or a translocation intermediate (IM), respectively. All samples were analyzed by SDS–PAGE followed by autoradiography. Lane 1 shows 10% of the input sample. The gel is representative of three replicates.
- A translocation intermediate (IM) was formed with dN20::H6 SecA using substrate with a C‐terminal disulfide bridge. Imidazole and WT or mutant SecA was then added, as indicated. After 10 min of incubation, DTT was added and the samples further incubated for the indicated times. All samples were treated with proteinase K (Prot. K) and analyzed by SDS–PAGE and autoradiography.
- Quantification of (C). Points show band intensities normalized to the 0‐min time point for each replicate. Lines show average of replicates. The intensities of both the IM (dark colors) and FL (light colors) bands were measured.
Source data are available online for this figure.
The Y794G THF mutant also maintained the intermediate in the presence of ATP, albeit with slightly lower efficiency. This mutant also completed the translocation of stalled substrate upon addition of DTT (Fig EV3C and D). Both observations are consistent with previous results showing that the mutant THF still interacts with a subset of amino acids in the polypeptide substrate (Bauer et al, 2014). More severe THF mutants, including a double mutant of two basic residues at the finger's tip (R792G/K797G) and a triple mutant of all three conserved amino acids (R792G/Y794G/K797G) (Fig 3D) (Erlandson et al, 2008a; Bauer et al, 2014), were unable to keep the stalled substrate from backsliding (Fig 3B and C). Thus, these mutants have lost their grip on the polypeptide chain and are unable to reinsert the substrate into the SecY channel when the polypeptide backslides during the ADP‐bound phase of SecA. Rapid backsliding was also observed with the clamp mutant (Fig 3B and C).
Although the Y794G mutant is functional in the presence of ATP, it proved defective if supplied with the slowly hydrolyzing ATPγS. When wild‐type SecA was added together with ATPγS and imidazole to the translocation intermediate, backsliding was prevented and the substrate was pushed into the channel (Fig 3E and F), although not quite as much as in experiments with wild‐type SecA alone (Fig 1C and D). In contrast, the Y794G THF mutant failed to shift the polypeptide at all (Fig 3E and F). In fact, the combination of a weakened interaction of the THF with the polypeptide and infrequent movements of the THF in the presence of ATPγS seems to allow effective backsliding, an effect exacerbated by the more severe THF mutants, as well as the clamp mutant (Fig 3E and F). Under these conditions, the THF is inserted into the SecY channel and the clamp is closed (Catipovic et al, 2019), so it seems that both domains are necessary to prevent backsliding in the ATP‐bound state of SecA.
The Y794G THF mutant was also defective in completing translocation in the presence of ATPγS. Translocation intermediates generated with dN20::H6 SecA were incubated with imidazole and either WT or mutant SecA for 10 min, a period long enough to completely displace the His‐tagged SecA (Fig 3B and C). When the disulfide in the intermediate was then reduced, wild‐type SecA completed translocation in the presence of ATPγS (Fig 3G and H). As before, the substrate took an initial step upon ATPγS binding and then slowly proceeded over subsequent hydrolysis events until the full‐length substrate was protected from proteolysis. Under the same conditions, the Y794G THF mutant could neither produce this initial step nor complete translocation of the stalled intermediate (Fig 3G and H).
Our data provide strong evidence for a power stroke during SecA‐mediated translocation. Previous single‐molecule experiments showed that the THF moves toward the SecY channel upon ATP binding (Catipovic et al, 2019). Our results now show that this movement is directly coupled to the forward translocation of the polypeptide substrate. The binding of ATP to SecA causes the THF to push a segment of the polypeptide into the channel. Furthermore, we show that closure of the SecA clamp cooperates with the THF to restrict substrate sliding before ATP hydrolysis has occurred. These results were obtained with proOmpA as a model substrate, so further experiments with other substrates are necessary to test the universality of these conclusions.
Our results argue against a proposed Brownian ratcheting mechanism of translocation (Allen et al, 2016; Corey et al, 2019). Contrary to this model, ATP binding does not permit passive polypeptide sliding, but rather pushes the translocation substrate into the channel, where it is held in place until hydrolysis is complete. Instead, sliding of the polypeptide substrate occurs predominantly during the ADP‐bound state of the ATPase.
Because the size of the protease‐protected polypeptide fragment increases by ~1 kDa upon ATP binding, it seems that SecA moves ~10 amino acids with each power stroke. This large movement would be consistent with the magnitude of the FRET change observed when the THF inserts into the SecY channel (Catipovic et al, 2019). However, the exact step size cannot be determined by protease protection experiments, as proteases do not cleave each peptide bond with equal efficacy (Keil, 1992). The occlusion of a cleavage site by a small movement could appear as a much larger step if the next available cleavage site is several residues away. The step size of SecA is also not constant, as it depends on the amino acid sequence of the substrate. For example, when the THF encounters a stretch of glycines, there would be no forward movement by power strokes (Bauer et al, 2014).
In summary, the presented protease protection experiments prove two crucial points of the power stroke model: (i) the polypeptide chain is pushed by the THF into the SecY channel when SecA binds ATP and (ii) the clamp plays an important role in holding the polypeptide chain before SecA converts into the ADP‐bound state. Together with previous results, including those from single‐molecule FRET experiments (Catipovic et al, 2019), our data provide compelling evidence for the push‐and‐slide model of protein translocation by SecA.
Materials and Methods
Protein expression and purification
SecA, including all non‐WB SecA mutants, was purified essentially as previously described (Bauer et al, 2014). Specifically, a construct encoding cysteine‐free SecA N95 (lacking the non‐essential C‐terminus (Matsuyama et al, 1990)) with a C‐terminal His‐6 tag and 3C protease cleavage site was cloned into the pET30b vector (EMD Millipore, Burlington, Massachusetts) and expressed in BL21(DE3) E. coli cells (New England Biolabs, Ipswich, Massachusetts) for 4 h at 37°C after induction at OD600 0.8 with 1 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG). Cells were collected by centrifugation for 10 min at 4,000 × g, resuspended in buffer A (50 mM HEPES/KOH pH 7.5, 300 mM NaCl, 15 mM imidazole, 20 mM β‐mercaptoethanol (BME)), and lysed by two passes through an EmulsiFlex‐C3 instrument (Avestin, Ottawa, Canada) at 20,000 psi. The homogenate was centrifuged at 110,000 × g for 45 min. The supernatant was bound to 2 ml Ni2+ resin, washed with 50 ml buffer B (50 mM HEPES/KOH pH7.5, 100 mM NaCl), and incubated overnight in 5 ml buffer B at 4°C with 5 μM 3C protease. The flow‐through was collected and subjected to anion exchange chromatography (HiTrap Q FF, GE Healthcare Life Sciences, Marlborough, Massachusetts) followed by size exclusion chromatography (Superdex 200 10/300GL, GE Healthcare Life Sciences) in buffer C (50 mM HEPES/KOH pH 7.5, 50 mM KCl).
DNA encoding cysteine‐free D209N WB SecA N95 with a C‐terminal His‐6 tag was cloned into the pET30b vector (EMD Millipore, Burlington, Massachusetts) and expressed in BL21(DE3) E. coli cells (New England Biolabs, Ipswich, Massachusetts) for 4 h at 37°C after induction at OD600 0.8 with 1 mM IPTG. Cells were collected by centrifugation for 10 min at 4,000 × g, resuspended in buffer A, and lysed by two passes through an EmulsiFlex‐C3 instrument (Avestin, Ottawa, Canada) at 20,000 psi. The resulting lysate was centrifuged for 45 min at 110,000 × g, and the insoluble pellet containing WB SecA was collected. The pellet was incubated in buffer A with 6 M urea for 1 h at 23°C and then centrifuged again at 110,000 × g for 45 min. The supernatant was bound to 2 ml Ni2+ resin and washed 4 times with 10 ml of buffer A containing 6, 4, 2, and finally 0 M urea. The protein was eluted in 5 ml of buffer B containing 250 mM imidazole. The eluate was collected and subjected to anion exchange chromatography (HiTrap Q FF, GE Healthcare Life Sciences, Marlborough, Massachusetts) followed by size exclusion chromatography (Superdex 200 10/300GL, GE Healthcare Life Sciences) in buffer C.
Genes encoding the three E. coli SecYEG protein components, with an N‐terminal His‐6 tag on SecE, were cloned into the pBAD22 vector (ATCC, Manassas, Virginia) under a single l‐arabinose‐inducible promoter. The cells were grown to OD600 0.8 and induced for 4 h at 37°C by addition of 10 ml 20% l‐arabinose. The cells were collected, lysed, and fractionated in the same manner as for SecA. The membrane fraction was solubilized for 90 min in buffer D (buffer A, 10% glycerol) with 1% n‐dodecyl β‐D‐maltoside (DDM, Anatrace Inc., Maumee, Ohio). The extract was subjected to high speed centrifugation at 110,000 × g for 45 min. Subsequent steps were carried out in buffers containing 0.03% DDM. The protein was bound to 1 ml Ni2+ resin in buffer D, washed with 50 ml buffer D, and eluted in 5 ml buffer E (buffer B, 10% glycerol) with 250 mM imidazole. The eluate was then subjected to cation exchange chromatography (HiTrap SP FF, GE Healthcare Life Sciences) and size exclusion chromatography (Superdex 200 10/300, GE Healthcare Life Sciences) in buffer F (buffer C, 10% glycerol).
Liposome preparation and membrane protein reconstitution
Liposomes were prepared from E. coli Polar Lipid Extract (Avanti Polar Lipids, Alabaster, Alabama). 2 mg of lipids from a 25 mg/ml chloroform stock were dried under nitrogen stream, resuspended in 500 μl diethyl ether, dried again, and stored under vacuum overnight to remove all solvent traces. The resulting lipid film was hydrated in 500 μl of buffer C by vortexing, followed by shaking for 1 h at 750 rpm at 23°C. This suspension was then subjected to 10 freeze/thaw cycles. Finally, the liposomes were passed 21 times through a 400 nm polycarbonate filter (Avestin) in a Mini‐Extruder (Avanti Polar Lipids). Liposomes intended for use with dN20::H6 SecA included 10%, by mass, of 18:1 DGS‐NTA(Ni) (Avanti Polar Lipids, Alabaster, Alabama) as well as 200 mM sucrose in all buffers.
To reconstitute SecYEG into these liposomes, 100 μl of 4 mg/ml liposomes were mixed with 0.4% Triton X‐100 and 1 nmol of purified protein. The reconstitution volume was brought to 200 μl by addition of buffer C and the mixture was incubated for 30 min at 4°C. Detergent was then removed by 4 sequential batches of SM‐2 Biobeads (Bio‐Rad Laboratories, Hercules, California) for 1, 4, 12, and 2 h. The final proteoliposomes were centrifuged for 5 min at 14,000 × g to remove any insoluble material before use. New proteoliposome preparations were used for each experiment. SecYEG reconstituted in this manner shows no orientation bias. Half of all complexes face outwards, with their cytosolic interface accessible to SecA and substrate (Allen et al, 2016).
Protein labeling
500 μl of 10 μM purified SecA L285C protein was incubated with 40 μM of tris(2‐carboxyethyl)phosphine (TCEP) for 20 min on ice. 100 μM maleimide‐conjugated Cyanine5 (Cy5, Lumiprobe, Hunt Valley, Maryland) was added to the SecY and SecA samples, respectively, from a 10 mM stock in dimethyl sulfoxide (DMSO) and rotated overnight at 4°C. Labeling was quenched by the addition of 10 mM 1,4‐dithiothreitol (DTT, GoldBio, St. Louis, MO). Dye excess was then removed via gel filtration through a 30 cm column packed with Superfine G50 Sephadex (GE Healthcare Life Sciences) equilibrated in buffer C. The first visible dye peak was collected and further purified by size exclusion chromatography (Superdex 200 10/300, GE Healthcare Life Sciences). Labeling efficiencies were generally around 80%.
Translocation assays
35S‐Met‐labeled proOmpA containing a truncation from position 176 to 294, 3 additional methionine residues at positions 47–49, and cysteines at the resulting positions 167 and 205 (proOmpA‐3M‐167/205C) was generated by in vitro translation. mRNA was transcribed from a linearized template with an SP6 promoter followed by the Kozak consensus ribosome‐binding site directly 5′ to the proOmpA gene start codon using a RiboMax SP6 in vitro transcription kit (Promega, Madison, Wisconsin). 2 μg of transcription product RNA was mixed with 35 μl of nuclease‐treated rabbit reticulocyte lysate (Promega), 1 μl of 1 mM amino acid mixture minus methionine (Promega), and 2 μl (0.022 μCi) of EasyTag Express S35 Labeling Mix (PerkinElmer, Waltham, Massachusetts). Translation product was precipitated by the addition of 150 μl saturated ammonium sulfate and resuspended in 50 μl 6 M urea pH 6.8.
Reactions were assembled in buffer G (50 mM Hepes/KOH pH 7.5, 50 mM KCl, 5 mM MgCl2, 0.2 mg/ml BSA). 200 nM SecA and 200 nM SecYEG proteoliposomes were mixed with 1 μl of the in vitro translation products in a 50 μl total volume. When indicated, 400 μM sodium tetrathionate (NaTT, Sigma‐Aldrich, St. Louis, MO) or 10 mM DTT was added to the reaction. Reactions were initiated by the addition of 1 mM ATP and incubated for 10 min at 37°C while shaking at 650 rpm. ATP‐free reactions were performed in the presence of 1 U of hexokinase (Sigma‐Aldrich) and 20 mM glucose in place of ATP. For specified reactions, 5 mM ATPγS (Jena Bioscience, Jena, Germany), 1 mM ADP•BeFx, or 1 μM WB SecA were added and incubated for an additional 5 min at 37°C. For specific experiments, the concentration added or the incubation length was varied, as noted in the corresponding figures. In some cases, 10 mM DTT was added after the initial incubation with NaTT to release the disulfide and observe the completion of translocation.
For experiments performed with dN20::H6 SecA, reactions were assembled with SecYEG proteoliposomes containing 10% DGS‐NTA(Ni) lipids. After incubation for 10 min at 37°C, 250 mM imidazole, along with 500 nM of the indicated SecA mutants, was added to the reaction. At this point, either 5 mM ATPγS or 1 mM ATP was also added and incubations were continued for the indicated time. In some cases, 10 mM DTT was added along with additional nucleotides 10 min after addition of wild‐type or mutant SecA.
Reactions were terminated by transfer to ice and addition of 0.4 mg/ml proteinase K and, where indicated, 0.2% Triton X‐100. Digests were continued for 45 min and quenched with 2 mM phenylmethylsulfonyl fluoride (PMSF). Reactions for protease resistance experiments were incubated at 23°C for 1 to 60 min. Reactions were then precipitated in 10% trichloroacetic acid (TCA) and resuspended in 1× Laemmli buffer with 300 mM Tris base. Samples were then analyzed by SDS–PAGE. The gels were vacuum dried, exposed to autoradiography screens overnight, and imaged by a Personal Molecular Imager (Bio‐Rad). Fluorescent imaging was performed with an Odyssey CLx Imaging System (LI‐COR Biosciences, Lincoln, Nebraska) using the 700 nm excitation channel.
Quantifications
Band intensity in autoradiographs was measured using ImageJ (ImageJ, RRID:SCR_003070). Background was subtracted from raw images using a 50 pixel rolling ball radius and the band intensity was extracted using the build‐in “Gel Analyzer” tools. The intensities of both FL and IM bands were normalized for each experiment to the intensity of the IM band at the 0‐min time point. Plots were generated in MATLAB (MathWorks, Natick, Massachusetts).
Flotation assay
40 μg 400 nm liposomes containing either 100% E. coli Polar Lipids (ecPL, Avanti Polar Lipids) or 90% ecPL and 10% DGS‐NTA(Ni) (Avanti Polar Lipids) was mixed with either 2.5 μM WT SecA or 2.5 μM dN20::H6 SecA in buffer C, including 250 mM imidazole where indicated, in a 20 μl volume. This sample was mixed with 20 μl 80% Nycodenz (Alere Technologies AS, Oslo, Norway) in buffer C in a 175 μl open top polypropylene ultracentrifuge tube (Part #: 342630, Beckman Coulter, Indianapolis, IN). Above this 40 μl layer were stacked 4 more 40 μl layers containing 30, 20, 10, and 0% Nycodenz in buffer C, either with or without imidazole. The tubes were centrifuged at 200,000 × g for 1 h at 25°C in a TLS‐55 rotor (Beckman Coulter). After centrifugation, five 40 μl fractions were taken from the top. 15 μl of each fraction was mixed with 5 μl of 4× reducing Laemmli buffer, heated to 65°C for 10 min, and analyzed by SDS–PAGE with Coomassie‐blue staining.
Author contributions
MAC: Conception and design, Acquisition of data, Analysis and interpretation of data, and Drafting the article. TAR: Analysis and interpretation of data and Drafting the article.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Expanded View
Review Process File
Acknowledgements
We thank B. Bauer and S. Shao for critical reading of the manuscript. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. This paper was supported by the following grants: NIGMS grant (R01GM052586) to Tom A. Rapoport. Howard Hughes Medical Institute to Tom A. Rapoport.
EMBO Reports (2020) 21: e50905
Data availability
Source data for all figures are available with the manuscript online. No data have been deposited in public databases.
References
- Allen WJ, Corey RA, Oatley P, Sessions RB, Baldwin SA, Radford SE, Tuma R, Collinson I (2016) Two‐way communication between SecY and SecA suggests a Brownian ratchet mechanism for protein translocation. Elife 5: e15598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer BW, Shemesh T, Chen Y, Rapoport TA (2014) A “Push and Slide” mechanism allows sequence‐insensitive translocation of secretory proteins by the SecA ATPase. Cell 157: 1416–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar N, Rapoport T (2017) Toward an understanding of the Cdc48/p97 ATPase. F1000 Res 6: 1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton RE, Baker TA, Sauer RT (2003) Energy‐dependent degradation: linkage between ClpX‐catalyzed nucleotide hydrolysis and protein‐substrate processing. Protein Sci 12: 893–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catipovic MA, Bauer BW, Loparo JJ, Rapoport TA (2019) Protein translocation by the SecA ATPase occurs by a power‐stroke mechanism. EMBO J 38: e101140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bauer BW, Rapoport TA, Gumbart JC (2015) Conformational changes of the clamp of the protein translocation ATPase SecA. J Mol Biol 427: 2348–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey RA, Ahdash Z, Shah A, Pyle E, Allen WJ, Fessl T, Lovett JE, Politis A, Collinson I (2019) ATP‐induced asymmetric pre‐protein folding as a driver of protein translocation through the Sec machinery. Elife 8: e41803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economou A, Pogliano JA, Beckwith J, Oliver DB, Wickner W (1995) SecA membrane cycling at SecYEG is driven by distinct ATP binding and hydrolysis events and is regulated by SecD and SecF. Cell 83: 1171–1181 [DOI] [PubMed] [Google Scholar]
- Erlandson KJ, Miller SBM, Nam Y, Osborne AR, Zimmer J, Rapoport TA (2008a) A role for the two‐helix finger of the SecA ATPase in protein translocation. Nature 455: 984–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlandson KJ, Or E, Osborne AR, Rapoport TA (2008b) Analysis of polypeptide movement in the SecY channel during SecA‐mediated protein translocation. J Biol Chem 283: 15709–15715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil B (1992) Specificity of proteolysis. Berlin Heidelberg: Springer; [Google Scholar]
- Ma C, Wu X, Sun D, Park E, Catipovic MA, Rapoport TA, Gao N, Li L (2019) Structure of the substrate‐engaged SecA‐SecY protein translocation machine. Nat Commun 10: 2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Kimura E, Mizushima S (1990) Complementation of two overlapping fragments of SecA, a protein translocation ATPase of Escherichia coli, allows ATP binding to its amino‐terminal region. J Biol Chem 265: 8760–8765 [PubMed] [Google Scholar]
- Mitchell C, Oliver D (1993) Two distinct ATP‐binding domains are needed to promote protein export by Escherichia coli SecA ATPase. Mol Microbiol 10: 483–497 [DOI] [PubMed] [Google Scholar]
- Rapoport TA, Li L, Park E (2017) Structural and mechanistic insights into protein translocation. Annu Rev Cell Dev Biol 33: 369–390 [DOI] [PubMed] [Google Scholar]
- Sauer RT, Baker TA (2011) AAA+ proteases: ATP‐fueled machines of protein destruction. Annu Rev Biochem 80: 587–612 [DOI] [PubMed] [Google Scholar]
- Schiebel E, Driessen AJM, Hartl FU, Wickner W (1991) ΔμH+ and ATP function at different steps of the catalytic cycle of preprotein translocase. Cell 64: 927–939 [DOI] [PubMed] [Google Scholar]
- Seinen A‐B, Driessen AJM (2019) Single‐molecule studies on the protein translocon. Annu Rev Biophys 48: 185–207 [DOI] [PubMed] [Google Scholar]
- van der Wolk JP, Klose M, de Wit JG, den Blaauwen T, Freudl R, Driessen AJ (1995) Identification of the magnesium‐binding domain of the high‐affinity ATP‐binding site of the Bacillus subtilis and Escherichia coli SecA protein. J Biol Chem 270: 18975–18982 [DOI] [PubMed] [Google Scholar]
- Zhao C, Slevin JT, Whiteheart SW (2007) Cellular functions of NSF: not just SNAPs and SNAREs. FEBS Lett 581: 2140–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer J, Nam Y, Rapoport TA (2008) Structure of a complex of the ATPase SecA and the protein‐translocation channel. Nature 455: 936–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Expanded View
Review Process File
Data Availability Statement
Source data for all figures are available with the manuscript online. No data have been deposited in public databases.