

Abstract
Prominin‐1 (Prom1) is a major cell surface marker of cancer stem cells, but its physiological functions in the liver have not been elucidated. We analyzed the levels of mRNA transcripts in serum‐starved primary WT (Prom1 +/+) and KO (Prom1 −/−) mouse hepatocytes using RNA sequencing (RNA‐seq) data, and found that CREB target genes were downregulated. This initial observation led us to determine that Prom1 deficiency inhibited cAMP response element‐binding protein (CREB) activation and gluconeogenesis, but not cyclic AMP (cAMP) accumulation, in glucagon‐, epinephrine‐, or forskolin‐treated liver tissues and primary hepatocytes, and mitigated glucagon‐induced hyperglycemia. Because Prom1 interacted with radixin, Prom1 deficiency prevented radixin from localizing to the plasma membrane. Moreover, systemic adenoviral knockdown of radixin inhibited CREB activation and gluconeogenesis in glucagon‐treated liver tissues and primary hepatocytes, and mitigated glucagon‐elicited hyperglycemia. Based on these results, we conclude that Prom1 regulates hepatic PKA signaling via radixin functioning as an A kinase‐anchored protein (AKAP).
Keywords: cAMP signaling, gluconeogenesis, Prominin‐1, protein kinase A, radixin
Subject Categories: Cancer, Metabolism, Signal Transduction
Prominin‐1 in the hepatocytes interacts with radixin at the plasma membrane and regulates hepatic gluconeogenesis by augmenting PKA signaling via radixin as a PKA‐anchoring protein.
Introduction
In well‐understood glucagon‐elicited signaling pathway, glucagon binds to and stimulates the glucagon receptor, a G protein‐coupled receptor, in the plasma membrane of hepatocytes. The stimulated glucagon receptor in turn activates Gsα and adenylyl cyclase (AC). The cyclic AMP (cAMP) produced by the activated AC binds to the regulatory domain of protein kinase A (PKA) to liberate the catalytic domain of PKA from the regulatory domain and to activate the catalytic domain. Not only does activated PKA induces glycogenolysis by consecutively phosphorylating and activating phosphorylase kinase and glycogen phosphorylase b, but also stimulates gluconeogenesis by phosphorylating and activating cAMP response element‐binding protein (CREB). Subsequently, activated CREB induces the transcriptional activation of gluconeogenesis‐related genes such as phosphoenolpyruvate carboxykinase (Pck) and glucose‐6‐phosphatase (G6p) (Meinkoth et al, 1993; Altarejos & Montminy, 2011).
Detergent‐resistant lipid rafts which are composed of cholesterol and glycolipids and serve as a signaling center facilitate the cascade of glucagon signaling by organizing glucagon receptor, Gsα, AC, A kinase‐anchored proteins (AKAPs), PKA, and PKA substrates in close proximity (Head et al, 2006; Kim et al, 2010; Delint‐Ramirez et al, 2011). AKAPs are scaffolding proteins that bind to PKA and its substrates. Thus, AKAPs allow PKA and its substrates to coexist in the same place, such as the plasma membrane, mitochondria, or nucleus, enabling effective signal transduction by cAMP (Wong & Scott, 2004; Dema et al, 2015).
The penta‐transmembrane glycoprotein prominin‐1, also called CD133, is associated with unique detergent‐resistant membrane rafts, bound to cholesterol (Roper et al, 2000), localized in membrane protrusions such as microvilli, filopodia, and primary cilia in epithelial cells (Corbeil et al, 1999); membrane expansions in the myelin sheath originating from oligodendrocytes (Corbeil et al, 2009); membrane invaginations in the outer segment of rod photoreceptor cells (Zacchigna et al, 2009); and the midbody in epithelial cells (Dubreuil et al, 2007), and expressed in various epithelial cells in the brain, kidney, digestive track, and liver (Corbeil et al, 2013).
Mice with a systemic deficiency in Prom1 exhibit disk dysmorphogenesis and photoreceptor degeneration along with a complete loss of vision, indicating that Prom1 might be necessary for the formation of membrane protrusions (Zacchigna et al, 2009). Indeed, it was reported recently that silencing Prom1 from non‐epithelial cells such as hematopoietic stem cells shortened microvilli length (Thamm et al, 2019). In addition, Prom1 has been extensively studied as a major cancer stem cell marker in human brain, colon, ovarian, and liver tumors (Singh et al, 2003; O'Brien et al, 2007). The Prom1‐positive cell population in these tumors has characteristics of self‐renewal, differentiation potential and resistance to chemo‐ and/or radiotherapy as well as tumor development after xenograft transplantation in immunocompromised mice (Dalerba et al, 2007; Li et al, 2007; Krishnan et al, 2013). Because phosphatidylinositide 3‐kinases (PI3K) interacts with Prom1 and Akt is highly phosphorylated in Prom1‐positive cell populations, the Prom1‐PI3K‐Akt signaling pathway may be required for the maintenance of cancer stem cells (Wei et al, 2013). In the aforementioned study by Thamm et al (Thamm et al, 2019), mutation on the conserved tyrosine phosphorylation sites of Prom1 reduced the interaction with p85 subunit of PI3K supporting Prom1‐PI3K signaling pathway. However, this pathway has not been verified in a Prom1‐deficient animal model.
It has been reported that Prom1 expression is not limited to stem cells and found in luminal differentiated epithelial cells in human liver samples using a specific antibody (Karbanová et al, 2008) or in mouse liver using Prom1 LacZ/+ mice (Shmelkov et al, 2008). Prom1 is also expressed at low level in the normal liver of Prom1 cre‐ert2-nlacz mice (Zhu et al, 2009) and associated with the progression of liver fibrosis (Nguyen et al, 2017). However, the physiological functions of Prom1 in the normal liver are not understood clearly. To gain insight into Prom1 function, we examined mRNA levels in primary hepatocytes from the livers of Prom1‐deficient mice and identified the downregulation of gluconeogenic genes. In particular, we found that hepatic control of glucagon or adrenergic stimuli‐elicited gluconeogenesis was impaired in Prom1‐deficient mice. Mechanistically, we showed that Prom1 directly interacted with radixin and facilitated PKA activation through its C‐terminal tail. Prom1 regulates hepatic PKA signaling via recruiting radixin, an A kinase‐anchored protein (AKAP) which brings PKA to membrane rafts where G protein‐coupled receptor (GPCR), AC, and PKA substrates are in close proximity.
Results
Prom1 is expressed in mouse primary hepatocyte
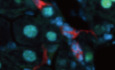
To gain insight into Prom1 function in the liver, we first examined the expression of Prom1 in mouse primary hepatocytes by correlative light and electron microscopy (CLEM) using the anti‐mouse Prom1 monoclonal antibody (13A4, Weigmann et al, 1997). Prom1 antibody detected the Prom1 expression in WT mouse hepatocytes (Fig 1A–F), but not in KO hepatocytes (Fig 1G–I). As reported by others (Corbeil et al, 1999), expression of Prom1 was observed in the microvilli (Fig 1B and C) as well as the planar regions of plasma membrane of hepatocytes (Fig 1E and F). We confirmed the expression of Prom1 in hepatocytes by comparing two serial tissue sections immunostained with Prom1 antibody or antibodies for cell lineage markers (Fig 1J). Because Prom1 antibody (13A4) and antibodies (K9218 for HNF4α and EP1580Y for CK19) for hepatocyte marker (HNF4α) and cholangiocyte marker (CK19), respectively, use incompatible antigen‐retrieval procedures, we immunostained Prom1 or HNF4α/CK19 on separate serial sections. Prom1 was co‐localized with HNF4α in many hepatocytes (triangles in the lower panel of Fig 1J) and with CK19 in the epithelium of bile ducts (asterisks in the upper panel of Fig 1J).
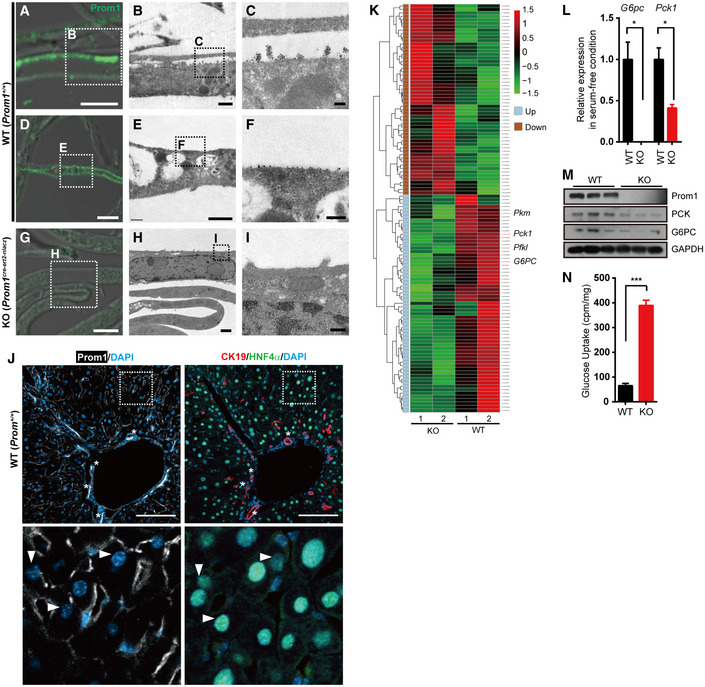
Figure 1. Prom1 is expressed in mouse primary hepatocytes and its deficiency prevents glucagon‐induced gluconeogenesis.
-
A–ICorrelative light electron microscopy (CLEM) showing Prom1 at the plasma membrane of primary hepatocytes. Primary hepatocytes were isolated from 12‐week-old male WT (Prom1 +/+) or KO (Prom1 cre‐ert2-nlacz) mice. The boxed areas in panels (A, D, G) and (B, E, H) were magnified and presented as panels (B, E, H) and (C, F, I), respectively. Scale bar; 5 μm (A, D and G), 1 μm (B, E and H), and 0.2 μm (C, F and I).
-
JImmunofluorescent staining of Prom1, CK19, and HNF4α in two serial cryosections of mouse liver. The boxed area in the top panel was magnified and presented as the bottom panel. Asterisks (*) in the top panel indicate cholangiocytes of the bile ducts and triangles ((Δ) in the bottom panel indicate hepatocytes. Scale bar, 100 μm.
-
K–NPrimary hepatocytes were isolated from 12‐week-old male Prom1 +/+ (WT) or Prom1 −/− (KO) mice and serum‐starved. (K) Differentially expressed genes showing more than a 2‐fold change with P < 0.05 (118 DEGs) induced (n = 55) or repressed (n = 63) by Prom1 knockout (n = 2/group). G6pc, glucose‐6-phosphatase catalytic subunit; Pck1, phosphoenolpyruvate carboxykinase 1. (L) Relative expression levels of G6pc and Pck1 mRNAs after serum starvation for 16 h were determined by qRT–PCR (n = 3/group). (M) Levels of the Prom1, PCK, G6PC, and GAPDH proteins were determined by immunoblotting. (N) Prom1 +/+ or Prom1 −/− hepatocytes serum‐starved for 8 h were incubated with 2‐[3H]deoxyglucose for 10 min. Glucose uptake was assessed by calculating the amount of cell‐associated radioactivity normalized to the amount of protein (n = 3 mice/group).
Prom1 −/− mouse used in this study to determine Prom1 functions in the liver did not show any apparent phenotype, as reported (Zhu et al, 2009), except Prom1 −/− primary hepatocytes were slightly larger than Prom1 +/+ hepatocytes. FACS analysis showed that 18% more Prom1 −/− primary hepatocytes belonged to the area larger than the mean of the forward scatter (FSC) of WT primary hepatocytes (Fig EV1A), which resulted in larger mean of the forward scatter of Prom1 −/− primary hepatocytes than that of WT primary hepatocytes (Fig EV1B). Because this mouse has the integrated nLacZ gene (where nlacZ indicates nuclear lacZ) in the Prom1 locus under the control of the Prom1 promoter, we further examined the hepatic Prom1 expression by means of β‐galactosidase assay. The strong nLacZ expression was observed in bile ducts and several patches of hepatocytes (Fig EV1C).
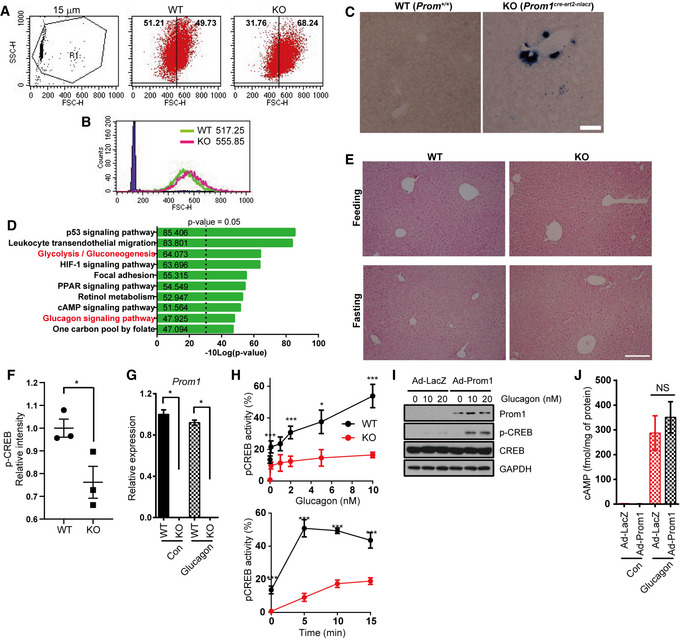
Figure EV1. Prom1 disruption prevents glucagon‐induced gluconeogenesis in primary mouse hepatocytes.
-
ATypical plot of side scatter versus forward scatter for 15 μm beads, WT (Prom1 +/+), and KO (Prom1 −/−) primary hepatocytes.
-
BThe forward scatter distribution of 15 μm beads, WT, and KO hepatocytes is presented as the superimposed histograms, and the means of forward scatter of WT and KO hepatocytes were labeled.
-
Cβ‐galactosidase expression pattern in WT (Prom1 +/+) and KO (Prom1 cre‐ert2-nlacz) liver using X‐gal and nitroblue tetrazolium. Scale bar, 200 μm.
-
DEnriched KEGG pathway terms for differentially expressed genes. P‐value was calculated by gProfileR in R package using g:SCS algorithm.
-
EH&E stain (Hematoxylin and eosin stain) of the liver of mouse in each condition. Scale bar, 500 μm.
-
FRelative level of p‐CREB in WT and KO mouse livers after fasting (Fig 2B, n = 3 mice/group).
-
GPrimary mouse hepatocytes were prepared from 12‐week-old male WT and KO mice and serum‐starved for 16 h. Relative expression levels of Prom1 mRNAs were determined using qRT–PCR (n = 3/group).
-
HThe percentage of cells with nuclear p‐CREB staining among more than 300 cells from each group was statistically determined after stimulation with the indicated concentrations of glucagon for 10 min (left panel) or stimulation with 10 nM glucagon for the indicated times (right panel) (n = 3 replicates/group).
-
I, JProm1 overexpression in KO hepatocytes restores glucagon‐induced CREB phosphorylation. Primary hepatocytes were isolated from 12‐week-old male KO mice, infected with an adenovirus harboring Prom1 for 24 h, serum‐starved for 18 h, and then stimulated with glucagon (10 nM). (I) Levels of p‐CREB and CREB were determined by immunoblotting 0, 10, and 20 min after glucagon (10 nM) stimulation. (J) The cAMP concentration was determined using the cAMP assay kit 10 min after glucagon stimulation (10 nM) in the presence of 10 μM IBMX (n = 3 mice/group).
Prom1 is required for glucagon‐ and cAMP‐elicited gluconeogenesis in mouse primary hepatocytes
After confirming Prom1 expression in mouse liver as well as primary hepatocytes, we analyzed the levels of mRNA transcripts in serum‐starved primary Prom1 +/+ and Prom1 −/− mouse hepatocytes using RNA sequencing (RNA‐seq) data to evaluate the transcriptional effects of Prom1 deficiency in the liver. We focused on the genes that were induced (n = 55) or repressed (n = 63) more than 2‐fold with P < 0.05 (Fig 1K). Singular enrichment analysis of those 118 differentially expressed genes (DEG) for KEGG pathways revealed that pathways such as glycolysis/gluconeogenesis, glucagon, and cAMP signaling pathway were highly enriched in the DEGs by Prom1 deficiency (Fig EV1D). Because the liver plays a central role in the regulation of glucose homeostasis, we focused on glycolysis/gluconeogenesis. Identified genes in these pathways included G6pc (glucose‐6‐phosphatase catalytic subunit), Pck1 (phosphoenolpyruvate carboxykinase 1), Pfkl (phosphofructokinase), and Pkm (pyruvate kinase), which were consistently downregulated when Prom1 was deficient (Fig 1K). These observations were further confirmed by qRT–PCR (Fig 1L), immunoblotting (Fig 1M) and glucose uptake assays (Fig 1N). These results led us to suspect that Prom1 may be involved in regulating hepatic gluconeogenesis.
To confirm the initial observations in mouse primary hepatocytes, we examined the livers of Prom1 +/+ and Prom1 −/− mice in the fasting state, which mimicks the serum‐free condition for hepatocytes (Fig 2A versus Fig 1L). The expression of G6pc and Pck1 was decreased in Prom1 −/− mouse liver compared with the levels in Prom1 +/+ mouse liver during fasting (Fig 2A). Fasting‐induced CREB phosphorylation was also decreased by 24% in Prom1 −/− mouse livers (Figs 2B and EV1F). Prom1 −/− mouse had lower blood glucose level without a difference in blood glucagon levels (Fig 2C and D). Hepatic glycogen breakdown during fasting was reduced in Prom1 −/− mice (Fig 2E), although livers of both groups did not show any histological difference (Fig EV1E). These results suggested that the loss of Prom1 interfered with the activation of hepatic gluconeogenesis.
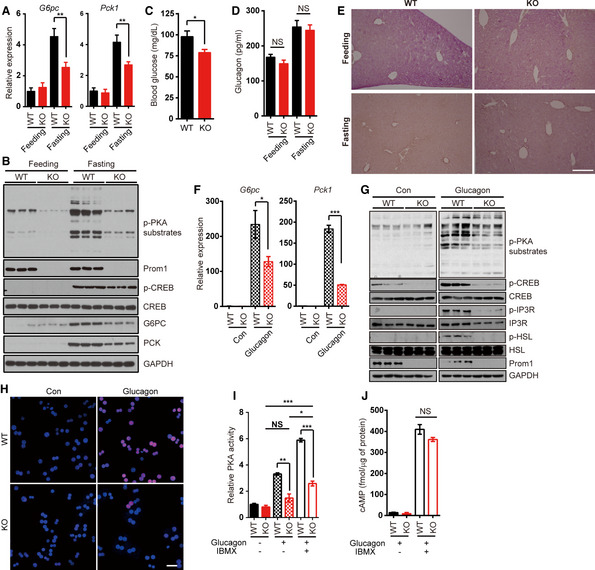
Figure 2. Prom1 deficiency defects glucagon‐induced gluconeogenesis.
-
ALevels of G6pc and Pck1 mRNAs in the liver of mice after 18 h fasting were determined by qRT–PCR. The level of each mRNA was normalized to the level of 18s rRNA (n = 3/fed, 7 or 8/fasted).
-
BLevels of phospho‐PKA substrates (p‐PKA substrates), phospho‐CREB (p‐CREB), CREB, G6pc Pck, Prom1, and GAPDH were determined after 18 h fasting. Band intensity for p‐CREB was quantified using Photoshop software.
-
CBlood glucose levels (mg/dl) of mice after fasted for 24 h (n = 12 or 14/group).
-
DSerum glucagon level in the fed or fasted mice (n = 3/fed, 7 or 8/fasted).
-
EGlycogen contents in the liver of mouse in each condition were shown by PAS (periodic acid‐schiff) staining. Scale bar, 500 μm.
-
F–JPrimary hepatocytes were isolated from 12‐week-old male Prom1 +/+ (WT) or Prom1 −/− (KO) mice, serum‐starved, and stimulated with glucagon. (F) Levels of the G6pc and Pck1 mRNAs 2 h after glucagon stimulation (10 nM) were determined by qRT–PCR. The level of each mRNA was normalized to the level of the 18S rRNA. For these experiments, primary hepatocytes were cultured under 3D conditions using a Matrigel matrix (n = 3 mice/group). (G) Levels of phospho‐PKA substrates (p‐PKA substrates), phospho‐CREB (p‐CREB), CREB, phospho‐inositol trisphosphate receptor (p‐IP3R), IP3R, phospho‐Hormone‐Sensitive Lipase (p‐HSL), HSL, Prom1, and Gapdh 10 min after glucagon stimulation (10 nM) were determined by immunoblotting. (H) The nuclear localization of p‐CREB 10 min after glucagon stimulation (10 nM) was determined by immunofluorescence staining. Scale bar, 20 μm. (I) Relative PKA activities 10 min after glucagon stimulation (10 nM) were determined using the PKA assay kit in the absence or presence of 10 μM IBMX (n = 3 mice/group). (J) The cAMP concentration 10 min after glucagon stimulation (10 nM) was determined using the cAMP assay kit in the absence or presence of 10 μM IBMX (n = 3 mice/group).
To further confirm the involvement of Prom1 in hepatic gluconeogenesis, we determined the expression levels of hepatic gluconeogenic genes in Prom1 +/+ and Prom1 −/− mouse hepatocytes after glucagon stimulation. Prom1 deficiency interfered with the upregulation of G6pc and Pck1 expression in glucagon‐stimulated primary hepatocytes (Figs 2F and EV1G). Because glucagon induces the upregulation of gluconeogenic genes by activation of CREB, we measured glucagon‐induced CREB phosphorylation. In Prom1‐deficient hepatocytes, glucagon‐induced CREB phosphorylation was significantly decreased (Fig 2G) and nuclear localization of CREB was also decreased as a result (Figs 2H and EV1H).
The decreased CREB phosphorylation and the reduced upregulation of gluconeogenic genes in glucagon‐induced Prom1 −/− mouse hepatocytes led us to examine the effect of Prom1 deficiency on the glucagon receptor signaling pathway. Prom1 deficiency reduced glucagon‐induced phosphorylation of PKA substrates (Fig 2G and I). The PKA activity and cAMP level in the presence or absence of IBMX (3‐isobutyl‐1‐methylxanthine), a phosphodiesterase (PDE) inhibitor, were measured in glucagon‐stimulated Prom1 +/+ and Prom1 −/− primary hepatocytes to investigate the abilities of the AC and PDE enzymes required to produce and degrade cAMP, respectively. Blocking cAMP degradation by IBMX failed to restore PKA activity in Prom1 −/− mouse hepatocytes completely, while glucagon and IBMX together slightly increased PKA activity (Fig 2I). Prom1 deficiency did not change glucagon‐induced cAMP production or degradation (Fig 2J). We also examined the effect of Prom1 overexpression on glucagon‐induced CREB phosphorylation and cAMP production in Prom1 −/− primary hepatocytes. Adenoviral Prom1 overexpression restored glucagon‐induced CREB phosphorylation (Fig EV1I), but did not affect glucagon‐elicited cAMP production (Fig EV1J).
To corroborate our observations, we tested two different PKA activators, forskolin (an AC activator) and 8‐Br‐cAMP (8‐Bromo‐cAMP, a non‐degradable cAMP analogue) in Prom1 +/+ and Prom1 −/− primary hepatocytes. Consistent with the previous results, Prom1 deficiency prevented forskolin‐ and 8‐Br‐cAMP‐induced CREB phosphorylation (Fig EV2A and B). Two PKA activators could not induce PKA activation in Prom1 −/− cells as good as in Prom1 +/+ cells even in the presence of IBMX, although high concentrations of PKA activators increased PKA activity marginally in Prom1‐deficient cells (Fig EV2C) without changing glucagon‐induced cAMP production (Fig EV2D). Taken together, we concluded that Prom1 regulated cAMP‐induced PKA activation in primary hepatocytes.
Figure EV2. Prom1 deficiency prevents glucagon‐, forskolin‐, or 8‐Br‐cAMP‐induced PKA activation in primary mouse hepatocytes.
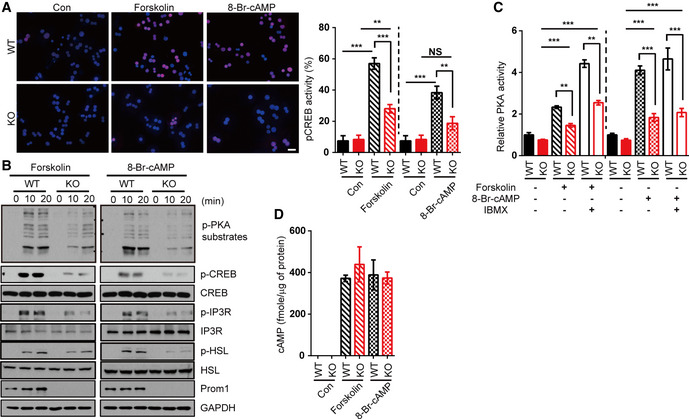
- Left panel, the nuclear localization of p‐CREB was determined by immunofluorescence staining after a 10‐min of forskolin (10 μM) or 8‐Br-cAMP stimulation (10 μM). Right chart, the percentage of cells with nuclear p‐CREB staining among more than 300 cells in each group was statistically determined in the images of immunofluorescence staining shown above. Scale bar, 20 μm.
- Levels of p‐PKA substrates, p‐CREB, CREB, p‐IP3R, IP3R, p‐HSL, HSL, and Prom1 after stimulation with forskolin (10 μM) or 8‐Br-cAMP (10 μM) for the indicated times were determined by immunoblotting.
- Relative PKA activities were determined using the PKA assay kit after a 10‐min stimulation with 8-Br‐cAMP (10 μM) or forskolin (10 μM) in the presence of 10 μM IBMX (n = 3/group).
- The cAMP concentration was determined using the cAMP assay kit after a 10‐min stimulation with 8-Br‐cAMP (10 μM) or forskolin (10 μM) in the presence of 10 μM IBMX (n = 3/group).
Prom1 is necessary for glucagon‐elicited hepatic gluconeogenesis in vivo
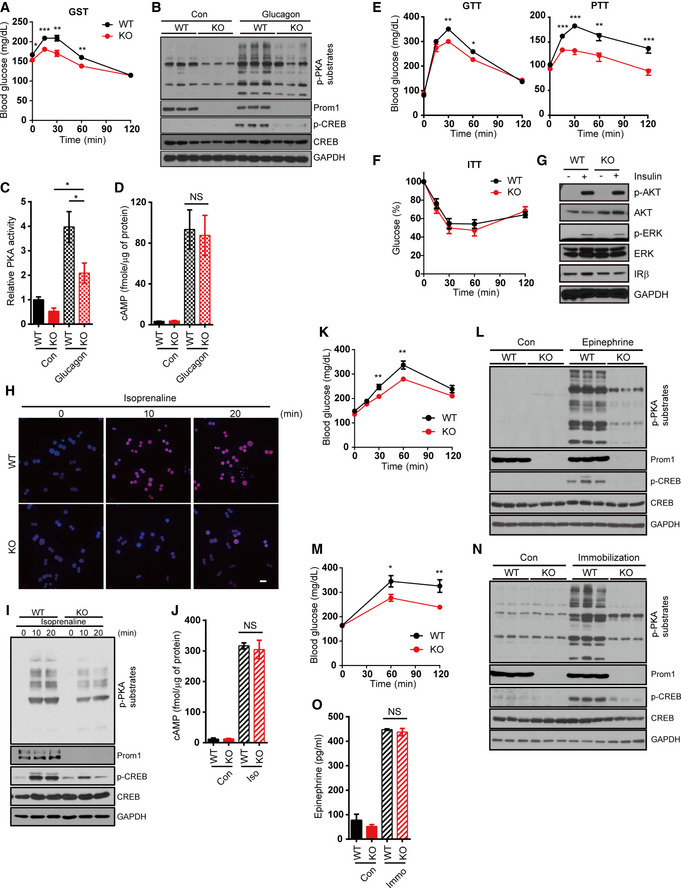
The requirement of Prom1 in glucagon‐elicited gluconeogenesis was further analyzed in vivo by treating Prom1 +/+ and Prom1 −/− mice with glucagon. We conducted glucagon challenge tests in 12‐week‐old mice. Glucagon‐elicited hyperglycemia was mitigated in Prom1 −/− mice compared to that in Prom1 +/+ mice (Fig 3A). The livers of Prom1‐deficient mice also exhibited reduced glucagon‐induced phosphorylation of CREB and PKA substrates (Fig 3B and C). However, Prom1‐deficient mice did not display a change in glucagon‐induced cAMP production in the liver (Fig 3D). Increased glucose internalization in Prom1 −/− mouse hepatocytes (Fig 1N) led us to hypothesize that Prom1‐deficient mice would show decreased blood glucose levels due to the reduced hepatic gluconeogenic capacity. To test this hypothesis, we performed glucose, pyruvate, and insulin tolerance tests in Prom1 +/+ and Prom1 −/− mice. Prom1‐deficient mice displayed improved glucose and pyruvate tolerance (Fig 3E). Prom1 deficiency did not change insulin tolerance (Fig 3F), as insulin‐induced signaling was not affected (Fig 3G). These results demonstrated that Prom1 deficiency prevented hepatic gluconeogenesis, but did not affect insulin signaling.
Figure 3. Prom1 deficiency prevents glucagon‐induced hyperglycemia in vivo and β‐adrenergic receptor‐mediated PKA activation.
-
A–D12-week‐old male WT (Prom1 +/+) and KO (Prom1 −/−) mice were fasted for 4 h and intraperitoneally injected with glucagon. (A) Blood glucose levels (mg/dL) were measured 0, 15, 30, 60, and 120 min after glucagon stimulation (200 μg/kg body weight, n = 10/group). (B) Levels of p‐PKA substrates, p‐CREB, CREB, and GAPDH in the liver after a 5 min of glucagon treatment were determined by immunoblotting (2 mg/kg body weight). (C) Relative PKA activities in the liver after a 5 min of glucagon treatment were determined using the PKA assay kit (2 mg/kg body weight, n = 3/group). (D) The cAMP concentration in the liver after a 5 min of gluc, agon treatment was determined using the cAMP assay kit (2 mg/kg body weight, n = 3/group).
-
E, FGlucose disposal rates were measured in 12-week‐old male mice using glucose, pyruvate (E), and insulin (F) tolerance tests (n = 10 mice/group).
-
GLevels of p‐AKT, AKT, p‐ERK, ERK, IRβ, and GAPDH were determined by immunoblotting after insulin stimulation (10 nM).
-
H–JPrimary hepatocytes were isolated from 12‐week-old male WT (Prom1 +/+) and KO (Prom1 −/−) mice, serum‐starved for 16 h, and stimulated with isoprenaline. (H) The nuclear localization of p‐CREB after isoprenaline stimulation (10 μM) for 10 min was determined by immunofluorescence staining. Scale bar, 20 μm. (I) Levels of p‐PKA substrates, p‐CREB, CREB, Prom1, and GAPDH after isoprenaline stimulation (10 μM) for 0, 10, or 20 min were determined by immunoblotting. (J) The cAMP concentration after isoprenaline stimulation (10 μM) for 10 min was determined using the cAMP assay kit in the presence of 10 μM IBMX (n = 3/group).
-
K, LWT (Prom1 +/+) and KO (Prom1 −/−) mice were fasted for 4 h and intraperitoneally injected with epinephrine (3 μg/10 g). (K) Blood glucose levels (mg/dl) were measured 0, 15, 30, 60, and 120 min after epinephrine stimulation (n = 13 or 14/group). (L) Levels of p‐PKA substrates, p‐CREB, CREB, and GAPDH in the liver 15 min after epinephrine treatment were determined by immunoblotting.
-
M–OWT (Prom1 +/+) and KO (Prom1 −/−) mice were fasted for 4 h and subjected to immobilization test. (M) Blood glucose levels (mg/dL) were measured 0, 60, and 120 min after immobilization (n = 10/group). (N) Levels of p‐PKA substrates, p‐CREB, CREB, and GAPDH in the liver after a 30 min of immobilization were determined by immunoblotting. (O) Serum epinephrine level of mice after a 30 min of immobilization was determined (n = 3/control group, 7 or 8/immobilized group).
Prom1 regulates β‐adrenergic receptor signaling in vivo
Because glucagon receptor is a member of the GPCR (G protein‐coupled receptor) family, we hypothesized that Prom1 would also regulate other GPCR signaling involved with PKA activation. To test this hypothesis, we investigated PKA signaling in Prom1 +/+ and Prom1 −/− primary hepatocytes treated with isoprenaline, a β‐adrenergic receptor agonist. Prom1 deficiency prevented phosphorylation of CREB and PKA substrates (Fig 3H and I), but not cAMP production (Fig 3J) in isoprenaline‐treated primary hepatocytes. To further validate that Prom1 regulates β‐adrenergic receptor signaling, we examined the blood glucose levels and PKA activation in Prom1 +/+ and Prom1 −/− mice after epinephrine injection. Epinephrine‐induced hyperglycemia was mitigated (Fig 3K), and PKA activation was reduced (Fig 3L) in Prom1 −/− mice, when compared to those in Prom1 +/+ mice. Because immobilization stress causes hyperglycemia via cholinergic muscarinic activation (Tajima et al, 1996), we performed immobilization test on Prom1 +/+ and Prom1 −/− mice to test the effect of Prom1 deficiency on endogenous epinephrine signaling. We found that immobilization stress‐induced hyperglycemia and PKA activation were decreased in Prom1 −/− mice, when compared to Prom1 +/+ mice (Fig 3M and N). However, the reduced responses of Prom1 −/− mice to immobilization stress were not due to the different epinephrine levels in the serum of Prom1 +/+ and Prom1 −/− mice (Fig 3O).
Radixin is the AKAP required for glucagon‐induced PKA activation
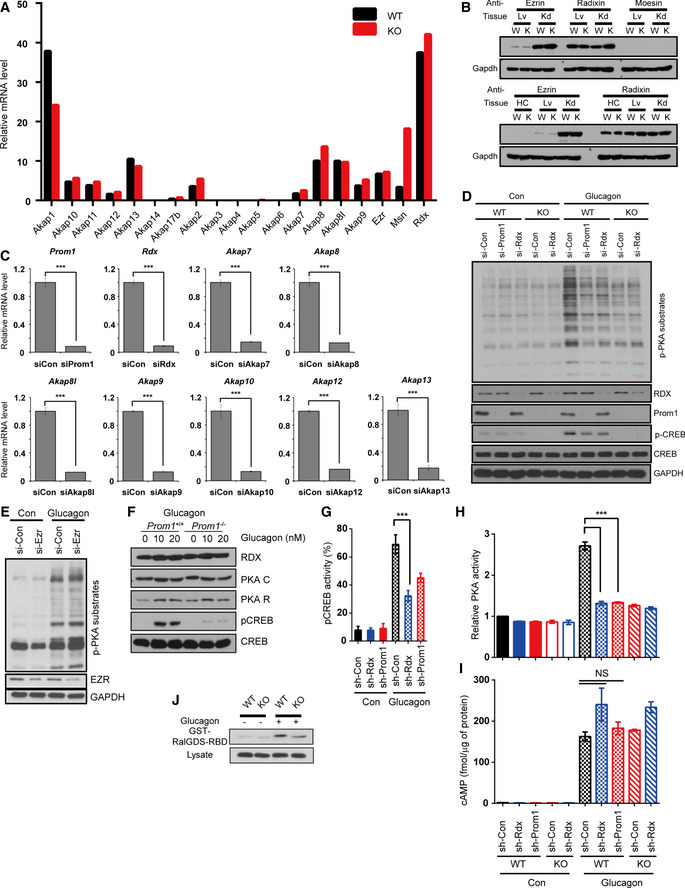
Because Prom1 deficiency decreased PKA activation in response to glucagon without either changing cAMP production (Figs 2 and 3) or increasing levels of protein kinase inhibitors, PKI (Appendix Fig S2), we postulated that Prom1 might regulate an AKAP which binds to the regulatory subunit of PKA and confines the PKA holoenzyme to a specific cellular location. We identified several AKAPs that are expressed in the liver using an RNA‐seq analysis (Fig EV3A). Among these AKAPs, AKAP7, AKAP8, AKAP8I, AKAP9, AKAP10, AKAP12, and AKAP13 were selected because they are cytoplasmic or plasma membrane‐bound proteins that may interact with Prom1. In addition, radixin was also selected as the AKAP in question among the ERM (ezrin, radixin, and moesin) protein family, because radixin is dominantly expressed in hepatocytes (Fig EV3B), links actin to the plasma membrane in the liver (Kikuchi et al, 2002), and is known to function as an AKAP (Gloerich et al, 2010; Hochbaum et al, 2011). We knocked down AKAPs expressed in the liver individually and examined their effects on glucagon‐induced phosphorylation of PKA substrates in primary hepatocytes to identify which AKAP was regulated by Prom1. Glucagon‐induced phosphorylation of PKA substrates was inhibited most markedly by small interfering RNAs (siRNAs) targeting Prom1 or radixin, suggesting that radixin might be an AKAP regulated by Prom1 (Figs 4A and EV3C and D). Because ezrin, a well‐studied AKAP, was marginally expressed in the liver (Fig EV3B), we tested the effect of ezrin knockdown by siRNA on glucagon‐induced phosphorylation of PKA substrates as a control. Ezrin deficiency did not change the phosphorylation of PKA substrates (Fig EV3E). Glucagon stimulation did not change the amount of radixin, PKA catalytic subunit, or regulatory subunit (Fig EV3F) as well. Similarly, adenoviral knockdown of radixin or Prom1 using short hairpin (sh) RNA in WT hepatocytes prevented glucagon‐induced phosphorylation of CREB and other PKA substrates (Figs 4B and C, and EV3G and H) while having no effect on Prom1 localization (Fig 4D). In contrast, radixin knockdown did not alter glucagon‐induced cAMP production (Fig EV3I). It has been reported that plasma membrane localization of Epac1, exchange factor directly activated by cAMP, and subsequent Rap1 activation are also mediated by radixin as an AKAP (Hochbaum et al, 2011). We determined Epac1 activity in Prom1 −/− primary hepatocytes treated with glucagon and observed that Prom1 deficiency decreased Rap1 activation as well (Fig EV3J). These results suggested that radixin functions as the AKAP regulated by Prom1 in primary mouse hepatocytes.
Figure EV3. Radixin functions as an AKAP in primary hepatocytes.
-
AThe relative expression levels of Akap1, Akap10, Akap11, Akap12, Akap13, Akap14, Akap17b, Akap2, Akap3, Akap4, Akap5, Akap6, Akap7, Akap8, Akap9, ezrin, radixin, and moesin mRNAs in hepatocytes isolated from 12‐week-old male WT (Prom1 +/+) and KO (Prom1 −/−) mice and serum‐starved for 16 h were determined using RNA‐seq. AKAP, A kinase‐anchored protein.
-
BThe expression of ezrin, radixin, and moesin in mouse primary hepatocyte, liver, and kidney was determined by immunoblotting. Lv, liver; Kd, kidney; HC, hepatocyte; W, wild type; K, knockout.
-
CThe expression of Prom1, radixin, AKAP7, AKAP8, AKAP8I, AKAP9, AKAP10, AKAP12, or AKAP13 was silenced in WT hepatocytes using siRNAs. Hepatocytes were then serum‐starved for 18 h and stimulated with glucagon (10 nM) for 10 min. Levels of the Prom1, radixin, Akap7, Akap8, Akap8I, Akap9, Akap10, Akap12, and Akap13 mRNA were determined using qRT–PCR (n = 3 replicated siRNA treatments/group).
-
DProm1 or radixin expression was silenced in WT hepatocytes, and radixin expression was silenced in KO hepatocytes. Hepatocytes were serum‐starved for 18 h and stimulated with glucagon (10 nM) for 10 min. Levels of p‐PKA substrates, p‐CREB, CREB, radixin, Prom1, and Gapdh were determined by immunoblotting.
-
ELevels of p‐PKA substrates, ezrin, and GAPDH in siRNA‐treated primary hepatocytes were determined by immunoblotting after stimulation with glucagon (10 nM) for 10 min.
-
FLevels of radixin, PKA catalytic subunit, regulatory subunit, p‐CREB, and CREB were determined by immunoblotting after glucagon stimulation (10 nM).
-
GThe percentage of cells with nuclear p‐CREB staining among more than 300 cells in each group was also statistically analyzed.
-
H, IProm1 and radixin expression was silenced in WT (Prom1 +/+) and KO (Prom1 −/−) hepatocytes by infection with an adenovirus harboring sh‐control (sh‐Con), sh‐radixin (sh‐Rdx), or sh‐Prom1 for 24 h. Hepatocytes were further serum‐starved for 18 h and stimulated with glucagon (10 nM) for 10 min. (H) Relative PKA activities were determined using the PKA assay kit after stimulation with glucagon for 10 min (n = 3 replicates/group). (I) The cAMP concentration was determined using the cAMP assay kit after a 10‐min glucagon stimulation in the presence of 10 μM IBMX (n = 3 replicates/group).
-
JPull‐down of activated Rap1 with GST‐RalGDS‐RBD from WT (Prom1 +/+) and KO (Prom1 −/−) primary mouse hepatocytes stimulated with glucagon (10 nM) for 10 min.
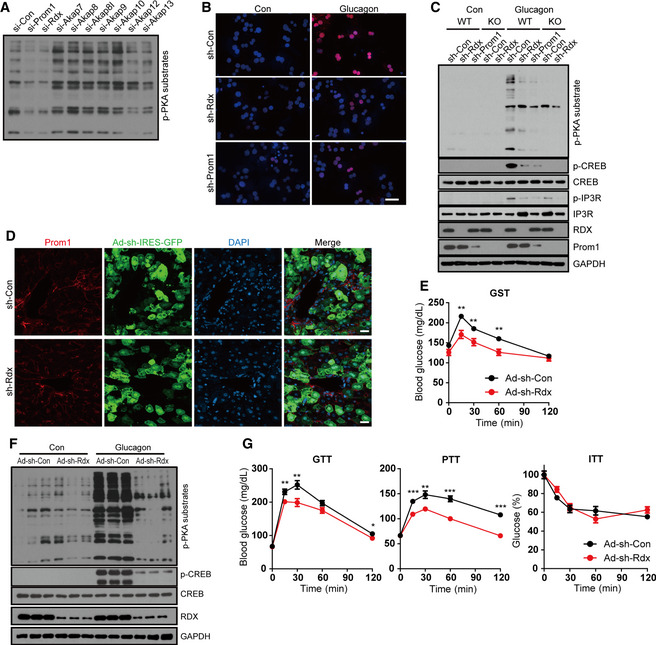
Figure 4. Radixin is the AKAP required for glucagon‐induced PKA activation and hyperglycemia in vivo .
-
AThe expression of Prom1, radixin, AKAP7, AKAP8, AKAP8I, AKAP9, AKAP10, AKAP12, or AKAP13 was silenced in WT hepatocytes using siRNAs. Hepatocytes were then serum‐starved for 18 h and stimulated with glucagon (10 nM) for 10 min. The phosphorylation of PKA substrates was determined by immunoblotting.
-
B, CProm1 and radixin expression was silenced in WT (Prom1 +/+) and KO (Prom1 −/−) hepatocytes by infection with an adenovirus harboring sh‐control (sh‐Con), sh‐radixin (sh‐Rdx), or sh‐Prom1 for 24 h. Hepatocytes were further serum‐starved for 18 h and stimulated with glucagon (10 nM) for 10 min. (B) The nuclear localization of p‐CREB after glucagon stimulation was determined by immunofluorescence staining. Blue; DAPI, Red; p‐CREB. The percentage of cells with nuclear p‐CREB staining among more than 300 cells in each group was also statistically analyzed (Fig EV3G). Scale bar, 20 μm. (C) Levels of p‐PKA substrates, p‐CREB, CREB, p‐IP3R, IP3R, Prom1, radixin, and GAPDH after glucagon stimulation (10 min) were determined by immunoblotting.
-
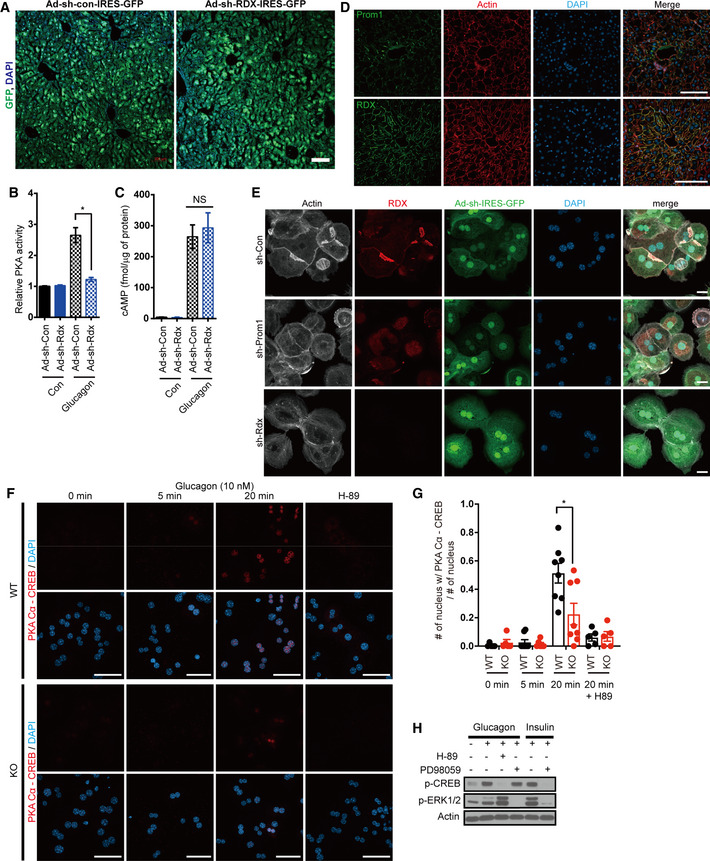
DImmunofluorescence staining of Prom1 in mouse liver transduced with adenoviral particles. Confocal microscope images of WT mouse liver infected with adenovirus harboring sh‐Con or sh‐Rdx linked with IRES‐GFP. Scale bar, 20 μm.
-
E–G12-week‐old male wild‐type mice were infected with an adenovirus harboring sh‐control (sh‐Con) or sh‐radixin (sh‐Rdx) for 3 days, fasted for 4 h, and intraperitoneally injected with glucagon. (E) Blood glucose levels (mg/dL) were measured 0, 15, 30, 60, and 120 min after glucagon administration (200 μg/kg body weight, n = 10 mice/group). (F) Levels of p‐PKA, p‐CREB, CREB, radixin, and GAPDH in the liver after a 10‐min of glucagon stimulation (2 mg/kg body weight) were determined by immunoblotting. (G) Glucose disposal rates in sh‐Con or sh‐Rdx (radixin) mice were measured using glucose, insulin, and pyruvate tolerance tests (n = 10 mice/group).
Next, to determine whether radixin functions as an AKAP during glucagon‐induced gluconeogenesis in vivo, we knocked down radixin in mice using adenoviral shRNA (short hairpin RNA) and performed the glucagon challenge test. The transduction efficiency of adenoviral particles was confirmed by bicistronic expression of GFP which was close to 100% in hepatocytes (Fig EV4A). We observed that radixin knockdown mitigated glucagon‐induced hyperglycemia (Fig 4E). Moreover, radixin knockdown reduced the phosphorylation of CREB and PKA substrates (Figs 4F and EV4B), but did not alter cAMP production in the livers of glucagon‐treated mice (Fig EV4C). Glucose, pyruvate, and insulin tolerance tests were performed on radixin knockdown mice to further investigate whether radixin regulates hepatic gluconeogenesis in vivo. Radixin knockdown improved glucose and pyruvate tolerance but not insulin tolerance (Fig 4G), suggesting that radixin knockdown prevented glucagon‐induced hepatic gluconeogenesis without affecting insulin signaling.
Figure EV4. Radixin functions as an AKAP required for glucagon‐induced PKA activation in primary hepatocytes and the liver.
-
AThe transduction efficiency of adenoviral particles was confirmed by bicistronic expression of GFP. Confocal microscope images of WT mice livers infected with adenovirus harboring sh‐Con or sh‐Rdx linked with IRES‐GFP. Scale bar, 100 μm.
-
B, C12-week‐old male wild‐type mice were infected with an adenovirus harboring sh‐control (sh‐Con) or sh‐radixin (sh‐Rdx) for 3 days, fasted for 4 h, and intraperitoneally injected with glucagon. (B) Relative PKA activities in the liver were determined using the PKA assay kit 10 min after glucagon (2 mg/kg body weight) stimulation (n = 3/group). (C) The cAMP concentration in the liver 10 min after glucagon (2 mg/kg body weight) stimulation was determined using the cAMP assay kit (n = 3/group).
-
DImmunofluorescent staining of Prom1 or radixin using actin staining by phalloidin as a reference in cryosections of WT mouse liver. Scale bar, 100 μm.
-
EThe cellular localization of radixin and actin in primary hepatocytes from 12‐week-old male wild‐type mice infected with an adenovirus harboring sh‐control (sh‐Con), sh‐Prom1, or sh‐radixin (sh‐Rdx) for 3 days was determined by immunofluorescence staining for radixin and phalloidin staining. Scale bar, 20 μm.
-
F, GThe molecular interaction between catalytic subunit of PKA and CREB in WT (Prom1 +/+) and KO (Prom1 −/−) hepatocytes was determined by a proximity ligation assay using anti‐PKA Cα and anti‐CREB antibodies. These hepatocytes were stimulated with glucagon (10 nM) for the indicated time or treated with H‐89, PKA inhibitor, prior to glucagon stimulation. Scale bar, 50 μm. (G) The portion of cells with nuclear PKA Cα‐CREB interaction (F) in more than 5 to 8 imaging area from each group was statistically determined.
-
HInhibition of ERK1/2 activity did not affect CREB phosphorylation. Levels of p‐CREB, p‐ERK1/2, and actin in mouse primary hepatocytes after glucagon stimulation (10 nM) or insulin stimulation (100 nM) for 15 min were determined by immunoblotting. PKA inhibitor, H‐89 or ERK1/2 inhibitor, PD98059 were added prior to glucagon/insulin treatment at 30 and 50 μM, respectively.
Prom1 is necessary to confine radixin to the plasma membrane
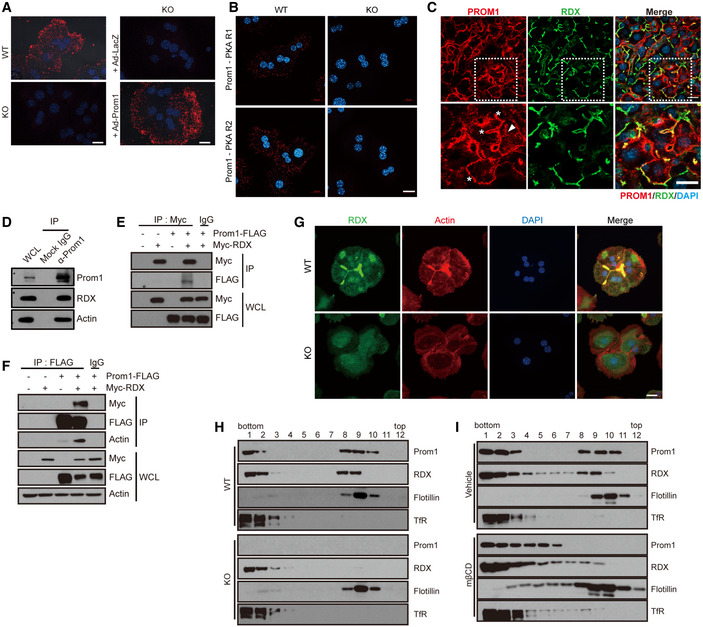
To determine the mechanism by which Prom1 regulates the AKAP activity of radixin, we first examined whether two proteins were located in close proximity using a proximity ligation assay. Fluorescence signals were observed in Prom1 +/+ but not in Prom1 −/− primary hepatocytes (Fig 5A, two left panels) and adenoviral overexpression of Prom1 drove the proximity‐based fluorescence to reappear in Prom1 −/− primary hepatocytes (Fig 5A, two right panels). PKA regulatory subunits were also in close proximity to Prom1 (Fig 5B), implying the formation of Prom1/radixin/PKA complex. We also determined the localization of PROM1 and radixin in human liver specimens by double immunofluorescence staining using anti‐human Prom1 monoclonal antibody (HB#7, Swaminathan et al, 2010) (Fig 5C). PROM1 and radixin were co‐localized at the plasma membrane and especially at the cell–cell contact sites called the canalicular membranes (asterisks in the lower panel of Fig 5C). PROM1 was also observed as intracellular foci (a triangle in the lower panel of Fig 5C). Co‐localization of Prom1 and radixin in mouse liver was also determined by indirect comparison of the immunostained slides using actin as a reference, because Prom1 antibody (13A4) and radixin antibody required incompatible antigen‐retrieval condition preventing double immunofluorescence staining. Similar to the observations in human liver samples, both Prom1 and radixin co‐localized with actin in cell–cell contact sites called the canalicular membranes (Fig EV4D). These observations suggested that Prom1 and radixin are co‐localized near the canalicular membranes.
Figure 5. Prom1 is required to confine radixin and actin.
-
A, BPrimary hepatocytes were obtained from 12‐week-old male WT (Prom1 +/+) and KO (Prom1 −/−) mice or KO (Prom1 −/−) hepatocytes infected with an adenovirus harboring LacZ (Ad‐LacZ) or Prom1 (Ad‐Prom1) for 24 h. The molecular interaction between Prom1 and radixin (A) or Prom1 and PKA regulatory subunits (B) in these hepatocytes was determined by a proximity ligation assay using anti‐Prom1 and anti‐radixin antibodies (A) or anti‐Prom1 and anti‐PKA regulatory subunits (B), respectively. Scale bar, 20 μm.
-
CHuman liver specimens were analyzed by double immunostaining for PROM1 (red) and radixin (green). The lower panels represent the magnification of the square frame in the upper panels. Scale bar, 20 μm.
-
DThe molecular interaction between Prom1, radixin (RDX), and actin in WT mouse primary hepatocytes was determined by co‐immunoprecipitation with an anti‐Prom1 antibody or isotype control IgG. WCL; whole‐cell lysate.
-
E, FHEK293 cells were transiently transfected with the combination of Prom1‐FLAG and Myc‐radixin plasmids for 24 h. The molecular interaction between Prom1‐FLAG and Myc‐radixin was determined by co‐immunoprecipitation using isotype control IgG, anti‐Myc (E), anti‐FLAG (F) antibodies.
-
GThe cellular localization of radixin and actin in WT (Prom1 +/+) and KO (Prom1 −/−) hepatocytes was determined by immunofluorescence staining for radixin and phalloidin staining. Scale bar, 20 μm.
-
HDetergent‐resistant lipid rafts were isolated from WT (Prom1 +/+) and KO (Prom1 −/−) mouse liver samples. Levels of the Prom1, radixin (RDX), flotillin, and transferrin receptor (TfR) proteins were determined in each fraction after sucrose gradient ultracentrifugation using immunoblotting.
-
IDetergent‐resistant lipid rafts were isolated from WT (Prom1 +/+) and KO (Prom1 −/−) mouse primary hepatocytes after cholesterol depletion by methyl‐β‐cyclodextrin (mβCD, 30 mM) for 30 min at 4°C. Levels of the Prom1, radixin, flotillin, and transferrin receptor proteins were determined in each fraction after sucrose gradient ultracentrifugation using immunoblotting.
Source data are available online for this figure.
Endogenous Prom1 was co‐immunoprecipitated with radixin in primary mouse hepatocytes (Fig 5D), suggesting the molecular interaction between Prom1 and radixin. Actin was also co‐immunoprecipitated by Prom1 (Fig 5D), as demonstrated previously (Yang et al, 2008). Reciprocal immunoprecipitations of exogenously expressed PROM1 and radixin showed similar results in HEK293 cells (Fig 5E and F), and the interaction between PROM1 and actin was radixin‐dependent (Fig 5F). The molecular interaction between PROM1 and radixin prompted us to speculate that PROM1 is required to confine actin‐bound radixin to the plasma membrane. We determined the cellular localization of radixin and actin in Prom1 +/+ and Prom1 −/− primary hepatocytes to test this hypothesis. Radixin and actin were clearly observed at the canalicular membranes in Prom1 +/+ hepatocytes but not in Prom1 −/− hepatocytes (Fig 5G). Moreover, Prom1 deficiency prevented cortical actin from localizing to canalicular membranes, suggesting that Prom1 was required to confine radixin and actin to the plasma membrane. Similarly, adenoviral knockdown of Prom1 or radixin using short hairpin (sh) RNA in WT hepatocytes interfered with the sub‐canalicular localization of radixin and actin (Fig EV4E).
Because Prom1 and ERM proteins are present in detergent‐resistant lipid rafts, we speculated that radixin enrichment in detergent‐resistant lipid rafts may depend on Prom1 expression. We examined the presence of radixin in detergent (Lubrol WX)‐resistant lipid rafts from Prom1 +/+ and Prom1 −/− mouse liver samples to test this hypothesis. While flotillin, a raft marker protein, was well separated from transferrin receptor, a non‐raft marker protein, regardless of PROM1, radixin in detergent‐resistant lipid rafts was sensitive to the presence of Prom1, and disappeared from detergent‐resistant lipid rafts in Prom1‐deficient cells (Fig 5H). In addition, cholesterol depletion by methyl‐β‐cyclodextrin (mβCD) led to the solubilization of radixin as well as Prom1 (Fig 5I), which suggested that the association of radixin with lipid rafts was Prom1‐dependent. These results suggested that Prom1 is required to confine radixin to plasma membrane lipid rafts. It is of note that Prom1/radixin complex and flotillin were associated with distinct lipid rafts. Prom1 and radixin were enriched in fraction 8 and 9 and sensitive to mβCD, while flotillin was found mainly in fraction 9 and less sensitive to mβCD (Fig 5H and I).
How does the complex of Prom1, radixin and PKA holoenzyme at the plasma membrane regulate the phosphorylation of PKA substrates in the nucleus such as CREB? Because Dalle and colleagues have demonstrated that p44/p42 mitogen‐activated protein kinase (ERK1/2) phosphorylates CREB via glucagon‐induced PKA dependent manner in the MIN6 cell line and pancreatic islets (Dalle et al, 2004), we tested whether ERK1/2 acts as a bridge between the PKA holoenzyme at the membrane and CREB in the nucleus. In primary hepatocyte, glucagon or insulin stimulation induced CREB and ERK1/2 phosphorylation. However, while insulin‐induced CREB and ERK1/2 phosphorylation were efficiently blocked by a ERK1/2 inhibitor (PD98059), glucagon‐induced CREB phosphorylation was not suppressed by the inhibitor (Fig EV4H). This result indicated that at least in primary hepatocytes ERK1/2 was not involved in glucagon‐induced CREB phosphorylation. Thus, we followed the interaction of PKA catalytic subunit and CREB after glucagon stimulation using proximity ligation assay to address this question (Fig EV4F and G). After 20 min of glucagon stimulation, the interaction between PKA catalytic subunits and CREBs was observed mostly in the nucleus (the second panel from the right in Fig EV4F). This nuclear interaction of PKA catalytic subunit and CREB disappeared when cells were treated with H‐89, a PKA inhibitor, indicating the specificity of the assay. However, the interaction disappeared in Prom −/− hepatocytes (the two lower panels of Fig EV4F). The portion of cells with nuclear PKA Cα‐CREB interaction in more than 100 cells from each group was statistically determined (Fig EV4G). These results suggest that PKA catalytic subunit in PKA holoenzyme is released from the complex of Prom1, radixin, and PKA holoenzyme at the plasma membrane, and translocates into the nucleus when local cAMP concentration is high.
The FERM domain of radixin is necessary for Prom1‐dependent gluconeogenesis
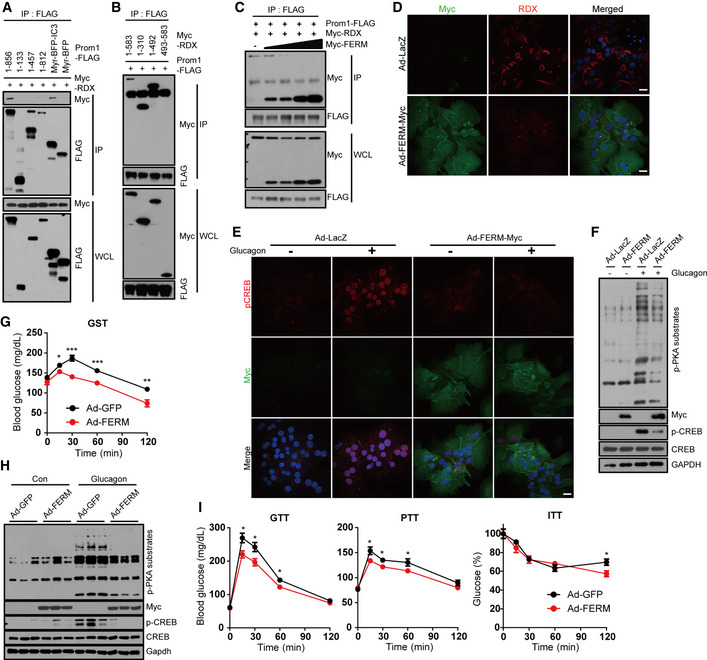
To determine which domain in each protein is required for the interaction, we constructed various deletion mutants of both PROM1 and radixin (Fig EV5A and B). Co‐immunoprecipitation after transient expression of both genes in HEK293 cells showed that the carboxy‐terminal tail of PROM1 (IC3) interacted with the FERM domain (1–310) of radixin, because the full‐length protein and carboxy‐terminal tail of Prom1 co‐immunoprecipitated the full‐length protein and FERM domain of radixin (Fig 6A and B). Competitive co‐immunoprecipitation showed a gradual decrease in the molecular interaction between PROM1 and radixin in proportion to the increasing expression level of the FERM domain in HEK293 cells (Fig 6C), indicating the specific interaction between two proteins through FERM domain. Indeed, canalicular localization of endogenous radixin disappeared in cells overexpressing the FERM domain (Fig 6D). Next, we monitored glucagon‐induced PKA activation after adenoviral overexpression of the FERM domain in primary mouse hepatocytes. Overexpression of the FERM domain prevented glucagon‐induced phosphorylation and nuclear localization of CREB (Figs 6E and EV5C). Overexpression of the FERM domain prevented glucagon‐induced phosphorylation of PKA substrates (Fig 6F), but did not affect glucagon‐elicited cAMP production (Fig EV5D). We further analyzed the dominant‐negative effect of the FERM domain on glucagon‐elicited gluconeogenesis in vivo. Systemic adenoviral overexpression of the FERM domain mitigated glucagon‐elicited hyperglycemia (Fig 6G). In addition, overexpression of the FERM domain prevented glucagon‐induced phosphorylation of CREB and PKA substrates in the livers of glucagon‐treated mice (Fig 6H), and improved glucose and pyruvate tolerance but not insulin tolerance (Fig 6I). Taken together, we concluded that the FERM domain of radixin is necessary for the interaction with PROM1 and its confinement to lipid rafts to facilitate radixin's function as an AKAP during hepatic gluconeogenesis.
Figure EV5. Prom1 and radixin interact via C‐terminal cytoplasmic domain of Prom1 and N‐terminal FERM domain of radixin.

-
A, BDomain structures of deletion mutants of human Prom1 (A) and human radixin (B). EX, extracellular domain; TM, transmembrane domain; IC, intracellular domain; BFP, blue fluorescence protein; Myr, myristoylation site; FERM, 4.1 protein–ezrin–radixin–moesin domain; ABD, actin‐binding domain.
-
C, DOverexpression of the FERM domain prevents glucagon‐induced PKA activation. WT primary hepatocytes were infected with an adenovirus harboring the FERM domain for 48 h, serum‐starved for 18 h, and stimulated with glucagon (10 nM). (C) The percentage of cells displaying nuclear p‐CREB staining among more than 300 cells from each group was statistically determined by analyzing the images of immunofluorescence staining shown in Fig 6E. (D) The cAMP concentration was determined using the cAMP assay kit 10 min after stimulation with glucagon (n = 3 replicates/group).
Figure 6. Overexpression of radixin mutant mitigates glucagon‐elicited hyperglycemia.
-
A, BThe molecular interactions between PROM1 and radixin mutants were determined by co‐immunoprecipitation with an anti‐FLAG antibody. HEK293 cells were co‐transfected with plasmids expressing Myc‐radixin and various deletion mutants of PROM1‐FLAG (A) or with plasmids expressing PROM1‐FLAG and various deletion mutants of Myc‐radixin (B) for 24 h.
-
CThe molecular interaction between PROM1‐FLAG and Myc‐radixin in the presence of increasing amounts of Myc‐FERM was determined by co‐immunoprecipitation with an anti‐FLAG antibody.
-
D–FWT primary hepatocytes were infected with an adenovirus harboring FERM‐Myc (Ad‐FERM-Myc) or LacZ (Ad‐LacZ) for 48 h. The cellular localization of radixin (D) and the nuclear localization of p‐CREB after glucagon stimulation for 10 min (E) was determined by immunofluorescence staining. Scale bar, 20 μm. (F) Ad‐FERM-Myc‐ or Ad‐LacZ-expressing WT hepatocytes were serum‐starved for 16 h and stimulated with glucagon (10 nM) for 10 min. Levels of p‐PKA substrates, p‐CREB, CREB, FERM‐Myc, and GAPDH were determined by immunoblotting.
-
G–ITwelve-week‐old male wild‐type mice were infected with an adenovirus harboring GFP or FERM for 3 days, fasted for 4 h, and intraperitoneally injected with glucagon. (G) Blood glucose levels (mg/dL) were measured 0, 15, 30, 60, and 120 min after glucagon stimulation (0.2 mg/kg body weight, n = 10 mice/group). (H) Levels of p‐CREB, CREB, p‐PKA substrates, and FERM in the liver 10 min after glucagon stimulation (2 mg/kg body weight) were determined by immunoblotting (n = 3 mice/group). (I) Glucose disposal rates in GFP‐ or FERM‐overexpressing mice were measured using glucose, pyruvate, and insulin tolerance tests (n = 10 mice/group).
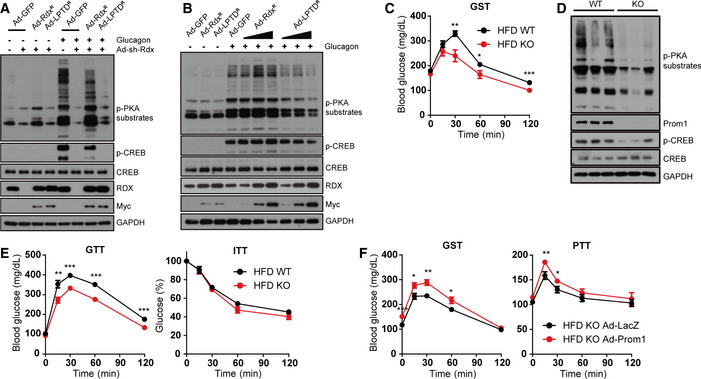
AKAP activity of radixin in glucagon‐elicited gluconeogenesis was further substantiated by a knockdown‐rescue experiment. We examined glucagon‐induced PKA activation in primary mouse hepatocytes in which endogenous radixin was knocked down by adenoviral shRNA, and shRNA‐resistant radixinR or LPTDR was re‐introduced by adenoviral transduction. We used the LPTD mutant of radixin, because L421P mutation disrupts the ability of radixin to bind to PKA, which prevents radixin from functioning as AKAP. T564D mutation is utilized because it is known to mimic phosphorylation in all ERM proteins and their open and active conformations (Deming et al, 2015). Ectopic expression of shRNA‐resistant radixinR rescued glucagon‐induced phosphorylation of PKA substrates, while radixin mutant LPTDR could not (Fig 7A). Overexpression of LPTDR mutant alone in primary mouse hepatocytes decreased the glucagon‐induced phosphorylation of CREB and PKA substrates (Fig 7B), which suggested the dominant‐negative effect of LPTDR mutant as well.
Figure 7. Prom1 disruption ameliorates glucagon‐induced hyperglycemia in DIO mice.
-
A, B(A) Wild‐type primary hepatocytes were infected with an adenovirus harboring sh‐control (sh‐Con), sh‐radixin (sh‐Rdx), GFP (Ad‐GFP), and/or sh‐resistant wild‐type radixin (Ad‐RdxR) or AKAP‐dead LPTD mutant of radixin (Ad‐LPTDR) for 24 h. (B) Increasing amount of Ad‐RdxR or Ad‐LPTDR was infected to WT primary hepatocytes for 24 h. Hepatocytes were further serum‐starved for 18 h and stimulated with glucagon (10 nM) for 10 min. Levels of p‐PKA substrates, p‐CREB, CREB, radixin, Myc‐radixin, and GAPDH were determined by immunoblotting.
-
C–FFour-week‐old male WT (Prom1 +/+) and KO (Prom1 −/−) mice were fed a high‐fat diet for 8 weeks (diet‐induced obesity; DIO), and fasted for 4 h before experiments. (C) DIO WT (Prom1 +/+) and KO (Prom1 −/−) mice were intraperitoneally injected with glucagon. Blood glucose levels (mg/dL) were measured 0, 15, 30, 60, and 120 min after glucagon (100 μg/kg body weight) stimulation (n = 10 mice/group). (D) Levels of p‐PKA substrates, p‐CREB, CREB, Prom1, and GAPDH were determined by immunoblotting 10 min after glucagon (2 mg/kg body weight) stimulation (n = 3 mice/group). (E) Glucose disposal rates in DIO WT (Prom1 +/+) and KO (Prom1 −/−) mice were measured using glucose and insulin tolerance tests (n = 10 mice/group). (F) DIO KO (Prom1 −/−) mice were infected with an adenovirus harboring LacZ or Prom1 for 24 h, fasted for 4 h, and intraperitoneally injected with glucagon (100 μg/kg body weight). Blood glucose levels (mg/dL) were measured 0, 15, 30, 60, and 120 min after glucagon stimulation (GST). Glucose disposal rates were measured using the pyruvate tolerance test (PTT) (n = 10 mice/group).
Because our results showed that Prom1‐deficient mice displayed improved glucose and pyruvate tolerance but not insulin tolerance (Fig 3E and F), we examined glucagon‐elicited gluconeogenesis in high‐fat diet‐induced obese (DIO) Prom1 +/+ and Prom1 −/− mice. DIO Prom1 −/− mice exhibited a mitigation of glucagon‐elicited hyperglycemia (Fig 7C). Prom1 deficiency also prevented glucagon‐induced phosphorylation of CREB and other PKA substrates (Fig 7D). In DIO Prom1 −/− mice, glucose tolerance, but not insulin tolerance, was improved (Fig 7E). Moreover, systemic adenoviral overexpression of Prom1 in DIO Prom1 −/− mice enhanced glucagon‐ and pyruvate‐induced hyperglycemia (Fig 7F). These in vivo results showed that the Prom1 deficiency protected mice from diet‐induced glucose intolerance.
Discussion
Prom1 is a cancer stem cell marker located in the plasma membrane detergent‐resistant lipid rafts, and its function as a regulator of membrane dynamics including the release of membrane vesicles from the apical surface (Marzesco et al, 2005), exosome‐mediated release of vesicles through exocytosis (Bauer et al, 2011), and primary cilia formation (Singer et al, 2019) during stem cell activation or differentiation have been elegantly demonstrated. Our study to delineate the function of Prom1 in normal liver provides new insights into the physiological role of Prom1. We examined the mechanism by which Prom1 regulates hepatic gluconeogenesis using Prom1‐deficient mice. The Prom1‐radixin axis is a key signaling pathway that regulates cAMP‐mediated PKA activation. We propose that Prom1 confines radixin to the plasma membrane and radixin recruits the PKA holoenzyme to the plasma membrane. Subsequently, glucagon‐G protein‐coupled receptor (GPCR)‐Gsα‐AC signaling pathway produces cAMP, liberating, and activating the PKA catalytic subunit from the PKA regulatory subunit attached to radixin. Because Prom1 deficiency interferes with the function of radixin as an AKAP, Prom1 deficiency prevents cAMP‐mediated PKA activation.
ERM family proteins (ezrin, radixin and moesin) act as AKAPs because they interact with the PKA regulatory subunit and various plasma membrane‐associated PKA substrates (Neisch & Fehon, 2011). Indeed, ERM proteins are required for PKA‐mediated phosphorylation of different membrane‐associated proteins, such as β‐adrenergic receptor, cystic fibrosis transmembrane conductance regulator (CFTR), Na+‐H+ exchanger 3 (NHE3), connexin 43 (Cx43), and exchange protein directly activated by cAMP (EPAC) (Sun et al, 2000; Fouassier et al, 2001; Bretscher et al, 2002; Weinman et al, 2003; Hochbaum et al, 2011; Pidoux et al, 2014). We focused on the Prom1‐radixin complex because among these ERM proteins radixin is the dominant ERM protein in hepatocytes (Tsukita et al, 1989; Kikuchi et al, 2002). Radixin knockout mice show hyperbilirubinemia due to loss of multidrug resistance protein 2 (MRP2) from canalicular membranes in the liver (Kikuchi et al, 2002), deafness associated with progressive degeneration of cochlear stereocilia in the inner ear (Kitajiri et al, 2004), and impaired reversal learning and short‐term memory by modulating inhibitory synapse transmission (Loebrich et al, 2006). Our study provides the first evidence to show that radixin is also involved in glucagon/β‐adrenergic receptor‐mediated gluconeogenesis via Prom1‐radixin axis. Prom1 deficiency disrupts the localization of radixin in canalicular membranes, prevents the formation of cortical actin (Figs 5F and EV4D), and inhibits glucagon‐, isoprenaline‐, or cAMP‐induced PKA activation (Figs 3 and EV2) without changing the amount of PKA catalytic subunit (Fig EV3F). These results demonstrate that Prom1 deficiency abrogates the function of radixin as an AKAP which allows the PKA holoenzyme and PKA substrates to assemble in the same place. Failing to confine the PKA holoenzyme and PKA substrates in close proximity due to Prom1 deficiency may explain the reduced phosphorylation level of PKA substrates even at the supra‐physiological concentration of activators used in our study. In addition, because PKA has a generous substrate specificity (Dalerba et al, 2007), it is crucial to tightly regulate PKA localization through its interaction with AKAPs (Beene & Scott, 2007) and activation through confined cAMP concentration (Rich et al, 2001; Zaccolo & Pozzan, 2002). Recently, Smith et al (Smith et al, 2017) have demonstrated that local PKA activation is regulated by the conformational change of holoenzyme, not by the complete dissociation of catalytic subunit at the physiological concentration of cAMP. In accordance with these reports our findings also demonstrated that local confinement of PKA to which cAMP is readily available by the interaction with radixin, a hepatocyte‐specific AKAP, and Prom1 complex is an important process that provides the specificity required for regulating glucagon‐induced gluconeogenesis. This may explain the lack of action from other AKAP proteins in the presence of glucagon or 8‐Br‐cAMP. More interestingly, we demonstrated the importance of Prom1‐radixin axis not only in glucagon‐elicited gluconeogenesis, but also in β‐adrenergic receptor‐mediated gluconeogenesis (Fig 3). These results suggest that Prom1‐AKAP interaction may participate in regulating GPCR signaling in other phenotypes or organs.
Membrane‐associated AKAPs are known to be involved in cAMP‐induced phosphorylation and the activation of nuclear CREB. Although membrane‐bound AKAP5 interacts with E‐cadherin, β‐adrenergic receptor, adenylyl cyclase, and the cytoskeleton, cAMP‐induced phosphorylation of nuclear CREB is increased by its overexpression but is reduced by its knockdown (Fraser et al, 2000; Altier et al, 2002; Gorski et al, 2005). cAMP‐induced phosphorylation of nuclear CREB is also reduced with a cell‐permeable peptide treatment that inhibits the molecular interaction between PKA and AKAP (Friedrich et al, 2010; Godbole et al, 2017). Similar to the observations in the above examples, the Prom1‐radixin complex in the plasma membrane regulates cAMP‐induced phosphorylation of nuclear CREB. Then, one may ask what connects these two events. Dalle and colleagues demonstrate that ERK1/2 mediates the activated PKA at the membrane and CREB phosphorylation in the nucleus in the MIN6 cell line and pancreatic islets (Dalle et al, 2004). However, we did not observe ERK1/2‐dependent phosphorylation of CREB when stimulated by glucagon in primary hepatocytes. On the other hand, Gervasi and colleagues have proposed two possibilities in discussing the relatively slow kinetics of PKA signaling in the cytosol and the nucleus compared to that at the membrane: Either the free PKA catalytic subunit moves out from the compartmentalized microdomain at the membrane or the substrates transport from the cytosol or the nucleus to the cAMP signaling domains (Gervasi et al, 2007). While supporting the former, our results showed that the interaction between PKA catalytic subunits and CREB took place in the nucleus in 20 min after glucagon stimulation (Fig EV4F). Additionally, Neary and Cho‐Chung have demonstrated that depletion of the PKA regulatory subunits induces nuclear translocation of the PKA catalytic subunit (Neary & Cho‐Chung, 2001). Therefore, it is possible that local high concentration of cAMP at the signaling microdomain releases the PKA catalytic subunit slowly from PKA holoenzymes, and free catalytic subunit translocates into the nucleus. We cannot rule out the other possibilities such as the presence of other cascading kinases between PKA at the membrane and the substrates or the translocation of the substrates to the membrane and moving back to their original locations. That remains to be answered elsewhere.
Even though Prom1 is known as a transmembrane glycoprotein, several studies have reported Prom1 in the nucleus of cultured cervical loop epithelium associated stem cells (CLESCs) (Singer et al, 2019) and hepatocellular carcinoma (HCC) with good prognosis (Chen et al, 2017; Singer et al, 2019) and the endocytic compartments of HCC cell lines (Izumi et al, 2019). Although the function of nuclear Prom1 in HCC is not clear, Prom1 in CLESC plays a key role in stem cell maintenance and activation through interaction with Glis2, a transcription factor, in the nucleus. Using the identical Prom1 antibody (13A4) used to detect nuclear Prom1 in CLESC, we have observed Prom1 both in the cytosolic vesicles and plasma membrane in the liver. It is possible that Prom1 in endocytic vesicles may be in endocytic or recycling route, and Prom1 in these routes may be associated with other signaling pathways.
It is of note that several Prom1 −/− mouse lines generated to understand the role of Prom1 in various organs (Nishide et al, 2009; Zacchigna et al, 2009; Zhu et al, 2009) show no apparent phenotypes other than the loss of vision (Zacchigna et al, 2009). In addition, individuals with a frame‐shift mutation (Maw et al, 2000), a non‐sense mutation (Zhang et al, 2007), or a missense mutation (Yang et al, 2008) in the PROM1 locus show retinal degeneration or sometimes polydactyly. Zacchigna et al (Zacchigna et al, 2009) suggested that lack of phenotype in Prom1‐deficient mice may have come from the expression of Prom2 with the redundant functions (Walker et al, 2013). Interestingly, retina and liver in which loss of Prom1 show phenotypes have no Prom2 expression (Fargeas et al, 2003).
Using Prom1‐deficient or radixin knockdown mice, we show that the Prom1‐radixin complex is required for hepatic gluconeogenesis. While our study shows that both lean and high‐fat diet‐fed mice lacking Prom1 or radixin exhibited decreased blood glucose level after fasting for 4 h, Karim et al (Karim et al, 2014) reported that the blood glucose level after fasting was 254 mg/dl in the Prom1 −/− mice compared to 158 mg/dl in the wild‐type mice. It is possible that the discrepancy may have come from the fact that mouse strain with a different genetic background, C57BL/6N, and 129SvEv, was used in all experiments, although the starting Prom1 −/− strain was identical.
Our observations, a decreased blood glucose level after a 4‐h fasting and improved glucose and pyruvate tolerance without changing the insulin sensitivity in both lean and high‐fat diet‐fed mice lacking Prom1 or radixin, suggest that Prom1 and radixin may be excellent target proteins for lowering blood glucose level in patients with diabetes. Therefore, a chemical drug that interferes with the molecular interaction between Prom1 and radixin might thus be useful as a treatment for hyperglycemia by inhibiting the gluconeogenic signaling pathway.
Materials and Methods
Animal studies
Prom1 knockout mice (Prom1 cre‐ert2-nlacz) were purchased from The Jackson Laboratory (Stock NO. 017743, Bar Harbor, ME, USA). The Prom1 −/− mice were backcrossed with C57BL/6N mice for five generations. Mice were genotyped and assigned to each experimental groups according to the genotype (Appendix Fig S1). All animal studies were conducted with the approval of the Korea University Institutional Animal Care and Use Committee and the Korean Animal Protection Law (KUIACUC‐2017‐14, ‐2018‐6 and ‐2019‐0111).
For glucagon stimulation tests, glucagon (200 μg/kg body weight for mice fed a normal chow diet, 100 μg/kg body weight for mice fed a high‐fat diet, Sigma‐Aldrich, St Louis, MO, USA) was intraperitoneally injected into male mice that had fasted for 4 h. For glucose tolerance tests, D‐glucose (2 g/kg body weight, Sigma‐Aldrich, St Louis, MO, USA) was intraperitoneally injected into male mice that had fasted overnight. For insulin tolerance test, insulin (0.75 U/kg body weight for mice fed a normal chow diet, 1.5 U/kg body weight for mice fed a high‐fat diet, Sigma‐Aldrich, St Louis, MO, USA) was intraperitoneally injected into male mice that had fasted for 4 h. For pyruvate tolerance test, pyruvate (2 g/kg body weight, Sigma‐Aldrich, St Louis, MO, USA) was intraperitoneally injected into male mice that had fasted overnight. For epinephrine stimulation test, epinephrine (3 μg/10 g, Sigma‐Aldrich, St Louis, MO, USA) was intraperitoneally injected into male mice that had fasted for 4 h. For immobilization stress test, male mice were restrained in a ventilated acrylic restrainers to allow the animal to breathe but not to move otherwise.
Preparation of mouse primary hepatocytes
Primary hepatocytes were isolated from 8‐week‐old C57BL/6 male mice as previously described (Koo et al, 2005). Briefly, mice were anesthetized with avertin (intraperitoneal injection of 250 mg/kg body weight), and livers were perfused with a pre‐perfusion buffer (140 mM NaCl, 6 mM KCl, 10 mM HEPES, and 0.08 mg/ml EGTA, pH 7.4) at a rate of 7 ml/min for 5 min, followed by a continuous perfusion with collagenase‐containing buffer (66.7 mM NaCl, 6.7 mM KCl, 5 mM HEPES, 0.48 mM CaCl2, and 3 g/ml collagenase type IV, pH 7.4) for 8 min. Viable hepatocytes were harvested and purified with a Percoll cushion. Then, hepatocytes were re‐suspended in complete growth medium, 199 medium containing 10% FBS, 23 mM HEPES, and 10 nM dexamethasone, and seeded on collagen‐coated plates at a density of 300,000 cells/ml. After a 4‐h attachment period, the medium was replaced with complete growth medium before use in any experiments and was changed daily. 3D culture of primary hepatocytes is described elsewhere (Swift & Brouwer, 2010).
Measurement of cAMP concentrations
Levels of cAMP were quantified using a cAMP ELISA kit (Applied Biosystems, Waltham, MA, USA) according to the manufacturer's protocol. For mouse primary hepatocytes, cells growing in 6‐well plates were lysed with IBMX and cAMP concentrations were quantified. Concentrations of cAMP in mouse liver samples were measured using ELISA and normalized to the weight.
Measurement of PKA activity
PKA activity was measured using a PKA kinase activity assay kit (Abcam, Cambridge, UK), according to the manufacturer's protocol. Briefly, whole‐cell lysates were incubated with a specific synthetic peptide as a substrate for PKA and a polyclonal antibody that recognizes the phosphorylated form of the substrate. Relative PKA activity was measured by determining the optical density.
Proximity ligation assay
In situ proximity ligation assay was used to detect protein–protein interactions in cells. Briefly, cells were cultured on collagen‐coated confocal dishes and fixed with 4% paraformaldehyde. Then, cells were permeabilized with 0.1% Triton X‐100 in phosphate buffer and incubated with a blocking buffer for 30 min to prevent nonspecific binding. Samples were sequentially incubated with a primary antibody, PLA probe, ligase, and polymerase, according to the manufacturer's instructions (Duolink, Sigma‐Aldrich, St Louis, MO, USA). PLA‐positive cells exhibited red fluorescent signals. The fluorescent signal was observed using a confocal laser scanning microscope (40× water objectives, LSM800, Carl Zeiss).
Quantitative real‐time PCR
RNA (2 μg) was reverse transcribed to cDNAs using random hexamer primers, oligo dT, and Reverse Transcription Master Premix (ELPIS Biotech, Daejeon, Korea). Quantitative real‐time PCR analyses were performed using the cDNAs from the reverse transcription reactions and gene‐specific oligonucleotides (Table 1) in the presence of TOPreal qPCR 2X premix (Enzynomics, Daejeon, Korea). The following PCR conditions were used as follows: an initial denaturation step at 95°C for 10 min, followed by 45 cycles of denaturation at 95°C for 10 s, annealing at 58°C for 15 s, and elongation at 72°C for 20 s. The melting curve for each PCR product was assessed for quality control..
Table 1.
List of primer sequences for quantitative real‐time polymerase chain reaction (qRT–PCR)
| Primers | Sequence (5′ > 3′) |
|---|---|
| G6pc | |
| F | TCTGTCCCGGATCTACCTTG |
| R | GTAGAATCCAAGCGCGAAAC |
| Pck1 | |
| F | TATGCTGATCCTGGGCATAA |
| R | CACGTTGGTGAAGATGGTGT |
| Prom1 | |
| F | CTCATGGCTGGGGTTGGATT |
| R | TGAGCAGATAGGGAGTGTCCA |
| Rdx | |
| F | GCTGCAGAAGAAGCCAAGTCT |
| R | AATCTTGGCAGTGAATTCGGC |
| Akap7 | |
| F | AAGCCAGGCTATCACATCGG |
| R | CAGCAAACGGCATGTCTGTC |
| Akap8 | |
| F | GTCGCAGACATGGAGCAAGG |
| R | GACACTGGTGTTCTGGGCAT |
| Akap8l | |
| F | GCGCTTAGATATGATGCCGC |
| R | ACCGGTAGAGGTCTCGTTCA |
| Akap9 | |
| F | TCCTCATGCAGCTGGAGATTC |
| R | TGGGCTGATCCCGAGTAGAT |
| Akap10 | |
| F | AAACAGCCAAGCCACATGGA |
| R | GGACAACAGTGTCACGCAAG |
| Akap12 | |
| F | GGACGAGCAGGAGGAAACAC |
| R | CTTGAAGCCGACGTCATTAGC |
| Akap13 | |
| F | AGAGAAGACGCACGAAGACAC |
| R | TTTGCCTTGCTTACCTGGTGC |
| 18s rRNA | |
| F | GTAACCCGTTGAACCCCATT |
| R | CCATCCAATCGGTAGTAGCG |
Measurement of hepatic glucose output
Glucose production was determined using the glucose assay kit from Sigma‐Aldrich according to the manufacturer's instructions. Primary hepatocytes were seeded on collagen‐coated 6‐well plates (0.8 × 106 cells/well). After 3 h, cells were infected with a virus overnight. After 24 h, the medium containing the virus was removed and replaced with fresh complete 199 medium. After 18 h, the medium was removed, and the cells were rinsed twice with PBS. Then, glucose production buffer (consisting of glucose‐free DMEM lacking phenol red, pH 7.4, 20 mM sodium pyruvate, 2 mM l‐glutamine and 15 mM HEPES) was added to the cells. After 4 h, the medium was collected and the amount of glucose in the medium was determined using the glucose assay kit (Sigma‐Aldrich, St Louis, MO, USA).
siRNA interference
All siRNAs (Table 2) were synthesized by Bioneer Inc. (Daejeon, Korea). Cells were transfected with 100 nM siRNA using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, USA).
Table 2.
List of siRNA sequences for RNA interference
| Gene | Sequence (5′ > 3′) |
|---|---|
| Control | CCUACGCCACCAAUUUCU |
| Prom1 | CAGAAACUGGCAAAGAGCAAUUUCA |
| Rdx | GCCAAUCAAUGUAAGAGUAACUACA |
| Akap7 | GAGGCGACAAGAATGATCATGTGAA |
| Akap8 | CAGGCCCTGTATACCTGAGCATAAT |
| Akap8l | CCCAGCCCACCTGTGATTATGGATA |
| Akap9 | CAGTGTGAAACAGACAACCTGATAA |
| Akap10 | CCCACTGCTTGTGACTCAATCAGAA |
| Akap12 | ACGTCGGCTTCAAGAAGGTATTTAA |
| Akap13 | CAGCTGTACTGGACATATTTCTAAA |
Adenovirus preparation and infection
Adenoviruses harboring sh‐Con, sh‐Rdx (radixin), sh‐Prom1, GFP, and FERM were produced as previously described (Yi et al, 2013). AD293 cells were re‐infected with viral stocks to amplify the viruses, and viruses were purified by double cesium chloride‐gradient ultracentrifugation. Infectious viral particles in the cesium chloride gradient (density = ~1.345) were collected, dialyzed against a 10 mM Tris (pH 8.0), 2 mM MgCl2, and 5% sucrose solution, and stored at −80°C. Recombinant adenovirus (0.5 × 109 pfu) was injected into the tail veins of mice. Four days after the injection, the mice were subjected to blood glucose metabolism tests.
Isolation of detergent‐resistant lipid rafts
Livers from wild‐type and Prom1 knockout mice or primary hepatocytes after cholesterol depletion by methyl‐β‐cyclodextrin (mβCD, 30 mM) for 30 min at 4°C were lysed in 1 ml of lysis buffer (0.5% Lubrol WX, 25 mM HEPES, pH 6.5, 150 mM NaCl, protease inhibitor cocktail, and protease inhibitor cocktail) and subjected to discontinuous sucrose gradient ultracentrifugation (40, 30 and 5%) using a SW41 Ti rotor (287,000 g) for 18 h at 4°C. After centrifugation, the sucrose solutions were fractionated into 12 fractions. An opaque buoyant band corresponding to the lipid rafts was collected at the interface between the 30 and 5% sucrose solutions.
Plasmid construction and transient transfection
Deletion mutants of FLAG‐tagged human PROM1 transcript variant 3(PROM1‐FLAG) and Myc‐radixin (Table 3) were generated by reverse PCR using the primer sets (Table 4). mTagBFP2‐Farnesyl‐5 was a gift from Michael Davidson (Addgene plasmid # 55295). The Myr‐BFP‐IC3 construct of Prom1 was generated as follows: First, the myristoylation sequence was added at the N‐terminus of pmTagBFP2 using reverse PCR; second, Prom1‐tail‐3XFLAG was added at the C‐terminus of pmTagBFP2 using the DNA assembly method (#E2621, NEB, Ipswich, MA, USA). Myr‐BFP was generated by removing the Prom1‐tail from Myr‐BFP‐IC3 by reverse PCR. DNA transfection was performed using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, USA), according to the manufacturer's protocol.
Table 3.
List of DNA constructs used in the study
| Plasmid construct | Backbone plasmid | Inserted gene | Database | Source |
|---|---|---|---|---|
| Prom1‐Flag | pCMV‐3Tag‐3A | Full‐length Human PROM1 1‐856 | GenBank: NM_001145847.1 | |
| 1‐133‐Flag | pCMV‐3Tag‐3A | Human PROM1 1‐133 | ||
| 1‐459‐Flag | pCMV‐3Tag‐3A | Human PROM1 1‐459 | ||
| 1‐812‐Flag | pCMV‐3Tag‐3A | Human PROM1 1‐812 | ||
| Myr‐BFP‐IC3‐Flag | pCMV‐3Tag‐3A | Myristoylation sequence‐BFP‐ PROM1 813‐856 | ||
| Myr‐BFP‐Flag | pCMV‐3Tag‐3A | Myristoylation sequence‐BFP | ||
| Myc‐Radixin | pCMV‐myc | Full‐length Human Radixin 1‐583 | GenBank: NM_001260494 | 21C Frontier Human Gene Bank (KRIBB, Daejeon, Republic of Korea) |
| Myc‐1‐310 | pCMV‐myc | Human Radixin 1‐310 | ||
| Myc‐1‐492 | pCMV‐myc | Human Radixin 1‐492 | ||
| Myc‐493‐583 | pCMV‐myc | Human Radixin 493‐583 |
Table 4.
List of primers used for Mutant constructions
| Deletion mutant | Sequence (5′ > 3′) | |
|---|---|---|
| PROM1 1‐133 | F | AAGCTTATCGATACCGTC |
| R | TTCTCCACCACATTTGTTAC | |
| PROM1 1‐457 | F | AAGCTTATCGATACCGTCG |
| R | CCTGTCATAGCCGCACAC | |
| PROM1 1‐812 | F | AAGCTTATCGATACCGTC |
| R | TCGACGATAGTACTTAGC | |
| Radixin 1‐310 | F | CTTATGGCCATGGAGGCCCGAATGCCGAAACCAATTAACGTG |
| R | CGAGAGATCTCGGTCGACCGTTACCTAGCCTGAGCCTTCATCTG | |
| Radixin 1‐492 | F | CTTATGGCCATGGAGGCCCGAATGCCGAAACCAATTAACGTG |
| R | CGAGAGATCTCGGTCGACCGTTAATTCTCATCGTGTTCATCATGTTC | |
| Radixin 493‐583 | F | CTTATGGCCATGGAGGCCCGAAATGCTGAAGCTAGTGCTGAATTATC |
| R | CGAGAGATCTCGGTCGACCGTTACATTGCTTCAAACTCATCGATAC | |
| Myr‐BFP‐IC3 | ||
| + Myristoylation sequence | F | AAGGACCCAAGCCAGAGAAGGGTGTCTAAGGGCGAAGAGC |
| R | TGGTTTGGACTTGCTAGATCCCATGGTGGCGACCGGTAG | |
| + CD133‐tail‐3xTail | F | CAAGCTTAATTCCGGACTCAGATCTATGGATTCGGAGGACGTG |
| R | CATCAGGAGGGTTCAGCTTAGATCTATTTATCGTCATCATCTTTGTAGTC | |
| Myr‐BFP | ||
| ‐ PROM1 ‐tail | F | AAGCTTATCGATACCGTCGACCT |
| R | AGATCTGAGTCCGGAATTAAGCTTGTG | |
Immunoblotting and immunoprecipitation
Cells were lysed with the following lysis buffer: 50 mM Tris‐Cl, pH 8.0, 150 mM NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% SDS, protease inhibitor mixture, and phosphatase inhibitor mixture (Sigma‐Aldrich, St Louis, MO, USA). Whole‐cell lysates (WCL) obtained from the supernatant after microcentrifugation at 20,000 g for 15 min at 4°C were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). The separated proteins were transferred to a nitrocellulose membrane and incubated with specific primary antibodies (Table 5) and horseradish peroxidase (HRP)‐conjugated secondary antibodies. Antigens were visualized using an enhanced chemiluminescence substrate kit (Thermo Fisher Scientific, Waltham, MA, USA). For immunoprecipitation, cells were lysed in buffer containing 20 mM Tris–HCl (pH 7.4), 137 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, and a protease inhibitor cocktail (Sigma‐Aldrich, St Louis, MO, USA). Whole‐cell lysates (500 μg of protein, WCL) were incubated with specific antibodies overnight and then with 60 μl of Protein A‐ or Protein G‐agarose bead slurry (Roche, Mannheim, Germany) for 3 h. Immunoprecipitates were analyzed by immunoblotting.
Table 5.
List of antibodies for immunoblotting (IB), immunofluorescence (IFA) and immunoprecipitation (IP)
| Antigen name | Company | Catalog number | Experiments (dilution factor) |
|---|---|---|---|
| Prom1 | Miltenyi Biotec | 130‐092‐442 | IB (1:200), IP (2ug) |
| Prom1 | Sigma | MAB4310 | IB (1:200), IP (2ug), IFA (1:200) |
| Prom1 | Abcam | Ab19898 | IB (1:200), IP (2ug), IFA (1:200) |
| Prom1 | eBioscience | 14‐1331‐82 | IB (1:200), IP (2ug), IFA (1:100) |
| PCK | Santa Cruz Biotechnology | 32879 | IB (1:2,000) |
| G6PC | Santa Cruz Biotechnology | 25840 | IB (1:2,000) |
| p‐CREB | Cell Signaling Technology | 9198 | IB (1:1,000), IFA (1:200) |
| CREB | Cell Signaling Technology | 9197 | IB (1:1,000), IFA (1:200) |
| p‐PKA substrate | Cell Signaling Technology | 9624 | IB (1:2,000) |
| HSL | Cell Signaling Technology | 4107 | IB (1:2,000) |
| p‐HSL | Cell Signaling Technology | 4139 | IB (1:2,000) |
| IP3R | Cell Signaling Technology | 8568 | IB (1:2,000) |
| p‐IP3R | Cell Signaling Technology | 3760 | IB (1:2,000) |
| RDX | Cell Signaling Technology | 2636 | IB (1:2,000), IFA (1:200) |
| RDX | Invitrogen | MA5‐17245 | IB (1:1,000), IFA (1:200) |
| Myc | Santa Cruz Biotechnology | sc‐40 | IB (1:1,000), IP (2ug) |
| Myc | EMD Millipore | 05‐724 | IB (1:1,000), IP (2ug), IFA (1:200) |
| Actin | Sigma‐Aldrich | A2066 | IB (1:3,000) |
| FLAG | Sigma‐Aldrich | F1804 | IB (1:1,000), IP (2ug) |
| Flotillin | Santa Cruz Biotechnology | sc‐25506 | IB (1:2,000) |
| TFR | Invitrogen | 13‐6800 | IB (1:2,000) |
| GAPDH | Santa Cruz Biotechnology | sc‐32233 | IB (1:1,000) |
| Prom1 | Miltenyi Biotec | 130‐092‐442 | IB (1:100) |
| p‐ERK | Santa Cruz Biotechnology | 7383 | IB (1:2,000) |
| ERK | Santa Cruz Biotechnology | 94 | IB (1:2,000) |
| p‐AKT | Cell Signaling Technology | 9271 | IB (1:2,000) |
| AKT | EMD Millipore | 05‐591 | IB (1:2,000) |
| IR β | BD | 611277 | IB (1:2,000) |
| Ezrin | Cell Signaling Technology | 3145 | IB (1:2,000) |
| Moesin | Cell Signaling Technology | 3150 | IB (1:2,000) |
| PKA C | Cell Signaling Technology | 4782 | IB (1:2,000) |
| PKA R | Cell Signaling Technology | 3927 | IB (1:2,000) |
Immunofluorescence staining
For immunofluorescence staining of cells, cells were fixed with 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4) for 15 min, permeabilized with 0.1% Triton X‐100 for 10 min, and blocked with blocking solution (2.5% normal horse serum in PBS) for 30 min. Cells were then incubated with primary antibody at their working concentrations in blocking buffer for 1 h at room temperature or overnight at 4°C, followed by 1 h of incubation with fluorescent dye‐labeled secondary antibodies (Thermo Fisher Scientific, Waltham, MA, USA). The primary antibodies at their working concentrations are as follows : rabbit anti‐phospho‐CREB (CST, 5 μg/ml), rabbit anti‐radixin (CST, 5 μg/ml), mouse anti‐radixin (BD, 5 μg/ml), and rabbit anti‐Myc (Sigma, 2.5 μg/ml). Confocal images were captured on an LSM 800 META confocal microscope (Zeiss, Oberkochen, DE). For immunofluorescence staining of tissues for Prom1, fresh‐frozen tissues were cut into 2‐μm thin sections using a Leica ultramicrotome. The sections were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer for 1 h at 37°C. The tissues were then blocked with blocking solution (2.5% normal horse serum in PBS) for 30 min at room temperature, and followed by incubation with the primary antibody (rat anti‐Prom1 antibody, clone, 13A4, 10 μg/ml, Thermo Fisher Scientific, Waltham, MA, USA) for overnight at 4°C. The slides were then incubated with fluorescence‐conjugated secondary antibody (Thermo Fisher, Waltham, MA, USA) for 1 h at room temperature. For CK‐19, HNF4α, radixin, and human PROM1 immunocytochemistry, heat‐induced antigen‐retrieval using hot steam in sodium citrate solution (pH = 6.0) was performed on thin cryosections, followed by blocking in blocking buffer, incubation with the primary antibody for 1 h, and the fluorescent dye‐labeled secondary antibodies (Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at room temperature. The primary antibodies at their working concentrations are as follows : rabbit anti‐cytokeratin 19 (Abcam, 2.5 μg/ml), mouse anti‐HNF4a (Abcam, 2.5 μg/ml), rabbit anti‐radixin (CST, 1/200), and mouse anti‐human PROM1 (DSHB, 1 μg/ml). After mounting with fluorescence mounting medium (Agilent, Santa Clara, CA, USA), the images were captured using an LSM 800 META confocal microscope (Zeiss, Oberkochen, DE).
Immunohistochemistry
Each paraformaldehyde‐fixed samples were either embedded in paraffin or frozen in OCT compound and cut into 5 μm‐thick sections. Tissue samples were then stained with hematoxylin–eosin (HE) or periodic acid‐schiff (PAS) according to standard protocol, and the images were captured using a light microscope (Leica).
β‐galactosidase staining of liver tissues
Improved β‐galactosidase staining of liver tissues was previously described (Trifonov et al, 2016). Briefly, livers were fixed in 2% paraformaldehyde in 0.1 M phosphate buffer for 4 h. They were then washed three times in 0.1 M phosphate buffer for 20 min each. The tissues were then incubated in 20% sucrose in phosphate buffer at 4°C overnight and finally embedded in the OCT compound. Frozen sections were then incubated in dark in staining solution at 37°C. The staining solution consisted of 1 mg/ml X‐gal (beamsbio, Korea) and 0.4 mM of NBT (4‐nitro blue tetrazolium chloride, Abcam, Cambridge, UK), in rinse solution; 0.1% sodium deoxycholate, 0.2% IGEPAL, 2 mM MgCl2, and 0.1 M phosphate buffer (pH 7.3) for overnight. During incubation in staining solution, the samples were wrapped in aluminum foil to protect them from light. The images were captured using a light microscope (Leica).
Correlative light and electron microscopy
For the correlative light and electron microscopy, primary hepatocytes from WT and KO mice were cryoprotected with 2.3 M sucrose in 0.1 M phosphate buffer and frozen in liquid nitrogen. Semi‐thin cryosection (1‐μm‐thick) was cut at −100°C with a glass knife in a Leica EM UC7 ultramicrotome equipped with an FC7 cryochamber (Leica). The sections were labeled at 4C overnight using a polyclonal antibody against Prom1 (1:200, clone:13A4, Thermo Fisher Scientific, Waltham, MA, USA). Antibody staining was visualized using an Alexa Fluor 488‐Fluoro Nanogold (1:100, Nanoprobes, Yaphank, NY, USA). Cover slipped sections were examined with a confocal microscope with a differential interference contrast setting to find specific areas for later examination by electron microscopy. After the coverslips had been floated off the sections, silver enhancement was performed using the HQ silver enhancement kit (Nanoprobes) for 3 min, and the tissues were prepared further for electron microscopy. After post‐fixation, dehydration, and embedding in Epon 812 (Polysciences, Warrington, PA, United States), areas of interest were excised and glued onto resin blocks. After being cut into ultrathin sections 70–90 nm thick, the samples were observed in an electron microscope (JEM 1010; JEOL, Tokyo, Japan) with slight uranyl acetate staining.
Human liver samples
Specimens of human liver were obtained from Dongguk University Hospital in Gyeongju, South Korea (Approval number: 110757‐201909‐HR‐06‐02). The patients had been diagnosed with mild liver fibrosis by histological examination at hospitals in South Korea.
Rap activation assay
Rap activity assays were performed according to the manufacture's protocol (Abcam; ab212011, Cambridge, UK). Briefly, primary mouse hepatocytes were lysed at 4°C in a buffer containing 50 mM Tris‐Cl, pH 8.0, 150 mM NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% SDS, protease inhibitor mixture, and phosphatase inhibitor mixture. Cells lysates were incubated for 30 min with agarose beads coupled to the Rap‐binding domain (RBD) of RalGDS (Ral Guanine Nucleotide Dissociation Stimulator), which bind specifically to the active form of Rap. Subsequently, the precipitated GTP‐Rap was detected by Western blot analysis using anti‐Rap1 antibody.
Statistical analysis
The number of mice used was determined on the basis of preliminary experiments in the same model and calculating the statistical power of each experiment. Data are presented as means ± SEM. Sample numbers and experimental repeats are indicated in the figure legends. A two‐tailed Student's t‐test was used to calculate the P‐values using the GraphPad Prism software. Significance levels are *P < 0.05; **P < 0.01; ***P < 0.001; and NS, non‐significant. P‐value of < 0.05 was considered statistically significant.
RNA sequencing analysis
RNA sequencing and analysis were performed by LAS (Gimpo, Korea) as followings.
Library preparation and sequencing
Total RNA was extracted using TRIzol® RNA Isolation Reagents (Life technologies). The quantity and quality of the total RNA were evaluated using Agilent 2100 bioanalyzer RNA kit (Agilent). The isolated total RNA was processed for preparing mRNA sequencing library using the Illumina TruSeq Stranded mRNA Sample Preparation kit (Illumina) according to the manufacturer's protocol. Quality and size of libraries were assessed using Agilent 2100 bioanalyzer DNA kit (Agilent). All libraries were quantified by qPCR using CFX96 Real‐Time System (Bio‐Rad) and sequenced on the NextSeq500 sequencers (Illumina) with a paired‐end 75 bp plus single 8 bp index read run.
Preprocessing and genome mapping
Potentially existing sequencing adapters and raw quality bases in the raw reads were trimmed by Skewer ver 0.2.2. The option –x AGATCGGAAGAGCACACGTCTGAACTCCAGTCA and ‐y AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT were used for the common adapter sequence of the Illumina TruSeq adapters, and the option ‐q 0 ‐l 25 ‐k 3 ‐r 0.1 ‐d 0.1 was used for trimming low‐quality 5′ and 3′ ends of the raw reads. The cleaned high‐quality reads after trimming the low‐quality bases and sequencing adapters were mapped to the reference genome by STAR ver 2.5 software. Since the sequencing libraries were prepared strand‐specifically by using Illumina's strand‐specific library preparation kit, the strand‐specific library option, –library‐type=fr‐firststrand was applied in the mapping process.
Quantifying gene expression and differential expressed gene analysis
To quantify the mapped reads on the reference genome in to the gene expression values, Cufflinks ver 2.2.1 with the strand‐specific library option, –library‐type=fr‐firststrand, and other default options were used. The gene annotation of the reference genome mm10 from UCSC genome (https://genome.ucsc.edu) in GTF format was used as gene models, and the expression values were calculated in Fragments Per Kilobase of transcript per Million fragments mapped (FPKM) unit. The differentially expressed genes between the two selected biological conditions were analyzed by Cuffdiff software in Cufflinks package with the strand‐specific library option, –library‐type=fr‐firststrand, and other default options was used. To compare the expression profiles among the samples, the normalized expression values of the selected a few hundred of the differentially expressed genes were unsupervised clustered by in‐house R scripts. The scatter plots for the gene expression values and the volcano plots for the expression‐fold changes and P‐values between the two selected samples also were drawn by in‐house R scripts.
Functional category analysis
To get the insights on the biological functional role of the differential gene expression between the compared biological conditions, gene set overlapping test between the analyzed differential expressed genes and functional categorized genes for KEGG pathways by g:Profiler ver 0.6.7.
Author contributions
HL, D‐MY, SL, and Y‐GK contributed to study concept and design; HL, D‐MY, JSP, HYL, and HLK involved in acquisition of data; HL, D‐MY, J‐SL, SL, and Y‐GK analyzed and interpreted the data; J‐SK and S‐HK contributed to material support; SL and Y‐GK drafted the manuscript. SL and Y‐GK is the guarantor of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Expanded View
Review Process File
Acknowledgements
We thank all members of our laboratory for their supports and intellectual inputs during the preparation of this manuscript and Dr. Ki Hoon Jung at Dongguk University Hospital for human liver specimens. Funding: This work was supported by grants from the National Research Foundation of Korea awarded to; Y.‐G. Ko (2015R1A5A1009024), H. Lee (2017R1A6A3A01009334) and S. Lee (2017R1D1A1A02017979), and partially by grant from Korea University to Y.‐G. Ko and H. Lee.
EMBO Reports (2020) 21: e49416
Contributor Information
Sungsoo Lee, Email: arrosung@korea.ac.kr.
Young‐Gyu Ko, Email: ygko@korea.ac.kr.
Data availability
The datasets produced in this study are available in the following databases: RNA‐seq data: Gene Expression Omnibus (GEO) as GSE144018 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144018).
References
- Altarejos JY, Montminy M (2011) CREB and the CRTC co‐activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol 12: 141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altier C, Dubel SJ, Barrere C, Jarvis SE, Stotz SC, Spaetgens RL, Scott JD, Cornet V, De Waard M, Zamponi GW et al (2002) Trafficking of L‐type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J Biol Chem 277: 33598–33603 [DOI] [PubMed] [Google Scholar]
- Bauer N, Wilsch‐Bräuninger M, Karbanová J, Fonseca A‐V, Strauss D, Freund D, Thiele C, Huttner WB, Bornhäuser M, Corbeil D (2011) Haematopoietic stem cell differentiation promotes the release of prominin‐1/CD133‐containing membrane vesicles—a role of the endocytic–exocytic pathway. EMBO Mol Med 3: 398–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beene DL, Scott JD (2007) A‐kinase anchoring proteins take shape. Curr Opin Cell Biol 19: 192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A, Edwards K, Fehon RG (2002) ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol 3: 586–599 [DOI] [PubMed] [Google Scholar]
- Chen Y‐L, Lin P‐Y, Ming Y‐Z, Huang W‐C, Chen R‐F, Chen P‐M, Chu P‐Y (2017) The effects of the location of cancer stem cell marker CD133 on the prognosis of hepatocellular carcinoma patients. BMC Cancer 17: 474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbeil D, Roper K, Hannah MJ, Hellwig A, Huttner WB (1999) Selective localization of the polytopic membrane protein prominin in microvilli of epithelial cells ‐ a combination of apical sorting and retention in plasma membrane protrusions. J Cell Sci 112(Pt 7): 1023–1033 [DOI] [PubMed] [Google Scholar]
- Corbeil D, Joester A, Fargeas CA, Jaszai J, Garwood J, Hellwig A, Werner HB, Huttner WB (2009) Expression of distinct splice variants of the stem cell marker prominin‐1 (CD133) in glial cells. Glia 57: 860–874 [DOI] [PubMed] [Google Scholar]
- Corbeil D, Karbanová J, Fargeas CA, Jászai J (2013) Prominin‐1 (CD133): molecular and cellular features across species In Prominin‐1 (CD133): new insights on stem & cancer stem cell biology, Corbeil D. (ed) pp. 3–24. New York, NY: Springer New York; [DOI] [PubMed] [Google Scholar]
- Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM et al (2007) Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA 104: 10158–10163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle S, Longuet C, Costes S, Broca C, Faruque O, Fontes G, Hani EH, Bataille D (2004) Glucagon promotes cAMP‐response element‐binding protein phosphorylation via activation of ERK1/2 in MIN6 cell line and isolated islets of Langerhans. J Biol Chem 279: 20345–20355 [DOI] [PubMed] [Google Scholar]
- Delint‐Ramirez I, Willoughby D, Hammond GR, Ayling LJ, Cooper DM (2011) Palmitoylation targets AKAP79 protein to lipid rafts and promotes its regulation of calcium‐sensitive adenylyl cyclase type 8. J Biol Chem 286: 32962–32975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dema A, Perets E, Schulz MS, Deak VA, Klussmann E (2015) Pharmacological targeting of AKAP‐directed compartmentalized cAMP signalling. Cell Signal 27: 2474–2487 [DOI] [PubMed] [Google Scholar]
- Deming PB, Campbell SL, Stone JB, Rivard RL, Mercier AL, Howe AK (2015) Anchoring of protein kinase A by ERM (ezrin‐radixin‐moesin) proteins is required for proper netrin signaling through DCC (deleted in colorectal cancer). J Biol Chem 290: 5783–5796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil V, Marzesco AM, Corbeil D, Huttner WB, Wilsch‐Brauninger M (2007) Midbody and primary cilium of neural progenitors release extracellular membrane particles enriched in the stem cell marker prominin‐1. J Cell Biol 176: 483–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fargeas CA, Florek M, Huttner WB, Corbeil D (2003) Characterization of prominin‐2, a new member of the prominin family of pentaspan membrane glycoproteins. J Biol Chem 278: 8586–8596 [DOI] [PubMed] [Google Scholar]
- Fouassier L, Duan CY, Feranchak AP, Yun CH, Sutherland E, Simon F, Fitz JG, Doctor RB (2001) Ezrin‐radixin‐moesin‐binding phosphoprotein 50 is expressed at the apical membrane of rat liver epithelia. Hepatology 33: 166–176 [DOI] [PubMed] [Google Scholar]
- Fraser ID, Cong M, Kim J, Rollins EN, Daaka Y, Lefkowitz RJ, Scott JD (2000) Assembly of an A kinase‐anchoring protein‐beta(2)‐adrenergic receptor complex facilitates receptor phosphorylation and signaling. Curr Biol 10: 409–412 [DOI] [PubMed] [Google Scholar]
- Friedrich MW, Aramuni G, Mank M, Mackinnon JA, Griesbeck O (2010) Imaging CREB activation in living cells. J Biol Chem 285: 23285–23295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervasi N, Hepp R, Tricoire L, Zhang J, Lambolez B, Paupardin‐Tritsch D, Vincent P (2007) Dynamics of protein kinase A signaling at the membrane, in the cytosol, and in the nucleus of neurons in mouse brain slices. J Neurosci 27: 2744–2750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloerich M, Ponsioen B, Vliem MJ, Zhang Z, Zhao J, Kooistra MR, Price LS, Ritsma L, Zwartkruis FJ, Rehmann H et al (2010) Spatial regulation of cyclic AMP‐Epac1 signaling in cell adhesion by ERM proteins. Mol Cell Biol 30: 5421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbole A, Lyga S, Lohse MJ, Calebiro D (2017) Internalized TSH receptors en route to the TGN induce local Gs‐protein signaling and gene transcription. Nat Commun 8: 443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Gomez LL, Scott JD, Dell'Acqua ML (2005) Association of an A‐kinase‐anchoring protein signaling scaffold with cadherin adhesion molecules in neurons and epithelial cells. Mol Biol Cell 16: 3574–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head BP, Patel HH, Roth DM, Murray F, Swaney JS, Niesman IR, Farquhar MG, Insel PA (2006) Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J Biol Chem 281: 26391–26399 [DOI] [PubMed] [Google Scholar]
- Hochbaum D, Barila G, Ribeiro‐Neto F, Altschuler DL (2011) Radixin assembles cAMP effectors Epac and PKA into a functional cAMP compartment: role in cAMP‐dependent cell proliferation. J Biol Chem 286: 859–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi H, Li Y, Shibaki M, Mori D, Yasunami M, Sato S, Matsunaga H, Mae T, Kodama K, Kamijo T et al (2019) Recycling endosomal CD133 functions as an inhibitor of autophagy at the pericentrosomal region. Sci Rep 9: 2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbanová J, Missol‐Kolka E, Fonseca A‐V, Lorra C, Janich P, Hollerová H, Jászai J, Ehrmann J, Kolář Z, Liebers C et al (2008) The stem cell marker CD133 (Prominin‐1) is expressed in various human glandular epithelia. J Histochem Cytochem 56: 977–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim BO, Rhee K‐J, Liu G, Yun K, Brant SR (2014) Prom1 function in development, intestinal inflammation, and intestinal tumorigenesis. Front Oncol 4: 323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi S, Hata M, Fukumoto K, Yamane Y, Matsui T, Tamura A, Yonemura S, Yamagishi H, Keppler D, Tsukita S et al (2002) Radixin deficiency causes conjugated hyperbilirubinemia with loss of Mrp2 from bile canalicular membranes. Nat Genet 31: 320 [DOI] [PubMed] [Google Scholar]
- Kim BW, Lee CS, Yi JS, Lee JH, Lee JW, Choo HJ, Jung SY, Kim MS, Lee SW, Lee MS et al (2010) Lipid raft proteome reveals that oxidative phosphorylation system is associated with the plasma membrane. Expert Rev Proteomics 7: 849–866 [DOI] [PubMed] [Google Scholar]
- Kitajiri S‐i, Fukumoto K, Hata M, Sasaki H, Katsuno T, Nakagawa T, Ito J, Tsukita S, Tsukita S (2004) Radixin deficiency causes deafness associated with progressive degeneration of cochlear stereocilia. J Cell Biol 166: 559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P et al (2005) The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437: 1109–1111 [DOI] [PubMed] [Google Scholar]
- Krishnan H, Ochoa‐Alvarez JA, Shen Y, Nevel E, Lakshminarayanan M, Williams MC, Ramirez MI, Miller WT, Goldberg GS (2013) Serines in the intracellular tail of podoplanin (PDPN) regulate cell motility. J Biol Chem 288: 12215–12221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM (2007) Identification of pancreatic cancer stem cells. Cancer Res 67: 1030–1037 [DOI] [PubMed] [Google Scholar]
- Loebrich S, Bähring R, Katsuno T, Tsukita S, Kneussel M (2006) Activated radixin is essential for GABAA receptor α5 subunit anchoring at the actin cytoskeleton. EMBO J 25: 987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzesco A‐M, Janich P, Wilsch‐Bräuninger M, Dubreuil V, Langenfeld K, Corbeil D, Huttner WB (2005) Release of extracellular membrane particles carrying the stem cell marker prominin‐1 (CD133) from neural progenitors and other epithelial cells. J Cell Sci 118: 2849 [DOI] [PubMed] [Google Scholar]
- Maw MA, Corbeil D, Koch J, Hellwig A, Wilson‐Wheeler JC, Bridges RJ, Kumaramanickavel G, John S, Nancarrow D, Röper K et al (2000) A frameshift mutation in prominin (mouse)‐like 1 causes human retinal degeneration. Hum Mol Genet 9: 27–34 [DOI] [PubMed] [Google Scholar]
- Meinkoth JL, Alberts AS, Went W, Fantozzi D, Taylor SS, Hagiwara M, Montminy M, Feramisco JR (1993) Signal transduction through the cAMP‐dependent protein kinase. Mol Cell Biochem 127–128: 179–186 [DOI] [PubMed] [Google Scholar]
- Neary CL, Cho‐Chung YS (2001) Nuclear translocation of the catalytic subunit of protein kinase A induced by an antisense oligonucleotide directed against the RIα regulatory subunit. Oncogene 20: 8019–8024 [DOI] [PubMed] [Google Scholar]
- Neisch AL, Fehon RG (2011) Ezrin, Radixin and Moesin: key regulators of membrane‐cortex interactions and signaling. Curr Opin Cell Biol 23: 377–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MV, Zagory JA, Dietz WH, Park A, Fenlon M, Zhao M, Xu J, Lua I, Mavila N, Asahina K et al (2017) Hepatic Prominin‐1 expression is associated with biliary fibrosis. Surgery 161: 1266–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishide K, Nakatani Y, Kiyonari H, Kondo T (2009) Glioblastoma formation from cell population depleted of prominin1‐expressing cells. PLoS ONE 4: e6869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien CA, Pollett A, Gallinger S, Dick JE (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445: 106–110 [DOI] [PubMed] [Google Scholar]
- Pidoux G, Gerbaud P, Dompierre J, Lygren B, Solstad T, Evain‐Brion D, Tasken K (2014) A PKA‐ezrin‐Cx43 signaling complex controls gap junction communication and thereby trophoblast cell fusion. J Cell Sci 127: 4172–4185 [DOI] [PubMed] [Google Scholar]
- Rich TC, Fagan KA, Tse TE, Schaack J, Cooper DM, Karpen JW (2001) A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc Natl Acad Sci USA 98: 13049–13054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper K, Corbeil D, Huttner WB (2000) Retention of prominin in microvilli reveals distinct cholesterol‐based lipid micro‐domains in the apical plasma membrane. Nat Cell Biol 2: 582–592 [DOI] [PubMed] [Google Scholar]
- Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T, St.Clair R, Baljevic M, White I, Jin DK et al (2008) CD133 expression is not restricted to stem cells, and both CD133+ and CD133– metastatic colon cancer cells initiate tumors. J Clin Investig 118: 2111–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer D, Thamm K, Zhuang H, Karbanová J, Gao Y, Walker JV, Jin H, Wu X, Coveney CR, Marangoni P et al (2019) Prominin‐1 controls stem cell activation by orchestrating ciliary dynamics. EMBO J 38: e99845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63: 5821–5828 [PubMed] [Google Scholar]
- Smith FD, Esseltine JL, Nygren PJ, Veesler D, Byrne DP, Vonderach M, Strashnov I, Eyers CE, Eyers PA, Langeberg LK et al (2017) Local protein kinase A action proceeds through intact holoenzymes. Science 356: 1288–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Hug MJ, Bradbury NA, Frizzell RA (2000) Protein kinase A associates with cystic fibrosis transmembrane conductance regulator via an interaction with ezrin. J Biol Chem 275: 14360–14366 [DOI] [PubMed] [Google Scholar]
- Swaminathan SK, Olin MR, Forster CL, Cruz KSS, Panyam J, Ohlfest JR (2010) Identification of a novel monoclonal antibody recognizing CD133. J Immunol Methods 361: 110–115 [DOI] [PubMed] [Google Scholar]
- Swift B, Brouwer KLR (2010) Influence of seeding density and extracellular matrix on bile Acid transport and mrp4 expression in sandwich‐cultured mouse hepatocytes. Mol Pharm 7: 491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima T, Endo H, Suzuki Y, Ikari H, Gotoh M, Iguchi A (1996) Immobilization stress‐induced increase of hippocampal acetylcholine and of plasma epinephrine, norepinephrine and glucose in rats. Brain Res 720: 155–158 [DOI] [PubMed] [Google Scholar]
- Thamm K, Šimait≐ D, Karbanová J, Bermúdez V, Reichert D, Morgenstern A, Bornhäuser M, Huttner WB, Wilsch‐Bräuninger M, Corbeil D (2019) Prominin‐1 (CD133) modulates the architecture and dynamics of microvilli. Traffic 20: 39–60 [DOI] [PubMed] [Google Scholar]
- Trifonov S, Yamashita Y, Kase M, Maruyama M, Sugimoto T (2016) Overview and assessment of the histochemical methods and reagents for the detection of β‐galactosidase activity in transgenic animals. Anat Sci Int 91: 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Hieda Y, Tsukita S (1989) A new 82‐kD barbed end‐capping protein (radixin) localized in the cell‐to‐cell adherens junction: purification and characterization. J Cell Biol 108: 2369–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker TL, Wierick A, Sykes AM, Waldau B, Corbeil D, Carmeliet P, Kempermann G (2013) Prominin‐1 allows prospective isolation of neural stem cells from the adult murine hippocampus. J Neurosci 33: 3010–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Jiang Y, Zou F, Liu Y, Wang S, Xu N, Xu W, Cui C, Xing Y, Liu Y et al (2013) Activation of PI3K/Akt pathway by CD133‐p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc Natl Acad Sci USA 110: 6829–6834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigmann A, Corbeil D, Hellwig A, Huttner WB (1997) Prominin, a novel microvilli‐specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non‐epithelial cells. Proc Natl Acad Sci USA 94: 12425–12430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinman EJ, Steplock D, Shenolikar S (2003) NHERF‐1 uniquely transduces the cAMP signals that inhibit sodium‐hydrogen exchange in mouse renal apical membranes. FEBS Lett 536: 141–144 [DOI] [PubMed] [Google Scholar]
- Wong W, Scott JD (2004) AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol 5: 959–970 [DOI] [PubMed] [Google Scholar]
- Yang Z, Chen Y, Lillo C, Chien J, Yu Z, Michaelides M, Klein M, Howes KA, Li Y, Kaminoh Y et al (2008) Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Investig 118: 2908–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JS, Park JS, Ham YM, Nguyen N, Lee NR, Hong J, Kim BW, Lee H, Lee CS, Jeong BC et al (2013) MG53‐induced IRS‐1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat Commun 4: 2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacchigna S, Oh H, Wilsch‐Brauninger M, Missol‐Kolka E, Jaszai J, Jansen S, Tanimoto N, Tonagel F, Seeliger M, Huttner WB et al (2009) Loss of the cholesterol‐binding protein prominin‐1/CD133 causes disk dysmorphogenesis and photoreceptor degeneration. J Neurosci 29: 2297–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M, Pozzan T (2002) Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 295: 1711–1715 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Zulfiqar F, Xiao X, Riazuddin SA, Ahmad Z, Caruso R, MacDonald I, Sieving P, Riazuddin S, Hejtmancik JF (2007) Severe retinitis pigmentosa mapped to 4p15 and associated with a novel mutation in the PROM1 gene. Hum Genet 122: 293–299 [DOI] [PubMed] [Google Scholar]
- Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW, Gilbertson RJ (2009) Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 457: 603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Expanded View
Review Process File
Data Availability Statement
The datasets produced in this study are available in the following databases: RNA‐seq data: Gene Expression Omnibus (GEO) as GSE144018 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144018).