

Abstract
Signaling via the intracellular pathogen receptors nucleotide‐binding oligomerization domain‐containing proteins NOD1 and NOD2 requires receptor interacting kinase 2 (RIPK2), an adaptor kinase that can be targeted for the treatment of various inflammatory diseases. However, the molecular mechanisms of how RIPK2 contributes to NOD signaling are not completely understood. We generated FLAG‐tagged RIPK2 knock‐in mice using CRISPR/Cas9 technology to study NOD signaling mechanisms at the endogenous level. Using cells from these mice, we were able to generate a detailed map of post‐translational modifications on RIPK2. Similar to other reports, we did not detect ubiquitination of RIPK2 lysine 209 during NOD2 signaling. However, using site‐directed mutagenesis we identified a new regulatory region on RIPK2, which dictates the crucial interaction with the E3 ligase XIAP and downstream signaling outcomes.
Keywords: inflammation, NOD signaling, RIPK2, ubiquitin, XIAP
Subject Categories: Immunology; Post-translational Modifications, Proteolysis & Proteomics
Polyubiquitination on multiple lysine residues of RIPK2 is required for anti‐bacterial responses mediated by the pathogen recognition receptor NOD2. This study identifies a regulatory region on the C‐lobe of the kinase domain that controls binding of the E3 ligase XIAP, RIPK2 ubiquitination and downstream signalling events.

Introduction
Nucleotide‐binding oligomerization domain‐containing (NOD) proteins NOD1 and NOD2 are intracellular pathogen recognition receptors (PRRs) that sense the bacterial peptidoglycan (PGN) fragments γ‐D‐Glu‐m diaminopimelic acid (DAP) and muramyl dipeptide (MDP), respectively (Girardin et al, 2003a,b). NOD1 and NOD2 play an important role in the clearance of bacterial pathogens, including Mycobacterium tuberculosis (Lee et al, 2016), Listeria monocytogenes (Jeong et al, 2014), and multiple Chlamydiae species (Zou et al, 2016). Aberrant NOD signaling has long been associated with a range of inflammatory disorders (Caruso et al, 2014; Philpott et al, 2014), and recent findings suggest that inhibition of the NOD pathways could be beneficial in the treatment of allergic asthma (Miller et al, 2018) and type 2 diabetes mellitus (T2DM) (Amar et al, 2011; Schertzer et al, 2011; Denou et al, 2015; Cavallari et al, 2017).
Binding of the respective ligands to NOD1 and NOD2 leads to their self‐oligomerization (Maharana et al, 2015) and the recruitment of receptor‐interacting serine/threonine‐protein kinase 2 (RIPK2) via homotypic caspase recruitment domain (CARD)–CARD interactions (Inohara et al, 2000). RIPK2 is the essential adaptor kinase in the NOD signaling pathway and drives nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) and mitogen‐activated protein (MAP) kinase activation (Chin et al, 2002; Park et al, 2007). The kinase activity of RIPK2 was initially reported to be required for signal transduction and for critical autophosphorylation of RIPK2 on S176 in the activation loop of the kinase domain (Dorsch et al, 2006) and Y474 in its CARD (Tigno‐Aranjuez et al, 2010). However, recent studies suggest that RIPK2 kinase activity is dispensable for NF‐κB activation and cytokine production (Goncharov et al, 2018; Hrdinka et al, 2018). Furthermore, it has been established that NOD signaling relies on ubiquitination of RIPK2 (Tigno‐Aranjuez et al, 2013). This process is coordinated by multiple ubiquitin E3 ligases, including X‐linked inhibitor of apoptosis protein (XIAP) (Krieg et al, 2009; Damgaard et al, 2012; Heim et al, 2019). XIAP binds to the kinase domain of RIPK2 via its baculovirus IAP repeat 2 (BIR2) domain (Krieg et al, 2009; Bertrand et al, 2011; Nachbur et al, 2015) and generates K63‐linked polyubiquitin chains on multiple lysine residues (Goncharov et al, 2018). This leads to the recruitment of the linear ubiquitin chain assembly complex (LUBAC) (Damgaard et al, 2012) and the generation of M1‐linked polyubiquitin chains on RIPK2 that serve as binding platforms for IκB kinase (IKK) and transforming growth factor beta‐activated kinase 1 (TAK1) complexes.
While the importance of XIAP and LUBAC for immune responses mediated by NOD1 and NOD2 has been demonstrated in vitro and in vivo, not much is known about the function of individual ubiquitination sites on RIPK2. A putatively ubiquitinated lysine residue on RIPK2 (K209) was discovered more than 10 years ago in a systematic screening of lysine to arginine mutations (K/R) that disrupted NF‐κB activation in overexpression experiments (Hasegawa et al, 2008). A subsequent study showed that the K209R mutation blocked RIPK2 ubiquitination and signaling (Tigno‐Aranjuez et al, 2013), and it was concluded that K209 is directly ubiquitinated and is indispensable for NOD2 responses. Nevertheless, ubiquitination of RIPK2 on K209 has not been demonstrated experimentally. Intriguingly, a recently published proteomics experiment reported multiple ubiquitination sites on RIPK2, but the authors did not identify K209 (Goncharov et al, 2018). Instead, they found ubiquitination sites on the C terminus of RIPK2 and generated a K410R/K538R double mutation that reduced MDP‐dependent responses of THP‐1 cells. Altogether, this highlights that our understanding of how post‐translational modifications (PTMs) of RIPK2 regulate NOD signaling is incomplete.
Due to the association with inflammatory diseases, pharmaceutical companies have invested in the development of inhibitors for the NOD signaling pathway. RIPK2 has been established as a potential drug target, particularly in inflammatory bowel disease, and RIPK2‐targeting kinase inhibitors have been developed (Damgaard et al, 2012; Tigno‐Aranjuez et al, 2014; Nachbur et al, 2015). Recent studies showed that the inhibition of NOD signaling is not directly due to the inhibition of the kinase function of RIPK2, but rather by the disruption of the RIPK2‐XIAP interaction (Nachbur et al, 2015; Goncharov et al, 2018; Hrdinka et al, 2018). This has led to the hypothesis that protein–protein interaction inhibitors could be used to treat NOD‐driven diseases and highlights the need for a detailed understanding of post‐translational modifications on RIPK2.
A significant issue that has hindered our understanding of such mechanisms of the NOD signaling pathway is the lack of specific biochemical tools. Most studies have been limited to overexpression experiments in cancer cell lines, which has many drawbacks including the formation of artefactual interactions or altered protein activities (von Mering et al, 2002; Aebersold & Mann, 2003). In the context of NOD signaling, it was shown that ectopic overexpression of NOD receptor complex components leads to pathway activation independent of PGN binding (McCarthy et al, 1998; Thome et al, 1998; Ogura et al, 2001). To study NOD2 signaling mechanisms and investigate the molecular determinants of RIPK2 activation, we established a new mouse strain with endogenously FLAG‐tagged RIPK2. This allowed us to isolate RIPK2 from primary tissues and cells and to characterize the PTMs on RIPK2 that occur during MDP‐induced signaling at endogenous levels. While we did not identify ubiquitination of the putative ubiquitination site K209, we identified a novel regulatory region that controls XIAP binding and is required for signal transduction.
Results and Discussion
FLAG‐RIPK2 knock‐in mice represent a novel tool to study endogenous NOD signaling mechanisms
To study NOD signaling at the endogenous level and to investigate how RIPK2 regulates signal transduction, we generated N‐terminally FLAG‐tagged RIPK2 knock‐in mice by microinjection of single guide RNAs (sgRNAs), recombinant Cas9 protein, and a dsDNA oligonucleotide encoding the FLAG‐tagged sequence of RIPK2 with homologous arms upstream and downstream of the sgRNA targeted region into wild‐type C57Bl/6 embryos (Fig EV1). Using this process, we generated mice harboring the desired FLAG‐tagged version of RIPK2, as well as mice with defined insertions and deletions. After backcrossing to C57Bl/6 mice, we established a FLAG‐RIPK2 knock‐in mouse strain as well as a new RIPK2 knockout strain.
Figure EV1. CRISPR/Cas9 targeting of Ripk2 to generate FLAG knock‐in mice.
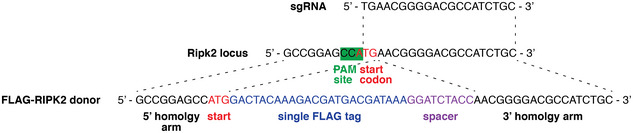
Schematic of the strategy to generate FLAG‐Ripk2 knock‐in mice using CRISPR/Cas9 technology. A sgRNA targeting the PAM site at the start codon was injected into fertilized one‐cell stage embryos together with recombinant Cas9 protein and a donor dsDNA oligonucleotide encoding N‐terminally FLAG‐tagged RIPK2, flanked with 60 bp‐long homology arms. In this illustration, only the first few nucleotides of the homology arms are shown.
First, we explored the tissue distribution of RIPK2 by testing homogenates from organs of FLAG‐RIPK2 and wild‐type mice by Western blot. As the expression levels of RIPK2 in these organs were too low for detection using anti‐FLAG antibodies, we subjected organ homogenates to anti‐FLAG immunoprecipitation (Fig 1A) and probed the supernatants from boiled beads for the presence of RIPK2. We found high levels of FLAG‐RIPK2 in homogenates from the lung and the spleen, and lower amounts in the brain, colon, small intestine, skin, and liver. We did not detect FLAG‐RIPK2 in the kidney.
Figure 1. Flag‐RIPK2 knock‐in mice as a tool to study endogenous NOD signaling mechanisms.
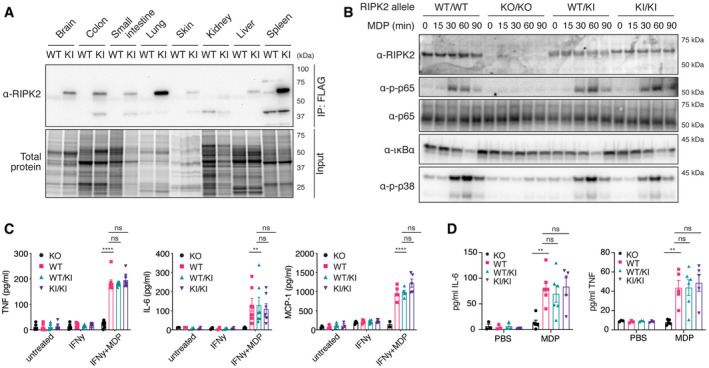
- Tissue distribution of RIPK2 determined by anti‐FLAG immunoprecipitation. Organ homogenates from WT (wild‐type) and KI (homozygous FLAG‐RIPK2) mice were subjected to anti‐FLAG immunoprecipitation and immunoblotting.
- Inflammatory signaling in wild‐type (WT/WT), RIPK2 CRISPR KO (KO/KO), and FLAG‐RIPK2 heterozygous (WT/KI) and homozygous (KI/KI) BMDMs. BMDMs were primed with IFNγ, stimulated with MDP for indicated times, and analyzed by immunoblotting.
- Cytokine production of RIPK2 CRISPR KO (KO), wild‐type (WT), and FLAG‐RIPK2 heterozygous (WT/KI) and homozygous (KI/KI) BMDMs in response to MDP. BMDMs were left untreated or treated with IFNγ alone or IFNγ and MDP overnight and cytokines were measured by ELISA. N = 5–8 mice. Shown is average ± SEM. ns P > 0.05; **P ≤ 0.01; ****P ≤ 0.0001; two‐way ANOVA.
- Serum cytokines in RIPK2 CRISPR KO (KO), wild‐type (WT), and FLAG‐RIPK2 heterozygous (WT/KI) or homozygous (KI/KI) mice after i.p. MDP administration. Mice were injected i.p. with PBS or MDP, sacrificed after 4 h and serum cytokines were measured by ELISA. N = 3–6 mice. Shown is average ± SEM. ns P > 0.05; **P ≤ 0.01; two‐way ANOVA.
We then tested the functionality of FLAG‐RIPK2 using IFNγ‐primed bone marrow‐derived macrophages (BMDMs). Priming with IFNγ is required for the BMDMs to take up and respond to MDP (Fekete et al, 2017). Upon MDP stimulation, cells generated from homozygous (KI/KI) or heterozygous (WT/KI) FLAG‐RIPK2 mice induced NF‐κB and MAP kinase pathways equivalent to wild‐type cells (Fig 1B). Western blot for RIPK2 also revealed even expression levels between wild‐type and FLAG‐tagged versions of RIPK2, which was particularly clear in the heterozygote samples (Fig 1B). After stimulation with MDP, ubiquitination of FLAG‐RIPK2 in BMDMs from knock‐in mice was comparable to ubiquitination of RIPK2 in cells from wild‐type mice (Fig EV2). As expected, cells generated from our new strain of RIPK2 knockout mice were unresponsive to MDP and did not express detectable RIPK2. Cytokine production of BMDMs after MDP stimulation was then measured by ELISA (Fig 1C). In IFNγ‐primed wild‐type and FLAG‐RIPK2 BMDMs, treatment of MDP induced the secretion of TNF, IL‐6, and MCP‐1 at equal levels, while RIPK2 KO BMDMs were unresponsive. To confirm that NOD2‐dependent responses in FLAG‐RIPK2 mice were indistinguishable from wild‐type mice in vivo, we intraperitoneally (i.p.) injected MDP or PBS into wild‐type, FLAG‐RIPK2 and RIPK2 knockout mice and measured cytokine levels in the serum by ELISA (Fig 1D). MDP challenge caused a reproducible increase in the levels of IL‐6 and TNF in the serum of wild‐type and knock‐in mice. In RIPK2 deficient mice, MDP injection did not result in an increase of cytokines. Altogether these data show that FLAG‐RIPK2 mice and primary cells from these mice responded normally to NOD2 stimulation.
Figure EV2. Ubiquitination of FLAG‐RIPK2 after MDP stimulation.
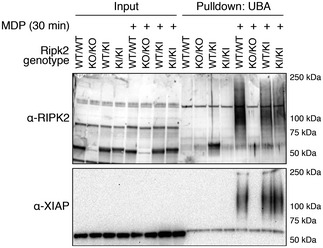
BMDMs from wild‐type (WT/WT), RIPK2 CRISPR KO (KO/KO), and FLAG‐RIPK2 heterozygous (WT/KI) and homozygous (KI/KI) mice were primed with IFNγ and stimulated with MDP for 30 min or left unstimulated. Cell lysates were subjected to UBA pulldown, separated by SDS–PAGE, and analyzed by Western blotting.
Post‐translational modifications on RIPK2
Initially, it was thought that the kinase activity of RIPK2 was required for signaling down stream of NOD receptors (Nembrini et al, 2009). Recently, however, two groups independently showed that it is dispensable for NOD mediated NF‐κB activation and cytokine production (Goncharov et al, 2018; Hrdinka et al, 2018), although it is an open question whether the kinase activity is important in other RIPK2‐regulated cellular processes, such as autophagy (Cooney et al, 2010; Homer et al, 2010; Anand et al, 2011; Lupfer et al, 2013). These studies indicate that it is rather post‐translational modification, particularly ubiquitination of the adaptor protein RIPK2, that is the critical mediator of NOD/RIPK2 signaling. Although it has been demonstrated that RIPK2 is post‐translationally modified during signaling, we did not observe ubiquitination of RIPK2, as characterized by a high molecular weight smear on Western blots, in whole‐cell lysates of MDP‐stimulated BMDMs (Fig 1B). We also did not observe ubiquitination sites on RIPK2 when we performed anti‐FLAG immunoprecipitation on MDP‐stimulated FLAG‐RIPK2 BMDMs followed by mass spectrometry analysis (Dataset EV1). These data indicate that the majority of cellular RIPK2 is not ubiquitinated and not part of the NOD signaling complex, even after stimulation, and suggests that an additional purification step is required to investigate RIPK2 in its activated state.
Therefore, we established a sequential pulldown protocol to enrich for the RIPK2 pool that participates in NOD2 signaling complexes. IFNγ‐primed BMDMs were stimulated with MDP for 30 min and lysates were first enriched for ubiquitinated proteins using glutathione S‐transferase (GST)‐ubiquitin associated domain (UBA) bound to Sepharose beads (Hjerpe et al, 2009; Fiil et al, 2013). Bound proteins were then eluted with Glutathione, and eluates were subjected to anti‐FLAG immunoprecipitation, followed by elution with 3x‐FLAG peptide (Fig 2A). The first pulldown (UBA, lane B) yielded a sample containing readily detectable levels of a ladder of RIPK2 species suggestive of ubiquitination, as well as many other ubiquitinated proteins (anti‐ubiquitin, bottom panel). The sample obtained by sequential pulldown with anti‐FLAG (lane C) also contained modified RIPK2; however, the background of ubiquitinated proteins was significantly reduced, further suggesting that this approach resulted in purification of activated RIPK2.
Figure 2. Identification of RIPK2 PTMs during NOD signaling by mass spectrometry.
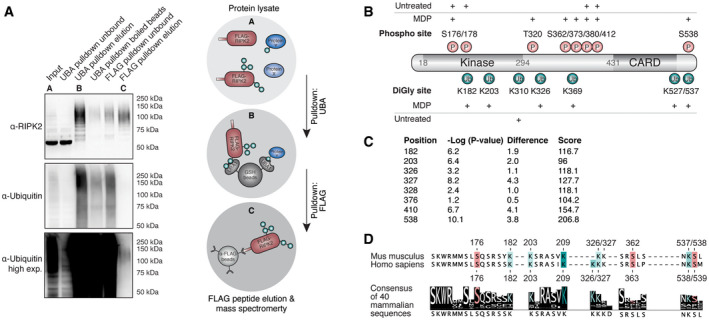
- Two‐step enrichment to isolate ubiquitinated RIPK2 from BMDMs. Protein lysates from FLAG‐RIPK2 BMDMs (A) were sequentially subjected to ubiquitin enrichment (UBA, B) and FLAG pulldown (C) prior to protein elution and subsequent mass spectrometry analysis.
- Schematic representation of RIPK2 PTMs detected in MDP‐stimulated vs. unstimulated BMDMs. Red: phosphorylation, green: ubiquitination. N = 3 experiments.
- DiGly modifications on RIPK2 in L18‐MDP-stimulated vs. L18‐MDP-unstimulated THP‐1 cells determined by diGly proteomics. Shown are P values and log2 differences of modified peptides based on imputed values; t‐test. Score: peptide identification score as determined by MaxQuant.
- Sequence conservation of stimulation‐dependent modified serine (red) and lysine (green) residues among mammals. The degree of conservation is indicated by color saturation.
Tryptic digests of these samples were then generated and analyzed by mass spectrometry, revealing a substantial enrichment of ubiquitin and RIPK2 peptides. To specifically determine stimulation‐dependent PTMs on RIPK2, this dataset was compared with datasets obtained from FLAG pulldowns of unstimulated BMDMs. In unstimulated BMDMs, only one single K‐ɛ‐diglycine site (diGly, diglycine remnant on lysine after tryptic digestion of ubiquitinated proteins) on RIPK2 was observed at the C‐terminal end of the kinase domain (K310). In contrast, MDP‐stimulated BMDMs revealed multiple RIPK2 ubiquitination sites within the kinase domain (K182, K203), in the intermediate region (K326, K369) and in the CARD (K527, K537) (Fig 2B). The function of most of the detected sites is still uncharacterized; only one recent study showed that human THP‐1 cells expressing a K410R/K538R double mutant (human K538 corresponds to K537 in mice) display reduced NF‐κB activation and cytokine production. We were not able to detect the previously described ubiquitination site K209 (Hasegawa et al, 2008) using our stringent protocol.
Phosphopeptides on RIPK2 were detected in both unstimulated and MDP‐stimulated BMDMs. Stimulation‐dependent phosphorylation was detected in the intermediate region (T320, S362, S373) and the CARD (S539). The activation loop of RIPK2 was phosphorylated on two residues (S176, S178); however, these phosphopeptides were discovered in stimulated as well as in unstimulated cells. This was surprising since phosphorylation of the kinase activation loop is associated with RIPK2 activation (Dorsch et al, 2006; Rahman et al, 2014).
To further confirm the physiological importance of the RIPK2 ubiquitination sites, we examined the ubiquinome of the human monocytic cell line THP‐1 employing diGly proteomics. In unstimulated cells, we did not detect any diGly sites on RIPK2 but we consistently observed several diGly marks after L18‐MDP stimulation (Fig 2C, Dataset EV2). The sites identified in THP‐1 cells reflected our results obtained using the sequential pulldown protocol in mouse cells, validating our initial approach. All stimulation‐dependent diGly modifications that we detected on murine RIPK2 residues, which are also conserved in humans, were detected in L18‐MDP‐stimulated THP‐1 cells (K182, K203, K326, K538). Additionally, two diGly‐modified lysines (K376, K410) that are not conserved in mice were found. Again, with this second protocol using human cells, we were not able to detect ubiquitination of K209. As expected, we did not detect any phosphorylation events on RIPK2 using this approach, indicating that there are no simultaneous ubiquitination and phosphorylation events on a single peptide after tryptic digest.
Most PTMs on RIPK2 are redundant for NOD2 signaling
While multiple ubiquitin E3 ligases and deubiquitinases (DUBs) have been suggested to regulate RIPK2 ubiquitination, the contribution of individual ubiquitination sites on RIPK2 has not been characterized in endogenous systems so far. Here, we demonstrated that RIPK2 is ubiquitinated on multiple lysine residues. This is not unusual as many proteins become ubiquitinated on multiple sites during signaling (Ball et al, 2016; Wagner et al, 2016). Typically, there appears to be flexibility in the lysine residues that can be ubiquitinated and often mutation of a single lysine has little impact on signaling. This is believed to be because E3 ligases are not usually restricted to a specific motif, in the way that for example kinases or caspases are, and can therefore be promiscuous in the lysine that they modify (Petroski & Deshaies, 2003; Wu et al, 2003).
To assess the impact of individual RIPK2 PTMs on NOD signaling, we generated mutants of lysine and serine residues that we found to be modified upon NOD2 stimulation and are highly conserved among mammals (Fig 2D). Additionally, we included conserved sites that have previously been associated with RIPK2 activation (Fig 2D; Dorsch et al, 2006; Hasegawa et al, 2008; Tigno‐Aranjuez et al, 2010). These corresponded to K182, K203, K326, K327, S363, K538, S539, and K209 in humans.
We used these RIPK2 variants to test whether single ubiquitination and phosphorylation sites on RIPK2 are critical for its function in a close‐to‐endogenous setting. For this, we generated RIPK2‐deficient THP‐1 cells by transient transfection with Cas9 and RIPK2 sgRNA encoding plasmids and confirmed the knockout of RIPK2 by Western blot. These knockout cells were then transduced with doxycycline‐inducible RIPK2 constructs to express RIPK2 at levels similar to wild‐type THP‐1. In contrast to overexpression studies, expression of RIPK2 alone did not autoactivate NF‐κB, but additional treatment with L18‐MDP‐induced transient phosphorylation of p65 and IκBα and degradation of IκBα (Fig 3A). Upon MDP stimulation, cells reconstituted with all mutant forms of RIPK2 activated NOD signaling normally, except the previously described K209R mutant.
Figure 3. Characterization of RIPK2 diGly‐ and phosphosite mutations.
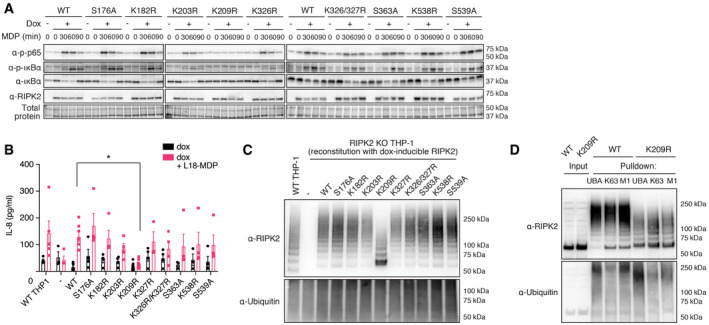
- Activation of the NF‐κB pathway by RIPK2 Lysine‐ and phosphosite mutants. RIPK2‐deficient THP‐1 cells reconstituted with wild‐type RIPK2 or RIPK2 mutants were stimulated with L18‐MDP, harvested at indicated time points and activation of the NF‐κB pathway was analyzed by immunoblotting.
- IL‐8 production of wild‐type THP‐1 and RIPK2‐deficient THP‐1 cells reconstituted with wild‐type RIPK2 or RIPK2 mutants and stimulated with L18‐MDP was assessed by ELISA. N = 4–8 experiments. Shown is average ± SEM. *P ≤ 0.05; two‐way ANOVA.
- RIPK2 ubiquitination determined by UBA pulldown in RIPK2‐deficient cells reconstituted with wild‐type or mutant RIPK2 after stimulation with L18‐MDP.
- Detection of K63‐ and M1‐linked ubiquitin chains by UBA pulldown or pulldown with ubiquitin chain type‐specific antibodies in RIPK2‐deficient cells reconstituted with wild‐type or K209R RIPK2 after stimulation with L18‐MDP.
After L18‐MDP stimulation, we observed significantly reduced levels of IL‐8 in RIPK2‐deficient cells reconstituted with the K209R mutant, while all other mutants produced equal amounts of IL‐8 compared with either wild‐type THP‐1 cells or RIPK2‐deficient cells reconstituted with wild‐type RIPK2 (Fig 3B).
Notably, RIPK2 K209R was the only form of RIPK2 which seemed to present in a second, higher molecular weight band after stimulation, similar to recently described Riposomes (Ellwanger et al, 2019). To test the impact of each individual modified site on the ubiquitination pattern on RIPK2 during NOD signaling, cells were subjected to UBA pulldowns and analyzed by Western blot. Stimulation with L18‐MDP led to distinct RIPK2 polyubiquitination (Fig 3C) and the removal of single ubiquitination or phosphorylation sites, besides K209, did not affect RIPK2 ubiquitination.
It was previously reported that K209 is critical for RIPK2 ubiquitination and NOD signaling (Hasegawa et al, 2008). However, we observed that residual ubiquitination of RIPK2 K209R was still present. Since RIPK2 K209R failed to activate NF‐κB and to produce cytokines, and displayed reduced ubiquitination, we hypothesized that this mutation led to a loss of the critical K63‐ or M1‐linked ubiquitin chains. To test this hypothesis, cells reconstituted with wild‐type RIPK2 or K209R RIPK2 were stimulated with L18‐MDP and subjected to either UBA pulldown or to pulldowns with K63‐ or M1‐chain‐specific antibodies (Newton et al, 2008; Matsumoto et al, 2012). Compared to wild‐type RIPK2, the ubiquitination of K209R was reduced in all pulldowns; however, K63‐ and M1‐linked chains were still detected, indicating that the K209R mutant is still able to be ubiquitinated on other lysine residues (Fig 3D). These reduced chains are, however, unable to induce downstream signaling which might explain the lack of global ubiquitination events after MDP stimulation in cells harboring the K209R mutation (Panda & Gekara, 2018).
Our results clearly indicate that most single phosphorylation and ubiquitination events on RIPK2 are redundant. While this is the case for many signaling proteins, there are some exceptions to this general rule. For example, mutation of K377 in RIPK1 has a profound effect on RIPK1 ubiquitination and TNFR1 induced activation of NF‐κB (O'Donnell et al, 2007). Our data as well as previous studies could lead to the assumption that K209 is another similarly special residue because the K209R mutation blocked overexpression‐induced NF‐κB activation and RIPK2 ubiquitination (Hasegawa et al, 2008) and the mutant has a loss of function phenotype when overexpressed together with ubiquitin in HEK 293T cells (Tigno‐Aranjuez et al, 2013). However, unlike K377 in RIPK1, K209 is on the C‐lobe of the kinase domain of RIPK2, which is already indicative of a different function of this site.
K209 and I212 form a regulatory region that influences signal transduction
Since neither we nor others were able to detect ubiquitination of K209 by mass spectrometry analysis (Goncharov et al, 2018), and mutation of K209 reduced, but did not completely abolish K63 and M1 linked ubiquitination of RIPK2 yet this same mutation stopped NF‐κB activation and cytokine production, we hypothesized that K209 is not a critical ubiquitination site but serves a different function. K209 is located at the N‐terminal end of αE helix in the C‐lobe of the kinase domain (Fig 4A; PDB 4C8B; (Canning et al, 2015)). It is part of a hydrophobic pocket formed by amino acids of helix αE and amino acids in the loop between helix αE and αEF, which are suggestive of a regulatory interface. To test the hypothesis that disruption of this region prevents NOD2 signaling, we generated mutations of amino acids K209 and I208 that contribute to the making of this pocket. While K209 is invariant among vertebrates, I208 is replaced by valine in most vertebrate RIPK2 sequences (Fig 4B). We also mutated I212, which is part of the αE helix with its side chain sitting deep within the pocket. I212 is highly conserved and only replaced by other hydrophobic amino acids, such as valine or methionine, in some vertebrate species.
Figure 4. Functional studies of RIPK2 mutations introduced in close proximity to K209R.
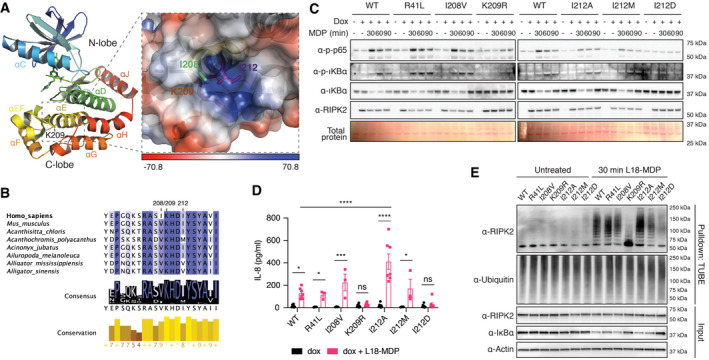
- Structural features of the RIPK2 kinase domain (left) and location of K209 within a hydrophobic pocket between helices αEF and αE (right). Shown is chain B of the RIPK2 kinase in complex with ponatinib from PDB:4C8B. The electrostatic interaction potential is shown as a blue to red gradient.
- Conservation of amino acids creating a hydrophobic pocket between helices αEF and αE. Degree of conservation among 227 vertebrate species indicated by background color saturation.
- Activation of the NF‐κB pathway by RIPK2 pocket mutants. RIPK2‐deficient THP‐1 cells were reconstituted with wild‐type RIPK2 or RIPK2 mutants, stimulated with L18‐MDP, harvested at indicated time points, and analyzed by immunoblotting.
- IL‐8 production of RIPK2‐deficient THP‐1 cells reconstituted with wild‐type RIPK2 or RIPK2 mutants and stimulated with L18‐MDP was assessed by ELISA. N = 3–8 experiments. Shown is average ± SEM. ns P > 0.05; *P ≤ 0.05; ***P ≤ 0.001; ****P ≤ 0.0001; two‐way ANOVA.
- Ubiquitination of RIPK2 pocket mutants. RIPK2‐deficient THP‐1 cells were reconstituted with wild‐type or mutant RIPK2, left unstimulated or stimulated with L18‐MDP and subjected to UBA pulldown and immunoblotting.
We reconstituted RIPK2‐deficient THP‐1 cells with the new RIPK2 mutants we generated and tested them for NF‐κB activation, RIPK2 ubiquitination, and cytokine secretion. The expression levels of these RIPK2 mutants seemed equivalent (Fig 4C). Conservative I208V and I212M mutations did not prevent activation of NF‐κB or the production of IL‐8 after MDP stimulation (Fig 4D). In contrast, cells that expressed the RIPK2 I212A mutant produced significantly more IL‐8 than cells expressing wild‐type RIPK2, and markers of NF‐κB activation were similar or enhanced (Fig 4C and E). We also observed enhanced ubiquitination after MDP stimulation of RIPK2 I212A compared with wild‐type or other mutant forms of RIPK2 (Fig 4E). Finally, the substitution of isoleucine 212 with an aspartic acid (I212D) completely blocked RIPK2 ubiquitination and NF‐κB activation. These results suggest that the C‐lobe pocket is critical in the regulation of RIPK2 activity and may act to recruit an E3 ligase, such as XIAP, to the NOD signaling complex.
While these data confirm previous reports that K209 is indispensable for NOD mediated NF‐κB activation and cytokine production, and that RIPK2 ubiquitination is impaired when K209 is mutated to an arginine, the absence of direct ubiquitination of K209 indicated a different mechanism than previously reported. The reason for this is twofold: Firstly, we used two multi‐replicate complementary experimental approaches to determine the phosphorylation‐ and ubiquitination signature on RIPK2 upon activation. The fact that we identified identical diGly sites on RIPK2 in mouse (BMDMs) and in human (THP‐1) gives high confidence in our datasets. Despite the consistency of the PTM data across the two cell lines, we, like others in the field, failed to observe a diGly site corresponding to K209. Secondly, mutation of a residue in close proximity, but not directly affecting K209, resulted in an even more dramatic impact on RIPK2 ubiquitination than mutation of K209 itself. This suggests that structural integrity of this region is critical for signal transduction. To this end, we cannot exclude that our I212D mutation rendered K209 inaccessible for ubiquitination, but further studies will be required to examine this experimentally.
It should be noted that we not only failed to detect ubiquitinated K209, but we also did not detect unmodified K209 in our datasets on MDP‐stimulated BMDMs or THP‐1 cells. Nevertheless, when analyzing other datasets that identified RIPK2 in deep proteome and pan‐kinome experiments (utilizing broad specificity kinase inhibitors to enrich for kinases), this region was readily identifiable as an unmodified tryptic peptide (Creixell et al, 2015; Ruprecht et al, 2015; Slany et al, 2016; Klaeger et al, 2017). This could be because we specifically enriched for activated RIPK2 in our protocols and we did not use kinase inhibitors in our approaches. We therefore cannot definitively refute the idea of K209 ubiquitination, although the current evidence suggests that there may be as‐yet other unidentified post‐translational modifications hindering its identification. Lastly, the fact that a third method did not lead to the detection of a diGly site on RIPK2 K209 (Goncharov et al, 2018) hints to the direction that this particular portion of RIPK2 is more complex than currently understood.
Mutation of K209 and I212 disrupts XIAP binding
An alternative explanation for the loss of the potential to activate NF‐κB and cytokine production of the K209R and I212D mutations is instability due to misfolding or impairment of the dimerization potential. The equivalent expression levels of the RIPK2 mutants suggested that the structural integrity of RIPK2 was not overtly compromised. However, to exclude a trivial explanation for the complete lack of signaling of the I212D mutant in particular, and more rigorously show that the mutants retained their structural integrity, we devised an intracellular thermal stability test (Fig 5A) based on reports that kinase inhibitors can increase the thermal stability of their targeted kinases (Martinez Molina et al, 2013; Jafari et al, 2014; Alshareef et al, 2016; Martinez Molina & Nordlund, 2016; Seashore‐Ludlow et al, 2018). We modified this assay to examine the effects of mutation on protein stability as a marker for structural integrity, and we think that it could be more widely used to determine whether a particular mutation affects structural integrity in a semi‐quantitative manner.
Figure 5. RIPK2 K209 and I212 mediate XIAP binding.
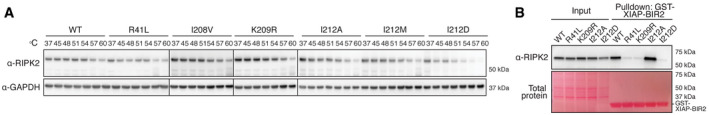
- Thermal stability assay using selected RIPK2 mutants. THP‐1 cells expressing RIPK2 were subjected to heat treatment, and non‐denatured fractions were analyzed by immunoblotting.
- Binding of RIPK2 to XIAP‐BIR2. Lysates from RIPK2‐deficient THP‐1 cells reconstituted with WT or mutant RIPK2 were subjected to pulldown experiments with recombinantly expressed XIAP‐BIR2 and analyzed by immunoblotting.
At the physiological temperature of 37°C, wild‐type RIPK2 and all RIPK2 mutants were expressed at equal levels. With increased temperature, all mutants displayed similar stability compared with wild‐type RIPK2. In particular, all I212 mutants, I212A (activating), I212M (residue in Damselfish), and I212D (inactivating) had an almost identical thermal stability profile, strongly suggesting that the mutations of this pocket had not caused major structural disruption. To estimate the effects of these mutations on the structural integrity of RIPK2, we employed molecular modeling using the DynaMut software (Rodrigues et al, 2018). This software predicted that all the mutants increase rigidity of the αG region, the activation loop, and a portion of the N‐lobe αC helix with K209R(1.411 kcal/mol) > I212A(0.764 kcal/mol) > I212D(0.38 kcal/mol) (Fig EV3). Since this increased rigidity is predicted for both activating (I212A) and inactivating (K209R, I212D) mutations, it is unlikely to be the cause of the loss of function.
Figure EV3. Molecular modeling for structural changes in the RIPK2 kinase domain.
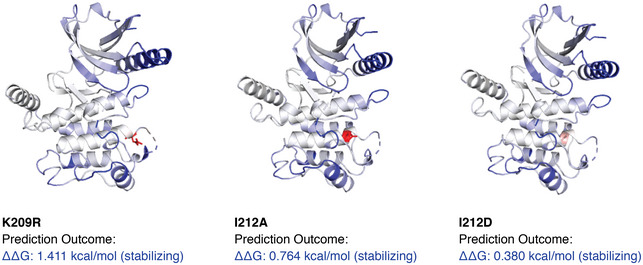
The impact of K209R, I212D, and I212A mutations on the structural integrity of the RIPK2 kinase domain was estimated utilizing the DynaMut software (Rodrigues et al, 2018) using the kinase domain of RIPK2 (PDB: 4C8B) as the input. Dark blue regions indicate the prediction of more rigid sections.
The ubiquitin E3 ligase XIAP is indispensable for NOD2 responses in vitro and in vivo (Bauler et al, 2008; Krieg et al, 2009; Damgaard et al, 2012; Stafford et al, 2018) and binds to the RIPK2 kinase domain via its Baculovirus IAP Repeat 2 (BIR2) domain (Krieg et al, 2009; Damgaard et al, 2012). The structural determinants underlying the interaction between the two proteins remain unclear. The XIAP‐BIR2 domain contains a deep and distinctive hydrophobic cleft that typically mediates binding to proteins harboring an IAP‐binding motif (IBM) as present in caspases or second mitochondria‐derived activator of caspases (SMAC) (Wu et al, 2000; Verhagen et al, 2007). However, RIPK2 does not contain such a motif. It is possible that XIAP binds to RIPK2 in a non‐canonical fashion, independently of an IBM. Such a mechanism has been shown with the caspase Dronc and the XIAP homolog DIAP1 in Drosophila melanogaster. Binding of Dronc to DIAP1 is mediated via a short 12‐residue peptide located between the CARD and the protease domain of Dronc that binds into the hydrophobic cleft of the DIAP in a similar fashion as observed in IBM‐containing proteins. Therefore, one explanation could be that the RIPK2‐XIAP interaction occurs in a similar, non‐canonical fashion; however, the region around K209 and I212 has the shape of a pocket. The binding mode of RIPK2 and XIAP‐BIR2, therefore, would be completely different from the previously observed interactions. Alternatively, XIAP could bind somewhere else, and mutation of K209 and I212 might lead to conformational changes that have an impact on the structure or orientation of the interaction interface. A region in the N‐lobe of RIPK2, in particular residues R36 and R41, was identified as a critical interaction region for the BIR2 of XIAP (Hrdinka et al, 2018). However in all reported crystal structures, the RIPK2 kinase domain is organized in head‐to‐tail dimers (PDB: 4C8B, 5AR4, 6ES0, 6FU5, 5J79) and the regions around R36 and R41 are quite far apart from K209 (approximately 40 Å). According to these structures, it is unlikely that the BIR2 of XIAP binds both areas simultaneously, but the reason for reduced binding of the K209R and I212D mutant could be due to conformational impairment or that it may have influenced binding to XIAP indirectly.
To test whether mutation of K209 and I212 disrupted XIAP binding, a purified, recombinant, GST‐coupled BIR2 domain of XIAP was used to precipitate RIPK2 from THP‐1 lysates (Fig 5B). Wild‐type RIPK2 was strongly enriched by XIAP‐BIR2 pulldown, and I212A was even more abundant, correlating with the increased stimulus‐dependent ubiquitination of this mutant and increased cytokine secretion compared with wild‐type RIPK2. Binding of K209R and I212D to the BIR2 of XIAP was, however, drastically reduced and comparable to R41L, a RIPK2 mutant previously shown to have impaired binding to XIAP (Hrdinka et al, 2018).
In recent studies, higher order intracellular signaling platforms consisting of RIPK2 and NOD receptors were described (Gong et al, 2018; Pellegrini et al, 2018; Ellwanger et al, 2019). In particular, inhibition of XIAP by either siRNA or by SMAC mimetic compounds led to RIPK2‐containing speck‐like structures in cells, termed Riposomes (Ellwanger et al, 2019). Here, we described two mutations that perturbed the interaction between RIPK2 and XIAP. On close examination, RIPK2 K209R seems to accumulate in a higher order band in a Western blot after stimulation (Fig 3A), consistent with the hypothesis that in the absence of XIAP, RIPK2 is moved to Triton‐insoluble Riposomes. However, the RIPK2 I212D or the RIPK2 R41L mutations, which reduce RIPK2‐XIAP interaction to a similar level to the RIPK2 K209R mutation, did not lead to an equivalent band in Western blots after stimulation. Therefore, these new XIAP binding mutants will provide novel insight into the mechanism and function of Riposome formation and their relevance for NOD signaling.
Taken together, our work provides a detailed and systematic map of post‐translational modifications on RIPK2 during NOD signaling, and we provide evidence that most single phosphorylation and ubiquitination events on RIPK2 are redundant in systems with close‐to‐endogenous levels of RIPK2. We identified a regulatory region on RIPK2 which influences the crucial interaction with XIAP. This region includes a pocket‐shaped region around residues I212 and K209 on the C‐lobe of RIPK2's kinase domain. Our findings give an explanation to the conundrum that has plagued the field to date: why mutations of K209 reduce RIPK2 ubiquitination and block NOD signaling, yet ubiquitination of K209 has never been detected experimentally. As interfering with the RIPK2‐XIAP interaction has emerged as a strategy to inhibit NOD signaling, it is tempting to speculate that this region could be targeted with small molecules for the treatment of diseases with increased NOD signaling. Although we cannot conclusively demonstrate that mutations in the region of this pocket do not disrupt RIPK2 in a manner which may be important for its function (kinase activity, autophagy, dimerization), our data collectively suggest that site‐directed mutagenesis of either K209 or I212 blocks RIPK2 ubiquitination and inflammatory signaling by displacing XIAP.
Materials and Methods
Generation of FLAG‐RIPK2 CRISPR knock‐in mice
The FLAG‐RIPK2 mice were generated by the MAGEC laboratory (WEHI) as previously described (Kueh et al, 2017) on a C57BL/6J background. To generate FLAG‐RIPK2 mice, 20 ng/μl of Cas9 mRNA, 10 ng/μl of sgRNA (TGAACGGGGACGCCATCTGC;) and 40 ng/μl of oligo donor (GCCGCCCCGGGACCTAGCGCCGCGGCCAGGGTCGGGCGGAGCCGCCGCGCAGCCGGAGCCATGGACTACAAAGACGATGACGATAAAGGATCTACCAACGGGGACGCCATCTGCAGCGCGCTACCCCCCATCCCGTACCACAAGCTCGCCGACCTG) were injected into the cytoplasm of fertilized one‐cell stage embryos. Twenty‐four hours later, two‐cell stage embryos were transferred into the oviducts of pseudo‐pregnant female mice. Viable offspring were genotyped by next‐generation sequencing.
Cell culture, generation of BMDMs, and stimulation protocols
Wild‐type THP‐1 cells and 293T cells were sourced from ATCC™. THP‐1 cells were cultured in RPMI supplemented with 8% FBS and antibiotics (penicillin, streptomycin, GIBCO) at 37°C with 10% CO2 in a humidified incubator. 293T cells were cultured in DMEM (GIBCO) with 8% FBS in the same conditions. BMDMs were generated from the femur and tibiae of mice and cultured for 6 days in DMEM (InvivoGen) supplemented with 8% FBS (GIBCO) and 20% L929 supernatant and antibiotics (penicillin, streptomycin). Cells were then detached using trypsin‐EDTA and replated in 12‐ and 24‐well tissue culture plates. Replated cultures of BMDMs were primed with murine interferon‐γ (5 ng/ml, R&D Systems) overnight and again 2 h before stimulation with MDP (10 μg/ml, InvivoGen). THP‐1 cells were stimulated with L18‐MDP (200 ng/ml, Bachem).
Generation of RIPK2 CRISPR knockout THP‐1 cells
RIPK2‐deficient THP‐1 cells were generated using a CRISPR/Cas9‐based knockout workflow as previously described (Schmid‐Burgk et al, 2014). Briefly, a sgRNA (GACCTGCGCTACCTGAGCCGCGG) targeting RIPK2 was designed. THP‐1 cells were nucleofected with one plasmid expressing sgRNA and one expressing mCherry‐Cas9 (pLK0.1‐ gRNA‐CMV‐GFP, CMV‐mCherry‐Cas9) using the SG Cell Line 4D‐Nucleofector™ X Kit S and a 4D‐Nucleofector X unit. mCherry‐positive cells were sorted and cloned by limiting dilution. After identifying clones, cells were replated and grown to identify RIPK2 knockout clones by assessing RIPK2 expression on Western blot.
Intraperitoneal MDP injections
All in vivo experiments were performed according to the guidelines of the animal ethics committee of WEHI, ethics approval (2011.014, 2014.004 and 2017.004). Sex‐ and age‐matched littermate controls were used within each experiment. For in vivo MDP challenge: wild‐type, RIPK2 knockout, and FLAG‐RIPK2 knock‐in (hetero‐ and homozygous) mice were administered MDP (5 mg/kg, i.p. in 200 μl PBS, Bachem) or PBS and sacrificed after 4 h. Peripheral blood was collected by cardiac puncture.
Western blotting
Following stimulation, cells were lysed in 2 × SDS lysis buffer (126 mM Tris–HCl pH 8, 20% v/v glycerol, 4% w/v SDS, 0.02% w/v Bromophenolblue, 5% v/v 2‐mercaptoethanol) and subjected to repeated freeze/boil cycles. Samples were separated using SDS–PAGE and transferred to polyvinylidene fluoride (PVDF) membranes. The following antibodies were used for probing: rabbit anti‐RIPK2 (4142S, Cell Signaling Technology), rabbit anti‐RIPK2 (SC 22763, Santa Cruz), mouse anti‐FLAG (F1804, Sigma), anti‐β actin (A‐1978, Sigma), rabbit anti‐p65 (631213, Upstate), rabbit anti‐phospho p65 (3033, Cell Signaling Technology), rabbit anti‐phospho p38 (9211, Cell Signaling Technology), mouse anti‐phospho IκBɑ (9246, Cell Signaling Technology), rabbit anti‐IκBɑ (9242, Cell Signaling Technology), mouse anti‐ubiquitin (3936, Cell Signaling Technology), rabbit anti‐GAPDH (2118, Cell Signaling Technology), human anti‐K63‐linked ubiquitin (Apu3.A8, Genentech), human anti‐M1‐linked ubiquitin (1F11, Genentech), anti‐K27‐linked ubiquitin (ab18153, abcam), goat anti‐mouse Ig (1010‐05), goat anti‐rabbit Ig (4010‐05) and goat anti‐rat Ig HRP (horseradish peroxidase, 3010‐05, Southern Biotech), and goat anti‐human Ig (109‐035‐003, Jackson ImmunoResearch).
Cytokine measurement by ELISA
Cytokines from mouse serum or cell culture supernatant were measured by ELISA kits for IL‐6, IL‐8, TNF, and MCP‐1, respectively (Invitrogen), according to the manufacturer's instructions. Sera and supernatants were diluted 1:10 for MCP‐1 measurements.
Immunoprecipitation of RIPK2 from mouse tissues
Organs from 6 weeks old wild‐type C57BL/6 mice and FLAG‐RIPK2 knock‐in mice were lysed in IP buffer (150 mM NaCl, 50 mM Tris pH 7.5, 10% glycerol, 1% Triton X‐100, 2 mM EDTA, all from SIGMA) supplemented with protease inhibitors (cOmplete protease inhibitor cocktail, Roche) using a TissueLyser II (Qiagen). Samples were clarified by centrifugation at 17,000 × g for 30 min, the protein concentration was assessed using a BCA assay (Thermo Fisher) and 2 mg of protein per lysate were subjected to anti‐FLAG immunoprecipitation using 15 ul of packed magnetic anti‐FLAG beads (M2, Sigma) for 4 h. Beads were washed three times in IP buffer, eluted with 2× SDS sample buffer, and subjected to immunoblotting.
Ubiquitin enrichments (UBA, TUBE and pulldowns with K63‐ and M1‐specific ubiquitin antibodies)
20*106 THP‐1 cells were treated with doxycycline (200 ng/ml) for 5 h and stimulated with L18‐MDP (200 ng/ml, Invitrogen) for 30 min, washed in PBS, and lysed in 1–2 ml IP buffer (150 mM NaCl, 50 mM Tris pH 7.5, 10% glycerol, 1% Triton X‐100, 2 mM EDTA) with protease and phosphatase inhibitors and 5 mM n‐ethylmaleimide (NEM). Samples were clarified by centrifugation at 17,000 × g for 15 min and added directly to 20 μl packed glutathione sepharose beads pre‐bound with 100 μg GST‐TUBE (Ubiquillin‐UBA4x) or GST‐UBA (Ubiquillin‐UBA1x) (Hjerpe et al, 2009). Beads were incubated on a rotating wheel at 4°C for at least 2 h, washed three times with IP buffer, and eluted with 2× SDS sample buffer.
To enrich for K63‐ and M1‐linked ubiquitin species, 20*106 THP‐1 cells were treated with doxycycline (200 ng/ml) for 5 h and stimulated with L18‐MDP (200 ng/ml, Invitrogen) for 30 min, washed with PBS, and lysed in 1–2 ml IP buffer, as above, supplemented with 6 M Urea (for anti‐K63‐linked ubiquitin pulldowns) or 8 M Urea (for anti M1‐linked ubiquitin pulldowns). Samples were clarified by centrifugation at 17,000 × g for 15 min and 4 μg of anti‐K63 or anti‐M1‐linked ubiquitin antibodies [Genentech; (Matsumoto et al, 2012; Newton et al, 2008)] were added, followed by incubation on a rotating wheel at 4°C for at least 2 h. Antibodies were precipitated with 10 μl of equilibrated protein G agarose (Thermo), washed three times in IP buffer without Urea, and eluted with 2× SDS sample buffer.
Two‐step enrichment of modified RIPK2
Twelve dishes of confluent FLAG‐RIPK2 BMDMs (equivalent of approximately 25*107 cells or 25 mg of total protein) were primed with IFNγ (5 ng/ml) overnight and fresh IFNγ was added the next morning for another 2 h before stimulation with MDP (10 μg/ml) for 30 min. Cells were harvested, washed in PBS, and lysed in 2 ml IP buffer per dish (150 mM NaCl, 50 mM Tris pH 7.5, 10% glycerol, 1% Triton X‐100, 2 mM EDTA; supplemented with protease and phosphatase inhibitors and 5 mM n‐ethylmaleimide (NEM)). Lysates were clarified by centrifugation at 17,000 × g for 15 min, and supernatants were directly added to 100 μl packed glutathione sepharose beads pre‐bound with 1 mg GST‐UBA. Beads were incubated on a rotating wheel at 4°C overnight, washed three times with IP buffer, and eluted twice with two volumes of IP buffer supplemented with 20 mM reduced glutathione (pHed to 7.5). Elutions were combined, diluted with an equal volume of IP buffer, and added to 50 μl of packed magnetic anti‐FLAG beads (M2, Sigma) and incubated on a rotating wheel at 4°C for 4 h. Beads were washed three times with IP buffer and eluted twice with two volumes of 3x‐FLAG peptide (1 mg/ml) in TBS pH 7.5.
BMDM diGly proteomics
For the two‐step protocol in BMDMs, eluted protein material from pulldowns of FLAG‐RIPK2 expressing BMDMs was subjected to tryptic digestion using the FASP method as previously described (Wisniewski et al, 2009). Peptides were lyophilized using CentriVap (Labconco) prior to reconstituting in 80 μl 0.1% FA/2% acetonitrile (ACN). Peptide mixtures were analyzed by nanoflow reversed‐phase liquid chromatography tandem mass spectrometry (LC‐MS/MS) on an M‐Class HPLC (Waters) coupled to a Q Exactive Orbitrap mass spectrometer (Thermo Fisher). Peptide mixtures were loaded in buffer A (0.1% formic acid, 2% acetonitrile, Milli‐Q water) and separated by reverse‐phase chromatography using C18 fused silica column (packed emitter, I.D. 75 μm, O.D. 360 μm × 25 cm length, IonOpticks, Australia) using flow rates and data‐dependent methods as previously described (Delconte et al, 2016; Kedzierski et al, 2017).
THP‐1 diGly proteomics
Frozen pellets from 50 × 106 WT THP‐1 cells were lysed in 1% sodium deoxycholate (SDC), 100 mM Tris–HCL pH 8.5, immediately boiled for 5 min at 95°C and sonication for 30 s (Branson Sonifierer). Protein concentrations were estimated by tryptophan assay. For protein reduction and alkylation, samples were incubated for 5 min at 45°C after addition of tris(2‐carboxyethyl)phosphine (TCEP) and 2‐chloroacetamide (CAA) to a final concentration of 10 mM and 40 mM, respectively. Samples were digested using trypsin (1:20 w/w, Sigma‐Aldrich) in combination with LysC (1/100 w/w, Wako) at 37°C overnight. Protease activity was quenched by addition of four‐sample volumes 1% trifluoroacetic acid (TFA) in isopropanol. Quenched samples were loaded onto SDB‐RPS cartridges (Strata™‐X‐C, 30 mg/3 ml, Phenomenex Inc), pre‐equilibrated with 4 ml 30% methanol (MeOH)/1% TFA, and washed with 4 ml 0.2% TFA. After two washes with 4 ml 1%TFA in isopropanol and 1 wash with 0.2% TFA/2% acetonitrile (can), samples were eluted twice with 2 ml 1.25% ammonium hydroxide(NH4OH)/80% ACN. Eluted samples were diluted with ddH2O to a final ACN concentration of 35%, snap frozen, and dried by lyophilization.
Lyophilized peptides were reconstituted in IAP buffer (50 mM MOPS, pH 7.2, 10 mM Na2HPO4, 50 mM NaCl), and the peptide concentration was estimated by tryptophan assay. For proteome analysis, 10 μg of peptide material was taken and desalted on SDB‐RPS StageTips (Empore) (Kulak et al, 2014). Peptides were diluted to a final volume of 200 μl with 0.2% TFA and loaded onto StageTips, followed by a wash with 200 μl of 0.2% TFA and 200 μl of 0.2%TFA/2%ACN, respectively. Captured peptides were eluted with 60 μl of 1.25% Ammonium hydroxide(NH4OH)/80% ACN and dried using a SpeedVac centrifuge (Eppendorf, Concentrator plus). Dried peptides were resuspended in buffer A* (2% ACN/0.1% TFA).
K‐ɛ‐GG remnant containing peptides were enriched using the PTMScan® Ubiquitin Remnant Motif (K‐ɛ‐GG) Kit (Cell Signaling Technology). Crosslinking of antibodies to beads and subsequent immunopurification was performed with slight modifications as previously described (Udeshi et al, 2013). Briefly, cross‐linked beads were split equally into eight tubes (~ 31 μg of antibody per tube), gently mixed with 1 mg peptide material (1 mg/ml) and incubated for 1 h at 4°C. Beads were washed twice with cold IAP and five times with cold ddH2O, and peptides were eluted twice with 50 μl 0.15% TFA. Eluted peptides were desalted and dried as described above and resuspended in 5 μl buffer A* for LC/MS‐MS analysis.
For the THP‐1 diGly‐enrichment analysis, samples were loaded onto a 50 cm reversed‐phase column [75 μm inner diameter, packed in house with ReproSil‐Pur C18‐AQ 1.9 μm resin (Dr. Maisch GmbH)]. The column temperature was maintained at 60°C using a homemade column oven. Peptides were separated with a binary buffer system of buffer A (0.1% formic acid (FA)) and buffer B (80% acetonitrile plus 0.1% FA), at a flow rate of 300 nl/min. Nano flow Liquid chromatography was performed with an EASY‐nLC 1200 system (Thermo Fisher Scientific), which was directly coupled online with the mass spectrometer (Q Exactive HF‐X, Thermo Fisher Scientific) via a nano‐electrospray source. For proteome measurements, 500 ng were loaded and eluted with a gradient starting at 5% buffer B and stepwise increased to 30% in 95 min, 60% in 5 min, and 95% in 5 min. The mass spectrometer was operated in Top15 data‐dependent mode (DDA) with a full scan range of 300–1,650 m/z at 60,000 resolution with an automatic gain control (AGC) target of 3e6 and a maximum fill time of 20 ms. Precursor ions were isolated with a width of 1.4 m/z and fragmented by higher‐energy collisional dissociation (HCD) (NCE 27%). Fragment scans were performed at a resolution of 15,000, an AGC of 1e5, and a maximum injection time of 28 ms. Dynamic exclusion was enabled and set to 30 s.
For K‐ɛ‐GG peptide samples, 2 μl were loaded and eluted with a gradient starting at 3% buffer B and stepwise increased to 7% in 6 min, 20% in 49 min, 36% in 39 min, 45% in 10 min, and 95% in 4 min. The mass spectrometer was operated in Top12 data‐dependent mode (DDA) with a full scan range of 250–1,350 m/z at 60,000 resolution with an automatic gain control (AGC) target of 3e6 and a maximum fill time of 20 ms. Precursor ions were isolated with a width of 1.4 m/z and fragmented by higher‐energy collisional dissociation (HCD) (NCE 28%). Fragment scans were performed at a resolution of 30,000, an AGC of 1e5 and a maximum injection time of 110 ms. Dynamic exclusion was enabled and set to 15 s.
MS data processing
For BMDM data sets, raw files consisting of high‐resolution MS/MS spectra were processed with MaxQuant (version 1.5.8.3) for feature detection and protein identification using the Andromeda search engine (Cox et al, 2011). Extracted peak lists were searched against the UniProtKB/Swiss‐Prot Mus musculus database (October 2016) and a separate reverse decoy database to empirically assess the false discovery rate (FDR) using strict trypsin specificity allowing up to two missed cleavages. The minimum required peptide length was set to seven amino acids. The mass tolerance for precursor ions and fragment ions were 20 ppm and 0.5 Da, respectively. The search included variable modifications of oxidation (methionine), amino‐terminal acetylation, carbamidomethyl (cysteine), GlyGly or ubiquitination (lysine), phosphorylation (serine, threonine, or tyrosine), and N‐ethylmaleimide (cysteine). Raw MS data were also searched with PEAKS, version 8 (Bioinformatics Solutions) using a Swiss‐Prot Human database and the same variable and fixed modifications as described above. A 0.1% and 1% FDR cutoff were applied at the PSM and peptide/protein levels, respectively.
Raw MS data from THP‐1 cells were searched against the UniProt Human FASTA (21,051 sequences) using MaxQuant (version 1.6.2.10) with a 1% FDR at peptide and protein levels. The match and alignment time window for the match between run (MBR) algorithm were set to 0.7 min and 20 min, respectively. A ratio count of two was used for the MaxLFQ algorithm. Cysteine carbamidomethylation was defined as fixed, protein N‐terminal acetylation and methionine oxidation as variable modification. In case of K‐ɛ‐GG samples, “GlyGly (K)” was additionally selected as variable modifications. Enzyme specificity was set to trypsin and two missed cleavages were allowed, while permitting a maximum of five modifications per peptide.
Molecular modeling
The effects of K209R, I212D, and I212A mutations on the structural integrity were modeled using the DynaMut software (Rodrigues et al, 2018) using the kinase domain of RIPK2 (PDB: 4C8B) as the input.
Recombinant protein purification
pGEX‐6 P‐1 or pGEX‐6 P‐3 plasmids encoding XIAP‐BIR2, Ubiquillin‐UBA1x (UBA), or Ubiquillin‐UBA4x (TUBE; Hjerpe et al, 2009) were transformed into BL21 (DE3þ) bacteria and grown in Super broth overnight at 37°C. Overnight culture was diluted 1:10 and grown until OD595 was 0.8. Isopropylthiogalactoside (IPTG) (0.3 mM) was added for 4 h at 30°C. Cells were pelleted and resuspended in Buffer A (50 mM Tris pH 8.0, 50 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol, all from SIGMA) and sonicated. After centrifugation at 21,000 g for 30 min, the supernatant was incubated with glutathione sepharose4B (GE Healthcare) for 4 h, washed five times with Buffer A, and eluted 2 × 45 min with 10 mM reduced glutathione in Buffer A at 4°C.
Generation of doxycycline‐inducible cell lines
Sequences of full length hsRIPK2 with an N‐terminal 3x‐Flag tag were synthesized by Genscript and cloned into doxycycline‐inducible lentiviral expression vectors (pF_TRE3G_rtTAAd_puro (Takara Bio)). For lentiviral transfections, 2.5 μg of the plasmid of choice was transfected into HEK293T cells together with 1 μg pVSV‐G and 1.5 μg pCMVΔR8.2 using an Effectene transfection kit (Qiagen). Twenty‐four hours after transfection, the media was changed and virus was harvested after another 24 h. Media was filtered and supplemented with polybrene (4 μg/ml). Viral media was then applied to cell lines, centrifuged for 45 min at 1,000 g at 30°C. After 2 days of incubation, cells were selected using 2.5 μg/ml puromycin (Sigma).
Thermal shift assay
5*106 THP‐1 cells were treated with doxycycline (200 ng/ml, Sigma) for 5 h, washed in PBS, and resuspended in PBS supplemented with protease inhibitors. Cell suspensions were transferred into PCR tubes and incubated for 3 min at a temperature gradient (37–60°C) in a PCR machine. Samples were cooled to room temperature, lysed by repeated freeze‐thawing, and centrifuged at 20,000 g for 20 min at 4°C. Supernatants were harvested and 2× SDS sample buffer was added, before analysis by Western blot.
XIAP‐BIR2 binding assay
20*106 THP‐1 cells were treated with doxycycline (200 ng/ml, Sigma) for 5 h, washed in PBS, and lysed in 1–2 ml IP buffer (150 mM NaCl, 50 mM Tris pH 7.5, 10% glycerol, 1% Triton X‐100, 2 mM EDTA) with cOmplete protease inhibitor cocktail, Roche, and phosphatase inhibitors (5 mM β‐Glycerophosphate, 1 mM Sodium molybdate, 2 mM Sodium pyrophosphate, 10 mM Sodium fluoride) and 5 mM n‐ethylmaleimide (NEM, Sigma). Lysates were clarified by centrifugation at 17,000 × g for 15 min and added directly to 20 μl packed glutathione sepharose beads pre‐bound with 100 μg GST‐XIAP‐BIR2. Beads were incubated on a rotating wheel at 4°C for at least 2 h, washed three times with IP buffer, and eluted with 2× SDS sample buffer.
Statistical analysis
The P values were calculated using two‐way ANOVA using Prism v.8 (GraphPad). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001; P values > 0.05 are indicated as not significant (ns). Normal distribution was confirmed using the D'Agostino–Pearson test (Fig 3B), respectively, the Shapiro–Wilk test (Fig 4D) using GraphPad PRISM software.
Author contributions
VJH, JS, AIW, and UN developed the concept for this work. VJH, LFD, CAS, FMH, EC, AB, and ISL performed experimental work. AIW oversaw the mass spectrometry work and ISL the biophysical characterization of generated RIPK2 mutants. VJH, JS, and UN wrote and revised the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Review Process File
Acknowledgements
We thank Prof. C. Day for the GST‐BIR2 constructs and C. Gatt, K.McKenzie, T. Ballinger, and C.Epifanio for assistance with animal work and the MAGEC facility of WEHI for the generation of affinity‐tagged RIPK2 mice. The work was supported by NHMRC grants 1046986, 1057888 and fellowships, 541901, 1058190 to JS and an ARC fellowship, FT130100166, to UN. This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS (#9000220). This project has received funding from the European Union's Framework Programme for Research and Innovation Horizon 2020 (2014–2020) under the Marie Skłodowska‐Curie Grant Agreement No. 754388 and from LMU Munich's Institutional Strategy LMUexcellent within the framework of the German Excellence Initiative (No. ZUK22).
EMBO Reports (2020) 21: e50400
Contributor Information
John Silke, Email: silke@wehi.edu.au.
Ueli Nachbur, Email: nachbur@wehi.edu.au.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Perez‐Riverol et al, 2019) with the dataset identifier PXD017741 (http://www.ebi.ac.uk/pride/archive/projects/PXD017741).
References
- Aebersold R, Mann M (2003) Mass spectrometry‐based proteomics. Nature 422: 198–207 [DOI] [PubMed] [Google Scholar]
- Alshareef A, Zhang HF, Huang YH, Wu C, Zhang JD, Wang P, El‐Sehemy A, Fares M, Lai R (2016) The use of cellular thermal shift assay (CETSA) to study Crizotinib resistance in ALK‐expressing human cancers. Sci Rep 6: 33710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermudez‐Humaran LG, Smirnova N, Berge M, Sulpice T, Lahtinen S et al (2011) Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med 3: 559–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand PK, Tait SW, Lamkanfi M, Amer AO, Nunez G, Pages G, Pouyssegur J, McGargill MA, Green DR, Kanneganti TD (2011) TLR2 and RIP2 pathways mediate autophagy of Listeria monocytogenes via extracellular signal‐regulated kinase (ERK) activation. J Biol Chem 286: 42981–42991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball KA, Johnson JR, Lewinski MK, Guatelli J, Verschueren E, Krogan NJ, Jacobson MP (2016) Non‐degradative ubiquitination of protein kinases. PLoS Comput Biol 12: e1004898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauler LD, Duckett CS, O'Riordan MX (2008) XIAP regulates cytosol‐specific innate immunity to Listeria infection. PLoS Pathog 4: e1000142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand MJM, Lippens S, Staes A, Gilbert B, Roelandt R, De Medts J, Gevaert K, Declercq W, Vandenabeele P (2011) cIAP1/2 are direct E3 ligases conjugating diverse types of ubiquitin chains to receptor interacting proteins kinases 1 to 4 (RIP1‐4). PLoS ONE 6: e22356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning P, Ruan Q, Schwerd T, Hrdinka M, Maki JL, Saleh D, Suebsuwong C, Ray S, Brennan PE, Cuny GD et al (2015) Inflammatory signaling by NOD‐RIPK2 is inhibited by clinically relevant type ii kinase inhibitors. Chem Biol 22: 1174–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso R, Warner N, Inohara N, Nunez G (2014) NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 41: 898–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallari JF, Fullerton MD, Duggan BM, Foley KP, Denou E, Smith BK, Desjardins EM, Henriksbo BD, Kim KJ, Tuinema BR et al (2017) Muramyl dipeptide‐based postbiotics mitigate obesity‐induced insulin resistance via IRF4. Cell Metab 25: 1063–1074 e1063 [DOI] [PubMed] [Google Scholar]
- Chin AI, Dempsey PW, Bruhn K, Miller JF, Xu Y, Cheng G (2002) Involvement of receptor‐interacting protein 2 in innate and adaptive immune responses. Nature 416: 190–194 [DOI] [PubMed] [Google Scholar]
- Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A (2010) NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16: 90–97 [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Creixell P, Schoof EM, Simpson CD, Longden J, Miller CJ, Lou HJ, Perryman L, Cox TR, Zivanovic N, Palmeri A et al (2015) Kinome‐wide decoding of network‐attacking mutations rewiring cancer signaling. Cell 163: 202–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard RB, Nachbur U, Yabal M, Wong WW‐L, Fiil BK, Kastirr M, Rieser E, Rickard JA, Bankovacki A, Peschel C et al (2012) The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell 46: 746–758 [DOI] [PubMed] [Google Scholar]
- Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, Stannard K, Zhang JG, Teh C, Firth M et al (2016) CIS is a potent checkpoint in NK cell‐mediated tumor immunity. Nat Immunol 17: 816–824 [DOI] [PubMed] [Google Scholar]
- Denou E, Lolmede K, Garidou L, Pomie C, Chabo C, Lau TC, Fullerton MD, Nigro G, Zakaroff‐Girard A, Luche E et al (2015) Defective NOD2 peptidoglycan sensing promotes diet‐induced inflammation, dysbiosis, and insulin resistance. EMBO Mol Med 7: 259–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsch M, Wang A, Cheng H, Lu C, Bielecki A, Charron K, Clauser K, Ren H, Polakiewicz RD, Parsons T et al (2006) Identification of a regulatory autophosphorylation site in the serine–threonine kinase RIP2. Cell Signal 18: 2223–2229 [DOI] [PubMed] [Google Scholar]
- Ellwanger K, Briese S, Arnold C, Kienes I, Heim V, Nachbur U, Kufer TA (2019) XIAP controls RIPK2 signaling by preventing its deposition in speck‐like structures. Life Sci Alliance 2: e201900346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekete T, Koncz G, Szabo B, Gregus A, Rajnavolgyi E (2017) Interferon gamma boosts the nucleotide oligomerization domain 2‐mediated signaling pathway in human dendritic cells in an X‐linked inhibitor of apoptosis protein and mammalian target of rapamycin‐dependent manner. Cell Mol Immunol 14: 380–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiil BK, Damgaard RB, Wagner SA, Keusekotten K, Fritsch M, Bekker‐Jensen S, Mailand N, Choudhary C, Komander D, Gyrd‐Hansen M (2013) OTULIN restricts Met1‐linked ubiquitination to control innate immune signaling. Mol Cell 50: 818–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, Tedin K, Taha MK, Labigne A, Zahringer U et al (2003a) Nod1 detects a unique muropeptide from gram‐negative bacterial peptidoglycan. Science 300: 1584–1587 [DOI] [PubMed] [Google Scholar]
- Girardin SE, Travassos LH, Herve M, Blanot D, Boneca IG, Philpott DJ, Sansonetti PJ, Mengin‐Lecreulx D (2003b) Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem 278: 41702–41708 [DOI] [PubMed] [Google Scholar]
- Goncharov T, Hedayati S, Mulvihill MM, Izrael‐Tomasevic A, Zobel K, Jeet S, Fedorova AV, Eidenschenk C, deVoss J, Yu K et al (2018) Disruption of XIAP‐RIP2 association blocks NOD2‐mediated inflammatory signaling. Mol Cell 69: 551–565.e557 [DOI] [PubMed] [Google Scholar]
- Gong Q, Long Z, Zhong FL, Teo DET, Jin Y, Yin Z, Boo ZZ, Zhang Y, Zhang J, Yang R et al (2018) Structural basis of RIP2 activation and signaling. Nat Commun 9: 4993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Núñez G, Inohara N (2008) A critical role of RICK/RIP2 polyubiquitination in Nod‐induced NF‐kappaB activation. EMBO J 27: 373–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim VJ, Stafford CA, Nachbur U (2019) NOD signaling and cell death. Front Cell Dev Biol 7: 208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe R, Aillet F, Lopitz‐Otsoa F, Lang V, England P, Rodriguez MS (2009) Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin‐binding entities. EMBO Rep 10: 1250–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C (2010) ATG16L1 and NOD2 interact in an autophagy‐dependent antibacterial pathway implicated in Crohn's disease pathogenesis. Gastroenterology 139: 1630–1641, 1641 e1631–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdinka M, Schlicher L, Dai B, Pinkas DM, Bufton JC, Picaud S, Ward JA, Rogers C, Suebsuwong C, Nikhar S et al (2018) Small molecule inhibitors reveal an indispensable scaffolding role of RIPK2 in NOD2 signaling. EMBO J 37: e99372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF, Ogura Y, Nunez G (2000) An induced proximity model for NF‐kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem 275: 27823–27831 [DOI] [PubMed] [Google Scholar]
- Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P, Martinez Molina D (2014) The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 9: 2100–2122 [DOI] [PubMed] [Google Scholar]
- Jeong YJ, Kang MJ, Lee SJ, Kim CH, Kim JC, Kim TH, Kim DJ, Kim D, Nunez G, Park JH (2014) Nod2 and Rip2 contribute to innate immune responses in mouse neutrophils. Immunology 143: 269–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedzierski L, Tate MD, Hsu AC, Kolesnik TB, Linossi EM, Dagley L, Dong Z, Freeman S, Infusini G, Starkey MR et al (2017) Suppressor of Cytokine Signaling (SOCS)5 ameliorates influenza infection via inhibition of EGFR signaling. Elife 6: e20444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig PA, Reinecke M, Ruprecht B, Petzoldt S, Meng C et al (2017) The target landscape of clinical kinase drugs. Science 358: eaan4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z, Knoefel WT, Reed JC (2009) XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA 106: 14524–14529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh AJ, Pal M, Tai L, Liao Y, Smyth GK, Shi W, Herold MJ (2017) An update on using CRISPR/Cas9 in the one‐cell stage mouse embryo for generating complex mutant alleles. Cell Death Differ 24: 1821–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M (2014) Minimal, encapsulated proteomic‐sample processing applied to copy‐number estimation in eukaryotic cells. Nat Methods 11: 319–324 [DOI] [PubMed] [Google Scholar]
- Lee JY, Hwang EH, Kim DJ, Oh SM, Lee KB, Shin SJ, Park JH (2016) The role of nucleotide‐binding oligomerization domain 1 during cytokine production by macrophages in response to Mycobacterium tuberculosis infection. Immunobiology 221: 70–75 [DOI] [PubMed] [Google Scholar]
- Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H et al (2013) Receptor interacting protein kinase 2‐mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol 14: 480–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharana J, Sahoo BR, Bej A, Jena I, Parida A, Sahoo JR, Dehury B, Patra MC, Martha SR, Balabantray S et al (2015) Structural models of zebrafish (Danio rerio) NOD1 and NOD2 NACHT domains suggest differential ATP binding orientations: insights from computational modeling, docking and molecular dynamics simulations. PLoS ONE 10: e0121415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P (2013) Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341: 84–87 [DOI] [PubMed] [Google Scholar]
- Martinez Molina D, Nordlund P (2016) The cellular thermal shift assay: a novel biophysical assay for in situ drug target engagement and mechanistic biomarker studies. Annu Rev Pharmacol Toxicol 56: 141–161 [DOI] [PubMed] [Google Scholar]
- Matsumoto ML, Dong KC, Yu C, Phu L, Gao X, Hannoush RN, Hymowitz SG, Kirkpatrick DS, Dixit VM, Kelley RF (2012) Engineering and structural characterization of a linear polyubiquitin‐specific antibody. J Mol Biol 418: 134–144 [DOI] [PubMed] [Google Scholar]
- McCarthy JV, Ni J, Dixit VM (1998) RIP2 is a novel NF‐kappa B‐activating and cell death‐inducing kinase. J Biol Chem 273: 16968–16975 [DOI] [PubMed] [Google Scholar]
- von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, Bork P (2002) Comparative assessment of large‐scale data sets of protein‐protein interactions. Nature 417: 399–403 [DOI] [PubMed] [Google Scholar]
- Miller MH, Shehat MG, Alcedo KP, Spinel LP, Soulakova J, Tigno‐Aranjuez JT (2018) Frontline Science: RIP2 promotes house dust mite‐induced allergic airway inflammation. J Leukoc Biol 104: 447–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachbur U, Stafford CA, Bankovacki A, Zhan Y, Lindqvist LM, Fiil BK, Khakham Y, Ko H‐J, Sandow JJ, Falk H et al (2015) A RIPK2 inhibitor delays NOD signalling events yet prevents inflammatory cytokine production. Nat Commun 6: 6442 [DOI] [PubMed] [Google Scholar]
- Nembrini C, Kisielow J, Shamshiev AT, Tortola L, Coyle AJ, Kopf M, Marsland BJ (2009) The kinase activity of Rip2 determines its stability and consequently Nod1‐and Nod2‐mediated immune responses. J Biol Chem 284: 19183–19188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA et al (2008) Ubiquitin chain editing revealed by polyubiquitin linkage‐specific antibodies. Cell 134: 668–678 [DOI] [PubMed] [Google Scholar]
- O'Donnell MA, Legarda‐Addison D, Skountzos P, Yeh WC, Ting AT (2007) Ubiquitination of RIP1 regulates an NF‐kappaB‐independent cell‐death switch in TNF signaling. Curr Biol 17: 418–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Núñez G (2001) Nod2, a Nod1/Apaf‐1 family member that is restricted to monocytes and activates NF‐κB. J Biol Chem 276: 4812–4818 [DOI] [PubMed] [Google Scholar]
- Panda S, Gekara NO (2018) The deubiquitinase MYSM1 dampens NOD2‐mediated inflammation and tissue damage by inactivating the RIP2 complex. Nat Commun 9: 4654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J‐H, Kim Y‐G, McDonald C, Kanneganti T‐D, Hasegawa M, Body‐Malapel M, Inohara N, Núñez G (2007) RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol 178: 2380–2386 [DOI] [PubMed] [Google Scholar]
- Pellegrini E, Desfosses A, Wallmann A, Schulze WM, Rehbein K, Mas P, Signor L, Gaudon S, Zenkeviciute G, Hons M et al (2018) RIP2 filament formation is required for NOD2 dependent NF‐kappaB signalling. Nat Commun 9: 4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ (2003) Context of multiubiquitin chain attachment influences the rate of Sic1 degradation. Mol Cell 11: 1435–1444 [DOI] [PubMed] [Google Scholar]
- Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE (2014) NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol 14: 9–23 [DOI] [PubMed] [Google Scholar]
- Rahman MA, Sundaram K, Mitra S, Gavrilin MA, Wewers MD (2014) Receptor interacting protein‐2 plays a critical role in human lung epithelial cells survival in response to Fas‐induced cell‐death. PLoS ONE 9: e92731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues CH, Pires DE, Ascher DB (2018) DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res 46: W350–W355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht B, Zecha J, Heinzlmeir S, Medard G, Lemeer S, Kuster B (2015) Evaluation of kinase activity profiling using chemical proteomics. ACS Chem Biol 10: 2743–2752 [DOI] [PubMed] [Google Scholar]
- Schertzer JD, Tamrakar AK, Magalhaes JG, Pereira S, Bilan PJ, Fullerton MD, Liu Z, Steinberg GR, Giacca A, Philpott DJ et al (2011) NOD1 activators link innate immunity to insulin resistance. Diabetes 60: 2206–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid‐Burgk JL, Schmidt T, Gaidt MM, Pelka K, Latz E, Ebert TS, Hornung V (2014) OutKnocker: a web tool for rapid and simple genotyping of designer nuclease edited cell lines. Genome Res 24: 1719–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seashore‐Ludlow B, Axelsson H, Almqvist H, Dahlgren B, Jonsson M, Lundback T (2018) Quantitative interpretation of intracellular drug binding and kinetics using the cellular thermal shift assay. Biochemistry 57: 6715–6725 [DOI] [PubMed] [Google Scholar]
- Slany A, Bileck A, Kreutz D, Mayer RL, Muqaku B, Gerner C (2016) Contribution of human fibroblasts and endothelial cells to the hallmarks of inflammation as determined by proteome profiling. Mol Cell Proteomics 15: 1982–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford CA, Lawlor KE, Heim VJ, Bankovacki A, Bernardini JP, Silke J, Nachbur U (2018) IAPs Regulate distinct innate immune pathways to co‐ordinate the response to bacterial peptidoglycans. Cell Rep 22: 1496–1508 [DOI] [PubMed] [Google Scholar]
- Thome M, Hofmann K, Burns K, Martinon F, Bodmer JL, Mattmann C, Tschopp J (1998) Identification of CARDIAK, a RIP‐like kinase that associates with caspase‐1. Curr Biol 8: 885–888 [DOI] [PubMed] [Google Scholar]
- Tigno‐Aranjuez JT, Asara JM, Abbott DW (2010) Inhibition of RIP2's tyrosine kinase activity limits NOD2‐driven cytokine responses. Genes Dev 24: 2666–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigno‐Aranjuez JT, Bai X, Abbott DW (2013) A discrete ubiquitin‐mediated network regulates the strength of NOD2 signaling. Mol Cell Biol 33: 146–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigno‐Aranjuez JT, Benderitter P, Rombouts F (2014) In vivo inhibition of RIPK2 kinase alleviates inflammatory disease. J Biol Chem 289: 29651–29664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udeshi ND, Mertins P, Svinkina T, Carr SA (2013) Large‐scale identification of ubiquitination sites by mass spectrometry. Nat Protoc 8: 1950–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen AM, Kratina TK, Hawkins CJ, Silke J, Ekert PG, Vaux DL (2007) Identification of mammalian mitochondrial proteins that interact with IAPs via N‐terminal IAP binding motifs. Cell Death Differ 14: 348–357 [DOI] [PubMed] [Google Scholar]
- Wagner SA, Satpathy S, Beli P, Choudhary C (2016) SPATA2 links CYLD to the TNF‐alpha receptor signaling complex and modulates the receptor signaling outcomes. EMBO J 35: 1868–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362 [DOI] [PubMed] [Google Scholar]
- Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y (2000) Structural basis of IAP recognition by Smac/DIABLO. Nature 408: 1008–1012 [DOI] [PubMed] [Google Scholar]
- Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP (2003) Structure of a beta‐TrCP1‐Skp1‐beta‐catenin complex: destruction motif binding and lysine specificity of the SCF(beta‐TrCP1) ubiquitin ligase. Mol Cell 11: 1445–1456 [DOI] [PubMed] [Google Scholar]
- Zou Y, Lei W, He Z, Li Z (2016) The role of NOD1 and NOD2 in host defense against chlamydial infection. FEMS Microbiol Lett 363: fnw170 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Review Process File
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Perez‐Riverol et al, 2019) with the dataset identifier PXD017741 (http://www.ebi.ac.uk/pride/archive/projects/PXD017741).