

Abstract
Lipid droplets (LDs) are dynamic cytoplasmic organelles present in most eukaryotic cells. The appearance of LDs in neurons is not usually observed under physiological conditions, but is associated with neural diseases. It remains unclear how LD dynamics is regulated in neurons and how the appearance of LDs affects neuronal functions. We discovered that mutations of two key lipolysis genes atgl‐1 and lid‐1 lead to LD appearance in neurons of Caenorhabditis elegans. This neuronal lipid accumulation protects neurons from hyperactivation‐triggered neurodegeneration, with a mild decrease in touch sensation. We also discovered that reduced biosynthesis of polyunsaturated fatty acids (PUFAs) causes similar effects and synergizes with decreased lipolysis. Furthermore, we demonstrated that these changes in lipolysis and PUFA biosynthesis increase PUFA partitioning toward triacylglycerol, and reduced incorporation of PUFAs into phospholipids increases neuronal protection. Together, these results suggest the crucial role of neuronal lipolysis in cell‐autonomous regulation of neural functions and neurodegeneration.
Keywords: Caenorhabditis elegans, lipid droplet, lipolysis, neurodegeneration, polyunsaturated fatty acid
Subject Categories: Membrane & Intracellular Transport, Neuroscience
This study reveals the role of neuronal lipolysis in cell‐autonomous regulation of neural functions and neurodegeneration.

Introduction
Lipid droplets (LDs) are dynamic cytoplasmic organelles which are present in most, if not all, eukaryotic cells and many prokaryotic cells. By storing excess lipids in the form of neutral lipids including triacylglycerol (TAG) and sterol ester (SE), LDs maintain cellular lipid homeostasis, along with the coordinated actions of lipogenesis and lipolysis (Chen et al, 2019; Olzmann & Carvalho, 2019). The nervous system, including neurons and glia, has a high concentration of lipids. However, LDs in the nervous system are generally found in glial cells but not in neurons under normal conditions in vivo (Kis et al, 2015).
The origin and the role of glial LDs have been investigated only in recent years. The formation of LDs in glia, which act as a niche for neuroblasts, preserves Drosophila larval neuroblast proliferation under ROS‐inducing stress conditions such as hypoxia. It is postulated that the incorporation of polyunsaturated fatty acids (PUFAs) into neutral lipids, and their storage in LDs, reduces the ROS insult and the toxic peroxidation of PUFAs in neuroblasts (Bailey et al, 2015). Several other studies reported the neuronal origin of glial LDs. When Drosophila neurons are under ROS insult or have mitochondrial dysfunction, neuronal lipid production is increased through SREBP‐mediated lipogenesis. Interestingly, instead of forming LDs in neurons, lipids are transferred to neighboring glia through fatty acid transfer protein (FATP) or apolipoprotein to form LDs (Liu et al, 2015, 2017). In cultured hippocampal neurons, hyperactivated neurons also produce excess fatty acids, which are transferred, via lipid particles associated with ApoE, to astrocytes and are incorporated into LDs. The storage of fatty acids in astrocyte LDs and their subsequent β‐oxidation in mitochondria protects neurons during periods of enhanced activity (Ioannou et al, 2019). These findings suggest that LD formation in glia plays a role in protecting neurons from stress insults. However, it is unknown why neurons do not form LDs in an autonomous fashion to protect themselves under stress conditions.
Although neurons do not normally have LDs, some neuronal diseases are associated with LD biology and neuronal LDs have been reported in some disease models. The Parkinson's disease protein α‐Synuclein is located on the surface of LDs in lipid‐loaded primary hippocampal neurons and α‐Synuclein expression is correlated with LD accumulation in yeast (Cole et al, 2002; Outeiro & Lindquist, 2003). Huntington's disease cells, including primary striatal neurons and glia in Huntington's disease mice, have dramatically increased LDs (Martinez‐Vicente et al, 2010). Several hereditary spastic paraplegia (HSP) proteins affect LD dynamics, such as Spartin, spastin, atlastin‐1, seipin, and REEP1 (Eastman et al, 2009; Klemm et al, 2013; Ebihara et al, 2015; Papadopoulos et al, 2015; Renvoise et al, 2016; Ding et al, 2018). Despite the apparent association between LD and neuronal diseases, the causal link between neuronal LD dynamics and neuronal disorders remains largely elusive.
In this study, we explored the dynamics and the physiological role of LDs in neurons. Using Caenorhabditis elegans as a model, we found that ATGL‐1/LID‐1‐mediated lipolysis autonomously regulates neuronal LD dynamics. ATGL‐1 is the C. elegans homolog of mammalian ATGL, the rate‐limiting enzyme of TAG hydrolysis, and LID‐1 is the C. elegans homolog of mammalian CGI‐58 (also named as ABHD5), which is the best known co‐activator of ATGL (Lass et al, 2006). Mutations in either CGI‐58 or ATGL lead to neutral‐lipid storage disease (NLSD) with neurological abnormalities in human (Schweiger et al, 2009; Massa et al, 2016). Importantly, defective neuronal lipolysis reduces PUFA‐mediated touch sensation and protects neurons from hyperactivation‐triggered neurodegeneration. The neuronal protective effect of atgl‐1 mutants is significantly enhanced by reduction of de novo PUFA synthesis and/or incorporation of PUFAs into phospholipids. Together, our results show that neuronal LDs participate in PUFA‐mediated neural functions and neurodegeneration.
Results
LDs accumulate in neurons of both atgl‐1 and lid‐1 mutants
To observe LDs in worm neurons, we generated a neuron‐specific GFP reporter line xdIs109[P unc‐119 ::PLIN1::GFP/rol-6] expressing the LD surface protein PLIN1. PLIN1::GFP forms ring‐like structures when expressed in tissues with LDs (Bi et al, 2012; Liu et al, 2014). In xdIs109 animals, we mainly focused on the head region, which is the location of most neuronal soma. We found that there are very few GFP rings in xdIs109 young adults (Fig 1A). This indicates that similar to mammals, there are few LDs in C. elegans neurons under normal conditions. It has been reported that both aging and general obesity may increase LD accumulation in non‐adipose tissues (Zhou et al, 2000; Shimabukuro et al, 2016; Palikaras et al, 2017). To explore the influence of aging and overall fat content increase on neuronal LDs, we examined the xdIs109 GFP pattern in 8‐day‐old wild‐type adults, daf‐2(e1370) mutants and glp‐1(e2141) mutants. It is well known that the latter two mutants have an overall increase in lipid storage (O'Rourke et al, 2009). We found that compared with young xdIs109 controls, there is no significant increase of LD number in 8‐day‐old xdIs109, daf‐2(e1370); xdIs109 and glp‐1(e2141); and xdIs109 (Fig 1B). This indicates that aging and general obesity do not necessarily result in neuronal LD accumulation.
Figure 1. lid‐1(xd288) and atgl‐1(xd314) display ectopic LD accumulation in neurons.

-
Alid‐1(xd288) and atgl‐1(xd314) mutants show LD accumulation in most neurons in Caenorhabditis elegans, including neurons in the head region, tail, and ventral nerve cord. xdIs109 is a stable transgenic line with pan‐neuronal expression of the LD marker PLIN1::GFP. The white arrows indicate PLIN1::GFP‐positive LDs in the ventral nerve cord of mutants. Scale bar: 20 μm. The right panels depict the head region. Scale bar: 5 μm. The insets in the right panels are enlarged views of PLIN1::GFP rings, representing LDs.
-
BQuantification of the numbers of LDs in head region neurons (each dot represents one worm). The data were analyzed using one‐way ANOVA with Dunnett's multiple comparison test. Asterisks denote significant differences as compared to the control xdIs109. **P < 0.01, ***P < 0.001, ****P < 0.0001. ns: not statistically significant. Data show mean ± SEM. n ≥ 9.
-
C, DThe genetic loci of lid‐1 and atgl‐1. The coding regions are in light blue boxes, and the noncoding regions are shown as lines. The UTRs are in red boxes. xd288 is a point mutant of lid‐1 and causes a missense S111L mutation in the α/β hydrolase domain. xd314 and xd310 are point mutants of atgl‐1. xd314 harbors a missense G19R mutation, and xd310 harbors a missense G210E mutation.
-
EOil red O staining show dramatically increased neutral lipids in the head region of lid‐1(xd288) and atgl‐1(xd314) compared with N2. Scale bar: 10 μm.
-
F–HEM images of neurons. The red dashed line marks the outline of neurons. The red arrows mark LDs. Scale bar: 0.5 μm. The scale bar in enlarged images is 100 nm.
-
ISerial sections of atgl‐1(xd314) show two LDs (indicated by blue and red arrows) in a neuron cell body. Scale bar: 0.5 μm.
To reveal the underlying mechanism(s) that control LD dynamics in neurons, we performed an EMS screen using the xdIs109 marker to search for mutants with neuronal LD accumulation. We isolated the xd288, xd310, and xd314 mutants, which show LDs in many neurons in the head region, ventral nerve cord and tail (Fig 1A and Appendix Fig S1). We also quantified LDs in the head region and found that the number of LDs is increased significantly in these mutants (Fig 1B).
Complementation tests divided these three mutants into two complementation groups. xd288 complements both xd310 and xd314, while xd310 fails to complement xd314. Through SNP mapping, we narrowed down the region of the xd288 mutation to chromosome I between +3.85 cM and +5.05 cM. Through fosmid transgenic rescue assays, we found that the WRM0632aE01 fosmid fully rescues the neuronal LD phenotype of xd288. The gene lid‐1 (lipid droplet protein 1), which encodes one of the C. elegans homologs of mammalian CGI‐58, is found in this fosmid (Lee et al, 2014). Importantly, we found a C332T point mutation in lid‐1, and this mutation leads to a missense S111L mutation of LID‐1 (Fig 1C). S111 is located in the putative α/β hydrolase domain of LID‐1 and is conserved in C. elegans, Drosophila, mouse, and human (Appendix Fig S2). Interestingly, S111 of LID‐1 corresponds to S115 in human, which is mutated in NLSD (Ben Selma et al, 2007).
A previous report showed that LID‐1 binds to the C. elegans ATGL homolog, ATGL‐1, and promotes ATGL‐1‐dependent lipolysis during fasting conditions (Lee et al, 2014). As expected, we found that xd310 and xd314 are mutants of C. elegans atgl‐1. The xd310 mutation causes a missense G210E change, while xd314 causes a missense G19R change in ATGL‐1 (Fig 1D). G19 of ATGL‐1 is conserved in C. elegans, Drosophila, mouse, and human (Appendix Fig S2B). The G19R and G210E mutations are located in an active (α/β/α) sandwich domain of ATGL‐1, which is responsible for its enzymatic activity. Oil red O staining shows that both lid‐1(xd288) and atgl‐1(xd314) have more fat than controls, especially in the head region (Fig 1E) and intestine (Appendix Fig S2). This suggests that, just like in NLSD, the overall fat content is increased in lid‐1 and atgl‐1 mutants. The recessive nature of the xd288, xd310, and xd314 mutations and the lipolytic function of ATGL and CGI‐58 suggest that defective lipolysis leads to LD accumulation in neurons.
These mutants were isolated based on the appearance of neuronal LDs in the xdIs109 background. To avoid potential interference from the overexpression of PLIN1::GFP, we also used BODIPY, a neutral‐lipid dye, combined with a pan‐neuron marker P rab‐3 ::mCherry to observe LDs in the ventral nerve cord of lid‐1(xd288) and atgl‐1(xd314) mutants that do not carry the xdIs109 marker. BODIPY‐positive LDs are found in neurons of lid‐1(xd288) and atgl‐1(xd314) but not N2 control (Appendix Fig S3). In addition, we used electron microscopy (EM) to observe whether there are LDs in lid‐1(xd288) and atgl‐1(xd314) neurons without the xdIs109 marker. LDs were easily found in the neuronal soma of xd288 and xd314 mutants, but not in wild type (Fig 1F–H). We could even trace whole LDs in mutants from continuous EM serial sections (Fig 1I). This demonstrates that atgl‐1(xd314) and lid‐1(xd288) mutants indeed have ectopic LDs in neurons.
The above results indicate that ATGL‐1/LID‐1‐mediated lipolysis prevents the appearance of visible LDs in neurons. In contrast to lipolysis, lipogenesis promotes lipid storage. We wondered whether overexpression of lipogenesis‐related genes in neurons could cause LD accumulation. DGAT1 and DGAT2 are key enzymes in the synthesis of TAG from diacylglycerol (DAG) (Fig 2A). DGAT1 has only one homolog, MBOA‐2, in C. elegans. DGAT2 has four homologs (DGAT‐2/F59A1.10, K07B1.4, DGTR‐1/W01A11.2, and Y53G8B.2) in C. elegans (Fig 2B). We pan‐neuronally overexpressed mboa‐2, F59A1.10, and K07B1.4, respectively. In these transgenic animals, there are ectopic LDs in neurons as revealed by the xdIs109 reporter (Fig 2C). BODIPY staining also confirms the presence of LDs in mboa‐2 or F59A1.10 overexpressing neurons labeled by P rab‐3 ::mCherry marker (Appendix Fig S3). Therefore, either elevating lipogenesis or reducing lipolysis leads to LD accumulation in neurons. Together, these results indicate that neurons have the ability to form and hydrolyze LDs. The lack of visible LDs in neurons reflects the dominance of lipolysis over lipogenesis under normal conditions.
Figure 2. LID‐1/ATGL‐1‐mediated lipolysis and DAGT1/2‐mediated lipogenesis regulate LD dynamics in neurons.
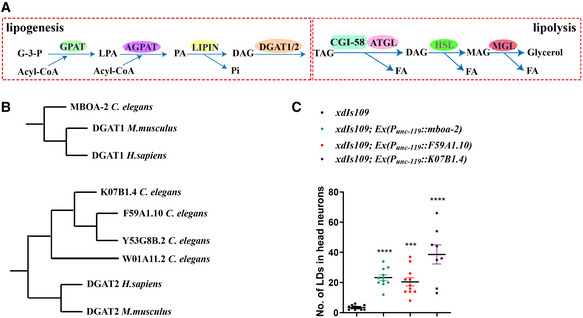
- The pathways of lipolysis and lipogenesis. G‐3-P: glycerol‐3-phosphate. LPA: lysophosphatidic acid. PA: phosphatidic acid. DAG: diacylglycerol. TAG: triacylglycerol. MAG: monoacylglycerol. FA: fatty acid.
- The evolutionary phylogenetic trees of DGAT1 and DGAT2.
- Overexpressing the homologs of DGAT1/2 in neurons of Caenorhabditis elegans leads to LD accumulation in neurons. Each dot represents one worm. The data were analyzed using one‐way ANOVA with Dunnett's multiple comparison test. Asterisks denote significant differences as compared to the control xdIs109. ***P < 0.001, ****P < 0.0001. Data show mean ± SEM. n ≥ 8.
atgl‐1 autonomously regulates neuronal LDs
ATGL‐1 is expressed strongly in worm intestine (Lee et al, 2014), but its expression in neurons has not been reported in detail. To observe the expression pattern of ATGL‐1, we first mapped the promotor of atgl‐1. The excess neuronal LD phenotype of atgl‐1(xd314) was fully rescued by several overlapping atgl‐1(+) fosmids. The overlapping region of the rescuing fosmids highlights a 3.4 Kb promoter region which may be important for the rescuing activity. We further found that an atgl‐1 genomic fragment with a 3 Kb promoter fully rescues the neuronal LD phenotype of atgl‐1(xd314) (Appendix Fig S4). Using this 3 Kb promoter region to drive a GFP reporter, we examined the expression pattern of atgl‐1 in detail. The GFP fluorescence is bright in the intestine, consistent with a previous report (Lee et al, 2014). We also found weak expression in neurons in the nerve ring, ventral nerve cord, and tail (Fig 3A).
Figure 3. atgl‐1 autonomously regulates neuronal LD dynamics.

- The expression pattern of atgl‐1 viewed by P atgl‐1 ::GFP transcriptional fusion reporter. P atgl‐1 ::GFP expression is high in intestine. Relatively low fluorescent signals are found in nerve ring, motor neuron commissure, ventral nerve cord, and tail. Scale bar: 20 μm.
- atgl‐1 is expressed in neurons. There is no fluorescent signal when P atgl‐1 ::GFP1‐10 and P rab‐3 ::GFP11 are expressed separately. When P atgl‐1 ::GFP1‐10 and P rab‐3 ::GFP11 are co‐expressed, the GFP signals are found in the head region, tail, and ventral nerve cord, where most neuronal cell bodies are located. The white lines mark the outline of worms. Scale bar: 50 μm.
- atgl‐1 is expressed widely in neurons. The left panels depict the whole worm. Scale bar: 50 μm. The insets in the right panels are enlarged views of the ventral nerve cord in the white dotted boxes of left panels. The signals of mCherry and GFP merge very well. Scale bar: 20 μm.
- Visualizing LDs in head neurons with the xdIs109 reporter in different genetic backgrounds. The accumulation of LDs in head neurons of atgl‐1(xd314) mutants can be rescued by expressing wild‐type atgl‐1 in neurons (P unc‐119 ::atgl‐1), but not in intestine (P vha‐6 ::atgl‐1), glia (P hlh‐17 ::atgl‐1), or hypodermis (P ajm‐1 ::atgl‐1). Scale bar: 5 μm.
- Quantification of the number of LDs in head neurons in different genetic backgrounds. Each dot represents one worm. The data were analyzed using one‐way ANOVA with Dunnett's multiple comparison test. Asterisks denote significant differences as compared to the control xdIs109. ns: not statistically significant. ****P < 0.0001. Data show mean ± SEM. n ≥ 7.
- Neuron‐specific knockout of atgl‐1 causes LD accumulation in neurons, similar to atgl‐1(xd314) mutants. Scale bar: 5 μm.
- Quantification of the number of LDs in head neurons in different genetic backgrounds. Each dot represents one worm. The data were analyzed using one‐way ANOVA with Dunnett's multiple comparison test. Asterisks denote significant differences as compared to the control xdIs109. t‐Test was used between the groups under the crossbars. ns: not statistically significant. ****P < 0.0001. Data show mean ± SEM. n ≥ 9.
To specifically analyze ATGL‐1 expression in neurons, we used the self‐complementing split GFP system. Split GFP is composed of two separate GFP fragments (a 15‐amino acid fragment, GFP11, and a 215‐amino acid fragment, GFP1‐10), which are expressed separately and are able to associate spontaneously to form fluorescent GFP (Cabantous et al, 2005). As controls, transgenic animals expressing either P atgl‐1 ::GFP1‐10 or pan‐neuronal P rab‐3 ::GFP11 do not exhibit GFP fluorescence (Fig 3B). The GFP fluorescence is clearly seen in neurons marked by pan‐neuronal P rab‐3 ::mCherry, including head region, ventral nerve cord, and tail neurons, in animals carrying both P atgl‐1 ::GFP1‐10 and P rab‐3 ::GFP11 (Fig 3B and C). This result provides further evidence for the neuronal expression of ATGL‐1.
To investigate whether atgl‐1 functions autonomously or non‐autonomously, we performed tissue‐specific rescue and tissue‐specific knockout experiments. We found that neuronal P unc‐119 ::atgl‐1 expression, but not glial P hlh‐17 ::atgl‐1, hypodermal P ajm‐1 ::atgl‐1 or intestinal P vha‐6 ::atgl‐1 expression, rescues the neuronal LD phenotype of atgl‐1(xd314); xdIs109 (Fig 3D and E). We also generated a neuron‐specific knockout of atgl‐1 using the Cre‐loxP system. We first used CRISPR‐Cas9 to insert two loxP sites flanking the atgl‐1 coding region, and then we expressed Cre in neurons to specifically knock out atgl‐1 in neurons (Appendix Fig S4). We found that similar to atgl‐1(xd314) mutants, neuronal‐specific deletion of atgl‐1 [atgl‐1-loxP(xd426); xdIs182 (P rab‐3 ::Cre/P odr‐1 ::RFP)] causes LD accumulation in neurons (Fig 3F and G). Together, these results demonstrate that ATGL‐1 acts autonomously in neurons to prevent neuronal LD accumulation.
Paralogs of ATGL‐1 and LID‐1 do not affect neuronal LD dynamics
There are two LID‐1 paralogs (C37H5.2 and C37H5.3) and two ATGL‐1 paralogs (B0524.2 and D1054.1) in C. elegans (Fig EV1A and B). Although LID‐1 is reported to regulate lipolysis by interacting with ATGL‐1 (Lee et al, 2014), C37H5.3, also named as cgi‐58, had been shown to promote ATGL‐1 activity at the dauer stage when the protein stability of ATGL‐1 is negatively regulated via phosphorylation by AMPK (Narbonne & Roy, 2009; Xie & Roy, 2015a,b). To test whether ATGL‐1 and LID‐1 paralogs affect LD homeostasis in neurons, we examined the neuronal LD phenotypes of C37H5.2(ok3722), C37H5.3(ok3245), D1054.1(tm3111), and B0524.2(tm6739) mutants with the xdIs109 marker. Similar to wild type, there are almost no LDs in neurons in these mutants (Fig EV1C). These mutants are all deletion alleles (Fig EV1D), and even though we are not sure whether they are null alleles, their phenotypes indicate that the ATGL‐1/LID‐1 pair plays the main role in regulating neuronal lipolysis and neuronal LD dynamics.
Figure EV1. The paralogs of ATGL‐1 and LID‐1 do not affect neuronal LD dynamics.
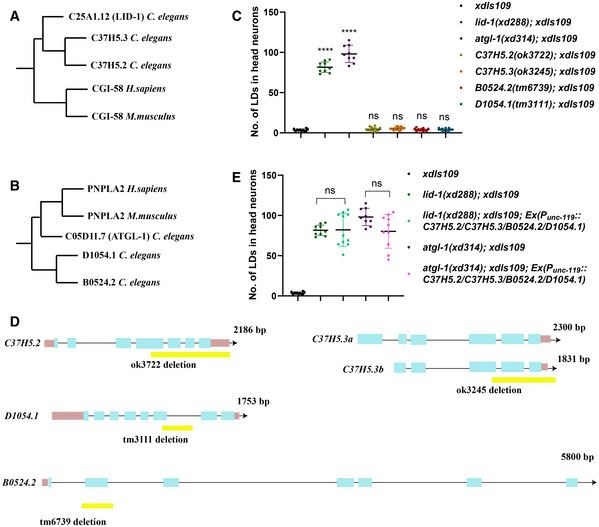
-
A, BThe evolutionary phylogenetic trees of LID‐1 and ATGL‐1.
-
CMutations in genes encoding LID‐1 or ATGL‐1 paralogs do not affect LD dynamics in neurons. Each dot represents one worm. The data were analyzed using one‐way ANOVA with Dunnett's multiple comparison test. Asterisks denote significant differences as compared to the control xdIs109. ns: not statistically significant. ****P < 0.0001. Data show mean ± SEM. n ≥ 9.
-
DSchematic diagrams of alleles of LID‐1/ATGL‐1 paralogs. The coding regions are in blue boxes and the noncoding regions are shown as lines. The UTRs are in pink boxes. The yellow boxes show the regions deleted in the corresponding alleles.
-
ENeuronal expression of LID‐1 and ATGL‐1 paralogs does not rescue lid‐1(xd288) or atgl‐1(xd314). Each dot represents one worm. The data were analyzed using one‐way ANOVA with Tamhane's T2 multiple comparison test. ns: not statistically significant. Data show mean ± SEM. n ≥ 9.
Next, we explored whether these paralogs can substitute for ATGL‐1 or LID‐1 in neurons. We overexpressed these paralogs in neurons specifically to examine whether they could rescue the neuronal LD phenotype of atgl‐1(xd314); xdIs109 or lid‐1(xd288); xdIs109. Because these paralogs may function in pairs, like ATGL‐1 and LID‐1, we overexpressed all four paralogs together in neurons. We found that they could not rescue the neuronal LD phenotype of either atgl‐1(xd314); xdIs109 or lid‐1(xd288); xdIs109 (Fig EV1E). Together, these results indicate the importance and the specificity of the ATGL‐1/LID‐1 partnership in regulating neuronal LD dynamics.
atgl‐1 and lid‐1 affect gentle touch sensation and genetically interact with the de novo PUFA biosynthesis pathway
The above results show that lipolysis restricts the appearance of visible LDs in neurons. We next examined the neuronal consequence of defective lipolysis. LDs are temporary storage places for excess lipids. Since lipids are important building blocks for membranes, we initially focused on examining whether atgl‐1 affects neuronal morphology. The PVD neuron has very complex neurites, especially dendrites, which may require lots of lipids for membrane biogenesis. We did not find any morphological difference between wild type and atgl‐1(xd314) or lid‐1(xd288) mutants based on observations with the PVD marker wdIs52(P F49H12.4 ::GFP) (Appendix Fig S5). This suggested that the appearance of LDs may not affect the gross morphology of neurons.
PUFAs affect the gentle touch sensation and mechanoelectrical transduction in C. elegans touch receptor neurons (Kahn‐Kirby et al, 2004; Vasquez et al, 2014). A previous report showed that when PUFAs are diverted away from membranes to the core of LDs, the expansion of LDs inhibits the oxidation of PUFAs and reduces the toxic effect of PUFAs (Bailey et al, 2015). Therefore, the presence of LDs in neurons may limit the availability of PUFAs and affect PUFA‐associated neuronal events. There are ectopic LDs in touch neurons in atgl‐1(xd314) as revealed by non‐invasive Stimulated Raman Scattering (SRS) microscopy (Fig 4A). To examine whether the gentle touch sensation is affected in atgl‐1(xd314) and lid‐1(xd288) mutants, we measured the touch sensitivity by a ten‐trial touch assay (Hart, 2006). mec‐4(u253) and N2 were used as the positive control and negative control, respectively. Compared with the strong touch sensation defect in mec‐4(u253) mutants, touch sensation is partially impaired in atgl‐1(xd314) and lid‐1(xd288) mutants. Importantly, expression of neuronal‐specific P unc‐119 ::atgl‐1 and P unc‐119 ::lid‐1 fully rescued the touch sensation defect in atgl‐1(xd314) and lid‐1(xd288), respectively (Fig 4B). This suggests that neuronal lipolysis is required for normal gentle touch sensation.
Figure 4. atgl‐1(xd314) and lid‐1(xd288) show a gentle touch sensation defect.

- LDs accumulate in touch neurons in atgl‐1(xd314) visualized by SRS (Stimulated Raman Scattering). The zdIs5 reporter marks touch neurons. The white arrows indicate LDs in touch neurons in atgl‐1(xd314). Scale bar: 10 μm.
- atgl‐1(xd314) and lid‐1(xd288) show a gentle touch sensation defect, which is rescued by the neuron‐specific expression of atgl‐1 and lid‐1, respectively. The fat‐4 mutation enhances the touch sensation defect in atgl‐1(xd314) and lid‐1(xd288). Wild type and mec‐4(u253) act as the negative control and positive control, respectively. The data were analyzed using Kruskal–Wallis test with Dunn's test. Asterisks denote significant differences as compared to wild type. ns: not statistically significant. **P < 0.01, ***P < 0.001, ****P < 0.0001. Data show mean ± SEM. # signify significant differences between the groups under the crossbars. ## P < 0.01. Data show mean ± SEM. Number of worms analyzed for each strain n = 25.
- The pathway of PUFA synthesis in Caenorhabditis elegans. LA: linoleic acid. ALA: alpha‐linolenic acid. GLA: gamma‐linoleic acid. STA: stearidonic acid. DGLA: dihomo‐gamma‐linolenic acid. ETA: eicosatetraenoic acid. AA: arachidonic acid. EPA: eicosapentaenoic acid. Fatty acids are also indicated by chemical abbreviations. For example, the abbreviation C18:2n−6 means 18 carbons with two double bonds and the first double bond is located at ω−6.
In the de novo PUFA biosynthetic pathway, fat‐4 is responsible for the synthesis of arachidonic acid (AA, C20:4 (20 carbons with four double bonds)) and eicosapentaenoic acid (EPA, C20:5) (Fig 4C; Watts & Browse, 2002). Mutation of fat‐4 leads to mild gentle touch sensation defects similar to those in atgl‐1(xd314) and lid‐1(xd288) mutants (Fig 4B; Vasquez et al, 2014). We then investigated the genetic relationship between ATGL‐1/LID‐1‐mediated lipolysis and de novo synthesis of PUFAs by double mutant analysis. fat‐3 or fat‐4 mutants do not affect neuronal LDs and neuronal LDs are either unchanged or slightly reduced in atgl‐1 or lid‐1 and fat‐3 or fat‐4 double mutants compared with atgl‐1 or lid‐1 single mutants (Fig EV2). Interestingly, the fat‐4(wa14) mutation enhances the touch sensation defect of both atgl‐1(xd314) and lid‐1(xd288) single mutants (Fig 4B), which suggests that ATGL/LID‐1‐regulated neuronal lipolysis participates in PUFA‐mediated touch sensation.
Figure EV2. PUFA synthesis slightly affects LD levels in atgl‐1 or lid‐1 mutant neurons.
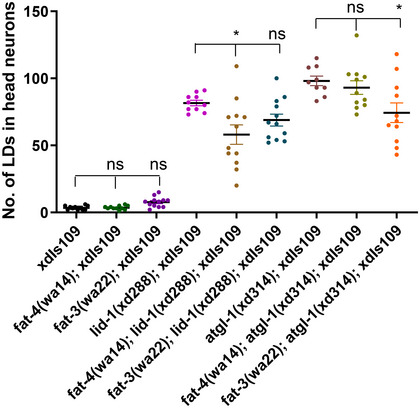
fat‐3 or fat‐4 single mutations do not affect the LD levels, but they slightly reduce LDs in neurons of atgl‐1 or lid‐1 mutants. Each dot represents one worm. The data were analyzed using one‐way ANOVA with Bonferroni's multiple comparison test. * signifies significant differences between the groups under the crossbars. ns: not statistically significant. *P < 0.05. Data show mean ± SEM. n ≥ 9.
atgl‐1(xd314) and lid‐1(xd288) mutants have reduced neuron hyperactivation‐triggered neurodegeneration
Glial LDs are reported to be involved in neurodegeneration in Drosophila (Liu et al, 2015, 2017). In particular, to avoid fatty acid toxicity in hyperactivated neurons, fatty acids are transported from neurons into glia and stored in LDs before detoxification through mitochondrial oxidation (Ioannou et al, 2019). These findings prompted us to investigate whether the appearance of neuronal LDs in atgl‐1 and lid‐1 mutants alleviates neurodegeneration. mec‐4 encodes the ion‐channel protein MEC‐4 and dominant negative mutants of mec‐4, often referred to as mec‐4(d), have been widely used as models of neuron hyperactivation‐triggered neurodegeneration (Driscoll & Chalfie, 1991; Calixto et al, 2012). The neurodegeneration in mec‐4(d) occurs as early as the embryonic stage, and by the L4 stage, the majority (63 ± 2%) of mec‐4(d) animals only have two touch neurons left as viewed by the zdIs5 marker, which labels six touch sensory neurons in wild type (Fig 5A). We found that atgl‐1 mutation reduces the neuronal degeneration of mec‐4(d) (Fig 5A and B). Specifically, the proportion of animals with three or more surviving touch neurons increased from 6 ± 1% in mec‐4(d) to 28 ± 1% in atgl‐1(xd314); mec‐4(d) worms (Fig 5C). Similarly, lid‐1 mutation also reduced the neuronal loss of mec‐4(d). The proportion of animals with three or more surviving touch neurons increased to 23 ± 2% in lid‐1(xd288); mec‐4(d) worms (Fig 5C). Moreover, the reduction of mec‐4(d)-triggered neurodegeneration by atgl‐1 and lid‐1 mutations can be reversed by neuronal‐specific expression of wild‐type atgl‐1 and lid‐1, respectively (Fig 5C). These data indicate that defective neuronal lipolysis reduces neuron hyperactivation‐triggered neurodegeneration.
Figure 5. PUFA synthesis defect significantly enhances the beneficial effect of atgl‐1 mutants in preventing the neurodegeneration of mec‐4(d) .

- Image of touch neurons viewed by zdIs5 in different genetic backgrounds. Scale bar: 20 μm.
- Quantification of the percentage of animals with 0, 1, 2, 3, 4, 5, and 6 surviving touch neurons in different genetic backgrounds. Data show mean ± SEM. At least 50 animals per strain were analyzed in each experiment. Number of experiments n = 5.
- The percentage of worms that have three or more surviving touch neurons in different genetic backgrounds in (B). The lid‐1(xd288) and atgl‐1(xd314) mutations significantly increase the number of surviving neurons. Neuron‐specific expression of atgl‐1 or lid‐1 reverses the protective effect of atgl‐1 or lid‐1 mutation. The data were analyzed using one‐way ANOVA with Turkey's multiple comparison test. Asterisks signify significant differences between the groups under the crossbars. ***P < 0.001, ****P < 0.0001. Data show mean ± SEM. Number of experiments n = 5, with at least 50 animals per strain analyzed in each experiment.
- The percentage of worms that have three or more surviving touch neurons in different genetic backgrounds. Mutations of fat‐1, fat‐3 and fat‐4 significantly enhance the suppression effect of atgl‐1(xd314) in preventing the neurodegeneration of mec‐4(d). The data were analyzed using one‐way ANOVA with Bonferroni's multiple comparison test. ### and #### denote significant differences as compared to atgl‐1(xd314); mec‐4(d). Asterisks signify significant differences as compared to mec‐4(d). ns: not statistically significant. ****P < 0.0001. ### P < 0.001, #### P < 0.0001. Data show mean ± SEM. Number of experiments n ≥ 3, with at least 50 animals per strain analyzed in each experiment.
- The percentage of worms that have three or more surviving touch neurons in different genetic backgrounds supplemented with different fatty acids. AA and EPA but not LA significantly enhance the neurodegeneration triggered by mec‐4(d). The data were analyzed using one‐way ANOVA with Bonferroni's multiple comparison test. Asterisks signify significant differences between the groups under the crossbars. ns: not statistically significant. ***P < 0.001, ****P < 0.0001. Data show mean ± SEM. Number of experiments n ≥ 3, with at least 50 animals per strain analyzed in each experiment.
Defects in neuronal lipolysis and de novo PUFA biosynthesis synergistically decrease the neuronal loss caused by mec‐4(d)
Hyperactivated neurons are vulnerable because the peroxidation of fatty acids, in particular PUFAs, can result in neurodegeneration (Ioannou et al, 2019). fat‐1, fat‐3, and fat‐4 encode key enzymes for de novo synthesis of different PUFAs in C. elegans (Watts & Browse, 2002; Watts & Ristow, 2017). Consistent with the potential involvement of PUFAs in neuron hyperactivation‐triggered neurodegeneration, we found that mec‐4(d)-triggered neurodegeneration was significantly reduced in fat‐4(wa14) and fat‐3(wa22) mutants but not fat‐1(wa9) (Fig 5D). The proportion of animals with three or more surviving touch neurons increased from 6 ± 1% in mec‐4(d) to 35 ± 1% in fat‐4(wa14); mec‐4(d) and 43 ± 3% in fat‐3(wa22); mec‐4(d) worms. These results indicate that reducing the biosynthesis of PUFAs alleviates the neuronal loss of mec‐4(d).
Since ATGL‐1/LID‐1‐mediated neuronal lipolysis is involved in PUFA‐mediated touch sensation (Fig 4A and B), and both atgl‐1 and lid‐1 mutants have reduced mec‐4(d)‐induced neurodegeneration (Fig 5B), we next explored the neuroprotective effect of double mutations in neuronal lipolysis and de novo synthesis of PUFAs. Interestingly, atgl‐1(xd314); fat‐1(wa9), atgl‐1(xd314); fat‐3(wa22) and atgl‐1(xd314); fat‐4(wa14) double mutants all have significantly reduced mec‐4(d)-triggered neurodegeneration compared with single mutants (Fig 5D). The proportion of animals with three or more surviving touch neurons increased from 6 ± 1% in mec‐4(d) to 52 ± 3% in fat‐1(wa9); atgl‐1(xd314); mec‐4(d), 78 ± 2% in fat‐4(wa14); atgl‐1(xd314); mec‐4(d) and 77 ± 1% in fat‐3(wa22); atgl‐1(xd314); mec‐4(d) worms. mboa‐2 overexpression also reduced mec‐4(d) neurodegeneration, although it did not exhibit a synergistic effect with fat‐4(wa14) (Appendix Fig S6). Therefore, blocking de novo PUFA synthesis and neuronal lipolysis together greatly protects neurons from degeneration caused by mec‐4(d).
The PUFAs AA and EPA promote the neurodegeneration of mec‐4(d)
Since fat‐3(wa22) and fat‐4(wa14) show similar neural protection phenotypes (Fig 5D), we reasoned that PUFAs, such as AA (C20:4, n−6) and EPA (C20:5, n−3) or their derivatives, may promote mec‐4(d)-induced neurodegeneration. We fed worms on NGM medium supplemented with different fatty acids. fat‐4(wa14); mec‐4(d) and fat‐4(wa14); atgl‐1(xd314); mec‐4(d) animals show a significant increase of neurodegeneration when grown in AA‐supplemented NGM compared with normal NGM (Fig 5E). Feeding worms with EPA, but not linoleic acid (LA, C18:2), had a similar effect (Fig 5E). In contrast, feeding worms with a mixture containing two saturated fatty acids (palmitic acid (PA, C16:0) and stearic acid (SA, C18:0)) and one monounsaturated fatty acid (oleic acid (OA, C18:1)) did not affect the neurodegeneration phenotype in fat‐4(wa14); mec‐4(d) and fat‐4(wa14); atgl‐1(xd314); mec‐4(d) (Appendix Fig S6). These results indicate that the PUFAs AA and EPA promote the neurodegeneration of mec‐4(d).
PUFAs promote neurodegeneration through incorporation into phospholipids
Elevated ROS and subsequent lipid peroxidation are proposed to be a key event for neurodegeneration triggered by neuron hyperactivation in both C. elegans and mammals (Calixto et al, 2012; Sangaletti et al, 2017; Ioannou et al, 2019). We then tested whether ROS contributes to PUFA‐mediated neurodegeneration. We fed mec‐4(d), fat‐4(wa14); mec‐4(d) or atgl‐1(xd314); mec‐4(d) mutants with the antioxidants N‐acetylcysteine (NAC), vitamin C or vitamin E. The survival of neurons was slightly increased in NAC‐treated mec‐4(d) worms and vitamin C‐treated fat‐4(wa14); mec‐4(d) worms, but not in worms subjected to other treatments (Fig EV3A). We introduced mutation of sod‐2, which encodes a ROS scavenger, and found that it did not significantly decrease the number of surviving neurons in atgl‐1(xd314); mec‐4(d), fat‐4(wa14); mec‐4(d), or fat‐4(wa14); atgl‐1(xd314); mec‐4(d) (Fig EV3B). Furthermore, overexpression of SOD‐1, SOD‐2, SOD‐3, or SOD‐4 in touch neurons did not affect mec‐4(d)‐triggered neurodegeneration (Fig EV3C). Lipid peroxides produced by PUFAs can be cleared with glutathione peroxidase. We found that overexpression of GPX‐1, GPX‐2, and GPX‐6, which are homologs of human phospholipid hydroperoxide glutathione peroxidase (GPx4), do not affect the neurodegeneration of mec‐4(d) and fat‐4(wa14); mec‐4(d) (Fig EV3D and E). Therefore, ROS may play a role, although probably not a major role, in neuronal lipolysis and PUFA‐mediated neuronal loss caused by mec‐4(d).
Figure EV3. Manipulating ROS scavengers slightly influences the protective effect of lipolysis mutations or PUFA mutations on neurodegeneration caused by mec‐4(d) .
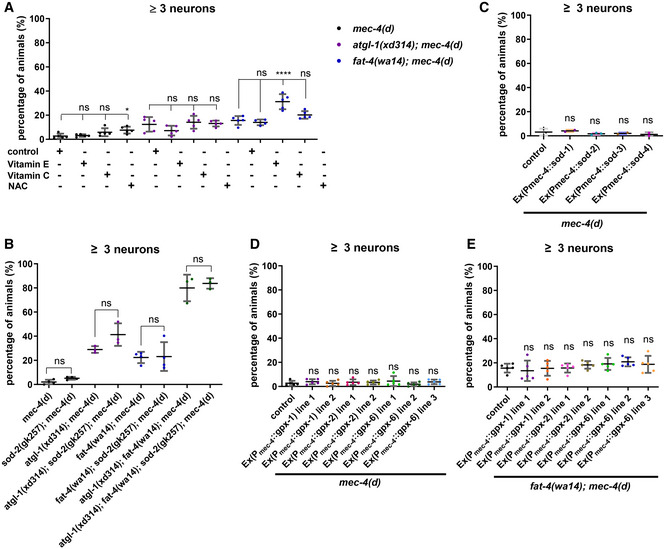
-
AThe percentage of worms that have three or more surviving touch neurons in different genetic backgrounds under different conditions. Feeding with the antioxidant NAC slightly increases the survival of neurons in mec‐4(d) worms. Feeding with the antioxidant vitamin C significantly increases the survival of neurons in fat‐4(wa14); mec‐4(d) worms. The data were analyzed using one‐way ANOVA with Bonferroni's multiple comparison test. Asterisks signify significant differences between the groups under the crossbars. ns: not statistically significant. *P < 0.05, ****P < 0.0001. Data show mean ± SEM. Number of experiments n ≥ 4, with at least 50 animals per strain analyzed in each experiment.
-
BThe percentage of worms that have three or more surviving touch neurons in different genetic backgrounds. sod‐2 mutation does not enhance neurodegeneration in mec‐4(d), atgl‐1(xd314); mec‐4(d), fat‐4(wa14); mec‐4(d) and atgl‐1(xd314); fat‐4(wa14); mec‐4(d). The data were analyzed using one‐way ANOVA with Bonferroni's multiple comparison test. Data show mean ± SEM. ns: not statistically significant. Number of experiments n ≥ 5, with at least 50 animals per strain analyzed in each experiment.
-
CThe percentage of worms that have three or more surviving touch neurons in different genetic backgrounds. Using the touch neuron‐specific promoter Pmec‐4 to drive sod‐1, sod‐2, sod‐3, or sod‐4 overexpression in touch neurons does not significantly affect neurodegeneration in mec‐4(d). The data were analyzed using one‐way ANOVA with Bonferroni's multiple comparison test. Data show mean ± SEM. ns: not statistically significant. Each dot stands for one experiment, with at least 50 animals per strain analyzed in each experiment.
-
D, EThe percentage of worms that have three or more surviving touch neurons in different genetic backgrounds. Overexpression of glutathione peroxidases in touch neurons does not generally increase the survival of neurons. The data were analyzed using one‐way ANOVA with Bonferroni's multiple comparison test. Data show mean ± SEM. ns: not statistically significant. Number of experiments n ≥ 5, with at least 50 animals per strain analyzed in each experiment.
To further investigate how PUFAs promote neurodegeneration, we performed a lipidomic analysis of mec‐4(d), atgl‐1(xd314); mec‐4(d), fat‐4(wa14); mec‐4(d) and fat‐4(wa14); atgl‐1(xd314); mec‐4(d) to find correlations between the level of PUFA‐containing lipids and the severity of neurodegeneration. In fat‐4 mutants, C20:3‐ or C20:4‐containing lipids are increased significantly while C20:5‐containing lipids are decreased dramatically when normalized to total polar lipids (Fig 6A–C). This is consistent with the role of FAT‐4 in generating EPA. Furthermore, although our analysis cannot distinguish between eicosatetraenoic acid (ETA, C20:4 n−3) and AA (C20:4 n−6), the increased C20:4 in fat‐4 mutants is ETA, as revealed in a previous study (Watts & Browse, 2002). Moreover, despite the increased level of C20:4‐containing lipids, the percentage of C20:4‐containing phospholipids is reduced in fat‐4 mutants, while the percentage of C20:4‐containing TAG is increased significantly (Fig 6D and E). Therefore, fat‐4 mutation reduces the levels of some PUFAs and may also alter the partitioning of PUFAs between phospholipids and TAG.
Figure 6. PUFA‐containing phospholipids regulate neurodegeneration of mec‐4(d) .
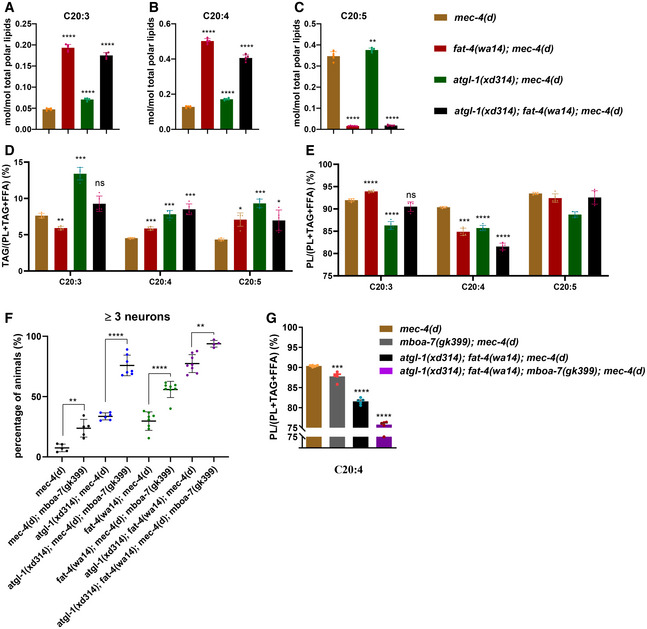
- Lipidomic data show that the relative content of total C20:3 is increased in atgl‐1(xd314); mec‐4(d), fat‐4(wa14); mec‐4(d) and atgl‐1(xd314); fat‐4(wa14); mec‐4(d). The increase is greatest in mutants with the fat‐4 mutation.
- Lipidomic data show that the relative content of total C20:4 is increased in atgl‐1(xd314); mec‐4(d), fat‐4(wa14); mec‐4(d) and atgl‐1(xd314); fat‐4(wa14); mec‐4(d). The increase is greatest in mutants with the fat‐4 mutation.
- Lipidomic data show that the relative content of total C20:5 is slightly increased in atgl‐1(xd314); mec‐4(d) but is dramatically decreased in fat‐4(wa14); mec‐4(d) and atgl‐1(xd314); fat‐4(wa14); mec‐4(d), which matches the role of FAT‐4 in synthesizing C20:5 (EPA).
- The percentage of C20:3‐, C20:4‐, or C20:5‐containing TAGs to total lipids containing the same PUFA in different genetic backgrounds. The percentage of C20:4‐containing TAGs to total lipids containing C20:4 is significantly increased in atgl‐1 and fat‐4 single mutants. The percentage is further increased in atgl‐1 and fat‐4 double mutant.
- The percentage of C20:3‐, C20:4‐, or C20:5‐containing phospholipids to total lipids containing the same PUFA in different genetic backgrounds. The percentage of C20:4‐containing phospholipids to total lipids containing C20:4 is significantly decreased in atgl‐1 and fat‐4 single mutants. The percentage is further decreased in atgl‐1 and fat‐4 double mutant.
- The percentage of worms that have three or more surviving touch neurons in different genetic backgrounds. mboa‐7 mutation significantly increases the percentage of three or more surviving touch neurons in mec‐4(d), atgl‐1(xd314); mec‐4(d), fat‐4(wa14); mec‐4(d) and atgl‐1(xd314); fat‐4(wa14); mec‐4(d). The data were analyzed using one‐way ANOVA with Bonferroni's multiple comparison test. Asterisks signify significant differences between the groups under the crossbars. **P < 0.01, ****P < 0.0001. Data show mean ± SEM. Number of experiments n ≥ 4, with at least 50 animals per strain analyzed in each experiment.
- The percentage of C20:4‐containing phospholipids to total lipids that have C20:4 is dramatically decreased in mboa‐7(gk399); atgl‐1(xd314); fat‐4(wa14); mec‐4(d). The data were analyzed using one‐way ANOVA with Dunnett's multiple comparison test. Asterisks signify significant differences as compared to mec‐4(d). ***P < 0.001, ****P < 0.0001. Data show mean ± SEM. Number of experiments n = 5, with about 10,000 animals per strain analyzed in each experiment.
Compared with fat‐4 mutants, the levels of C20:3‐, C20:4‐, or C20:5‐containing lipids are slightly increased in atgl‐1 mutants, when normalized to total polar lipids (Fig 6A–C). Interestingly, consistent with a storage role for LDs, the percentages of C20:3, C20:4, or C20:5‐containing TAGs are significantly increased, while the percentages of C20:3, C20:4, or C20:5‐containing phospholipids are significantly decreased in atgl‐1 mutants (Fig 6D and E). Therefore, atgl‐1 mutation alters the partitioning of PUFAs into phospholipids and TAG.
In atgl‐1; fat‐4 double mutants, there were similar changes of C20:3, C20:4, and C20:5 fatty acids as in fat‐4 single mutants. However, in the double mutants, we also noticed persistent and further changes in PUFA partitioning between phospholipids and TAG, in particular the partitioning of C20:4. There is a further reduction of the percentage of C20:4‐containing phospholipids in atgl‐1; fat‐4 double mutants compared with either single mutant (Fig 6D and E). The altered partitioning of PUFAs between phospholipids and TAG correlates well with the moderate neuroprotective effects in atgl‐1 and fat‐4 single mutants and the strong neuroprotective effects in atgl‐1; fat‐4 double mutants. These results suggest that PUFA‐containing phospholipids may promote neurodegeneration.
We further explored the hypothesis that PUFA‐containing phospholipids contribute to mec‐4(d)‐induced neurodegeneration. MBOA‐7 incorporates PUFAs into phospholipids, especially PI (Lee et al, 2008). We found that mboa‐7 mutation alone significantly increased the percentage of mec‐4(d) animals with ≥ 3 surviving touch neurons (from ~ 5% to ~ 20%) (Fig 6F). Moreover, mboa‐7 mutation further increases neuron survival in atgl‐1(xd314); mec‐4(d), fat‐4(wa14); mec‐4(d) and atgl‐1(xd314); fat‐4(wa14); mec‐4(d) (Figs 6F, and EV4A and B). The enhancement of the neuroprotective effect is probably because the mboa‐7 mutation reduces PUFA incorporation into phospholipids. Consistent with that, the percentage of C20:4‐containing phospholipids is further decreased in atgl‐1(xd314); fat‐4(wa14); mboa‐7(gk399); mec‐4(d) mutants compared with atgl‐1(xd314); fat‐4(wa14); mec‐4(d) mutants (Fig 6G). Together, these results indicate that PUFAs promote neuron hyperactivation‐mediated neurodegeneration of mec‐4(d) through their incorporation into phospholipids.
Figure EV4. mboa‐7 mutation significantly affects neurodegeneration caused by mec‐4(d) .
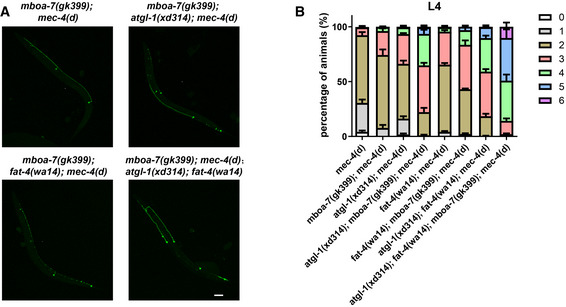
- Image of touch neurons in worms viewed by zdIs5(P mec‐4 ::GFP) in different genetic backgrounds. Scale bar: 20 μm.
- Quantification of the percentage of animals with 0, 1, 2, 3, 4, 5, and 6 surviving touch neurons in different genetic backgrounds. The distribution of the number of surviving neurons is further improved when mboa‐7 is mutated in atgl‐1 and fat‐4 single mutants or the double mutant. Data show mean ± SEM. At least 50 animals per strain were analyzed in each experiment. Number of experiments was 5.
Discussion
This study addresses two questions: Why are there normally no LDs in neurons, and what are the consequences of LD accumulation in neurons? We reveal that the balance of lipolysis and lipogenesis regulates neuronal LD dynamics. In particular, we show that defective ATGL‐1/LID‐1‐mediated lipolysis causes LD accumulation autonomously in C. elegans neurons. Defective neuronal lipolysis affects normal gentle touch sensation, while importantly, it alleviates neurodegeneration triggered by neuron hyperactivation. The neuroprotective effect is synergistically enhanced when de novo biosynthesis of PUFAs is blocked. Lastly, the incorporation of PUFAs into phospholipids likely underlies neuronal lipolysis and PUFA‐mediated neurodegeneration. Therefore, both neuronal LD dynamics and de novo PUFA synthesis regulate the availability of PUFAs, which may be incorporated into phospholipids to maintain the proper function of neurons under normal conditions and to promote neurodegeneration under stress conditions (Fig 7).
Figure 7. Neuronal lipolysis participates in the homeostasis of PUFAs, which regulates neuronal functions and neurodegeneration through incorporation of PUFAs into phospholipids.
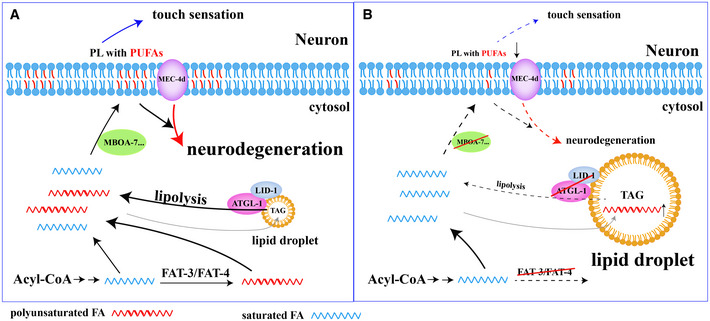
-
A, BThe homeostasis of PUFAs is determined by PUFA de novo synthesis and the balance of neuronal lipolysis and lipogenesis. The incorporation of PUFAs into phospholipids regulates normal neuronal functions (such as touch sensation) and affects neurodegeneration of mec‐4(d).
LDs in neuron: a tug‐of‐war between lipogenesis and lipolysis
Neurons mainly use glucose to generate energy instead of lipids to avoid extra ROS production by lipid β‐oxidation. Although normally there are no LDs in neurons, our results demonstrate that neurons have the ability to form and hydrolyze LDs. Both overexpression of lipogenesis genes and loss of the key lipolysis genes atgl‐1 and lid‐1 lead to LD accumulation in neurons. It is possible that the lipogenesis in neurons is limited and lipolysis is kept at a relatively high level, which promotes a fast turnover of LDs to meet the lipid requirements of neurons. Thus, there is a tug‐of‐war between lipogenesis and lipolysis in neurons.
Both atgl‐1 and lid‐1 are highly conserved from worm to mammals. Previous studies on Drosophila and cultured hippocampal neurons show that instead of forming LDs, neurons transfer the excess lipids into glial cells to form LDs under stress conditions (Bailey et al, 2015; Liu et al, 2015, 2017; Ioannou et al, 2019). If neurons have the ability to form and hydrolyze LDs, it is puzzling why they do not form LDs themselves. Could the formation of LDs in neurons be worm‐specific because most neurons, including touch neurons, in C. elegans are not surrounded by glial cells? The appearance of LDs in DDHD2 −/− mice, which have a defect in a lipase, strongly argues against this possibility (Inloes et al, 2014). Alternatively, it is possible that under conditions of intact neuronal lipolysis, excess lipids can still engage in generating toxic hyperoxidated PUFAs in neurons. Therefore, to avoid neuronal toxicity, exporting excess lipids to glia is a better choice. Examining the flux of lipid export and lipid incorporation into neutral lipids may provide a further explanation.
Interestingly, mice with deficiencies of ATGL or CGI‐58 accumulate massive amounts of neutral lipids in many tissues/organs including brain (Radner et al, 2010; Etschmaier et al, 2011). Similarly, lipid accumulation in brain was clearly demonstrated in CGI‐58 human patients (Huigen et al, 2015). However, in both mouse and human studies, there is no direct evidence of LD accumulation in neurons. Compared with LDs in glia or blood vessels, neuronal LDs may be too small to observe and may therefore be neglected. Specific examination of neuronal LDs in these lipolysis‐defective mutants will be required to characterize the neuronal defects.
Neuronal lipolysis and neuronal normal function
Here, we show that intact neuronal lipolysis is important for maintaining normal neuron function in worm. Currently, there is no study on neurological function of ATGL‐mediated lipolysis in mice. In patients with mutated CGI‐58, neurological symptoms, including cognitive impairment and psychiatric disorders, are a frequent characteristic (Schweiger et al, 2009). A human ATGL mutation case with global cognitive impairment was also reported (Massa et al, 2016). Therefore, the critical role of neuronal lipolysis in neurons is likely conserved. The detail neuron‐specific pathological phenotype and its underlying mechanism remain to be elucidated in ATGL or CGI‐58 mouse mutant models and more importantly in human patients.
Our results show that neuronal lipolysis participates in PUFA‐mediated touch sensation. Through incorporation into membrane‐forming phospholipids or as signals regulating neuron activity, PUFAs and their derivatives play important roles in neurons (Watts & Ristow, 2017). In C. elegans mechanical and touch sensation, PUFA‐containing phospholipids modulate the activity of particular channels through membrane remodeling and changes of membrane fluidity (Matsuda et al, 2008; Vasquez et al, 2014). In addition, PUFA depletion causes defects in neurotransmission (Lesa et al, 2003; Marza & Lesa, 2006). It remains to be explored whether the neurological defects in ATGL or CGI‐58 human patients are linked to altered PUFA‐mediated neuronal functions.
Neuronal LDs and neurodegeneration
In this study, we found that LDs have a neuroprotective effect in the neuron hyperactivation‐triggered neurodegeneration model. Interestingly, neuronal LDs also show a protective effect in another neurodegenerative disease, Parkinson's disease. The expression of the Parkinson's disease protein α‐Synuclein alters the cellular lipid profile, notably by elevating DAG and monounsaturated fatty acid OA. By partitioning excess DAG and OA into TAG, the formation of LDs reduces neuron death triggered by expression of α‐Synuclein (Fanning et al, 2019). In these two neurodegeneration models, LD acts as a harbor to store excess and/or toxic fatty acids which promote neurodegeneration, although the detail mechanisms of neuroprotective effect of LDs are different. Therefore, the presence of LDs may be beneficial for neurodegenerative diseases in general.
Besides Parkinson's disease, many other neuronal diseases, in particular the HSP diseases, are reported to be accompanied by abnormal levels of neutral lipids, and the disease genes are related to LD dynamics. Knockdown of Spartin, also known as SPG20, increases the number and size of LDs in cells loaded with oleic acid (Papadopoulos et al, 2015). In another HSP model, DDHD2 −/− mice exhibit LD accumulation in neurons (Inloes et al, 2014). Despite the alteration of LD homeostasis, the role of LD accumulation in neurons under these disease conditions has not been investigated. Our findings also raise the possibility that the accumulation of LDs is a compensatory response to relieve neuronal stress in some HSP diseases. Further studies will be required to explore this possibility.
PUFAs and neurodegeneration
We show that reducing PUFA incorporation into phospholipids has a beneficial effect in alleviating neuronal loss in a neurodegeneration model. The mechanism underlying this effect is not fully clear. Although we only observed a marginal effect of NAC and vitamin C treatment, it is still possible that lipid peroxides produced from PUFAs contribute to the neuronal degeneration. It is possible that these antioxidants do not show a strong effect on neurodegeneration because only limited amounts of the small antioxidant molecules actually reach the neuronal cells. Phospholipids are building blocks of all membranes in mammalian cells, providing the structural integrity that is necessary for protein function. They also serve as precursors for various second messengers such as AA, DHA, ceramide, 1,2‐diacylglycerol, IP3, phosphatidic acid, and lyso‐phospholipids (O'Donnell et al, 2018). Generally, phospholipids with PUFA chains are more flexible. The cis‐double bond lowers the packing density of the acyl chains, which increases membrane fluidity, bending stiffness, and effective viscosity (Holthuis & Menon, 2014; Vasquez et al, 2014). One possibility is that a reduced level of PUFA‐containing phospholipids impacts membrane fluidity and in turn reduces ion‐channel activity. For example, AA and DHA can regulate channels such as K+ channels and TRPV4 (Villarroel & Schwarz, 1996; Horimoto et al, 1997; Caires et al, 2017). PUFA synthesis mutations also decreased neuronal Ca2+ transients in C. elegans (Kahn‐Kirby et al, 2004). Thus, it is possible that PUFA‐containing phospholipids regulate the constitutively active MEC‐4(d) ion channel through Ca2+ transients. Moreover, phospholipids with PUFAs can be degraded and produce the second messengers 2‐AG, 1,2‐diacylglycerol and IP3. Therefore, another possibility is that reduced levels of phospholipids with PUFAs affect downstream signals that execute neurodegeneration triggered by ion‐channel hyperactivation.
Our study indicates that reducing the influx of PUFAs into neuron phospholipids protects neurons from degeneration triggered by hyperactivation. This suggests a potential strategy for treating neurodegeneration caused by ion hyperactivation.
Materials and Methods
Strains
Worms were cultured on OP50‐seeded nematode growth medium (NGM) plates at 22°C (Brenner, 1974). Wild‐type (N2), CB4856, VC3025 C37H5.2(ok3722), RB2386 C37H5.3(ok3245), TU253 mec‐4(u253), CB1370 daf‐2(e1370), CB4037 glp‐1(e2141), CB1611 mec‐4(e1611), VC942 mboa‐7(gk399), and GA184 sod‐2(gk257) were obtained from the Caenorhabditis Genetics Center (CGC). B0524.2(tm6739) and D1054.1(tm3111) were obtained from the NBRP (Japan). Mutant strains BX24 fat‐1(wa9), BX30 fat‐3(wa22), and BX17 fat‐4(wa14) were kindly provided by Dr. Bin Liang. lid‐1(xd288), atgl‐1(xd310), and atgl‐1(xd314) were generated by EMS. xdIs182 (P rab‐3 ::Cre/P odr‐1 ::RFP) was generated by integration of Ex(P rab‐3 ::Cre/P odr‐1 ::RFP). atgl-1‐loxP(xd426) was generated by CRISPR‐Cas9‐mediated genome editing. All the transgenic worms were generated by microinjection of the respective plasmids or fosmids with co‐injection markers. All the fosmids were kindly provided by Dr. Xiaochen Wang.
Molecular biology
The promoters of P unc‐119 (1,235 bp), P vha‐6 (1,808 bp), P hlh‐17 (2,049 bp), P ajm‐1 (3,500 bp), P rab‐3 (1,207 bp), P atgl‐1 (3,004 bp), and P mec‐4 (206 bp) were amplified from N2 genomic DNA. The coding regions of lid‐1, atgl‐1, C37H5.2, C37H5.3, B0524.2, D1054.1, mboa‐2, F59A1.10, K07B1.4, sod‐1, sod‐2, sod‐3, sod‐4, gpx‐1, gpx‐2, and gpx‐6 were amplified from cDNA. The promotor and target cDNA were inserted into plasmid Δpsm. The split GFP1‐10 and GFP11 were inserted into pPD95.75. P atgl‐1 and P rab‐3 were inserted upstream of GFP1‐10 and GFP11, respectively. P rab‐3 ::Cre was generated by replacing the eft‐3 promotor in pDD104, which was kindly provided by Dr. Shiqing Cai. atgl‐1‐sgRNAs for loxP insertion were designed using the Zhang laboratory's CRISPR design tool at http://crispr.mit.edu to select the target sites. The sgRNAs were inserted into pDD162, which was kindly provided by Dr. Guangshuo Ou. All the constructed plasmids were verified by sequencing.
EMS screen
We treated L4 and young adult xdIs109 (P unc‐119 ::PLIN1::GFP) worms with ethyl methanesulfonate (EMS). About 2,500 F1 were screened. We observed LDs using a Zeiss compound microscope. Mutants were selected that showed LD accumulation in neurons.
SNP mapping and fosmid rescue
We used rapid single nucleotide polymorphism (SNP) mapping (Davis et al, 2005). Briefly, we crossed the mutant with CB4856 and picked F2 animals with LD accumulation. We amplified some SNPs using F2 or F3 lysates as templates, and we digested the PCR products with enzymes. After chromosome mapping and interval mapping, we identified a narrow region. Then, we did the fosmid rescue assay. Fosmids were injected individually into mutant worms at 5–10 ng/μl together with 50 ng/μl P odr‐1 ::RFP which acts as the co‐injection marker.
Oil red O staining
Oil red O was acquired from Sigma‐Aldrich. Oil red O staining was conducted as previously reported (O'Rourke et al, 2009).
BODIPY staining
Worms were fixed as for Oil red O staining (O'Rourke et al, 2009). Worms were washed three times with 1× PBS to remove paraformaldehyde. After that, worms were incubated in PBS with 2 μg/ml BODIPY (Invitrogen) for 30 min in the dark with gentle rocking. Then worms were washed with 1× PBS and imaged directly by confocal microscopy (Leica SP8) (Tian et al, 2011).
Behavioral assays
Gentle touch sensitivity was tested and scored as described (Hart, 2006; Vasquez et al, 2014). Briefly, we performed ten‐trial touch assays. We scored the touch response percentage by stroking an eyebrow hair across the anterior and posterior body. Twenty‐five animals were tested in each trial, and results were compared across three trials. All assays were performed by investigators blinded to genotype and/or treatment.
Quantification of neuron loss
Animals were mounted on thin agarose pads and immobilized by 1‐phenoxy‐2‐propanol (10 μl/ml). Animals were visualized under a 20× objective. GFP‐expressing touch neurons were counted in synchronized L4 animals.
Fatty acid, NAC, vitamin C, and vitamin E supplementation
AA (arachidonic acid), NAC (N‐Acetyl‐L‐cysteine), vitamin C, and vitamin E were acquired from Aladdin. SA (stearic acid), OA (oleic acid), PA (palmitate acid), EPA (eicosapentaenoic acid), LA (linoleic acid), and Tergitol (70%) were acquired from Sigma‐Aldrich. FAs, dissolved in ethanol with 0.1% Tergitol, were added to NGM agar to reach a final concentration of approximately 200 μM. NAC and vitamin C was dissolved in ddH2O and added into NGM agar to reach a final concentration of approximately 10 mM, respectively. Vitamin E was dissolved in methanol and added into NGM agar to reach a final concentration of approximately 200 μg/ml. Plates were seeded with Escherichia coli OP50 and kept at room temperature (Vasquez et al, 2014). For each strain, about 10 L4 worms were placed on the plate. We quantified the phenotype in the next generation.
Stimulated Raman Scattering microscopy
To quantify lipid droplet metrics in the touch neurons, 1‐day adult worms were imaged using Stimulated Raman Scattering (SRS) microscopy. Worms were mounted onto 2% agarose pads with 0.5% NaN3 as anesthetic on glass microscope slides and imaged using a 60× water objective (UPlanAPO/IR; 1.2 N.A.; Olympus). A femtosecond‐pulsed laser and picosecond‐pulsed laser were used for simultaneously imaging label‐free lipids (SRS channel) and GFP‐labeled neurons (fluorescence channel). Images were analyzed using ImageJ software (NIH). The neuronal area was selected using the fluorescent signal based on several Z‐projected stacks to confirm the presence of lipid droplets within the cell and to exclude extracellular signal.
EM analysis
Young adult worms were collected for high‐pressure freezing and freeze‐substitution, embedding and sectioning following a procedure essentially as described by (Weimer, 2006). Then, the sections were visualized with a Hitachi HT7700 and pictures were recorded on a 4,008 × 2,672 CCD camera.
Lipidomic analysis
Worms were washed 9–10 times in M9. More than 10,000 worms per genotype were collected per sample and five samples were analyzed per genotype. Lipid extraction and analysis were conducted as previously reported (Lam et al, 2014). The lipid content was normalized by the mole fraction of each lipid to total polar lipids.
Statistical analyses
All data are presented as mean ± SEM. The data were first analyzed normal distribution. When the data meet the normal distribution, we used one‐way ANOVA or two‐way ANOVA for statistically significant overall results. When the data do not meet the normal distribution, we used Kruskal–Wallis test. Post hoc multiple comparison tests were carried out to determine significant differences. Significant difference is noted with a number sign (#) or an asterisk (*). ns represents not statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. # P < 0.05, ## P < 0.01, ### P < 0.001, #### P < 0.0001.
Author contributions
LY directed the project and her work includes EMS screen, molecular biology, SNP mapping and fosmid rescue, Oil red O staining, behavioral assays, neuronal loss analysis, fatty acid, NAC, vitamin C and vitamin E supplementation and analyzing the results. JL and MD did the EM imaging and analysis. SML and GS did the lipidomic analysis. AY did the SRS imaging. LY and XH wrote the manuscript. XH guided the project and edited the manuscript.
Conflict of interest
All authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Drs. Shiqing Cai, Bin Liang, Pingsheng Liu, Guangshuo Ou, Meng C. Wang, and Xiaochen Wang for providing reagents and helpful discussions. We thank the Caenorhabditis Genetics Center (CGC) and National BioResource Project (NBRP) for providing strains. This research was supported by grants 31630019, 2018YFA0506902, 91954207, and 2016YFA0500100 from the National Natural Science Foundation of China and National Key R&D program of China.
EMBO Reports (2020) 21: e50214
Data availability
This study includes no data deposited in external repositories.
References
- Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, Postle AD, Gould AP (2015) Antioxidant role for lipid droplets in a stem cell niche of Drosophila . Cell 163: 340–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Selma Z, Yilmaz S, Schischmanoff PO, Blom A, Ozogul C, Laroche L, Caux F (2007) A novel S115G mutation of CGI‐58 in a Turkish patient with Dorfman‐Chanarin syndrome. J Invest Dermatol 127: 2273–2276 [DOI] [PubMed] [Google Scholar]
- Bi J, Xiang Y, Chen H, Liu Z, Gronke S, Kuhnlein RP, Huang X (2012) Opposite and redundant roles of the two Drosophila perilipins in lipid mobilization. J Cell Sci 125: 3568–3577 [DOI] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous S, Terwilliger TC, Waldo GS (2005) Protein tagging and detection with engineered self‐assembling fragments of green fluorescent protein. Nat Biotechnol 23: 102–107 [DOI] [PubMed] [Google Scholar]
- Caires R, Sierra‐Valdez FJ, Millet JRM, Herwig JD, Roan E, Vasquez V, Cordero‐Morales JF (2017) Omega‐3 fatty acids modulate TRPV4 function through plasma membrane remodeling. Cell Rep 21: 246–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calixto A, Jara JS, Court FA (2012) Diapause formation and downregulation of insulin‐like signaling via DAF‐16/FOXO delays axonal degeneration and neuronal loss. PLoS Genet 8: e1003141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen X‐W, Huang X, Song B‐L, Wang Y, Wang Y (2019) Regulation of glucose and lipid metabolism in health and disease. Sci China Life Sci 62: 1420–1458. [DOI] [PubMed] [Google Scholar]
- Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL (2002) Lipid droplet binding and oligomerization properties of the Parkinson's disease protein alpha‐synuclein. J Biol Chem 277: 6344–6352 [DOI] [PubMed] [Google Scholar]
- Davis MW, Hammarlund M, Harrach T, Hullett P, Olsen S, Jorgensen EM (2005) Rapid single nucleotide polymorphism mapping in C. elegans . BMC Genom 6: 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Yang X, Tian H, Liang J, Zhang F, Wang G, Wang Y, Ding M, Shui G, Huang X (2018) Seipin regulates lipid homeostasis by ensuring calcium‐dependent mitochondrial metabolism. EMBO J 37: e97572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll M, Chalfie M (1991) The mec‐4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature 349: 588–593 [DOI] [PubMed] [Google Scholar]
- Eastman SW, Yassaee M, Bieniasz PD (2009) A role for ubiquitin ligases and Spartin/SPG20 in lipid droplet turnover. J Cell Biol 184: 881–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara C, Ebihara K, Aizawa‐Abe M, Mashimo T, Tomita T, Zhao M, Gumbilai V, Kusakabe T, Yamamoto Y, Aotani D et al (2015) Seipin is necessary for normal brain development and spermatogenesis in addition to adipogenesis. Hum Mol Genet 24: 4238–4249 [DOI] [PubMed] [Google Scholar]
- Etschmaier K, Becker T, Eichmann TO, Schweinzer C, Scholler M, Tam‐Amersdorfer C, Poeckl M, Schuligoi R, Kober A, Chirackal Manavalan AP et al (2011) Adipose triglyceride lipase affects triacylglycerol metabolism at brain barriers. J Neurochem 119: 1016–1028 [DOI] [PubMed] [Google Scholar]
- Fanning S, Haque A, Imberdis T, Baru V, Barrasa MI, Nuber S, Termine D, Ramalingam N, Ho GPH, Noble T et al (2019) Lipidomic analysis of alpha‐synuclein neurotoxicity identifies stearoyl CoA Desaturase as a target for parkinson treatment. Mol Cell 73: 1001–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart AC (2006) Behavior. WormBook, 10.1895/wormbook.1891.1887.1891 [DOI] [Google Scholar]
- Holthuis JC, Menon AK (2014) Lipid landscapes and pipelines in membrane homeostasis. Nature 510: 48–57 [DOI] [PubMed] [Google Scholar]
- Horimoto N, Nabekura J, Ogawa T (1997) Arachidonic acid activation of potassium channels in rat visual cortex neurons. Neuroscience 77: 661–671 [DOI] [PubMed] [Google Scholar]
- Huigen MC, van der Graaf M, Morava E, Dassel AC, van Steensel MA, Seyger MM, Wevers RA, Willemsen MA (2015) Cerebral lipid accumulation in Chanarin‐Dorfman Syndrome. Mol Genet Metab 114: 51–54 [DOI] [PubMed] [Google Scholar]
- Inloes JM, Hsu KL, Dix MM, Viader A, Masuda K, Takei T, Wood MR, Cravatt BF (2014) The hereditary spastic paraplegia‐related enzyme DDHD2 is a principal brain triglyceride lipase. Proc Natl Acad Sci USA 111: 14924–14929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, Pasolli HA, Xu CS, Pang S, Matthies D et al (2019) Neuron‐astrocyte metabolic coupling protects against activity‐induced fatty acid toxicity. Cell 177: 1522–1535 [DOI] [PubMed] [Google Scholar]
- Kahn‐Kirby AH, Dantzker JL, Apicella AJ, Schafer WR, Browse J, Bargmann CI, Watts JL (2004) Specific polyunsaturated fatty acids drive TRPV‐dependent sensory signaling in vivo . Cell 119: 889–900 [DOI] [PubMed] [Google Scholar]
- Kis V, Barti B, Lippai M, Sass M (2015) Specialized cortex glial cells accumulate lipid droplets in Drosophila melanogaster . PLoS ONE 10: e0131250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm RW, Norton JP, Cole RA, Li CS, Park SH, Crane MM, Li L, Jin D, Boye‐Doe A, Liu TY et al (2013) A conserved role for atlastin GTPases in regulating lipid droplet size. Cell Rep 3: 1465–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SM, Wang Y, Duan X, Wenk MR, Kalaria RN, Chen CP, Lai MK, Shui G (2014) Brain lipidomes of subcortical ischemic vascular dementia and mixed dementia. Neurobiol Aging 35: 2369–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R (2006) Adipose triglyceride lipase‐mediated lipolysis of cellular fat stores is activated by CGI‐58 and defective in Chanarin‐Dorfman Syndrome. Cell Metab 3: 309–319 [DOI] [PubMed] [Google Scholar]
- Lee HC, Inoue T, Imae R, Kono N, Shirae S, Matsuda S, Gengyo‐Ando K, Mitani S, Arai H (2008) Caenorhabditis elegans mboa‐7, a member of the MBOAT family, is required for selective incorporation of polyunsaturated fatty acids into phosphatidylinositol. Mol Biol Cell 19: 1174–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Kong J, Jang JY, Han JS, Ji Y, Lee J, Kim JB (2014) Lipid droplet protein LID‐1 mediates ATGL‐1‐dependent lipolysis during fasting in Caenorhabditis elegans . Mol Cell Biol 34: 4165–4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesa GM, Palfreyman M, Hall DH, Clandinin MT, Rudolph C, Jorgensen EM, Schiavo G (2003) Long chain polyunsaturated fatty acids are required for efficient neurotransmission in C. elegans . J Cell Sci 116: 4965–4975 [DOI] [PubMed] [Google Scholar]
- Liu Z, Li X, Ge Q, Ding M, Huang X (2014) A lipid droplet‐associated GFP reporter‐based screen identifies new fat storage regulators in C. elegans . J Genet Genomics 41: 305–313 [DOI] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A et al (2015) Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160: 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, MacKenzie KR, Putluri N, Maletic‐Savatic M, Bellen HJ (2017) The glia‐neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab 26: 719–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D et al (2010) Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci 13: 567–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marza E, Lesa GM (2006) Polyunsaturated fatty acids and neurotransmission in Caenorhabditis elegans . Biochem Soc Trans 34: 77–80 [DOI] [PubMed] [Google Scholar]
- Massa R, Pozzessere S, Rastelli E, Serra L, Terracciano C, Gibellini M, Bozzali M, Arca M (2016) Neutral lipid‐storage disease with myopathy and extended phenotype with novel PNPLA2 mutation. Muscle Nerve 53: 644–648 [DOI] [PubMed] [Google Scholar]
- Matsuda S, Inoue T, Lee HC, Kono N, Tanaka F, Gengyo‐Ando K, Mitani S, Arai H (2008) Member of the membrane‐bound O‐acyltransferase (MBOAT) family encodes a lysophospholipid acyltransferase with broad substrate specificity. Genes Cells 13: 879–888 [DOI] [PubMed] [Google Scholar]
- Narbonne P, Roy R (2009) Caenorhabditis elegans dauers need LKB1/AMPK to ration lipid reserves and ensure long‐term survival. Nature 457: 210–214 [DOI] [PubMed] [Google Scholar]
- O'Donnell VB, Rossjohn J, Wakelam MJ (2018) Phospholipid signaling in innate immune cells. J Clin Invest 128: 2670–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Carvalho P (2019) Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol 20: 137–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke EJ, Soukas AA, Carr CE, Ruvkun G (2009) C. elegans major fats are stored in vesicles distinct from lysosome‐related organelles. Cell Metab 10: 430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S (2003) Yeast cells provide insight into alpha‐synuclein biology and pathobiology. Science 302: 1772–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Mari M, Petanidou B, Pasparaki A, Filippidis G, Tavernarakis N (2017) Ectopic fat deposition contributes to age‐associated pathology in Caenorhabditis elegans . J Lipid Res 58: 72–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Orso G, Mancuso G, Herholz M, Gumeni S, Tadepalle N, Jungst C, Tzschichholz A, Schauss A, Honing S et al (2015) Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet 11: e1005149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radner FP, Streith IE, Schoiswohl G, Schweiger M, Kumari M, Eichmann TO, Rechberger G, Koefeler HC, Eder S, Schauer S et al (2010) Growth retardation, impaired triacylglycerol catabolism, hepatic steatosis, and lethal skin barrier defect in mice lacking comparative gene identification‐58 (CGI‐58). J Biol Chem 285: 7300–7311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renvoise B, Malone B, Falgairolle M, Munasinghe J, Stadler J, Sibilla C, Park SH, Blackstone C (2016) Reep1 null mice reveal a converging role for hereditary spastic paraplegia proteins in lipid droplet regulation. Hum Mol Genet 25: 5111–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangaletti R, D'Amico M, Grant J, Della‐Morte D, Bianchi L (2017) Knock‐out of a mitochondrial sirtuin protects neurons from degeneration in Caenorhabditis elegans . PLoS Genet 13: e1006965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R (2009) Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI‐58/ABHD5. Am J Physiol Endocrinol Metab 297: E289–E296 [DOI] [PubMed] [Google Scholar]
- Shimabukuro MK, Langhi LG, Cordeiro I, Brito JM, Batista CM, Mattson MP, Mello Coelho V (2016) Lipid‐laden cells differentially distributed in the aging brain are functionally active and correspond to distinct phenotypes. Sci Rep 6: 23795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Bi J, Shui G, Liu Z, Xiang Y, Liu Y, Wenk MR, Yang H, Huang X (2011) Tissue‐autonomous function of Drosophila seipin in preventing ectopic lipid droplet formation. PLoS Genet 7: e1001364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez V, Krieg M, Lockhead D, Goodman MB (2014) Phospholipids that contain polyunsaturated fatty acids enhance neuronal cell mechanics and touch sensation. Cell Rep 6: 70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroel A, Schwarz TL (1996) Inhibition of the Kv4 (Shal) family of transient K+ currents by arachidonic acid. J Neurosci 16: 2522–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JL, Browse J (2002) Genetic dissection of polyunsaturated fatty acid synthesis in Caenorhabditis elegans . Proc Natl Acad Sci USA 99: 5854–5859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JL, Ristow M (2017) Lipid and carbohydrate metabolism in Caenorhabditis elegans . Genetics 207: 413–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer RM (2006) Preservation of C. elegans tissue via high‐pressure freezing and freeze‐substitution for ultrastructural analysis and immunocytochemistry. Methods Mol Biol 351: 203–221 [DOI] [PubMed] [Google Scholar]
- Xie M, Roy R (2015a) AMP‐activated kinase regulates lipid droplet localization and stability of adipose triglyceride lipase in C. elegans Dauer Larvae. PLoS ONE 10: e0130480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Roy R (2015b) The causative gene in Chanarian Dorfman Syndrome regulates lipid droplet homeostasis in C. elegans . PLoS Genet 11: e1005284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH (2000) Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci USA 97: 1784–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
This study includes no data deposited in external repositories.