

Abstract
The latent HIV‐1 reservoir is a major barrier to viral eradication. However, our understanding of how HIV‐1 establishes latency is incomplete. Here, by performing a genome‐wide CRISPR‐Cas9 knockout library screen, we identify phosphatidylethanolamine‐binding protein 1 (PEBP1), also known as Raf kinase inhibitor protein (RKIP), as a novel gene inducing HIV latency. Depletion of PEBP1 leads to the reactivation of HIV‐1 in multiple models of latency. Mechanistically, PEBP1 de‐phosphorylates Raf1/ERK/IκB and IKK/IκB signaling pathways to sequestrate NF‐κB in the cytoplasm, which transcriptionally inactivates HIV‐1 to induce latency. Importantly, the induction of PEBP1 expression by the green tea compound epigallocatechin‐3‐gallate (EGCG) prevents latency reversal by inhibiting nuclear translocation of NF‐κB, thereby suppressing HIV‐1 transcription in primary CD4+ T cells isolated from patients receiving antiretroviral therapy (ART). These results suggest a critical role for PEBP1 in the regulation of upstream NF‐κB signaling pathways governing HIV transcription. Targeting of this pathway could be an option to control HIV reservoirs in patients in the future.
Keywords: CRISPR‐Cas9, genome‐wide screening, HIV latency, NF‐κB, PEBP1
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
The latent HIV‐1 reservoir prevents efficient viral eradication. The phosphatase PEBP1 plays a critical role in the regulation of NF‐κB‐dependent pathways governing HIV‐1 transcription, and its targeting could control HIV reservoirs in patients.

Introduction
Current antiretroviral therapy (ART) has succeeded in reducing human immunodeficiency virus type 1 (HIV‐1) to undetectable levels in HIV‐1‐infected patients (Ruelas & Greene, 2013; Margolis et al, 2016). However, ART alone cannot cure AIDS. The viral reservoirs comprised of latently infected and long‐lived resting CD4+ T cells can survive for decades, thereby preventing our current efforts to cure HIV (Finzi et al, 1997; ;Chun et al, 1998; Richman et al, 2009; ;Barouch & Deeks, 2014). These latent HIV reservoirs are considered as the main barrier for viral eradication (Ho et al, 2013; Kim et al, 2018; Pitman et al, 2018; Rojas et al, 2019). In the last few decades, progress has been made to elucidate the molecular mechanisms underlying the establishment of HIV‐1 latency, mostly acting at the level of transcriptional suppression of the viral promoter‐long terminal repeats (LTR) (Verdin et al, 1993; Bieniasz et al, 1999; Blazkova et al, 2009; Archin et al, 2014; Kumar et al, 2015; Elsheikh et al, 2019). It has been shown that transcriptional blocks to productive HIV‐1 replication are associated with multiple layers of regulation, including epigenetic modifications at the HIV‐1 LTR, inadequate availability of transcription factors at the HIV LTR, such as NF‐κB, positive transcription elongation factor b (P‐TEFb or CDK9/CycinT1), HIV‐1 Tat, and among others (Mancebo et al, 1997; Bieniasz et al, 1999; Fiume et al, 2012). Many small molecule compounds to target these signaling pathways have been tested to directly reactivate latent HIV‐1 (Alexaki et al, 2007; Imai et al, 2010; Beans et al, 2013; Li et al, 2013; Spivak et al, 2014; Søgaard et al, 2015; Boehm et al, 2017; Wang et al, 2017). Unfortunately, none of these latency reversal agents (LRAs) can effectively reduce the reservoir size in patients although latent HIV‐1 can be disrupted in vivo (Archin et al, 2012; Søgaard et al, 2015). While an opposing intervention strategy has been proposed to deeply silence the HIV reservoirs (Mousseau et al, 2015; Elsheikh et al, 2019), an effective prevention of viral rebound has not yet been achieved in a clinical or pre‐clinical setting (Kessing et al, 2017), indicating that our current understanding of how HIV‐1 establishes and maintains its latency remains limited.
In recent years, the methodology behind genome editing has greatly expanded with the emergence of the CRISPR/Cas9 system. In 2014, Dr. Zhang’s laboratory constructed a lentivirus library to target human genome using array libraries (Shalem et al, 2014). By using the CRISPR/Cas9 library, researchers successfully identified the host factors necessary for viral infection, such as flavivirus, Zika virus, and HIV‐1, to invade host cells for their own replication (Ma et al, 2015; Marceau et al, 2016; Savidis et al, 2016; Park et al, 2017; Jin et al, 2018; Huang et al, 2019; Li et al, 2019), which greatly helped us understand the host–pathogen interaction during viral infection.
Here, we carried out a CRISPR‐based genetic screen in a latently HIV‐infected CD4+ T‐cell model of latency using a high complexity and whole genome‐wide sgRNA library. Among the enriched genes, we identified that PEBP1 or RKIP is associated with the suppression of HIV replication and promotes the establishment of HIV latency. The knockout of PEBP1 gene reactivated latent HIV‐1 by inducing Raf1/ERK/IκB and IKK/IκB/NF‐κB signaling pathways in several HIV latency models, including a primary CD4+ T‐cell model of HIV latency. Importantly, PEBP1 can directly inhibit HIV‐1 infection and induce HIV latency in primary CD4+ T cells. When PEBP1 was induced by a small molecule extracted from Chinese green tea, epigallocatechin‐3‐gallate (EGCG), the reactivation of HIV latency was effectively blocked in the primary CD4+ T cells isolated from HIV‐positive individuals receiving suppressive ART. To our knowledge, this is the first report to elucidate how upstream signaling of the NF‐κB pathway is controlled by PEBP1 or RKIP during the establishment of HIV latency. Our study has discovered mechanistically novel insights of HIV latency. The EGCG compound identified in this study could be further investigated as a new tool for therapeutic intervention of HIV latency in the future.
Results
Genome‐wide CRISPR/Cas9 library screening enriches host factors associated with the establishment of HIV latency
To identify host factors associated with HIV‐1 latency, we conducted a GECKO library screen (Shalem et al, 2014) in the CD4+ T model of HIV latency, C11, which was previously established in our laboratory (Qu et al, 2013; Wang et al, 2017). This GECKO library contains over 120,000 gRNAs targeting 19,050 human genes. The C11 cell line of HIV latency model is derived from CD4+ T cells (Jurkat), which harbors an HIV‐1 proviral DNA with a reporter gene encoding green fluorescent protein (GFP). In the latent state, HIV‐1 expression in C11 cells is silenced with an expression level of GFP below 2% (Qu et al, 2013; Wang et al, 2017). We prepared the GECKO lentivirus library to infect the C11 HIV latency model with a multiplicity of infection (MOI) of 0.2. The cell line was screened with puromycin (2 μg/ml) selection for 14 days (Fig 1A). Then, roughly 10% of GFP‐positive C11 cells were enriched after two rounds of cell sorting (Fig 1A and B). For the positive knockout cells, the targeted sgRNA sequence was confirmed by PCR. Our data showed that the integrated sgRNA was found in both the unselected group and the positive cells sorted in the first or second round of screening (Fig 1C). Under fluorescence microscopy, we confirmed that the sorted C11 cells were GFP positive after gene knockout (Fig 1D). Following genomic DNA (gDNA) extraction from both sorted and unsorted control cells and PCR amplification of each sgRNA sequence, we performed Illumina sequencing to generate read counts for each gene‐targeting GECKO construct. The gene of interest was compared with the distribution of the log2 enrichment values of the negative control sgRNAs and the initial control sgRNAs. We found that several genes such as UBB, SERBP1, ZDHHC1,CNTNAP1, and PEBP1 (Beshir et al, 2010; Lin et al, 2010; Oh et al, 2013; Laquerriere et al, 2014; Bolger, 2017) are among the hotspot candidate genes with high abundance of sgRNAs, along with BRD2 and BRD4 genes which are known as HIV latency‐associated genes (Boehm et al, 2013) (Fig 1E and Dataset EV1).
Figure 1. A pooled, genome‐wide CRISPR screen for candidate genes involved in HIV‐1 latency.
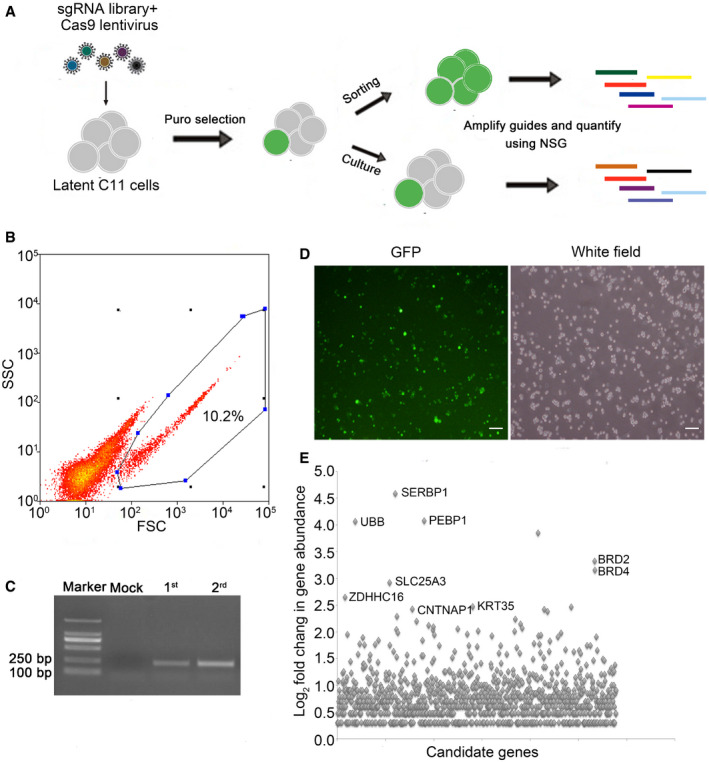
- The outline of the genome‐wide CRISPR screen strategy. C11 cells were infected with a lentiviral library containing Cas9 proteins and sgRNAs that target 19,050 human genes. After fourteen days of puromycin selection, genomic DNA was extracted from GFP‐positive cells after two rounds of sorting. The candidate genes were then identified by next generation sequencing.
- Flow cytometry of cells infected with the lentiCRISPR v2.0 library where the expression of GFP indicates latency reactivation. Target genes were enriched through two‐round sorting. Continuously cultured C11 latent cells infected with the lentiCRISPRv2.0 served as control. Blue dots and the encircled area represent the GFP‐positive cells for flow cytometry analysis.
- Validation of sgRNAs in unsorted and sorted C11 cells by PCR after lentiCRISPR v2.0 library infection.
- Before next generation sequencing, the GFP expression in two‐round sorted C11 cells was confirmed by fluorescence microscopy. Scale bar, 100 μm.
- Fold change (Log2) of the abundance of target genes in sorted C11 cells. Enriched sgRNA genes are highlighted.
Validation of candidate HIV latency inducing genes
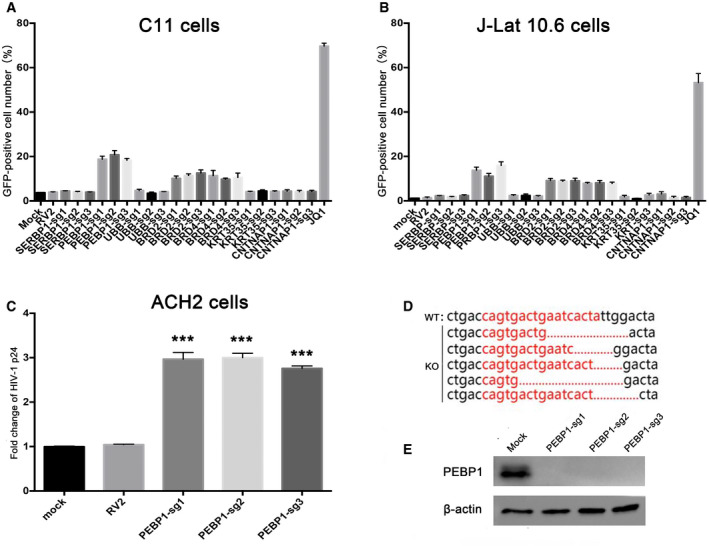
To validate whether these candidate genes are related to HIV‐1 transcription, we directly infected HIV latency model of C11 cells with CRISPR/Cas9 lentiviruses containing sgRNAs which target these candidate genes, followed by clone selection with puromycin (2 μg/ml) for 14 days. Among these top‐hit genes, we found that the knockout of PEBP1 gene significantly induced the reactivation of latent HIV‐1 (roughly 20%), which was higher than the cells with the known HIV latency‐related gene BRD2 or BRD4 knockout (Fig 2A), supporting our idea that PEBP1 gene is associated with HIV latency. A similar effect was observed after PEBP1 was knocked out in two other HIV latency models, J‐Lat 10.6, and ACH2 cells (Fig 2B and C). To prove that PEBP1 gene was indeed knocked out in these latency disrupted C11 cells, we sequenced the genomic targeting sites in these PEBP1 knockout cell clones by genomic DNA sequencing. We found that PEBP1 gene was deleted in the target sites of PEBP1 sgRNA1 with different forms of indels (Fig 2D). The PEBP1 protein was nearly undetectable after the knockout of PEBP1 gene targeted with PEBP1‐specific guide RNAs (Fig 2E). In order to further determine the effect of PEBP1 on HIV‐1 latency, a random PEBP1 monoclonal cell line was obtained by flow sorting (Streaming data is not shown). We sequenced the genomic target sites of five different monoclonal cell lines by genomic DNA sequencing and found that PEBP1 gene was successfully knocked out in all five monoclonal cell lines (Fig EV1A). With Western blot, we confirmed that PEBP1 protein expression significantly decreased (Fig EV1B). The levels of HIV‐1 transcription were similar among these monoclonal cell lines and cells without clone screening after gene knockout (Figs 2A and EV1C). Cell proliferation and apoptosis were also evaluated in PEBP1‐KO‐C11 cells and mock C11 cells by CCK8 assay and TUNEL staining. We found the proliferation rate was slightly higher in PEBP1‐KO‐C11 cells than that of mock knockout C11; however, PEBP1 knockout did not affect apoptosis (Fig EV2A and B). Taken together, our data suggest that PEBP1 is a new HIV latency‐associated gene.
Figure 2. Validation of the candidate genes screened from the lentiCRISPR v2.0 library in HIV‐1 latently infected cell lines.
-
AValidation of the top candidate genes in the C11 cell line. C11 cells were infected by lentiCRISPR v2.0 packaged lentiviruses with sgRNA following by screening for 14 days with 2 μg/ml puromycin. The percentage of GFP‐positive cells was measured by flow cytometry to determine the level of HIV‐1 reactivation.
-
B, CThe effect of candidate genes on HIV latency was further verified in J‐Lat 10.6 (B) and ACH2 (C) models of HIV latency. GFP expression in J‐Lat 10.6 cells and p24 in ACH2 cells was analyzed by flow cytometry and ELISA, respectively.
-
DPEBP1 was deleted after CRISPR/Cas9 knockout. PCR products related to PEBP1 from control or PEBP1 knockout cells were cloned and then sequenced. PEBP1‐sg1 target sites are shown in red letters. Dashes indicate the deleted bases relative to the wild‐type PEBP1 gene sequence.
-
EPEBP1 protein levels were measured by Western blot after knock out in C11 cells by LvPEBP1‐sg1. Mock C11 cells served as control.
Figure EV1. Monoclonal analysis of PEBP1‐KO‐C11 cell line.
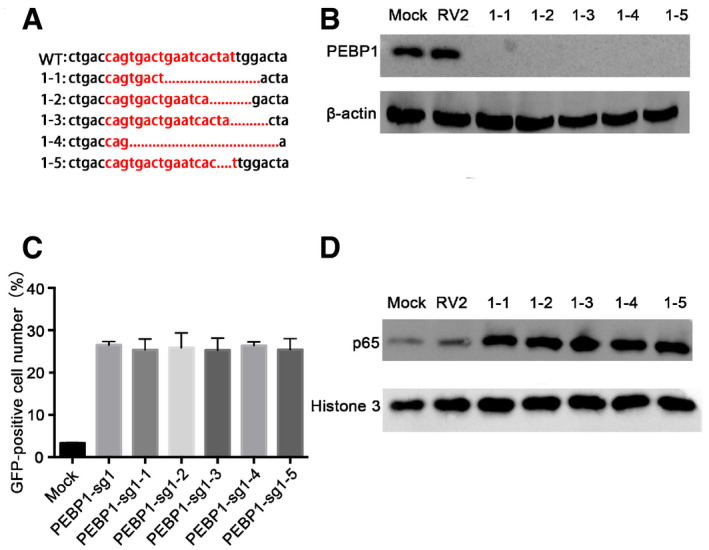
- Sequencing of PEBP1 PCR products after monoclonal sorting. The PCR products of PEBP1 gene were cloned then sequenced. PEBP1‐sg1 target sites were shown in red letters. Dashes indicate the deleted bases relative to the wild‐type sequence. 1‐1, 1-2, 1‐3, 1-4, and 1‐5 represent different monoclonal cell lines, respectively.
- The expression of PEBP1 was detected by Western blot in individual monoclonal cell lines.
- The proportion of GFP‐positive cells was detected by flow cytometry after PEBP1 knockout.
- The levels of nuclear NF‐κB/p65 protein were analyzed by Western blot in different monoclonal cell lines and the mock C11 cells.
Figure EV2. Effect of PEBP1 knockout on the proliferation and apoptosis of latently infected C11 cells. The NF‐κB nuclear entry inhibitor SC75741 prevents the reactivation of HIV‐1 after PEBP1 knockout in C11 cell model of latency.
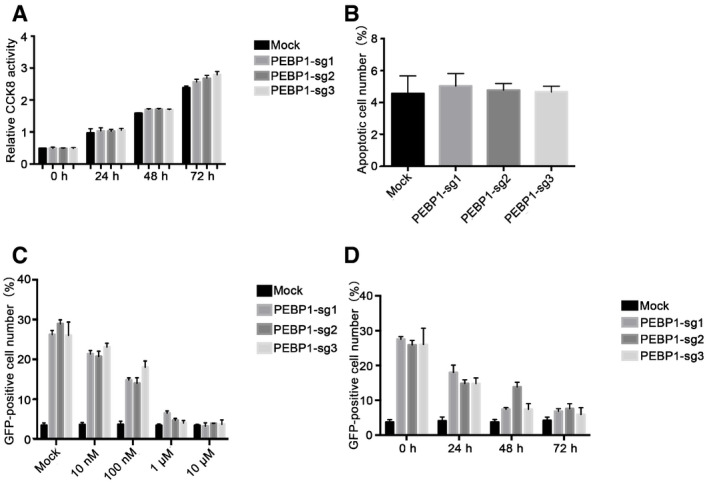
- Cell proliferation of PEBP1‐KO-C11 cells and mock C11 cells was analyzed by CCK‐8 levels.
- Apoptosis of PEBP1‐KO-C11 cells and mock C11 cells was measured by TUNEL staining followed by flow cytometry.
- The expression of GFP in the cells was measured by flow cytometry in C11‐PEBP1‐KO and mock C11 cells after treatment with NF‐κB inhibitor SC75741 for 48 h.
- The expression of GFP in the cells was detected by flow cytometry in C11‐PEBP1‐KO and mock C11 cells after treatment with 1 μM of NF‐κB inhibitor SC75741 at different time points.
The PEBP1/Raf1 protein complex promotes HIV‐1 latency through inactivation of MAPK and NF‐κB signaling pathways in CD4+ T cells
Previous studies indicated that PEBP1, also known as Raf1 kinase inhibitor protein RKIP, is involved in MAPK and NF‐κB signaling pathways via interaction with Raf1 and IKK in cancer cells (Yeung et al, 2001; Lee et al, 2006; Tavel et al, 2012; Wei et al, 2015). When PEBP1 is defective, the MAPK and NF‐κB signaling pathways are activated, leading to the translocation of NF‐κB from the cytoplasm to the nucleus (Lin et al, 2010). It has been demonstrated that inactivation of NF‐κB signaling is essential for the establishment of HIV latency while activation of NF‐κB signaling pathway disrupts latent HIV‐1 (Fiume et al, 2012). However, it is not clear how cytoplasmic sequestration of NF‐κB is regulated during the establishment of HIV latency in CD4+ T cells. We hypothesized that PEBP1 gene is essential for the upstream signaling of NF‐κB by preventing its translocation into the nucleus, therefore turning off the transcription of HIV for transcriptional silence. We found that both the expression of PEBP1 mRNA and protein in the C11 HIV latent cell line were significantly higher than its parental Jurkat cell line (Fig 3A). To see whether the expression of PEBP1 causes silencing of HIV by inactivating NF‐κB signaling pathway and whether this is through its interacting with Raf1 and IKK, we performed a co‐immunoprecipitation (Co‐IP) assay and revealed that PEBP1 not only interacted with Raf1 but also with IKK in C11 HIV latency cell model (Fig 3B). After PEBP1 was knocked out, the expression levels of MEK1/2, RSK, ERK1/2, IKKβ, and IKBα was marginally changed; however, their phosphorylation levels increased significantly, indicating that the Raf1/ERK/IκB and IKK/IκB/NF‐κB signaling pathways were indeed activated when PEBP1 gene was knocked out (Fig 3C). The level of NF‐κB/p65 significantly increased in the nucleus when PEBP1 gene was knocked out (Fig 3D), leading to a significant binding of NF‐κB/p65 to HIV‐1 LTR in C11‐PEBP1‐KO cells but not in the control C11 cells (Fig 3E). In addition, more NF‐κB/p65 entered the nucleus after PEBP1 was knocked out in monoclonal knockout cell lines (Fig EV1D).
Figure 3. PEBP1 inhibits HIV‐1 reactivation by inactivating Raf1/IKK/NF‐κB signaling pathways.
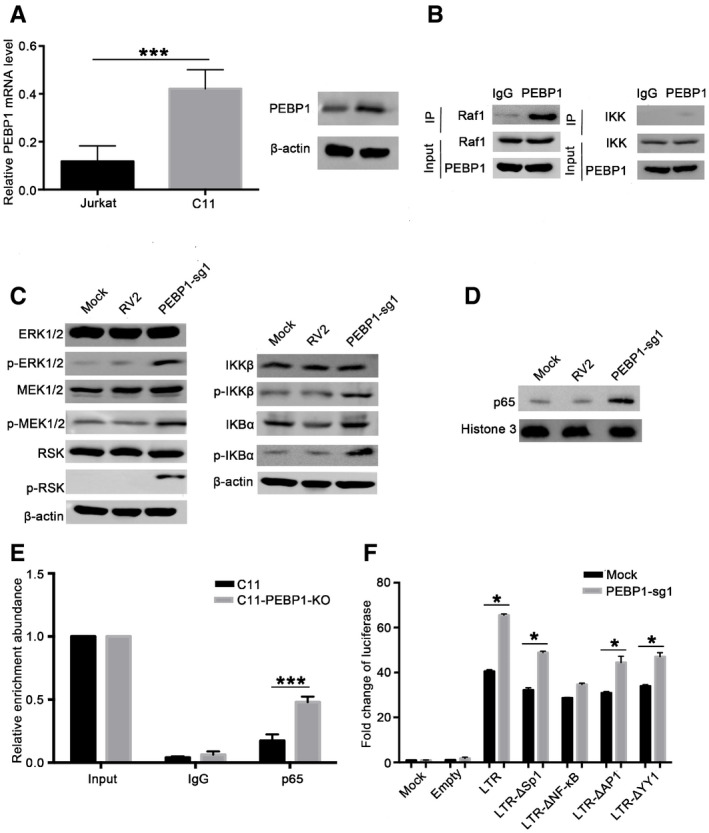
-
APEBP1 expression was measured by qPCR (left panel) and Western blot (right panel) in the latently infected C11 cells and the parental Jurkat cells.
-
BThe immunoprecipitation assay was performed in C11 cell lysates with anti‐PEBP1 antibody followed by Western blot with anti‐Raf1 or anti‐IKK antibodies.
-
C, DEffect of PEBP1 on the Raf1/ERK/IκB and IKK/IκB/NF-κB signaling pathways. The levels of indicated proteins in total protein lysates (C) or the levels of NF‐κB/p65 protein in nucleus (D) were analyzed by Western blot in C11‐PEBP1‐KO cells and mock C11 cells.
-
ENF‐κB/p65 protein recruitment into HIV LTR was analyzed by ChIP‐qPCR with specific primers targeting the HIV LTR after normalization to the input.
-
FThe impact of PEBP1 on the activation of the HIV LTR was explored by luciferase reporter assay in 293T cells. 293T cells were co‐transfected with PEBP1‐sg1 alone or PEBP1‐sg1 with HIV‐1 LTR‐empty plasmids, HIV‐1 wild‐type LTR‐luciferase plasmids, HIV‐1 LTR‐Δsp1‐luciferase, HIV‐1 LTR‐ΔNF‐κB‐luciferase, HIV‐1 LTR‐ΔAp1‐luciferase, or HIV‐1 LTR‐ΔYY1‐luciferase. Transcription of HIV‐1 was determined by luciferase reporter assay.
To see whether PEBP1 regulates HIV transcription through its interaction with NF‐κB signaling, 293T cells were transiently transfected with wild‐type or mutated LTR‐driven luciferase reporter plasmids with deletion mutations in YY1‐binding site, Sp1‐binding site, Ap1‐binding site, or NF‐κB‐binding site at the HIV LTR (Wang et al, 2005; Gary et al, 2008), after PEBP gene knockout. We found that there were significant differences in luciferase expression among cells transfected with all other deletion mutations except cells transfected with NF‐κB‐binding site‐deleted HIV LTR‐driven luciferase reporter plasmids when PEBP1 was knocked out (Fig 3F). When NF‐κB nuclear entry was pharmacologically inhibited by NF‐κB inhibitor SC75741, the GFP expression level of PEBP1‐KO‐C11 cells decreased in a dose or time‐dependent manner (Fig EV2C and D). These observations further supported our hypothesis that the regulation of PEBP1 in HIV transcription is directly through NF‐κB signaling.
The induction of PEBP1 inhibits HIV‐1 replication during viral infection
Next, we wanted to determine whether PEBP1 can directly inhibit the viral transcription in the early stage of infection. To test this idea, PEBP1 was overexpressed in TZM‐bl cell line (Fig 4A). Then, the PEBP1 overexpressing TZM‐bl cells were infected with HIV‐1 in serum supernatants isolated from patient peripheral blood. Patient serum supernatants were collected by low‐speed centrifugation from blood containing the virus with a viral load of 129 copies/ml. Both TZM‐bl cells and supernatants were harvested 72 h post‐infection where HIV‐1 transcription was measured by luciferase activity and p24 expression, respectively. Our data showed that the overexpression of PEBP1 significantly inhibited the transcription and replication of HIV‐1 (Fig 4B and C). It has been reported that EGCG and Dihydroartemisinin (DHA) are PEBP1 inducers (Kim & Kim, 2013; Hu et al, 2014). In HIV latency model of C11 cells, both EGCG and DHA effectively induced PEBP1 protein expression (Fig 4D). Importantly, EGCG or DHA can also induce PEBP1 in primary CD4+ T cells (Fig 4E), which significantly reduced the level of p24 in the culture supernatants compared with mock cells (Fig 4F). These data indicate that the induction of PEBP1 expression directly restricts HIV‐1 transcription during the early stage of viral infection.
Figure 4. PEBP1 restricts HIV‐1 transcription to induce HIV latency.
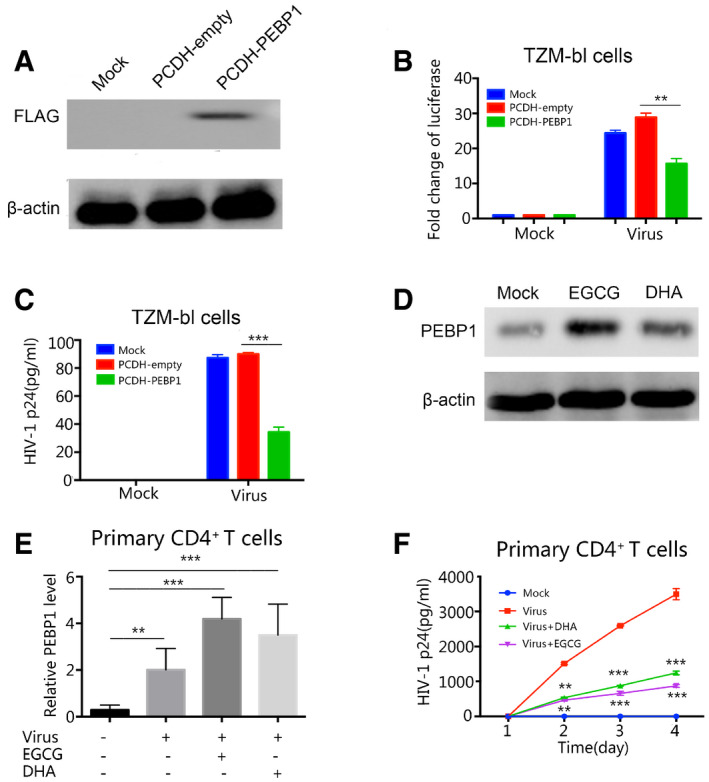
-
AThe expression of PEBP1 was analyzed by Western blot in TZM‐bl cells infected with mock, Lv‐PCDH-empty, or Lv‐PCDH-PEBP1 plasmids.
-
B, COverexpression of PEBP1 suppressed HIV‐1 replication. TZM‐bl cells were transfected with Lv-PCDH‐empty or Lv‐PCDH-PEBP1 followed by infection of HIV derived from patient plasma whose viral load is 129 copies/ml. The transcription of HIV‐1 was evaluated 72 h post‐infection by luciferase activity (B) and levels of p24 in the supernatants (C).
-
DThe expression level of PEBP1 was detected by Western blot using the whole cell lysate of C11 cells treated with 10 μM EGCG or DHA for 24 h.
-
EInduction of PEBP1 by EGCG or DHA in HIV‐1 infected primary CD4+ T cells. Primary CD4+ T cells from healthy donors were treated with 10 μM EGCG or DHA during HIV‐1 infection. Similar to Panel B, HIV‐1 was isolated from the blood supernatants of patients receiving ART with a viral load of 129 copies/ml. Seventy‐two hours post‐treatment with 10 μM EGCG or DHA, the expression of PEBP1 was detected by qPCR.
-
FThe induction of PEBP1 by EGCG or DHA suppressed HIV replication in the primary CD4+ T cells. The primary CD4+ T cells from healthy donors were treated with 10 μM EGCG or DHA during HIV‐1 infection. Similar to Panel B and E, HIV‐1 was isolated from the blood supernatants of patients receiving ART with a viral load of 129 copies/ml. The supernatants from HIV‐1-infected CD4+ T cell were collected 1, 2, 3 or 4 days post‐infection. Replication of HIV‐1 was analyzed by p24 ELISA.
PEBP1 prevents HIV latency reversal in primary CD4+ T cells isolated from HIV‐positive individuals receiving ART
When tested in primary CD4+ T cells purified from PBMCs from patients under ART (Shan et al, 2013), we found that EGCG, the PEBP1 agonist, significantly prevented the reactivation of latent HIV under α‐CD3/CD28 stimulation in four out of five patients examined (Fig 5A; Table EV1). In addition, the gene expression of PEBP1 from reactivation‐restricted samples was significantly induced (Fig 5B). As an example, EGCG significantly induced protein expression of PEBP1 in cells from patient 1 compared with control treatment (Fig 5C), along with a reduction of nuclear protein NF‐κB/p65 induced by α‐CD3/CD28 stimulation (Fig 5D). Taken together, these data further validate that PEBP1 suppresses HIV transcription and prevents the reactivation of latent HIV by inactivating the NF‐κB signaling pathway ex vivo.
Figure 5. PEBP1 induction by EGCG inhibits the reactivation of latent HIV in primary CD4+ T cells isolated from patients receiving ART .
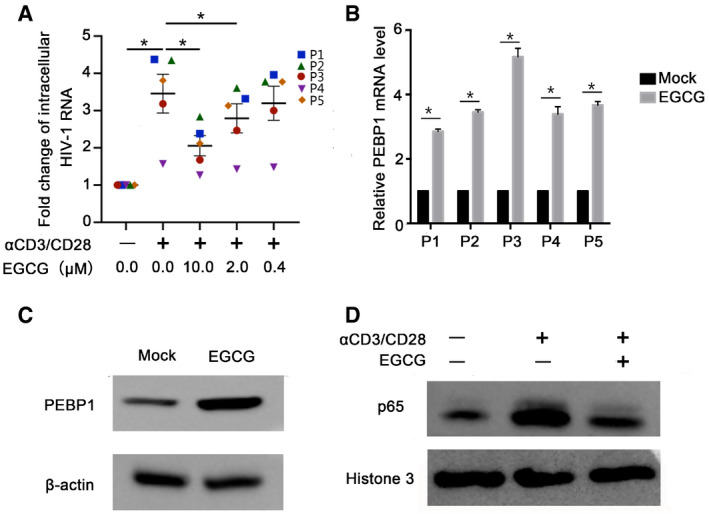
- EGCG inhibited α‐CD3/CD28 reactivation of latent HIV‐1. Primary CD4+ T cells were isolated from HIV‐1-positive patients on antiretroviral therapy (n = 5, P1–P5). Primary CD4+ T cells were treated with α‐CD3/CD28 alone or α‐CD3/CD28 and 10 μM EGCG. Cell‐associated RNA was extracted 7 days post‐treatment. The transcription of HIV‐1 was determined by real‐time qPCR.
- PEBP1 mRNA was induced by EGCG in patient primary CD4+ T cells. Primary CD4+ T cells were treated with 10 μM EGCG. The expression of PEBP1 in the cells was measured by qPCR.
- PEBP1 protein was induced by EGCG in patient primary CD4+ T cells, which was determined by Western blot of whole cell lysate from primary CD4+ T cells.
- EGCG‐induced PEBP1 suppressed nuclear entry of NF‐κB/p65. This was measured by Western blot of nuclear protein of primary CD4+ T cells treated with α‐CD3/CD28 or α‐CD3/CD28 plus 10 μM EGCG.
PEBP1 induces HIV latency in primary CD4+ T cells
The data above supports our hypothesis that PEBP1 is a latency inducing gene. To directly test whether PEBP1 promotes HIV‐1 latency in the primary human CD4+ T lymphocyte, we established a primary HIV‐1 latent infection model as described before (Bosque & Planelles, 2011; Kim et al, 2014; Pandeló José et al, 2014) (Fig 6A). Nanoluc‐luciferase expression was detected 2 days post‐HIV‐1 infection to indicate active HIV replication. Twelve days post‐gradual decreasing of IL‐2, a significant reduction of nanoluc‐luciferase was observed (Fig 6B), indicating that HIV‐1 was in a latent state. Importantly, PEBP1 gene expression decreased during early active viral infection (day 3 post‐infection). However, during the latency stage induced by a continuous decrease of IL‐2 concentration, PEBP1 gene expression significantly increased until latency was established at day 12 post‐infection (Fig 6C). These data indicates that PEBP1 gene expression is positively related to latency induction in primary CD4+ T‐cell model of latency.
Figure 6. PEBP1 reactivates latent HIV‐1 in the primary CD4+ T model of latency.
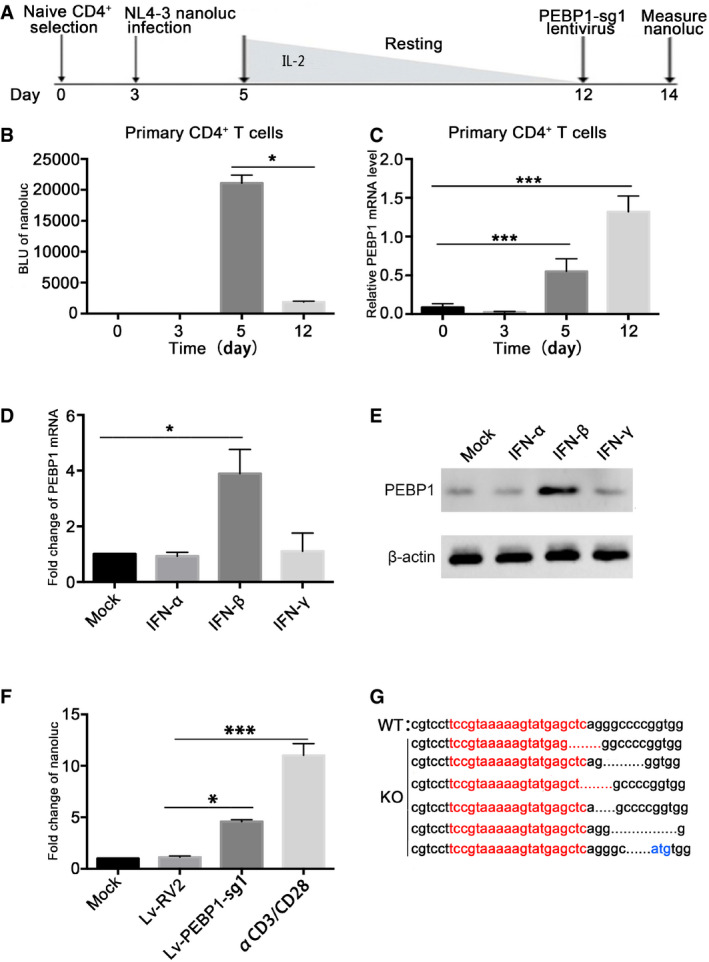
-
AOutline of latency establishment in the primary CD4+ T cells. Human primary CD4+ T cells were activated and expanded with α‐CD3/CD28 beads at day 1. The α‐CD3/CD28 beads were removed at day 3. Cells were then infected with HIV‐1 NL4.3‐nanoluc at 3rd day after expansion and maintained over 7 days with a decreasing concentration of IL‐2 to establish latency until day 12. At day 12, cells were infected with CRISPR/Cas9 and PEBP1‐sgRNA lentivirus.
-
BTranscription of HIV‐1 in the primary CD4+ T cells was determined by nanoluc‐luciferase assays during HIV‐1 infection.
-
CAfter infection with VSVG pseudotyped HIV‐1 NL4.3‐nanoluc, the mRNA expression of PEBP1 was measured by qPCR.
-
D, EJurkat CD4+ T cells were treated with IFN‐α, IFN‐β, and IFN‐γ for 24 h. The mRNA (A) or protein (B) expression levels of PEBP1 were measured by qPCR or Western blot.
-
FPEBP1 knockout enhanced HIV‐1 transcription in the primary CD4+ T‐cell model of latency. The expression of HIV‐1 was measured by nanoluc after gene knockout where α‐CD3/CD28 stimulation served as a positive control.
-
GPEBP1 gene deletion after Lv‐PEBP1‐sg1 knockout in the primary CD4+ T cells. The PCR products of PEBP1 were cloned and then sequenced. PEBP1‐sg1 target gene sequences are shown in red letters. Dashes indicate deleted bases relative to the wild‐type sequence.
It has been shown that many factors involved with the restriction of HIV‐1 transcription are related to interferon signaling (Hotter et al, 2019; Liu et al, 2019). We hypothesize that PEBP1 induction is related to interferon signaling during HIV‐1 infection of primary T lymphocytes. We found that both mRNA and protein levels of PEBP1 were increased significantly after treatment of Jurkat CD4+ T cells with IFN‐β, but not IFN‐α or IFN‐γ, for 48 h (Fig 6D and E). These observations indicate HIV‐1 infection‐triggered innate immunity in T lymphocytes may promote the expression of PEBP1 through IFN‐β. Enhanced PEBP1 expression inhibits HIV‐1 transcription and promotes HIV latency. In contrast, when PEBP1 was knocked out, the expression of HIV‐1 was increased (Fig 6F and G), further supporting our hypothesis that PEBP1 is involved in HIV latency.
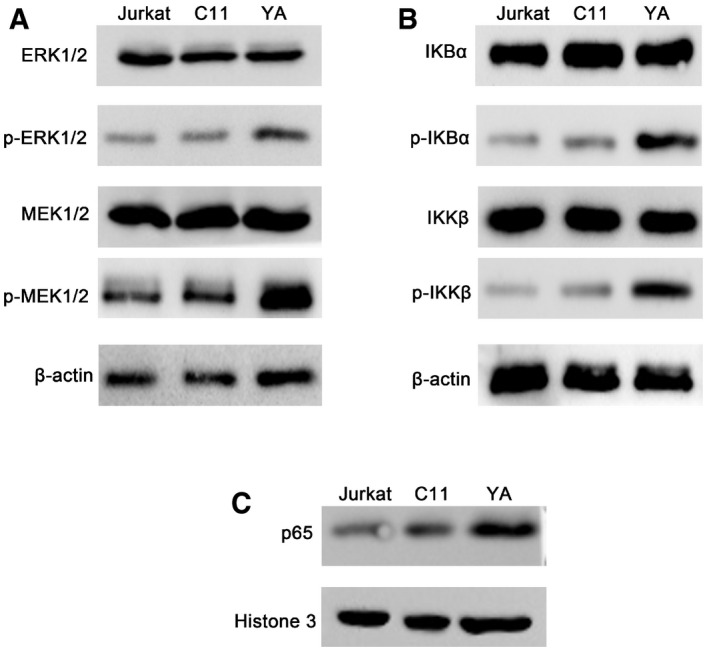
We want to know whether the activation of MAPK and IKK signaling pathways was different in CD4+ T cells with or without infection. While both of the pathways were highly active in HIV‐1 replicating YA CD4+ T cells, MAPK or IKK signaling was mostly inactive in the HIV latently infected C11 cell line (Fig EV3A and B). Interestingly, the nuclear level of p65 in HIV‐1 latently infected C11 cells was slightly lower than Jurkat cells but significantly lower than YA CD4+ T cells where HIV‐1 was highly replicated (Fig EV3C). Considering PEBP1 was highly expressed in C11 latent cells (Fig 3A) and knockout of PEBP1 enhanced the activation of MAPK and IKK signaling pathways by phosphorylation of ERK1/2, MEK1/2, RSK, IKKβ, and IκBα (Fig 3C), these data indicate that PEBP1 inhibits the activation of MAPK and IKK signaling pathways and prevents NF‐κB from entering the nucleus to induce HIV latency in CD4+ T cells.
Figure EV3. Differential activation of MAPK, IKK, and NF‐κB signaling pathways in uninfected Jurkat CD4+ T cells, HIV‐1 latently infected C11 CD4+ T cells, and HIV‐1 actively infected YA CD4+ T cells.
-
A, BActivation of Raf1/ERK/IκB (A) or IKK/IκB (B) signaling pathway was analyzed in total protein lysates by Western blot in Jurkat, C11, and YA cells.
-
CNuclear levels of NF‐κB/p65 protein were analyzed by Western blot in Jurkat, C11, and YA cells.
Source data are available online for this figure.
Discussion
In this study, we conducted a CRISPR genome‐wide knockout library screen in an HIV latently infected cell line model. We discovered a previously unrecognized PEBP1/IKK/IκB/NF‐κB signaling and PEBP1/Raf1/ERK/IκB signaling in the suppression of HIV transcription to induce the establishment of HIV latency. PEBP1 prevents NF‐κB from translocation into the nucleus to induce HIV latency in resting CD4+ T cells by directly regulating the upstream signaling of NF‐κB. Accordingly, we proposed a working model that PEBP1 restricts HIV‐1 transcription and is associated with HIV‐1 latency (Fig 7). In the transcription initiation stage, protein level of PEBP1 is low; therefore, PEBP1 is relieved from its inhibitory function of Raf1 or IKK, leading to the phosphorylation of IKKβ and IκBα, the degradation of IKBα, and the translocation of NF‐κB into the nucleus followed by its recruitment into the HIV LTR to activate HIV transcriptional machinery. On the contrary, when HIV‐1 enters the latency state, PEBP1 protein is induced. A high level of PEBP1 protein interacts with both Raf1 and IKK to inhibit the NF‐κB signaling cascade, resulting in the sequestration of p65/p50 heterodimer in the cytoplasm, thereby silencing HIV‐1 transcription. It has been reported that PEBP1 forms a protein complex with Raf1 or IKKβ (Yeung et al, 2001; Granovsky et al, 2009). Our data also supported that PEBP1 formed a protein complex with Raf1 and/or IKK to suppress NF‐κB signaling and induce HIV latency in CD4+ T cells. However, it is not clear how the Raf1 or IKK phosphorylation signaling is blocked. It could be that within a protein complex with PEBP1, the kinase enzyme sites in these two proteins are blocked; therefore, the sites are inaccessible by their downstream target proteins. Importantly, we found that PEBP1 protein level can be induced in the primary CD4+ T cells by pharmacologic small molecule EGCG, a compound discovered in Chinese green tea, and DHA. The induction of PEBP1 indeed prevents the reactivation of latent HIV‐1 in resting CD4+ T cells isolated from ART‐suppressed patients. It has been reported that PEBP1 is also essential for type I interferon production in its anti‐viral innate immunity (Gu et al, 2016). Our data are consistent with these observations and showed that IFN‐β innate immune response may induce PEBP1 early in HIV‐1 infection. Our study uncovered a previously unrecognized novel molecular mechanism of how the upstream signaling of canonical NF‐κB pathway is finely regulated by PEBP1/Raf1/IKK signaling during the establishment of HIV latency. Our findings fill the knowledge gap regarding how NF‐κB is sequestered into cytoplasm to inactivate initiation of HIV‐1 transcription during the establishment of HIV latency in resting CD4+ T cells, which has not been understood for many years. Interestingly, it has been reported that PEBP1 interacts with Nef by a yeast two‐hybrid screen assay (Kammula et al, 2012). Whether HIV‐1 Nef directly impacts PEBP1/Raf1 signaling for its own replication and whether PEBP1 affects other steps of HIV replication warrants further investigation in the future.
Figure 7. A working model of the role of PEBP1 in the establishment of HIV latency.
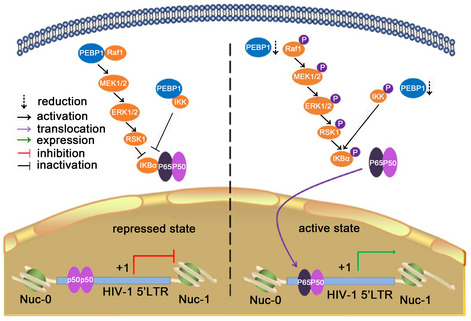
PEBP1 interacts and inhibits the activity of Raf1 and IKK kinases. This blocks the phosphorylation of downstream proteins ERK1, IKK, and IκBα, resulting in the sequestration of p65/p50 heterodimer in the cytoplasm, thereby silencing HIV‐1 transcription. When PEBP1 is knocked out, Raf1 and IKK signaling pathways are activated to phosphorylate IκBα, thereby releasing p65/p50 heterodimer to translocate into the nucleus to drive HIV‐1 transcription.
ShRNA libraries have been previously used to screen host genes related to HIV latency. Unfortunately, among the genes with the most significant changes in abundance, no HIV latency‐related genes were discovered (Besnard et al, 2016). Compared with shRNA knockout libraries, CRISPR technology can completely disrupt gene expression while shRNAs can only temporarily reduce gene expression levels. Therefore, CRISPR technology was proposed as a better alternative for the identification of genes that play a functional role in a low abundance (Shalem et al, 2014). With this technology, tyrosylprotein sulfotransferase 2 (TPST2), solute carrier family 35 member B2 (SLC35B2) and activated leukocyte cell adhesion molecule (ALCAM) were identified as genes related to HIV‐1 entry into CD4+ T cells (Park et al, 2017). Similarly, histone demethylase MINA53 and proteasome signaling pathways were recently reported as potential novel HIV‐1 latency‐promoting genes (Huang et al, 2019; Li et al, 2019). While it is not known whether these newly discovered HIV latency‐related genes have any functional links with each other, these studies demonstrate that CRISPR/Cas9 library screening is a powerful tool for HIV latency study.
In summary, by high‐throughput CRIPR/Cas9 library screening, we have discovered that PEBP1 suppresses HIV‐1 transcription and is essential for the establishment of HIV latency in CD4+ T cells by regulating upstream signaling of the NF‐κB pathway. NF‐κB signaling is among one of the most important targets currently investigated in the development of HIV cure strategies. The finding that the induction of PEBP1 by EGCG or DHA inhibits NF‐κB function to suppress latency reversal indicates that upstream signaling of NF‐κB pathways could be exploited to enforce HIV latency for a functional HIV cure.
Materials and Methods
HIV‐1 latent cell lines
The HIV‐1 latent infection model C11 cell line (constructed in our laboratory) (Qu et al, 2013; Wang et al, 2017) and J‐Lat 10.6 cell line (obtained from NIH AIDS Reagent Program; Jordan et al, 2001, 2003) contain a single integrated latent HIV‐GFP reporter genome. ACH2 is a clone of HIV‐1 latently infected CD4+ CEM cells that contains a single copy of proviral DNA per cell (obtained from NIH AIDS Reagent Program). HeLa‐based TZM‐bl cells contain an integrated HIV LTR‐luciferase construct (obtained from NIH AIDS Reagent Program).
Cell culture
C11, J‐Lat 10.6, ACH2, Jurkat, and YA (Qu et al, 2013) cells were cultured in RPMI1640 (Gibco, C11875500BT) with 10% fetal bovine serum (FBS) (Gibco, 10110154) and 1% penicillin/streptomycin (Gibco, 15140‐122) in a 37 °C incubator containing 5% CO2. TZM‐bl and 293T cells were cultured in DMEM (Gibco, C11995500BT) and supplemented with 10% fetal calf serum (Lonsera, S711‐001S), and 1% penicillin/streptomycin (Gibco) in a 37 °C incubator containing 5% CO2.
Antibodies and reagents
The following antibodies were used throughout this study: Anti‐PEBP1 (ab76582) and anti‐Histone 3 (ab1791), anti‐NF‐κB p65 (ab16502), and anti‐IKK (ab178870) were purchased from Abcam (Cambridge, UK). Anti‐IKKβ (8943), anti‐Phospho‐IKKβ (Ser176/180) (14938), anti‐IκBα (9242), anti‐Phospho‐IκBα (Ser32) (2859), anti‐RSK (9533), anti‐Phospho‐RSK (Thr359/Ser363) (9341), anti‐MEK (9126), anti‐Phospho‐MEK1/2 (Ser217/221) (9154), anti‐ERK1/2 (4695), anti‐Phospho‐ERK1/2 (Thr202/Tyr204) (9101), and anti‐β‐Actin (4970) were purchased from Cell Signaling Technology (MA, US). 2 × Taq Master Mix (P112) and High fidelity PCR enzyme‐ 2 × Phanta Max Master Mix (P515) were purchased from Vazyme (Nanjing, China). PMD18‐T (6011) was purchased from Takara (Beijing, China). FastFire qPCR PreMix (SYBR Green) (FP208), Cell Genome Extraction Kit (DP304), and Plasmid Extraction Kit (DP103, DP108, DP117) were purchased from Tiangen (Beijing, China). Gel Extraction Kit (CW2302) was purchased from CWBIO (Nanjing, China). Luciferase and nanoluc detection kit (E6110, N1110) was purchased from Promega (Madison, USA). Recombinant human IFN‐alpha (11200‐1), recombinant human IFN‐beta (8499‐IF), and recombinant human IFN‐gamma (285‐IF) were purchased from R&D Systems (UN). Cell Counting Kit‐8 (CCK‐8) and TUNEL Apoptosis Detection Kit (FITC) were purchased from Yeasen Biotechnology Co., Ltd (Shanghai, China). NF‐κB inhibitor SC74751 (HY‐10496) was purchased from MCE (NJ, USA).
Pooled genome‐wide CRISPR screen
The lentivirus library was produced by co‐transfection of GECKO library plasmid, ∆8.91 and VSVG plasmid into HEK293T cells at a 1000‐fold concentration to increase the viral titer. A total of 1 × 107 C11 cells were infected at a low multiplicity of infection (MOI = 0.2), to ensure that most cells received only one viral construct. After 72 h, C11 cells were selected with 2 μg/ml puromycin for 14 days. After that, 108 puromycin‐resistant C11 cells were sorting by FACS to obtain GFP+ C11 cells. Sorted C11 cells were cultured for 1 week, and cell sorting was performed again. The gDNA was extracted by a genome extraction kit. PCR was performed with the indicated primers to confirm that the selected cell genome contains sgRNA targeting different genes.
Screen analysis
Sequencing reads were aligned to the sgRNA library, and the abundance of each sgRNA was calculated. SgRNAs with less than 25 counts in the initial set were removed from downstream analyses. The log2 fold change in abundance of each sgRNA was calculated for the sorted and unsorted final population samples.
Vector construction
Individual sgRNA constructs targeting SERBP1, PEBP1, UBB, BRD2, BRD4, KRT35, and CNTNAP1 were cloned into lentiCRISPR v2.0 (addgene 52961). For all other experiments, PEBP1‐sg1 was used.
For cDNA expression vectors, a linearized lentiviral backbone was generated from PCDH (YouBio, Hunan, China). Protein‐coding plasmids were gifts from Professor Han Jiahuai laboratory. All the constructed plasmids were confirmed by restriction enzyme digestion and DNA sequencing.
Cas9‐mediated gene knockout and cDNA overexpression
C11, J‐Lat 10.6, ACH2, and TZM‐bl cells were infected with lentivirus at an MOI of 1 and then selected with 2 μg/ml puromycin for 14 days. The knockout efficiency was analyzed using Sanger DNA sequencing. Single knockout cells were sorted by flow cytometry, and knockout efficiency was detected by Western blot (WB) analysis.
Visualization of GFP and flow cytometry assay
Green fluorescent protein (GFP), a marker for the activation of HIV‐1 in infected cells, was visualized by fluorescence microscopy after cell sorting. The cells were collected and washed with phosphate‐buffered saline (PBS). Cells were kept in PBS before analysis on a BD LSRII flow cytometer for enhanced GFP expression. FlowJo software (FlowJo LLC, Ashland, OR) was used to perform the flow cytometry analysis.
ELISA detection of antigen p24 levels
TZM‐bl and ACH2 cells were each seeded at a density of 1 × 106 on a 6‐well plate. After 48 h of culture, HIV‐1 production was measured via quantification of p24 in culture supernatant using p24 ELISA kit (R&D System, Minnesota, USA).
ChIP experiments
ChIP experiments were performed according to protocol provided by EZ‐ChIP chromatin immunoprecipitation kit (Millipore). Briefly, C11 and C11‐PEBP1‐KO cells were cross‐linked with 1% formaldehyde for 10 min at room temperature and quenched with 0.125 M glycine for 5 min. After lysis, nuclear extracts were separated and chromatin was sheared by sonicator (Bioruptor UCD‐200; Diagenode) for 10 min (10 s on and 10 s off) on ice to obtain DNA fragments of 200–1,000 bp in length. One percent of total sheared chromatin DNA was used as the input. Nuclear extracts were incubated with the indicated antibodies at 4°C overnight. Protein G/A‐labeled Dynabeads were added to each sample at 4°C for 2 h for immunoprecipitation. The immunoprecipitated DNA was analyzed by real‐time PCR with Thunderbird SYBR qPCR mix (Toyobo).The NF‐κB‐binding sites of HIV LTR was amplified using the following PCR primer pairs: 5′‐AGGTTTGACAGCCGCCTA‐3′ and 5′‐AGAGACCCAGTACAGGCAAAA‐3′.
Luciferase reporter assay
PEBP1‐sg1 (600 ng) was co‐transfected with LTR‐driven luciferase reporter plasmids containing the deletion of YY1‐binding site, Sp1‐binding site, Ap1‐binding site or NF‐κB‐binding site (100 ng) and internal mock pcDNA3.1 (100 ng) (as empty) using Hieff Trans™ Liposomal Transfection Reagent (Yeasen Biotechnology Co., Ltd., Shanghai, China) according to the manufacturer's instructions. The cells were harvested and lysed 72 h post‐transfection where luciferase activity of the lysates was then measured by Dual‐Luciferase Reporter Assay System (Promega). Each experiment was performed in triplicate. The genome of the TZM‐bl cells is integrated with a luciferase reporter driven by the HIV‐1 5′‐LTR promoter. Cells were harvested at 72 h post‐infection, and the lysate was assayed for luciferase activity. Triplicate cultures were measured for each experiment.
Cell proliferation by CCK‐8 assay
Control KO C11 and PEBP1‐KO‐C11 cells were seeded at a density of 0.5 × 104 on 96‐well plate. 10% CCK‐8 solution was added to fresh culture medium. The cells were incubated at 37°C for 1 h. The OD 450 nanometer value was measured to determine cell proliferation.
Apoptosis detected by TUNEL staining
A total of 1 × 106 cells were collected in a 1.5‐ml tube and centrifuged at 300 g for 5 min. The cells were washed twice with 500 μl PBS and analyzed with TUNEL‐FITC apoptosis detection kit according to the manufacturer's instructions. The proportion of FITC‐positive cells was assayed by flow cytometer and analyzed by FlowJo software (FlowJo LLC, Ashland, OR).
Western blot
A total of 1 × 106 cells were seeded in a 10‐cm dish and cultured for 24 h. Then, the cells were harvested, lysed, and subjected to Western blot. Membranes were visualized using the Immun‐Star WesternC Chemiluminescence Kit (Bio‐Rad), and images were captured using the ChemiDoc XRS+ System and processed using ImageLab software (Bio‐Rad).
Isolation of primary CD4+ T cells
Peripheral blood mononuclear cells (PBMCs) isolated from healthy donors were purchased from the Shanghai Blood Center (Shanghai, China). Naive CD4+ T cells were further purified from peripheral blood mononuclear cells by negative selection according to the manufacturer's instructions (Thermo). The Naive CD4+ T cells were maintained in serum‐free medium supplemented with 1% penicillin‐streptomycin and 5 ng/ml recombinant human interleukin‐2 (R&D) and 10 ng/ml recombinant human interleukin‐7 (R&D) at 37°C in 5% CO2.
Treatment of patient CD4+ T cells with EGCG
Shanghai Public Health Clinical Center approved this study, and the methods were carried out in accordance with the guidelines of Bullen CK’ laboratory (Kim et al, 2014). All research participants in this study gave written informed consent. HIV‐1‐infected individuals were enrolled under the criteria of suppression of viremia to undetectable levels (< 50 copies/ml) on ART for at least six months. Peripheral blood mononuclear cells (PBMCs) were purified using density gradient centrifugation from whole blood. CD4+ T lymphocytes were enriched by negative depletion (CD4+ T‐cell Isolation Kit, Miltenyi Biotec). EGCG was added at the time of α‐CD3/CD28 stimulation. Cells were treated with EGCG and stimulated with α‐CD3/CD28 for 72 h. HIV‐specific qPCR was conducted as described (Shan et al, 2013).
Statistical analyses
Data are representative of three independent experiments, and error bars represent standard errors (SD). Paired samples t‐tests were performed with use of SPSS version 13.0 (SPSS Inc., Chicago), and statistical significance was indicated at *P < 0.05, **P < 0.01, or ***P < 0.001.
Author contributions
HZ conceived and designed the experiments. XY carried out most experiments. YW, PL, YS, XZ, YZhu, ZJ, HY, HP, LZ, YZho, JW, ZL, and XS participated in some of the experiments. HZ, GJ, HL, HW, JX, SJ, and DL directed and supervised the experiments and interpretation of data. HZ and GJ generated the initial concepts of this study. XY, GJ, and HZ wrote the paper. All authors approved the publication.
Conflict of Interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Ms. Lilly M. Wong for her critical reading and editing. This work was supported by National Natural Science Foundation of China (31771484, 81761128020), National Grand Program on Key Infectious Disease (2017ZX10202102002). GJ is supported by Qura Therapeutics funding 2019‐01, University of North Carolina at Chapel Hill Center for AIDS Research (P30AI50410), and NIAID/CARE (1UM1AI126619). The funders have no roles in experimental design, data collection and analysis, interpretation of the data, or writing of this paper.
EMBO Reports (2020) 21: e49305
Data availability
No primary datasets have been generated or deposited.
References
- Alexaki A, Quiterio SJ, Liu Y, Irish B, Kilareski E, Nonnemacher MR, Wigdahl B (2007) PMA‐induced differentiation of a bone marrow progenitor cell line activates HIV‐1 LTR‐driven transcription. DNA Cell Biol 26: 387–394 [DOI] [PubMed] [Google Scholar]
- Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC et al (2012) Administration of vorinostat disrupts hiv‐1 latency in patients on antiretroviral therapy. Nature 487: 482–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archin NM, Sung JM, Garrido C, Soriano‐Sarabia N, Margolis DM (2014) Eradicating HIV‐1 infection: seeking to clear a persistent pathogen. Nat Rev Microbiol 12: 750–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barouch DH, Deeks SG (2014) Immunologic strategies for HIV‐1 remission and eradication. Science 345: 169–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beans EJ, Fournogerakis D, Gauntlett C, Heumann LV, Kramer R, Marsden MD, Murray D, Chun TW, Zack JA, Wender PA (2013) Highly potent, synthetically accessible prostratin analogs induce latent HIV expression in vitro and ex vivo . Proc Natl Acad Sci USA 110: 11698–11703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beshir AB, Ren G, Magpusao AN, Barone LM, Yeung KC, Fenteany G (2010) Raf kinase inhibitor protein suppresses nuclear factor‐kappaB‐dependent cancer cell invasion through negative regulation of matrix metalloproteinase expression. Cancer Lett 299: 137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard E, Hakre S, Kampmann M, Lim HW, Hosmane NN, Martin A, Bassik MC, Verschueren E, Battivelli E, Chan J et al (2016) The mTOR complex controls HIV latency. Cell Host Microbe 20: 785–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniasz PD, Grdina TA, Bogerd HP, Cullen BR (1999) Recruitment of cyclin T1/P‐TEFb to an HIV type 1 long terminal repeat promoter proximal RNA target is both necessary and sufficient for full activation of transcription. Proc Natl Acad Sci USA 96: 7791–7796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazkova J, Trejbalova K, Gondois‐Rey F, Halfon P, Philibert P, Guiguen A, Verdin E, Olive D, Van Lint C, Hejnar J et al (2009) CpG methylation controls reactivation of HIV from latency. PLoS Pathog 5: e1000554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, Aull K, Li P‐C, Planelles V, Bradner JE et al (2013) BET bromodomain‐targeting compounds reactivate HIV from latency via a tat‐independent mechanism. Cell Cycle 12: 452–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm D, Jeng M, Camus G, Gramatica A, Schwarzer R, Johnson JR, Hull PA, Montano M, Sakane N, Pagans S et al (2017) SMYD2‐mediated histone methylation contributes to HIV‐1 latency. Cell Host Microbe 21: 569–579.e566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger GB (2017) The RNA‐binding protein SERBP1 interacts selectively with the signaling protein RACK1. Cell Signal 35: 256–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque A, Planelles V (2011) Studies of HIV‐1 latency in an ex vivo model that uses primary central memory T cells. Methods 53: 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Engel D, Berrey MM, Shea T, Corey L, Fauci AS (1998) Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV‐1 infection. Proc Natl Acad Sci USA 95: 8869–8873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsheikh MM, Tang Y, Li D, Jiang G (2019) Deep latency: a new insight into a functional HIV cure. EBioMedicine 45: 624–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R et al (1997) Identification of a reservoir for HIV‐1 in patients on highly active antiretroviral therapy. Science 278: 1295–1300 [DOI] [PubMed] [Google Scholar]
- Fiume G, Vecchio E, De Laurentiis A, Trimboli F, Palmieri C, Pisano A, Falcone C, Pontoriero M, Rossi A, Scialdone A et al (2012) Human immunodeficiency virus‐1 Tat activates NF‐kappaB via physical interaction with IkappaB‐alpha and p65. Nucleic Acids Res 40: 3548–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gary DB, Joe W, Nathan C, Brett S, Stacey AR, Sergey AT, Andrew DB (2008) Infected cell killing by HIV‐1 protease promotes NF‐kB dependent HIV‐1 replication. PLoS ONE 3: e2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granovsky AE, Clark MC, McElheny D, Heil G, Hong J, Liu XD, Kim Y, Joachimiak G, Joachimiak A, Koide S et al (2009) Raf kinase inhibitory protein function is regulated via a flexible pocket and novel phosphorylation‐dependent mechanism. Mol Cell Biol 29: 1306–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Liu Z, Lai R, Liu S, Lin W, Ouyang C, Ye S, Huang H, Wang X (2016) RKIP and TBK1 form a positive feedback loop to promote type I interferon production in innate immunity. EMBO J 35: 2553–2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, Siliciano RF (2013) Replication‐competent noninduced proviruses in the latent reservoir increase barrier to HIV‐1 cure. Cell 155: 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotter D, Bosso M, Jønsson KL, Krapp C, Stürzel CM, Das A, Littwitz‐Salomon E, Berkhout B, Russ A, Wittmann S et al (2019) IFI16 targets the transcription factor Sp1 to suppress HIV‐1 transcription and latency reactivation. Cell Host Microbe 25: 858–872.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CJ, Zhou L, Cai Y (2014) Dihydroartemisinin induces apoptosis of cervical cancer cells via upregulation of RKIP and downregulation of bcl‐2. Cancer Biol Ther 15: 279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Kong W, Jean M, Fiches G, Zhou D, Hayashi T, Que J, Santoso N, Zhu J (2019) A CRISPR/Cas9 screen identifies the histone demethylase MINA53 as a novel HIV‐1 latency‐promoting gene (LPG). Nucleic Acids Res 47: 7333–7347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Togami H, Okamoto T (2010) Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV‐1 latency and its reactivation by BIX01294. J Biol Chem 285: 16538–16545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Liao Q, Chen J, Zhang L, He Q, Zhu H, Zhang X, Xu J (2018) TSC1 and DEPDC5 regulate HIV‐1 latency through the mTOR signaling pathway. Emerg Microbes Infect 7: 138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Defechereux P, Verdin E (2001) The site of HIV‐1 integration in the human genome determines basal transcriptional activity and response to Tat, transactivation. EMBO J 20: 1726–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Bisgrove D, Verdin E (2003) HIV reproducibly establishes a latent infection after acute infection of T cells in vitro . EMBO J 22: 1868–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammula EC, Mötter J, Gorgels A, Jonas E, Hoffmann S, Willbold D (2012) Brain transcriptome‐wide screen for HIV‐1 Nef protein interaction partners reveals various membrane‐associated proteins. PLoS ONE 7: e51578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessing CF, Nixon CC, Li C, Tsai P, Takata H, Mousseau G, Ho PT, Honeycutt JB, Fallahi M, Trautmann L (2017) In vivo suppression of HIV rebound by Didehydro‐Cortistatin A, a “Block‐and‐Lock” strategy for HIV‐1 treatment. Cell Rep 21: 600–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SO, Kim MR (2013) (‐)‐Epigallocatechin 3‐gallate inhibits invasion by inducing the expression of Raf kinase inhibitor protein in AsPC1 human pancreatic adenocarcinoma cells through the modulation of histone deacetylase activity. Int J Oncol 42: 349–358 [DOI] [PubMed] [Google Scholar]
- Kim M, Hosmane NN, Bullen CK, Capoferri A, Yang HC, Siliciano JD, Siliciano RF (2014) A primary CD4(+) T cell model of HIV‐1 latency established after activation through the T cell receptor and subsequent return to quiescence. Nat Protoc 9: 2755–2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Anderson JL, Lewin SR (2018) Getting the “Kill” into “Shock and Kill”: strategies to eliminate latent HIV. Cell Host Microbe 23:14–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Darcis G, Van Lint C, Herbein G (2015) Epigenetic control of HIV‐1 post integration latency: implications for therapy. Clin Epigenetics 7: 103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laquerriere A, Maluenda J, Camus A, Fontenas L, Dieterich K, Nolent F, Zhou J, Monnier N, Latour P, Gentil D et al (2014) Mutations in CNTNAP1 and ADCY6 are responsible for severe arthrogryposis multiplex congenita with axoglial defects. Hum Mol Genet 23: 2279–2289 [DOI] [PubMed] [Google Scholar]
- Lee HC, Tian B, Sedivy JM, Wands JR, Kim M (2006) Loss of Raf kinase inhibitor protein promotes cell proliferation and migration of human hepatoma cells. Gastroenterology 131: 1208–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Guo J, Wu Y, Zhou Q (2013) The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat‐transactivation. Nucleic Acids Res 41: 277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wu J, Chavez L, Hoh R, Deeks SG, Pillai SK, Zhou Q (2019) Reiterative enrichment and authentication of CRISPRi targets (REACT) identifies the proteasome as a key contributor to HIV‐1 latency. PLoS Pathog 15: e1007498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Baritaki S, Militello L, Malaponte G, Bevelacqua Y, Bonavida B (2010) The role of B‐RAF mutations in melanoma and the induction of EMT via dysregulation of the NF‐kappaB/Snail/RKIP/PTEN circuit. Genes Cancer 1: 409–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fu Y, Wang Q, Li M, Zhou Z, Dabbagh D, Fu C, Zhang H, Li S, Zhang T et al (2019) Proteomic profiling of HIV‐1 infection of human CD4+ T cells identifies PSGL‐1 as an HIV restriction factor. Nat Microbiol 4: 813–825 [DOI] [PubMed] [Google Scholar]
- Ma H, Dang Y, Wu Y, Jia G, Anaya E, Zhang J, Abraham S, Choi JG, Shi G, Qi L et al (2015) A CRISPR‐based screen identifies genes essential for west‐nile‐virus‐induced cell death. Cell Rep 12: 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancebo HS, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, Price D et al (1997) P‐TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro . Genes Dev 11: 2633–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P et al (2016) Genetic dissection of Flaviviridae host factors through genome‐scale CRISPR screens. Nature 535: 159–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DM, Garcia JV, Hazuda DJ, Haynes BF (2016) Latency reversal and viral clearance to cure HIV‐1. Science 353: aaf6517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousseau G, Kessing CF, Fromentin R, Trautmann L, Chomont N, Valente ST (2015) The tat inhibitor didehydro‐cortistatin a prevents hiv‐1 reactivation from latency. mBio 6: e00465‐15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh C, Park S, Lee EK, Yoo YJ (2013) Downregulation of ubiquitin level via knockdown of polyubiquitin gene Ubb as potential cancer therapeutic intervention. Sci Rep 3: 2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandeló José D, Bartholomeeusen K, da Cunha RD, Abreu CM, Glinski J, da Costa TB, Bacchi Rabay AF, Pianowski Filho LF, Dudycz LW, Ranga U et al (2014) Reactivation of latent HIV‐1 by new semi‐synthetic ingenol esters. Virology 462–463: 328–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park RJ, Wang T, Koundakjian D, Hultquist JF, Lamothe‐Molina P, Monel B, Schumann K, Yu H, Krupzcak KM, Garcia‐Beltran W et al (2017) A genome‐wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat Genet 49: 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman MC, Lau JSY, McMahon JH, Lewin SR (2018) Barriers and strategies to achieve a cure for HIV. Lancet HIV 5: e317–e328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Wang P, Ding D, Li L, Wang H, Ma L, Zhou X, Liu S, Lin S, Wang X et al (2013) Zinc‐finger‐nucleases mediate specific and efficient excision of HIV‐1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res 41: 7771–7782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ (2009) The challenge of finding a cure for HIV infection. Science 323: 1304–1307 [DOI] [PubMed] [Google Scholar]
- Rojas CV, Valiente‐EF Soto RR, Toro AD (2019) New challenges of HIV‐1 infection: how HIV‐1 attacks and resides in the central nervous system. Cell 8: 1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruelas DS, Greene WC (2013) An integrated overview of HIV‐1 latency. Cell 155: 519–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, John SP, Aker AM, Renzette N, Robbins DR et al (2016) Identification of Zika virus and dengue virus dependency factors using functional genomics. Cell Rep 16: 232–246 [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG et al (2014) Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L, Rabi SA, Laird GM, Eisele EE, Zhang H, Margolick JB, Siliciano RF (2013) A novel PCR assay for quantification of HIV‐1 RNA. J Virol 87: 6521–6525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søgaard OS, Graversen ME, Steffen L, Rikke O, Brinkmann CR, Nissen SK, Kjaer AS, Schleimann MH, Denton PW, Hey‐Cunningham WJ et al (2015) The depsipeptide romidepsin reverses hiv‐1 latency in vivo . PLoS Pathog 11: e1005142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak AM, Andrade A, Eisele E, Hoh R, Bacchetti P, Bumpus NN, Emad F, Buckheit R III, McCance‐Katz EF, Lai J et al (2014) A pilot study assessing the safety and latency‐reversing activity of disulfiram in HIV‐1‐infected adults on antiretroviral therapy. Clin Infect Dis 58: 883–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavel L, Jaquillard L, Karsisiotis AI, Saab F, Jouvensal L, Brans A, Delmas AF, Schoentgen F, Cadene M, Damblon C (2012) Ligand binding study of human PEBP1/RKIP: interaction with nucleotides and Raf‐1 peptides evidenced by NMR and mass spectrometry. PLoS ONE 7: e36187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Paras P Jr, Van Lint C (1993) Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J 12: 3249–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang FX, Xu Y, Sullivan J, Souder E, Argyris EG, Acheampong EA, Fisher J, Sierra M, Thomson MM, Najera R et al (2005) IL‐7 is a potent and proviral strain‐specific inducer of latent HIV‐1 cellular reservoirs of infected individuals on virally suppressive HAART. J Clin Invest 115: 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Lu P, Qu X, Shen Y, Zeng H, Zhu X, Zhu Y, Li X, Wu H, Xu J et al (2017) Reactivation of HIV‐1 from latency by an ingenol derivative from euphorbia kansui. Sci Rep 7: 9451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Gao HQ, Li HB, Qi SJ, Liu WL, Xu L, Li H, Liu JX, Dong ZM (2015) Correlation among RKIP expression, NF‐κB p65 levels, and T‐lymphocyte subsets in gastric cardia adenocarcinoma. Genet Mol Res 14: 16491–16496 [DOI] [PubMed] [Google Scholar]
- Yeung KC, Rose DW, Dhillon AS, Yaros D, Gustafsson M, Chatterjee D, McFerran B, Wyche J, Kolch W, Sedivy JM (2001) Raf kinase inhibitor protein interacts with NF‐kappaB‐inducing kinase and TAK1 and inhibits NF‐kappaB activation. Mol Cell Biol 21: 7207–7217 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
No primary datasets have been generated or deposited.