

Abstract
Kidney failure (KF) is associated with cardiac fibrosis and significantly increased mortality in heart failure. Thrombospondin-1 (TSP1), a key regulator of latent transforming growth factor-β1 (L-TGF-β1) activation, is a predicted target of miR-221. We hypothesized miR-221 attenuates severe KF-associated cardiac fibrosis via targeting of Thbs1 with subsequent inhibition of L-TGF-β1 activation. Rat cardiac fibroblasts (cFB) were isolated and transfected with microRNA-221 (miR-221) mimics or mimic control (miR-221 and MC) with or without exposure to L-TGF-β1. We demonstrate miR-221 downregulates Thbs1 via direct 3′ untranslated region (3′ UTR) targeting with consequent inhibition of L-TGF-β1 activation in cFB as proven by the significant reduction of myofibroblast activation, collagen secretion, TGF-β1 signaling, TSP1 secretion, and TGF-β1 bioactivity measured by Pai1 promoter reporter. The 5/6 nephrectomy (Nx) model of cardiac fibrosis was used to test the in vivo therapeutic efficacy of miR-221 (i.v. 1 mg/kg ×3). miR-221 significantly inhibited Nx-induced upregulation of TSP1 and p-SMAD3 in the heart at day-7 and reduced cardiac fibrosis (picro-sirius), improved cardiac function (±dP/dt), and improved 8-week survival rate (60% versus 36%; p = 0.038). miR-221 mimic treatment improved survival and reduced cardiac fibrosis in a model of severe KF. miR-221 is a therapeutic target to address cardiac fibrosis originating from renal disease and other causes.
Keywords: microRNA, thrombospondin-1, cardiac fibrosis, kidney failure, latent TGF-β, heart failure
Graphical Abstract
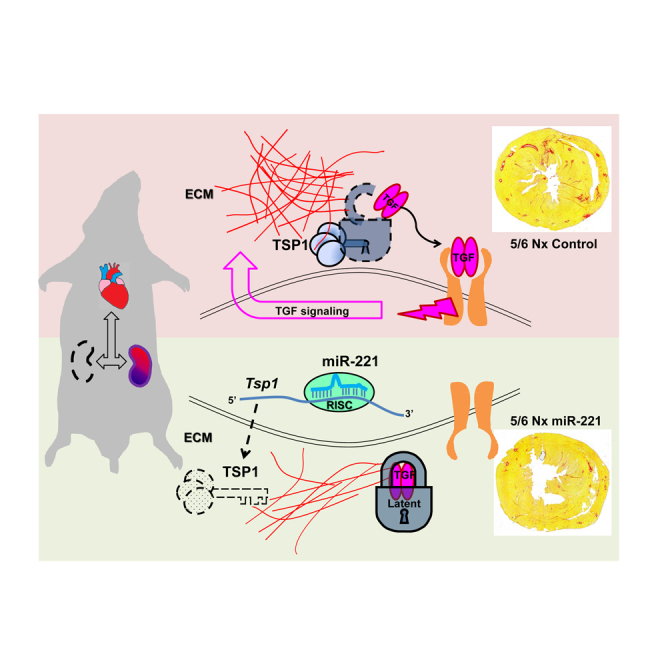
Kidney failure (KF) increases cardiac fibrosis and mortality in heart failure patients. Via targeting thrombospondin-1, miR-221 inhibits TGF-β1 activation. Zhou et al. demonstrated that miR-221 mimic reduces cardiac fibrosis and improves cardiac function and 8-week survival of severe KF. miR-221 is a candidate therapeutic target in treatment of cardiac fibrosis.
Introduction
Population-based data indicates 41% of women and 36% of men aged 70 years and above have impaired kidney function.1 Twenty percent of adults aged 40 years are destined to incur heart failure in later life. Cardiac and kidney disease commonly co-exist. Interactions generated by concurrent impairment of these two organs accelerate pathologic processes and lead to significantly increased mortality.2 In clinical post-mortem studies, the severity of kidney failure (KF) is strongly and directly related to the extent of cardiac fibrosis.3,4 The mechanisms underlying cardio-renal interactions are incompletely understood. Excess collagen deposition in extracellular matrix (ECM) is a key determinant of impaired cardiac systolic and diastolic function.5 The development of fibrosis occurs independent of hypertension. Neurohormonal imbalance and altered cell signaling appear central to the pathophysiology of cardiac fibrosis.6, 7, 8 Transforming growth factor-β1 (TGF-β1) signaling activation is believed to be a master regulator and final common signaling pathway leading to fibrosis.9, 10, 11 The 5/6 nephrectomy (Nx) model is a well-established surgical model of KF characterized by hypertension, neurohormonal derangement, and ultimately cardiac hypertrophy and fibrosis.12,13
KF-induced neurohormonal changes activate TGF-β1 signaling, triggering cardiac fibroblasts (cFB) to differentiate into “myofibroblasts” (myoFB) with increased collagen synthesis and deposition in the ECM.14 TGF-β1 is secreted by almost all cell types in a latent form and stored as a latent complex in the ECM.15,16 Latency is an important feature in TGF-β1 biology, which allows efficient and precise regulation in response to stimuli. Activation of latent TGF-β1 (L-TGF) is a critical step in the development of fibrosis.17 In brief, TGF-β1 is synthesized as a precursor protein with 390-412 amino acids. It is cleaved by furin intracellularly into N-terminal latency-associated peptide (LAP) and C-terminal peptide mature TGF-β1.18 Both dimerize and together form a small latent complex. The small latent complex then binds to latent TGF-β1 binding protein (LTBP) to form the large latent complex. L-TGF-β1 activation is tightly regulated with thrombospondin1 (TSP1) recognized as one of the most important L-TGF-β1 activators in vitro and in vivo.15,19 The basal expression of TSP1, a matricellular protein, in the healthy heart is very low but is greatly upregulated by an array of adverse stimuli including acute and chronic ischemia, oxidative stress, excess hemodynamic load, inflammation, cytokines, and neuroendocrine factors.20 TSP1 expression is closely associated with the development of cardiac fibrosis.15 miR-221 mimics suppress human THBS1 mRNA and TSP1 protein in LN-18, a human malignant glioma cell line, and bovine luteal endothelial cells, respectively.21,22 In silico, human THBS1 is a predicted target of miR-221 (http://www.targetscan.org/). Dogar et al.21 confirmed direct binding of miR-221 to human THBS1 3′ UTR. The role of TSP1 in the context of miR-221-mediated effects upon cardiac fibrosis has not been previously investigated.
The study of miR-221 in the heart was initially driven by discovery of its pro-survival properties.23 We found miR-221 was consistently upregulated in the heart by selected cardioprotective drugs and our subsequent in vitro studies demonstrated miR-221 protected cardiomyocytes via both anti-apoptotic and anti-autophagic mechanisms.24,25 Recently, we reported an anti-fibrotic effect of miR-221 in a rodent model of myocardial infarction (MI). miR-221 reduced the α-SMA positive myoFB population, TGF-β1 signaling and left ventricular (LV) fibrosis in both the infarct and remote myocardium.26 Clinically, miR-221 is downregulated in the myocardium of patients with severe fibrosis and dilated cardiomyopathy or aortic stenosis. The degree of downregulation correlates to the extent of myocardial fibrosis and LV stiffness.27 On this background, we hypothesized that microRNA-221 (miRNA-221) inhibits L-TGF-β1 activation via targeting of Thbs1 thus attenuating the development of cardiac fibrosis and preserving cardiac function. We tested the efficacy of miR-221 mimics in the treatment of KF-induced (5/6-Nx model) cardiac fibrosis.
We demonstrate the anti-fibrotic effects and underlying mechanisms of miR-221 in vitro and in vivo. In addition, anti-apoptotic effects were re-evaluated in the infarcted kidney. The beneficial effects of miR-221 in the heart, and maybe in the kidney, may synergistically ameliorate both cardiac and renal aspects of the cardio-renal syndrome. These results add new dimensions to the potential therapeutic applications of miR-221.
Results
miR-221 Displays Anti-fibrotic Effects against Latent, but Not Active, TGF-β1
Our previous work indicated that miR-221 is anti-fibrotic in a rodent myocardial infarction model.26 However, we found miR-221 could not block the pro-fibrotic effects of TGF-β1 in cFB (serum free), as assessed by de novo α-SMA expression and phalloidin stress fiber formation; collagen gel contraction, α-SMA protein expression and SMAD3 phosphorylation (p-SMAD3; Figures S1A–S1C). Nevertheless, miR-221 did have significant anti-fibrotic effects when cFB cultured with 10% FBS as shown by significant inhibition of α-SMA expression and p-SMAD3, and reduced gel contraction (Figures S1D and S1E).
Fetal bovine serum (FBS) is rich in growth factors, including TGF-β1 mainly in the latent form.28,29 Therefore we suspected that miR-221 blocks L-TGF-β1 activation rather than directly opposing TGF-β1 signaling. L-TGF-β1 significantly increased phalloidin stress fiber formation, collagen secretion, α-SMA expression, and p-SMAD3, similar to the effects of active TGF-β1 but with higher dosage and longer incubation time (80 ng/mL versus 10 ng/mL, 5-day versus 2-day) reflecting L-TGF-β1 activation by cFB. miR-221 dramatically inhibited all of these L-TGF-β1-related effects (Figures 1A–1C).
Figure 1.

miR-221 Inhibits Latent TGF-β1 Mediated Cardiac Fibroblast Activation
Adult rat cardiac fibroblasts (cFB) were serum starved for 24 h and then treated with 80 ng/mL L-TGF-β1 for 5-day with transfections of miR-221 mimic (miR-221) or mimic control (MC). (A) Immunofluorescence images of cFB stained with α-SMA (green), phalloidin (red) and DAPI (blue). Scale bar = 100 μm. (B) Collagen secretion in cFB culture supernatant measured by Sircol assay. (C) Western blot measurement for α-SMA, phosphorylated (P-), and total (T-) SMAD3, with β-actin as loading control. Values were expressed as mean ± SD. #p < 0.05, ##p < 0.01, ###p < 0.001 versus L-TGF-β1+MC; ∗∗p < 0.01, ∗∗∗p < 0.001 versus Ctrl by by one-way ANOVA and Bonferroni post hoc analysis. Experiments performed three times in triplicates.
miR-221 Targets Thbs1 mRNA through Direct 3′ UTR Binding
The expression of Thbs1 mRNA in cFB and TSP1 contents in cell lysate and secreted in culture supernatant were significantly decreased by miR-221 (Figures 2A and 2B). L-TGF-β1 significantly induced TSP1 secretion without increasing Thbs1 mRNA expression. Similar results were observed in active TGF-β1 treatment (Figure S2). This miR-221 mediated downregulation of Thbs1 implied direct targeting and was further validated using 3′ UTR luciferase reporter assay. Human THBS1 3′ UTR is 3,561 bp long with 2 conventional binding sites (BS) for miR-221. In contrast, the rat Thbs1 mRNA is not a predicted miR-221 target. Close analysis revealed that rat Thbs1 3′ UTR is incomplete with only 186 bp (Refseq).30 On cross-species genomic DNA sequence alignment (Refseq and BLAST),31 we found that the coding sequences of Thbs1 between rat and human are highly conserved and extended an additional 1,827 bp 3′ UTR in rat (Figure 2C). However, with extended 3′ UTR, the 2 human conventional miR-221 BSs are not found in the rat due to seed region nucleotide mismatch. We then did RNA-RNA interaction energy score calculation. Thbs1-1 and Thbs1-2 (∼1 kbp each), contained putative unconventional BSs (predicted with energy scores −10.84, −14.87, and −13.12, −12.96 kcal/mol, respectively)32 (Figure 2C). Thbs1-1 and Thbs1-2 were cloned using rat cDNA templates. Luciferase reporter assay studies showed a significant inhibition by miR-221 mimics of the Thbs1-1 clone, while Thbs1-2 was unaffected. The two putative BSs within Thbs1-1 were then subjected to mutagenesis. Mutation of BS2 but not BS1 caused the loss of miR-221 inhibition, indicating that BS2 mediated the interaction of miR-221 with Thbs1 (Figures 2D and 2E).
Figure 2.

miR-221 Downregulates Thbs1 via Direct 3′ UTR Binding
Adult rat cFB were serum starved for 24 h and then treated with 80 ng/mL L-TGF-β1 for 5-day with transfections of miR-221 or MC. Expressions of (A) Thbs1 by qRT-PCR and (B) TSP1 by western blot. (C) Diagram of the alignment of human THBS1 and rat Thbs1 and the indications of miR-221 binding sites (BS). Human conventional miR-221 BSs shown as hollow arrowheads, rat unconventional miR-221 BSs shown as solid arrowheads. (D) Schematic of rat Thbs1 3′ UTR binding sites sequence and mutations (from shaded to red). (E) 3′ UTR luciferase reporter assay using a 221 sensor (psiCHECK2 vector carrying perfect complimentary sequence to miR-221) as a positive control. Values were expressed as mean ± SD. ##p < 0.01, ###p < 0.001 versus L-TGF-β1+MC; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus Ctrl by by one-way ANOVA and Bonferroni post hoc analysis (A and B) and unpaired t test (E). Experiments performed three times in triplicate.
miR-221 Inhibits L-TGF-β1 Activation
Transformed mink lung epithelial cells (TMLC) with Pai1 promoter luciferase reporter were used to measure TSP1-mediated L-TGF-β1 activation, and the effect of miR-221, in cFB.33 Sensitivity and specificity of TMLC were confirmed as (1) dose-dependent response to active TGF-β1; (2) negligible response to L-TGF-β1; (3) cumulative cFB endogenous L-TGF-β1 production and activation with cFB culture (2 days versus 7 days) and co-culture (18 h versus 48 h; Figures S3A–S3C). TSP1 secretion in the cFB supernatant was readily detectable from 24 h and increased proportionally in 2- and 7-day cultures (Figure 3A). Total endogenous TGF-β1 production (assessed by acid activation) in both the supernatant and cell lysates was detected at low levels at day 2 in culture but significant accumulation was evident by 7-day (Figure 3B). At 7-days cFB/TMLC co-culture, endogenous L-TGF-β1 activation was significantly inhibited by miR-221 (Figures 3C and 3D). Interestingly, total TGF-β1 and Tgfb1 mRNA were not reduced by miR-221 (Figures 3E and 3F). Conversely, nor did TGF-β1 regulate miR-221 expression (Figure 3G). Taken together, the data indicate that miR-221 inhibited L-TGF-β1 activation in cFB through downregulation of TSP1 without altering total TGF-β1 production.
Figure 3.

miR-221 Inhibits Latent TGF-β1 Activation
Adult rat cFB reaching 80% confluence were incubated in serum free medium for 2–7 days, with or without transfections of miR-221 or MC. (A) Western blot of TSP1 in 40× concentrated cFB supernatants from 1-, 2-, and 7-day culture. Samples loaded by equal volume. (B) Total TGF-β1 bioactivity in acid activated supernatant and cFB lysate assessed by TMLC. Results were expressed in relative luminescence units (RLUs). (C) Culture protocol to assess endogenous L-TGF-β1 activation. (D) Endogenous L-TGF-β1 activation assessed by cFB and TMLC co-culture. (E) Total endogenous TGF-β1 production in cFB. (F) Tgfb1 mRNA expression assessed by qRT-PCR. (G) miR-221 expression by stem-loop qPCR in TGF-β1 treated versus Ctrl. Values were expressed as mean ± SD. ∗p < 0.05, ∗∗p < 0.01 versus MC by by one-way ANOVA and Bonferroni post hoc analysis. Experiments performed three times in triplicates.
miR-221 Downregulates TSP1 and Inhibits Cardiac TGF-β1 Signaling in 5/6 Nx
Samples were collected serially to monitor the development of kidney failure and cardiac fibrosis in the rat 5/6 Nx model. Plasma creatinine and urea nitrogen were significantly elevated at day 3 and remained high thereafter (Figure S4A). Developing cardiac fibrosis was obvious by day 10 and fully established by week 3 post-Nx (Figure S4B). Thbs1 mRNA expression and protein levels were low in Sham animals but upregulated from day 3 and continuously increased to day 7 post-Nx (Figures 4A and 4B). Concurrently p-SMAD3 was significantly activated in Nx from day 7 (Figure 4C). Short and long-term of miR-221 treatment protocols were applied to verify the molecular and therapeutic effects respectively (Figure 4D). With a single injection, miR-221 expression in the heart increased by a 1.7-fold compared to Nx control (Figure 4E). Thbs1 mRNA and protein and p-SMAD3 were significantly reduced by miR-221 (Figures 4F–4H). By the end of 8 weeks post-Nx, no significant changes were detected (Figure S5).
Figure 4.

miR-221 Inhibits TSP1 Upregulation and Reduces TGF-β1 Signaling in the Heart
Heart tissues were harvested from Sham or Nx animals at day 3 and 7 post-Nx. (A) Thbs1 mRNA expression by qRT-PCR. Protein expressions of (B) TSP1 and (C) phosphorylated (P-) and total (T-) SMAD3. HSP90 and β-actin served as loading control, respectively. (D) Scheme of miR-221 treatment. Arrows indicating i.v. injections of 1 mg/kg mimics or PBS. Short-term: rats with Nx received 1 mg/kg miR-221 mimics or PBS injection i.v. at day-6 post-Nx and hearts were harvested 24 h later. (E) Stem-loop qPCR measurement of miR-221. (F) qRT-PCR measurement of Thbs1 mRNA. Western blot measurement of (G) TSP1 protein expression and (H) phosphorylated (P-) and total (T-) SMAD3 in the heart. Values were expressed as mean ± SD. #p < 0.05, ##p < 0.01 versus Nx Ctrl; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus Sham by by one-way ANOVA and Bonferroni post hoc analysis. n = 3–6 each group.
miR-221 Reduces Cardiac Fibrosis and Improves Survival and Cardiac Function in 5/6 Nx
The therapeutic effects of miR-221 were monitored for 8 weeks post-Nx. The miR-221 mimic treatment was designed to target the critical window of Thbs1 upregulation as early as day 3 and further strengthened at days 7 and 14 post-Nx. At week 8, picro-sirius red staining demonstrated that miR-221 significantly decreased cardiac fibrosis compared with Nx control (Figure 5A). The ratio of heart weight to tibial length was not changed by miR-221 in Nx and remained significantly higher than in Sham animals (Figure 5B). Immunofluorescence staining of α-SMA indicated myoFB activation in Nx, which was significantly reduced by miR-221 (Figure 5C).
Figure 5.

miR-221 Reduces Cardiac Fibrosis and Myofibroblast Activation in the Heart
Rats received 1 mg/kg miR-221 mimics (Nx miR-221) or PBS i.v. injection (Nx Ctrl) for 3 times at day 3, 7, and 14 post-Nx. Hearts were harvested by the end of 8 weeks. Middle sections of 1/3 the heart were used for histological analysis. (A) Picro-sirius red staining of the heart was performed as follows (i) cross-section, scale bar = 2 mm; (ii) zoom-in images, scale bar = 200 μm; (iii) quantified percentage of fibrosis with ImageJ. (B) Heart weight (HW) normalized by tibial length (TL). (C) (i) Immunofluorescence staining with α-SMA (green) and DAPI (blue), Scale bar = 100 μm; (ii) Quantification of the α-SMA+myoFBs expressed as percentage of cells. Values were expressed as mean ± SD. #p < 0.05 versus Nx Ctrl; ∗p < 0.05, ∗∗p < 0.01 versus Sham by by one-way ANOVA and Bonferroni post hoc analysis. n = 6 per group.
8-week survival rate was substantially improved by miR-221 from 36% to 60% (Nx Ctrl versus Nx miR-221; p = 0.0381; Figure 6A). Severe weight loss was observed 2–3 weeks post-Nx. However, all surviving animals recovered from weight loss with no significant difference between groups by week 8 (Figure 6B). Before sacrifice, LV function and BP were assessed. miR-221 treatment significantly improved LV contractility and relaxation as indicated by ± dP/dt, along with slight increases in left ventricular developed pressure (LVDP). BP, LV end diastolic pressure (EDP), and Tau were increased in Nx and not significantly altered by miR-221 (Figure 6C). Heart rate was similar across all the groups. Echocardiography demonstrated changes of cardiac structure and function in Nx including increased LV mass with progression through concentric (week 1–3) to eccentric (week 5–8) LV hypertrophy, reduced ejection fraction (EF), and chamber dilation by 7–8 weeks. miR-221 treatment reduced cardiac hypertrophy and reduced wall thickening (Figure S6).
Figure 6.

miR-221 Improves Survival and Cardiac Function In Vivo
Rats received 1 mg/kg Nx miR-221 or PBS i.v. injections (Nx Ctrl) 3 times on day 3, 7, and 14 post-Nx and were kept for 8 weeks. (A) A Kaplan-Meier curve of survival rate. n = 6 for Sham, n = 17 for Nx Ctrl, n = 10 for Nx miR-221. (B) Body weight measurement at 8 weeks. (C) Left ventricle (LV) function measured by Millar pressure sensor catheterized through right carotid artery into LV. BP, blood pressure; d, diastole, s, systole; LVDP, LV developed pressure; HR, heart rate; ± dP/dt, maximum and minimum rate changes of LV pressure; Tau, LV relaxation time constant; EDP, end diastolic pressure. Values were expressed as mean ± SD. ∗∗p < 0.01 versus Nx Ctrl by one-way ANOVA and Bonferroni post hoc analysis. n = 6 each group.
miR-221 Does Not Alter Kidney Function Despite Downregulation of TSP1 in the Kidney
With miR-221 mimic treatment, the level of miR-221 was increased by 7-fold in the renal infarct and 1.5-fold in tissue remote from the infarct at day 7 post-Nx (Figure 7A). Renal Thbs1 mRNA and protein greatly increased in the infarct and were downregulated by miR-221. TSP1 remained very low in remote tissue, with no difference from Sham, and was not affected by miR-221 (Figures 7B and 7C). Whether miR-221 downregulation of TSP1 had affected fibrosis development in the infarct remained unknown. The remote area (1/3 kidney) underwent a significant remodeling process of hypertrophy and fibrosis. miR-221 treatment did not significantly reverse this process (Figure S7C; Figure 7D). At week 8, the infarct had been completely absorbed (Figure 7B). Expression of miR-221 and p-SMAD3 did not differ between groups. TSP1 was not detectable (Figure S8). Kidney function, as indicated by levels of plasma creatinine and urea nitrogen and urine protein, was not improved by miR-221 (Figure 7E). This suggested the beneficial effects of miR-221 in the heart were independent of any change in kidney function.
Figure 7.

miR-221 Targets Thbs1 but Does Not Alter Kidney Fibrosis in the End of 8 Weeks Post-Nx
Rats with Nx received 1 mg/kg miR-221 mimics or PBS i.v. injection at day 6 post-Nx and kidneys were harvested 24 h later and dissected into infarct and remote region for RNA and protein analysis. (A) Kidney miR-221 levels were measured by stem-loop qPCR. Expressions of (B) Thbs1 mRNA by qRT-PCR and (C) TSP1 protein by western blot. Rats received 1 mg/kg Nx miR-221 or PBS i.v injections (Nx Ctrl). for 3 times on day 3, 7, and 14 post-Nx and were kept for 8 weeks. Plasma and urine were collected for kidney function analysis, kidneys for histology. (D) Picro-sirius red staining of kidney sections for assessment of fibrosis. Scale bar = 200 μm. (E) Measurements of plasma creatinine, urea nitrogen, and urine protein. Values were expressed as mean ± SD. #p < 0.05, ##p < 0.01 versus Nx Ctrl; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus Sham by one-way ANOVA and Bonferroni post hoc analysis. n = 3–6 each group.
We investigated apoptosis at day 7 in the kidney. Cleaved caspase-3 (CASP3) was strongly increased in the infarct region and significantly inhibited by miR-221. There was no change in cleaved CASP3 in remote non-infarct renal tissue (Figure S9A). The previously confirmed anti-apoptotic targets of P53 (phosphorylated not total) and BAK1 were dramatically reduced by miR-221 in the renal infarct (Figure S9A). In the heart this apoptosis pathway was not affected by Nx or miR-221 (Figure S9B).
Other Reported Fibrosis-Related Potential Targets of miR-221
A previous report indicated that Ets1, Jnk1, TGFbr1, and TGFbr2 are potential miR-221 targets contributing to anti-fibrotic properties.27 L-TGF did not upregulate any of these genes. miR-221 downregulated Tgfbr1 at mRNA level and ETS1 at protein level but did not affect TGFbr2 or Jnk1 (Figures S10A and S10B). Luciferase reporter assays indicated direct miR-221 binding to Ets1 and Tgfbr1 3′ UTR without significant inhibition of Jnk1 or Tgfbr2 (Figure S10C). However, none of these genes were dysregulated by Nx or inhibited by miR-221 in vivo (Figure S11). Taken together, our results suggest that Ets1 and TGFbr1 are direct targets of miR-221 but do not play a part in the anti-fibrotic effects of miR-221 in 5/6 Nx.
Discussion
From in vitro functional assessments of cardiac fibroblasts and observations in vivo from the 5/6 Nx model of severe KF-induced cardiac fibrosis, we demonstrate miR-221 exerts its anti-fibrotic effects by directly targeting TSP1, which in turn inhibits L-TGF-β1 activation and TGF-β1 downstream signaling. Hence, miR-221 treatment attenuates cardiac fibrosis and ameliorates KF-induced LV functional deterioration. miR-221 attenuates apoptosis in the infarct area of kidney through targeting of P53 and BAK1, the same mechanism as previously discovered in myocardial infarction.26 The beneficial effects of miR-221 in the heart and kidney may underpin improved survival. Other predicted mir-221 targets including Ets1, TGFbr1, TGFbr2, and Jnk1 do not contribute to the anti-fibrotic effects of miR-221 in KF-induced cardiac fibrosis.
Activation of TGF-β1 signaling is central to the development of fibrosis.10,11 In earlier work, we demonstrated miR-101a directly targets key components in the TGF-β1 signaling pathway34 and that miR-221 reduces post-myocardial infarction cardiac fibrosis in the infarct and in remote non-infarct zones of the left ventricle.26 Our in vitro investigation of the anti-fibrotic mechanisms of miR-221 revealed that miR-221 failed to block the pro-fibrotic effects of TGF-β1 (zero serum) in cFB. However, miR-221 exerted anti-fibrotic effects observed in the presence of 10% serum which led to our hypothesis that miR-221 acted on L-TGF-β1 activation rather than directly inhibiting TGF-β1 signaling.
TSP1 plays an important role in L-TGF-β1 activation and is upregulated by multiple stimuli.15 Our results clearly show that TSP1 is undetectable in the sham heart but greatly induced post-Nx. Therefore, the regulation of TSP1 offers efficient control of L-TGF-β1 activation, which may be exploited for therapeutic purposes. Human THBS1 as a target of miR-221 has been previously reported.21 Rat Thbs1 is not a predicated target of miR-221 as its 3′ UTR sequence is incomplete compared to human THBS1 3′ UTR; 186 bp versus 3,561 bp, respectively. We took a three-step approach: (1) cross-species genomic DNA sequence alignment to acquire additional 3′ UTR information; (2) unconventional target prediction by RNA-RNA interaction energy scores;31,32 (3) Thbs1 3′ UTR cloning and BS mutagenesis assessed by luciferase reporter assays. Successful application of this strategy has been reported previously.26,35 For the first time, we have confirmed that miR-221 downregulates rat Thbs1 via direct binding to the 3′ UTR of its mRNA.
We demonstrated that cFB produce a large amount of TSP1, which is markedly inhibited by miR-221. However, to prove miR-221 inhibits L-TGF-β1 activation in the ECM is challenging. With Dr. Rifkin’s kind gift of TMLC,36 we established reliable protocols. As the production of TSP1 and L-TGF-β1 in cFB is an accumulative process as is L-TGF-β1 activation, longer study times are required. In this study, 5-days cFB plus 2-days TMLC co-culture were applied. Our results clearly showed that endogenous L-TGF-β1 activation was directly related to TSP1 production and miR-221 significantly inhibited such activation. Tgfb1 gene expression and total TGF-β1 production were not changed by miR-221. Similarly, Dogar et al. reported that miR-221 did not affect Tgfb1 mRNA expression in HeLa cells.21 This finding is consistent with existing knowledge that (1) the reserve of L-TGF-β1 is very large and only a small fraction of it is activated to maintain cellular activity and (2) TGF-β1 bioactivity is primarily regulated by activation of L-TGF-β1 rather than the regulation of TGF-β1 production.37 Through targeting TSP1, miR-221 effectively regulates L-TGF-β1 activation. Previous reports suggested that TGF-β1 increases TSP1 production.38 L-TGF-β1 when activated seems to not only increase production but directly stimulates TSP1 release in cFB. Farberov et al.22 reported downregulation of miR-221 by TGF-β1 treatment in luteal endothelial cells (0.5% BSA and 0.1% Serum). Under similar condition, we found that miR-221 was not regulated by TGF-β1. Whether there is a feedback regulation between TGF-β1 and miR-221 remains uncertain and warrants further investigation.
The 5/6 Nx is a well-characterized surgical model of KF caused by renal mass reduction and infarction.39,40 It incorporates removal of one kidney plus ligation of two of the three renal artery branches (infarction) or resection of two poles (ablation) of the remaining kidney. This model was used in this study as it is characterized by pro-fibrotic activation of the renin-angiotensin-aldosterone system and severe hypertension.41,42 Conventional two-step surgery, i.e., right nephrectomy first and 1 week late for left kidney artery ligation, requires more than 4 weeks for development of kidney failure, hypertension, and cardiac remodeling.42, 43, 44 A one-time surgical procedure was used to minimize surgery induced suffering to rats and shorten the duration to develop pathological changes. We observed increases in plasma creatinine and urea nitrogen within 3 days post-Nx with initial cardiac fibrosis obvious at 10 days after surgery and well-established within 3 weeks. With these early and clear-cut changes in the heart, the therapeutic efficacy of miR-221 could be readily tested. The disadvantage of a one-step surgical procedure is higher mortality due to acute development of severe KF. In this study, ∼5% early death, within 3 days post-Nx, was observed and these rats were excluded from analysis.
The therapeutic time window and dosage for treatment are crucial. TSP1 is well known for its transient expression in the tissue in response to stress. Our results showed that TSP1 was not detected in the sham heart or kidney. In the heart, TSP1 was induced from day 3 post-Nx, peaked at day 7, and then gradually returned to normal. Changes in p-SMAD3 corresponded to those in TSP1. Cardiac fibrosis appeared at 1 week and was fully established within 3 weeks after Nx. These data suggested that TSP1 induction and TGF-β1 signaling activation are critical in the early stage of the development of fibrosis but might not be required for the maintenance of fibrosis in later time points. Early intervention could lead to a long-term beneficial outcome. Therefore, we carefully designed the experiment to suppress these early changes. Mimics at 1 mg/kg have proven to be effective in our earlier work.26,34 Similar or lower dosage has been reported by others as well.45,46 To verify mimic delivery and functional target gene knockdown, we injected a single dose at day 6 with hearts harvested 24 h later. In these samples, miR-221 was increased by 1.7-fold (compared with Nx Ctrl), Thbs1 gene and protein were markedly reduced and downstream TGF-β1 signaling was inhibited as well. To sustain the therapeutic effects, miR-221 treatments started at day-3, followed by additional injections at days 7 and 14 post-Nx to maintain the therapeutic effects. With 3 miR-221 treatments, cardiac fibrosis and cardiac function were significantly improved at 8 weeks. Systemically delivered, miR-221 mimics reached other organs including the kidney. The upregulation of miR-221 in infarct and remote tissue in the kidney was ∼7- and 1.5-fold respectively. This pattern is similar to that observed in cardiac tissue from our previous myocardial infarction model.26 Anti-apoptotic effects of miR-221 were assessed in the kidney. At day 7 post-Nx, cleaved CASP3 was significantly elevated in the infarct kidney. The increase was inhibited by miR-221 along with downregulation of BAK1 and P53 phosphorylation (s46). None of these changes were observed in non-infarct regions of kidney or heart. These results indicate the downregulation of apoptosis related targets p53 and Bak1 is confined to the infarct tissue while no effects in the non-infarct tissue.
Other predicted miR-221 gene targets of Ets1, TGFbr1, TGFbr2, and Jnk1 were examined. Previous reports were based on miR-221 in silico target prediction and mRNA regulation in vitro. Protein level changes and 3′ UTR luciferase responses were not measured.27 In this study, we took the investigation further by validating their protein expression in vitro and in vivo under pathological conditions and by testing their direct interactions with miR-221 using 3′ UTR luciferase reporter assay. Among them, 3′ UTR of Ets1 and TGFbr1 showed binding activity with miR-221 and genes were downregulated by miR-221 at either mRNA or protein level in vitro. TGFbr2 and Jnk1 were unchanged. However, none of these targets were affected by Nx or reduced by miR-221 in the heart. Therefore, we believe that direct targeting of TSP1 and inhibition of L-TGF-β1 activation is the predominant anti-fibrotic mechanism of miR-221. Ets1 and TGFbr1 are direct targets of miR-221 but they were not involved in the pathogenesis of KF-induced cardiac fibrosis nor the anti-fibrotic effect of miR-221.
In summary, through direct targeting on TSP1, miR-221 inhibits L-TGF-β1 activation and reduces cardiac fibrosis induced by kidney failure, severe hypertension, and neurohumoral derangement. As a result, it improves cardiac function and increases survival rate. Through targeting BAK1 and P53, miR-221 inhibits apoptosis in the kidney infarct. The possible protective and anti-fibrotic effects of miR-221 in the kidney warrant future investigation with appropriate models exhibiting less severe kidney damage. Together with previous reports, this study further demonstrates that miR-221 offers a unique combination of anti-fibrotic and anti-apoptotic actions. miR-221 mimic has promising therapeutic potential and warrants further study.
Materials and Methods
Animals
A total of 95 male Sprague-Dawley rats (170–220 g) were used for 5/6 Nx surgery and 35 rats for cFB isolation. All rats were kept two in a cage at 21°C ± 2°C with a 12 h light/dark cycle. Water and diet were available ad libitum. Protocols were approved by the Institutional Animal Care and Use Committee of the National University of Singapore and complied with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, Revised 1996).
Primary cFB Isolation and myoFB Activation Assessment
Adult cFB isolation and culture were conducted as reported previously.47 Sub-confluent cFBs were synchronized for 24 h in low glucose-serum free DMEM, then treated with: (1) 25 nM miR-221 mimic (miR-221) or mimic control (MC) via LipofectamineRNAiMax (Thermo Fisher Scientific, Singapore), (2) L-TGF (small latent complex, 80 ng/mL, 299-LT-025, R&D Systems, Singlab Technologies). Recombinant active TGF-β1 was used as needed (10 ng/mL, H8541, Sigma-Aldrich, MERCK, Singapore). Serum free was used during treatment unless specifically mentioned. Stress fiber formation was assessed by α-SMA and Phalloidin immunofluorescence staining, collagen secretion by Sircol assay.47
TGF-β1 Activity Assay
TMLC was a kind gift from Dr. Daniel B. Rifkin. This cell line contains stably transformed luciferase reporter driven by plasminogen activator inhibitor-1 (Pai1) promoter.48 TMLC were cultured in DMEM, 10% FBS, penicillin (100 U/mL), and streptomycin (100 μg/mL; GIBCO, Thermo Fisher Scientific, Singapore), and supplemented with 200 μg/mL G418 (Sigma-Aldrich, MERCK, Singapore). For recombinant TGF-β1 dosage, cFB culture supernatant, or cFB lysate, TMLC were seeded at 20,000/well in 96-well plate and allowed to attach for 3–4 h, replaced medium with test samples and incubated for 18 h. Acid activation was performed by adding 20% volume of 1N HCL, incubating at room temperature for 20 min, followed by neutralization with 20% volume of 1N NaOH. To assess endogenous L-TGF-β1 activation, we seeded cFB at 4,000/well in 96-well plate, followed by miRNA mimic transfection for 5-day, then TMLC was seeded at 20,000/well into the same plate, for 48 h co-culture (7-day protocol). Finally, luciferase activity was measured in TMLC lysates (Promega, Singapore).
miRNA Stem-Loop qPCR and mRNA qRT-PCR
Manufacturer-suggested protocols were used. RNA was extracted with TRIzol Reagent followed by DNase I treatment (Invitrogen, Life Technologies, Thermo Fisher Scientific, Singapore). miRNA expression was determined by stem-loop qPCR (TaqMan MicroRNA Assay, QuantStudio 7 Flex Real-Time PCR System, Thermo Fisher Scientific, Singapore) and normalized to U6B. Gene expressions were measured by universal RT and real-time PCR (iScript RT Supermix, iTaq Universal SYBR Green Supermix, Bio-Rad Laboratories, Singapore), and normalized by β-actin. Primers are listed in Table S1.
Western Blot
Protein was extracted from cell or tissue lysates and analyzed using SDS-PAGE.49 Antibodies used were α-SMA (Sigma-Aldrich, MERCK, Singapore), cleaved caspase-3, phospho-SMAD3 (Ser423/425), SMAD3, phospho-JNK1 (Thr183/Tyr185), JNK1, TGFbr2 (Cell Signaling Technology, Research Biolabs, Singapore), BAK1, P53, TSP1, ETS1, TGFbr1, β-actin, HSP90 (Santa Cruz Biotechnology, Axil Scientific, Singapore) and phospho-P53 (Ser46) (GeneTex, Axil Scientific, Singapore). Chemiluminescence was imaged on ChemiDoc Touch Imaging System and analyzed with Image Lab Software V5.2.1 (Bio-Rad Laboratories, Singapore).
Sample preparation for TSP1 was modified: (1) 100 mM DTT used in sample loading buffer to break TSP1 homotrimer into monomer of 180 kDa; (2) methanol-free transfer buffer with 0.01% sodium dodecyl sulfate; (3) HSP90 was used as a high MW loading control. Culture supernatants were concentrated 40-fold using Amicon Ultra Centrifugal filters with a 50 kDa cutoff (Millipore, MERCK, Singapore) and loaded by equal volumes.
Luciferase Reporter Assays
For the 3′ UTR luciferase reporter assay, target gene 3′ UTR cDNA sequences were cloned into psiCHECK-2 dual luciferase reporter plasmid (Promega, Singapore). miR-221 sensor, the complementary binding sequence to miR-221, served as a positive control. Non-conventional binding sites were discovered by analyzing potential RNA-RNA interactions based on binding energy.32 Q5 Site-Directed Mutagenesis Kit (New England Biolabs, Singapore) was used to confirm binding site specificity. Primers used for cloning are listed in Table S2. The luciferase reporter plasmid(s) was co-transfected with the miR-221 or mimics control for 24 h. Renilla and Firefly luciferase activities were measured using Dual-Glo luciferase assay system (Promega, Singapore).
5/6 Nx Surgery and Cardiac Function Assessments
Rats were anesthetized with ketamine and xylazine (75 and 10 mg/kg respectively, i.p.). Briefly, a mid-ventral abdominal incision was made, the left kidney was exposed and the posterior and anterior lower renal artery branches were isolated and ligated separately with 6/0 suture. Ischemia of 2/3 of the kidney was visually confirmed by color change. The right kidney was then removed. The peritoneum and skin were sutured. Rats undergoing the identical procedure without nephrectomy were used as sham controls (Sham). Total of 9 out of 91 rats were excluded due to early death within 3-day post-Nx (3 rats); unsuccessful surgery, e.g., bleeding or small infarction (4 rats); sutures open by scratching (2 rats). All rats at day 3 post-Nx were randomly assigned to each experimental groups on any given day of operation.
miR-221 was pre-incubated with Invivofectamine 3.0 (Thermo Fisher Scientific, Singapore). A complex of 1 mg/kg miR-221 in 800 μL volume or equal volume of PBS was given via tail vein injection. Short term study: rats received one injection at day 6 post-Nx and kept for another 24 h. Long term study: rats each received three injections at days 3, 7, and 14 post-Nx. Rats were kept for 8 weeks or till death. Echocardiography (Echo; Vevo 2100, FUJIFILM VisualSonics, ITS Science, and Medical, Singapore) was monitored before and every week post-Nx. Rats were anesthetized with inhaled isoflurane (1.5%–2%) and 1 L/min oxygen for Echo and injections. Before sacrifice rats were anesthetized with pentobarbital (60 mg/kg, i.p.) for right carotid artery and left ventricular (LV) catheterization to measure blood pressure and assess LV function (Powerlab data acquisition system, ADInstruments, Advanced Tech, Singapore). Euthanasia was performed by opening the chest and abdomen and samples were collected, e.g., the heart, kidney, blood and urine. Plasma creatinine and urea nitrogen levels (Arbor Assays, Gene-Ethics, Singapore) and urine protein concentration (Bradford assay, Bio-Rad Laboratories, Singapore) were measured as indicators of kidney function.
Histology
Mid-ventricular section of the heart and mid-longitudinal section of the kidney were fixed in 10% neutral buffered formalin and paraffin embedded for histology. Tissue sections of 5 μm thickness were subject to picro-sirius red (Sigma-Aldrich, Singapore) or immunofluorescence staining for α-SMA and 4’,6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific, Singapore). Bright field and fluorescent images were captured (Nikon Eclipse Ti inverted microscope with C-HGFIE pre-centered fiber illuminator, Nikon, Singapore) and analyzed using ImageJ 1.45 s.50 MyocFB was indicated by percent of α-SMA+ cells.51
Statistics
Data were compared for differences by one-way ANOVA followed Bonferroni post hoc analysis or unpaired two-tail t test as appropriate. Two-way ANOVA repeated measurement with Bonferroni adjustment was used for analysis of serial measurement of echocardiography. Kaplan-Meier analysis was used to calculate the survival rate. All statistics was done using a software of GraphPad Prism (La Jolla, CA, USA). Values were expressed as mean ± SD. Differences between groups were regarded as significant at the p < 0.05 probability level.
Author Contribution
Y.Z. designed and conducted ex vivo and in vivo experiments, analyzed data, and prepared manuscript. D.Y.E.N. conducted ex vivo experiments. P.W. designed research studies, conducted in vivo experiments, analyzed data, prepared and finalized manuscript. A.M.R. provided funds for this research and revised and edited manuscripts.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Dr. Daniel Rifkin (New York University, US) for the kind gift of TMLCs. This work was supported by grants from National Medical Research Council Center Grant (Ministry of Health, Singapore, A.M.R.) and CVRI Operating Fund (National University of Singapore, A.M.R.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.09.041.
Supplemental Information
References
- 1.O’Callaghan C.A., Shine B., Lasserson D.S. Chronic kidney disease: a large-scale population-based study of the effects of introducing the CKD-EPI formula for eGFR reporting. BMJ Open. 2011;1:e000308. doi: 10.1136/bmjopen-2011-000308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palmer S.C., Yandle T.G., Frampton C.M., Troughton R.W., Nicholls M.G., Richards A.M. Renal and cardiac function for long-term (10 year) risk stratification after myocardial infarction. Eur. Heart J. 2009;30:1486–1494. doi: 10.1093/eurheartj/ehp132. [DOI] [PubMed] [Google Scholar]
- 3.Mall G., Huther W., Schneider J., Lundin P., Ritz E. Diffuse intermyocardiocytic fibrosis in uraemic patients. Nephrol. Dial. Transplant. 1990;5:39–44. doi: 10.1093/ndt/5.1.39. [DOI] [PubMed] [Google Scholar]
- 4.Charytan D.M., Padera R., Helfand A.M., Zeisberg M., Xu X., Liu X., Himmelfarb J., Cinelli A., Kalluri R., Zeisberg E.M. Increased concentration of circulating angiogenesis and nitric oxide inhibitors induces endothelial to mesenchymal transition and myocardial fibrosis in patients with chronic kidney disease. Int. J. Cardiol. 2014;176:99–109. doi: 10.1016/j.ijcard.2014.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piek A., de Boer R.A., Silljé H.H. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016;21:199–211. doi: 10.1007/s10741-016-9536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennedy D.J., Vetteth S., Periyasamy S.M., Kanj M., Fedorova L., Khouri S., Kahaleh M.B., Xie Z., Malhotra D., Kolodkin N.I. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension. 2006;47:488–495. doi: 10.1161/01.HYP.0000202594.82271.92. [DOI] [PubMed] [Google Scholar]
- 7.Elkareh J., Kennedy D.J., Yashaswi B., Vetteth S., Shidyak A., Kim E.G., Smaili S., Periyasamy S.M., Hariri I.M., Fedorova L. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension. 2007;49:215–224. doi: 10.1161/01.HYP.0000252409.36927.05. [DOI] [PubMed] [Google Scholar]
- 8.Burchill L., Velkoska E., Dean R.G., Lew R.A., Smith A.I., Levidiotis V., Burrell L.M. Acute kidney injury in the rat causes cardiac remodelling and increases angiotensin-converting enzyme 2 expression. Exp. Physiol. 2008;93:622–630. doi: 10.1113/expphysiol.2007.040386. [DOI] [PubMed] [Google Scholar]
- 9.Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 10.Khalil H., Kanisicak O., Prasad V., Correll R.N., Fu X., Schips T., Vagnozzi R.J., Liu R., Huynh T., Lee S.J. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Invest. 2017;127:3770–3783. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pohlers D., Brenmoehl J., Löffler I., Müller C.K., Leipner C., Schultze-Mosgau S., Stallmach A., Kinne R.W., Wolf G. TGF-beta and fibrosis in different organs - molecular pathway imprints. Biochim. Biophys. Acta. 2009;1792:746–756. doi: 10.1016/j.bbadis.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy D., Omran E., Periyasamy S.M., Nadoor J., Priyadarshi A., Willey J.C., Malhotra D., Xie Z., Shapiro J.I. Effect of chronic renal failure on cardiac contractile function, calcium cycling, and gene expression of proteins important for calcium homeostasis in the rat. J. Am. Soc. Nephrol. 2003;14:90–97. doi: 10.1097/01.asn.0000037403.95126.03. [DOI] [PubMed] [Google Scholar]
- 13.Tian J., Shidyak A., Periyasamy S.M., Haller S., Taleb M., El-Okdi N., Elkareh J., Gupta S., Gohara S., Fedorova O.V. Spironolactone attenuates experimental uremic cardiomyopathy by antagonizing marinobufagenin. Hypertension. 2009;54:1313–1320. doi: 10.1161/HYPERTENSIONAHA.109.140038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fedulov A.V., Ses T.P., Gavrisheva N.A., Rybakova M.G., Vassilyeva J.G., Tkachenko S.B., Kallner A., MacMillan J.C. Serum TGF-beta 1 and TNF-alpha levels and cardiac fibrosis in experimental chronic renal failure. Immunol. Invest. 2005;34:143–152. [PubMed] [Google Scholar]
- 15.Murphy-Ullrich J.E., Suto M.J. Thrombospondin-1 regulation of latent TGF-β activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018;68-69:28–43. doi: 10.1016/j.matbio.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rifkin D.B., Rifkin W.J., Zilberberg L. LTBPs in biology and medicine: LTBP diseases. Matrix Biol. 2018;71-72:90–99. doi: 10.1016/j.matbio.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayashi H., Sakai T. Biological Significance of Local TGF-β Activation in Liver Diseases. Front. Physiol. 2012;3:12. doi: 10.3389/fphys.2012.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubois C.M., Blanchette F., Laprise M.H., Leduc R., Grondin F., Seidah N.G. Evidence that furin is an authentic transforming growth factor-beta1-converting enzyme. Am. J. Pathol. 2001;158:305–316. doi: 10.1016/s0002-9440(10)63970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crawford S.E., Stellmach V., Murphy-Ullrich J.E., Ribeiro S.M., Lawler J., Hynes R.O., Boivin G.P., Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- 20.Stenina-Adognravi O. Invoking the power of thrombospondins: regulation of thrombospondins expression. Matrix Biol. 2014;37:69–82. doi: 10.1016/j.matbio.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dogar A.M., Semplicio G., Guennewig B., Hall J. Multiple microRNAs derived from chemically synthesized precursors regulate thrombospondin 1 expression. Nucleic Acid Ther. 2014;24:149–159. doi: 10.1089/nat.2013.0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farberov S., Meidan R. Fibroblast growth factor-2 and transforming growth factor-beta1 oppositely regulate miR-221 that targets thrombospondin-1 in bovine luteal endothelial cells. Biol. Reprod. 2018;98:366–375. doi: 10.1093/biolre/iox167. [DOI] [PubMed] [Google Scholar]
- 23.Hu S., Huang M., Nguyen P.K., Gong Y., Li Z., Jia F., Lan F., Liu J., Nag D., Robbins R.C., Wu J.C. Novel microRNA prosurvival cocktail for improving engraftment and function of cardiac progenitor cell transplantation. Circulation. 2011;124(11, Suppl):S27–S34. doi: 10.1161/CIRCULATIONAHA.111.017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Y., Chen Q., Lew K.S., Richards A.M., Wang P. Discovery of Potential Therapeutic miRNA Targets in Cardiac Ischemia-Reperfusion Injury. J. Cardiovasc. Pharmacol. Ther. 2016;21:296–309. doi: 10.1177/1074248415604463. [DOI] [PubMed] [Google Scholar]
- 25.Chen Q., Zhou Y., Richards A.M., Wang P. Up-regulation of miRNA-221 inhibits hypoxia/reoxygenation-induced autophagy through the DDIT4/mTORC1 and Tp53inp1/p62 pathways. Biochem. Biophys. Res. Commun. 2016;474:168–174. doi: 10.1016/j.bbrc.2016.04.090. [DOI] [PubMed] [Google Scholar]
- 26.Zhou Y., Richards A.M., Wang P. MicroRNA-221 Is Cardioprotective and Anti-fibrotic in a Rat Model of Myocardial Infarction. Mol. Ther. Nucleic Acids. 2019;17:185–197. doi: 10.1016/j.omtn.2019.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verjans R., Peters T., Beaumont F.J., van Leeuwen R., van Herwaarden T., Verhesen W., Munts C., Bijnen M., Henkens M., Diez J. MicroRNA-221/222 Family Counteracts Myocardial Fibrosis in Pressure Overload-Induced Heart Failure. Hypertension. 2018;71:280–288. doi: 10.1161/HYPERTENSIONAHA.117.10094. [DOI] [PubMed] [Google Scholar]
- 28.Oida T., Weiner H.L. Depletion of TGF-β from fetal bovine serum. J. Immunol. Methods. 2010;362:195–198. doi: 10.1016/j.jim.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan S.A., Joyce J., Tsuda T. Quantification of active and total transforming growth factor-β levels in serum and solid organ tissues by bioassay. BMC Res. Notes. 2012;5:636. doi: 10.1186/1756-0500-5-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 32.Mann M., Wright P.R., Backofen R. IntaRNA 2.0: enhanced and customizable prediction of RNA-RNA interactions. Nucleic Acids Res. 2017;45(W1):W435–W439. doi: 10.1093/nar/gkx279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wipff P.J., Rifkin D.B., Meister J.J., Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Y., Shiok T.C., Richards A.M., Wang P. MicroRNA-101a suppresses fibrotic programming in isolated cardiac fibroblasts and in vivo fibrosis following trans-aortic constriction. J. Mol. Cell. Cardiol. 2018;121:266–276. doi: 10.1016/j.yjmcc.2018.07.251. [DOI] [PubMed] [Google Scholar]
- 35.Mortimer S.A., Doudna J.A. Unconventional miR-122 binding stabilizes the HCV genome by forming a trimolecular RNA structure. Nucleic Acids Res. 2013;41:4230–4240. doi: 10.1093/nar/gkt075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abe M., Ho C.H., Kamm K.E., Grinnell F. Different molecular motors mediate platelet-derived growth factor and lysophosphatidic acid-stimulated floating collagen matrix contraction. J. Biol. Chem. 2003;278:47707–47712. doi: 10.1074/jbc.M306228200. [DOI] [PubMed] [Google Scholar]
- 37.Annes J.P., Munger J.S., Rifkin D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 38.Takekawa M., Tatebayashi K., Itoh F., Adachi M., Imai K., Saito H. Smad-dependent GADD45beta expression mediates delayed activation of p38 MAP kinase by TGF-beta. EMBO J. 2002;21:6473–6482. doi: 10.1093/emboj/cdf643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bongartz L.G., Braam B., Gaillard C.A., Cramer M.J., Goldschmeding R., Verhaar M.C., Doevendans P.A., Joles J.A. Target organ cross talk in cardiorenal syndrome: animal models. Am. J. Physiol. Renal Physiol. 2012;303:F1253–F1263. doi: 10.1152/ajprenal.00392.2012. [DOI] [PubMed] [Google Scholar]
- 40.Perez-Ruiz L., Ros-Lopez S., Cardús A., Fernandez E., Valdivielso J.M. A forgotten method to induce experimental chronic renal failure in the rat by ligation of the renal parenchyma. Nephron, Exp. Nephrol. 2006;103:e126–e130. doi: 10.1159/000092198. [DOI] [PubMed] [Google Scholar]
- 41.Griffin K.A., Picken M., Bidani A.K. Method of renal mass reduction is a critical modulator of subsequent hypertension and glomerular injury. J. Am. Soc. Nephrol. 1994;4:2023–2031. doi: 10.1681/ASN.V4122023. [DOI] [PubMed] [Google Scholar]
- 42.Griffin K.A., Picken M.M., Churchill M., Churchill P., Bidani A.K. Functional and structural correlates of glomerulosclerosis after renal mass reduction in the rat. J. Am. Soc. Nephrol. 2000;11:497–506. doi: 10.1681/ASN.V113497. [DOI] [PubMed] [Google Scholar]
- 43.Fleck C., Appenroth D., Jonas P., Koch M., Kundt G., Nizze H., Stein G. Suitability of 5/6 nephrectomy (5/6NX) for the induction of interstitial renal fibrosis in rats--influence of sex, strain, and surgical procedure. Exp. Toxicol. Pathol. 2006;57:195–205. doi: 10.1016/j.etp.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 44.van Dokkum R.P., Eijkelkamp W.B., Kluppel A.C., Henning R.H., van Goor H., Citgez M., Windt W.A., van Veldhuisen D.J., de Graeff P.A., de Zeeuw D. Myocardial infarction enhances progressive renal damage in an experimental model for cardio-renal interaction. J. Am. Soc. Nephrol. 2004;15:3103–3110. doi: 10.1097/01.ASN.0000145895.62896.98. [DOI] [PubMed] [Google Scholar]
- 45.Trang P., Wiggins J.F., Daige C.L., Cho C., Omotola M., Brown D., Weidhaas J B, Bader A.G., Slack F.J. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther. 2011;19:1116–1122. doi: 10.1038/mt.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Potus F., Ruffenach G., Dahou A., Thebault C., Breuils-Bonnet S., Tremblay È., Nadeau V., Paradis R., Graydon C., Wong R. Downregulation of MicroRNA-126 Contributes to the Failing Right Ventricle in Pulmonary Arterial Hypertension. Circulation. 2015;132:932–943. doi: 10.1161/CIRCULATIONAHA.115.016382. [DOI] [PubMed] [Google Scholar]
- 47.Zhou Y., Richards A.M., Wang P. Characterization and Standardization of Cultured Cardiac Fibroblasts for Ex Vivo Models of Heart Fibrosis and Heart Ischemia. Tissue Eng. Part C Methods. 2017;23:422–433. doi: 10.1089/ten.TEC.2017.0169. [DOI] [PubMed] [Google Scholar]
- 48.Abe M., Harpel J.G., Metz C.N., Nunes I., Loskutoff D.J., Rifkin D.B. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- 49.Zhou Y., Wang D., Gao X., Lew K., Richards A.M., Wang P. mTORC2 phosphorylation of Akt1: a possible mechanism for hydrogen sulfide-induced cardioprotection. PLoS ONE. 2014;9:e99665. doi: 10.1371/journal.pone.0099665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ivey M.J., Kuwabara J.T., Pai J.T., Moore R.E., Sun Z., Tallquist M.D. Resident fibroblast expansion during cardiac growth and remodeling. J. Mol. Cell. Cardiol. 2018;114:161–174. doi: 10.1016/j.yjmcc.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu X., Khalil H., Kanisicak O., Boyer J.G., Vagnozzi R.J., Maliken B.D., Sargent M.A., Prasad V., Valiente-Alandi I., Blaxall B.C., Molkentin J.D. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Invest. 2018;128:2127–2143. doi: 10.1172/JCI98215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.