

Abstract
Although mitochondrial DNA (mtDNA) haplotype variation is often applied for estimating population dynamics and phylogenetic relationships, economical and generalized methods for entire mtDNA genome enrichment prior to high-throughput sequencing are not readily available. This study demonstrates the utility of differential centrifugation to enrich for mitochondrion within cell extracts prior to DNA extraction, short-read sequencing, and assembly using exemplars from eight maternal lineages of the insect species, Ostrinia nubilalis. Compared to controls, enriched extracts showed a significant mean increase of 48.2- and 86.1-fold in mtDNA based on quantitative PCR, and proportion of subsequent short sequence reads that aligned to the O. nubilalis reference mitochondrial genome, respectively. Compared to the reference genome, our de novo assembled O. nubilalis mitochondrial genomes contained 82 intraspecific substitution and insertion/deletion mutations, and provided evidence for correction of mis-annotated 28 C-terminal residues within the NADH dehydrogenase subunit 4. Comparison to a more recent O. nubilalis mtDNA assembly from unenriched short-read data analogously showed 77 variant sites. Twenty-eight variant positions, and a triplet ATT codon (Ile) insertion within ATP synthase subunit 8, were unique within our assemblies. This study provides a generalizable pipeline for whole mitochondrial genome sequence acquisition adaptable to applications across a range of taxa.
Subject terms: Biological techniques, Computational biology and bioinformatics, Genetics
Introduction
Insect mitochondrial genomes are relatively small (range: 14 to 40 kbp) and encode 13 protein-coding genes, 22 transfer RNAs, and two ribosomal RNAs1 in a highly conserved order and orientation2. Due to higher rates of sequence evolution3,4, low recombination rates5, and lower effective population size of the maternally-inherited molecule compared to biparental nuclear loci, mitochondrial DNA (mtDNA) is often used to predict evolutionary relationships and estimate species divergence times6–11, albeit for shallow time scales due to accumulating effects of homoplasy12. Additionally, intraspecies variation can shed light on population genetics, demographics, and female dispersal13. Although the majority of studies are based on haplotype variation estimated from a relatively small number of mitochondrial genes (typically one or a few), whole mtDNA genome analyses offer advantages for phylogenetic, population genetic, and functional genomic studies14–16.
Ostrinia nubilalis is a lepidopteran pest that feeds on and causes yield loss to cultivated maize and other crops across its native range of Europe and western Asia, as well as within introduced and invasive areas of North America17. Feeding damage was reduced in the United States following the widespread adoption of transgenic maize encoding insecticidal Bacillus thuringiensis (Bt) toxins18, yet O. nubilalis remains a model for the study of sympatric population divergence and incipient species formation19. Although low-levels of mtDNA cytochrome c oxidase subunit I (coxI) variation were significant between sympatric and allopatric O. nubilalis ecotypes differing in the number of annual reproductive generations20, mtDNA variation is generally considered to be low and uninformative within this species21,22. This is in stark contrast to the greater inter-individual (haplotype) variation reported in the related species, O. furnacalis23,24, and other species of Lepidoptera25,26. The evolutionary rate of nucleotide change varies across mitochondrial genome regions for animals27, including insects7, indicating studies based on short mtDNA gene fragments may be skewed by genome sampling ascertainment bias14.
The number of sequenced whole mitochondrial genomes has greatly increased in recent years28. This increase has facilitated mitochondrial genome assemblies directly from high-throughput short-read sequencing of shotgun total genomic libraries8, or libraries enriched for mtDNA using commercial miniprep-29, rolling circle-30, or probe-based methods31. Regardless, many of these methods co-enrich nuclear-fragments with integrated mitochondrial DNA fragments (NUMTs)32–36, or arguably require expensive commercial reagents. The current study establishes a relatively rapid, less expensive, and portable enrichment method, which, when applied to samples prior to high-throughput DNA sequence library construction and sequencing, allows for downstream assessment of full mtDNA genome variation. Differential centrifugation, adapted from prior methods37, is used to obtain mitochondrial- and nuclear-enriched fractions from Ostrinia nubilalis thorax homogenates. This enrichment method, which minimized the number of reads derived from NUMTs, may be especially useful for assembly of novel reference mitochondrial genomes. The de novo mitochondrial genome assembly and annotation are described, and we report two major differences compared to the reference assemblies (AF442957.1 and MN793322.1). This study demonstrates the utility of mitochondrion enrichments within a full mtDNA genome sequencing protocol, and how detected variation can be used in population and phylogenetic studies.
Results
Optimization of differential centrifugation and DNA extraction
Following initial centrifugation at 1000 relative centrifugation force (rcf) to remove cell debris, a second centrifugation at 6000 rcf optimally separated intact nuclei from mitochondria within thorax cellular homogenates. This combination resulted in the greatest sedimentation of nuclei (chromosomal DNA) and retention of mitochondria (mtDNA) in the supernatant. Subsequent centrifugation of the supernatant at 13,000 rcf provided a precipitate rich in mitochondria, as estimated by Janus Green B stained organelles (Fig. 1a). Enrichment was also demonstrated by the ratio of amplified fragment intensities by semi-quantitative PCR of mitochondrial (coxI) compared to the apn1 nuclear target gene (Fig. 1b, S1). By comparison, DNA extracts centrifuged at 2000 and 4000 rcf resulted in either no or inconsistent nuclear amplification, respectively, indicating ineffective separation of nuclei and mitochondria (data not shown). Mitochondrial coxI amplification was also inconsistent or non-existent within DNA extracts obtained from fractions pelleted at 2000 and 4000 rcf. Nuclear and mitochondrial target genes were both PCR amplified from fractions collected at 8000 rcf, suggesting co-precipitation of both organelles (data not shown).
Figure 1.

Representative example of differentially fractionated samples of Ostrinia nubilalis homogenized thorax tissue to acquire nuclear and mitochondrial components at 6000 and 13,000 rcf, respectively. Janus Green B staining of the mitochondrial fraction indicated abundant mitochondria (a). Gel electrophoresis of serial-diluted DNA extracted from the mitochondrial and nuclear fractions amplified in duplicate with mitochondrial primers (HCO/LCO;59) and nuclear primers (Onapn1-F and -R;46) (b). Mitochondrial fraction (13,000 rcf) was PCR amplified with mitochondrial primers (i) and nuclear primers (ii). Nuclear fraction (6000 rcf) was PCR amplified with mitochondrial primers (iii) and nuclear primers (iv). Although nuclear DNA was present in both the mitochondrial and the nuclear fractions, mtDNA was only present in the mitochondrial fraction.
Quantification of mitochondrion enrichment at optimized parameters
Using mitochondrial coxI primers to amplify mitochondrial fractions as template with real-time quantitative PCR (qPCR) resulted in an estimated mean CT (14.83 ± 0.09) that was significantly lower compared to that estimated from nuclear fractions (17.43 ± 0.17) or unenriched controls (17.54 ± 0.41) (Fig. 2a; − 5.069 < Z > 3.205; df = 2, 34; p < 0.0039; Table S1). By comparison, when mitochondrial fractions, nuclear fractions, and unenriched control samples were amplified with nuclear apn1 primers, the mean CT values were 30.25 ± 0.21, 24.56 ± 0.29, and 26.58 ± 0.21, respectively, and were significantly different from each other (− 6.905 < Z > 3.763; df = 2, 34; p < 0.0005; Table S1).
Figure 2.
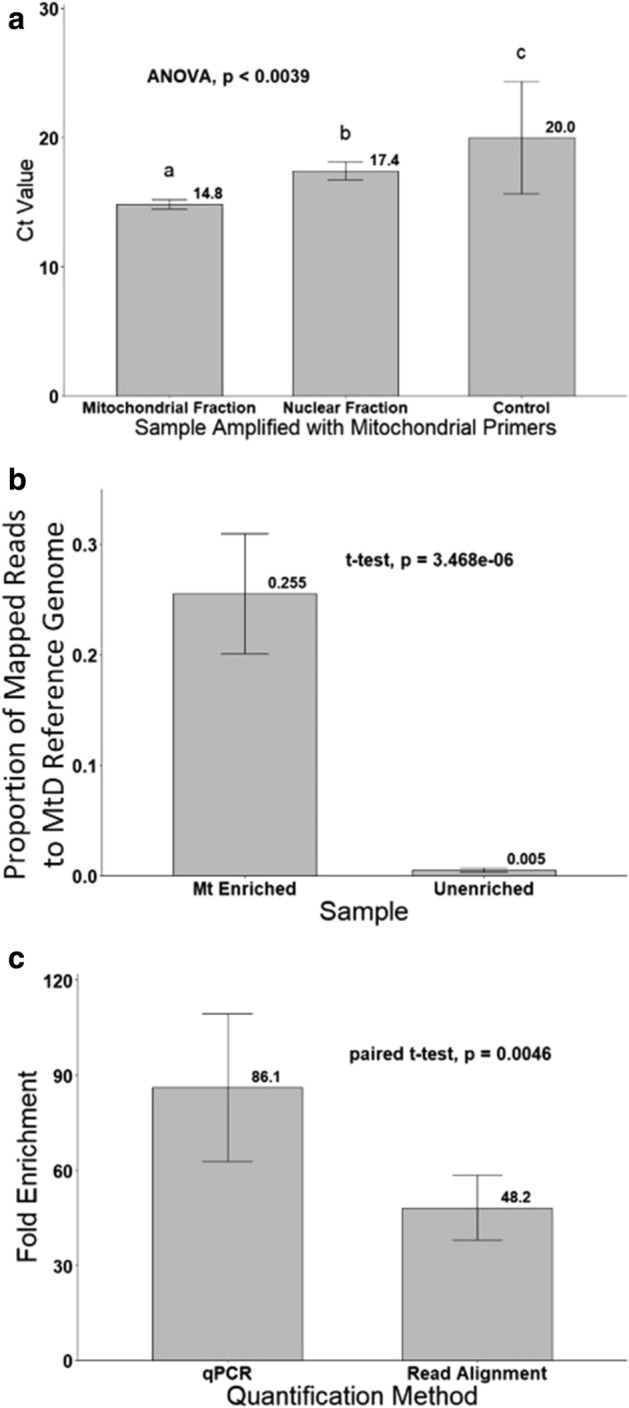
Estimates of mitochondrion enrichment of Ostrinia nubilalis from homogenized thorax tissue through differential fractionation prior to DNA extraction. (a) Significantly lower CT values were estimated by real-time PCR among mitochondrial fractions (14.83 ± 0.37) amplified with mitochondrial cytochrome c oxidase subunit I (coxI) primers (OnCOXIrtS and OnCOXIrtA) compared to the nuclear fractions (17.43 ± 0.67) or unenriched controls (20.0 ± 4.34) (ANOVA, p < 0.001). Different letters above bars represent a significant difference. (b) Significant increase in the ratio of reads from enriched mitochondrion samples (0.255 ± 0.054) that aligned to the O. nubilalis mitochondrial genome reference (AF442957.1) compared to unenriched controls (0.0005 ± 0.0017; two-sample t-test, p < 0.0001) (c) Difference in fold-enrichment estimates for mitochondrion fractions estimated by with mitochondrial target amplicons estimate enrichment by real-time quantitative PCR (mean 86.1 ± 22.3) and read alignment (mean 48.2 ± 10.2). Although significant (paired t-test, p = 0.0046), both estimation methods indicate consistent trends in enrichment.
A mean of 3,047,025 (± 162,751) reads was obtained across eight individual mitochondrion-enriched Illumina MiSeq libraries (BioProject: PRJNA604593; accessions SRR5182721-SRR5182724; Table S1). Among these reads, a mean of 781,200 (± 77,773) aligned to the O. nubilalis reference mitochondrial genome sequence (AF442957.1) with a Q-score > 30 (mean ~ 26.7 ± 2.0% alignment rate). The unenriched controls had a mean of 183,349 (± 34,965) mapped reads with Q-score > 30 from among a mean of 35,319,000 (± 4,800,041) total reads (~ 0.6 ± 0.1% mean alignment rate). The increase in the proportions of reads that aligned to the mitochondrial reference from libraries generated from mitochondrion enriched (~ 26.7%) compared to unenriched libraries (~ 0.6%) was significant (Fig. 2b; t = 13.065; df = 1, 11; p < 0.0001; Table S1).
The resulting ratio of CT values estimated from real-time qPCR amplification of mitochondrial-enriched extracts compared to the mean CT of unenriched extracts showed an estimated mean 86.1 ± 8.25-fold enrichment (Fig. 2c). Based on the ratio of aligned Illumina MiSeq reads from enriched to unenriched libraries, a mean enrichment of 48.2 ± 3.6-fold was estimated. Although there was a significant difference in the estimated fold-enrichments based on real-time qPCR compared to Illumina MiSeq read alignment (Fig. 2c, t = − 4.0958; df = 1, 7; p = 0.0046), a ≥ 29.8-fold mtDNA enrichment was predicted among all samples with both estimation methods (Table S1).
De novo mitochondrial genome assembly, annotation and variant prediction
Mitochondrion-enriched short read library data were de novo assembled, annotated from query results of O. nubilalis mitochondrial genome RefSeq models using the BLASTn algorithm, and submitted to NCBI GenBank (MT492030.1-MT492037.1; Table 1). Each assembly was annotated with 13 protein-coding genes (PCGs), 22 tRNAs, and two rRNAs typical of animal mtDNA genomes. Based on the invertebrate mitochondrial code, all PCG translations were initiated with Met start codon (ATA or ATG), with the exception of coxI that had an atypical Arg (CGA). Stop codons, TAA or TAG, were predicted for all PCGs, with the exception of coxII that ends in a single T nucleotide that putatively forms a functional TAA stop codon following polyadenylation during transcript maturation.
Table 1.
Ostrinia nubilalis mitochondrial genome sequence assembly accessions.
| Accession | Description | Assembly Len | Citation | Read type/platform |
|---|---|---|---|---|
| AF442957.1 | RefSeq NC_003367.1 | 14,535 bp | Coates et al. 200420 | Sanger/ABI3700 |
| MN792233.1 | Yili, Xinjiang Region | 15,248 bp | Zhou et al. 202035 | Illumina/HiSeq |
| MT492030.1 | F1_Family_ID_02 | 14,838 bp | This study | Illumina/MiSeq |
| MT492031.1 | F1_Family_ID_03 | 14,817 bp | This study | Illumina/MiSeq |
| MT492032.1 | F1_Family_ID_04 | 14,691 bp | This study | Illumina/MiSeq |
| MT492033.1 | F1_Family_ID_06 | 14,621 bp | This study | Illumina/MiSeq |
| MT492034.1 | F1_Family_ID_07 | 14,622 bp | This study | Illumina/MiSeq |
| MT492035.1 | F1_Family_ID_11 | 15,082 bp | This study | Illumina/MiSeq |
| MT492036.1 | F1_Family_ID_12 | 14,809 bp | This study | Illumina/MiSeq |
| MT492037.1 | F1_Family_ID_18 | 15,064 bp | This study | Illumina/MiSeq |
Our assemblies were compared to the Sanger sequence-based reference O. nubilalis mitochondrial genome assembly, AF442957.138 (RefSeq NC_003367.1), and a recent Illumina short shotgun read-based assembly39. For the former comparison, a total of 82 mutations were predicted among our de novo assemblies (MT492030.1–MT492037.1; Table 1) and the reference AF442957.1 (RefSeq NC_003367.1; Table S2). Because each of our de novo mitochondrial genomes had a different start position due to starts at random seeds, homologous nucleotide sites with respect to the reference were defined within each accession (Table S3). Among these predicted variants, 58 (69.8%) were within protein-coding sequences (CDS; coxI, coxII, atp8, atp6, coxIII, nd3, nd5, nd4, nd4L, cytb, or nd1; Table S2), for which 35 generated nonsynonymous (amino acid) changes (60.3% of CDS mutations; 42.2% of all mutations). Twenty-two of the remaining CDS mutations were synonymous (silent; 26.8%). In addition, an in-frame insertion of an intact triplet ATT (Ile) codon was predicted within atp8 from F1_Family_ID_04 (accession MT492032.1; Fig. 3a). This inserted ATT codon was not present in any other previously sequenced Ostrinia mitochondrial genome, but was found in the atp8 CDS of the butterfly species, Ochlodes venata (Lepidoptera: Hesperiidae). Moreover, the two adjacent Ile residues within this region of ATP8 showed variable presence among species of Lepidoptera, with both omitted from Parnassius epaphus (Lepidoptera: Papilionidae) (Fig. 3a). Our analyses also predicted a frameshift impacting three consecutive codons in coxI among 4 of our assemblies (MT492030.1, MT492035.1 to MT492037.1) compared to the reference, and shared with other Ostrinia including the recent Illumina sequence-base O. nubilalis assembly (MN793322.1;39) (Fig. 3b). This frameshift is predicted to result from upstream insertion and downstream deletion mutations that function in tandem to maintain the frame within the downstream CDS beyond the three affected codons (Fig. 3b). The remaining 25 predicted mutations compared to the reference were within rRNALSU (8 SNVs and three indels), rRNASSU (6 indels), and among tRNAs (5 SNVs and two indels; Table S2).
Figure 3.

In-frame and frameshift mutations predicted among assembled Ostrinia mitochondrial genomes. (a) Multiple sequence alignment of an ATP synthase subunit 8 (atp8) gene region showing a putative in-frame ATT nucleotide (Isoleucine, I) duplication within F1_Family_ID_07 (MT492034.1) compared to other Ostrinia. (b) Frameshift impacting three codons of the COI protein-coding sequence caused by flanking compensatory insertion/deletion mutations that return sequences to the same downstream frame. Genus species (Gsp) abbreviations: Onub, O. nubilalis; Ofur, O. furnacalis; Osca, O. scapulalis; Ozea, O. zealis; Opal, O. palustralis. Open, O. penitalis; Oven, Ochlodes venata (Lepidoptera: Hesperiidae); Pepa, Parnassius epaphus (Lepidoptera: Papilionidae). Conserved sites highlighted in grey.
The frequency of each variant site among the eight F1 family-specific assemblies compared to the reference ranged from 0.13 to 1.00, of which 20 of the 82 variable positions (22.4%; coxI frameshift not counted as variable position) were conserved across all eight F1 families (Freq = 1.00 in Table S2). All of the 20 fixed differences with respect to the reference were SNVs, with the exception of a deletion in nd4. For this nd4 deletion, all assemblies from this study lacked a C nucleotide reported for position 8200 in reference AF442957.1, which caused a discrepancy in predicted C-terminal amino acids starting at position 418 of nd4 (Fig. 4). Subsequent multiple nd4 protein sequence alignment and phylogenetic comparison showed ≥ 83.33% amino acid identity between the terminal 60 residues of nd4 from our O. nubilalis assemblies and other lepidopteran species (Fig. 4), whereas nd4 from AF442957.1 showed complete mismatch in the terminal 29 residues. Inspection of bam alignments of short reads from each F1 family-specific library confirmed a lack of this C compared to the O. nubilalis reference (Fig. S2). Seven substitutions predicted in coxI and coxII genes among our assemblies were previously observed in a prior population genetics study, of which mutations at reference positions 2,554 and 3,050 were validated using HaeIII and Sau3AI PCR-restriction fragment length polymorphism (PCR–RFLP) assays within that study (Table S2)20.
Figure 4.

Alignment of 60 C-terminal residues from representative NADH Dehydrogenase subunit 4 (ND4) protein sequence accessions. Conserved residues (highlighted) showed ≥ 83.33% identity between species, with the exception of the 29 terminal residues from O. nubilalis AAL66246.1 predicted in the reference mitochondrial genome (AF442957.1). Frameshift in ND4 accession AAL66246.1 predicted from the reference AF442957.1 putatively caused by insertion of a cytosine (C) nucleotide at position 1254 of the ND4 CDS (genome position 8013; Table S2). Companion Maximum-Likelihood (ML) constructed based on the mtREV + F + G model of sequence evolution that maximized the Bayesian Information Criterion (BIC) score at 4929.63, and used an empirically-derived discrete gamma distribution (shape parameter; G) of 0.3988 that minimized the log-likelihood score (-2330.81). Total branch length of 0.56 amino acid changes per site. Species abbreviations: Sfru, Spodoptera frugiperda; Tni; Trichoplusia ni; Epos, Epiphyas postvittana; Mvit, Maruca vitrata; Ostrinia species abbreviations as in Fig. 3).
Analogously, there were a total of 77 variant sites, plus an Ile (ATT) triplet codon insertion within atp8 and a frameshift mutation in coxI identified within the multiple sequence alignment among our eight assemblies and a previous unenriched Illumina HiSeq read data-assembled mitochondrial genome sequence39 (MN792233.1; Fig. S2). Among these 77 variant sites, 43 (55.1%) were within CDS, of which 24 (55.8%) were nonsynonymous and predicted to cause an amino acid change (Table S4; Fig S3). Eight mutations were fixed differently between MT492031.1 to MT492034.1 compared to MN792233.1. Identical to comparison with AF442957.1, the Ile codon insertion within our F1 Family_ID_04 (MT492032.1) was not present with the atp8 CDS of MN792233.1 (Fig. 3a; Fig S3). Additionally, compared to reference AF442957.1 (RefSeq NC_003367.1), the cox1 frameshift identified within four of our eight assemblies was also present in MT492032.1 (Fig. 3b; Fig S3). The remaining 35 mutations were within the rRNALSU (n = 14), rRNASSU (n = 6), tRNAs (n = 14), and non-coding sequence (n = 1). Three of the substitutions within coxI and coxII genes were previously described by Coates et al.20, including two validated by HaeIII and Sau3AI PCR–RFLP assays (Table S4).
Comparison between enriched and unenriched reads for de novo mitochondrial genome assembly
Our use of reads from mitochondrion-enriched and unenriched libraries as input for SPAdes and SOAPdenovo2, respectively, resulted in assemblies that showed different levels of contiguity and computational efficiency. Specifically, library read data from enriched samples were assembled in 52 ± 8 s compared to 1380 ± 180 s among the unenriched libraries. Additionally, the total number of contigs within resulting assembles were lower among enriched (98 ± 41) compared to unenriched libraries (337,151 ± 154,655), wherein mean contig lengths were also greater for enriched (1110 ± 469 bp) compared to unenriched libraries (117 ± 992 bp) (remaining data not shown). Among the assembled contigs a mean of 31% and 0.0006% from enriched and unenriched libraries, respectively, showed homology to mtDNA based on BLASTn query with the mitochondrial reference genome sequence (Table S6). Moreover, among these mtDNA contigs, normalized read depth (RPKM) the estimated mean was ~ 65 times higher and significantly greater (t = 14.435; df = 1, 667; p < 2.2 × 10–16) among enriched samples compared to unenriched samples (Fig. S4). Furthermore, there was a distribution of contigs with both high RPKM estimate and length across all assemblies generated from enriched libraries that were of predicted to have mtDNA origin (Fig S5), and deemed to be of putative mitochondrial genome origin. In addition, the observed distribution of contigs of mtDNA origin with correspondingly low RPKM estimates and comparative bias toward short contig length were categorized at putative NUMTs. In contrast, contigs with high RPKM estimates in assemblies from each of the unenriched libraries were mostly of non-mtDNA origin (Fig. S5).
Discussion
Variation in mtDNA sequence data is frequently used within interspecies phylogenetic and intraspecies population genetic analyses. However, a vast majority of studies assess variation based on a few genes or gene fragments, which potentially impact estimates of genetic variation and bias results, leading to the possibility of arriving at specious conclusions. This premise resides in evidence that the mutation rates vary among regions of the mitochondrial genome within plant species40 and humans41, as well as between species42. Methods have recently been developed to more efficiently obtain sequence data from entire mtDNA genomes, capitalizing on advancements in DNA sequencing technologies. These include generation of low-pass short shotgun sequencing reads from total genomic DNA, from which success of mtDNA genome assemblies rely on the proportionally high copy number of mtDNA compared to nuclear DNA8,10, as recently reported for several species of Ostrinia39. Targeted mtDNA sequencing employs various enrichments prior to library construction, including those based on commercial reagents43, isolation of highly intact circular mtDNA30, the requirement to develop specific antibodies for organelle pull-down44, or necessitating prior knowledge of the mtDNA genome sequence for probe-based nucleotide capture31,32. Although differential centrifugation was analogously used in methods to isolate human mitochondrion prior to mtDNA genome sequencing, the commercial kit protocols used are tailored to specific species45 and not directly transferable to other organisms. Here, we developed and validated a relatively rapid and low-cost differential centrifugation method to prepare mitochondrion-rich fractions from O. nubilalis thorax homogenates prior to DNA extraction and subsequent high-throughput DNA sequence library construction and sequencing. Although not tested here, this enrichment method is likely transferrable to other non-model organisms where no prior genome assemblies are available, due to our evidence that enrichments lead to significant reductions in raw data input and computational time requirements, as well as increase contiguity of resulting mtDNA assemblies. Furthermore, the additive effect of these efficiencies likely may enable individual-wise assessment of mitochondrial genome-wide haplotype variation on a population scale.
Our protocol development empirically determined that a second centrifugation step of 6000 rcf maximized the separation of mitochondria and nuclei in thorax homogenates based on qualitative Janus-Green visualization and semi-quantitative PCR comparisons between enriched and unenriched extracts (Fig. 1). Subsequent RT-qPCR and read alignment analyses indicated that while our differential centrifugation did not provide pure organelle fractions (Fig. 2a), it did provide for subsequent extracts enriched with mtDNA (Fig. 2b). Although these two methods lead to significantly different estimates of enrichment, a mean fold-enrichment of ≥ 48.2 was estimated among the libraries (Fig. 2c). These results suggest that secondary enrichment steps, such as the use of equilibrium density-gradient centrifugation46, could likely be incorporated into our protocol in cases where higher purity isolations are desired. While the level of enrichment and purity of our mitochondrion fractions were lower compared to methods employing mitochondrion surface protein-specific antibody capture44,47 or probe-assisted mtDNA isolation31,32, our method has the advantage of not requiring upfront development of custom biological reagents or reliance upon tailored commercial kits. Our differential centrifugation achieved 26% of reads aligning to a reference mitochondrial genome, which is comparable to a 22% alignment rate obtained following a two-step protocol that used a commercial circular DNA miniprep followed by antibody tagged paramagnetic bead isolation43. Regardless, due to variation in the mass of organelles, it may be anticipated that rcf values will need to be optimized for application to different species. Since mitochondrion enrichment prior to sequencing produced at least a 29.8-fold greater read alignment rate to the mitochondrial reference compared to unenriched controls (Table S1), showing that our optimized protocol facilitates a significant reduction in short sequence read input required for downstream assemblies.
Comparison of processes involved in the assembly of enriched compared to unenriched libraries, showed is a significant (~ 27-fold) reduction in computation time and lower required CPU node count and random access memory. Moreover, although it was possible to de novo assemble the mitochondrial genome from unenriched samples, the resulting assemblies were less contiguous compared to those from enriched libraries (Table S6). Specifically, the mean size of mitochondrial-derived contigs among unenriched assemblies were nearly ten-fold shorter and more numerous compared to contigs assembled from enriched libraries (Fig. S4, S5). Interestingly, these increases were achieved despite a 11.7- to 65.7-fold reduction in input reads among enriched libraries (Table S1) compared to that within unenriched SRA data files (SRX2498822-SRX2498825). Furthermore, due to lower sequencing costs and computational time, as well as increased contiguity, we suggest that enrichment provides greater accessibility to the potential for higher throughput assessment of haplotype variation among haplotypes of a species or among species. This may provide for downstream large-scale population and phylogenetic analyses that arguably remain out of reach using other methods.
In this study, we applied the full mtDNA genome assemblies derived from our enrichment method to assess variation across O. nubilalis maternal lineages. The genus Ostrinia is a model for the study of sympatric population divergence and incipient species formation19,48 and comprises two corn borer species, O. nubilalis, and O. furnacalis, which are major pest insects that feed on cultivated maize in North America, Europe and/or Asia. Compared to prior O. nubilalis mtDNA genome assemblies AF442957.120 and MN792233.139, our eight F1 families showed 82 and 77 SNVs, respectively (Table S2, S4). These comparisons also predicted a single insertion and frameshift mutation. Among the mutations we predicted across all eight F1 family-based assemblies, 20 and 8 were fixed differently compared to the reference AF442957.120 and MN792233.139, respectively. Fixed differences could represent natural intraspecies haplotype differences, but given the small sample size of the eight F1 families, some of the fixed differences could be a consequence of sampling bias from a laboratory colony that has undergone a genetic bottleneck and influenced by random genetic drift for > 12 generations. Alternatively, some of these SNVs could have resulted from Sanger sequencing errors incorporated into the reference assembly AF442957.120. The latter might be concluded, for a C nucleotide inserted within nd4 at position 8200 from the Sanger reference as compared to all Illumina-based assemblies (Fig. S2; MN792032.1), which caused a fixed discrepancy in the C-terminal 29 residues of nd4 compared to assemblies from other Ostrinia and lepidopteran species generated from short-read data (Fig. 4). This putative correction suggests that the high read depths obtained from short read sequencing of enriched mtDNA libraries provides superior error correction capabilities compared to dual-pass Sanger read data.
The structural gene annotation of our eight assembles showed two major differences compared to the reference. Firstly, our results also predicted a novel ATT insertion within F1 family 4 (MT492032.1) residing at reference position 3,953 within atp8, causing a putative in-frame Ile codon duplication at atp8 amino acid position 52 (Fig. 3a). This insertion is novel compared to all other exemplar sequences within the genus Ostrinia and other species of Lepidoptera, with the exception of a Leu codon (TTA) found at the orthologous position of the Ochlodes venata mitochondrial genome (Fig. 3b). Moreover, the two adjacent Ile residues are deleted at orthologous sites from the butterfly Parnassius epaphus. These comparisons suggest that sites orthologous to Ostrinia atp8 amino acid positions 52 and 53 may show relaxed functional constraint, and thus indels involving these amino acids might putatively be impacted by purifying selection to a lesser degree. Although intriguing, testing this hypothesis is beyond the scope of the current study. Secondly, out of eight assemblies, four show a pair of indels that cause a frameshift over three amino acids of coxI (Fig. 3b). Specifically, compensation for the upstream deletion by a 10 bp downstream insertion retains the predicted downstream coding frame. Typically, mitochondrial frameshifts are highly deleterious49, but can result in apparently non-deleterious changes depending upon position and context50,51. In other instances, suppressor tRNA mutations or “leaky” ribosomal frameshift compensation mechanisms have been proposed52,53. Regardless, the frameshift described here, resulting in the putative alteration of 3 amino acids of coxI, has an unknown functional consequence and was not tested further. Due to presence within reads at this position, the indel is likely valid, but may be unique among individuals within a relatively inbred laboratory colony and not representative of wild individuals under the influence of selective forces in field conditions.
The variation we detected cannot be interpreted as representing population frequencies, but would require large-scale screening and comparison of random samples from wild populations. Regardless, prior population screening of 1,414 O. nubilalis individuals from North American populations determined that 89.6% were comprised of a single haplotype based on PCR–RFLP, wherein the sequencing of a 2,156 bp fragment comprised of coxI and coxII from 14 individuals predicted 26 polymorphic positions20 (≤ 0.8% variation). Interestingly, of the 82 variant sites predicted in the current study, seven were identical to the set of 26 previously predicted by Coates et al.20. Furthermore, substitutions validated by HaeIII and Sau3AI PCR–RFLP were predicted within comparisons of our assemblies to the Sanger sequence assembled reference38 and the more recent short read-based assembly39, suggesting variants are real as opposed to sequencing or assembly artifacts.
Prior phylogenetic reconstructions differ between Ostrinia species when using variation in sequence data from coxII54 coxI55 gene fragment data, as compared to full mitochondrial genome alignments39. Specifically, full genome analysis by Zhou et al.39 predicted that corn borer sister species, O. nubilalis, and O. furnacalis, were within a clade along with O. scapulalis. In comparison, an O. nubilalis and O. scapulalis clade were separated from O. furnacalis by O. zealis based on coxII gene54 or coxI sequence data55. Although not investigated further for Ostrinia, a disparity in the evolutionary rate between mitochondrial genome regions (genes or gene fragments) is described across taxa7,27, suggesting that phylogenetic relationships could be biased when applying a subset of mitochondrial haplotype variation within short fragments. Therefore, phylogenetic relationships predicted based on the full mtDNA genome sequence might arguably be more reflective of evolutionary divergence patterns, at least over appropriate timespans14.
Prior to our analysis, O. nubilalis mtDNA variation was generally estimated to be low and uninformative in a population context21,22. By implementing a mitochondrial enrichment method and predicting variation across the full mitochondrial genome sequences, we identified a greater number of variant sites within O. nubilalis than previously reported. The future application of analogous full mtDNA sequencing to population or phylogenetic studies will contribute to unbiased estimates of inter- and intra-species variation. This study demonstrated that a differential centrifugation method could be a component of this goal, due to demonstrated enrichment of cell homogenates for mitochondrion and correspondingly removing nuclei prior to DNA extraction. This method provides a low-cost option for organelle enrichment, resulting in samples with a high proportion of mtDNA for library construction and sequencing. Undoubtedly further cost efficiencies could be implemented through indexing of a greater number of sample libraries within higher capacity flow cells compared to the MiSeq that was used here. Furthermore, we demonstrate increased downstream computational efficiencies and resulting assembly contiguities achieved during assembly of reads from mitochondrion-enriched libraries compared to those obtained from unenriched libraries. Although we applied these methods to O. nubilalis, the protocol could be adapted and optimized for other species, thereby facilitating a higher throughput of mtDNA genome sequencing for application in unbiased population and phylogenetic studies, where additive effects of time and cost saving may facilitate performance on larger scales.
Methods
Specimens and sampling
Z-pheromone race Ostrinia nubilalis pupae were obtained from the United States Department of Agriculture, Agriculture Research Service, Corn Insects and Crop Genetics Unit (USDA-ARS, CICGRU) in Ames, IA. Pupae were individually maintained in a Percival incubator (Percival, Boone, IA) (16:8 [L:D] h; 26 °C; 40 to 60% RH). Upon eclosion, single mate pairs were initiated to create F1 families (maternal-specific haplotype lineages). Pairs were maintained in wire mesh cages with wax paper for oviposition substrate, as previously described56. Mated pairs were monitored, and eggs were collected daily for seven days. Wax papers with egg masses were stored in the incubator (16:8 [L:D] h; 26 °C; 40–60% RH) until near hatch. Egg masses from each F1 family were separately placed into individual 10-cm diameter plastic containers with approximately 50-ml of standard O. nubilalis meridic diet57. Pupae were collected from each family and placed into individual paper cups until eclosion. Adults were either live-dissected or frozen at − 20 °C.
Optimization of differential centrifugation and DNA extraction
Thorax tissue containing flight muscle was dissected from three live O. nubilalis adults per F1 family. Since mitochondria are maternally inherited without recombination, tissues were pooled by family in 2.0-mL glass Dounce homogenizers (Corning Inc., Corning, NY) and homogenized on ice in 1.0-mL of a homogenization medium (0.32 M sucrose, 1.0 mM EDTA, 10.0 mM Tris–HCl, 4 °C, pH 7.8). Each F1 family homogenate was transferred to a separate 1.5 mL microcentrifuge tube.
Differential centrifugation was used to fractionate extracts into nuclear and non-nuclear fractions based on molecular mass. The insoluble exoskeleton, cell debris, and other particles were removed from samples at 1,000 relative centrifugal force (rcf) for 10 min at 4 °C in an Eppendorf 5417R centrifuge (Eppendorf, Hauppauge, NY). Aqueous supernatant was transferred to new 1.5-mL microcentrifuge tubes. Replicate samples within each F1 family were centrifuged at 2000, 4000, 6000, and 8000 rcf at 4 °C for 10 min to optimize the force that most effectively pelleted nuclei (referred to as nuclear fraction) while retaining mitochondria in the supernatant. The remaining supernatants of each replicate (F1 family pool) were transferred to new 1.5-mL microcentrifuge tubes and centrifuged at 4 °C for 10 min at 13,000 rcf to pellet the remaining organelles (referred to as mitochondrial fraction).
The optimal centrifugal force was determined using microscopy and the ratio of amplified DNA fragment intensities as determined by semi-quantitative/qualitative PCR of mitochondrial (coxI), and nuclear (apn1) indicator genes. Subsamples of the 13,000 rcf pellet were treated with Janus Green B cell normalization stain (Abcam Inc., Cambridge, MA); stained mitochondria were observed by light microscopy (Olympus Microscopy, model BH-2, Tokyo, Japan)58. DNA was then extracted from each of the fractionated replicates within F1 family pools using the DNEasy Blood and Tissue Extraction Kit according to manufacturer directions (Qiagen, Hilden, Germany), except DNA was eluted from silica columns using 50.0 µL of Elution Buffer. Samples were serially diluted using nuclease-free water, and 1.0 µL from each dilution and was PCR amplified in duplicate using mitochondrial primers (HCO/LCO)59 and nuclear primers (OnFLWA-01) in separate reactions as described earlier60. Entire products were separated by 2% agarose gel electrophoresis with the Lambda HindIII and EcoRI ladder. Based on staining and PCR results, the following method was employed: (1) initial pelleting of cell debris at 1000 rcf; (2) resultant supernatant centrifuged at 6000 rcf to obtain nuclear fraction; (3) resultant supernatant centrifuged at 13,000 rcf to obtain the mitochondrial fraction. All centrifugation steps were conducted for 10 min at 4 °C.
The empirically-determined optimal rcf was employed to pellet nuclei with three frozen/preserved adult individuals of each F1 family. Fractions were subsequently extracted with DNEasy extraction kits, as described above. Unenriched controls were prepared by identical DNA extraction from dissected thorax tissue without nuclear or mitochondrion fractionation from individual Z-strain O. nubilalis field samples collected at South Shore, SD (SDSS ♀1 and SDSS ♀2; contributed by Dr. Micheal Catangui, South Dakota State University).
Quantification of mitochondrion enrichment at optimized parameters
Using the optimized centrifugation forces (6000 and 13,000 rcf), the fold-enrichment of mitochondrion compared to nuclei was estimated by two different methods: (1) analysis of cycle threshold (CT) obtained through real-time quantitative PCR (qPCR) with nuclear and mitochondrial primers, and (2) high-throughput read alignment rates to a mitochondrial reference genome of enriched and unenriched samples.
For real-time qPCR, DNA was quantified from the nuclear and mitochondrial fractions of the eight F1 families and the two unenriched controls on a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Concentrations were adjusted to ~ 1.0-ng/μl with nuclease-free water. All samples were PCR amplified in duplicate on an MX3000P real-time qPCR system (Stratagene, La Jolla, CA) with 2.0-ng of the template along with 10.0-nM of each forward and reverse primer in iQsupermix reactions per to manufacturer instructions (BioRad, Hercules, CA). Mitochondrial DNA-specific primers OnCOXIrtS, 5′-CCT GAT ATA GCA TTC CCA CGA ATA-3′, and OnCOXIrtA, 5′-AAC CAG TTC CTG CTC CAT TT-3′, were designed using the RealTime qPCR Assay tool61. Single locus nuclear primers, APN1RT-1F and APN1RT-1R, amplifying the aminopeptidase N1 gene (apn1), were designed and applied as previously described56. Statistical significance of differences in CT values from mitochondrial and nuclear primer amplification reactions of the mitochondrial and nuclear fractions, and unenriched controls, were analyzed with a general linearized model, one-way ANOVA in RStudio version 1.0.15362,63 with the Estimated Marginal Means (emmeans) package64. Summaries of paired comparisons were made with Tukey’s HSD method using the Visualizations of Paired Comparisons (multcompView) package65.
DNA extracts from each mitochondrial-enriched fraction for each F1 family were submitted to the Iowa State University (ISU) DNA Facility (≤ 100-ng each). Individual indexed short insert libraries were generated using AmpliSeq Library Plus methods according to manufacturer instructions (Illumina, San Diego, CA). All indexed libraries were sequenced in a single lane of a MiSeq system (Illumina, San Diego, CA). Data were received in fastq format and submitted to the National Center for Biotechnology Information (NCBI) GenBank short read archive (SRA). Additionally, four previously generated Illumina HiSeq reads from the whole genomic sequence of pooled O. nubilalis population samples were downloaded from the NCBI SRA database (Accessions SRX2498822-SRX249882566).
Reads from each of our mitochondrion-enriched libraries (n = 8) and whole genomic libraries (n = 4) were aligned to the 14,535 nucleotides O. nubilalis mitochondrial genome reference sequence (NCBI GenBank accession AF442957.1; RefSeq NC_003367.1) assembled previously from Sanger sequencing data from overlapping PCR amplicons38. Nucleotide quality was initially assessed and visualized with fastqc67 (fastqc/0.11.7-d5mgqc7). Reads were trimmed for those with a Phred quality score (q) ≥ 20 over a 4-nucleotide sliding window: the first 15 nucleotides (HEADCROP:15) and the last ten nucleotides (TAILCROP:10) were removed using trimmomatic68 (trimmomatic/0.36-lkktrba). Paired reads were aligned to the reference sequence using bowtie269 (bowtie2/2.3.4.1-py2-jl5zqym) in unpaired mode (-U). Resulting sequence alignment and map (.sam) files were converted to binary alignment and map (.bam) format, and inclusive reads filtered for those that mapped to the reference sequence (view –bhF 2) and had a mapping quality score (Q) ≥ 30 (view –bhq 30) using SAMtools70 (SAMtools/1.9-k6deoga). All bioinformatic operations were performed on the ISU Pronto server (See Table S5 for an example of Linux shell commands). The proportion of reads to meet filtering criteria in the mitochondrial-enriched and unenriched whole genomic libraries were calculated, and the statistical significance of the difference in the proportion of aligned reads was estimated using a two-sample t-test at a threshold α = 0.0562,63.
Mitochondrial fold-enrichment was calculated for real-time qPCR and MiSeq read alignment methods. For qPCR, the relative difference in copy number between CT values from enriched and unenriched fractions was used to calculate relative fold enrichments. Delta CT (ΔCT) values were calculated by subtracting the CT value produced with mitochondrial primers from the CT value produced from the same sample with nuclear primers. For each family, mitochondrial fold-enrichment was calculated with the formula: 2(mitochondrial fraction ΔCT – unenriched control ΔCT). Likewise, nuclear fold-enrichment was calculated with 2(nuclear fraction ΔCT – unenriched control ΔCT). In the read alignment method, mitochondrial fold-enrichment was calculated as the proportion of reads from each enriched sample that aligned to the reference divided by the mean proportion of reads that analogously aligned from the unenriched samples. Statistical significance in fold enrichment estimations was calculated using paired t-tests at a threshold α = 0.0562,63.
De novo mitochondrial genome assembly, annotation, and variant prediction
Alignments for mitochondrion-enriched reads to the O. nubilalis mitochondrial genome reference (AF442957.1; RefSeq NC_003367.1) were sorted (SAMtools -sort), and the wrapper script plasmidspades.py was used to de novo assemble reads with SPAdes71 (spades/3.11.1-py2-arfy7sc; default parameters) to increase the coverage of the mitochondrial genome in comparison to the reference genome (See Table S5 for an example of Linux shell commands). Resulting Kmer contigs of the mitochondrion-enriched de novo assembled reads were used as subjects against the mitochondrial reference genome query38 (AF442957.1; RefSeq NC_003367.1) with the BLASTn algorithm72 (ncbi-rmblastn/2.6.0-2kyyml7; default parameters) because the de novo assembly increased the coverage of the mitochondrial genome in comparison to the reference genome. Kmer contigs from each F1 family that generated BLASTn hits with E-values ≥ 10–90 were assembled separately with the Sequence Assembly Program, CAP373 (cap3/2015-02-11-2jwa5sb; default parameters). Gene features in each assembled CAP3 contig (mitochondrial genome) were annotated by queries with protein and RNA coding sequences from the reference assembly (AF442957.1; RefSeq NC_003367.1) using the BLASTn algorithm. Any discrepancies were corrected based on evidence from corresponding bowtie2-generated bam alignments visualized in Integrative Genome Viewer (IGV)74. Final annotated assemblies were submitted to the NCBI GenBank nr database. Variable nucleotide positions among the reference and F1 family-specific mitochondrial genomes were identified from corresponding filtered .bam files with bcftools75 (bcftools/1.9-womp5gh) using the call and varFilter options (See Table S5 for an example of Linux shell commands; minimum read depth ≥ 2,000). Results were output in VCF format v4.276. Bedtools v 2.29.277 was implemented to retrieve genome sequence features (CDS, rRNA, and tRNAs). Variations between our submitted annotated genomes and the reference genome were manually verified in Microsoft Word.
Additionally, a multiple sequence alignment was generated between our assembled mitochondrial genomes and the 15,248 bp O. nubilalis mitochondrial genome assembled from unenriched Illumina HiSeq read data39 (MN793322.1). This O. nubilalis sample was collected from the Yili area of the Xinjiang Autonomous region of western China. Nucleotide sequences for our eight assembles and MN793322.1 were loaded into the MEGA8.0 alignment utility78, and aligned using the ClustalW algorithm79 (default parameters) with adjustments to codon frame made manually. Wrapping to a width of 100 bp and demarcation of variant sites was performed with the Multiple Alignment Viewer80; alignment was manually decorated with corresponding gene intervals. The position of variant sites within each corresponding assembly and the consensus were retrieved from the multiple sequence alignment NCBI MSA viewer81. The location and impact of variants on codon use within protein CDS were identified manually and verified by the alignment of corresponding nucleotide and protein sequences retrieved from GenBank accessions.
A discrepancy in the putative translation of ND4 was predicted between the reference (AF442957.1; RefSeq NC_003367.1) and all subsequent Illumina short read-based sequence assemblies (MT793322.1 and this study). To investigate this discrepancy further, a multiple protein sequence alignment was generated among ND4 orthologs from Ostrinia species (O. nubilalis, O. furnacalis, O. scapulalis, O. zealis, O. penitalis, and O. palustralis) and a subset of related lepidopteran species using the ClustalW algorithm79 within the MEGA8.0 alignment utility78 (default parameters; accessions provided in Fig. 4). The general reversible mitochondrial model of protein sequence evolution82 (mtREV24) with among site frequency variation (F) and empirically-derived gamma distribution (G) maximized the Bayesian Information Criterion (BIC) and was chosen as the most appropriate model. The mtREV24 + G + F model was subsequently applied within a Maximum-Likelihood (ML) approach to reconstruct an unrooted phylogeny with a consensus tree constructed from 1,000 iterative bootstrap pseudo-replicate sampling steps.
Comparison between enriched and unenriched reads for de novo mitochondrial genome assembly
Short Illumina reads from four unenriched libraries Accessions SRX2498822-SRX249882566, that contained from 44.6 to 157.7 million reads, were de novo assembled with SOAPdenovo283 (soapdenovo2/240-bg2qxy6; default parameters). Time to complete assemblies and total contigs assembled were quantified, and compared to those for our eight libraries constructed from mitochondrion-enriched extracts. Read depth among contigs within each resulting SOAPdenovo2 assembly were determined by realigning component reads using bowtie269 (bowtie2/2.3.4.1-py2-jl5zqym) in unpaired mode (-U) as described above, and subsequent .bam file conversion, indexing, and retrieval of read counts performed using Samtools70-view, -index, and -idxstats commands respectively. Normalized read counts for contig length was performed by calculation of reads per kilo base per million mapped reads (RPKM; number of reads / (contig length/1000 * total number of reads/1,000,000). RPKM was analogously estimated from .bam output from SPAdes assemblies performed above for enriched library read data, and significant of comparative difference in assembled contig length and read depth estimates using paired t-tests within Rstudio62,63 and evaluated at a threshold α = 0.05. The SOAPdenovo2 assembled contigs assembled from each of the unenriched library reads were loaded into separate local BLAST databases. Each database was then queried with the entire reference O. nubilalis mitochondrial genome sequence (AF442957.1) using the BLASTn algorithm72, with results filtered by an E-value cutoff of ≤ 10–40 and sorted by query start position. The number and percent identity of MtD fragments within each assembly were compared to that obtained from separate assemblies derived from enriched libraries (see above).
Ethics declarations
This research was not conducted on human subjects and was consistent with the United States Animal Welfare Act.
Supplementary information
Acknowledgments
This work is supported by the United States Department of Agriculture (USDA), Agricultural Research Service (ARS) (CRIS Project 5030-22000-018-00D), and the Iowa Agriculture and Home Economics Experiment Station, Ames, IA (Project 3543). Any mention of commercial products in this work does not represent an endorsement or recommendation by USDA for its use. We thank Dr. Andrew Severin and Dr. Maryam Sayadi at the Iowa State University Genomic Informatics Facility for guidance in computational methods. We also thank Keith Binde from the USDA-ARS Corn Insects and Crop Genetics Research Unit, Ames, IA, for maintaining and providing insects used in this study. USDA is an equal opportunity employer and provider.
Author contributions
All authors conceived ideas and designed the methods. K.F. performed laboratory methods under the guidance of B.C. K.F. and B.C. analyzed data. K.F. and B.C. led manuscript writing. All authors contributed critically to manuscript drafts and gave approval for publication.
Data availability
Illumina HiSeq3000 reads can be found on National Center for Biotechnology Information (NCBI) BioProject PRJNA604593 under Short Read Archive (SRA) accession numbers SRR11007774-SRR11007781. Annotated mitochondrial genome sequence assemblies are submitted to the NCBI non-redundant (nr) database under accessions MT492030.1-MT492037.1. The Ostrinia nubilalis mitochondrial genome used as a reference in this study can be found under NCBI GenBank accession AF442957.1. O. nubilalis population Pool-seq reads that were considered unenriched controls are available in the SRA database (Accessions SRX249882-SRX249882566).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-76088-0.
References
- 1.Crozier RH, Crozier YC. The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics. 1993;133:97–117. doi: 10.1093/genetics/133.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolstenholme DR. Genetic novelties in mitochondrial genomes of multicellular animals. Curr. Opin. Genet. Dev. 1992;2:918–925. doi: 10.1016/S0959-437X(05)80116-9. [DOI] [PubMed] [Google Scholar]
- 3.Brown WM, George M, Wilson AC. Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci U S A. 1979;76:1967–1971. doi: 10.1073/pnas.76.4.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moritz C, Dowling TE, Brown WM. Evolution of animal mitochondrial Dna: relevance for population biology and systematics. Annu. Rev. Ecol. Syst. 1987;18:269–292. doi: 10.1146/annurev.es.18.110187.001413. [DOI] [Google Scholar]
- 5.Piganeau G, Gardner M, Eyre-Walker A. A broad survey of recombination in animal mitochondria. Mol. Biol. Evol. 2004;21:2319–2325. doi: 10.1093/molbev/msh244. [DOI] [PubMed] [Google Scholar]
- 6.Avise JC. Gene trees and organismal histories: a phylogenetic approach to population biology. Evolution. 1989;43:1192–1208. doi: 10.1111/j.1558-5646.1989.tb02568.x. [DOI] [PubMed] [Google Scholar]
- 7.Cameron SL. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 2014;59:95–117. doi: 10.1146/annurev-ento-011613-162007. [DOI] [PubMed] [Google Scholar]
- 8.Coates BS. Assembly and annotation of full mitochondrial genomes for the corn rootworm species, Diabrotica virgifera virgifera and Diabrotica barberi (Insecta: Coleoptera: Chrysomelidae), using next generation sequence data. Gene. 2014;542:190–197. doi: 10.1016/j.gene.2014.03.035. [DOI] [PubMed] [Google Scholar]
- 9.Douglas DA, Gower DJ. Snake mitochondrial genomes: phylogenetic relationships and implications of extended taxon sampling for interpretations of mitogenomic evolution. BMC Genom. 2010;11:14. doi: 10.1186/1471-2164-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun W, et al. Comparison of complete mitochondrial DNA sequences between old and new world strains of the cowpea aphid, Aphis craccivora (Hemiptera: Aphididae) Agri Gene. 2017;4:23–29. doi: 10.1016/j.aggene.2017.03.003. [DOI] [Google Scholar]
- 11.Yu L, Li Y-W, Ryder OA, Zhang Y-P. Analysis of complete mitochondrial genome sequences increases phylogenetic resolution of bears (Ursidae), a mammalian family that experienced rapid speciation. BMC Evol. Biol. 2007;7:198. doi: 10.1186/1471-2148-7-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Archie JW. Homoplasy excess ratios: new indices for measuring levels of homoplasy in phylogenetic systematics and a critique of the consistency index. Syst. Biol. 1989;38:253–269. [Google Scholar]
- 13.Harrison RG. Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol. Evol. (Amst.) 1989;4:6–11. doi: 10.1016/0169-5347(89)90006-2. [DOI] [PubMed] [Google Scholar]
- 14.Boore JL, Macey JR, Medina M. Sequencing and comparing whole mitochondrial genomes of animals. Meth. Enzymol. 2005;395:311–348. doi: 10.1016/S0076-6879(05)95019-2. [DOI] [PubMed] [Google Scholar]
- 15.Bevers, R. P. J. et al. Extensive mitochondrial population structure and haplotype-specific phenotypic variation in the Drosophila genetic reference panel. bioRxiv. 466771 (2018), 10.1101/466771.
- 16.Eimanifar A, Kimball RT, Braun EL, Ellis JD. Mitochondrial genome diversity and population structure of two western honey bee subspecies in the Republic of South Africa. Sci. Rep. 2018;8:1333. doi: 10.1038/s41598-018-19759-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mason CE, et al. European Corn Borer—Ecology and Management and Association with other Corn Pests. Iowa: Iowa State University Extension and Outreach; 2018. [Google Scholar]
- 18.Hutchison WD, et al. Areawide suppression of european corn borer with Bt maize reaps savings to non-Bt maize growers. Science. 2010;330:222–225. doi: 10.1126/science.1190242. [DOI] [PubMed] [Google Scholar]
- 19.Coates BS, Dopman EB, Wanner KW, Sappington TW. Genomic mechanisms of sympatric ecological and sexual divergence in a model agricultural pest, the European corn borer. Curr. Opin. Insect. Sci. 2018;26:50–56. doi: 10.1016/j.cois.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Coates BS, Sumerford DV, Hellmich RL. Geographic and voltinism differentiation among North American Ostrinia nubilalis (European corn borer) mitochondrial cytochrome c oxidase haplotypes. J. Insect Sci. 2004;4:35. doi: 10.1093/jis/4.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoshizaki S, et al. Limited variation in mitochondrial DNA of maize-associated Ostrinia nubilalis (Lepidoptera: Crambidae) in Russia Turkey and Slovenia. EJE. 2013;105:545–552. [Google Scholar]
- 22.Marçon PC, Taylor DB, Mason CE, Hellmich RL, Siegfried BD. Genetic similarity among pheromone and voltinism races of Ostrinia nubilalis (Hübner) (Lepidoptera: Crambidae) Insect. Mol. Biol. 1999;8:213–221. doi: 10.1046/j.1365-2583.1999.820213.x. [DOI] [PubMed] [Google Scholar]
- 23.Li J, et al. The genetic structure of Asian corn borer, Ostrinia furnacalis, populations in China: haplotype variance in northern populations and potential impact on management of resistance to transgenic maize. J. Hered. 2014;105:642–655. doi: 10.1093/jhered/esu036. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, et al. Introgression between divergent corn borer species in a region of sympatry: implications on the evolution and adaptation of pest arthropods. Mol. Ecol. 2017;26:6892–6907. doi: 10.1111/mec.14387. [DOI] [PubMed] [Google Scholar]
- 25.Sperling FA, Hickey DA. Mitochondrial DNA sequence variation in the spruce budworm species complex (Choristoneura: Lepidoptera) Mol. Biol. Evol. 1994;11:656–665. doi: 10.1093/oxfordjournals.molbev.a040144. [DOI] [PubMed] [Google Scholar]
- 26.Ciminera M, et al. Genetic variation and differentiation of Hylesia metabus (Lepidoptera: Saturniidae): moths of public health importance in French Guiana and in Venezuela. J. Med. Entomol. 2019;56:137–148. doi: 10.1093/jme/tjy167. [DOI] [PubMed] [Google Scholar]
- 27.Eo SH, DeWoody JA. Evolutionary rates of mitochondrial genomes correspond to diversification rates and to contemporary species richness in birds and reptiles. Proc. R. Soc. B Biol. Sci. 2010;277:3587–3592. doi: 10.1098/rspb.2010.0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith DR. The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs? Brief Funct. Genom. 2016;15:47–54. doi: 10.1093/bfgp/elv027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mascolo C, et al. Comparison of mitochondrial DNA enrichment and sequencing methods from fish tissue. Food Chem. 2019;294:333–338. doi: 10.1016/j.foodchem.2019.05.026. [DOI] [PubMed] [Google Scholar]
- 30.Marquis J, et al. MitoRS, a method for high throughput, sensitive, and accurate detection of mitochondrial DNA heteroplasmy. BMC Genom. 2017;18:326. doi: 10.1186/s12864-017-3695-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richards SM, et al. Correction: Low-cost cross-taxon enrichment of mitochondrial DNA using in-house synthesised RNA probes. PLoS ONE. 2019;14:e0213296. doi: 10.1371/journal.pone.0213296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li M, Schroeder R, Ko A, Stoneking M. Fidelity of capture-enrichment for mtDNA genome sequencing: influence of NUMTs. Nucleic Acids Res. 2012;40:e137. doi: 10.1093/nar/gks499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grau ET, et al. Survey of mitochondrial sequences integrated into the bovine nuclear genome. Sci. Rep. 2020;10:2077. doi: 10.1038/s41598-020-59155-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lenglez S, Hermand D, Decottignies A. Genome-wide mapping of nuclear mitochondrial DNA sequences links DNA replication origins to chromosomal double-strand break formation in Schizosaccharomyces pombe. Genome Res. 2010;20:1250–1261. doi: 10.1101/gr.104513.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srinivasainagendra V, et al. Migration of mitochondrial DNA in the nuclear genome of colorectal adenocarcinoma. Genome Med. 2017;9:31. doi: 10.1186/s13073-017-0420-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hazkani-Covo E, Zeller RM, Martin W. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 2010;6:e1000834. doi: 10.1371/journal.pgen.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graham JM. Isolation of mitochondria from tissues and cells by differential centrifugation. Curr. Prot. Cell Biol. 1999;4:331–3315. doi: 10.1002/0471143030.cb0303s04. [DOI] [PubMed] [Google Scholar]
- 38.Coates BS, Sumerford DV, Hellmich RL, Lewis LC. Partial mitochondrial genome sequences of Ostrinia nubilalis and Ostrinia furnicalis. Int. J. Biol. Sci. 2005;1:13–18. doi: 10.7150/ijbs.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou N, Dong Y, Qiao P, Yang Z. Complete mitogenomic structure and phylogenetic implications of the genus Ostrinia (Lepidoptera: Crambidae) Insects. 2020;11:332. doi: 10.3390/insects11060332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barr CM, Keller SR, Ingvarsson PK, Sloan DB, Taylor DR. Variation in mutation rate and polymorphism among mitochondrial genes of Silene vulgaris. Mol. Biol. Evol. 2007;24:1783–1791. doi: 10.1093/molbev/msm106. [DOI] [PubMed] [Google Scholar]
- 41.Hagelberg E. Recombination or mutation rate heterogeneity? Implications for mitochondrial eve. Trends Genet. 2003;19:84–90. doi: 10.1016/S0168-9525(02)00048-3. [DOI] [PubMed] [Google Scholar]
- 42.Martin AP, Palumbi SR. Body size, metabolic rate, generation time, and the molecular clock. Proc. Natl. Acad. Sci. U. S. A. 1993;90:4087–4091. doi: 10.1073/pnas.90.9.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quispe-Tintaya W, White RR, Popov VN, Vijg J, Maslov AY. Rapid mitochondrial DNA isolation method for direct sequencing. Methods Mol. Biol. 2015;1264:89–95. doi: 10.1007/978-1-4939-2257-4_9. [DOI] [PubMed] [Google Scholar]
- 44.Franco LM, et al. Integrative genomic analysis of the human immune response to influenza vaccination. eLife. 2013;2:e00299. doi: 10.7554/eLife.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gould MP, et al. PCR-free enrichment of mitochondrial DNA from human blood and cell lines for high quality next-generation DNA sequencing. PLoS ONE. 2015;10:e0139253. doi: 10.1371/journal.pone.0139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lodish H, et al. Molecular Cell Biology. W. H: Freeman; 2000. [Google Scholar]
- 47.Hornig-Do H-T, et al. Isolation of functional pure mitochondria by superparamagnetic microbeads. Anal. Biochem. 2009;389:1–5. doi: 10.1016/j.ab.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 48.Dopman EB, Robbins PS, Seaman A. Components of reproductive isolation between North American pheromone strains of the European corn borer. Evolution. 2010;64:881–902. doi: 10.1111/j.1558-5646.2009.00883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kytövuori L, et al. Case report: a novel frameshift mutation in the mitochondrial cytochrome c oxidase II gene causing mitochondrial disorder. BMC Neurol. 2017;17:1–5. doi: 10.1186/s12883-017-0883-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beckenbach AT, Robson SKA, Crozier RH. Single nucleotide +1 frameshifts in an apparently functionalmitochondrial cytochrome b gene in ants of the genus polyrhachis. J. Mol. Evol. 2005;60:141–152. doi: 10.1007/s00239-004-0178-5. [DOI] [PubMed] [Google Scholar]
- 51.Russell RD, Beckenbach AT. Recoding of translation in turtle mitochondrial genomes: programmed frameshift mutations and evidence of a modified genetic code. J. Mol. Evol. 2008;67:682–695. doi: 10.1007/s00239-008-9179-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Atkins JF, Björk GR. A gripping tale of ribosomal frameshifting: extragenic suppressors of frameshift mutations spotlight P-site realignment. Microbiol. Mol. Biol. Rev. 2009;73:178–210. doi: 10.1128/MMBR.00010-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fox TD, Weiss-Brummer B. Leaky +1 and −1 frameshift mutations at the same site in a yeast mitochondrial gene. Nature. 1980;288:60–63. doi: 10.1038/288060a0. [DOI] [PubMed] [Google Scholar]
- 54.Kim C, Hoshizaki S, Huang Y, Tatsuki S, Ishikawa Y. Usefulness of mitochondrial COII gene sequences in examining phylogenetic relationships in the Asian corn borer, Ostrinia furnacalis, and allied species (Lepidoptera: Pyralidae) Appl. Entomol. Zool. 1999;34:405–412. doi: 10.1303/aez.34.405. [DOI] [Google Scholar]
- 55.Ruisheng Y, Zhenying W, Kanglai H. Genetic diversity and phylogeny of the genus Ostrinia (Lepidoptera: Crambidae) inhabiting China inferred from mitochondrial COI gene. J. Nanjing Agric. Univ. 2011;5:13. [Google Scholar]
- 56.Coates BS, Sumerford DV, Siegfried BD, Hellmich RL, Abel CA. Unlinked genetic loci control the reduced transcription of aminopeptidase N 1 and 3 in the European corn borer and determine tolerance to Bacillus thuringiensis Cry1Ab toxin. Insect. Biochem. Mol. Biol. 2013;43:1152–1160. doi: 10.1016/j.ibmb.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 57.Lewis LC, Lynch RE. Rearing the European corn borer, Ostrinia nubilalis (Hubner), on diets containing corn leaf and wheat germ. Iowa State J. Sci. 1969;44:9–14. [Google Scholar]
- 58.Lazarow A, Cooperstein SJ. Studies on the mechanism of Janus green B staining of mitochondria. I. Review of the literature. Exp. Cell Res. 1953;5:56–69. doi: 10.1016/0014-4827(53)90094-9. [DOI] [PubMed] [Google Scholar]
- 59.Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994;3:294–299. [PubMed] [Google Scholar]
- 60.Coates BS, et al. Frequency of hybridization between Ostrinia nubilalis E-and Z-pheromone races in regions of sympatry within the United States. Ecol. Evol. 2013;3:2459–2470. doi: 10.1002/ece3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Integrated DNA Technologies. https://www.idtdna.com/scitools/Applications/RealTimePCR/.
- 62.RStudio Team. RStudio: Integrated Development for R. (RStudio, PBC, 2020).
- 63.R Core Team. R: A language and environment for statistical computing. (R Foundation for Statistical Computing, 2019).
- 64.Lenth, R., Singmann, H., Love, J., Buerkner, P. & Herve, M. emmeans: Estimated Marginal Means, aka Least-Squares Means. (2020).
- 65.Graves S, Dorai-Raj H-PP. and L. S. with help from S. multcompView: Visualizations of Paired Comparisons; 2019. [Google Scholar]
- 66.Kozak GM, et al. A combination of sexual and ecological divergence contributes to rearrangement spread during initial stages of speciation. Mol. Ecol. 2017;26:2331–2347. doi: 10.1111/mec.14036. [DOI] [PubMed] [Google Scholar]
- 67.Andrews, S. Babraham Bioinformatics—FastQC A Quality Control tool for High Throughput Sequence Data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
- 68.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bankevich A, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 73.Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Robinson JT, et al. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Narasimhan V, et al. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics. 2016;32:1749–1751. doi: 10.1093/bioinformatics/btw044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kumar S, Nei M, Dudley J, Tamura K. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008;9:299–306. doi: 10.1093/bib/bbn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thompson, J. D., Gibson, T. J. & Higgins, D. G. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinform. Chapter 2, Unit 2.3 (2002). [DOI] [PubMed]
- 80.MView < Multiple Sequence Alignment < EMBL-EBI. https://www.ebi.ac.uk/Tools/msa/mview/.
- 81.NCBI Multiple Sequence Alignment Viewer 1.15.0. https://www.ncbi.nlm.nih.gov/projects/msaviewer/.
- 82.Adachi J, Hasegawa M. Model of amino acid substitution in proteins encoded by mitochondrial DNA. J. Mol. Evol. 1996;42:459–468. doi: 10.1007/BF02498640. [DOI] [PubMed] [Google Scholar]
- 83.Luo R, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Illumina HiSeq3000 reads can be found on National Center for Biotechnology Information (NCBI) BioProject PRJNA604593 under Short Read Archive (SRA) accession numbers SRR11007774-SRR11007781. Annotated mitochondrial genome sequence assemblies are submitted to the NCBI non-redundant (nr) database under accessions MT492030.1-MT492037.1. The Ostrinia nubilalis mitochondrial genome used as a reference in this study can be found under NCBI GenBank accession AF442957.1. O. nubilalis population Pool-seq reads that were considered unenriched controls are available in the SRA database (Accessions SRX249882-SRX249882566).