

Abstract
Alpha-synuclein SNCA has been implicated in the etiology of Parkinson’s disease (PD); however, the normal function of alpha-synuclein protein and the pathway that mediates its pathogenic effect is yet to be discovered. We investigated the mechanistic role of SNCA in the nucleus utilizing isogenic human-induced pluripotent stem cells-derived neurons from PD patients with autosomal dominant mutations, A53T and SNCA-triplication, and their corresponding corrected lines by genome- and epigenome-editing. Comparisons of shape and integrity of the nuclear envelope and its resistance to stresses found that both mutations result in similar nuclear envelope perturbations that were reversed in the isogenic mutation-corrected cells. Further mechanistic studies showed that SNCA mutation has adverse effects on the nucleus by trapping Ras-related nuclear protein (RAN) and preventing it from transporting key nuclear proteins such as, DNMT3A, for maintaining normal nuclear function. For the first time, we proposed that α-syn interacts with RAN and normally functions in the nucleocytoplasmic transport while exerts its pathogenic effect by sequestering RAN. We suggest that defects in the nucleocytoplasmic transport components may be a general pathomechanistic driver of neurodegenerative diseases.
Introduction
Alpha-synuclein (α-syn) protein, encoded by the SNCA gene, is a small (14 kDa) presynaptic nerve terminal protein, abundant in the brain, that was originally identified as a precursor protein for the non-β-amyloid component (NAC) of Alzheimer disease amyloid plaques (1). A few years later, the SNCA gene was the first gene implicated in familial Parkinson’s disease (fPD) (2). The earlier studies, identified missense mutations in the SNCA gene that caused an early onset, Lewy body-positive, autosomal dominant Parkinson’s Disease (PD) in a few rare families of Mediterranean and German origin, Ala53Thr (hereafter, A53T) and Ala30Pro, respectively (2, 3). Subsequently, copy number variations (CNV) in the SNCA gene were identified in few other families with an early onset, autosomal dominant form of PD (4–9). While it has been suggested that overexpression of the SNCA gene mediates the pathogenic effect in fPD patients carrying the CNV of SNCA genomic region, triplication (4, 10, 11) and duplication (5–8), the molecular perturbation underpinning the pathogenic effect of the dominant missense mutations are yet to be deciphered.
Over the ensuing years from the original discovery of SNCA as a genetic factor (12) for fPD, the role of α-syn protein in health and disease has been extensively studied, however, its normal function and the pathway that mediates its pathogenic effect in disease state are still poorly understood (13). α-syn has been suggested to be involved in various cellular pathways including, membrane interactions, protein degradation, maintaining a supply of synaptic vesicles, regulation of dopamine release and transport, mitochondrial dysfunction and autophagy-lysosome pathway (14, 15). Although α-syn was historically identified as a neuron-specific protein, localized to a region of the nuclear envelope (16), its role in relation to the nucleus has been understudied. To date, only few reports examined α-syn in the context of the nuclear localization (17–20) and function such as chromatin organization (21–23) and transcription (20, 24). Furthermore, we recently described a pathological disruption of the nuclear envelope shape, loss of heterochromatin markers and substantial DNA damage in human-induced pluripotent stem cells (hiPSC)-derived neurons from a patient with triplication of the SNCA locus (25).
Here, we investigated the mechanistic role of SNCA in the nucleus utilizing isogenic hiPSC-derived neurons from PD patients and their corresponding genome-edited lines with the corrected mutations. We compared the effects of two fPD mutations, A53T, and triplication, on the architecture of the nuclear envelope and its resistance to heat and osmotic stresses. We found that both mutations similarly perturbed the shape and the integrity of the nuclear envelope and these phenotypic perturbations were reversed in the corresponding isogenic mutation-corrected cells. We then used an isogenic hiPSC-derived system to further investigate the mechanism underlying nuclear envelope abnormalities induced by SNCA mutation. Additional mechanistic studies showed that SNCA mutation has an adverse effect on the nucleus by trapping Ras-related nuclear protein (RAN) and preventing it from transporting key nuclear proteins needed to maintain normal nuclear function. Here, for the first time, we proposed a normal function of α-syn in nucleocytoplasmic transport via interaction with RAN and that the pathogenic effect of overexpressed/mutated SNCA is mediated by sequestering RAN.
Results
Description of the SNCA mutated and corrected isogenic hiPSC-derived neuronal lines
Previously, we reported that SNCA-triplication perturbed the architecture of the nuclear envelope (25). Here, we aimed to extend on these observations and to test whether the deficits in the shape of the nuclear envelope are shared phenotypes caused by a broader range of fPD mutations in SNCA, i.e. a missense coding and a CNV mutation, and to define the underpinning mechanism mediating the SNCA mutations-induced impairment of the nuclear envelope architecture. Toward this goal, we characterized hiPSC-derived dopaminergic progenitor neurons (MD NPC) lines from a patient with the missense A53T coding mutation (157G > A, Ala53Thr; hereafter A53T) and a patient with the triplication of the SNCA locus (hereafter, SNCA-Tri) compared with their corresponding isogenic mutation-corrected hiPSC-derived lines (hereafter, A53T-corrected and SNCA-Tri-corrected, respectively). The SNCA A53T corrected line was generated by CRISPR/Cas9 genome editing, and the SNCA-Tri corrected line was generated previously by DNA-methylation editing of SNCA-intron 1 resulting in ~30% reduction in SNCA-mRNA and protein expression levels, as published (26). The successful differentiation each of the four hiPSC lines into MD NPC cells was validated by analysis of markers specifically expressed in MD NPCs, Nestin and Forkhead box protein A2 (FOXA2) using immunocytochemistry (Supplementary Material, Fig. S1).
SNCA A53T mutation and triplication induce nuclear envelope abnormalities
First, we examined in the effects of the A53T and SNCA-Tri mutations on the nuclear architecture. We evaluated changes in the nuclear envelope shape by immunostaining with Lamin B1 marker to measure the nuclear circularity (27) and with Lamin A/C to determine the percentage of abnormal nuclei (28). Lamin B1 and Lamin A/C are the major components of the nuclear lamina and contribute to the structural integrity of the nuclear shape and the nuclear envelope, respectively (29, 30). Staining with Lamin B1 showed a significant decrease in nuclear circularity in the A53T hiPSC-derived MD NPCs compared with the isogenic corrected line (P < 0.0001, Fig. 1A, B and I). Similarly, we found a significant loss in nuclear circularity in the SNCA-Tri hiPSC-derived MD NPCs in comparison with the SNCA-Tri-corrected line (P < 0.0001, Fig. 1C, D and J). The extent of decreased nuclear circularity in the SNCA-Tri MD NPCs was comparable with that observed for the A53T (Fig. 1I and J). Next, we stained with Lamin A/C and measured the percentage of abnormal nuclei (folded and ‘blebbed’). Both the A53T and the SNCA-Tri hiPSC-derived MD NPCs showed a significant increase in abnormal nuclei compared with their corresponding isogenic corrected lines (~1.5-fold increase in abnormal nuclei P = 0.02 in the comparison of the isogenic A53T lines, Fig. 1K, L and S; 2-fold increase P = 0.04 in the comparison of the isogenic SNCA-Tri lines, Fig. 1M, N and T). These results were consistent with our previous findings that showed impaired nuclear envelope architecture in SNCA-Tri hiPSC-derived dopaminergic and cholinergic neurons compared with that of hiPSC-derived neurons from a healthy donor (25). In addition, we demonstrated that the isogenic corrections of the SNCA mutations rescued these nuclear envelope abnormalities (Fig. 1B-F, D-H, I, J, L-P, N-R, S and T). Last, here we showed that these nuclear shape abnormalities were induced to approximately the same extent by both SNCA overexpression mediated by SNCA-Tri and a missense coding mutation in the gene. Thus, suggesting a common mechanism via which SNCA mutations induce defects in the nuclear envelope.
Figure 1.
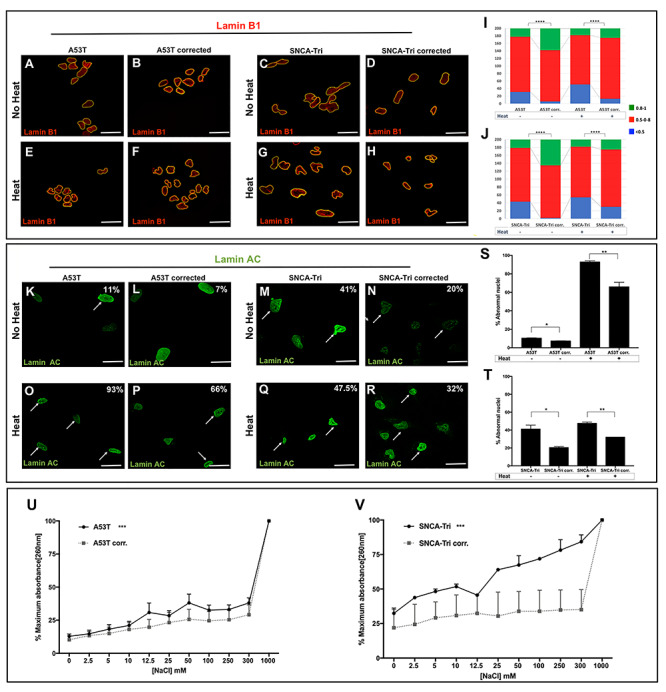
Alpha-synuclein A53T mutation and triplication induce nuclear envelope abnormalities and sensitivity to stressors. (A-H) Immunocytochemistry for Lamin B1 in A53T and SNCA-Tri hiPSC-derived MD NPCs and their isogenic corrected controls in regular conditions and upon heat-shock. (A, B) A53T and (C, D) SNCA-Tri hiPSC-derived NPCs showed a loss in the nuclear envelope circularity compared with their isogenic controls. (E, F) A53T and (G-H) SNCA-Tri hiPSC-derived MD NPCs showed a higher susceptibility to heat-shock demonstrated by greater loss in the nuclear envelope circularity compared with their isogenic controls. Quantification of the nuclear envelope circularity in the (I) A53T and (J) SNCA-Tri hiPSC-derived MD NPCs demonstrated loss in circularity in the mutated lines compared with the isogenic corrected line and increased susceptibility to heat-shock in the mutated lines. The data are plotted as frequency distributions for 200 cells. n = 2 independent differentiation protocols. (K-R) Immunocytochemistry for Lamin A/C in A53T and SNCA-Tri hiPSC-derived MD NPCs and their isogenic corrected controls in regular conditions and upon heat-shock. Percentages indicate the proportion of cells with folded/blebbed nuclear morphology, white arrows denote abnormal nuclei. (K, L) A53T and (M, N) SNCA-Tri hiPSC-derived MD NPCs showed higher percentage of abnormal nuclei compared with their isogenic controls. (O, P) A53T and (Q, R) SNCA-Tri hiPSC-derived MD NPCs showed a higher susceptibility to heat-shock demonstrated by higher percentage of abnormal nuclei compared with their isogenic controls. (U-V) Absorption260 curve for supernatant from nuclei isolated from (U) A53T hiPSC-derived MD NPCs compared with the corresponding isogenic corrected line, and (V) SNCA-Tri hiPSC-derived MD NPCs compared with the corresponding isogenic corrected line. A53T and SNCA-Tri showed higher susceptibility to osmotic stress compared with their corresponding isogenic corrected lines (n = 3). (I, J) ****P < 0.0001, Kolmogorov–Smirnov test; (S-V) *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test. Scale bars: 25 μm.
The SNCA A53T mutation and the triplication increase the sensitivity of the nuclear envelope integrity to stress conditions
Exposure to stress conditions compromises the integrity of the nuclear envelope (31, 32). We evaluated the resistance of the nuclear envelope to two stressors, heat-shock (31) and osmotic (33). The four studied hiPSC-derived NPCs, A53T, SNCA-Tri and the corresponding corrected MD NPCs were exposed to the stress conditions and the integrity of the nuclear envelope was measured before and after treatments. To evaluate the heat-shock effect, we used the two phenotypes described above, nuclear circularity and percentage of abnormal nuclei (Fig. 1). After heat treatment, each MD NPCs line demonstrated a decrease in nuclear circularity (p(A53T) = 0.0002, p(A53T-corrected) < 0.0001, p(SNCA-Tri) = 0.0097, p(SNCA-Tri corrected) < 0.0001, Fig. 1E-H, I and J) and an increase in abnormal nuclei (p(A53T) = 0.0003, p(A53T-corrected) = 0.007, p(SNCA-Tri) = 0.31, p(SNCA-Tri corrected) = 0.01, Fig. 1O-R, S and T) compared with untreated cells, providing validation for the heat-induced condition. Next, we compared the A53T and the SNCA-Tri hiPSC-derived MD NPCs with their corresponding isogenic corrected lines. After heat-shock, the nuclear circularity was significantly decreased in the MD NPCs with both mutations in comparison with the corresponding corrected lines (4-folds more imperfect circularity P < 0.0001 in the A53T compared with the isogenic corrected line and 2-folds P < 0.0001 in the SNCA-Tri compared with the isogenic corrected lines, Fig. 1E-H, I and J) and the percentage of abnormal nuclei was significantly increased in the mutated compared with the corrected lines (~1.5-folds more abnormal nuclei P = 0.0343 in the A53T compared with the isogenic corrected lines, and 1.5-folds P = 0.0092 in the SNCA-Tri compared with the isogenic corrected lines, Fig. 1O-R, S and T). These results showed that the nuclear envelope was highly susceptible to heat treatment for both mutations and the isogenic correction rescued the hypersensitivity of the nuclear envelope. Noteworthy, while the A53T mutation showed a larger increase in the number of nuclei with imperfect circularity and in the percentage of abnormal nuclei after heat shock relative to the baseline (no heat) than that observed for the corrected line, this was not the case for the SNCA-Tri possibly, in part, because the SNCA-Tri cells exhibited more severe effects on the nuclear envelope already at baseline. To complement the assessment of the nuclear envelope integrity, we measured nuclear fragility in response to high salt conditions (33). Consistently, following osmotic stress by exposure to increasing salt (NaCl) concentrations, we found that both the A53T and the SNCA-Tri hiPSC-derived MD NPCs were more susceptible to high salt conditions compared with the corresponding isogenic corrected lines (Fig. 1U and V). Collectively, the results of these experiments demonstrated the effect of SNCA mutations on the hypersensitivity of the nuclear envelope to stress conditions. Furthermore, the A53T and the SNCA-Tri mutations have the same effects on the structure, integrity and sensitivity of the neuronal nuclear envelope.
RAN protein interacts with alpha-synuclein
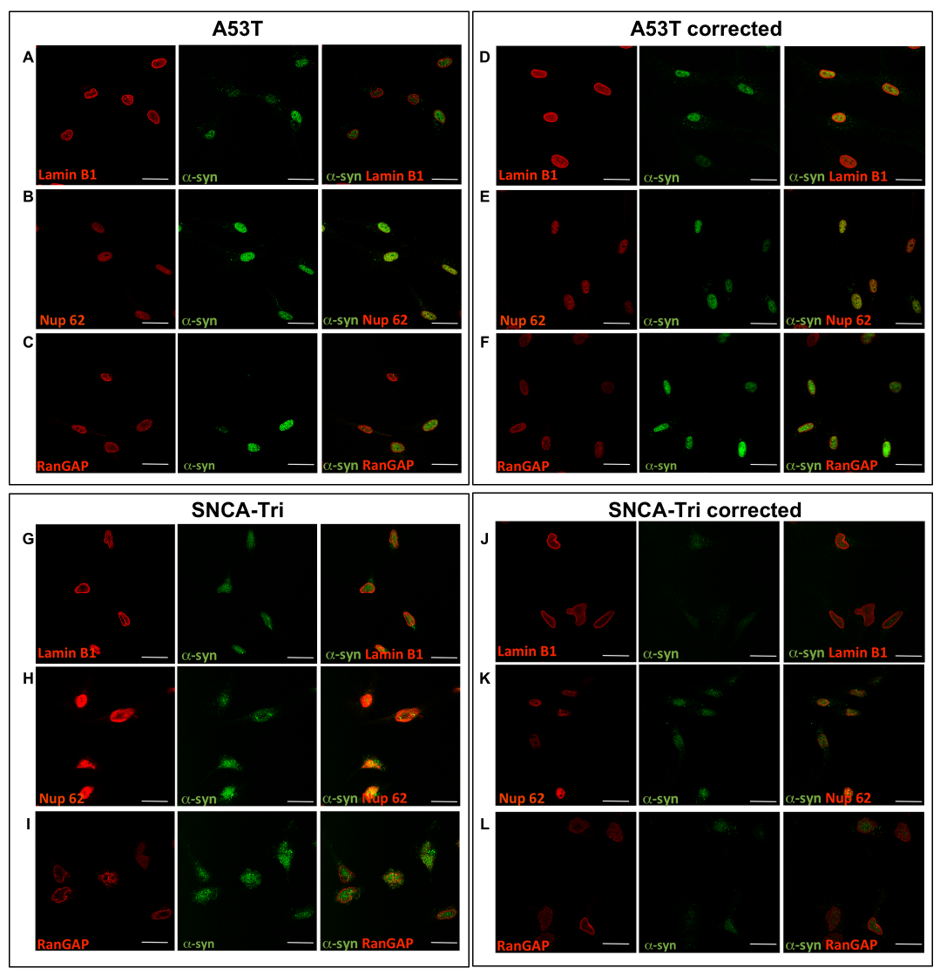
Next, we aimed to uncover the plausible pathways underlying the alterations in the nuclear envelope structure and integrity. Identification of nuclear proteins that interact with α-syn will provide clues regarding its putative function in the nucleus. To this end, we first analyzed four proteins, that have known roles in the nucleus related to the maintenance and function of the nuclear envelope, by co-immunostaining experiments to identify which may co-localize with α-syn: (i) Lamin B1, an integral component of the nuclear lamina (34, 35), (ii) Nucleoporin 62 (Nup62), a component of the central channel of the nuclear pore complex and is directly involved in nuclear import, transcription and chromatin organization (36), (iii) RAN is a GTP-binding protein involved in nucleocytoplasmic transport into and out of the nucleus (37) (38) and its activator, (iv) RAN GTPase activating protein (RANGAP), a cytoplasmatic protein involved in proteins transport from the cytosol to the nucleus by maintaining the RAN gradient (37, 38). In all hiPSC-derived MD NPCs lines, A53T, SNCA-Tri and their isogenic corrected lines, α-syn signal was detected predominantly in the nucleus. Lamin B1 and Nup62 proteins were exclusively localized to the nucleus and showed no co-localization with α-syn signal (Supplementary Material, Fig. S2A, B, D, E, G, H, J and K). RANGAP showed a predominant perinuclear signal in all lines, however, also was not co-localized with the α-syn signal (Supplementary Material, Fig. S2C, F, I and L). RAN was abundant and localized throughout the nucleus in all the isogenic lines, while a lower intensity signal was observed in the cell soma (Fig. 2A-D). The results showed that RAN signals co-localized with α-syn in the nucleus in all four MD NPCs lines (Fig. 2A-D), suggesting that RAN and α-syn may interact and possibly act jointly in the same pathway. Next, we further explored the nature of the interaction between α-syn and RAN and whether they interact in a complex. Towards this goal, we performed co-immunoprecipitation (Co-IP) assay in SH-SY5Y cells ectopically expressing the Myc-tagged human α-syn and the 3xFlag-tagged human RAN. This system confirmed, in both directions, the complex interaction between α-syn and RAN (Fig. 2E). While Co-IP experiment in the nuclear and cytosolic compartments separately poses technical challenges, collectively, the results of both the Co-IP and the co-immunostaining experiments suggested that α-syn and RAN interact, and that this interaction occurs predominantly in the nucleus.
Figure 2.
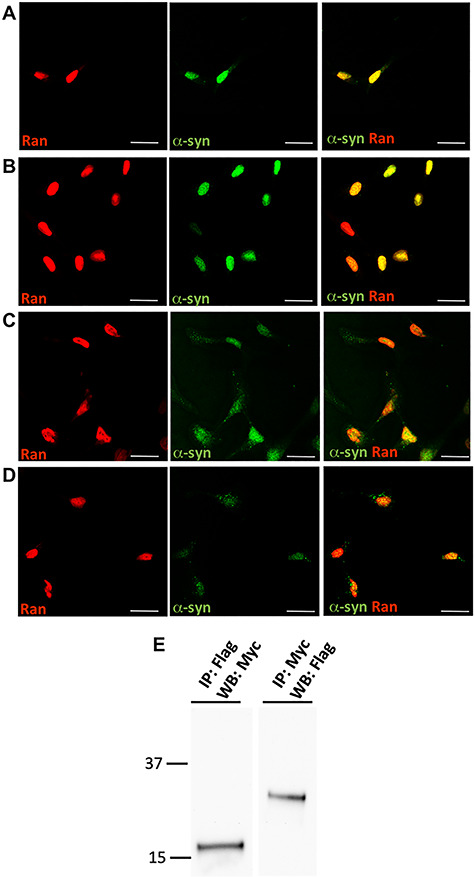
RAN protein interacts with α-synuclein. (A-D) hiPSC-derived NPCs were labeled with anti-RAN and anti-α-syn antibodies. RAN protein co-localized with α-syn signal in the nucleus in all cell-lines, the A53T (A) and SNCA-Tri (C) hiPSC-derived MD NPCs and their isogenic corrected controls, A53T corrected (B) and SNCA-Tri corrected (D). Scale bars: 10 μm. (E) Western blot analysis of homogenates prepared from SH-SY5Y cells co-infected with LV-myc-α-syn and LV-3xFlagRAN. Anti-Flag immunoprecipitates were analyzed by western blotting using anti-myc antibody and anti-myc immunoprecipitates were analyzed by western blotting using anti-Flag antibody. No-antibody Co-IP was performed as controls to ruled out non-specific binding (not shown). Co-IP of the endogenous proteins is demonstrated in Figure 5.
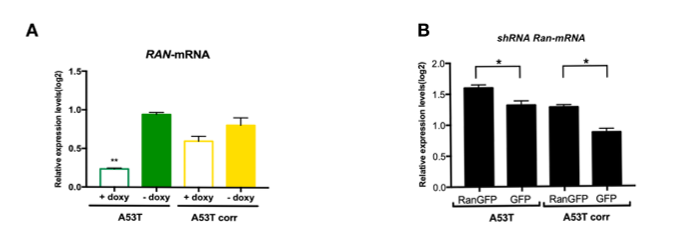
Proteins larger than 40 kDa are subjected to active nucleocytoplasmic transport via RAN gradient, while smaller molecules passively transit between the cytoplasm and nucleoplasm. α-syn is a small protein (14 kDa) protein that may not require active transport; therefore, the interaction with RAN implied a putative novel role for α-syn in the nucleocytoplasmic transport. Thus, we ought to explore the hypothesis that the interaction of α-syn with RAN has a biological role important to maintain normal nuclear function and to provide evidence that the interaction of α-syn and RAN is not required for the transportation of α-syn to the nucleus. We employed a tetracycline-inducible shRNA system to downregulate RAN and evaluated the effect of RAN downregulation on the levels of α-syn in the nucleus by immunocytochemistry. The doxycycline induced A53T and the isogenic corrected hiPSC-derived NPCs lines expressing RAN–shRNA in comparison with the respective control uninduced lines displayed lower levels of RAN–mRNA (4- and 1.3-fold reduction in the A53T and isogenic A53T corrected line, respectively, between induced RAN–shRNA versus the uninduced control, lines, Supplementary Material, Fig. S3A) and protein (1.7- and 1.3-fold reduction P = 0.04 in the A53T and isogenic A53T corrected line, respectively, between induced RAN–shRNA versus the uninduced control lines, Fig. 3A and B). Comparisons between the induced RAN–shRNA to the uninduced respective lines showed similar α-syn levels (P = 0.4637, Fig. 3C), indicating that downregulation of RAN did not affect the localization of α-syn to the nucleus. Thus, α-syn nuclear import is passive and independent of RAN. This result suggested the involvement of α-syn in nucleocytoplasmic transport via its interaction with RAN.
Figure 3.
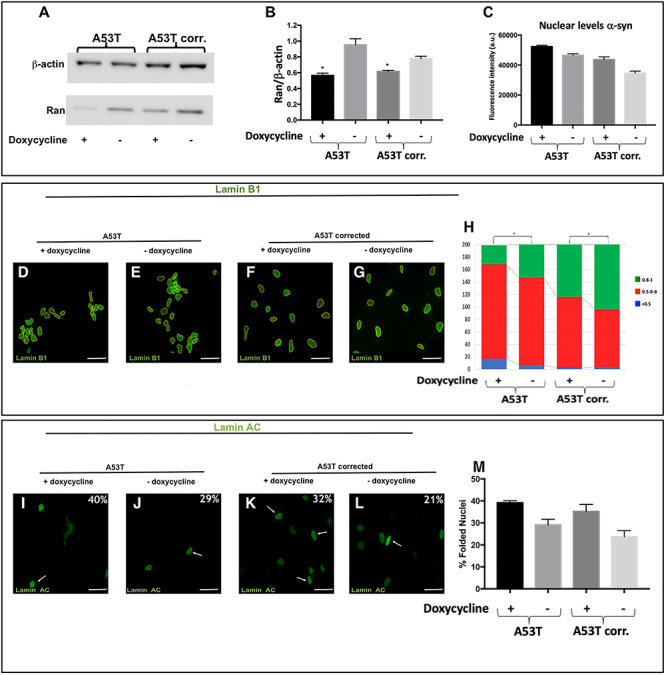
The effects of RAN downregulation on the integrity and function of the nuclear envelope. A53T hiPSC-derived MD NPCs and the isogenic corrected control were transduced with lentiviral vector harboring shRNA–RAN followed by 72 h induction with doxycycline. (A) Downregulation of RAN was confirmed by western-blot analysis using anti-RAN antibody (25 kDa) normalized to β-actin (42 kDa). (B) RAN protein levels were quantified in two independent western-blot experiments. Lower levels of RAN protein were detected in the A53T hiPSC-derived MD NPCs and the isogenic corrected control. (C) The levels of α-syn in the nucleus were measured by immunocytochemistry and no significant changes were observed upon downregulation of RAN protein in both the A53T hiPSC-derived MD NPCs and the isogenic corrected control. (D-M) The effect of downregulation of RAN on the integrity of the nuclear envelope was evaluated by immunocytochemistry for Lamin B1 (D-H) and Lamin A/C (I-M) in the A53T hiPSC-derived MD NPCs and the isogenic corrected control. The expression of shRNA-RAN was validated using dsRed reporter; thus, for these set of experiments, we used green fluorescent conjugated secondary antibody to detect the staining of Lamin B1 (D-G) and Lamin A/C (I-L). Percentages indicate the proportion of cells with folded/blebbed nuclear morphology, white arrows denote abnormal nuclei (I-L). (H) The nuclear circularity data are plotted as frequency distributions of for 250 cells (n = 2). (M) Percentages indicate the proportion of cells with folded/blebbed nuclear morphology. Bars represents mean ± SEM (n = 2). (A-B) *P < 0.05, **P < 0.001, Student’s t-test. Scale bars: 10 μm; (H) *P < 0.05, Kolmogorov–Smirnov test.
The effects of RAN on nuclear envelope integrity
To explore the effect of RAN levels on nuclear envelope architecture in the context of SNCA mutation, we downregulated and overexpressed RAN using the A53T and the corrected hiPSC-derived NPCs lines and characterized the nuclear envelope phenotypes. First, we utilized the tetracycline-inducible shRNA–RAN system described above to investigate RAN downregulation (Fig. 3A and B). The tetracycline-induced RAN–shRNA compared with the respective uninduced lines showed significant loss in the nuclear circularity in both the A53T and the corrected hiPSC-derived MD NPCs lines (A53T, P = 0.02; A53T-corrected, P = 0.04 Fig. 3D-H) and increased abnormal nuclei (A53T, ~1.4-fold; A53T corrected, 1.5-fold, Fig. 3I-M) measured by staining with Lamin B1 and Lamin A/C, respectively, as described above. These results suggested that downregulation of RAN led to nuclear envelope impairment and demonstrated the essential role of RAN and the robust deleterious effect of its downregulation.
Next, we assessed the effect of RAN overexpression using a lentiviral vector expressing the RAN–GFP fusion protein compared with GFP only. The isogenic A53T and the corrected hiPSC-derived MD NPCs transduced with the RAN–GFP relative to the GFP only control showed overexpression of RAN–mRNA (~1.25-fold increased RAN expression in the A53T lines, P = 0.04 and ~1.5-fold in the isogenic A53T corrected lines, P = 0.01, Supplementary Material, Fig. S3B) and protein (5-fold higher RAN levels in the A53T lines, P = 0.03, and 3-fold in the isogenic A53T corrected lines, P = 0.0006, Fig. 4A and B). The A53T hiPSC-derived MD NPCs with overexpression of RAN–GFP compared with GFP-only showed a significant improvement of nuclear circularity measured by staining with Lamin B1 (P = 0.03, Fig. 4C, D and K). However, staining with Lamin A/C showed no significant differences in the percentage of the abnormal nuclei in the A53T line with overexpression of RAN–GFP compared with GFP only (Fig. 4M, N and U). As expected, the A53T-corrected MD NPCs overexpressing RAN–GFP showed the same distribution of nuclear circularity as the GFP only cells, where most cells exhibited complete circled nuclei (Fig. 4E, F and L), and also a similar low percentage of abnormal nuclei (~10%, Fig. 4O, P and V). We further examined whether overexpression of RAN can also rescue the vulnerability to stress. Heat-shock resulted in a significant loss in nuclear circularity (P < 0.0001, Fig. 4G-J, K and L) and increased abnormal nuclei (Fig. 4Q-T, U and V) in all four lines, i.e. A53T and the corrected overexpressing RAN–GFP or GFP only recombinant proteins, which validated the induction of stress condition. The A53T-hiPSC-derived MD NPCs overexpressing RAN–GFP was significantly less susceptible to heat-shock compared with the control GFP only as demonstrated by the significant increase in nuclear circularity (P = 0.0017, Fig. 4G, H and K) and significant decrease in the percentage of abnormal nuclei (1.5-fold decrease, P = 0.02, Fig. 4Q, R and U). Furthermore, while the control A53T line (GFP only) exhibited a larger increase in the number of nuclei with imperfect circularity (several-folds) and in the percentage of abnormal nuclei (~2.5-folds) after heat shock relative to the baseline (no heat), the A53T mutation line with RAN overexpression showed only slight increase in the number of nuclei with imperfect circularity and much lesser extent of increase in the percentage of abnormal nuclei upon heat shock relative to the baseline (no heat). As expected, the A53T corrected line overexpressing RAN–GFP showed a similar magnitude of sensitivity to heat-shock compared with the respective control line (GFP only), as measured by nuclear circularity (Fig. 4I, J and L) and abnormal shape, (Fig. 4S, T and V). Collectively, our results showed that overexpression of RAN reversed the A53T-induced nuclear envelope perturbations. RAN effect was specifically robust under stress condition resulted in the rescue of the hypersensitivity to heat-shock that was comparable with the effect of the correction of the A53T mutation to the normal coding sequence. In addition, as expected the correction of the SNCA A53T coding mutation was sufficient to rescue the perturbations to the nuclear envelope shape, integrity and sensitivity to stress, thus, RAN overexpression did not exert an effect on nuclear structure in the A53T corrected line.
Figure 4.
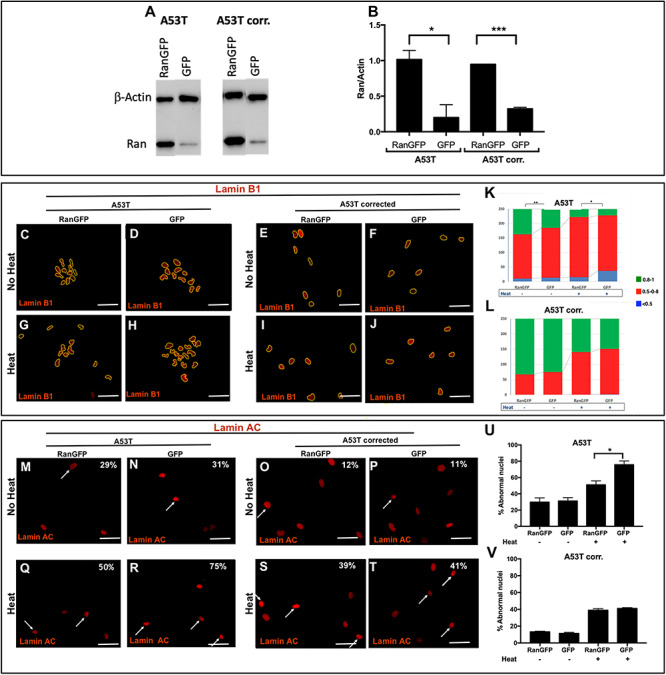
The effects of RAN overexpression on the integrity and function of the nuclear envelope. A53T hiPSC-derived MD NPCs and the isogenic corrected control were transduced with pLenti-IRES-GFP and pLenti-RAN-IRES-GFP. (A) Overexpression of RAN was confirmed by western blot analysis using anti-RAN antibody (25 kDa) normalized to β-actin (42 kDa). (B) RAN protein levels were quantified in two independent western-blot experiments. Higher levels of RAN protein were detected in the A53T hiPSC-derived MD NPCs and the isogenic corrected control. (C-V) The effect of RAN overexpression on the integrity of the nuclear envelope and its sensitivity to heat-shock was evaluated by immunocytochemistry for Lamin B1 (C-J) and Lamin A/C (M-T) in the A53T hiPSC-derived MD NPCs and the isogenic corrected control in regular conditions (C-F, M-P) and upon heat-shock (G-J, Q-T). The overexpression of RAN was visualized using GFP reporter; thus, for these set of experiments, we used red fluorescent conjugated secondary antibody to detect the staining of Lamin B1 (C-J) and Lamin A/C (M-T). Percentages indicate the proportion of cells with folded/blebbed nuclear morphology, white arrows denote abnormal nuclei (M-T). (K, L) The nuclear circularity data are plotted as frequency distributions of for 250 cells (n = 2). (U, V) Percentages indicate the proportion of cells with folded/blebbed nuclear morphology. Bars represents mean ± SEM, n = 2. (K, L) *P < 0.05, **P < 0.001, Kolmogorov–Smirnov test; (B, U, V) *P < 0.05, ***P < 0.001 Student’s t-test. Scale bars: 10 μm.
The effects of the A53T mutation on the interactionof alpha-synuclein with RAN
We investigated the endogenous interaction between α-syn and RAN proteins by Co-IP and tested the effect of the A53T mutation on the strength of the interaction by comparing the A53T hiPSC-derived MD NPCs with their isogenic corrected control. Co-IP in both lines demonstrated the interaction between the endogenous α-syn and RAN proteins (Fig. 5A) confirming our observation with the ectopically expressed tagged proteins (Fig. 2E). We then characterized the effect of the A53T mutation on the endogenous interaction between α-syn and RAN. Quantification analysis of the Co-IP fractions (α-syn—RAN bound normalized to unbound RAN referenced to actin protein expression) showed a stronger intensity ratio for the A53T line compared with the isogenic corrected line, amounting to 35% increase in binding (P = 0.03, Fig. 5B). This data indicated a stronger interaction between the A53T mutated α-syn and RAN in comparison with the interaction of the normal α-syn with RAN, thus, the A53T mutation exerts a stronger binding affinity with RAN compared with the wild-type α-syn. Collectively, our results suggested that the A53T-induced hyperfunction of α-syn sequestered RAN such that RAN is prevented from carrying out its normal physiological function, which in turn results in perturbations to the nuclear envelope structure, integrity and resistance to stress condition.
Figure 5.
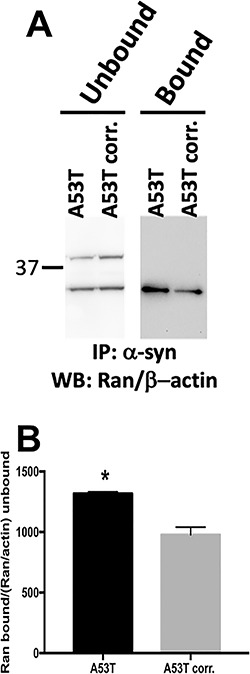
Alpha-synuclein A53T mutation affects the strength of the interaction of α-syn with RAN. (A) Western-blot analysis of homogenates from A53T and A53T-corrected hiPSC-derived MD NPC lines. Anti-syn immunoprecipitates were analyzed by western blot using anti-RAN antibody in unbound and bound fractions. The immunostaining with β-actin antibody was used as the reference for the unbound fractions. The results were calculated as the ratio between bound and unbound fractions for RAN protein and normalized to β-actin expression in the same lane. RAN bands visualized at 25 kDa, β-actin (42 kDa) was used as internal control for normalization in the unbound fraction. (B) Ratios of the bound compared with the unbound levels of RAN showed stronger interaction for the A53T mutation compared with the isogenic mutation corrected control (*P < 0.05, Student’s t-test).
DNA methylation and DNMT3A
RAN plays a key role in transport of large cargo including high mass proteins through the nuclear pore. Thus, it is important to examine the consequences of SNCA A53T-induced RAN sequestering on the nucleocytoplasmic transport of essential large nuclear proteins. A reduction in global DNA-methylation was reported in brains from PD patients (39, 40); however, the underpinning mechanism has yet to be discovered. We aimed to explore the mechanistic link between PD associated loss in DNA methylation and our observation of A53T-induced sequestered RAN and focused on testing the hypothesis that the A53T α-syn may hijack RAN, compromising its function to efficiently transport of DNA methyltransferase (DMT) machinery. DNA methyltransferase 3 subunit alpha (DNMT3A) is a de novo methyltransferase highly abundant in adult brains. (41). DNMT3A is a large protein (130 kDa), and has been shown to be transported to the nucleus via the RAN gradient (41). Thus, as the first step, we focused on DNMT3A as an example and studied the effect of the A53T mutation on the active transport of DNMT3A to the nucleus. We measured the cytoplasmatic and total levels of DNMT3A using western blotting and found that the relative expression levels of the cytoplasmatic DNMT3A from the total DNMT3A (Fig. 6A and B, Supplementary Material, Fig. S4A and B) was significantly higher in the A53T hiPSC-derived MD NPCs compared with the isogenic corrected control (1.5-fold, P = 0.046 Fig. 6B). We confirmed these results using immunocytochemistry experiments and measured the levels of nuclear DNMT3A. We found that the nuclear levels of DNMT3A were significantly lower in the A53T compared with the isogenic corrected NPCs (64% decrease, P < 0.0001, Fig. 6 C-E, Supplementary Material, Fig. S4C-H). Overall, these data indicated a mis-localization of the DNMT3A to the cytoplasm.
Figure 6.
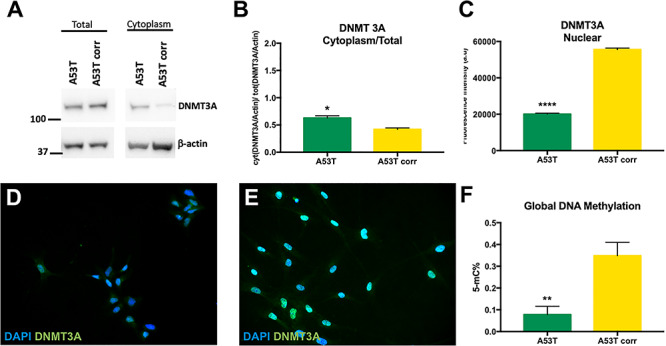
Alpha-synuclein A53T mutation affects DNMT3A localization in the nucleus and global DNA methylation. (A) Levels of total and cytoplasmic DNMT3A (130 kDa) were assayed using western blot and normalized to β-actin (42 kDa). (B) Ratios of cytoplasmic DNMT3A/total DNMT3A showed a mislocalization of DNMT3A in the cytoplasm for the A53T hiPSC-derived MD NPCs compared with the isogenic mutation corrected control. (C) Quantification of the nuclear levels of DNMT3A in hiPSC-derived MD NPCs by immunocytochemistry. Each column represents the mean of nuclear DNMT3A measured in 150 cells in two independent experiments. The nuclear levels of DNMT3A were significantly lower A53T hiPSC-derived MD NPCs compared with the isogenic mutation corrected control. (D, E) Representative immunocytochemistry of DNMT3A signal in (D) A53T and (E) isogenic A53T corrected hiPSC-derived MD NPCs. (F) A53T hiPSC-derived MD NPCs showed a loss in global DNA methylation compared with the isogenic corrected control. Each column represents the mean of global 5 mC%. The error bars represent the SEM (n = 2). *P < 0.05, **P < 0.01, ****P < 0.0001, Student’s t-test.
DNMTs play an important role in 5-mC methylation. Thus, to study the consequences of lower nuclear DNMT3A levels in the A53T line, we determined the levels of 5-methyl-cytosine (5-mC%) in the isogenic A53T and corrected hiPSC-derived NPCs lines. We observed a significant reduction in 5-mC% in the A53T line compared with the isogenic control (4.5-fold, P = 0.01, Fig. 6F). This observation was in agreement with the association of PD with reduced global DNA-methylation (39, 40). Here, we established the correlation between depletion of DNMT3A from the nucleus and reduction in global DNA-methylation in the context of the PD A53T mutation. Altogether, these results suggested that loss in global DNA methylation caused by the A53T mutation is plausibly mediated, at least in part, by mis-localization of the DNMT3A. These outcomes exemplified the biological significance of A53T-induced RAN sequestering, i.e. preventing RAN from transporting essential protein such as DNMT3A to the nucleus.
Discussion
In this manuscript, we investigated a role of SNCA in the nucleus utilizing isogenic hiPSC-derived neurons from PD patients with autosomal dominant mutations, A53T and SNCA-triplication, and the corresponding corrected lines. Using these models, we demonstrated that that two autosomal dominant PD mutations, have similar perturbed effects on the shape, integrity and vulnerability to stress conditions of the nuclear envelope, and corrections of the mutations in isogenic hiPSC-derived neuronal lines rescued these phenotypic defects. In addition, our mechanistic experiments demonstrated, for the first time, the involvement of α-syn in nucleocytoplasmic transport via interaction with RAN. We found that the A53T mutation exerts its adverse effect by trapping RAN, consequently preventing it from transporting key nuclear proteins, such as DMNT3A, and by that leading to nuclear dysfunction. Overexpression of RAN ameliorated the SNCA A53T-associated nuclear dysfunction, while downregulation of RAN worsened the A53T-associated nuclear envelope abnormalities and induced impaired nuclear structure in the corrected line. Multiple hypotheses have been proposed regarding the function of SNCA and its role in disease. The results of this study generated a new hypothesis about the role of SNCA in health and disease. Specifically, our findings provide novel insights into the normal function of α-syn in nucleocytoplasmic transport and interpret that α-syn acquires hyperfunction in disease state resulted from either overexpression or dominant-negative type of missense coding mutations. We further suggest that mutated as well as overexpressed SNCA induced nuclear deficits mediated by sequestering RAN, lead, at least in part, to neurodegeneration.
Our results that demonstrated the adverse effect of SNCA mutations on the nuclear envelope integrity are consistent with previous studies that described the relation between SNCA aggregations and mutations and loss of nuclear envelope integrity (20, 25, 42). Here, we propose a mechanistic explanation for the genotype–phenotype observation. For the first time, we suggest that α-syn is involved in the nucleocytoplasmic transport across the nuclear envelope and that the mutated α-syn disrupts the active transportation of nuclear proteins. It is important to note that our study was performed using MD NPC, which may not fully represent mature dopaminergic neuron in their brain microenvironment. Thus, these results warrant further investigations using additional model systems such as mature neuronal cultures and transgenic animal carrying the SNCA mutations.
Nucleocytoplasmic transport is a fundamental, though complex biological process involves multiple factors and the asymmetric gradient of RAN–GTP and RAN–GDP between the nucleus and cytoplasm that determines the unidirectional transport between the nuclear and cytoplasmic compartments (38, 43–45). The nuclear transport events of large molecules (>40 kDa) require RAN, while smaller molecules are shuttled by passive diffusion (46–48). In accord, we showed that reduction in RAN did not affect α-syn localization in the nucleus, implying that the transportation of α-syn itself to the nucleus does not depend on RAN, as expected for small protein. However, α-syn complexing with RAN is essential for the transport of larger proteins such as, DNMT3A.
SNCA mutations cause autosomal dominant PD. It was shown that the mutated α-syn protein (A30P, A53T and G51D) exhibit increased nuclear targeting and more rapid shuttling between the nucleus/cytoplasm (17, 20, 49). Here, we showed that the interaction of the A53T mutated α-syn with RAN is stronger compared with the wild-type protein. The stronger affinity of the mutated α-syn to the key component of the active nuclear transport machinery may explain, at least in part, the previous observations. Furthermore, our results suggest that SNCA coding mutations exert their pathogenic effect by sequestering RAN such that RAN is prevented from carrying out its normal physiological function. Mutations-induced hyperfunction of α-syn exemplified an enhancement of the normal protein function, as a molecular mechanism underlying the pathogenic of SNCA coding mutations, which could potentially correspond to the mechanistic effect of SNCA overexpression.
Accumulating evidence have demonstrated dysregulation nucleocytoplasmic transport, disruption of the nuclear pore complex and defects in the nuclear envelope shape and integrity in several other neurodegenerative diseases (50) including, Huntington disease (HD) (51, 52), amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD) (53–58) and Alzheimer’s disease (ad) and other tauopathies (59–62). Similarly, recently we and others described defects in the nuclear envelope architecture in PD with the SNCA triplication and LRRK2 G2019S dominant mutation, respectively (25, 27, 63). In this study, we strengthen the role of the nuclear envelope dysfunction in PD and extended the phenomenon to an additional SNCA dominant mutation. Furthermore, the involvement of RAN in neurodegenerative diseases was investigated most extensively in the context of TDP-43 pathology in ALS and FTD (55, 64, 65), and deficits in RAN and RanGAP1 were also implicated in HD (51, 52). Our results demonstrated the contribution of RAN in PD as well, and by that extending the importance role of RAN in a broader range of neurodegenerative disorders. Collectively, these studies suggested that impairment of the nucleocytoplasmic transport is a pathogenic driver of neurodegeneration and a possibly general pathomechanism underpinning multiple neurodegenerative diseases characterized by protein aggregation (66). Nonetheless, whether impaired nucleocytoplasmic transport is a consequence, secondary effect, of such aggregates or rather it is the actual initial cause and subsequently contributes to their formation is still mostly unclear. Here, we provide mechanistic insights suggesting that sequestering the gradient nucleocytoplasmic transportation machinery is the primary event; however, further studies warrant to reveal the conformation of α-syn protein that exhibits the strong binding to RAN.
It is clear that RAN sequestering may have detrimental effect on many proteins that depend on RAN for their nuclear transportation. (51, 52, 55, 64, 65). Here, we demonstrated an example of DNMT3A enzyme that catalyzes de-novo DNA-methylation. Compromising DNMT3A transportation to the nucleus, whereas it functions caused perturbations in the major epigenetic pathway sequential to the RAN malfunction. In this regard, our results underscore a novel mechanistic explanation for epigenetic dysregulation in PD, which may have a broader implication in other Lewy body diseases and more generally in neurodegenerative diseases and conditions (26, 39).
In conclusion, the results of this work provide a mechanistic understanding for the dominant negative effect of SNCA mutations on PD pathogenesis. This knowledge is translational for the development of new therapeutic interventions for PD based on restoration of RAN function and for a broader range of neurodegenerative diseases by targeting the pertinent components of the nucleocytoplasmic transportation machinery.
Materials and Methods
Cell culture and hiPSC-derived NPC differentiation
hiPSCs from a patient with the SNCA A53T mutation (ND50050) and the corresponding isogenic mutation-corrected line (ND50085) and a patient with the triplication of the SNCA locus (ND34391) were purchased from the NINDS Human Cell and Data Repository (https://nindsgenetics.org). hiPSCs were cultured under feeder-independent conditions in mTeSR™1 medium (StemCell Technologies) onto hESC-qualified matrigel-coated plates. Cells were passaged using Gentle Cell Dissociation Reagent (StemCell Technologies) according to the manufacturer’s manual.
hiPSCs were differentiated into NPCs using optimized protocols, previously described (67). Briefly, differentiation into NPCs was performed using a monolayer culture protocol. Single-cell hiPSCs were seeded in Neural Induction Medium (NIM—Stem Cell Technologies) supplemented with Y27632 (10 μM) and plated onto matrigel-coated plates in NIM. On day 6, NIM was supplemented with 200 ng/mL SHH (Peprotech) leading to the formation of single cells NPCs. On day 12, the cells are replated in matrigel-coated plates in N2B27 medium supplemented with 3 μM CHIR99021, 2 μM SB431542, 5 μg/ml BSA, 20 ng/ml bFGF and 20 ng/ml EGF, leading to the formation of NPCs. NPCs were passaged every 2 days in their respective medium with Accutase (StemCell Technologies) and plated on matrigel-coated plates (2.5 × 104 cells/cm2). Phenotypic analyses were performed on NPCs at passages P14–P16. To evaluate the effects of A53T mutations and SNCA-Tri, four different lines were used (Table 1).
Table 1.
Isogenic hiPSC lines used in this study
Immunocytochemistry and imaging
Prior immunostaining, cells were plated onto matrigel-coated cells imaging coverglasses (Eppendorf, 0030742060). Cells were fixed in 4% paraformaldehyde and permeabilized in 0.1% Triton-X100. Immunocytochemistry was performed as follows; cells were blocked in 5% goat serum for 1 h before incubating with primary antibodies overnight at 4°C (Supplementary Material, Table S1). Secondary antibodies (Alexa fluor, Life Technologies) were incubated for 1 h at room temperature. Nuclei were stained with NucBlue® Fixed Cell ReadyProbes® Reagent (ThermoFisher), according to the manufacturers’ instructions. Images were acquired on the Leica SP5 confocal microscope using a 40X objective.
Immunocytochemistry quantification of the nuclear envelope markers
Nuclear folding was analyzed using the Lamin A/C marker, and folded nuclear envelope shape was considered as abnormal. Nuclear circularity was quantified using the built-in ImageJ circularity plugin and assessed based on the Lamin B1 marker. A circularity value of 1.0 indicates a perfect circle. A value approaching 0 indicates an increasingly elongated polygon (68). A total of 100 cells per staining were analyzed for two independent experiments.
Heat-shock treatment
Before heat-shock treatment, cells were plated onto matrigel-coated cells imaging coverglasses (Eppendorf, 0030742060). Cells were transferred from 37 to 45°C for 30 min, according to (31). Cells were used for immunocytochemistry as previously described. After staining with Lamin A/C and Lamin B1 antibodies, at least 100 nuclei for each of the two independent experiments were analyzed.
Nuclear membrane integrity assay
Nuclei were isolated from MD NPCs according to established protocols, with slight modifications (69–71). Briefly, 1 × 106 cells were dissolved in lysis buffer (0.32 M Sucrose, 5 mM CaCl2, 3 mM Mg (Acetate)2, 0.1 mM EDTA, 10 mM Tris–HCl, pH 8, 1 mM DTT, 0.1% Triton X-100). The lysate was transferred to a 14 × 89 mm polypropylene ultracentrifuge tube, carefully underlaid with sucrose solution (1.8 M Sucrose, 3 mM Mg (Acetate)2, 1 mM DTT, 10 mM Tris–HCl, pH 8), and subjected to ultracentrifugation at 107 000 RCF for 15 min at 4°C. Supernatant and the debris interphase were carefully aspirated, and resuspended in STM buffer (250 mM sucrose, 50 mM Tris (pH 7.4), 5 mM MgCl2) was added to the nuclei pellet. The nuclear membrane integrity assay was conducted according to (33). Briefly, after 5 min incubation on ice, nuclei were gently resuspended and prepared for the nuclear membrane integrity assay. Aliquots of NaCl-STM were added to each tube such that a typical assay would have tubes containing 0, 25, 50, 75, 100, 125, 150, 175, 200, 250, 300 or 1000 mM NaCl (final concentrations). Nuclei were incubated for 30 min on ice. After incubation, nuclei were centrifuged at 7500 g for 10 min. The supernatants were collected, and the absorbance was measured at 260 nm. The absorbance was normalized to the maximum absorbance measured (usually at 1000 mM NaCl) and expressed as a percentage of maximum absorbance260 (A260). Analyses were carried in triplicates for each of the three independent experiments.
Plasmid design and construction
pLenti- dCas9-DNMT3A vector was described in (72). The vector carried gRNA4 sequence, 5′-CTGCTCAGGGTAGATAGCTG-3′ targeting SNCA-intron1 gene is pBK500 (26).
The pBK1082 (pLenti-CMV-3xFLAG-RAN-SV40-Hygro-WPRE), and pBK1013 (pLenti-CMV-c-Myc-Alpha-SynucleinWT-IRES-Puro-WPRE) represent tagged and over-expressed RAN and alpha-syn, respectively were generated as follows:
pBK1082 was derived from pET11d-RAN (gift from Larry Gerace’s lab obtained from addgene plasmid # 108919) was digested with XbaI-BamHI and purified. The insert was then cloned into pBK134 vector (pLenti-IRES-GFP) (unpublished) digested with XbaI-BamHI. The resulted plasmid, pLenti- RAN-IRES-GFP was named pBK958. Next, the RAN gene was PCR amplified with primers 958RUN-PmeI/R: gaaggagtttaaacccatggctg and 958RUN-HpaII/L: gatccaggttaacattctcacagg, then digested with enzymes PmeI and HpaI, purified and ligated with a vector that was generated by digesting pBK953 (pLenti-CMV-3xHAtag-SV40-Hygro-WPRE) with PmeI and MscI. Next, the 3xHAtag was replaced by 3xFLAG via PmeI-XbaI digestion. 3xFLAG was prepared by phosphorylating and annealing oligos CTAGAatgGACTACAAAGACCATGACGGTGATTATAAAGATCATGACATCGATTACAAGGATGACGATGACAAGggcggaggctcGTTT and AAACgagcctccgccCTTGTCATCGTCATCCTTGTAATCGATGTCATGATCTTTATAATCACCGTCATGGTCTTTGTAGTCcatT.
pBK1013 harboring alpha-synuclein-WT was a gift from David Rubinsztein (Addgene plasmid #40822). Alpha-synuclein gene was amplified using primers NheI927Syn/R: caagtccggctagcagatctcgagc and SpeI927Syn/L: gcccactagtaccgtcgactgcag. Resultant PCR product was digested by enzymes NheI and SpeI and ligated with NheI and SpeI digested vector pBK921 (pLenti-CMV-IRES-Puro-WPRE). Next cMYC tag was introduced upstream the a-syn gene via NheI site. The tag was created by phosphorylating and annealing the oligos CTAGATGGAGCAGAAACTCATCTCTGAAGAGGATCTGGGAGGCGGAGGATCCT and CTAGAGGATCCTCCGCCTCCCAGATCCTCTTCAGAGATGAGTTTCTGCTCCAT.
To generate RAN overexpressing construct, pET11d-RAN (obtained from Larry Gerace (Addgene plasmid # 108919) was digested with XbaI-BamHI and purified. The insert then was cloned into pBK134 vector (pLenti-IRES-GFP) (unpublished) digested with XbaI-BamHI restriction enzymes. The resulted plasmid, pLenti-RAN-IRES-GFP was named pBK958. The pLenti- RAN-IRES-GFP, pBK958 was amplified with 958RUN-PmeI/R primer: gaaggagtttaaacccatggctg; 958RUN-HpaII/L gatccaggttaacattctcacagg.
The inducible shRNA clones were derived from Inducible Dharmacon™ TRIPZ™ Lentiviral shRNA library. The following clone was selected after being screened for the knockdown efficiency: V3THS_395424 carried a mature antisense, ACAAGTTTGAACTGGACCT, and the resulted plasmid was named pBK1019.
Vector production
Lentiviral vectors were generated using the transient transfection protocol, as described in (72). Briefly, 15 μg vector plasmid, 10 μg psPAX2 packaging plasmid (Addgene #12260 generated in Dr Didier Trono’s lab, EPFL, Switzerland), 5 μg pMD2.G envelope plasmid (Addgene #12259, generated in Dr Trono’s lab) and 2.5 μg pRSV-Rev plasmid (Addgene #12253, generated in Dr Trono’s lab) were transfected into 293 T cells. Vector particles were collected from the filtered conditioned medium at 72 h post-transfection. The particles were purified using the sucrose-gradient method64 and concentrated >250-fold by ultracentrifugation (2 h at 20 000 rpm). Vector and viral stocks were aliquoted and stored at −80°C.
Tittering vector preparations
Titers were determined for the vectors expressing puromycin or hygromycin B selection markers by counting puromycin-resistant or hygromycin B-resistant colonies as described in (72) and by p24gag ELISA method equating 1 ng p24gag to 1 × 104 viral particles. The MOI was calculated by the ratio of the number of viral particles to the number of cells. The p24gag ELISA was carried out as per the instructions in the HIV-1 p24 antigen capture assay kit (NIH AIDS Vaccine Program). Briefly, high-binding 96-well plates (Costar) were coated with 100 μL monoclonal anti-p24 antibody (NIH AIDS Research and Reference Reagent Program, catalog 3537) diluted 1:1500 in PBS. Coated plates were incubated at 4°C overnight, then blocked with 200 μL 1% BSA in PBS and washed three times with 200 μL 0.05% Tween 20 in cold PBS. Next, plates were incubated with 200 μL samples, inactivated by 1% Triton X-100 for 1 h at 37°C. HIV-1 standards (catalog SP968F) were subjected to a 2-fold serial dilution and applied to the plates at a starting concentration equal to 4 ng/mL. Samples were diluted in RPMI 1640 supplemented with 0.2% Tween 20 and 1% BSA, applied to the plate and incubated at 4°C overnight. Plates were then washed six times and incubated at 37°C for 2 h with 100 μL polyclonal rabbit anti-p24 antibody (catalog SP451T), diluted 1:500 in RPMI 1640, 10% fetal bovine serum, 0.25% BSA and 2% normal mouse serum (NMS; Equitech-Bio). Plates were then washed as above and incubated at 37°C for 1 h with goat anti-rabbit horseradish peroxidase immunoglobulin G (IgG) (Santa Cruz Biotechnology), diluted 1:10 000 in RPMI 1640 supplemented with 5% normal goat serum (NGS; Sigma), 2% NMS, 0.25% BSA and 0.01% Tween 20. Plates were washed as above and incubated with 3,3′,5,5′-tetramethylbenzidine peroxidase substrate (KPL) at room temperature for 10 min. The reaction was stopped by adding 100 μL 1 N HCL. Plates were read by Microplate Reader (The iMark Microplate Absorbance Reader, Bio-Rad) at 450 nm and analyzed in Excel. All experiments were performed in triplicates.
Viral transduction
Transductions of cells were performed in complete culture medium as follows: SH-SY5Y cells were transduced with LV-myc and LV-3xFlag vectors at the MOIs = 2. Sixteen hours post-transduction the media was replaced, and 2 days later the cells were used for Co-IP experiments.
For the RAN overexpression experiments, A53T and A53T corrected hiPSC-derived NPCs were transduced with LV vectors at the MOIs = 3. At 16 h post-transduction, the media was replaced, and once confluent, cells were replated. After 24 h, boost of MOIs = 3 was applied to the cells. Medium was changed after 16 h, and cells were used 72 h post-transduction for further analyses.
For downregulation experiments, cells were transduced with MOIs = 1. At 16 h post-transduction, medium was replaced. At 48 h post-transduction, 1 μg/mL puromycin was applied. Cells were maintained under puromycin selection. Expression of shRNA was induced by treating cells with 0.2 μg/mL doxycycline hyclate (Sigma). Full medium changed was performed every day for three consecutive days prior to further analyses.
Western blotting
About 20 μg of each sample was run on 12% Tris-glycine SDS–PAGE gels. Proteins were transferred to nitrocellulose membranes, and blots were blocked with 5% milk PBS Tween 20. Primary antibodies were incubated at 4°C overnight (Supplementary Material, Table S3). Horseradish peroxidase-conjugated secondary antibodies were incubated for 1 h at room temperature (Abcam; 1:10 000). For Co-IP experiments, signal was detected using SuperSignal™ West Femto (ThermoFisher, Cat#34094). For western-blotting experiment, signal was detected with HyGLO Quick Spray (Denville Scientific). Immunoblots were imaged using ChemiDoc MP Imaging System (Bio-Rad). The densitometry was measured using ImageJ software, and protein levels were normalized to β-actin expression in the same lane. Each experiment was repeated twice and represents two independent biological replicates.
Total protein lysate
Cells were scraped from the dish and homogenized in 10× volume of 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, in the presence of a protease and phosphatase inhibitor cocktail (Sigma, St. Louis, MO). Samples were centrifugated at >10 000 g at 4°C for times for 15 min, and protein quantification was performed using DC™ Protein Assay (BioRad, Cat#5000113).
Isolation of nuclear and cytoplasmic extracts
Nuclear and cytoplasmic extracts were prepared using the NE-PER cytoplasmic and nuclear extraction kit (Thermo Scientific, USA) according to the manufacturer’s instruction. Briefly, cells were detached with Accutase (StemCell Technologies), washed with PBS and centrifuged at 500 g for 3 min. The cell pellet was resuspended in Cytoplasmic Extraction Reagent I (CER I), and vortexed vigorously. The suspension was incubated on ice for 10 min followed by the addition of CER II. The suspension was vortexed and centrifuged for 5 min at 16 000 g. The supernatant (cytoplasmic extract) was collected in a pre-chilled tube. The insoluble fraction was resuspended in Nuclear Extraction Reagent (NER) and vortexed every 10 min for 40 min. After centrifugation, the supernatant, constituting the nuclear extract, was collected and stored at 80°C until use.
RNA extraction and gene expression analysis
Total RNA was extracted using TRIzol reagent (Invitrogen) followed by purification with a RNeasy kit (QIAGEN), used per the manufacturer’s protocol. The RNA concentration was determined spectrophotometrically at 260 nm, while the quality of the purification was determined by a 260: a 280-nm ratio that showed values between 1.9 and 2.1, indicating high RNA quality. cDNA was synthesized using MultiScribe RT enzyme (Applied Biosystems) using the following conditions: 10 min at 25°C and 120 min at 37°C.
Real-time PCR was used to quantify gene expression levels. Briefly, duplicates of each sample were assayed by relative real-time qPCR using TaqMan expression assays and the ABI QuantStudio 7 (Supplementary Material, Table S4). ABI minor groove binder probe and primer set assays (Applied Biosystems) that were used are listed in Table S2. Each cDNA (20 ng) was amplified in duplicate in at least two independent runs for two independent experiments (overall ≥8 repeats), using TaqMan Universal PCR master mix reagent (Applied Biosystems) and the following conditions: 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C and 1 min at 60°C. As a negative control for the specificity of the amplification, we used RNA control samples that were not converted to cDNA (no RT) and no-cDNA/RNA samples (no template) on each plate. No amplification product was detected in control reactions. Data were analyzed with a threshold set in the linear range of amplification. The cycle number at which any particular sample crossed that threshold (Ct) was then used to determine fold difference, whereas the geometric mean of the two control genes served as a reference for normalization. Fold difference was calculated as 2 − ΔΔCt;67 ΔCt = [Ct(target) − Ct (geometric mean of reference)] and ΔΔCt = [ΔCt(sample)] − [ΔCt(calibrator)]. The calibrator was a particular RNA sample, obtained from the control MD NPCs, used repeatedly in each plate for normalization within and across runs. The variation of the ΔCt values among the calibrator replicates was smaller than 10%.
Co-immunoprecipitation
Co-IP was performed using Dynabeads Co-Immunoprecipitation Kit (Cat#14321D, ThermoFisher, CA) per manufacturers’ instructions. Briefly, 1 mg of Dynabeads M-270 Epoxy was coupled with 7 μg of antibody (Supplementary Material, Table S2). For western-blotting analyses, 1.5 mg of antibody-coupled beads. A total of 25–50 mg of cells were used for each experiment. Purified protein complexes were eluted in 60 μl of the elution buffer (EB), and immediately used for western-blotting analysis. The unbound fractions were collected in 2 mL. The signal was detected using SuperSignal™ West Femto (ThermoFisher, Cat#34094). Immunoblots were imaged using ChemiDoc MP Imaging System (Bio-Rad). The densitometry was measured using ImageJ software. The results were calculated as the ratio between bound and unbound fractions for RAN protein and normalized to β-actin expression in the same lane (unbound fractions). Each experiment was repeated twice and represents two independent biological replicates.
Global DNA methylation
DNA from each cell line was extracted using the DNeasy Blood and Tissue Kit (Qiagen). Global DNA methylation was assessed using a commercially available 5-methylcytosine (5-mC)-based immunoassay platform (MethylFlash™ Global DNA Methylation (5-mC) ELISA Kit, Epigentek), according to the manufacturer’s instructions. Briefly, purified DNA (100 ng) and unmethylated (negative) control DNA (10 ng) were incubated in strip wells with a solution to promote DNA binding and adherence to the well. The samples in the strip-wells were treated with solutions containing the diluted 5-mC capture and the detection antibodies. The methylated fraction of DNA was quantified colorimetrically by absorbance readings using a FLUOstar Optima, BMG. The percentage of methylated DNA was calculated as a proportion of the optical density (OD), according to manufacturers’ instructions using the formula:
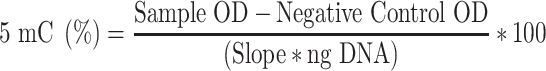 |
The percentage of 5-mC was determined using two replicates in each of the two independent experiments.
Statistical analyses
The differences between the isogenic lines were analyzed statistically using the following data comparisons tests (GraphPad Prism7): (i) Analysis of corresponding cumulative distributions using the Kolmogorov–Smirnov test and (ii) two-group comparisons using Student’s t-tests.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was funded in part by the National Institutes of Health/National Institute of Neurological Disorders and Stroke – NIH/NINDS (R01 NS085011 to O.C.-F.) and (R01 NS113548-01A1 to O.C.-F.).
Conflict of Interest Statement. Duke University filed a provisional patent application related to this study.
Contributor Information
Vivian Chen, Division of Translational Brain Sciences, Department of Neurology, Duke University Medical Center, Durham, NC 27710, USA; Center for Genomic and Computational Biology, Duke University Medical Center, Durham, NC 27710, USA.
Malik Moncalvo, Viral Vector Core, Duke University Medical Center, Durham, NC 27710, USA; Department of Neurobiology, Duke University Medical Center, Durham, NC 27710, USA.
Dominic Tringali, Division of Translational Brain Sciences, Department of Neurology, Duke University Medical Center, Durham, NC 27710, USA; Center for Genomic and Computational Biology, Duke University Medical Center, Durham, NC 27710, USA.
Lidia Tagliafierro, Division of Translational Brain Sciences, Department of Neurology, Duke University Medical Center, Durham, NC 27710, USA; Center for Genomic and Computational Biology, Duke University Medical Center, Durham, NC 27710, USA.
Ahila Shriskanda, Division of Translational Brain Sciences, Department of Neurology, Duke University Medical Center, Durham, NC 27710, USA; Center for Genomic and Computational Biology, Duke University Medical Center, Durham, NC 27710, USA.
Ekaterina Ilich, Viral Vector Core, Duke University Medical Center, Durham, NC 27710, USA; Department of Neurobiology, Duke University Medical Center, Durham, NC 27710, USA.
Wendy Dong, Viral Vector Core, Duke University Medical Center, Durham, NC 27710, USA; Department of Neurobiology, Duke University Medical Center, Durham, NC 27710, USA.
Boris Kantor, Viral Vector Core, Duke University Medical Center, Durham, NC 27710, USA; Department of Neurobiology, Duke University Medical Center, Durham, NC 27710, USA.
Ornit Chiba-Falek, Division of Translational Brain Sciences, Department of Neurology, Duke University Medical Center, Durham, NC 27710, USA; Center for Genomic and Computational Biology, Duke University Medical Center, Durham, NC 27710, USA.
References
- 1. Ueda K., Fukushima H., Masliah E., Xia Y., Iwai A., Yoshimoto M., Otero D.A., Kondo J., Ihara Y. and Saitoh T. (1993) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A., 90, 11282–11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Polymeropoulos M.H., Lavedan C., Leroy E., Ide S.E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R. et al. (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science, 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- 3. Kruger R., Kuhn W., Muller T., Woitalla D., Graeber M., Kosel S., Przuntek H., Epplen J.T., Schols L. and Riess O. (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat. Genet., 18, 106–108. [DOI] [PubMed] [Google Scholar]
- 4. Singleton A.B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R. et al. (2003) Alpha-Synuclein locus triplication causes Parkinson's disease. Science (New York, N.Y), 302, 841. [DOI] [PubMed] [Google Scholar]
- 5. Fuchs J., Nilsson C., Kachergus J., Munz M., Larsson E.M., Schule B., Langston J.W., Middleton F.A., Ross O.A., Hulihan M. et al. (2007) Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology, 68, 916–922. [DOI] [PubMed] [Google Scholar]
- 6. Ross O.A., Braithwaite A.T., Skipper L.M., Kachergus J., Hulihan M.M., Middleton F.A., Nishioka K., Fuchs J., Gasser T., Maraganore D.M. et al. (2008) Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann. Neurol., 63, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chartier-Harlin M.C., Kachergus J., Roumier C., Mouroux V., Douay X., Lincoln S., Levecque C., Larvor L., Andrieux J., Hulihan M. et al. (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet, 364, 1167–1169. [DOI] [PubMed] [Google Scholar]
- 8. Ibanez P., Bonnet A.M., Debarges B., Lohmann E., Tison F., Pollak P., Agid Y., Durr A. and Brice A. (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet, 364, 1169–1171. [DOI] [PubMed] [Google Scholar]
- 9. Ibanez P., Lesage S., Janin S., Lohmann E., Durif F., Destee A., Bonnet A.M., Brefel-Courbon C., Heath S., Zelenika D. et al. (2009) Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch. Neurol., 66, 102–108. [DOI] [PubMed] [Google Scholar]
- 10. Farrer M., Kachergus J., Forno L., Lincoln S., Wang D.S., Hulihan M., Maraganore D., Gwinn-Hardy K., Wszolek Z., Dickson D. et al. (2004) Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol., 55, 174–179. [DOI] [PubMed] [Google Scholar]
- 11. Miller D.W., Hague S.M., Clarimon J., Baptista M., Gwinn-Hardy K., Cookson M.R. and Singleton A.B. (2004) Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology, 62, 1835–1838. [DOI] [PubMed] [Google Scholar]
- 12. Nussbaum R.L. (2017) The identification of alpha-synuclein as the first Parkinson disease gene. J. Parkinsons Dis., 7, S45–S51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goedert M., Jakes R. and Spillantini M.G. (2017) The synucleinopathies: twenty years on. J. Parkinsons Dis., 7, S53–S71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Villar-Pique A., Lopes da Fonseca T. and Outeiro T.F. (2016) Structure, function and toxicity of alpha-synuclein: the Bermuda triangle in synucleinopathies. J. Neurochem., 139, 240–255. [DOI] [PubMed] [Google Scholar]
- 15. Stefanis L. (2012) Alpha-synuclein in Parkinson's disease. Cold Spring Harb. Perspect. Med., 2, a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maroteaux L., Campanelli J.T. and Scheller R.H. (1988) Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci., 8, 2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goncalves S. and Outeiro T.F. (2013) Assessing the subcellular dynamics of alpha-synuclein using photoactivation microscopy. Mol. Neurobiol., 47, 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McLean P.J., Ribich S. and Hyman B.T. (2000) Subcellular localization of alpha-synuclein in primary neuronal cultures: effect of missense mutations. J. Neural Transm. Suppl., 53–63. [DOI] [PubMed] [Google Scholar]
- 19. Mori F., Tanji K., Yoshimoto M., Takahashi H. and Wakabayashi K. (2002) Immunohistochemical comparison of alpha- and beta-synuclein in adult rat central nervous system. Brain Res., 941, 118–126. [DOI] [PubMed] [Google Scholar]
- 20. Pinho R., Paiva I., Jercic K.G., Fonseca-Ornelas L., Gerhardt E., Fahlbusch C., Garcia-Esparcia P., Kerimoglu C., Pavlou M.A.S., Villar-Pique A. et al. (2019) Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet., 28, 31–50. [DOI] [PubMed] [Google Scholar]
- 21. Goers J., Manning-Bog A.B., McCormack A.L., Millett I.S., Doniach S., Di Monte D.A., Uversky V.N. and Fink A.L. (2003) Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry, 42, 8465–8471. [DOI] [PubMed] [Google Scholar]
- 22. Kontopoulos E., Parvin J.D. and Feany M.B. (2006) Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet., 15, 3012–3023. [DOI] [PubMed] [Google Scholar]
- 23. Sugeno N., Jackel S., Voigt A., Wassouf Z., Schulze-Hentrich J. and Kahle P.J. (2016) Alpha-synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Sci. Rep., 6, 36328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iwata A., Miura S., Kanazawa I., Sawada M. and Nukina N. (2001) Alpha-synuclein forms a complex with transcription factor Elk-1. J. Neurochem., 77, 239–252. [DOI] [PubMed] [Google Scholar]
- 25. Tagliafierro L., Zamora M.E. and Chiba-Falek O. (2019) Multiplication of the SNCA locus exacerbates neuronal nuclear aging. Hum. Mol. Genet., 28, 407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kantor B., Tagliafierro L., Gu J., Zamora M.E., Ilich E., Grenier C., Huang Z.Y., Murphy S. and Chiba-Falek O. (2018) Downregulation of SNCA expression by targeted editing of DNA methylation: a potential strategy for precision therapy in PD. Mol. Ther., 26, 2638–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu G.H., Qu J., Suzuki K., Nivet E., Li M., Montserrat N., Yi F., Xu X., Ruiz S., Zhang W. et al. (2012) Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature, 491, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller J.D., Ganat Y.M., Kishinevsky S., Bowman R.L., Liu B., Tu E.Y., Mandal P.K., Vera E., Shim J.W., Kriks S. et al. (2013) Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell, 13, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Robin J.D. and Magdinier F. (2016) Physiological and pathological aging affects chromatin dynamics, structure and function at the nuclear edge. Front. Genet., 7, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burke B. and Stewart C.L. (2006) The laminopathies: the functional architecture of the nucleus and its contribution to disease. Annu. Rev. Genomics Hum. Genet., 7, 369–405. [DOI] [PubMed] [Google Scholar]
- 31. Vigouroux C., Auclair M., Dubosclard E., Pouchelet M., Capeau J., Courvalin J.C. and Buendia B. (2001) Nuclear envelope disorganization in fibroblasts from lipodystrophic patients with heterozygous R482Q/W mutations in the lamin A/C gene. J. Cell Sci., 114, 4459–4468. [DOI] [PubMed] [Google Scholar]
- 32. Romero-Bueno R., Cruz Ruiz P., Artal-Sanz M., Askjaer P. and Dobrzynska A. (2019) Nuclear organization in stress and aging. Cells, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Czubryt M.P., Ramjiawan B. and Pierce G.N. (1997) The nuclear membrane integrity assay. Mol. Cell. Biochem., 172, 97–102. [PubMed] [Google Scholar]
- 34. Burke B. and Stewart C.L. (2002) Life at the edge: the nuclear envelope and human disease. Nat. Rev. Mol. Cell Biol., 3, 575–585. [DOI] [PubMed] [Google Scholar]
- 35. Hutchison C.J. (2002) Lamins: building blocks or regulators of gene expression? Nat. Rev. Mol. Cell Biol., 3, 848–858. [DOI] [PubMed] [Google Scholar]
- 36. Liang Y. and Hetzer M.W. (2011) Functional interactions between nucleoporins and chromatin. Curr. Opin. Cell Biol., 23, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fried H. and Kutay U. (2003) Nucleocytoplasmic transport: taking an inventory. Cell. Mol. Life Sci., 60, 1659–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Izaurralde E., Kutay U., Kobbe C., Mattaj I.W. and Gorlich D. (1997) The asymmetric distribution of the constituents of the ran system is essential for transport into and out of the nucleus. EMBO J., 16, 6535–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Desplats P., Spencer B., Coffee E., Patel P., Michael S., Patrick C., Adame A., Rockenstein E. and Masliah E. (2011) Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem., 286, 9031–9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chouliaras L., Mastroeni D., Delvaux E., Grover A., Kenis G., Hof P.R., Steinbusch H.W., Coleman P.D., Rutten B.P. and Hove D.L. (2013) Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients. Neurobiol. Aging, 34, 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mastroeni D., Chouliaras L., Grover A., Liang W.S., Hauns K., Rogers J. and Coleman P.D. (2013) Reduced RAN expression and disrupted transport between cytoplasm and nucleus: a key event in Alzheimer's disease pathophysiology. PLoS One, 8, e53349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jiang P., Gan M., Yen S.H., Moussaud S., McLean P.J. and Dickson D.W. (2016) Proaggregant nuclear factor(s) trigger rapid formation of alpha-synuclein aggregates in apoptotic neurons. Acta Neuropathol., 132, 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nachury M.V. and Weis K. (1999) The direction of transport through the nuclear pore can be inverted. Proc. Natl. Acad. Sci. U.S.A., 96, 9622–9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kalab P., Pralle A., Isacoff E.Y., Heald R. and Weis K. (2006) Analysis of a RanGTP-regulated gradient in mitotic somatic cells. Nature, 440, 697–701. [DOI] [PubMed] [Google Scholar]
- 45. Gorlich D., Seewald M.J. and Ribbeck K. (2003) Characterization of ran-driven cargo transport and the RanGTPase system by kinetic measurements and computer simulation. EMBO J., 22, 1088–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Naim B., Zbaida D., Dagan S., Kapon R. and Reich Z. (2009) Cargo surface hydrophobicity is sufficient to overcome the nuclear pore complex selectivity barrier. EMBO J., 28, 2697–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mohr D., Frey S., Fischer T., Guttler T. and Gorlich D. (2009) Characterisation of the passive permeability barrier of nuclear pore complexes. EMBO J., 28, 2541–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Naim B., Brumfeld V., Kapon R., Kiss V., Nevo R. and Reich Z. (2007) Passive and facilitated transport in nuclear pore complexes is largely uncoupled. J. Biol. Chem., 282, 3881–3888. [DOI] [PubMed] [Google Scholar]
- 49. Fares M.B., Ait-Bouziad N., Dikiy I., Mbefo M.K., Jovicic A., Kiely A., Holton J.L., Lee S.J., Gitler A.D., Eliezer D. et al. (2014) The novel Parkinson's disease linked mutation G51D attenuates in vitro aggregation and membrane binding of alpha-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet., 23, 4491–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Patel V.P. and Chu C.T. (2011) Nuclear transport, oxidative stress, and neurodegeneration. Int. J. Clin. Exp. Pathol., 4, 215–229. [PMC free article] [PubMed] [Google Scholar]
- 51. Grima J.C., Daigle J.G., Arbez N., Cunningham K.C., Zhang K., Ochaba J., Geater C., Morozko E., Stocksdale J., Glatzer J.C. et al. (2017) Mutant huntingtin disrupts the nuclear pore complex. Neuron, 94, 93–107 e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gasset-Rosa F., Chillon-Marinas C., Goginashvili A., Atwal R.S., Artates J.W., Tabet R., Wheeler V.C., Bang A.G., Cleveland D.W. and Lagier-Tourenne C. (2017) Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron, 94, 48–57 e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim H.J. and Taylor J.P. (2017) Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron, 96, 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang K., Daigle J.G., Cunningham K.M., Coyne A.N., Ruan K., Grima J.C., Bowen K.E., Wadhwa H., Yang P., Rigo F. et al. (2018) Stress granule assembly disrupts nucleocytoplasmic transport. Cell, 173, 958–971 e917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chou C.C., Zhang Y., Umoh M.E., Vaughan S.W., Lorenzini I., Liu F., Sayegh M., Donlin-Asp P.G., Chen Y.H., Duong D.M. et al. (2018) TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci., 21, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ferreira P.A. (2019) The coming-of-age of nucleocytoplasmic transport in motor neuron disease and neurodegeneration. Cell. Mol. Life Sci., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Woerner A.C., Frottin F., Hornburg D., Feng L.R., Meissner F., Patra M., Tatzelt J., Mann M., Winklhofer K.F., Hartl F.U. et al. (2016) Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science, 351, 173–176. [DOI] [PubMed] [Google Scholar]
- 58. Paonessa F., Evans L.D., Solanki R., Larrieu D., Wray S., Hardy J., Jackson S.P. and Livesey F.J. (2019) Microtubules deform the nuclear membrane and disrupt nucleocytoplasmic transport in tau-mediated frontotemporal dementia. Cell Rep., 26, 582–593 e585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Frost B. (2016) Alzheimer's disease: an acquired neurodegenerative laminopathy. Nucleus (Calcutta), 7, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eftekharzadeh B., Daigle J.G., Kapinos L.E., Coyne A., Schiantarelli J., Carlomagno Y., Cook C., Miller S.J., Dujardin S., Amaral A.S. et al. (2019) Tau protein disrupts nucleocytoplasmic transport in Alzheimer's disease. Neuron, 101, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lester E. and Parker R. (2018) The tau of nuclear-cytoplasmic transport. Neuron, 99, 869–871. [DOI] [PubMed] [Google Scholar]
- 62. Sheffield L.G., Miskiewicz H.B., Tannenbaum L.B. and Mirra S.S. (2006) Nuclear pore complex proteins in Alzheimer disease. J. Neuropathol. Exp. Neurol., 65, 45–54. [DOI] [PubMed] [Google Scholar]
- 63. Shani V., Safory H., Szargel R., Wang N., Cohen T., Elghani F.A., Hamza H., Savyon M., Radzishevsky I., Shaulov L. et al. (2019) Physiological and pathological roles of LRRK2 in the nuclear envelope integrity. Hum. Mol. Genet., 28, 3982–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ward M.E., Taubes A., Chen R., Miller B.L., Sephton C.F., Gelfand J.M., Minami S., Boscardin J., Martens L.H., Seeley W.W. et al. (2014) Early retinal neurodegeneration and impaired ran-mediated nuclear import of TDP-43 in progranulin-deficient FTLD. J. Exp. Med., 211, 1937–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang K., Donnelly C.J., Haeusler A.R., Grima J.C., Machamer J.B., Steinwald P., Daley E.L., Miller S.J., Cunningham K.M., Vidensky S. et al. (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature, 525, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hutten S. and Dormann D. (2020) Nucleocytoplasmic transport defects in neurodegeneration - cause or consequence? Semin. Cell Dev. Biol., 99, 151–162. [DOI] [PubMed] [Google Scholar]
- 67. Tagliafierro L., Glenn O.C., Zamora M.E., Beach T.G., Woltjer R.L., Lutz M.W. and Chiba-Falek O. (2017) Genetic analysis of alpha-synuclein 3′ untranslated region and its corresponding microRNAs in relation to Parkinson's disease compared to dementia with Lewy bodies. Alzheimers Dement., 13, 1237–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Marzluff W.F. (1990) Preparation of active nuclei. Methods Enzymol., 181, 30–36. [DOI] [PubMed] [Google Scholar]
- 70. Matevossian A. and Akbarian S. (2008) Neuronal nuclei isolation from human postmortem brain tissue. J. Vis. Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jiang Y., Matevossian A., Huang H.S., Straubhaar J. and Akbarian S. (2008) Isolation of neuronal chromatin from brain tissue. BMC Neurosci., 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tagliafierro L., Ilich E., Moncalvo M., Gu J., Sriskanda A., Grenier C., Murphy S.K., Chiba-Falek O. and Kantor B. (2019) Lentiviral vector platform for the efficient delivery of epigenome-editing tools into human induced pluripotent stem cell-derived disease models. J. Vis. Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.