

Abstract
Loss of UBE3A expression, a gene regulated by genomic imprinting, causes Angelman syndrome (AS), a rare neurodevelopmental disorder. The UBE3A gene encodes an E3 ubiquitin ligase with three known protein isoforms in humans. Studies in mouse suggest that the human isoforms may have differences in localization and neuronal function. A recent case study reported mild AS phenotypes in individuals lacking one specific isoform. Here we have used CRISPR/Cas9 to generate isogenic human embryonic stem cells (hESCs) that lack the individual protein isoforms. We demonstrate that isoform 1 accounts for the majority of UBE3A protein in hESCs and neurons. We also show that UBE3A predominantly localizes to the cytoplasm in both wild type and isoform-null cells. Finally, we show that neurons lacking isoform 1 display a less severe electrophysiological AS phenotype.
Introduction
Angelman syndrome (AS) is a rare neurodevelopmental disorder that affects 1 in 15 000 individuals and is characterized by severe seizures, intellectual disability, absent speech, ataxia and happy affect (1). AS is caused by loss of function from the maternally inherited copy of UBE3A. UBE3A is regulated by tissue-specific genomic imprinting—it is expressed exclusively from the maternal allele in neurons and is expressed biallelically in other cell types (2,3). UBE3A encodes an E3 ubiquitin ligase that forms polyubiquitin chains to substrates, targeting them for degradation by the 26S proteasome (4). In humans, there are three known UBE3A protein isoforms (5), all of which include the Homologous to E6AP Carboxy Terminus (HECT) domain and are thus capable of functioning as an E3 ligase. Human isoforms 2 and 3 only differ from human isoform 1 by 23 and 20 amino acids, respectively, at their N termini (6) (Fig. 1A). The mouse only has two protein isoforms of UBE3A that retain ubiquitin ligase activity, with the shorter of these two (termed isoform 3 in mouse) being equivalent to human isoform 1 and the longer (termed isoform 2 in mouse) being most similar to human isoform 3 (Fig. 1A). Mouse isoform 1 is predicted to lack ubiquitin ligase activity, and an analogous isoform does not exist in humans, while an analogous form of human isoform 2 does not exist in mouse (Fig. 1A). While there has been little published research examining the human protein isoforms in human cells, human isoforms ectopically expressed in mouse indicate that the isoforms likely have differences in localization and function (7–9). Furthermore, it is currently unknown whether the different human isoforms have different abundances or functions. This knowledge is important as some therapeutic avenues currently being explored for AS involve delivery and expression of exogenous UBE3A transgenes using vector-based delivery (10). For these approaches, knowledge of which of the protein isoforms need to be replaced in AS is essential.
Figure 1.
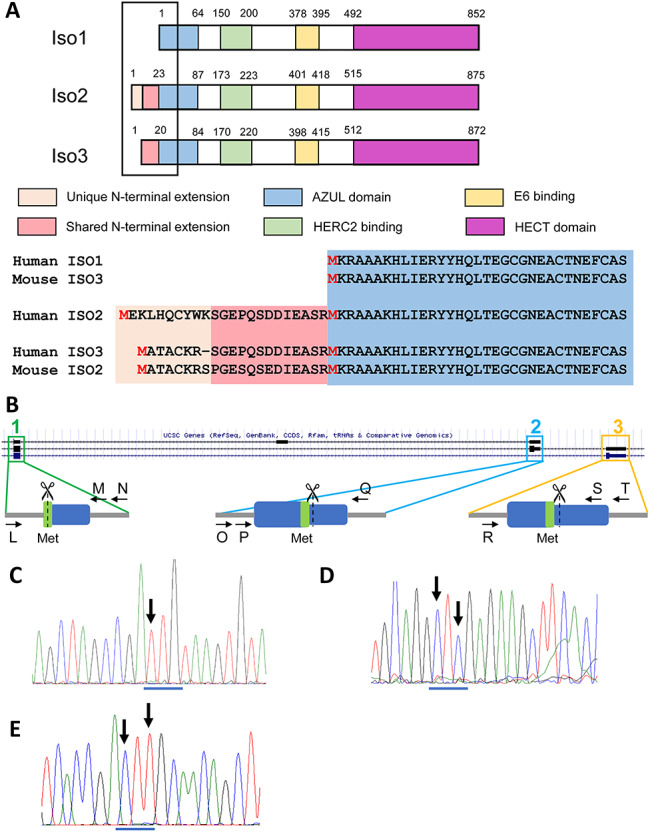
Generation of isogenic isoform-null hESC lines. (A) Schematic of the three UBE3A protein isoforms with color coding of known protein domains. The amino acid sequence of the N-terminus is shown for each isoform along with a comparison to the mouse protein isoforms. (B) Schematic illustrating isoform translational start sites and proposed genome editing. The UCSC Genome browser track depicting the UCSC gene annotation for a portion of UBE3A is shown. Arrows indicate primers used for screening. Scissors indicate CRISPR cut sites. Blue box = exon; grey line = intron; green = methionine used as start site. Numbers refer to relevant UBE3A protein isoform. (C-E) Sanger sequencing traces of genomic DNA showing homozygous mutations of the translational start site (ATG) for isoform 1 (ATG → TTG; c), isoform 2 (ATG → CTC; d) and isoform 3 (ATG → CTT; e). Arrows indicate locations of changed nucleotides. The original ATG start site is underlined in blue. The edited nucleotides changed the original methionine to a leucine.
Recently, Sadhwani (11) published three cases of AS in two different families caused by missense mutations at the isoform 1 translational start site. Interestingly, these patients present with milder phenotypes than AS patients with 15q11-q13 deletions or other UBE3A loss of function mutations, including normal gait and use of syntactic speech. These data, coupled with the fact that the human and mouse isoforms are not entirely conserved, illustrate the need to study the human UBE3A protein isoforms specifically, as they may play a role in both normal neuronal function and in disease. Here, we have used human embryonic stem cells (hESCs) and their neuronal derivatives to examine the abundance and localization of the three human isoforms. We have also examined whether neurons lacking individual protein isoforms recapitulate any AS phenotypes are previously seen in induced pluripotent stem cell (iPSC)-derived neurons. We show that in both hESCs and hESC-derived neurons, all three UBE3A protein isoforms are predominantly localized to the cytoplasm, with low levels of expression in other cellular compartments. We also show that protein isoform 1 is the predominant protein isoform in human cells. Finally, we show that neurons lacking isoform 1 show some, but not all, of the phenotypes displayed by AS iPSC-derived neurons, while loss of isoform 2 or 3 does not produce any phenotypic changes. These results not only provide useful insight into the human UBE3A isoforms but also provide information important for the development of potential AS therapies.
Results
Generation of isogenic hESC lines lacking individual protein isoforms
To study the abundance and localization of the UBE3A protein isoforms, we first generated isogenic hESC lines lacking the individual protein isoforms. Isogenic hESCs and neurons would allow us to minimize molecular and phenotypic differences caused by normal human genetic variation. All three human protein isoforms are full-length versions of UBE3A: isoforms 2 and 3 have an additional 23 and 20 amino acids, respectively, at their N terminus (12). Because of this, the RNAs encoding the isoforms are predicted to be nearly identical aside from the exons that encode for these additional amino acids (6). The three UBE3A protein isoforms are, however, translated from unique translational start sites (13). Using CRISPR/Cas9 and single-stranded olignonucleotide (ssODN) templates, we generated homozygous mutations for each of the three translational start sites, changing them from methionines to leucines, to prevent the translation of the isoform of interest (Fig. 1). Because the methionine that serves as the isoform 1 translational start site is present in the other two protein isoforms, we used multiple protein prediction softwares (14,15) to find an amino acid substitute (leucine) that was predicted to have a benign effect on the other two isoforms (Supplementary Material, Fig. S1). Indeed, all three isoform-null hESC lines made normal levels of UBE3A mRNA (Supplementary Material, Fig. S2a).
Isoform 1 is the most abundant UBE3A protein isoform
To determine the relative abundance of the human UBE3A protein isoforms in hESCs and hESC-derived neurons, we examined total UBE3A protein levels in whole-cell lysates prepared from the isoform knockout and isogenic control lines. Our data indicate that isoform 1 is the predominant isoform in both cell types—loss of this isoform produced a significant reduction (84–88%) in total UBE3A levels in whole-cell lysates prepared from both hESCs (Fig. 2A,B) and neurons (Fig. 3A,B). Loss of isoform 2 or isoform 3, however, did not produce any significant changes in total UBE3A levels. These data indicate that isoform 1 is the most abundant of the three human protein isoforms in hESCs and hESC-derived neurons.
Figure 2.
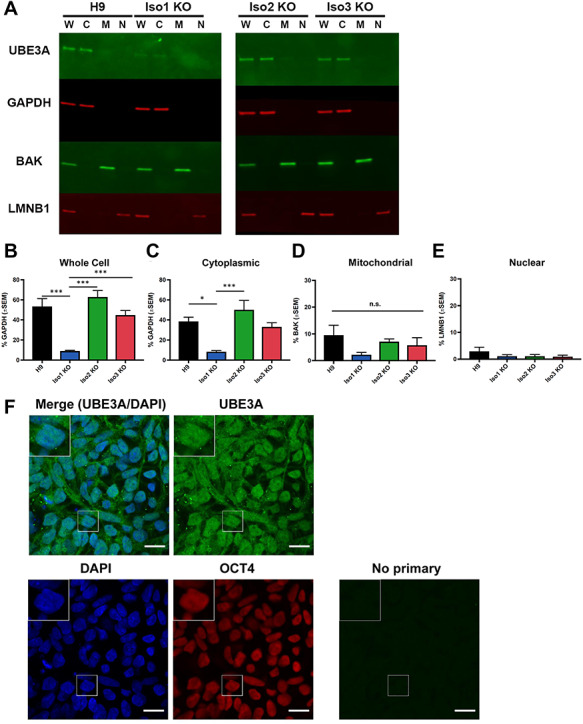
Abundance and localization of UBE3A isoforms in hESCs. (A) Western blot showing UBE3A levels in subcellular fractions from H9 and isoform-null hESC lines. W = whole-cell lysate, C = cytoplasmic fraction, M = mitochondrial fraction, N = nuclear fraction. (B–E) Quantification of western blots for each fraction. Graphs show the average percentage of appropriate loading control (n = 4 fractionations). Error bars: standard error of the mean. *P < 0.05; ***P < 0.005 (univariate ANOVA) (F) immunocytochemistry for UBE3A in H9 hESCS shows that UBE3A is cytoplasmic, in agreement with above fractionation results. Scale bar 20 μm.
Figure 3.
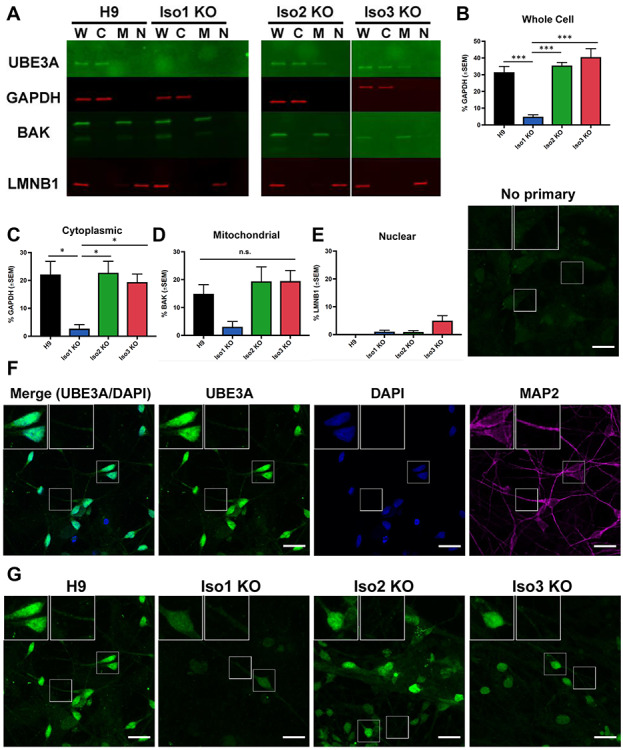
Abundance and localization of UBE3A isoforms in neurons. (A) Western blot showing UBE3A levels in subcellular fractions from H9 and isoform-null neurons. W = whole-cell lysate, C = cytoplasmic fraction, M = mitochondrial fraction, N = nuclear fraction. (B-E) Quantification of western blots for each fraction. Graphs show the average percentage of appropriate loading control (n = 3 fractionations). Error bars: standard error of the mean. *P < 0.05; ***P < 0.005 (univariate ANOVA) (F–G) immunocytochemistry for UBE3A in H9 neurons shows that UBE3A is highly concentrated in the nucleus but also is expressed in the cytoplasm. (F) Single plane image, (G) maximum projections of confocal z-stacks. Scale bar 20 μm.
Subcellular localization of the three UBE3A isoforms
Because studies of the mouse protein isoforms indicate differences in isoform localization (7,9), we sought to determine whether the human protein isoforms are also localized differentially. We examined UBE3A abundance in cytoplasmic, mitochondrial and nuclear fractions in both hESCs and neurons. In both cell types, the bulk of UBE3A protein is found in the cytoplasm, both in normal and isoform-null lines (Figs 2A–E and 3A–E). Consistent with our whole-cell results, loss of isoform 1 produced a significant reduction of UBE3A from the cytoplasmic fraction (Figs 2C and 3C), but not the mitochondrial or nuclear fractions (Figs 2D,E and 3D,E). Loss of isoform 2 or isoform 3 did not cause a significant change in the abundance of UBE3A protein in any cellular fractions when compared to the H9 parent line (Figs 2A–E and 3A–E). Since UBE3A in the nuclear fractions was difficult to see, we ran a western blot using a larger concentration of lysates from wildtype and isoform-null hESCs and neurons that were intentionally exposed to the point of saturation (3 min; Supplementary Material, Fig. S5b). This showed a faint signal, confirming the presence of UBE3A protein in the nucleus.
We confirmed the fractionation results using wildtype hESCs and neurons along with a second primary antibody against UBE3A, which reacts to a different portion of the protein (N terminus versus HECT domain; Supplementary Material, Fig. S5a). A third primary antibody (the same antibody used for immunocytochemistry, see below) similarly showed an almost undetectable nuclear band in both wildtype and Isoform1-null hESCs (Supplementary Material, Fig. S5c) as well as in wildtype neurons (Supplementary Figure 5d). Specific anti-UBE3A antibodies used for each figure are described in Supplementary Material, Table S4. Fractionation was also carried out on hESCs harboring a homozygous insertion of a hemagglutinin (HA)-tag into the isoform 1 translational start site of UBE3A. Western blot performed using an anti-HA antibody further confirmed our previous observations (Supplementary Material, Fig. S7b).
We then examined UBE3A localization via an orthologous method: immunocytochemistry. We first stained hESCs for UBE3A and OCT3/4, a transcription factor and pluripotency marker that localizes to the nucleus (Fig. 2F and Supplementary Material, Fig. S3). We included an isogenic UBE3A KO hESC line to serve as a negative control (Supplementary Material, Fig. S2c-f). Expression of UBE3A in hESCs appears to be distributed across the cytoplasm and nucleus in both normal and isoform-null hESCs, as there is the expression of UBE3A inside and outside of the area positive for OCT3/4 staining. We next stained hESC-derived neurons for UBE3A and MAP2, a microtubule protein that localizes to the cytoplasm and is a marker for postmitotic neurons (Fig. 3F and Supplementary Material, Fig. S4a). Although the strongest UBE3A signal appears in the nucleus, there is a clear UBE3A signal in the neuronal processes and in the area of the soma outside of the nucleus (Supplementary Material, Fig. S4a,b). This is especially apparent in maximum intensity projections of confocal z-stacks from normal and isoform-null neurons (Fig. 3G).
Immunocytochemistry was repeated in neurons derived from wildtype and isoform-null hESCs using a different primary antibody against UBE3A (Supplementary Material, Table S4). These results (Supplementary Figure S6a) confirmed our previous observations: while there was a strong UBE3A signal in the nucleus, UBE3A can also be seen outside of the nucleus in both the soma and processes. Wildtype neurons colabeled with antibodies against UBE3A and NeuN, which is expressed predominantly in the nucleus in mature neurons, showed intense nuclear UBE3A signal, as evidenced by colocalization with NeuN, but also revealed UBE3A signal in the soma and neuronal processes (Supplementary Material, Fig. S6b). Immunocytochemistry performed using anti-HA antibodies in neurons derived from UBE3A-HA-tagged hESCs similarly labeled the nucleus, soma and neuronal processes (Supplementary Material, Fig. S7c).
UBE3A is diffusely localized throughout nuclei and cytoplasm in stem cells and neurons
To make sense of seemingly disparate UBE3A localization results, we examined UBE3A localization in neurons by transmission electron microscopy (TEM). These results showed diffuse UBE3A puncta in the soma both within and outside of nucleus, as well as in the neuronal processes (Supplementary Material, Fig. S8).
Isoform 1-null neurons display mild AS electrical phenotypes
Previously, we established that AS iPSC-derived neurons exhibit a phenotype of impaired physiological maturation, as indicated by a more depolarized resting membrane potential (RMP), immature patterns of action potential (AP) firing, and decreased frequency of spontaneous excitatory synaptic activity (16). We last wanted to determine whether human neurons lacking the individual UBE3A protein isoforms displayed any of the same AS phenotypes. Based on our results examining the abundance of the isoforms (Fig. 3A,B), and our knowledge of the AS patients with isoform 1 translational start site mutations (11), we hypothesized that loss of isoform 1 would produce AS phenotypes in hESC-derived neurons while the loss of the other two isoforms would likely would not produce any phenotype. We examined these three phenotypes in our isoform-null and isogenic control neurons at 12 weeks of differentiation. Isoform 1 KO neurons displayed a more depolarized RMP compared to controls, while there were no differences in the isoform 2 KO or isoform 3 KO neurons (Fig. 4A). Interestingly, there were no statistically significant differences in AP firing (Fig. 4B) or synaptic activity (Fig. 4C) in the any of the isoform-null neurons, indicating that the isoform 1 KO neurons only display limited AS electrical phenotypes.
Figure 4.
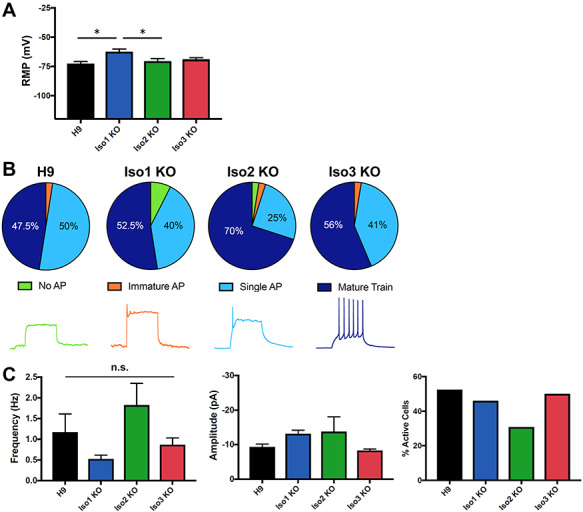
Isoform 1-null neurons partially recapitulate AS cellular phenotypes. (A) Isoform-1 null neurons have a more depolarized RMP. *P < 0.05 (univariate ANOVA) (B) no significant differences in AP firing upon loss of any UBE3A isoforms, Top: AP characterization in isoform-null neurons; Bottom: sample AP traces (C) isoform-null neurons do not have any significant differences in spontaneous excitatory synaptic activity (left, middle). n = 39 (Iso3 KO) or 40 (all other lines) cells on 3 coverslips per cell line; only active neurons were included in these analyses (right). All error bars: standard error of the mean.
Discussion
The human UBE3A gene encodes an E3 ubiquitin ligase that has three known protein isoforms (4,5). Surprisingly little is known about the human isoforms; however, studies in which human isoforms were ectopically expressed in mouse cells indicate that the isoforms may have differences in localization and function (7,8). Here we have used CRISPR/Cas9 to generate hESCs lacking these individual isoforms and studied their abundance and localization in both hESCs and neurons. We have also examined the phenotype of neurons lacking the individual isoforms. We have shown that human isoform 1 is the most abundant of the three isoforms, accounting for almost 90% of total UBE3A protein in these two cell types. We have also demonstrated that in both hESCs and neurons, the majority of UBE3A protein can be found in the cytoplasm, independent of which isoform is absent or present, although UBE3A is present in the nuclear and mitochondrial fractions, as well. Finally, we have shown that neurons lacking isoform 1, which accounts for ~84–88% of total UBE3A in both neurons and stem cells, display limited in vitro AS electrical phenotypes.
To examine the localization of the UBE3A isoforms, we used two approaches: subcellular fractionation and immunocytochemistry. Both approaches showed that isoform 1 is the predominant protein isoform in both hESCs and neurons. Loss of individual isoforms did not change the abundance or localization of remaining UBE3A RNA or protein, suggesting that each protein isoform may be translated from independent RNAs. Subcellular fractionation experiments revealed that the bulk of total UBE3A protein is found in the cytoplasmic portions of fractionated lysates and revealed that UBE3A is also located in the nuclear and mitochondrial fractions (which may also include some endoplasmic reticulum). Interestingly, immunocytochemistry experiments seemingly show that UBE3A is overwhelmingly localized to the nucleus of normal and isoform-null neurons. Rigorous experiments to confirm both fractionation and immunocytochemistry experiments with additional antibodies confirmed these findings. The distribution of UBE3A throughout normal neurons by the TEM suggests that the protein may be distributed somewhat evenly across all subcellular compartments. These seemingly disparate results may be explained by the relative proportion of cytoplasm versus nucleus in neurons. In most neurons, the nucleus occupies less than a tenth of the total cell volume (17). We would then expect that an evenly distributed protein would be 10-fold more enriched in the cytoplasm versus the nucleus of neurons. Based on quantification of UBE3A and GAPDH protein levels in fractionated H9 neurons, we estimate that ~70% of UBE3A protein is found in the cytoplasm, leaving the remaining 30% to be localized to the nucleus and mitochondria, which accounts for ~ 10% of the cellular volume and may explain the apparent enrichment in the nucleus. Together, our data strongly suggest that in human neurons, UBE3A protein—including isoform 1—is found in all cellular compartments assayed.
Murine UBE3A has been shown to become increasingly nuclear during neuronal maturation as determined by immunocytochemistry (9,18). Furthermore, a recent publication reports that the short UBE3A isoform, which is most similar to human isoform 1, is predominantly nuclear (9). However, these studies only used immunocytochemistry to assess localization, and in many cases involved over-expressing UBE3A isoforms in wildtype mouse neurons, and thus may not accurately reflect the localization of endogenous isoforms. Fractionation data from our isoform 1 knockout neurons show loss of cytoplasmic UBE3A, strongly suggesting that human isoform 1 is localized to both the nucleus and cytoplasmic compartments (Fig. 3C). We cannot rule out the possibility that our hESC-derived neurons are too developmentally immature to display these changes in localization. However, a recent publication reporting the UBE3A distribution in human postmortem brain sample shows both nuclear and cytoplasmic localization of UBE3A (19). Moreover, our isoform 1 null hESC-derived neurons appear to have UBE3A staining in the nucleus (Fig. 3G and Supplementary Figures 4 and 6), suggesting that the remaining two longer UBE3A isoforms are also localized in both cytoplasm and nucleus.
Isoform 1-null neurons lack ~84–88% of the total UBE3A protein and recapitulate some, but not all AS electrophysiological phenotypes. Isoform 1-null neurons have a more depolarized RMP compared to controls or the other two isoform-null neurons (Fig. 4A). It is possible that the difference in RMP between isoform 1-null neurons and their isogenic controls would be even more pronounced at an earlier age, such as 9 to 10 weeks in vitro. Although isoform 1-null neurons have a more depolarized RMP, nearly all cells fired single mature or mature trains of APs (Fig. 4B). It is possible that a more subtly delayed development of AP firing would be seen if we assayed this over a developmental time course in isoform 1-null neurons. It is interesting that we observe a milder physiological phenotype in neurons lacking isoform 1. This is consistent with the milder AS phenotype seen in patients with an analogous mutation in UBE3A (11), and suggests that isoforms 2 and 3 are contributing important UBE3A function in neurons, despite their low abundance relative to isoform 1. It is not clear whether ~ 12–16% of normal UBE3A protein levels are sufficient to improve physiology, or whether isoforms 2 and/or 3 have increased or specialized activity that contributes to neuronal physiology. Future studies are needed to examine the effects of the combined loss of two isoforms at a time to further elucidate their role in the electrical maturation of normal and AS neurons, as well as examine these phenotypes at various stages of neuronal development in our isoform-null neurons.
We have also shown here that isoform 1 accounts for the majority of UBE3A protein in hESCs and neurons, while isoforms 2 and 3 appear to account for very little protein at this stage. This is consistent with the relative abundances of long versus short isoforms of UBE3A in the mouse (9). This knowledge of the relative abundance of the isoforms is important for the development of AS therapies. One promising therapeutic avenue currently being explored for AS is the introduction of a UBE3A transgene through vector-based therapies (10), which typically can only contain one cDNA at a time. Replacement of all three human isoforms would likely require the development of three separate vectors, and is not feasible for current therapeutic approaches. Based on the results shown here and in recent publications (9,11), it is possible that delivery of isoform 1 alone may provide substantial therapeutic benefit, as it accounts for the majority of total UBE3A protein in human neurons in vitro. However, it is not known whether isoforms 2 and 3 have a specialized function not yet described, and it is possible that the different UBE3A isoforms have different ubiquitin ligase activities (i.e. more versus less active) or bind different cofactors. Future studies examining whether the introduction of individual UBE3A protein isoforms into human AS iPSC-derived neurons is capable of restoring their phenotypes are needed to determine whether all isoforms are functionally interchangeable.
Importantly, our results here replicate several major findings from recent UBE3A isoform studies in mouse (9) and most recent human isoform studies from the Elgersma/Distel lab (see paper in current issue). We found that human isoform 1 (equivalent to mouse isoform 3) accounts for ~85% of total UBE3A levels, similar to observations in the mouse (9). We further show that the loss of this isoform in human neurons is also able to recapitulate some AS physiological phenotypes (9). Finally, we also observe that UBE3A appears to be localized to the nucleus via ICC. However, there is a major discrepancy between our studies. We found the majority of UBE3A was localized in the cytoplasm when neurons were fractionated and protein was quantified by western blot. While it is possible that UBE3A is nucleosolic (unbound to chromatin or the nuclear membrane) and ‘leaks’ into the cytosolic fraction when neurons are fractionated, we think this is somewhat unlikely since the first step of fractionation, lysis of cells with digitonin, has been previously shown to leave nuclei intact and allow for later fractionation of the nucleosolic compartment (20). Furthermore, leaking of a nucleosolic protein would be more likely to result in even distribution of a protein to the nucleus and cytosol fractions, rather than a concentration in the cytosol. We note that our studies differed from those in the Elgersma lab by our approach. We examined the levels of endogenous UBE3A protein following mutation of the native UBE3A translational start sites, while the studies in the Elgersma/Distel lab examined isoform localization and abundance following overexpression of individual isoforms via plasmid transfection.
Regardless of whether UBE3A is predominantly nuclear or cytoplasmic, there are still important remaining questions about the human UBE3A isoforms that remain to be answered. It is still not known whether the two longer isoforms (human isoforms 2 and 3) are functionally equivalent to isoform 1, and whether these two isoforms have the same ubiquitin ligase activity level. This will be an important consideration when trying to determine how much total UBE3A is sufficient for neurotypical development, as the amount of necessary total UBE3A may differ depending on the isoform. Future studies should endeavor to answer these questions.
In summary, we have determined the relative abundance and localization of the three protein isoforms of UBE3A in human stem cells and neurons by genetic manipulation of the endogenous isoform translation start sites. We have shown that isoform 1 is the most abundant, and that UBE3A protein localizes to both the cytoplasm and nucleus in both cell types, independent of the specific isoform. We have also demonstrated that neurons lacking isoform 1 recapitulate some, but not all, AS physiological phenotypes. This knowledge is important for understanding the pathophysiology of AS cellular mechanisms and for the development of gene therapies to treat AS.
Materials and Methods
hESC culture and neural differentiation
All reagents for hESC culture and neuronal differentiation are Gibco products (ThermoFisher Scientific, Waltham, MA, USA) unless otherwise specified. hESCs were cultured on irradiated mouse embryonic fibroblasts and fed daily with hESC media [DMEM/F12 containing knockout serum replacement, L-glutamine + β-mercaptoethanol, non-essential amino acids and basic fibroblast growth factor (Millipore-Sigma, Billerica, MA, USA)]. hESCs were cultured at 37°C in a humid incubator at 5% CO2. Cells were manually passaged every 5–7 days. hESCs were differentiated into neurons using a modified version of the monolayer protocol. Neural induction was begun 2 days after passaging by culturing cells in N2B27 medium (Neurobasal medium, 1% N2, 2% B27, 2 mM L-glutamine, 0.5% penicillin/streptomycin, 1% insulin-transferrin-selenium). N2B27 medium was supplemented with fresh Noggin (500 ng/ml; R&D systems, Minneapolis, MN, USA) for the first 10 days of differentiation. Neural rosettes were manually passaged onto poly-D-lysine and laminin-coated plates using the Stem Pro EZPassage Tool (ThermoFisher Scientific) ~14 days after beginning neural induction. Neural progenitors were replated at a high density around 3 weeks of differentiation, switched to neural differentiation medium (NDM) around 4 weeks of differentiation, then plated sparsely for terminal differentiation at around 5 weeks. NDM consisted of neurobasal medium, 1% B27, 2 mM L-glutamine, 0.5% pen-strep, non-essential amino acids, 1 μM ascorbic acid, 200 μM cyclic AMP, 10 ng/ml brain-derived neurotrophic factor (Peprotech, Rocky Hill, NJ, USA), and 10 ng/ml glial-derived neurotrophic factor (Peprotech). Neurons were maintained in culture until 10–12 weeks of differentiation, with the exception of the neurons in Figure 3F,G and Supplementary Material, Figure S4, which were maintained until 15 weeks. The protocols for hESC maintenance and neuronal differentiation have been described previously (21,22).
CRISPR/Cas9-mediated genome editing
All CRISPRs were designed using MIT’s CRISPR design tool (http://crispr.mit.edu). sgRNAs were cloned into the pX459v2.0 vector (Addgene 62 988), as previously described (23,24), with one exception: the 3′ sgRNA used to generate the UBE3A KO line was designed by the hESC/iPSC Targeting Core at the University of Connecticut Health Center and was cloned into the pX330 vector. To generate the isoform-null hESCs, a single sgRNA was used to generate a double-stranded break, and a ssODN template harboring the desired edit was provided as a homology-directed repair template. For isoform 1 null hESCs, a single nucleotide was edited, in which the A nucleotide of the ATG codon was edited to a T. Note that for this isoform start site, AT is at the 3′ end of an exon, and the G for the starting methionine is on the next exon. For isoform 2 null hESCs, two nucleotides were altered to change ATG to CTC. For isoform 3 null hESCs, two nucleotides were altered to change ATG to CTT. All sgRNA sequences used in this work are included in Supplementary Material, Table S1. All ssODN sequences used to generate isoform KO lines and UBE3A HA-tagged lines are included in Supplementary Material, Table S2.
Prior to electroporation or nucleofection, hESCs were treated with ROCK inhibitor (Y-27632; Selleck Chemicals, Houston, TX, USA) for 24 h. hESCs were then singlized using Accutase (Millipore-Sigma) and electroporated using the Gene Pulser X Cell (Bio-Rad Laboratories, Hercules, CA, USA) or nucleofected using the Amaxa 4D Nucleofector (Lonza, Basal, Switzerland). For the generation of the Isoform1 and UBE3A KO lines, hESCs were electroporated in PBS with 10 μg of CRISPR plasmid and, for the Isoform1 KO line, 8 μL of single-stranded oligonucleotide (ssODN) template (100 μM). For the generation of the Isoform2 KO and Isoform3 KO lines, and the HA-tagged UBE3A lines, hESCs were nucleofected with 2 μg of CRISPR and 3 μL ssODN (100 μM) using the P3 Primary Cell Kit L (Lonza). hESCs were then plated onto puromycin-resistant (DR4) irrMEFs at low density, supplemented with ROCK inhibitor and L755507 (5 μM, Xcessbio, Chicago, IL, USA), which has been shown to improve efficiency of homology-directed repair (25). Twenty four hours after plating, cells underwent selection for 48 h with puromycin (0.5–1 μg/ml). Puromycin resistant colonies were screened by conventional PCR (UBE3A KO and UBE3A HA-tagged lines) or conventional PCR followed by restriction digest (Isoform KO lines) 11–14 days after plating. Putative clones were plated onto regular irrMEFs and successful genome editing was confirmed by Sanger sequencing. Correctly edited, homozygous clones were obtained following a single round of genome editing in each case. Primer sequences used to generate the hESC lines in this paper are listed in Supplementary Material, Table S3. All oligonucleotides were ordered from IDT (Newark, NJ, USA). A list of gene-edited cell lines generated for this paper can be found in Supplementary Material, Table S5.
Subcellular fractionation
Subcellular fractionation was performed using the Cell Fractionation Kit—Standard (Abcam, Cambridge, UK) according to the manufacturer’s instructions with the following modifications: 1) Protease Inhibitor Cocktail III (Millipore-Sigma) was added to 1× Buffer A at the beginning of the fractionation protocol at a 1:1000 dilution; 2) a whole-cell lysate was collected following the first lysis step in Buffer B by collecting 1/6 of the volume of lysate, and downstream volumes used in the protocol were adjusted accordingly; and 3) whole-cell and nuclear fractions were sonicated at the end of the protocol to sheer DNA using the following settings: [3 s on, 3 s off] × 2 at 30% amplitude. All fractions were stored at −80°C until use.
Western blot
Equal volumes of lysate from each fraction were separated by SDS-PAGE using 4–20% TGX Stain-Free mini gels (Bio-Rad). Protein was transferred to PVDF membrane using the TransBlot Turbo system (Bio-Rad). Membranes were blocked in TBS Odyssey Blocking Buffer (LI-COR, Inc., Lincoln, NE, USA) for 1 h at room temperature then incubated in a blocking buffer containing primary antibodies overnight at 4°C. Membranes were washed with TBS-T (Tris-buffered saline plus 0.1% Tween-20) at room temperature, incubated in a blocking buffer containing IRDye Secondary Antibodies (LI-COR, Inc.) for 1 h at room temperature, then washed again in TBS-T. All washes were done three times for 10–12 min each at room temperature. Membranes were imaged using the Odyssey imaging machine and software (LI-COR, Inc.). Images were quantified using Image Studio Lite (LI-COR, Inc.). The following primary antibodies were used: rabbit anti-UBE3A (1:3000; Bethyl Laboratories, Montomery, TX, USA; A300-351), rabbit anti-UBE3A (1:3000; Bethyl Laboratories A300–352), mouse anti-UBE3A (1:1000; Millipore-Sigma E8655), rabbit anti-LMNB1 (1:5000; Abcam), mouse anti-GAPDH (1:10000; Millipore-Sigma); rabbit anti-BAK (1:1000; Cell Signaling Technology, Danvers, MA, USA). The following secondary antibodies were used at a concentration of 1:10000: IRDye 800 CW Donkey anti-Rabbit, IRDye 680RD Goat anti-Rabbit and IRDye 680RD Goat anti-Mouse (LI-COR, Inc.). For Supplementary Material, Figure S5b, a western blot containing only nuclear fractions from all four hESC and neuron lines was performed with the following changes from above: blocking and antibody incubations were done in 5% BSA, anti-rabbit HRP-conjugated secondary antibody (1:3000; Cell Signaling Technology) was used, and the blot was imaged using Clarity Western ECL substrate (Bio-Rad) on the ChemiDoc Touch imaging system (Bio-Rad).
Immunocytochemistry
hESCs and neurons were grown on glass chamber slides (ThermoFisher Scientific) for immunocytochemistry. Cells were fixed using room temperature 4% paraformaldehyde for 10 min then permeabilized using 0.5% PBS-Triton X 100 (PBS-T) for 10 min at room temperature. Cells were blocked in 0.1% PBS-T containing 2% bovine serum albumin and 5% normal goat serum. Cells were incubated in primary antibody in blocking buffer overnight at room temperature then washed with PBS. Cells were then incubated in secondary antibody in blocking buffer for 3 h at room temperature then washed with PBS. Cells were mounted with ProLong Gold Anti-Fade Hard Set with DAPI (ThermoFisher) and allowed to set overnight at room temperature before imaging. The following primary antibodies were used: mouse anti-UBE3A (1:500; Millipore-Sigma SAB1404508 clone 3E5), mouse anti-UBE3A (1:500; Millipore-Sigma E8655 clone E6AP-330), rabbit anti-OCT4 (1:200; Stemgent, Cambridge, MA, USA; 09–0023), chicken anti-MAP2 (1:10000; Abcam ab3592), rabbit anti-NeuN (1:300; Abcam ab177487). The following secondary antibodies were used: anti-mouse Alexa Fluor 488 (1:400; ThermoFisher), anti-rabbit Alexa Fluor 594 (1:400; ThermoFisher), anti-chicken Alexa Fluor 647 (1:250, Abcam). Confocal images were acquired with a 63× oil immersion objective (NA 1.4) on a Zeiss 780 confocal system mounted on an inverted Axio Observer Z1 (Carl Zeiss, Oberkochen, Germany). Images were averaged twice, with pixel size of 0.26 μm, pixel dwell time of 1.27 s, and scan time of 2.34 s. Three randomly chosen areas of the chamber slide were imaged for each cell line. Z-series were done with a 0.2 μm interval and maximum intensity projection images were created using Fiji (ImageJ) software (26). Fiji was also used to quantify the integrated intensity per relative area for UBE3A in the soma, nuclei and neurites of single-plane images. Each quantified region was manually traced. Average background fluorescence was calculated by measuring three individual regions per image and the average subtracted from each measured region of interest, producing the corrected total cell fluorescence (CTCF; 26).
Electrophysiology
Neurons were plated onto glass coverslips around 5 weeks of differentiation. Whole-cell voltage and current-clamp recordings were performed at 12 weeks of differentiation as previously described (16).
Quantitative RT-PCR (qRT-PCR)
cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher) according to manufacturer’s instructions. Quantitative RT-PCR was performed using Taqman Gene Expression Assays and Mastermix (ThermoFisher) on the Step One Plus (ThermoFisher). Reactions were performed in technical duplicates, with GAPDH Endogenous Control Taqman Assay used as the housekeeping gene for normalization. Gene expression was quantified using the ΔΔCt method. The expression of UBE3A was detected using TaqMan assay Hs00963664_g1 (ThermoFisher). This assay spans exons 5 and 6 (Isoform 1), 9 and 10 (Isoform 2) and 8 and 9 (Isoform 3).
Immunogold TEM
Neurons were plated for terminal differentiation onto plastic 4 well plates at 6 weeks of differentiation. At 10 weeks, neurons were fixed using 4% paraformaldehyde at room temperature for 30 min. Cells were washed in PBS at room temperature, then blocked and permeabilized by incubating in 0.1% PBS-T containing 2% bovine serum albumin and 5% normal goat serum for 30 min at room temperature. Cells were incubated overnight at room temperature in blocking buffer containing primary antibody [mouse anti-UBE3A (Millipore-Sigma SAB1404508 clone 3E5)] at 1:250. Cells were washed at room temperature in PBS then incubated in secondary antibody for 1 h at room temperature. Secondary antibody (goat anti-mouse Nanogold Fab, Nanoprobes, Yaphank, NY, USA) was diluted 1:200 in 1% BSA. Cells were washed several times in PBS then washed in deionized water. Gold enhancement was performed for 4 min using GoldEnhance (Nanoprobes). Cells were rinsed in deionized water to stop enhancement reaction. To prepare cells for electron microscopy, cells were rinsed for 5 min in 0.1 M Cacodylate buffer, then postfixed in 1% OsO4, 0.8% Potassium Ferricyanide in 0.1 M Cacodylate buffer for 30 min at room temperature. Cells were then washed five times in deionized water for 5 min per wash, block stained in 1% Uranyl Acetate in deionized water for 30 min at room temperature, then washed again three times (5 min each) in deionized water. Next cells were dehydrated in ethanol (5 min in 50% EtOH, 5 min in 75% EtOH, 5 min in 95% EtOH, then three incubations in 100% EtOH for 5 min each), then infiltrated in 100% resin overnight at room temperature. The following day, fresh 100% resin was added and the cells were polymerized at 60°C for 48 h. Thin sections of 70–80 nm were cut on the Ultramicrotome Leica EM UC7 (Leica Microsystems, Buffalo Grove, IL, USA) and sections placed on Cu grids. Sections were counterstained with 6% Uranyl Acetate in 50% methanol for 4 min. Images were acquired with the Hitachi H-7650 Transmission Electron Microscope (Hitachi High-Technologies Corporation, Tokyo, Japan). Electron microscopy preparation and imaging was performed by the UConn Health Central Electron Microscopy Facility.
Statistics
Statistical analysis was performed used SPSS software (IBM, Armonk, NY, USA). Univariate ANOVA was performed to compare levels UBE3A protein levels in western blots (Figs 2B–E and 3B–E), to compare RMP (Fig. 4E) and spontaneous synaptic activity (Fig. 4C) between normal and isoform null cells, and to compare UBE3A fluorescence in confocal images (Supplementary Material, Figure 4Sb). Chi-square tests were performed to compare AP firing patterns (Fig. 4B) between normal and isoform null cells.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The authors would like to thank members of the Chamberlain and Levine labs for helpful discussions during the course of this project. This work was funded by NIH grants R21HD091823-02 and R01HD094953-01 and an AS Foundation grant to SJC, an AS Foundation fellowship to NDG, and a Connecticut DPH Regenerative Medicine Research Fund grant 14-SCDIS-UCHC-01 to SJC and ESL. Conflict of Interest Statement. None declared.
Contributor Information
Carissa L Sirois, Department of Genetics and Genome Sciences, University of Connecticut Health Center, Farmington, CT 06030, USA.
Judy E Bloom, Richard D. Berlin Center for Cell Analysis and Modeling, University of Connecticut Health Center, Farmington, CT 06030, USA; Department of Neuroscience, University of Connecticut Health Center, Farmington, CT 06030, USA.
James J Fink, Department of Neuroscience, University of Connecticut Health Center, Farmington, CT 06030, USA.
Dea Gorka, Department of Genetics and Genome Sciences, University of Connecticut Health Center, Farmington, CT 06030, USA.
Steffen Keller, Department of Biology, University of Konstanz, 78457 Konstanz, Germany.
Noelle D Germain, Department of Genetics and Genome Sciences, University of Connecticut Health Center, Farmington, CT 06030, USA.
Eric S Levine, Department of Neuroscience, University of Connecticut Health Center, Farmington, CT 06030, USA.
Stormy J Chamberlain, Department of Genetics and Genome Sciences, University of Connecticut Health Center, Farmington, CT 06030, USA; Institute for Systems Genomics, University of Connecticut, Storrs, CT 06269, USA.
Author Contributions
C.L.S. and S.J.C. conceived of and designed the study, collected and analyzed data, and wrote the manuscript. S.K. generated the UBE3A-HA tagged lines and performed characterization of said lines. J.E.B. performed all confocal microscopy except for Supplementary Material, Figure S7, which was performed by N.D.G. J.J.F. performed whole-cell electrical recordings and the data were analyzed by D.G. E.S.L. provided critical revision of the manuscript.
References
- 1. Dagli A.I. and Williams C.A. (1993) Angelman Syndrome In Pagon R.A. et al. (eds), GeneReviews(R). University of Washington, Seattle, NBK1144 [bookaccession]. [PubMed] [Google Scholar]
- 2. Rougeulle C., Glatt H. and Lalande M. (1997) The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet., 17, 14–15. [DOI] [PubMed] [Google Scholar]
- 3. Kishino T., Lalande M. and Wagstaff J. (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet., 15, 70–73. [DOI] [PubMed] [Google Scholar]
- 4. Scheffner M., Huibregtse J.M., Vierstra R.D. and Howley P.M. (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell, 75, 495–505. [DOI] [PubMed] [Google Scholar]
- 5. Yamamoto Y., Huibregtse J.M. and Howley P.M. (1997) The human E6-AP gene (UBE3A) encodes three potential protein isoforms generated by differential splicing. Genomics, 41, 263–266. [DOI] [PubMed] [Google Scholar]
- 6. Aken B.L., Achuthan P., Akanni W., Amode M.R., Bernsdorff F., Bhai J., Billis K., Carvalho-Silva D., Cummins C., Clapham P. et al. (2017) Ensembl 2017. Nucleic Acids Res., 45, D635–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miao S., Chen R., Ye J., Tan G.H., Li S., Zhang J., Jiang Y.H. and Xiong Z.Q. (2013) The Angelman syndrome protein Ube3a is required for polarized dendrite morphogenesis in pyramidal neurons. J. Neurosci., 33, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Valluy J., Bicker S., Aksoy-Aksel A., Lackinger M., Sumer S., Fiore R., Wüst T., Seffer D., Metge F., Dieterich C. et al. (2015) A coding-independent function of an alternative Ube3a transcript during neuronal development. Nat. Neurosci., 18, 666–673. [DOI] [PubMed] [Google Scholar]
- 9. Avagliano Trezza R., Sonzogni M., Bossuyt S.N.V., Zampeta F.I., Punt A.M., Berg M., Rotaru D.C., Koene L.M.C., Munshi S.T., Stedehouder J. et al. (2019) Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat. Neurosci., 22, 1235–1247. [DOI] [PubMed] [Google Scholar]
- 10. Daily J.L., Nash K., Jinwal U., Golde T., Rogers J., Peters M.M., Burdine R.D., Banko J.L. and Weeber E.J. (2011) Adeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS One, 6, e27221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sadhwani A., Sanjana N.E., Willen J.M., Calculator S.N., Black E.D., Bean L.J.H., Li H. and Tan W.H. (2018) Two Angelman families with unusually advanced neurodevelopment carry a start codon variant in the most highly expressed UBE3A isoform. Am. J. Med. Genet. A, 176, 1641–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. LaSalle J.M., Reiter L.T. and Chamberlain S.J. (2015) Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics, 7, 1213–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M. and Haussler D. (2002) The human genome browser at UCSC. Genome Res., 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yachdav G., Kloppmann E., Kajan L., Hecht M., Goldberg T., Hamp T., Hönigschmid P., Schafferhans A., Roos M., Bernhofer M. et al. (2014) PredictProtein—an open resource for online prediction of protein structural and functional features. Nucleic Acids Res., 42, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bendl J., Stourac J., Salanda O., Pavelka A., Wieben E.D., Zendulka J., Brezovsky J. and Damborsky J.P.S.N.P. (2014) Robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput. Biol., 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fink J.J., Robinson T.M., Germain N.D., Sirois C.L., Bolduc K.A., Ward A.J., Rigo F., Chamberlain S.J. and Levine E.S. (2017) Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat. Commun., 8, 15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kandel E., Schwartz J. and Jessel T. (2000) Principals of Neural Science. Health Professions Division, McGraw-Hill. [Google Scholar]
- 18. Judson M.C., Sosa-Pagan J.O., Del Cid W.A., Han J.E. and Philpot B.D. (2014) Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J. Comp. Neurol., 522, 1874–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burette A.C., Judson M.C., Li A.N., Chang E.F., Seeley W.W., Philpot B.D. and Weinburg R.J. (2018) Subcellular organization of UBE3A in human cerebral cortex. Mol. Autism., 9, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu X. and Fagotto F. (2011) A method to separate nuclear, cytosolic, and membrane-associated signaling molecules in cultured cells. Sci. Signal., 4, pl2. [DOI] [PubMed] [Google Scholar]
- 20. Chamberlain S.J., Chen P.F., Ng K.Y., Bourgois-Rocha F., Lemtiri-Chlieh F., Levine E.S. and Lalande M. (2010) Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl. Acad. Sci. USA, 107, 17668–17673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Germain N.D., Chen P.F., Plocik A.M., Glatt-Deeley H., Brown J., Fink J.J., Bolduc K.A., Robinson T.M., Levine E.S., Reiter L.T. et al. (2014) Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1. Mol. Autism, 5, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen P.-F., Hsiao J.S., Sirois C.L. and Chamberlain S.J. (2016) RBFOX1 and RBFOX2 are dispensable in iPSCs and iPSC-derived neurons and do not contribute to neural-specific paternal UBE3A silencing. Sci. Rep., 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Langouet M., Glatt-Deeley H.R., Chung M.S., Dupont-Thibert C.M., Mathieux E., Banda E.C., Stoddard C.E., Crandall L. and Lalande M. (2018) Zinc finger protein 274 regulates imprinted expression of transcripts in Prader-Willi syndrome neurons. Hum. Mol. Genet., 27, 505–515. [DOI] [PubMed] [Google Scholar]
- 24. Yu C., Liu Y., Ma T., Liu K., Xu S., Zhang Y., Liu H., La Russa M., Xie M., Ding S. et al. (2015) Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell, 16, 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCloy R.A., Rogers S., Caldon C.E., Lorca T., Castro A. and Burgess A. (2014) Partial inhibition of Cdk1 in G2 phase overrides the SAC and decouples mitotic events. Cell Cycle, 13, 1400–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.