

Abstract
Recent genome-wide association studies of age-at-onset in Huntington’s disease (HD) point to distinct modes of potential disease modification: altering the rate of somatic expansion of the HTT CAG repeat or altering the resulting CAG threshold length-triggered toxicity process. Here, we evaluated the mouse orthologs of two HD age-at-onset modifier genes, FAN1 and RRM2B, for an influence on somatic instability of the expanded CAG repeat in Htt CAG knock-in mice. Fan1 knock-out increased somatic expansion of Htt CAG repeats, in the juvenile- and the adult-onset HD ranges, whereas knock-out of Rrm2b did not greatly alter somatic Htt CAG repeat instability. Simultaneous knock-out of Mlh1, the ortholog of a third HD age-at-onset modifier gene (MLH1), which suppresses somatic expansion of the Htt knock-in CAG repeat, blocked the Fan1 knock-out-induced acceleration of somatic CAG expansion. This genetic interaction indicates that functional MLH1 is required for the CAG repeat destabilizing effect of FAN1 loss. Thus, in HD, it is uncertain whether the RRM2B modifier effect on timing of onset may be due to a DNA instability mechanism. In contrast, the FAN1 modifier effects reveal that functional FAN1 acts to suppress somatic CAG repeat expansion, likely in genetic interaction with other DNA instability modifiers whose combined effects can hasten or delay onset and other CAG repeat length-driven phenotypes.
Introduction
Huntington’s disease (HD) (OMIM #143100) is one of several inherited trinucleotide repeat disorders. An unstable expanded (>35) CAG repeat in HTT is the root genetic cause of a disease process that culminates in HD’s distinctive neurological, behavioral and cognitive signs (1). The strong inverse relationship between age-at-onset of motor signs and inherited HTT CAG repeat length pointed to CAG length as the primary determinant of the rate of HD pathogenesis leading to onset (2–6). However, modifier genes were also predicted to play a role due to the significant amount of heritable variation in age-at-onset not explained by individual CAG repeat size (6–8). Indeed, recent successive genome-wide association studies (GWAS) have identified multiple genes that influence the rate at which motor signs emerge (9–11). The first GWAS (~4000 individuals) identified FAN1 (FANCD2 and FANCI associated nuclease 1) on chromosome (Chr.)15, RRM2B (ribonucleotide reductase regulatory TP53 inducible subunit M2B) on Chr. 8 (9), and then, with additional subjects, the DNA repair gene MLH1 (MutL homolog 1), on Chr. 3 (10). Previously, the ortholog of MLH1 had been discovered in a linkage study to be a modifier of somatic Htt CAG repeat instability in HD knock-in mice (12) and this finding, together with the observation that HD individuals with the earliest onset had the highest degree of brain CAG repeat expansion (13), implicated somatic instability of the repeat in the rate of HD pathogenesis. However, it was unclear whether the driver of disease onset was the CAG repeat itself or the encoded polyglutamine tract in huntingtin, as assumed in the moniker ‘polyglutamine disease’ (14). The latest HD age-at-motor-onset GWAS (~9000 individuals) revealed HD individuals with rare polymorphisms that change the pair of CAACAG glutamine codons downstream of the glutamine-encoding pure CAG repeat. These variants permitted the demonstration that the timing of motor onset tracks better with length of the inherited pure CAG repeat than with the size of the encoded polyglutamine tract (11).
Providing further evidence for a property of the CAG repeat as the onset rate-driver, this larger GWAS identified additional loci with candidate modifier genes involved in DNA maintenance processes: PMS1 (post meiotic segregation increased 1 homolog; Chr. 2), MSH3 (MutS homolog 3; Chr. 5), PMS2 (post meiotic segregation increased 2 homolog; Chr. 7) and LIG1 (DNA Ligase 1; Chr. 19). Like MLH1, the orthologs of all except PMS1 have been reported previously to modulate somatic CAG repeat instability in mouse models (15–18). Indeed, the onset-hastening variant at MSH3 was associated with increased expression and with highest HTT CAG repeat expansion in HD blood DNA (11). Moreover, an haplotype associated with delayed onset, which is also tagged by a number of single nucleotide polymorphisms (SNPs) (e.g. rs1382539 (19) and rs1650742 (10)) that are in linkage disequilibrium with a repeat variant in MSH3 (20), has been associated with slower progression (20,21) and decreased blood cell CAG repeat instability (19,20). This haplotype was also associated with reduced expression of both MSH3 and its neighbor DHFR in cortex (10), so the actual source of the functional effect(s) is at present unclear, though could reflect the level of the protein, the sequence of the protein or both.
The recent GWAS also identified other modifier genes not known to be involved in DNA maintenance: CCDC82 (Chr. 11) and TCERG1 (Chr. 5). This apparent duality suggests distinct drivers of HD pathogenesis leading to the onset of motor signs: the timing/rate-driver, which is the rate at which the inherited CAG repeat length undergoes somatic increase in target cells, and a harm-inducing (toxicity) process that is triggered in target cells, above a certain CAG-length threshold, and culminates in disease (11), as proposed in a mathematical model of trinucleotide repeat disease onset (22).
Here we explore this two component hypothesis, by functional in vivo analysis of the murine homologs of HD modifier genes from the initial GWAS: Fan1 (FAN1), likely to be involved in somatic Htt CAG repeat instability in vivo from recent report of mild (3–8 repeat) HTT CAG expansion in cultured cells (23), and Rrm2b (RRM2B) involved in maintaining mitochondrial and cytosolic nucleotide pools (24) but not directly in DNA repair. Specifically, we evaluated their potential impact on instability of the inherited CAG repeat length by knocking-out Fan1 and Rrm2b in HD CAG repeat knock-in mice. For comparison, we also examined the impact of knocking-out the closely adjacent genes at each locus, Mtmr10, encoding a myotubularin family member involved in phosphatidylinositol phosphate metabolism (25) and Ubr5, specifying an E3 ubiquitin-protein ligase that targets specific proteins for ubiquitin-mediated proteolysis (26). Both have been discounted as the true HD modifiers by deeper GWAS analysis, eQTL analyses and variant analysis which found protein-altering changes, not in MTMR10 or UBR5, but only in FAN1 and RRM2B (11).
Results
We knocked-out Fan1, Mtmr10, Rrm2b and Ubr5, using CRISPR/Cas9, to generate lines of C57BL/6 J mice with targeted in/del mutations (Supplementary Material, Table S1; Fig. S1). Mtmr10 heterozygote and homozygote knock-out mice were fertile and without obvious phenotype. Fan1, Rrm2b and Ubr5 homozygote knock-out mice, but not heterozygote mice, exhibited previously reported phenotypes that confirmed loss of gene function. Matings of mice heterozygous for each mutant allele generated offspring at weaning (21 days) in the expected Mendelian genotype ratios, except for Ubr5, a known embryonic lethal (26), where homozygotes were not observed (Table 1). Fan1 homozygote knock-out mice showed karyomegaly in kidney (5 months of age) and embryonic fibroblast cultures exhibited increased sensitivity to mitomycin C (MMC), as reported (27–30) (Supplementary Material, Fig. S2). Rrm2b homozygote knock-out mice showed reported weight loss and pale kidney (2.5 months of age) (Supplementary Material, Fig. S3) and premature death at about 3 months of age (31).
Table 1.
Progeny testing for recovery of genotypes expected from intercross mating
| Line name intercross parents | Number breeding pairs | Total number litters | Total number progeny | Average number per litter | Number male progeny (%) | Number female progeny (%) | Progeny genotype | ||
|---|---|---|---|---|---|---|---|---|---|
| Number wild-type homozygote (%) | Number mutant heterozygote (%) | Number mutant homozygote (%) | |||||||
| Fan1_del10 X Fan1_del10 | 10 | 33 | 181 | 5.5 | 85 (47.0) | 96 (53.0) | 44 (24.3) | 98 (54.1) | 39 (21.5) |
| Fan1_ins1 X Fan1_ins1 | 4 | 9 | 49 | 5.4 | 26 (53.1) | 23 (46.9) | 10 (20.4) | 30 (61.2) | 9 (18.4) |
| Mtmr10_del5 X Mtmr10_del5 | 5 | 11 | 66 | 6.0 | 38 (57.6) | 28 (42.4) | 16 (24.2) | 35 (53.0) | 15 (22.7) |
| Rrm2b_del10 X Rrm2b_del10 | 6 | 21 | 111 | 5.3 | 63 (56.8) | 48 (43.2) | 23 (20.7) | 59 (53.2) | 29 (26.1) |
| Rrm2b_del24 X Rrm2b_del24 | 2 | 9 | 46 | 5.1 | 27 (58.7) | 19 (41.3) | 9 (19.6) | 24 (52.2) | 13 (28.3) |
| Ubr5_ins2 X Ubr5_ins2 | 2 | 2 | 9 | 4.5 | 5 (55.6) | 4 (44.4) | 0 (0) | 9 (100) | 0 (0) |
| Ubr5_del7 X Ubr5_del7 | 2 | 4 | 11 | 2.75 | 4 (36.4) | 7 (63.6) | 2 (18.2) | 9 (81.8) | 0 (0) |
Summary of breeding data revealed that mutant alleles at Fan1, Mtmr10 and Rrm2b were observed in approximately Mendelian ratios (25% homozygous wild-type, 50% heterozygous mutant, 25% homozygous mutant) in progeny of intercross mating genotyped at weaning, consistent with reported normal development for mice with other mutations inactivating Fan1 and Rrm2b. Mtmr10 null mice have not previously been reported but null mutations in other family members is also not lethal. Progeny homozygous for Ubr5 mutant alleles were not recovered, as this gene (previously Edd) is essential early in development. Mutant alleles were passed to males and females about equally, as expected.
To test whether inactivation of HD modifier gene homologs Fan1 and Rrm2b may alter CAG repeat instability compared with knock-out of the neighboring gene (Mtmr10 and Ubr5, respectively), we crossed mutant knock-out mice with Hdh CAG knock-in mice carrying expanded CAG repeats at the mouse Htt locus (32). The population of HdhQ111/+ CAG knock-in mice display a range of individual constitutional Htt CAG repeat lengths (from 105 to 135 in these experiments) and somatic instability of the CAG repeat leads over time to a gradual increase in the instability and expansion indices that is dramatic in striatum and liver (33,34). Inspection of the repeat lengths inherited by the progeny from these matings confirmed the reported frequent increases and decreases in CAG size in transmissions from males and females, respectively (32), and revealed little effect of heterozygous knock-out of Fan1, Mtmr10, Rrm2b or Ubr5 or by homozygous knock-out of Fan1 (Supplementary Material, Fig. S4). Somatic CAG repeat size variation in the progeny was examined at several ages using a previously established instability index assay, in which peaks from GeneMapper traces representing both shorter products and expanded alleles are taken into account (instability index) or, in some cases, for greater sensitivity, measuring only expansion peaks of sizes greater than the main allele (expansion index) (33,34).
Homozygous inactivation of Fan1 in HdhQ111/+ knock-in mice strikingly increased the levels of repeat expansion in striatum (P = 0.0003) and liver (P = 0.0003) assessed at 5 months of age (Fig. 1; example traces in Supplementary Material, Fig. S5), whereas homozygous inactivation of the adjacent gene Mtmr10 had no significant effect (Fig. 1; example traces in Supplementary Material, Fig. S6). Homozygous Ubr5 knock-out HdhQ111/+ mice were not recovered so could not be tested. Homozygous Rrm2b knock-out HdhQ111/+ mice are viable but die at ~3 months of age, so we assessed these at 2.5 months of age, in comparison with a smaller number of homozygous Fan1 knock-out HdhQ111/+ mice. The increase of instability index in the latter was detectable in the liver at 2.5 months (P = 0.01) (Fig. 2A; example traces in Fig. 2B). In contrast, the cohort of Rrm2b knockout HdhQ111/+ mice showed a slight, though statistically significant, decrease in the instability index in both the striatum (P < 0.0003) and the liver (P = 0.0011) (Fig. 2A; example traces in Fig. 2B). Assessment of somatic instability of the CAG repeat in heterozygous Fan1, Rrm2b, Mtmr10 and Ubr5 knock-out HdhQ111/+ mice at 5 months of age showed no effect of the latter two genes (Fig. 1; example traces in Supplementary Material, Fig. S6), but revealed an increase and decrease, respectively, in instability index for Fan1 (P = 0.0028 in striatum, P = 0.0002 in liver) and Rrm2b (P = 0.0198 in liver), albeit of lower magnitude than the effect of homozygous inactivation (Fig. 1; example traces, Supplementary Material, Figs S5 and S6).
Figure 1.
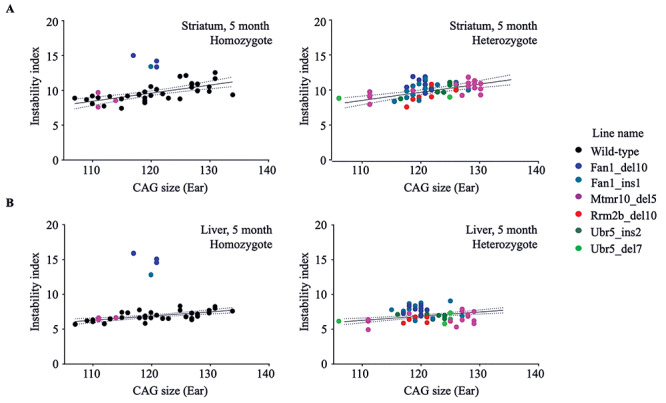
Fan1_del10 and Fan1_ins1 mutations enhance somatic instability of the HdhQ111/+ CAG repeat. The instability index (Y-axis), a quantitative measure of CAG repeat instability, calculated from ABI3730XL trace profiles (Methods) of PCR amplification products generated with DNA isolated from striatum (top panels) and liver (bottom panels) of genotyped progeny of matings between HdhQ111/+ and Fan1, Mtmr10, Rrm2b or Ubr5 mutant parental mice, is plotted against the size of the CAG repeat measured in the stable ear tissue of the same mouse at 3 weeks of age (X-axis). The instability index of mutant Fan1_del10 homozygote (n = 3), Fan1_ins1 homozygote (n = 1) and Mtmr10_del5 homozygote (n = 3) mice (left panel) and mutant Fan1_del10 heterozygote (n = 8), Fan1_ins1 heterozygote (n = 12), Mtmr10_del5 heterozygote (n = 16), Rrm2b_del10 heterozygote (n = 7), Ubr5_ins2 heterozygote (n = 7), Ubr5_del7 heterozygote (n = 5) mice (right panel) is shown relative to the instability index of wild-type littermates (n = 34). The regression trend line (solid line) and 95% confidence interval (dotted lines) illustrate the instability for a given inherited HdhQ111/+ CAG size in the absence of modifier gene mutation at 5 months of age.
Figure 2.
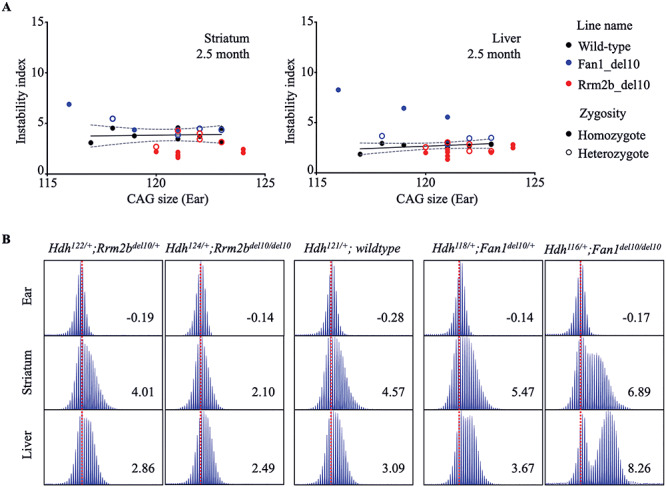
Contrasting effects of Fan1_del10 and Rrm2b_del10 mutation on somatic HdhQ111/+ CAG repeat instability at 2.5 months. (A) The plots show the relationship of instability index (Y-axis), determined at 2.5 months of age in striatum (left panel) and liver (right panel) of genotyped progeny of mating of HdhQ111/+ mice with Fan1_del10 or Rrm2b_del10 mutant parental mice, with the size of HdhQ111/+ CAG repeat measured at 3 weeks of age in the stable ear tissue of the same mouse (X-axis). The instability index of the Fan1_del10 heterozygote (n = 4) and Rrm2b_del10 heterozygote (n = 5) mice and Fan1_del10 homozygote (n = 3) and Rrm2b_del10 homozygote (n = 8) Fan1 and Rrm2b mutant mice are shown with instability index of their wild-type HdhQ111/+ littermates (n = 8). The regression trend line (solid line) and 95% confidence interval (dotted lines) illustrates the instability index for a given inherited HdhQ111/+ CAG size in the absence of modifier gene mutation at 2.5 months of age. (B) The ABI GeneMapper trace plots show the distribution of the PCR amplification products (peak height on the Y-axis and fragment size on the X-axis) generated with DNA isolated from liver and striatum of progeny of HdhQ111/+ and Rrm2b_del10 parents (left panels) or Fan1_del10 parents (right panels), compared with wild-type HdhQ111/+ parents (middle panels), with the calculated instability index value shown in each panel. Instability index is decreased for the Rrm2b_del10 heterozygote, and more so for the homozygote, compared with the wild-type HdhQ111/+ tissue but is enhanced in Fan1_del10 mutant tissue, especially in tissue from the Fan1_del10 homozygote mouse. The trace plots for each mouse are aligned relative to maximum peak in ear DNA (3 weeks of age) (dotted red line). The Hdh genotype for each mouse was based on the maximum peak CAG repeat in ear DNA.
The Fan1 findings were readily evident and consistent with increased somatic instability of the CAG repeat leading to longer mutant Htt alleles. A similar effect in humans would be expected to yield more rapid onset. However, our assessment utilized Htt CAG expansion alleles that correspond to the early juvenile onset CAG size range in humans, whereas the rate-driver hypothesis for HD onset would predict effects on instability even on the more modest inherited repeat sizes in the typical HD adult onset range. Therefore, we performed the same assessment, using expansion index as the instability metric, on HdhneoQ50/+ and HdhQ50/+ CAG knock-in mice with an adult-onset range CAG repeat (48 CAGs). By 8 months of age, homozygous and to a lesser degree heterozygous Fan1 mutant HdhneoQ50/+ mice exhibited increased CAG sizes in cortex, striatum and liver (Fig. 3), an expansion-enhancing effect also observed in HdhQ50/+ striatum and liver (Supplementary Material, Fig. S7).
Figure 3.
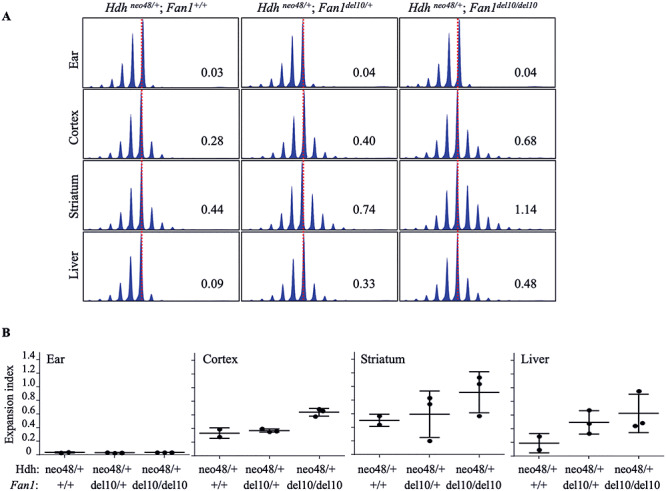
Fan1_del10 mutation enhanced somatic instability of an adult onset HD CAG repeat. (A) The ABI GeneMapper trace plots show age-related somatic instability of a 48 CAG repeat detected at 8 months of age, as evidenced by the broadened distribution of CAG repeat assay PCR amplification products (peak height on the Y-axis and fragment size on the X-axis) in liver, striatum and cortex, relative to ear tissue DNA (isolated at weaning) for each of three progeny of a mating between a HdhneoQ50 mouse, with a pure CAG tract of 48 units, and Fan1_del10 mouse. By 8 months of age the distribution of CAG sizes is further increased in the somatic tissues of Fan1_del10 mutant mice, compared with the wild-type Fan1 mouse, as revealed by the quantified CAG repeat expansion index, calculated from the trace profiles, given in each panel. The trace plots for each mouse are aligned relative to the maximum peak height for the ear DNA trace (dotted red lines). (B) The plots give the expansion index (Y-axis) calculated from CAG repeat assay ABI GeneMapper trace profiles, for cortex, striatum and liver of 8 month old HdhneoQ50 mice (2–3 mice/group) (Hdhneo48/+ genotype), and for their ear tissue DNA (isolated at weaning), that are Fan1 wild-type or are heterozygous or homozygous for the Fan1_del10 mutation (X-axis). Whiskers indicate standard deviations. The increased expansion index value, relative to Fan1 wild-type tissue, demonstrates that the Fan1_del10 mutation enhances instability of the 48 CAG repeat in cortex, liver and striatum. Notably, the Fan1_del10 mutation also enhanced expansion of the 48 CAG repeat in the HdhQ50 line, which lacks the upstream PGKneo-cassette (Supplementary Material, Fig. S7).
Since we have shown previously that homozygous Mlh1 knockout suppresses somatic expansion of the Htt CAG repeat (12) and that the MLH1 locus is significantly associated with HD age-at-onset (10,11), we also crossed Fan1 knock-out mice with Mlh1 knock-out mice (35) to determine the potential genetic interaction of these loci. As shown in Figure 4, HdhQ111/+ CAG knock-in mice with both Mlh1 and Fan1 inactivated showed no evidence of somatic repeat expansion at 3 months of age, indicating that the presence of functional MLH1 protein is required for the mechanism by which Fan1 inactivation enhances repeat instability. These observations strongly imply, therefore, that in HD individuals FAN1 normally acts to suppress the MLH1-dependent lengthening of the inherited expanded CAG repeat in brain cells that are relevant to the disease phenotypes.
Figure 4.
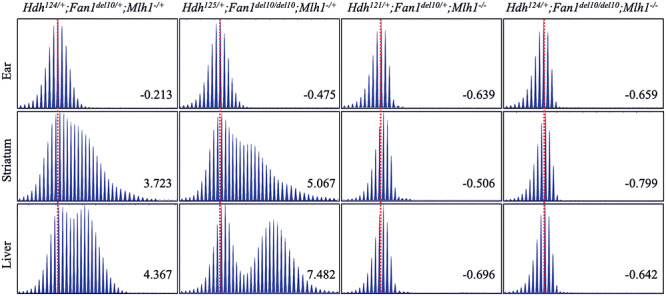
Mlh1 inactivation blocks the enhancing effect of Fan1_del10 mutation on somatic CAG repeat instability. The ABI GeneMapper trace plots show the patterns of age-related HdhQ111 CAG repeat instability in striatum and liver tissue of 3-month old mice, with combinations of Fan1 (Fan1_del10) and Mlh1 (Mlh1delex2 called Mlh1−) inactivating mutations. The distribution of the CAG repeat assay PCR amplification product peak heights (Y-axis) relative to fragment size (X-axis) is plotted for striatum and liver, and for stable ear tissue DNA of that mouse isolated at weaning (3 weeks of age). The trace plots for tissues from the same mouse are aligned relative to the maximum peak height in ear-DNA (dotted red lines) and the instability index value calculated for each trace profile is provided in each plot. Note that instability index is calculated using both the peaks for PCR products shorter than the main allele and PCR products with expanded CAG repeats larger than the main allele, so that positive instability index values denote a larger contribution of expanded alleles, whereas negative index values denote a relatively larger proportion of PCR products smaller than the main peak. The Hdh genotype of each mouse includes the maximum peak CAG repeat in ear DNA. Heterozygous and homozygous Fan1_del10 mutation enhances CAG repeat instability in striatum and liver (left panels), except in mice that are homozygous for the Mlh1 inactivating mutation (right panels), which exhibit an absence of somatic repeat instability.
Discussion
HD pathogenesis leading to onset of disease, as dissected by age-at-onset modifiers found by GWAS, is postulated to comprise two components in any given target cell: a CAG-expansion-based rate-driver mechanism and a toxicity mechanism triggered by further CAG expansion. Our functional assessment of the orthologs of modifier genes from our initial GWAS as potential DNA instability-based modulators provides clear support for FAN1 as a modifier of HD CAG repeat length change in somatic cells, though not of germline repeat instability, akin to observations in the Fmr1 CGG repeat knock-in mouse model of Fragile X disorders (36). FAN1 is involved in repair of interstrand cross-link (ICL)-induced DNA breaks, using its 5′-3′ exonuclease activity to cleave DNA at every third nucleotide from a cut end (37). Two FAN1 modifier variants represent damaging changes that both hasten HD onset: R377W annotated as deleterious and R507H associated with karyomegalic interstitial nephritis (OMIM #614817), a recessively inherited disease caused by loss of FAN1 function (38,39). The effect of Fan1 heterozygous and homozygous loss-of-function mutation on increasing somatic CAG expansion in our genetic HD mouse models provides an attractive explanation for how these variants increase the rate of HD pathogenesis leading to disease onset. Such a mechanism is also consistent with the known dependence on MLH1 function of somatic CAG expansion in the mouse and the emergence of MLH1 as another HD onset-modifier gene. In addition to onset-hastening protein altering variation, SNPs tagging onset-delaying modifier effects correspond with increased FAN1 expression in the cortex (11) suggesting that increased expression of FAN1 is likely to suppress HTT CAG repeat instability and thereby slow the rate of somatic CAG expansion in HD brain cells. Importantly, the conclusion that FAN1 likely influences the timing of HD pathogenesis via its effects on somatic instability of the HTT CAG repeat supports the potential of FAN1 as a modifier of other CAG repeat disorders (40) and, in conjunction with a report that Fan1 knock-out enhances somatic CGG repeat instability in the model of Fragile X disorders (36), implies a role for FAN1 more broadly in triplet repeat diseases.
The HD modifier RRM2B encodes a p53-inducible ribonucleotide reductase subunit involved in the conversion of ribonucleoside diphosphates to deoxyribonucleoside diphosphates required for DNA synthesis. The GWAS modifier haplotype carries a frameshift variant that prevents production of a minor RRM2B isoform from an alternative exon 1 (11), which the results of focused biochemical and cell biological studies demonstrate is involved in mitochondrial function (IS Seong, unpublished data). This primate-specific alternative exon 1 is not present in the mouse Rrm2b, so we could not generate an isoform specific knock-out. However, the possibility that the modifier effect detected in the GWAS may actually have its onset-hastening effects via an impact on the major RRM2B isoform, regardless of changes in the minor isoform, has not been ruled out. Complete knockout of this RRM2B homolog could only be tested at 2.5 months, shortly before premature death that is consistent with deleterious effects of RRM2B loss of function on cell viability, which can be sufficiently severe to cause several human mitochondrial disorders (mitochondrial depletion syndrome OMIM #612075; progressive external opthalmoplegia OMIM #613077). Rrm2b knockout showed a mild heterozygous and homozygous effect on suppressing somatic CAG repeat expansion, which however would predict that reduced RRM2B function would delay HD onset rather than producing the onset-hastening effect identified in the GWAS. This telling discrepancy implies that the human modifier effect is not due to reduced RRM2B function enhancing repeat instability (akin to FAN1 loss of function) unless an effect is specific to the minor primate isoform. If modification in HD is due to reduced RRM2B function, it might act instead via a non-DNA-instability process, perhaps involving mitochondrial function (41). Another possibility not tested in our study is that the human modifier effect may involve increased activity of the major RRM2B isoform, which could in principle enhance CAG repeat instability and hasten HD onset. Thus, current data remain inconclusive in distinguishing whether the RRM2B modifier haplotype acts via CAG repeat instability or, perhaps more likely, by influencing the toxicity component of HD pathogenesis, thought to be triggered in target cells above a repeat threshold. The rate with which this threshold is reached is influenced by CAG repeat rate-driver modifier genes such as FAN1 and MLH1.
Genetic interactions between the FAN1 and MLH1 modifiers are strongly predicted by our Fan1:Mlh1 double knock-out HdhQ111/+ mice. The HD modifier onset-delaying MLH1 I219V change is annotated as benign but its effects may reflect a suppression of the repeat instability mechanisms via altered interactions (10,11) or a functional deficiency specific to handling of expanded repeat structures. Though FAN1 and MLH1 were found to co-immunoprecipitate (28), the mechanisms by which FAN1 suppresses CAG repeat instability and MLH1 interacts with this process are not readily predicted based upon the activities of each protein in other DNA maintenance processes. Searches for gene–gene interactions between the orthologs of HD onset DNA-instability HD onset modifiers (FAN1, MLH1, PMS1, PMS2, MSH3, LIG1) in lines of CAG repeat knock-in mice may uncover effects not anticipated by current knowledge and could help to elucidate the mechanism of trinucleotide repeat, instability, thereby helping to dissect the molecular machinery of the driver of the timing of HD and the other triplet repeat disorders. Though detection of bona fide modifier gene–gene interactions in HD individuals remains challenging, given the complexity of the human genome and the comparatively small effects of DNA variants that become evident late in life, our observations indicate that this is an important area of research that in the future may help to accurately predict effects of modifier genes in individuals participating in clinical trials or genetic counseling.
Materials and Methods
Lines of mice and genotyping assays
All animal procedures were carried out to minimize pain and discomfort, under approved Institutional Animal Care and Use Committee (IACUC) protocol of the Massachusetts General Hospital. CRISPR/Cas9-mediated gene targeting in C57BL/6 J zygotes, to generate lines of C57BL/6 J mice with inactivating insertion/deletion mutations in Fan1, Mtmr10, Rrm2b and Ubr5, was accomplished by co-injection of sgRNA for each locus with Cas9 mRNA, essentially as reported (42) (Genome Modification Facility of Harvard University). Briefly, multiple sgRNA sequences for each gene (ENSEMBL/HAVANA database) were chosen using the MIT CRISPR Design tool (http://crispr.mit.edu/) (43), cloned into the pSpCas9(BB)-2A-Puro (PX459) V2.0 plasmid vector (a gift of Dr Feng Zhang, Addgene plasmid #62988) (44,45) and their endonuclease cutting efficiencies at targeted loci were compared in cultured NIH3T3 and STHdh cells by performing T7 Endonuclease Assay (New England Biolabs) (46). For each gene, the two most efficient sgRNAs were chosen and site-specific generation of insertion/deletion mutations was confirmed by Sanger sequencing of PCR amplification products from genomic DNA isolated from the transfected cells. For C57BL/6 J zygote injection, the sgRNA transcripts were generated from the pSpCas9(BB)-2A-Puro (PX459) V2.0 plasmids using the MEGAshortscript T7 kit protocol (Thermo Fisher Scientific), purified using the MEGAclear kit (Life Technologies), and subsequently co-injected with Cas9 mRNA (TriLink Biotech) into C57BL/6 J zygotes, which were implanted into pseudopregnant female mice and carried to term. At weaning, mice judged, by locus-specific PCR amplification assay (ABI3730XL DNA sequencer), to exhibit a high level of insertion/deletion mutation were identified and were later bred to wild-type C57BL/6 J mates, to attempt germ-line transmission of the mutant alleles to progeny. From this subsequent generation, mice carrying independent deleterious mutant alleles for each locus, confirmed by locus-specific PCR amplification genotyping assay and Sanger DNA sequencing (ABI3730XL DNA sequencer), were chosen as founders for breeding with wild-type C57BL/6 J mates to establish seven lines of HD modifier candidate gene mice utilized in this study. Sperm from each mutant line is cryopreserved at The Jackson Laboratories. Progeny mice, from crosses for line maintenance and experimental crosses were genotyped at weaning using DNA isolated from ear-clip tissue, utilizing the locus appropriate mutant allele specific genotyping PCR primers (Fan1, 6-FAM 5′-TACTACCTGCGGAGCTTCCT-3′ and 5′-CATGGCACACAAACCAGACG-3′; Mtmr10, 6-FAM 5′-GTGACAGGGCGGTGTTTGGA-3′ and 5′-TGGAAAGGGAAAGAGAACTACACAA-3′; Rrm2b, 6-FAM 5′-ACAGAAGAAAATGTGGTGAGGTC-3′ and 5′-TACCTCCTCTGCTGTCCAGA-3′; Ubr5, 6-FAM 5′-TGTTCCCTAGCCACCCACCT-3′ and 5′-TCTCAGTGCCTTCCCTTGACA-3′) on ABI3730XL DNA sequencer.
HdhQ111 knock-in mice, with an Htt CAG repeat knock-in allele (C57BL/6 J inbred background) have been described previously (33). HdhneoQ50 and HdhQ50 (CD1 genetic background) are Htt knock-in lines with 48 CAG repeats, with or without the PGKneo-cassette located ~900 bp upstream of the start ATG codon in exon1, respectively (47). Note that Hdh was the previous locus designation and HdhQ111, HdhneoQ50 and HdhQ50 were the line designations in the original reports (32,47). The ‘Q’ designation in these line names denotes that the humanized knock-in pure CAG tract is adjacent to a pair of glutamine codons (CAACAG), so that the encoded polyglutamine tract in the mutant endogenous huntingtin protein is two more than the size of the pure CAG tract. The original line names are used here to permit continuity with the literature. The HdhQ111 expanded CAG repeat exhibits striking intergenerational instability, so the actual size of the repeat is indicated in the identifier for each knock-in mouse as a CAG genotype (e.g. Hdh119/+ for 119 CAGs), whereas the CAG 48 allele is more stably inherited and actual size of the repeat in ear tissue is also given for each mouse (e.g. Hdh48/+). The size of the CAG repeat allele was estimated by specific PCR amplification product fragment size assay, using the forward primer 6-FAM-5′-ATGAAGGCCTTCGAGTCCCTCAAGTCCTT-3′ and reverse primer 5′-GGCGGCTGAGGAAGCTGAGGAC-3′ primer pair, on ABI3730XL DNA sequencer relative to sequenced DNA standards, as previously reported (15). Mlh1 knock-out mice (C57BL/6 J) with a targeted deletion in exon 2 (35) were obtained from the National Cancer Institute Mouse Repository and maintained on a C57BL/6 J background, and genotyped using a Mlh1 mutant allele specific PCR amplification genotyping assay (35). All genotyping assays were performed with genomic DNA isolated using DNeasy Blood and Tissue Kit (Qiagen) from ear clip tissue at weaning or from fresh tissue, snap frozen in liquid nitrogen, or fixed tissue, from mice that had been perfused transcardially with 2% paraformaldehyde, at ages ranging from 2.5 to 8 months of age.
To examine the impact of Fan1, Mtmr10, Rrm2b and Ubr5 mutations on Htt CAG instability, mice heterozygous for each of the targeted alleles were crossed with HdhQ111/+ mice, and, for Fan1, with HdhneoQ50 and HdhQ50 mice. Progeny intercrosses generated HdhQ111/+, or HdhneoQ50 and HdhQ50 littermates, as appropriate, that were homozygous (where viable) or heterozygous for the targeted allele or were wild-type. To test the genetic interaction between Fan1_del10 and Mlh1 mutant alleles, HdhQ111/+; Fan1_del10/+ mice were crossed with Mlh1+/− mice. Progeny heterozygous for both mutant alleles were then intercrossed to generate HdhQ111/+ mice with homozygous mutations in both Fan1 and Mlh1 and littermates that were heterozygous or wild-type for one or both of these alleles.
Inherited Htt CAG repeat instability
This study entailed matings designed to generate progeny for specific experiments and to maintain the mutant mouse lines, with mating data recorded in our database, including sex, genotype and HdhQ111 Htt CAG size. To assess intergenerational repeat instability, we examined the records of progeny whose repeats were inherited from 13 females and 26 males. Parental repeats ranged from 107 to 122 CAGs (mean 118 CAGs; mode 118 CAGs). The recorded CAG size of the HdhQ111 Htt repeat allele was determined in DNA isolated from ear-clip biopsy performed at 3 weeks of age for male and female parents and for progeny of matings in which the repeat was segregating. The change in repeat size inherited by the progeny, relative to the size of the parental repeat, was calculated and for each cross the absolute number of each size change as well as the proportion of inherited alleles with the change was calculated.
Htt CAG repeat somatic instability index and expansion index parameters
The instability of the HdhQ111 allele CAG repeat in tissues was determined by quantifying the fragment lengths produced in the Htt CAG repeat ABI3730 PCR amplification genotyping assay. The ABI3730XL GeneMapperV5 traces were used to calculate an instability index that includes both the PCR product peaks shorter than the maximum peak length, shown in single molecule amplification assays to mainly be PCR-stutter artifact, as well as the PCR product peaks that are longer than the maximum peak length, shown in single molecule assays to be products with expanded CAG repeats. Positive instability index values reflect the degree to which expanded allele peaks exceed the proportion of peaks shorter than the main peak. Negative instability index values indicate the degree to which PCR-stutter is greater than the proportion of expanded alleles (34). To accurately assess the mild degree of somatic instability of the 48 CAG repeat allele in HdhneoQ50 and HdhQ50 mice, we calculated expansion index in order to focus on expansion. The expansion index scores only peaks longer than the maximum peak length, as previously reported (34). Unless otherwise noted, a threshold of 5% relative to the maximum height peak was applied to all traces when calculating the instability index or expansion index.
Tissue sectioning and H&E staining
To test for karyomegaly, paraffin-embedded kidney tissue from mice fixed transcardially with 2% paraformaldehyde were transversely sectioned to 7 μm thickness and mounted onto glass slides for staining. Deparaffinization washes were as follows: 2 × 3 min xylenes, 3 min xylenes:ethanol (1:1), 3 min 95% ethanol, 3 min 80% ethanol, 3 min 70% ethanol, 3 min 50% ethanol, H2O rinse. Slides were stained with Hematoxylin QS (Vector) and Eosin stain (VWR).
Wild-type and Fan1_del10 HdhQ111 mouse embryonic fibroblast cell lines and MMC survival assays
A set of six mouse embryonic fibroblast (MEF) cell lines, with wild-type Fan1 and Fan1_del10 heterozygote or Fan1_del10 homozygote alleles, as well as matching Fan1 series of six MEFs that also had an HdhQ111 Htt CAG knock-in allele were generated from littermate E11 to E14 day embryos produced by mating between parental mice of the appropriate genotypes. Briefly, primary embryonic fibroblast cultures were prepared from dissected embryo tissue using the Primary Mouse Embryonic Fibroblast Isolation Kit (Thermo), according to manufacturer’s instructions and cells plated in complete DMEM plus 10% FBS (Gibco) plus 1% penicillin/streptomycin (Thermo). At subsequent passage, medium was changed to standard DMEM and ~10% of the cells were used for DNA extraction (Qiagen DNeasy kit) and locus specific genotyping assay. After ~3 passages, the primary fibroblast cultures representing each genotype were immortalized using the p1321 HPV-16 E6/E7 mammalian expression plasmid (a gift of Dr Peter Howley, Addgene plasmid #8641) (48) transfected using the Lipofectamine 3000 (Thermo) protocol. Transfected cells were passaged every 3–4 days and considered transformed when non-transfected cells grown in parallel ceased growing (typically passage 10). For MMC dose–response survival assays, MEFs were plated at 2000 cells/well of a 96-well plate in DMEM plus 10% FBS (Gibco) plus 1% of penicillin/streptomycin (Thermo Fisher Scientific) and the next day MMC was diluted in media to achieve the appropriate final concentration when added in equal volume to replicate wells. Standard media without MMC was added for untreated cell group. After, 5 days, cell viability was assessed using Cell Counting Kit-8 (Dojindo) according to manufacturer’s protocol. The surviving cells are reported as a percentage relative to non-treated cells.
Statistics
For mouse somatic instability, a linear regression model was built using CAG size (ear tissue at weaning) for HdhQ111 mice that were wild-type at a candidate modifier gene locus (X-axis), with instability parameters in their somatic tissue (Y-axis) (GraphPad Prism). Then, the CAG size in ear tissue of the HdhQ111; modifier gene mutant mice and instability parameter values for their somatic tissues were plotted. To generate residual instability parameter values, the instability index for a given mutant tissue was subtracted from the expected instability value derived from the regression model. P-values were generated using Mann–Whitney unpaired non-parametric test with the residual values for HdhQ111 modifier gene mutant tissue relative to values from tissue of HdhQ111 wild-type at modifier loci. Mann–Whitney unpaired non-parametric test was also used for determining P-values for inherited instability values and violin plots were made using GraphPad Prism.
Supplementary Material
Acknowledgements
The authors thank the Harvard Genome Modification core facility, Dr Lin Wu, Director, for generating the founder C57BL/6 J mice with CRISPR/Cas9 targeted modifier gene mutations that we used to establish the lines of modifier candidate gene mutant mice and Abigail Nowell for animal husbandry.
Conflict of Interest statement. M.E.M., S.K., D.H., R.L., I.-S.S., J.-M.L., T.G., K.-H.K., R.M.P., J.S.M., R.M., M.A.A., M.K. and J.M.L. have no conflicts of interest to declare. V.C.W. is a scientific advisory board member of Triplet Therapeutics, a company developing new therapeutic approaches to address triplet repeat disorders such Huntington’s disease and Myotonic Dystrophy and of LoQus23 Therapeutics and has provided paid consulting services to Alnylam. Her financial interests in Triplet Therapeutics were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. J.F.G. is a Scientific Advisory Board member and has a financial interest in Triplet Therapeutics, Inc. His NIH-funded project is using genetic and genomic approaches to uncover other genes that significantly influence when diagnosable symptoms emerge and how rapidly they worsen in Huntington Disease. The company is developing new therapeutic approaches to address triplet repeat disorders such Huntington’s Disease, Myotonic Dystrophy and spinocerebellar ataxias. His interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies.
Contributor Information
Jacob M Loupe, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02115, USA.
Ricardo Mouro Pinto, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02115, USA.
Kyung-Hee Kim, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02115, USA.
Tammy Gillis, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA.
Jayalakshmi S Mysore, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA.
Marissa A Andrew, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA.
Marina Kovalenko, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA.
Ryan Murtha, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA.
IhnSik Seong, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02115, USA.
James F Gusella, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02115, USA; Department of Genetics, Blavatnik Institute, Harvard Medical School, Boston, MA 02115, USA; Medical and Population Genetics Program, The Broad Institute of M.I.T. and Harvard, Cambridge, MA 02142, USA.
Seung Kwak, CHDI Foundation, Princeton, NJ 08540, USA.
David Howland, CHDI Foundation, Princeton, NJ 08540, USA.
Ramee Lee, CHDI Foundation, Princeton, NJ 08540, USA.
Jong-Min Lee, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02115, USA.
Vanessa C Wheeler, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02115, USA.
Marcy E MacDonald, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02115, USA; Medical and Population Genetics Program, The Broad Institute of M.I.T. and Harvard, Cambridge, MA 02142, USA.
Funding
National Institutes of Health [NINDS NS091161 to J.F.G., NS105709 to J.-M.L. and NS049206 to V.C.W.]; the CHDI Foundation [to J.F.G. and M.E.M.] and the Berman/Topper Family HD Career Development Fellowship from the Huntington’s Disease Society of America [to R.M.P.].
References
- 1. Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's disease collaborative research group. Cell, 72, 971–983. [DOI] [PubMed] [Google Scholar]
- 2. Andrew S.E., Goldberg Y.P., Kremer B., Telenius H., Theilmann J., Adam S., Starr E., Squitieri F., Lin B., Kalchman M.A. et al. (1993) The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat. Genet., 4, 398–403. [DOI] [PubMed] [Google Scholar]
- 3. Duyao M., Ambrose C., Myers R., Novelletto A., Persichetti F., Frontali M., Folstein S., Ross C., Franz M., Abbott M. et al. (1993) Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat. Genet., 4, 387–392. [DOI] [PubMed] [Google Scholar]
- 4. Persichetti F., Srinidhi J., Kanaley L., Ge P., Myers R.H., D'Arrigo K., Barnes G.T., MacDonald M.E., Vonsattel J.P., Gusella J.F. et al. (1994) Huntington's disease CAG trinucleotide repeats in pathologically confirmed post-mortem brains. Neurobiol. Dis., 1, 159–166. [DOI] [PubMed] [Google Scholar]
- 5. Snell R.G., MacMillan J.C., Cheadle J.P., Fenton I., Lazarou L.P., Davies P., MacDonald M.E., Gusella J.F., Harper P.S. and Shaw D.J. (1993) Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington's disease. Nat. Genet., 4, 393–397. [DOI] [PubMed] [Google Scholar]
- 6. Lee J.M., Ramos E.M., Lee J.H., Gillis T., Mysore J.S., Hayden M.R., Warby S.C., Morrison P., Nance M., Ross C.A. et al. (2012) CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology, 78, 690–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Djousse L., Knowlton B., Hayden M., Almqvist E.W., Brinkman R., Ross C., Margolis R., Rosenblatt A., Durr A., Dode C. et al. (2003) Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease. Am. J. Med. Genet. A, 119A, 279–282. [DOI] [PubMed] [Google Scholar]
- 8. Li J.L., Hayden M.R., Almqvist E.W., Brinkman R.R., Durr A., Dode C., Morrison P.J., Suchowersky O., Ross C.A., Margolis R.L. et al. (2003) A genome scan for modifiers of age at onset in Huntington disease: the HD MAPS study. Am. J. Hum. Genet., 73, 682–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium (2015) Identification of genetic factors that modify clinical onset of Huntington's disease. Cell, 162, 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee J.M., Chao M.J., Harold D., Abu Elneel K., Gillis T., Holmans P., Jones L., Orth M., Myers R.H., Kwak S. et al. (2017) A modifier of Huntington's disease onset at the MLH1 locus. Hum. Mol. Genet., 26, 3859–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium (2019) CAG repeat not polyglutamine length determines timing of Huntington's disease onset. Cell, 178, 887–900 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pinto R.M., Dragileva E., Kirby A., Lloret A., Lopez E., St Claire J., Panigrahi G.B., Hou C., Holloway K., Gillis T. et al. (2013) Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington's disease mice: genome-wide and candidate approaches. PLoS Genet., 9, e1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Swami M., Hendricks A.E., Gillis T., Massood T., Mysore J., Myers R.H. and Wheeler V.C. (2009) Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum. Mol. Genet., 18, 3039–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lieberman A.P., Shakkottai V.G. and Albin R.L. (2019) Polyglutamine repeats in neurodegenerative diseases. Annu. Rev. Pathol., 14, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dragileva E., Hendricks A., Teed A., Gillis T., Lopez E.T., Friedberg E.C., Kucherlapati R., Edelmann W., Lunetta K.L., MacDonald M.E. et al. (2009) Intergenerational and striatal CAG repeat instability in Huntington's disease knock-in mice involve different DNA repair genes. Neurobiol. Dis., 33, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gomes-Pereira M., Fortune M.T., Ingram L., McAbney J.P. and Monckton D.G. (2004) Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion. Hum. Mol. Genet., 13, 1815–1825. [DOI] [PubMed] [Google Scholar]
- 17. Tome S., Manley K., Simard J.P., Clark G.W., Slean M.M., Swami M., Shelbourne P.F., Tillier E.R., Monckton D.G., Messer A. et al. (2013) MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington's disease mice. PLoS Genet., 9, e1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tome S., Panigrahi G.B., Lopez Castel A., Foiry L., Melton D.W., Gourdon G. and Pearson C.E. (2011) Maternal germline-specific effect of DNA ligase I on CTG/CAG instability. Hum. Mol. Genet., 20, 2131–2143. [DOI] [PubMed] [Google Scholar]
- 19. Ciosi M., Maxwell A., Cumming S.A., Hensman Moss D.J., Alshammari A.M., Flower M.D., Durr A., Leavitt B.R., Roos R.A.C., team T.-H. et al. (2019) A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine, 48, 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Flower M., Lomeikaite V., Ciosi M., Cumming S., Morales F., Lo K., Hensman Moss D., Jones L., Holmans P., Investigators T.-H. et al. (2019) MSH3 modifies somatic instability and disease severity in Huntington's and myotonic dystrophy type 1. Brain, 142, 1876–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moss D.J.H., Pardinas A.F., Langbehn D., Lo K., Leavitt B.R., Roos R., Durr A., Mead S., investigators T.-H., investigators R. et al. (2017) Identification of genetic variants associated with Huntington's disease progression: a genome-wide association study. Lancet Neurol., 16, 701–711. [DOI] [PubMed] [Google Scholar]
- 22. Kaplan S., Itzkovitz S. and Shapiro E. (2007) A universal mechanism ties genotype to phenotype in trinucleotide diseases. PLoS Comput. Biol., 3, e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goold R., Flower M., Moss D.H., Medway C., Wood-Kaczmar A., Andre R., Farshim P., Bates G.P., Holmans P., Jones L. et al. (2019) FAN1 modifies Huntington's disease progression by stabilizing the expanded HTT CAG repeat. Hum. Mol. Genet., 28, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El-Hattab A.W., Craigen W.J. and Scaglia F. (2017) Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta Mol. basis Dis., 1863, 1539–1555. [DOI] [PubMed] [Google Scholar]
- 25. Amoasii L., Hnia K. and Laporte J. (2012) Myotubularin phosphoinositide phosphatases in human diseases. Curr. Top. Microbiol. Immunol., 362, 209–233. [DOI] [PubMed] [Google Scholar]
- 26. Saunders D.N., Hird S.L., Withington S.L., Dunwoodie S.L., Henderson M.J., Biben C., Sutherland R.L., Ormandy C.J. and Watts C.K. (2004) Edd, the murine hyperplastic disc gene, is essential for yolk sac vascularization and chorioallantoic fusion. Mol. Cell. Biol., 24, 7225–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kratz K., Schopf B., Kaden S., Sendoel A., Eberhard R., Lademann C., Cannavo E., Sartori A.A., Hengartner M.O. and Jiricny J. (2010) Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell, 142, 77–88. [DOI] [PubMed] [Google Scholar]
- 28. Smogorzewska A., Desetty R., Saito T.T., Schlabach M., Lach F.P., Sowa M.E., Clark A.B., Kunkel T.A., Harper J.W., Colaiacovo M.P. et al. (2010) A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell, 39, 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thongthip S., Bellani M., Gregg S.Q., Sridhar S., Conti B.A., Chen Y., Seidman M.M. and Smogorzewska A. (2016) Fan1 deficiency results in DNA interstrand cross-link repair defects, enhanced tissue karyomegaly, and organ dysfunction. Genes Dev., 30, 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lachaud C., Slean M., Marchesi F., Lock C., Odell E., Castor D., Toth R. and Rouse J. (2016) Karyomegalic interstitial nephritis and DNA damage-induced polyploidy in Fan1 nuclease-defective knock-in mice. Genes Dev., 30, 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kimura T., Takeda S., Sagiya Y., Gotoh M., Nakamura Y. and Arakawa H. (2003) Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat. Genet., 34, 440–445. [DOI] [PubMed] [Google Scholar]
- 32. Wheeler V.C., Auerbach W., White J.K., Srinidhi J., Auerbach A., Ryan A., Duyao M.P., Vrbanac V., Weaver M., Gusella J.F. et al. (1999) Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum. Mol. Genet., 8, 115–122. [DOI] [PubMed] [Google Scholar]
- 33. Lee J.M., Pinto R.M., Gillis T., St Claire J.C. and Wheeler V.C. (2011) Quantification of age-dependent somatic CAG repeat instability in Hdh CAG knock-in mice reveals different expansion dynamics in striatum and liver. PLoS One, 6, e23647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee J.M., Zhang J., Su A.I., Walker J.R., Wiltshire T., Kang K., Dragileva E., Gillis T., Lopez E.T., Boily M.J. et al. (2010) A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC Syst. Biol., 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Edelmann W., Cohen P.E., Kane M., Lau K., Morrow B., Bennett S., Umar A., Kunkel T., Cattoretti G., Chaganti R. et al. (1996) Meiotic pachytene arrest in MLH1-deficient mice. Cell, 85, 1125–1134. [DOI] [PubMed] [Google Scholar]
- 36. Zhao X.N. and Usdin K. (2018) FAN1 protects against repeat expansions in a fragile X mouse model. DNA Repair (Amst), 69, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang R., Persky N.S., Yoo B., Ouerfelli O., Smogorzewska A., Elledge S.J. and Pavletich N.P. (2014) DNA repair. Mechanism of DNA interstrand cross-link processing by repair nuclease FAN1. Science, 346, 1127–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou W., Otto E.A., Cluckey A., Airik R., Hurd T.W., Chaki M., Diaz K., Lach F.P., Bennett G.R., Gee H.Y. et al. (2012) FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet., 44, 910–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bastarache L., Hughey J.J., Hebbring S., Marlo J., Zhao W., Ho W.T., Van Driest S.L., McGregor T.L., Mosley J.D., Wells Q.S. et al. (2018) Phenotype risk scores identify patients with unrecognized Mendelian disease patterns. Science, 359, 1233–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bettencourt C., Hensman-Moss D., Flower M., Wiethoff S., Brice A., Goizet C., Stevanin G., Koutsis G., Karadima G., Panas M. et al. (2016) DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann. Neurol., 79, 983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cho E.C., Kuo M.L., Cheng J.H., Cheng Y.C., Hsieh Y.C., Liu Y.R., Hsieh R.H. and Yen Y. (2015) RRM2B-mediated regulation of mitochondrial activity and inflammation under oxidative stress. Mediat. Inflamm., 2015, 287345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang H., Wang H. and Jaenisch R. (2014) Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protoc., 9, 1956–1968. [DOI] [PubMed] [Google Scholar]
- 43. Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. et al. (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol., 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A. and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc., 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trettel F., Rigamonti D., Hilditch-Maguire P., Wheeler V.C., Sharp A.H., Persichetti F., Cattaneo E. and MacDonald M.E. (2000) Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet., 9, 2799–2809. [DOI] [PubMed] [Google Scholar]
- 46. Guschin D.Y., Waite A.J., Katibah G.E., Miller J.C., Holmes M.C. and Rebar E.J. (2010) A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol., 649, 247–256. [DOI] [PubMed] [Google Scholar]
- 47. White J.K., Auerbach W., Duyao M.P., Vonsattel J.P., Gusella J.F., Joyner A.L. and MacDonald M.E. (1997) Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nat. Genet., 17, 404–410. [DOI] [PubMed] [Google Scholar]
- 48. Munger K., Phelps W.C., Bubb V., Howley P.M. and Schlegel R. (1989) The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol., 63, 4417–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.