

Abstract
Autophagy is a functionally conserved self-degradation process that facilitates the survival of eukaryotic life via the management of cellular bioenergetics and maintenance of the fidelity of genomic DNA. The first known autophagy inducer was Beclin-1. Beclin-1 is expressed in multicellular eukaryotes ranging throughout plants to animals, comprising a nonmonophyllic group, as shown in this report via aggressive BLAST searches. In humans, Beclin-1 is a haploinsuffient tumor suppressor as biallelic deletions have not been observed in patient tumors clinically. Therefore, Beclin-1 fails the Knudson hypothesis, implicating expression of at least one Beclin-1 allele is essential for cancer cell survival. However, Beclin-1 is frequently monoallelically deleted in advanced human cancers and the expression of two Beclin-1 allelles is associated with greater anticancer effects. Overall, experimental evidence suggests that Beclin-1 inhibits tumor formation, angiogenesis, and metastasis alone and in cooperation with the tumor suppressive molecules UVRAG, Bif-1, Ambra1, and MDA-7/IL-24 via diverse mechanisms of action. Conversely, Beclin-1 is upregulated in cancer stem cells (CSCs), portending a role in cancer recurrence, and highlighting this molecule as an intriguing molecular target for the treatment of CSCs. Many aspects of Beclin-1’s biological effects remain to be studied. The consequences of these BLAST searches on the molecular evolution of Beclin-1, and the eukaryotic branches of the tree of life, are discussed here in greater detail with future inquiry focused upon protist taxa. Also in this review, the effects of Beclin-1 on tumor suppression and cancer malignancy are discussed. Beclin-1 holds significant promise for the development of novel targeted cancer therapeutics and is anticipated to lead to a many advances in our understanding of eukaryotic evolution, multicellularity, and even the treatment of CSCs in the coming decades.
1. GENERAL INTRODUCTION
Autophagy remains a topic of intensive investigation due to its affect upon cancer including cell survival, invasion, and metastasis; or conversely, the suppression of tumor growth, through lysing cells via type II programmed cell death (PCD; Kroemer et al., 2009). These distinct affects are thought to vary depending on the tissue, redox status, and other biochemical pathways which are activated concurrently with autophagy signaling. Generally, autophagy leads to the sequestration of organelles and long-lived proteins into autophagosomes, which later fuse with lysosomes to form the autophagolysosome, degrading those sequestered cellular components to produce energy and cellular monomers (Fig. 1; Ren & Taegtmeyer, 2015). The maintenance of autophagy flux is essential to successfully accomplish mammalian embryogenesis. As such, autophagy is evolutionarily conserved in eukaryotes, supporting the significance of this biochemical pathway. The term “autophagy” can refer to many different subclassifications of this process; for the purpose of this review, “autophagy” refers to macroautophagy.
Fig. 1.
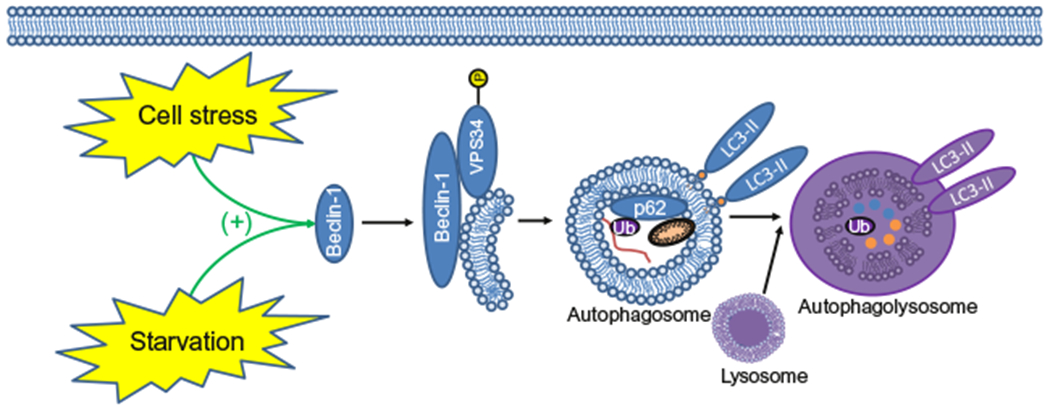
The effect of cell stress and starvation on the maturation of the auto-phagolysosomes and autophagy. This graphic depicts the extension of the autophagy membrane, the sequestration of ubiquitinated long-lived proteins, p62-mediated protein scavenging, and damaged organelles such as the mitochondria. Ultimately, these components are degraded following lysosomal fusion. Blue circles are representative of degraded proteins and orange circles are representative of the degradation of the mitochondria. Purple coloring is indicative of autophagosome acidification and the degradation of its components occurring during the final stages of autophagolysosome maturation.
As many as 30 autophagy-related genes (Atg) have been identified in Saccharomyces cerevisiae; please refer to Cao et al. and Klionsky et al. for a more complete review of these genes and their molecular mechanisms (Cao & Klionsky, 2007; Li et al., 2012). Beclin-1 was initially cloned in 1998 (Liang et al., 1998) and was the first identified mammalian autophagy inducing protein (Liang etal., 1999). Beclin-1 has now been shown to participate in many other biological processes including gametogenesis, neurodegeneration, apoptosis, and tumorigenesis (Mizushima, Levine, Cuervo, & Klionsky, 2008). Despite the well-characterized interaction of Beclin-1 and cancer, no prior review has comprehensively discussed the evolution and cancer therapeutic functions of Beclin-1, to our knowledge, at this time. Accordingly, to better understand the cancer therapeutic effects of Beclin-1, this review will discuss: (1) the molecular evolution of Beclin-1, (2) Beclin-1’s tumor suppressive mechanisms of action, (3) the effects of Beclin-1 expression upon tumors and cancer malignancy, (4) the effect of Beclin-1 upon conventional cancer therapeutics, and (5) the effect of Beclin-1 upon cancer stem cells (CSCs).
2. INTRODUCTION TO BECLIN-1
Following the discovery and characterization of Beclin-1, DNA sequencing analysis revealed that Beclin-1 expression is conserved in all animals and most other multicellular eukaryotes (Cuervo, 2004; Klionsky, 2007; Levine & Klionsky, 2004; Mizushima, 2007; Mizushima et al., 2008), indicating the significance of Beclin-1 at the earliest stages of eukaryotic divergence (Fig. 2). Interestingly, Beclin-1 preforms nearly identical functions over the diversity of animals, promoting their survival in response to stress via autophagy to degrade damaged cellular proteins and organelles. In this way, Beclin-1 plays a critical role in maintaining the complex balance of cellular bioenergetics during periods of stress and starvation in a wide range of organisms. Furthermore, Beclin-1 has also been shown to play a role during embryogenesis and reproduction of eukaryotic organisms ranging from plants (Singh et al., 2010) to mice (Gawriluk, Ko, Hong, Christenson, & Rucker, 2014). Therefore, Beclin-1 appears to be a key regulator of not only survival, but also the life cycle of most multicellular eukaryotes.
Fig. 2.
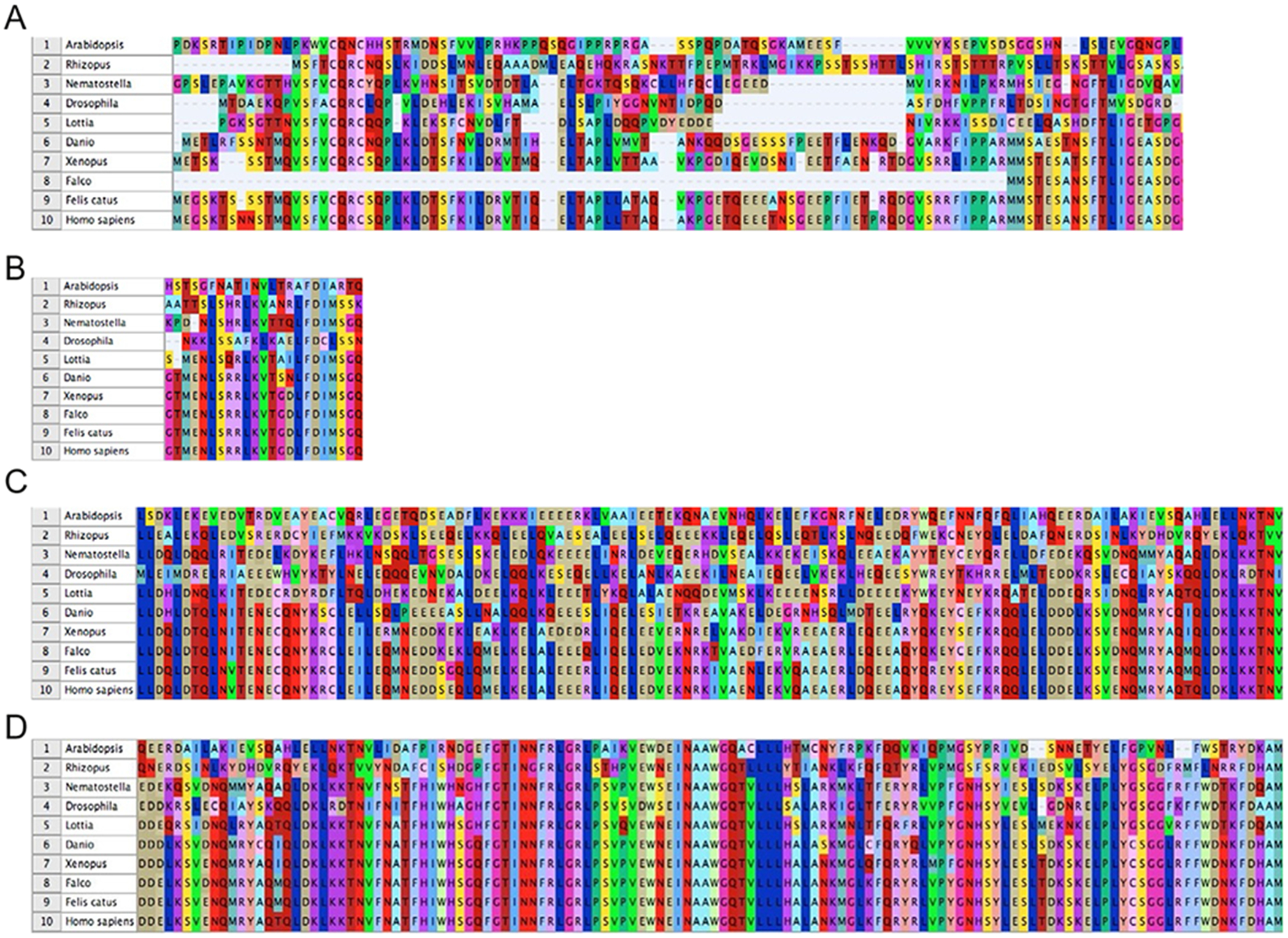
The conservation of Beclin-1 amino acid sequences between humans and 10 critical multicellular eukaryotic taxa. (A) The alignment of Beclin-1 5′ amino acids (1–113). (B) Alignment of the Beclin-1 Bcl-2 homology domain 3 (BH3; 114–130) amino acids. (C) Alignment of the Beclin-1 CCD region (144–269) amino acids. (D) The alignment of the evolutionarily conserved domain (ECD; 244–337 aa’s). The numbers 1–10 are indicative of the following taxa, respectively. 1 = Arabidopsis (plant), 2 = Rhizopus (fungi), 3 = Nematostella (Cnidaria), 4 = Drosophila (Protostome—Ecdysozoa), 5 = Lottia (Protostome—Lophotrochozoa), 6 = Danio (fish), 7 = Xenopus (amphibian), 8 = Falco (bird), 9 = Felis (cat), and 10 = Homo (human). BH3, Bcl-2 homology domain; CCD, coiled-coil domain; ECD, evolutionarily conserved domain.
Beclin-1 contains many domains with conserved amino acid sequences across eukaryotic species (Fig. 3). One of these evolutionarily conserved domains (ECD; 244–377 aa’s) is essential for mediating the protein—protein interaction of Beclin-1 and the class III phosphatidylinositol 3′-kinase (PI3K) Vps34, facilitating eukaryotic cell adaptation to stressful conditions (Furuya, Yu, Byfield, Pattingre, & Levine, 2005). In this manner, it is likely that Beclin-1 provided a selective advantage during the early stages of eukaryotic evolution as primitive eukaryotes biochemically diverged from prokaryotes, however, the point of this divergence remains unknown. For example, Beclin-1 has been shown to contribute to DNA stability, enhance DNA repair, and strengthen overall DNA genome integrity via the interaction with Topoisomerase-IIβ (Xu et al., 2017). For this interaction to occur, Beclin-1 must translocate to the nucleus. The nuclear translocation of Beclin-1 appears to occur early during murine development (15–20 days postnatal; Xu et al., 2017). As mammals, such as mice, mature Beclin-1 translocates back to the cytoplasm while some of these proteins (~50%) remain in the nucleus. However, Beclin-1 and Topoisomerase-IIβ are not the only DNA damage repair proteins expressed in eukaryotic cells. Other examples include heat shock proteins (HSPs), which are expressed in both prokaryotic and eukaryotic cells as an adaptation to cell stress (Finka & Goloubinoff, 2013). It is intriguing, however, that Beclin-1 appears to have been selectively advantageous to eukaryotic, but not prokaryotic cells. Furthermore, if Beclin-1 provided an advantage to prokaryotic cells, then it would have undoubtedly arisen in the prokaryotic cell lineage, indicating that there are unique aspects of eukaryotic cell biochemistry which led to the molecular evolution of Beclin-1. Some of these unique eukaryotic cell factors may have included the development of the nucleus and the longer-life spans observed in eukaryotic cells. It is possible that the expression of the HSPs alone were not sufficient for multicellular eukaryotes and additional biochemical pathways, such as Beclin-1, were necessary to ensure the fidelity of the eukaryotic DNA genome and cell viability in response to cell starvation and DNA-damaging stimuli compared to prokaryotes.
Fig. 3.
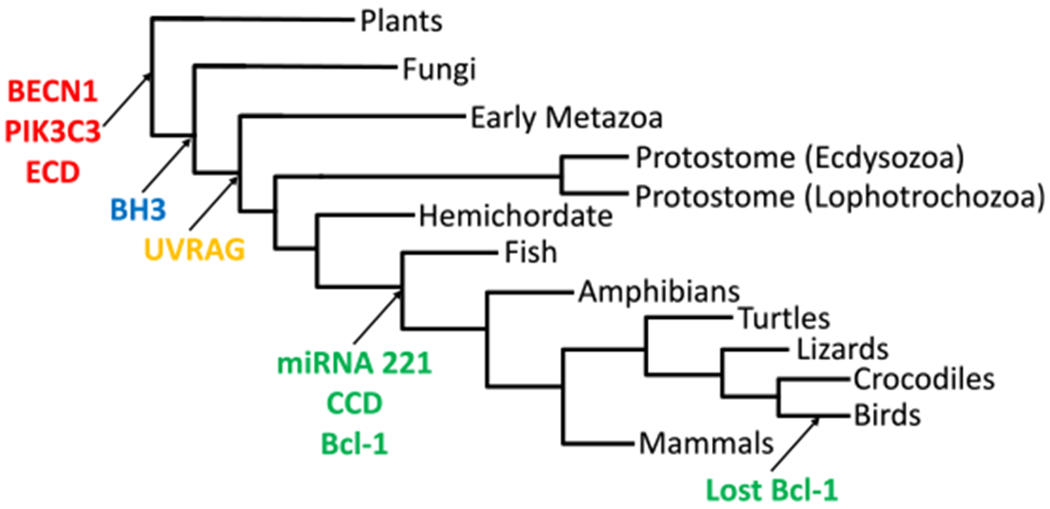
The evolutionary tree of the origins of Beclin-1 and several of its interaction partners in eukaryotes. Specific protein domains of BECN1 are also indicated. BECN1, Beclin-1; ECD, evolutionarily conserved domain; BH3, Bcl-2 homology domain; UVRAG, ultraviolet radiation resistance-associated gene; CCD, coiled-coil domain.
2.1. Molecular Evolution of Eukaryotic Beclin-1
To better understand the significance of Beclin-1 during eukaryotic evolution, we performed aggressive BLAST searches of genomic databases (see Supplementary file available on https://doi.org/10.1016/bs.acr.2017.11.002) using human Beclin-1 as a query sequence. The searches were performed locally using the approaches outlined in Rosenfeld and DeSalle (2012). Initially, human Beclin-1 was compared to the Beclin-1 peptide sequences of 10 critical taxa (Fig. 2A). These 10 critical taxa included Arabidopsis (plant), Rhizopus (fungi), Nematostella (Cnidaria), Drosophila (Protostome—Ecdysozoa), Lottia (Protostome—Lophotrochozoa), Danio (fish), Xenopus (amphibian), Falco (bird), Felis (cat), and Homo (human). Focusing upon the 1–133 5′ Beclin-1 amino acid sequences (Bcl-1), only mammalian and amphibian sequences were conserved while bird species (Falco) have lost Bcl-1. In the lower fungi, nonbilaterian animals, and protostomes (Rhizopus, Nematostella, Drosophila, and Lottia), 1–133 5′ Beclin-1 amino acid sequences displayed greater variability relative to mammalian Beclin-1. Much greater amino acid conservation was observed in the Beclin-1 Bcl-2 homology domain 3 (BH3; 114–130) amino acids as all animal taxa except for Drosophila contained the BH3 domain; BH3 was also not found in the plant taxa Arabidopsis (Fig. 2B). The evolutionary conservation of the Beclin-1 coiled-coil domain (CCD, 144–269 aa’s), revealed a similar pattern to that of BH3; vertebrate taxa displaying similarity while the Drosophila and Arabidopsis taxa diverged from human Beclin-1 amino acid sequences (Fig. 2C). It is interesting to note that fungi, Cnidaria and the other protostome (Lottia) in this smaller dataset do not show the same high degree of divergence as Arabidopsis and Drosophila (Fig. 2D). In contrast to the BH3 and CCD domains, the ECD (244–337 aa’s) was highly conserved between all studied taxa. While these 10 critical taxa were selected for their evolutionary relevance to mammalian evolution, additional sequence alignments were also performed from a richer diversity of 82 additional taxa for a total 92 studied taxa overall (Supplementary Fig. 1 in the online version at https://doi.org/10.1016/bs.acr.2017.11.002).
From these insights and deep sequencing data, an evolutionary tree of Beclin-1 proteins was constructed from plants, fungi, and animals (Fig. 3). This tree also shows where in the phylogeny interactors with Beclin-1 (PIK3C3, UVRAG, and miRNA-221) also arose. Starting from the earliest branch point, Beclin-1, ECD, and PIK3C3 appear to have arisen together during the first appearance of Beclin-1 in eukaryotes. Plant species do not have the Beclin-1 BH3 domain, while fungi do, which is interesting because Beclin-1 is not found in any known protist taxa. However, both plants and fungi do not display the Beclin-1 interacting protein ultraviolet radiation resistance-associated gene (UVRAG) which is interesting as plants and fungi are inundated with high doses of UV irradiation on average over their lifetimes. The Beclin-1 CCD domain, Bcl-1 domain, and miRNA-221, a negative regulator of Beclin-1, emerged in the common ancestor of vertebrates. The implications of these domains upon Beclin-1 function are summarized in Figure 4 (Fig. 4). The Bcl-1 domain was lost in the bird taxa later during vertebrate evolution (Fig. 3).
Fig. 4.
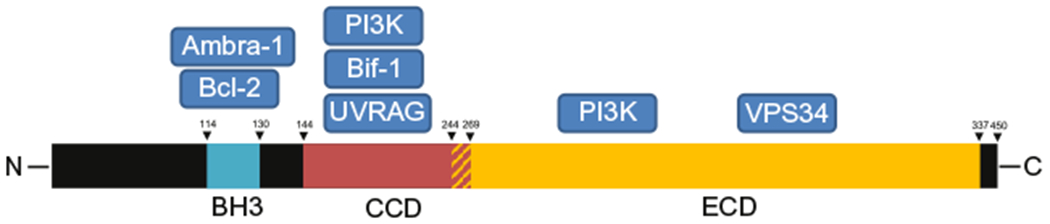
Schematic of the major Beclin-1 protein domains and their respective protein–protein interaction partners. The red–yellow shaded region indicates the overlap between the Beclin-1 CCD and ECD domains. BH3, Bcl-2 homology-3 domain; CCD, coiled-coil domain; ECD, evolutionary conserved domain of Beclin-1.
Beclin-1 was not found in prokaryotes (Bacteria, Archaea) as indicated in previous reports of Beclin-1 conservation in eukaryotic organisms (Wirawan et al., 2012). Interestingly however, Beclin-1 was also not found in protists like Chlamydomonas or algae, the common ancestors of plants, or in amoeba, Plasmodium or other ancestral protist lineages to fungi and animals. This occurs despite the fact that Beclin-1 is found in nearly all multicellular eukaryotes ranging from plants to fungi to sponges to cnidarians to bilateral animals (Supplementary Fig. 2 in the online version at https://doi.org/10.1016/bs.acr.2017.11.002). Furthermore, single celled eukaryotic organisms do not appear to require Beclin-1 expression, which makes the pattern of origin of Beclin-1 peculiar as multicellular eukaryotes (plants and animals) do not comprise a monophyletic group.
Considering these factors, there are at least three scenarios which could explain these patterns of Beclin-1 presence in the genomes of multicellular eukaryotes. (1) Beclin-1 may have been horizontally transferred from animals to plants or vice versa, which is very unlikely. (2) The common ancestor of all eukaryotes may have acquired Beclin-1. Subsequently, Beclin-1 may have been lost over time from these protist lineages which may have been lost to the fossil record. This scenario would be especially appealing if Beclin-1 is deleterious to single celled life. Additional support for this argument was discussed by Huettenbrenner et al. in which single celled organisms were strongly selected against acquiring cell death effectors such as Beclin-1 at all costs while PCD appears to have only become advantageous in multicellular organisms such that individual cells could be sacrificed for the benefit of the organism as a whole (Huettenbrenner et al., 2003). In this scenario, we hypothesize that such selective pressures, including the maintenance of DNA genome fidelity in longer-lived multicellular eukaryotes, may have been sufficient to select for Beclin-1 expression. This scenario implies a hypothetical ancestral single celled protist having Beclin-1 in its genome and would be known as “the master ancestor” of all Beclin-1 containing multicellular eukaryotes or dominus antecorris. The downside of this scenario is that it would require several parallel losses of Beclin-1 in protists as these early protist lineages are very deep and not monophyletic. (3) Strong selective pressures may have driven all animals, and most multicellular plants such as angiosperms and clubmosses (Beclin-1 is absent in mosses and gymnosperms) to acquire Beclin-1. Prior to this analysis, the authors postulated that Beclin-1 arose as the first eukaryotic cell evolved. What we can hypothesize given the distribution of Beclin-1 in these eukaryotic genomes, is that natural selection has been a major driver of the evolution of this gene. Since the horizontal gene transfer scenario is highly unlikely, this leaves the most likely scenario of single origin with strong selection eliminating Beclin-1 in the genomes of protists, and the two-origin scenario with strong selection for convergence of the gene in plants and fungi/animals.
2.2. Beclin-1 and Eukaryotic Cell Autophagy
One of the principal functions of Beclin-1 is to facilitate the formation of the autophagosomes in response to cell stress (Fig. 1). Beclin-1 is regulated via phosphorylation at many sites including Ser-234, Ser-295, and Ser-90. Beclin-1 phosphorylation at Ser-234 and Ser-295 inhibits Beclin-1 signaling, while its phosphorylation at Ser-90 activates Beclin-1 to induce autophagy (Fujiwara, Usui, Ohama, & Sato, 2016; Wang et al., 2012); Beclin-1 also appears to have three Tyr phosphorylation sites: 229, 233, and 352 associated with epidermal growth factor receptor (EGFR) while phosphorylation at Thr-388 is regulated by AMPK (Wei et al., 2013; Zhang et al., 2016). Furthermore, Beclin-1 is inhibited by complex formation with Bcl-2 (Klein et al., 2015). In this manner, Beclin-1 can be activated via the phosphorylation of Bcl-2 at the Ser-90 site, preventing the formation of the inhibitory Beclin-1/Bcl-2 complex via c-jun N-terminal kinase (JNK; Wei, Pattingre, Sinha, Bassik, & Levine, 2008; Zhou, Li, Jiang, & Zhou, 2015). AMP-activated protein kinase (AMPK) is one of the most well-known autophagy regulators; AMPK regulates autophagy by phosphorylating Beclin-1 at Thr-388. Thr-388 is required for autophagy to be induced during periods of cell starvation (Zhang et al., 2016). AMPK serves as an energy sensor to coordinate intracellular bioenergetic responses to cell starvation. When energy is low, AMPK activates Ulk1 via phosphorylation Ser-317 and Ser-777, inducing autophagy (Kim, Kundu, Viollet, & Guan, 2011). Beclin-1 is highly conserved with functional homology in yeast and higher-order eukaryotes including plants, reptiles, and humans, implicating its requirement for the growth and survival of eukaryotic cells as they complete their respective life cycles via the cooperation of these cell signaling pathways. In this review, the effects of Beclin-1 upon many aspects of cancer biology are discussed to illustrate the diverse roles of Beclin-1 to promote cancer therapeutic effects.
3. BECLIN-1 AND CANCER
Beclin-1 has been shown to be a haploinsufficient tumor suppressor in mice (Qu et al., 2003). Beclin-1 overexpression has also been shown in 85.2% of stage IIIB colon cancers (Li et al., 2009). Further analysis indicated that the overexpression of Beclin-1 in these colorectal tumors was associated with greater patient survival (Li et al., 2009). The controversial role of Beclin-1 upon cancer (summarized in Fig. 5) is primarily associated with its role as an autophagy inducer. Therefore, its influence upon oncogenesis and cancer therapy warrants further discussion of these phenomena. In this review, we will discuss the signaling pathways associated with Beclin-1 to determine how this complex tumor suppressive molecule affects oncogenesis, cancer prognosis, and cancer therapeutic responses clinically.
Fig. 5.
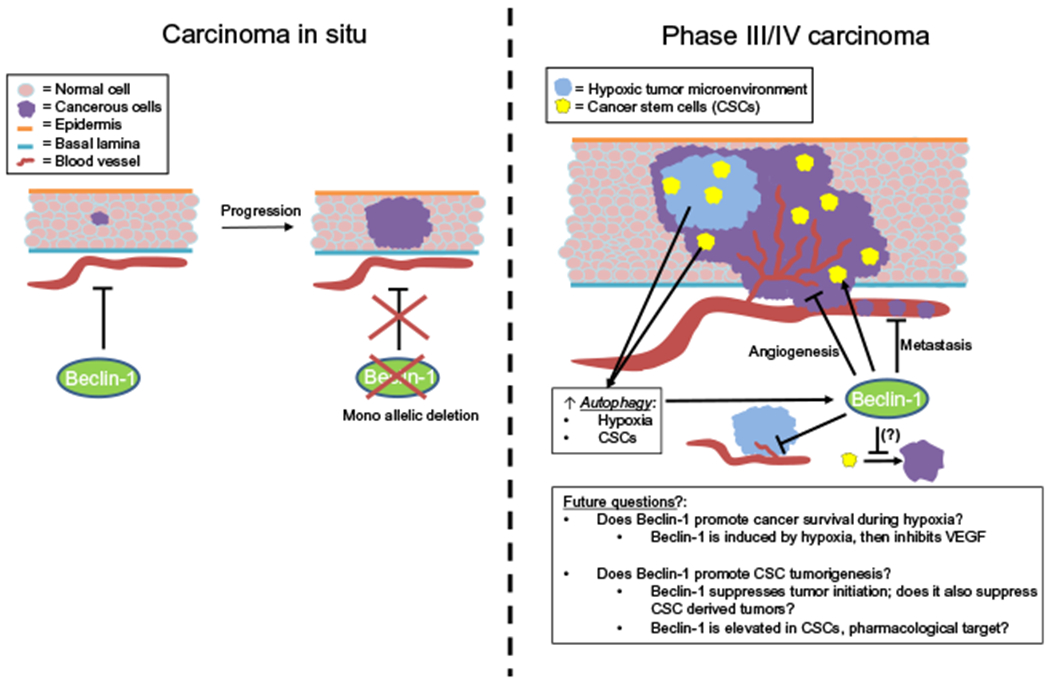
Schematic of the regulation of tumor growth, angiogenesis, and cancer stem cells by Beclin-1. Beclin-1 has been shown to inhibit tumor initiation in transgenic mice. Furthermore, Beclin-1 deletions are common in human cancers. When Beclin-1 is monoallelically deleted, tumor formation therefore becomes much more likely. In large (Phase III/IV) tumors, however, Beclin-1 and autophagy in general may play a role to enhance tumor malignancy. Interestingly however, Beclin-1 is associated with the inhibition of MMP-9 and VEGF to inhibit tumor angiogenesis and cancer cell metastasis despite the effects of tumor hypoxia to induce Beclin-1 and autophagy in vivo. Beclin-1 is also overexpressed in cancer stem cells (CSCs), however it remains unknown it Beclin-1 also inhibits tumor formation by CSCs as seen phenomenologically in primary tumors in transgenic mice. Perhaps Beclin-1 is an appealing target for the development of novel CSC targeted therapeutics; however, this will remain an area for further investigation.
3.1. Beclin-1 as a Tumor Suppressor
Beclin-1 has been implicated as a tumor suppressor, preventing tumor formation in many human tissues. However, the role of Beclin-1 as an autophagy inducer complicates this discussion as autophagy can be either tumor suppressive or oncogenic depending on the cellular context and genetic profile of each tumor (Bhutia et al., 2013; Liu & Debnath, 2016).
Clinically, Beclin-1 deletions are observed in approximately 40% of prostate, 50% of breast, and 75% of ovarian cancers clinically (Gao et al., 1995; Knudson, 1971; Qu et al., 2003). Decreased Beclin-1 expression is also frequently observed in human breast tumor tissue compared to patient matched normal controls (Liang et al., 1999). Furthermore, stable Beclin-1 expressing MCF-7 breast cancer cells were shown to display greater levels of autophagy, inhibiting colony formation in vitro, and decreased tumor growth in vivo (Gao et al., 1995). Beclin-1 heterozygous (+/−) mice were shown to display increased incidence of spontaneous tumor formation concurrent with decreased autophagy levels, establishing a clear role of autophagy, and Beclin-1 as a haploinsufficient tumor suppressor (Qu et al., 2003).
These phenomenological observations strongly implicate a tumor suppressive role of Beclin-1 in many differing tumor types. For the remainder of this review, when possible, Beclin-1 will be discussed within the context of breast cancer cells to focus upon Beclin-1 signal transduction. This is because Beclin-1 is a breast cancer tumor suppressor, its deletion enhances breast cancer malignancy (Liang et al., 1999), and Beclin-1 expression has been shown to be enhanced in breast CSC (Gong et al., 2013). Thus, breast cancer represents an excellent disease to understand the role of Beclin-1 upon malignancies in vitro and in vivo.
UVRAG contains four functional domains, a proline-rich domain, a lipid-binding domain, a C-terminal domain, and a Beclin-1-binding CCD (Gong et al., 2013; Zhao et al., 2012). UVRAG is an autophagy inducer and has been shown to be frequently deleted in breast, colorectal, and gastric cancers (Gong et al., 2013). Furthermore, the serine–threonine-specific protein kinase B (Akt) has been shown to inhibit UVRAG via mTOR (Yang et al., 2013); increased Akt/PI3K/mTOR signaling is thought to be oncogenic during breast tumorigenesis (Mohan et al., 2016). UVRAG is also inhibited by Rubicon (Kim et al., 2015), however, Rubicon has not been shown to promote tumorigenesis, to our knowledge, at this time. Considering these findings, UVRAG has been classified as a candidate haploinsufficient tumor suppressor in breast tissues (Zhao et al., 2012). Biochemical analysis of UVRAG-Beclin-1 binding indicates that UVRAG disrupts the inhibitory Bcl-2-Beclin-1 complex to induce autophagy (Noble, Dong, Manser, & Song, 2008). In this manner, the tumor suppressive effects of Beclin-1 may be suppressed indirectly by the deletion of UVRAG in Beclin-1 wild-type tumors of the breast. UVRAG also mediates the interaction of Beclin-1 with other downstream Beclin-1 signaling molecules to promote cancer therapeutic responses.
Bax-interacting factor-1 (Bif-1) is another tumor suppressor which has been shown to interact with Beclin-1 to inhibit breast cancer malignancy (Runkle, Meyerkord, Desai, Takahashi, & Wang, 2012). Bif-1 is also known as SH3GLB1 or Endophilin B1 (Cuddeback et al., 2001; Pierrat et al., 2001) and contains an N-terminal Bin–Amphiphysin–Rvs (BAR) domain and a C-terminal Src-homology 3 (SH3) domain. The Src oncogene has been shown to bind and phosphorylate Bif-1 on Tyr-80 to inhibit its cell signaling effects and the induction of apoptosis (Yamaguchi et al., 2008). The N-BAR domain is responsible for binding the plasma membrane, driving membrane curvature (Gallop et al., 2006; Masuda et al., 2006; Peter et al., 2004). Bif-1 has been shown to form a complex with Beclin-1 through UVRAG, regulating autophagosome formation (Takahashi et al., 2007). While the exact mechanism of Bif-1 and Beclin-1 interaction remains unknown, Bif-1 has been shown to suppress breast cancer cell migration by degrading EGFR (Runkle et al., 2012). Beclin-1 and EGFR have also been shown to directly interact with each other (Tan, Thapa, Sun, & Anderson, 2015). A survey of Bif-1 mutations in 284 cancerous tissue samples from various origins, revealed only one Bif-1 mutation (0.35%), indicating that Bif-1 mutations are rare in cancers (Kim, Yoo, & Lee, 2008). Interestingly however, Bif-1 expression has been shown to be downregulated in a subset of pancreatic adenocarcinomas (Coppola, Helm, Ghayouri, Malafa, & Wang, 2011) and in invasive and metastatic breast carcinoma in situ (Ho et al., 2009). Therefore, Bif-1 gene mutations may not contribute to tumorigenesis, however, it is likely that Bif-1 dysregulation may contribute to cancer progression via increased invasion and metastasis. Since Bif-1 mutations have not been observed in breast tumors, Bif-1 expression may also be silenced by gene methylation or epigenetic modifications in metastatic breast cancer cells. Even if such epigenetic changes in Bif-1 occurred it is unlikely such changes would establish a stable feedback-loop sufficient for malignant transformation, as shown by Hatziapostolou et al. in hepatocellular carcinoma (HCC; Hatziapostolou et al., 2011).
The activating molecule in Beclin-1-regulated autophagy protein 1 (Ambra1) is critical for autophagy induction and the regulation of Beclin-1 activity. Ambra1 also inhibits apoptosis driven cell death (Fimia, Corazzari, Antonioli, & Piacentini, 2013; Fimia et al., 2007; Gu et al., 2014). Ambra1 is expressed in the majority of pancreatic cancer patient tumors (~64%) and was significantly associated with poor overall survival (P = 0.032; Ko et al., 2013). This finding was surprising as Ambra1 has been described as a haploinsufficient tumor suppressor in MEF’s transformed with the RasV12/E1A oncogene. Furthermore, when mTOR was inhibited, Ambra1 enhanced the interaction of c-Myc and its phosphatase PP2A, inhibiting myc-induced oncogenesis (Cianfanelli et al., 2015). Consistent with these results, Ambra1 as an inducer of Beclin-1 and autophagy, is expected to stimulate cancer therapeutic responses via the Beclin-1 cell signaling axis alone or in combination with mTOR inhibitors such as the Rapalog family of drugs (Fig. 6).
Fig. 6.
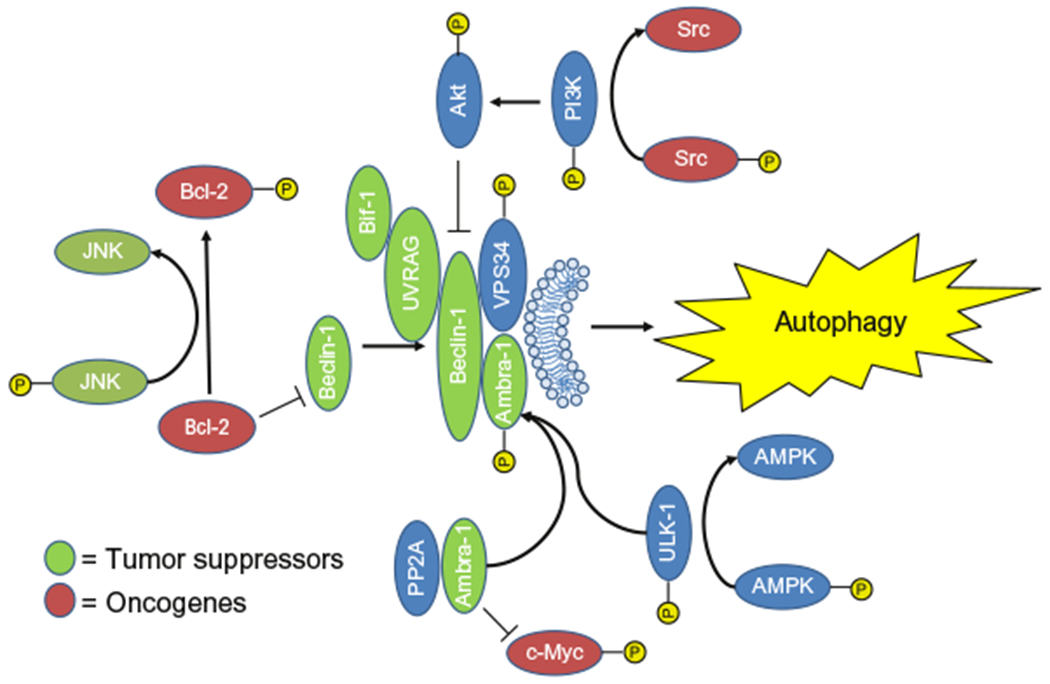
The regulation of Beclin-1 activity by a selection of oncogenes and tumor suppressive molecules. These molecules were colored green for tumor suppressors and red for oncogenes to indicate their interactions with Beclin-1 and autophagy. Molecules shown in blue are thought to be neutral in regard to oncogenesis.
Beclin-1 interacts with Ambra1 via its WD40 domain (Sun, 2016). In mice, the deficiency of Ambra1 results in significant neural tubular defects associated with autophagy impairment, accumulation of ubiquitinated proteins, increased apoptosis, and dysregulated cell division (Fimia et al., 2013). Conversely, the overexpression of Ambra1 in rapamycin-treated cells has been shown to significantly increase both rapamycin-induced and basal levels of autophagy in vitro (Fimia et al., 2013). Therefore, Ambra1 is clearly an autophagy inducer. Ambra1 is physiologically inactive when it associates with mTORC1. mTORC1 inhibits Ambra1 via phosphorylation at Ser-52, preventing the Ambra1 phosphorylation via ULK1 to promote Beclin-1-induced autophagy (Nazio et al., 2013). This study also demonstrated that the ULK1 and Beclin-1 complex crosstalk exists prior to autophagy induction, leading to autophagy-mediated cell signaling in response to cell starvation (Nazio et al., 2013). While the oncogenic effects of Ambra1 in pancreatic cancer requires further characterization, it is likely that Ambra1 is inactivated in pancreatic cancers which express high levels of activated KRAS and mTOR (Morran et al., 2014), which may account for these clinical observations. Additionally, Ambra1 is predominantly expressed in neural tissue so the effects of Ambra1 expression in glioma, and nonneurological malignances, remains an intriguing area for further study.
These tumor suppressive molecules, Beclin-1, UVRAG, Bif-1, and Ambra1, induce autophagy via the direct or in-direct stimulation of Beclin-1 to mediate their cancer therapeutic effects. As described with Ambra1, these molecules can be incriminated as tumor suppressors or oncogenic factors depending on the tissue or the activation of diverse cell signaling contexts, respectively. Before addressing the effects of these and other molecules on enhancing cancer malignancy, we will speak globally about these proteins and their interactions to induce cytotoxic autophagy-dependent cancer cell death. When Bcl-2 is phosphorylated and mTOR is inhibited, Beclin-1 forms an autophagy inducing complex, which together with UVRAG, Bif-1, and Ambra1, inhibits the activity of the oncogenes Bcl-2, Src, and myc to repress tumor malignancy and kill cancer cells (Fig. 6). Therapeutically, stimulating this pathway has been effective using the drug Bergapten in breast cancer cells (De Amicis et al., 2015). Bergapten is a natural psoralen derivative present in many fruits and vegetables. In this study, Bergapten was shown to induce cytotoxic autophagy via the upregulation of phosphatase and tensin homolog (PTEN), p38MAPK/NF-Y, Beclin-1, PI3KIII, UVRAG, and Ambra1, and downregulate mTOR signaling (De Amicis et al., 2015). PTEN is a tumor suppressive molecule which induces autophagy and arrests cell cycle progression (Arico et al., 2001), while suppressing the oncogenes PI3K and AKT, respectively (Arico et al., 2001; Petiot, Ogier-Denis, Blommaart, Meijer, & Codogno, 2000). For cancers with dysregulated mTOR and Bcl-2 expression, targeting the Beclin-1 pathway appears to induce profound cancer therapeutic effects for the treatment of cancer clinically.
3.2. The mda-7-miR-221-Beclin-1 Axis
One of the genes known to have antitumor activity in a broad range of malignancies is mda-7/IL-24 (Cunningham et al., 2005; Fisher, 2005; Fisher et al., 2003). This IL-10 gene family member (Dash et al., 2010) was discovered as a tumor suppressor using subtraction hybridization in terminally differentiated human melanoma cells (Jiang & Fisher, 1993; Jiang, Lin, Su, Goldstein, & Fisher, 1995). Overexpression of mda-7/IL-24 has been shown to sensitize cancer cells to conventional chemotherapeutic drugs (Chada et al., 2006; Emdad, Lebedeva, Su, Gupta, et al., 2007; Emdad, Lebedeva, Su, Sarkar, et al., 2007) and radiotherapeutics (Nishikawa, Ramesh, Munshi, Chada, & Meyn, 2004; Su et al., 2003; Yacoub et al., 2003). One of the most attractive features of mda-7/IL-24 is its capacity to induce cancer-selective cell death, as well as its role as an immune-stimulatory molecule killing distant metastasis via bystander effects (Gao et al., 2008; Sarkar et al., 2002; Sauane et al., 2008; Su et al., 2005, 1998). Furthermore, mda-7/IL-24 also blocks angiogenesis (Nishikawa et al., 2004; Ramesh et al., 2003) slowing tumor growth and decreasing the risk of cancer metastasis. The cancer-specific cell death effects of mda-7/IL-24 have been shown to be mediated through its interaction with the chaperone protein BiP/GRP78, initiating UPR or unfolded protein response (Gupta et al., 2006). Some of the other signaling pathways and molecules downstream of mda-7/IL-24 which have been described in the literature include AIF (Bhoopathi et al., 2016), miR-221 (Pradhan et al., 2017), and Beclin-1 (Bhutia et al., 2010; Pradhan et al., 2017). Owing to its in vitro and in vivo anticancer activity, mda-7/IL-24 has been successfully translated into early phase cancer clinical trials (Cunningham et al., 2005; Emdad et al., 2009; Fisher et al., 2003, 2007; Lebedeva et al., 2005; Tong et al., 2005).
MicroRNAs are noncoding small, 19–22 nucleotide RNA molecules which are key regulators of gene expression. MicroRNAs negatively regulate gene expression by selectively binding and degrading target messenger RNA (mRNA) or by inhibiting mRNA translation. Recent studies have shown that microRNAs are also regulated by mda-7/IL-24. In a human melanoma model, it was shown that mda7/IL-24 downregulates miR-221 (Das et al., 2010). Recent studies from our group also indicated that miR-221 transcriptionally targets Beclin-1 (Pradhan et al., 2017). Beclin-1 is also transcriptionally regulated by a number of other cellular pathways. p65, a NF-κB pathway family member, induces Beclin-1 (Copetti, Demarchi, & Schneider, 2009). 14-3-3 protein and E2F1 also transactivate Beclin-1 leading to the upregulation of autophagy (Wang, Ling, & Lin, 2010). Previous studies have also shown the role of microRNAs in Beclin-1 regulation. miR-30a targets Beclin-1 UTR and regulates the autophagy process (Zhu et al., 2009). Overexpression of miR-30a downregulates Beclin-1 transcripts as well as protein levels. MicroRNA-221 is one of the most frequently upregulated miRs in cancer. It targets a number of tumor suppressors including p27 (le Sage et al., 2007), PUMA (Zhang et al., 2010), PTEN (Garofalo et al., 2009), and p57 (Fornari et al., 2008). Beclin-1 can also be transcriptionally targeted by miR-221 (Pradhan et al., 2017), blocking toxic autophagic cell death. Overall this regulatory loop identified a new pathway of mda-7/IL-24-mediated cancer-specific toxic autophagy leading to cell death (Fig. 7).
Fig. 7.
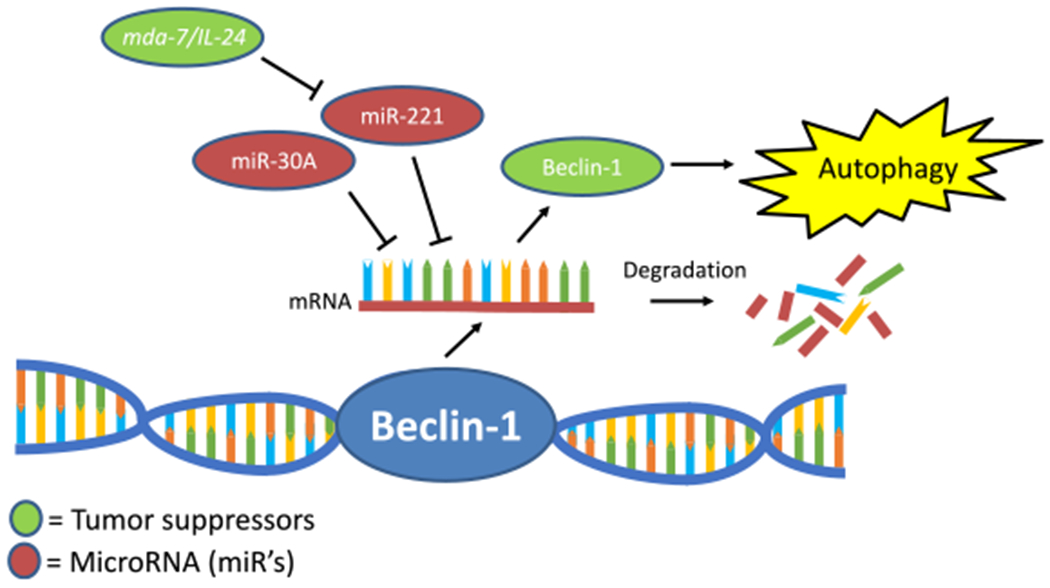
The regulation of Beclin-1 expression via microRNAs and the effect of mda-7/IL-24 to increase the activity of Beclin-1 to induce autophagy and lyse cancer cells. Green circles indicate tumor suppressive molecules. Red circles are indicative of microRNA. The blue double-stranded molecule is a cartoon of the Beclin-1 gene. The single-stranded molecule with the red backbone is indicative of the Beclin-1 mRNA transcript. The debris is indicative of mRNA degradation following the association of miRs with Beclin-1 mRNA transcripts.
3.3. Arguments for and Against the Oncogenicity of Beclin-1
Enhanced autophagy signaling via Beclin-1 may also promote cancer malignancy during periods of cell stress imposed by hypoxia and the metastatic process. The phenomenological observations from breast cancer patients indicate that Beclin-1 expression declines with increasing cancer stages, however, some reports suggest Beclin-1 and autophagy may contribute to resistance to cancer chemotherapy (Ying et al., 2015) and radiation (Apel, Herr, Schwarz, Rodemann, & Mayer, 2008). Beclin-1 may play a role in breast CSC maintenance which are refractory to chemotherapy and radiation treatment (Gong et al., 2013). These possibilities will be discussed and the contribution of Beclin-1 in promoting cancer oncogenesis and tumor malignancy in greater detail in this review.
3.3.1. Beclin-1 Expression and Cancer
Beclin-1 monoallelic deletions have been observed in many tumors implicating Beclin-1 as a tumor suppressive molecule. A more detailed description of Beclin-1 in cancer is shown in Table 1 (Ahn et al., 2007; Cliby et al., 1993; Ding et al., 2008; Eccles et al., 1992; Futreal et al., 1992; Gao et al., 1995; Koukourakis et al., 2010; Li et al., 2009; Li, Chen, et al., 2010; Lin et al., 2013; Miracco et al., 2010, 2007; Pirtoli et al., 2009; Radwan et al., 2016; Russell et al., 1990;Saito etal., 1993; Tangir et al., 1996; Zhang etal., 2009; Zhou et al., 2013). While most studies indicate that Beclin-1 is tumor suppressive, it is possible that the deletion of Beclin-1 may have led to the loss of autophagy signaling, and thereby stimulated many other pathways including necrosis and inflammation which are associated with tumorigenesis (Mathew & White, 2011; White & DiPaola, 2009). Interestingly, Beclin-1 biallelic deletions have not been observed in tumors clinically, failing the Knudson two-hit hypothesis required for genes to be characterized as true tumor suppressors (Knudson, 1971). Therefore, Beclin-1 is described as a haploinsufficient tumor suppressor instead as Beclin-1 heterozygous mice displayed greater tumor formation (Qu et al., 2003). The lack of biallelic Beclin-1 deletions in tumors clinically also indicates that expression of at least one functional Beclin-1 allele is required for cancer cell survival, or at least, for tumor growth.
Table 1.
The Expression of Beclin-1 in Cancers Clinically
| Cancer Types | Dysregulation of Beclin-1 Expression | Clinical Correlations | References |
|---|---|---|---|
| Acute Lymphocytic Leukemia (ALL) | Decreased Beclin-1 expression | Beclin-1 extended ALL patient survival seemingly by inhibiting HIF-1α | Radwan, Hamdy, Hegab, and El-Mesallamy (2016) |
| Brain Cancer | Expression of Beclin-1 mRNA is decreased in higher-grade tumors | Low Beclin-1 correlates with poor survival | Lin et al. (2013) and Miracco et al. (2007) |
| Brain Cancer | Cytoplasmic Beclin-1 was positively associated with apoptosis and the inhibition of cell proliferation | High cytoplasmic Beclin-1 expression is associated with better survival and high Karnofski classification values | Pirtoli et al. (2009) |
| Breast Cancer | The locus which harbors Beclin-1 is monoallelically deleted in ~50% of cases | ND | Futreal et al. (1992) and Saito et al. (1993) |
| Breast Cancer | Decreased Beclin-1 mRNA and protein expression, monoallellic deletions, promoter methylation, and occurred in breast cancer tissues | Higher-Beclin-1 mRNA expression in BRCA1-positive tumors | Li, Chen, et al. (2010) |
| Colorectal Cancer | Underexpresssion and extensive overexpression of Beclin-1 were observed in 15.5% and 21.3% of cases, respectively | Dysregulated Beclin-1 was associated with poorer survival; “normal-like” Beclin-1 expression improved survival | Koukourakis et al. (2010) |
| Colorectal Cancer | Beclin-1 expression was greater in cancerous than matched normal tissue in 95% of cases | ND | Ahn et al. (2007) |
| Colorectal Cancer | Strong expression of Beclin-1 was detected in stage IIIB tumors but not adjacent normal tissues | Higher-Beclin-1 expression is associated with longer survival | Li et al. (2009) |
| Gastric Cancer | Beclin-1 expression was greater in cancerous than matched normal tissue in 83% of cases | ND | Ahn et al. (2007) |
| Liver Cancer | A >twofold decrease in Beclin-1 mRNA was observed in 45.5% HCC tissues; decreased Beclin-1 protein expression was confirmed in six out of eight cases | Lower levels of Beclin-1 mRNA was correlated with recurrence. Decreased patient survival was observed in Beclin-1 negative, Bcl-xL-positive tumors | Ding et al. (2008) |
| Lung Cancer | Beclin-1 was significantly lower in NSCLC compared with adjacent normal controls | Decreased Beclin-1 lead to increased recurrence, decreased overall survival | Zhou et al. (2013) |
| Melanoma | Beclin-1 and LC3 are downregulated during disease progression | Beclin-1 and LC3 mRNA levels can discriminate between nonmalignant and malignant lesions | Miracco et al. (2010) |
| Osteosarcoma | Beclin-1 protein levels are lower (by IHC) in osteosarcoma than in normal bone | ND | Zhang et al. (2009) |
| Ovarian cancer | The locus harboring Beclin-1 is monoallelically deleted in ~75% of cases | ND | Cliby et al. (1993), Eccles et al. (1992), Russell, Hickey, Lowry, White, and Atkinson (1990), and Tangir et al. (1996) |
| Prostate cancer | The locus harboring Beclin-1 is monoallelically deleted in ~40% of cases | ND | Gao et al. (1995) |
Beclin-1 plays a significant role in the regulation of tumor growth in numerous malignancies. ND indicates not determined.
Qu et al. rigorously tested whether or not Beclin-1 functions as a tumor suppressor or an oncogene by crossing mice which express the oncogenic hepatitis B virus (HBV) large-envelope polypeptide with Beclin-1 wild-type or heterozygous mice, respectively (Qu et al., 2003). These data indicated that 15% of Beclin-1 heterozygous mice developed palpable tumors with confirmed histological malignancy vs only 1% of Beclin-1 wild-type littermates that formed tumors at 13–18 weeks of age. One hundred mice were examined in both the Beclin-1 wild-type and heterozygous groups, respectively. These malignancies were consistent with lymphoma, lung, and liver cancers. Beclin-1 heterozygote mice also displayed tissue hyperplasia in the mammary gland and in splenic germinal centers. Beclin-1 heterozygous tumors also displayed greater overall cell division and larger tumor formation (Qu et al., 2003). Finally, Beclin-1 heterozygous mice were shown to display less autophagy activity in bronchial epithelial cells and germinal B lymphocytes. Tumors were later observed to form from these tissues (bronchus and lymphocytes), indicating that autophagy induced by Beclin-1 was protective against lung and lymphoma oncogenesis in mice expressing the HBV oncogene (Qu et al., 2003). This study was carefully designed and categorically disproved the hypothesis that Beclin-1 promotes tumor oncogenesis, indicating that the downregulation of Beclin-1 expression both promoted tumor cell growth and enhanced tumor formation in vivo. Consistent with these observations, Beclin-1 may also serve as a tumor suppressor gene due to its ability to control the cell cycle (Sun et al., 2011) and stimulate apoptosis (Li et al., 2013).
Alternatively, when Beclin-1 is suppressed and autophagy is therefore dysregulated, tumorigenesis may occur directly via other autophagy-related molecules or indirectly via the formation of oxidative free radicals. Although the crosstalk between mitochondrial dysfunction, autophagy, and redox signaling are not well understood (Lee, Giordano, & Zhang, 2012), these concepts will be discussed further here. For example, p62/SQSTM1, which binds ubiquitinated proteins and sequesters them into the autophagolysosome for degradation, has been shown to be upregulated along with other autophagy genes by nuclear factor-erythroid 2-related factor 2 (Nrf2) nuclear translocation and redox signaling (Giles, Gutowski, Giles, & Jacob, 2003; Riley et al., 2010). By this mechanism, Nrf2 has been shown to increase p62 expression, leading to p62-mediated reactive oxygen species scavenger activity in models of neurodegeneration (Lee et al., 2012).
Relating these concepts back to cancer, it is hypothesized that deficiencies in autophagy which could lead to the accumulation of oxidative radicals, contributing to overall genome instability and tumorigenesis. Interestingly however, the upregulation of p62 was only shown to be oncogenic when autophagy is suppressed (Mathew et al., 2009). Furthermore, p62 is essential for HER2-driven oncogenic breast transformation via multiple signaling pathways, including the PTEN/PI3KIII/AKT axis, WNT/β-catenin, NF-κB, and NRF2-KEAP1 pathways, respectively (Cai-McRae, Zhong, & Karantza, 2015). For example, Beclin-1 has been shown to interact with Her2 and when this interaction is disrupted by the dual tyrosine kinase inhibitor, lapatinib, autophagy is induced to lyse Her2 expressing breast cancer cells via the phosphorylation of Akt (Han et al., 2013). These studies indicate that the disruption of p62-mediated oxidative scavenging is cancer therapeutic and suppresses the formation of breast tumors. The cancer therapeutic consequences of autophagy disruption via Beclin-1 and p62 deletion, suggests a dual role of autophagy during carcinogenesis such that autophagy suppresses tumor initiation (Kung, Budina, Balaburski, Bergenstock, & Murphy, 2011), but autophagy also may support the maintenance of established tumors and their respective microenvironments during hypoxia (Tan et al., 2016), further enhancing tumor invasion and metastasis (Mowers, Sharifi, & Macleod, 2017); these concepts are summarized as illustrated in Fig. 5.
3.4. Beclin-1 Expression and Its Effect Upon Cancer Progression
Overall, the expression of Beclin-1 is not clearly implicated as repressed or overexpressed in colorectal cancers clinically (Koukourakis et al., 2010). In T-cell lymphoma, however, decreased Beclin-1 expression associates with decreased overall and progression-free survival indicating tissue-specific consequences of Beclin-1 upon cancer malignancy (Huang et al., 2010). Therefore, to elucidate the effect of Beclin-1 upon colorectal tumors clinically, the effect of Beclin-1 expression upon colorectal adenocarcinoma prognosis was assessed in 155 patients treated with surgery alone (Koukourakis et al., 2010). Beclin-1 expression was shown to be at normal levels (n=62, 40%), under expressed (n=24, 15.5%), limited overexpression (n=36, 23.2%), and extensively overexpressed (n=33, 21.3%) in these 155 colorectal adenocarcinoma patients by tumor histopathological analysis. Patient 3-year overall survival was the greatest in the normal-like Beclin-1 expression group (89%) compared to Beclin-1 limited overexpression (77.7%), Beclin-1 extensive overexpression (43.3%), however, the lowest patient survival was shown in the Beclin-1 under expression group (31%). The authors indicated that two biologically distinct pathways of Beclin-1 activity, both linked with tumor aggressiveness and that the maintenance of physiological levels of Beclin-1 in cancers is relevant in colon cancer (Koukourakis et al., 2010). First, the loss of Beclin-1, by monoallelic gene deletion, was shown to stimulate breast oncogenesis (Aita et al., 1999). Second, as Beclin-1 interacts with the antiapoptotic Bcl-2 protein (Cao & Klionsky, 2007), the loss of Beclin-1 may have potentiated the antiapoptotic machinery which may account, at least in part, for the poorer prognosis of Beclin-1 negative colorectal tumors. Furthermore, Li et al. found that increased Beclin-1 expression is linked with better stage III colorectal cancer patient prognosis (Liang, Yu, Brown, & Levine, 2001). Beclin-1 was also shown to be a crucial regulator of colorectal cancer growth and metastasis upon further study (Koukourakis et al., 2010). Therefore, additional insights into the role of Beclin-1 to promote cancer malignancy are critical to understanding this process mechanistically.
3.4.1. Beclin-1 in Cancer Invasion, Metastasis, and Tumor Hypoxia
Autophagy is known to support the survival of cancer cells in hypoxic tumor microenvironments (Mathew & White, 2011). However, Beclin-1 has also been shown to inhibit the proliferation, invasion, and metastasis of CaSki cervical cancer cells in vivo (Sun et al., 2011). In this cancer model system, Beclin-1 expression was shown to arrest cancer cells in the G0/G1 phase of the cell cycle and to inhibit vascular endothelial growth factor (VEGF) and matrix metalloprotease 9 (MMP-9), which likely was responsible for the observed repression of invasion and metastasis in CaSki tumors which stably overexpress Beclin-1 relative to pcDNA3.1 (Sun et al., 2011). Beclin-1 overexpression was also shown to suppress angiogenesis, VEGF, and MMP-9 expression in vitro (Sun et al., 2011). Interestingly, however, in Beclin-1 heterozygous mice, differences in circulating VEGF were not observed whereas the production of EPO was markedly elevated in the circulation of Beclin-1 heterozygotes relative to Beclin-1 wild-type mice (Lee, Kim, Jin, Choi, & Ryter, 2011). It is possible therefore that Beclin-1 only suppresses VEGF in vitro or in the tumor microenvironment. Taken together, these data suggest that Beclin-1 plays a role in preventing cancer malignancy via the suppression of cancer cell growth, invasion, and metastasis, while simultaneously preventing tumor angiogenesis.
Consistent with these findings, heterozygous disruption of Beclin-1 accelerated tumor growth and angiogenesis under hypoxic conditions in melanoma bearing mice in vivo (Lee et al., 2011). Cells cultured from Beclin-1 wild-type and Beclin-1 heterozygous mice indicated that Beclin-1 expression inhibited angiogenesis via decreased hypoxia-inducible factor-2α (HIF-2α) expression, limiting endothelial cell proliferation, and tube formation in response to hypoxia (Lee et al., 2011). Furthermore, these data demonstrate that mice deficient in Beclin-1 displayed a proangiogenic phenotype associated with HIF-2α upregulation and increased erythropoietin production (Lee et al., 2011). Conversely, however, when colon cancer patient samples were analyzed histologically, these data suggested high levels of Beclin-1 expression was associated with improved survival (Li et al., 2009) , while an in depth metaanalysis of data from six clinical studies indicated that elevated Beclin-1 expression was associated with tumor metastasis and poor prognosis in colorectal cancer patients (Han et al., 2014). On a molecular level, Beclin-1 negatively regulates angiogenesis in vivo in murine model systems, with implications for the inhibition of tumor growth (Sun et al., 2011). While the role of Beclin-1 in colorectal cancer pathology remains controversial, it is clear that Beclin-1 is an intriguing molecular target for colorectal and many other human malignancies. These competing findings may also reflect the differential effects of Beclin-1 depending on its subcellular compartmentalization in these individual tumor cells or the expression of other Beclin-1 regulatory molecules.
3.5. Beclin-1 and Resistance to Cancer Therapy
3.5.1. The Effect of Beclin-1 Upon Cancer Chemotherapeutic Efficacy and Patient Survival
While Beclin-1 possesses numerous tumor suppressive mechanisms of action, the effects of Beclin-1 on autophagy induction may antagonize the efficacy of cytotoxic cancer therapeutics acting through apoptosis. These antagonistic effects of autophagy upon apoptosis are likely a consequence of numerous individual molecular interactions (Moreau, Luo, & Rubinsztein, 2010) . Two examples are the mutually inhibitory interactions between LC3 and Beclin-1 with the apoptosis inducing and effector caspase, caspase-3 (Ma, Zhang, Huang, Guo, & Hu, 2016). Interestingly, while autophagy has been implicated in enhancing cancer cell survival via the inhibition of apoptosis (Moreau et al., 2010), such antagonism has not be observed via the autophagy inducer Beclin-1; as determined via selectively silencing Beclin-1 expression with siRNA (Huang et al., 2014). In this manner, the role of Beclin-1 upon cancer therapeutic responses are predicated upon many factors, reflecting the Janus faces of autophagy and its effects upon Beclin-1 signaling.
The effects of Beclin-1 expression upon cancer therapeutic responses of colorectal and ovarian cancer patients treated with the apoptosis inducers cetuximab or carboplatin and paxilitaxel, respectively, were determined in the clinical setting. Colorectal cancer patients treated with cetuximab-containing or noncetuximab-containing chemotherapy were then evaluated for their respective cancer therapeutic responses and Beclin-1 expression in treated tumors (Guo et al., 2011). Cetuximab is a monoclonal antibody which inhibits the EGFR and has been approved for use in many human cancers. Cetuximab has been reported to induce autophagy via the suppression of the mammalian target of rapamycin (mTOR) (Li, Lu, Pan, & Fan, 2010), however, cetuximab has also been implicated as an apoptosis inducer via the inhibition of the mitogen-activated protein kinase (MAPK) and the Janus kinase/STAT3 pathways in head and neck squamous cell carcinoma cells (Bonner et al., 2000). In colorectal cancer patients treated with cetuximab, decreased Beclin-1 expression was associated with prolonged progression-free survival (Guo et al., 2011). The authors indicated that the role of autophagy in cetuximab treatment is controversial and that increased or decreased levels of Beclin-1 expression may not be indicative of the presence or absence of autophagy in treated tumors (Guo et al., 2011). However, decreased LC3 expression was correlated with greater overall response rates for cetuximab-treated colorectal cancer patients (Guo et al., 2011). In ovarian cancer patients treated with surgical resection followed by treatment with the cytotoxic cancer chemotherapeutics carboplatin and paclitaxel; patient cancer therapeutic responses and Beclin-1 expression in treated tumors were also evaluated (Valente et al., 2014). Carboplatin is a platinum-based chemotherapeutic and cell cycle non-selective inducer of DNA damage and apoptosis. Paclitaxel is a cell cycle selective inhibitor of cell division which stabilizes microtubules by binding β-tubulin polymers, preventing cell division, and inducing apoptosis via prolonged activation of the mitotic cell cycle checkpoint. No differences were observed at 5 years’ posttreatment, however, a greater proportion of ovarian cancer patients whose tumors expressed higher levels of Beclin-1 displayed significantly greater survival (n = 34, 83%; P-value < 0.03) relative to patients with tumors expressing less Beclin-1 (n = 7, 17%; Valente et al., 2014). Therefore, Beclin-1 appears to antagonize the cancer therapeutic efficacy of cetuximab in colorectal cancer cells while Beclin-1 expression appears to enhance the cancer therapeutic effects of the apoptosis inducers carboplatin and paclitaxel in ovarian cancer patients. Similarly, Beclin-1 overexpression enhanced the sensitivity of CaSki cervical cancer cells to apoptosis via cisplatin, paclitaxel, 5-FU, and epirubicin (Sun et al., 2010). Unfortunately, to the best of our knowledge, no studies have compared the same cancer chemotherapeutics upon the survival of cancer patients with differing tumor types investigating the effects of Beclin-1 as a clinical biomarker of overall cancer patient survival, indicating the need for further research in this area. Therefore, it cannot be concluded at this time if either fundamental differences exist between Beclin-1 expression and the oncogenesis of ovarian and colorectal cancers or if yet undiscovered molecular markers for response to these drugs may explain the differences observed between these cancer therapeutics, respectively.
3.5.2. The Effect of Beclin-1 Upon Radiation Therapy, Autophagy, and UVRAG
Growing evidence suggests that autophagy contributes to the resistance of cancers to radiation therapy (Gewirtz, 2014; Wilson et al., 2011; Yin et al., 2011), however, the effects of the Beclin-1 pathway upon radiation-resistance remains undefined. Beclin-1 has been shown to improve DNA stability by physical interactions with UVRAG (Park, Tougeron, Huang, Okamoto, & Sinicrope, 2014). Very recently, Beclin-1-mediated DNA stability was shown to occur independently of autophagy via interaction with UVRAG to enhance cellular DNA damage repair and maintain genome integrity (Xu et al., 2017; Zhao et al., 2012). Likewise, increased UVRAG expression has been shown to protect cancer cells against radiation treatment (Park et al., 2014). UVRAG also disrupts irradiation-induced apoptosis via altering Bax localization thereby inhibiting apoptosis (Yin et al., 2011), when Beclin-1 translocates to the nucleus. Consequently, the downregulation of UVRAG expression inhibits autophagy and sensitizes cancer cells to DNA damage-mediated cell death via the upregulation of serine–threonine-specific protein kinase B (Akt) and mTOR (Yang et al., 2013).
The clinical application of radiation therapy entails the targeted exposure of tumors to high doses to cytotoxic doses of ionizing radiation, leading to the formation of oxidative radicals and DNA damage and eventually apoptotic cell death via p53 (Cui et al., 2016). In K-ras-induced transgenic murine lung tumors, however, treatment with Beclin-1 aerosols was shown to sensitize these tumors to fractionated radiation treatment (Shin et al., 2012). This result was surprising considering the role of the autophagy proteins Beclin-1, Atg5, and Atg12 to protect against DNA damage (He, Dai, Jin, Liu, & Rent, 2012). However, if the effects of Beclin-1 and UVRAG upon autophagy are independent from its effects upon radiation-induced cell death, as recent reports suggest (Xu et al., 2017; Zhao et al., 2012), additional molecules may predict the consequences of Beclin-1 and UVRAG signaling in cancerous tissues.
Returning to the discussion of the K-ras-induced lung carcinogenic model (Shin et al., 2012), by supplementing these cells with Beclin-1 containing aerosols, the resistance of these tumors to radiation treatment inherent to K-ras expressing tumors was overcome (Bernhard et al., 2000). It is likely that Beclin-1 stimulated the death of tumors following radiation treatment by stimulating DNA damage responses in vivo. Phenomenologically, radiation treatment does not appear to depend upon autophagy. For example, while autophagy inhibition rendered some breast cancer cells such as MCF-7 and ZR-75 more sensitive to radiation (Bristol et al., 2012; Wilson et al., 2011), the inhibition of autophagy in 4H1 and HS578t breast cancer cells had no effect upon their viability posttreatment (Bristol et al., 2013; Gewirtz, 2014). Mechanistically, the inhibition of autophagy has only been shown to enhance radiation efficacy in cancer cells with functional P53 (Chakradeo et al., 2015). Beclin-1 and UVRAG have also been shown to protect cells against radiation treatment (Park et al., 2014), however, the antagonism of autophagy may only be therapeutic due to the inhibitory crosstalk of the downstream apoptosis autophagy-signaling molecules of the respective DNA damage response genes P53 and Beclin-1, respectively (Ma et al., 2016). Therefore, it appears that Beclin-1 expression supports the lysis of cancer cells in response to DNA damage, however, with careful consideration the expression profiles of p53 and UVRAG, the development of more personalized radiation treatment protocols is feasible to improve patient clinical outcomes from radiation treatment.
3.6. Beclin-1 and CSC Maintenance
One fundamental problem with cancer chemotherapy and radiation treatments is the inherent resistance of CSCs to these therapeutic approaches. Gong et al. created three-dimensional cultures of breast cancer cells called “mammospheres” to study the biology of breast CSC containing tumors in an easily manipulated model system in vitro. Using this approach, Beclin-1 expression and autophagic flux were enhanced in cancer cells grown in spherical vs monolayer cultures (Gong et al., 2013). Beclin-1 expression was enhanced further in mammosphere cultures in the presence of CSCs (Gong et al., 2013). Using models of Beclin-1 suppression (tet-off, shRNA), decreased Beclin-1 expression was shown to inhibit the formation of spherical cultures by cancer cells and CSCs in vitro; decreased Beclin-1 also decreased tumor volumes produced from implanted mammospheres in vivo (Gong et al., 2013). Taken together, these data indicated that Beclin-1 expression was required for breast CSC-induced tumorigenesis.
Considering that enhanced Beclin-1 expression was associated with spherical cancer cell growth and was upregulated in the presence of CSCs, additional insight is necessary to elucidate the cancer therapeutic or oncogenic effects of Beclin-1 expression upon CSCs. One of the best validated markers for stem/progenitor-like cells is the expression of aldehyde dehydrogenase 1 (ALDH1; Martin et al., 2016). High levels of ALDH1 expression can correctly identify tumorigenic cell populations prior to implantation and is correlated with poor overall prognosis in cancer patients (Martin et al., 2016). At this time, Beclin-1 has not been shown to interact with ALDH1 or other cancer stem cell markers mechanistically. However, the close association of enhanced Beclin-1 expression in ALDH1-positive cells indicates that a potential direct or indirect signaling axis may exist between these two molecules. Therefore, Beclin-1 appears to contribute to the maintenance of CSCs and may serve as an intriguing molecular target for their destruction.
4. CONCLUSIONS AND FUTURE DIRECTIONS
The tumor suppressive properties of Beclin-1 appear to be a result of complex series of interactions with autophagy and apoptotic cell death pathways in cooperation with other tumor suppressive molecules such as UVRAG, Bif-1, Ambra1, and mda-7/IL-24. Evolutionarily, Beclin-1 appears to have emerged as multicellular eukaryotes needed additional biochemical pathways by which to adapt to cell stress, maintain the integrity of their DNA genomes, or provide PCD mechanism to enhance the survival of multicellular organisms. In this review, we have also postulated the evolutionary events in the molecular evolution of Beclin-1. The distribution of Beclin-1 in eukaryotes requires parallel losses in protest lineages or parallel gains of the gene in plant and fungi/animal genomes. Which scenario is correct will require further investigation. In human cancers, Beclin-1 is frequently monoallelically deleted and, it is implied that, one functional copy is always present in human cells and this remaining allele must not be silenced via methylation or epigenetic modifications for the cancer cells to survive and grow. Furthermore, the deletion of both Beclin-1 alleles has not been observed in tumors, clinically supporting this claim. The overexpression of Beclin-1 has also been implicated in inhibiting angiogenesis and tumor metastasis. One of the most prominent and growing concerns in modern oncology is the elimination of CSC. Beclin-1 expression has been shown to be elevated in these cells, comprising an intriguing molecular target for the development of novel CSC treatment modalities. Although clearly complex, defining the precise role of Beclin-1 in mediating cancer cell development, response to therapy and progression will be pivotal in determining if manipulating this molecule can be used to enhance therapeutic outcomes.
Supplementary Material
ACKNOWLEDGMENTS
Support for our laboratories was provided in part by National Institutes of Health Grants R01 CA097318 (P.B.F.), R01 CA168517 (Maurizio Pellecchia and P.B.F.), GM093857 VCU IRACDA (P.B.F. and Joyce A. Lloyd), and P50 CA058326 (M.G.P. and P.B.F.); the Samuel Waxman Cancer Research Foundation (P.B.F. and D.S.); National Foundation for Cancer Research (P.B.F.); NCI Cancer Center Support Grant to VCU Massey Cancer Center P30 CA016059 (P.B.F.); and VCU Massey Cancer Center developmental funds (P.B.F.). P.B.F. and D.S. are SWCRF investigators. P.B.F. holds the Thelma Newmeyer Corman Chair in Cancer Research in the VCU Massey Cancer Center. R.D. thanks the Sackler Institute for Comparative Genomics, the Simons Foundation, and the Korein Foundation for continued support.
Footnotes
Conflict of interest: Dr. Fisher is a cofounder and has ownership interest in Cancer Targeting Systems (CTS), Inc.
SUPPLEMENTARY MATERIAL
Supplementary data to this article can be found online at https://doi.org/10.1016/bs.acr.2017.11.002.
REFERENCES
- Ahn CH,Jeong EG, Lee JW, Kim MS, Kim SH, Kim SS, et al. (2007). Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. APMIS, 115(12), 1344–1349. 10.1111/j.1600-0463.2007.00858.x. [DOI] [PubMed] [Google Scholar]
- Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, et al. (1999). Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics, 59(1), 59–65. 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- Apel A, Herr I, Schwarz H, Rodemann HP, & Mayer A (2008). Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Research, 68(5), 1485–1494. 10.1158/0008-5472.CAN-07-0562. [DOI] [PubMed] [Google Scholar]
- Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, et al. (2001). The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. The Journal of Biological Chemistry, 276(38), 35243–35246. 10.1074/jbc.C100319200. [DOI] [PubMed] [Google Scholar]
- Bernhard EJ, Stanbridge EJ, Gupta S, Gupta AK, Soto D, Bakanauskas VJ, et al. (2000). Direct evidence for the contribution of activated N-ras and K-ras oncogenes to increased intrinsic radiation resistance in human tumor cell lines. Cancer Research, 60(23), 6597–6600. [PubMed] [Google Scholar]
- Bhoopathi P, Lee N, Pradhan AK, Shen XN, Das SK, Sarkar D, et al. (2016). mda-7/IL-24 induces cell death in neuroblastoma through a novel mechanism involving AIF and ATM. Cancer Research, 76(12), 3572–3582. 10.1158/0008-5472.CAN-15-2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutia SK, Dash R, Das SK, Azab B, Su ZZ, Lee SG, et al. (2010). Mechanism of autophagy to apoptosis switch triggered in prostate cancer cells by antitumor cytokine melanoma differentiation-associated gene 7/interleukin-24. Cancer Research, 70(9), 3667–3676. 10.1158/0008-5472.CAN-09-3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutia SK, Mukhopadhyay S, Sinha N, Das DN, Panda PK, Patra SK, et al. (2013). Autophagy: Cancer’s friend or foe? Advances in Cancer Research, 118, 61–95. 10.1016/B978-0-12-407173-5.00003-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner JA, Raisch KP, Trummell HQ, Robert F, Meredith RF, Spencer SA, et al. (2000). Enhanced apoptosis with combination C225/radiation treatment serves as the impetus for clinical investigation in head and neck cancers. Journal of Clinical Oncology, 18(21 Suppl), 47S–53S. [PubMed] [Google Scholar]
- Bristol ML, Di X, Beckman MJ, Wilson EN, Henderson SC, Maiti A, et al. (2012). Dual functions of autophagy in the response of breast tumor cells to radiation: Cytoprotective autophagy with radiation alone and cytotoxic autophagy in radiosensitization by vitamin D 3. Autophagy, 8(5), 739–753. 10.4161/auto.19313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristol ML, Emery SM, Maycotte P, Thorburn A, Chakradeo S, & Gewirtz DA (2013). Autophagy inhibition for chemosensitization and radiosensitization in cancer: Do the preclinical data support this therapeutic strategy? The Journal of Pharmacology and Experimental Therapeutics, 344(3), 544–552. 10.1124/jpet.112.199802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai-McRae X, Zhong H, & Karantza V (2015). Sequestosome 1/p62 facilitates HER2-induced mammary tumorigenesis through multiple signaling pathways. Oncogene, 34(23), 2968–2977. 10.1038/onc.2014.244. [DOI] [PubMed] [Google Scholar]
- Cao Y, & Klionsky DJ (2007). Physiological functions of Atg6/Beclin 1: A unique autophagy-related protein. Cell Research, 17(10), 839–849. 10.1038/cr.2007.78. [DOI] [PubMed] [Google Scholar]
- Chada S, Mhashilkar AM, Liu Y, Nishikawa T, Bocangel D, Zheng M, et al. (2006). mda-7 gene transfer sensitizes breast carcinoma cells to chemotherapy, biologic therapies and radiotherapy: Correlation with expression of bcl-2 family members. Cancer Gene Therapy, 13(5), 490–502. 10.1038/sj.cgt.7700915. [DOI] [PubMed] [Google Scholar]
- Chakradeo S, Sharma K, Alhaddad A, Bakhshwin D, Le N, Harada H, et al. (2015). Yet another function of p53—The switch that determines whether radiation-induced autophagy will be cytoprotective or nonprotective: Implications for autophagy inhibition as a therapeutic strategy. Molecular Pharmacology, 87(5), 803–814. 10.1124/mol.114.095273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, et al. (2015). AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nature Cell Biology, 17(1), 20–30. 10.1038/ncb3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliby W, Ritland S, Hartmann L, Dodson M, Halling KC, Keeney G, et al. (1993). Human epithelial ovarian cancer allelotype. Cancer Research, 53(10 Suppl), 2393–2398. [PubMed] [Google Scholar]
- Copetti T, Demarchi F, & Schneider C (2009).p65/RelA binds and activates the beclin 1 promoter. Autophagy, 5(6), 858–859. [DOI] [PubMed] [Google Scholar]
- Coppola D, Helm J, Ghayouri M, Malafa MP, & Wang HG (2011). Down-regulation of Bax-interacting factor 1 in human pancreatic ductal adenocarcinoma. Pancreas, 40(3), 433–437. 10.1097/MPA.0b013e318205eb03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddeback SM, Yamaguchi H, Komatsu K, Miyashita T, Yamada M, Wu C, et al. (2001). Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. The Journal of Biological Chemistry, 276(23), 20559–20565. 10.1074/jbc.M101527200. [DOI] [PubMed] [Google Scholar]
- Cuervo AM (2004). Autophagy: In sickness and in health. Trends in Cell Biology, 14(2), 70–77. 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Cui L, Song Z, Liang B, Jia L, Ma S, & Liu X (2016). Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells. Oncology Reports, 35(6), 3639–3647. 10.3892/or.2016.4752. [DOI] [PubMed] [Google Scholar]
- Cunningham CC, Chada S, Merritt JA, Tong A, Senzer N, Zhang Y, et al. (2005). Clinical and local biological effects of an intratumoral injection of mda-7 (IL24; INGN 241) in patients with advanced carcinoma: A phase I study. Molecular Therapy, 11(1), 149–159. 10.1016/j.ymthe.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Das SK, Sokhi UK, Bhutia SK, Azab B, Su ZZ, Sarkar D, et al. (2010). Human polynucleotide phosphorylase selectively and preferentially degrades microRNA-221 in human melanoma cells. Proceedings of the National Academy of Sciences of the United States of America, 107(26), 11948–11953. 10.1073/pnas.0914143107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash R, Bhutia SK, Azab B, Su ZZ, Quinn BA, Kegelmen TP, et al. (2010). mda-7/IL-24: A unique member of the IL-10 gene family promoting cancer-targeted toxicity. Cytokine & Growth Factor Reviews, 21(5), 381–391. 10.1016/j.cytogfr.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Amicis F, Aquila S, Morelli C, Guido C, Santoro M, Perrotta I, et al. (2015). Bergapten drives autophagy through the up-regulation of PTEN expression in breast cancer cells. Molecular Cancer, 14, 130 10.1186/s12943-015-0403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z, et al. (2008). Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Research, 68(22), 9167–9175. 10.1158/0008-5472.CAN-08-1573. [DOI] [PubMed] [Google Scholar]
- Eccles DM, Russell SE, Haites NE, Atkinson R, Bell DW, Gruber L, et al. (1992). Early loss of heterozygosity on 17q in ovarian cancer. The Abe ovarian cancer genetics group. Oncogene, 7(10), 2069–2072. [PubMed] [Google Scholar]
- Emdad L, Lebedeva IV, Su ZZ, Gupta P, Sarkar D, Settleman J, et al. (2007). Combinatorial treatment of non-small-cell lung cancers with gefitinib and Ad.mda-7 enhances apoptosis-induction and reverses resistance to a single therapy. Journal of Cellular Physiology, 210(2), 549–559. 10.1002/jcp.20906. [DOI] [PubMed] [Google Scholar]
- Emdad L, Lebedeva IV, Su ZZ, Gupta P, Sauane M, Dash R, et al. (2009). Historical perspective and recent insights into our understanding of the molecular and biochemical basis of the antitumor properties of mda-7/IL-24. Cancer Biology & Therapy, 8(5), 391–400. [DOI] [PubMed] [Google Scholar]
- Emdad L, Lebedeva IV, Su ZZ, Sarkar D, Dent P, Curiel DT, et al. (2007). Melanoma differentiation associated gene-7/interleukin-24 reverses multidrug resistance in human colorectal cancer cells. Molecular Cancer Therapeutics, 6(11), 2985–2994. 10.1158/1535-7163.MCT-07-0399. [DOI] [PubMed] [Google Scholar]
- Fimia GM, Corazzari M, Antonioli M, & Piacentini M (2013). Ambra1 at the crossroad between autophagy and cell death. Oncogene, 32(28), 3311–3318. 10.1038/onc.2012.455. [DOI] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, et al. (2007). Ambra1 regulates autophagy and development of the nervous system. Nature, 447(7148), 1121–1125. 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- Finka A, & Goloubinoff P (2013). Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress & Chaperones, 18(5), 591–605. 10.1007/s12192-013-0413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher PB (2005). Is mda-7/IL-24 a “magic bullet” for cancer? Cancer Research, 65(22), 10128–10138. 10.1158/0008-5472.CAN-05-3127. [DOI] [PubMed] [Google Scholar]
- Fisher PB, Gopalkrishnan RV, Chada S, Ramesh R, Grimm EA, Rosenfeld MR, et al. (2003). mda-7/IL-24, a novel cancer selective apoptosis inducing cytokine gene: From the laboratory into the clinic. Cancer Biology & Therapy, 2(4 Suppl. 1), S23–37. [PubMed] [Google Scholar]
- Fisher PB, Sarkar D, Lebedeva IV, Emdad L, Gupta P, Sauane M, et al. (2007). Melanoma differentiation associated gene-7/interleukin-24 (mda-7/IL-24): Novel gene therapeutic for metastatic melanoma. Toxicology and Applied Pharmacology, 224(3), 300–307. 10.1016/j.taap.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornari F, Gramantieri L, Ferracin M, Veronese A, Sabbioni S, Calin GA, et al. (2008). MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene, 27(43), 5651–5661. 10.1038/onc.2008.178. [DOI] [PubMed] [Google Scholar]
- Fujiwara N, Usui T, Ohama T, & Sato K (2016). Regulation of Beclin 1 protein phosphorylation and autophagy by protein phosphatase 2A (PP2A) and death-associated protein kinase 3 (DAPK3). The Journal of Biological Chemistry, 291(20), 10858–10866. 10.1074/jbc.M115.704908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya N, Yu J, Byfield M, Pattingre S, & Levine B (2005). The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy, 1(1), 46–52. [DOI] [PubMed] [Google Scholar]
- Futreal PA, Soderkvist P, Marks JR, Iglehart JD, Cochran C, Barrett JC, et al. (1992). Detection of frequent allelic loss on proximal chromosome 17q in sporadic breast carcinoma using microsatellite length polymorphisms. Cancer Research, 52(9), 2624–2627. [PubMed] [Google Scholar]
- Gallop JL, Jao CC, Kent HM, Butler PJ, Evans PR, Langen R, et al. (2006). Mechanism of endophilin N-BAR domain-mediated membrane curvature. The EMBO Journal, 25(12), 2898–2910. 10.1038/sj.emboj.7601174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Sun X, Chen X, Wang Y, Foster BA, Subjeck J, et al. (2008). Secretable chaperone Grp170 enhances therapeutic activity of a novel tumor suppressor, mda-7/IL-24. Cancer Research, 68(10), 3890–3898. 10.1158/0008-5472.CAN-08-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Zacharek A, Salkowski A, Grignon DJ, Sakr W, Porter AT, et al. (1995). Loss of heterozygosity of the BRCA1 and other loci on chromosome 17q in human prostate cancer. Cancer Research, 55(5), 1002–1005. [PubMed] [Google Scholar]
- Garofalo M, Di Leva G, Romano G, Nuovo G, Suh SS, Ngankeu A, et al. (2009). miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell, 16(6), 498–509. 10.1016/j.ccr.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gawriluk TR, Ko C, Hong X, Christenson LK, & Rucker EB 3rd (2014). Beclin-1 deficiency in the murine ovary results in the reduction of progesterone production to promote preterm labor. Proceedings of the National Academy of Sciences of the United States of America, 111(40), E4194–4203. 10.1073/pnas.1409323111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirtz DA (2014). The autophagic response to radiation: Relevance for radiation sensitization in cancer therapy. Radiation Research, 182(4), 363–367. 10.1667/RR13774.1. [DOI] [PubMed] [Google Scholar]
- Giles NM, Gutowski NJ, Giles GI, & Jacob C (2003). Redox catalysts as sensitisers towards oxidative stress. FEBS Letters, 535(1–3), 179–182. [DOI] [PubMed] [Google Scholar]
- Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V, et al. (2013). Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene, 32(18), 2261–2272. 2272e.2261–2211. 10.1038/onc.2012.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Wan D, Qian Q, Yi B, He Z, Gu Y, et al. (2014). Ambra1 is an essential regulator of autophagy and apoptosis in SW620 cells: Pro-survival role of Ambra1. PLoS One, 9(2), e90151 10.1371/journal.pone.0090151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo GF, Jiang WQ, Zhang B, Cai YC, Xu RH, Chen XX, et al. (2011). Autophagy-related proteins Beclin-1 and LC3 predict cetuximab efficacy in advanced colorectal cancer. World Journal of Gastroenterology, 17(43), 4779–4786. 10.3748/wjg.v17.i43.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Walter MR, Su ZZ, Lebedeva IV, Emdad L, Randolph A, et al. (2006). BiP/GRP78 is an intracellular target for MDA-7/IL-24 induction of cancer-specific apoptosis. Cancer Research, 66(16), 8182–8191. 10.1158/0008-5472.CAN-06-0577. [DOI] [PubMed] [Google Scholar]
- Han J, Hou W, Lu C, Goldstein LA, Stolz DB, Watkins SC, et al. (2013). Interaction between Her2 and Beclin-1 proteins underlies a new mechanism of reciprocal regulation. The Journal of Biological Chemistry, 288(28), 20315–20325. 10.1074/jbc.M113.461350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Xue XF, Shen HG, Guo XB, Wang X, Yuan B, et al. (2014). Prognostic significance of Beclin-1 expression in colorectal cancer: A meta-analysis. Asian Pacific Journal of Cancer Prevention, 15(11), 4583–4587. [DOI] [PubMed] [Google Scholar]
- Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, et al. (2011). An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell, 147(6), 1233–1247. 10.1016/j.cell.2011.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WS, Dai XF,Jin M, Liu CW, & Rent JH. (2012). Hypoxia-induced autophagy confers resistance of breast cancer cells to ionizing radiation. Oncology Research, 20(5–6), 251–258. [DOI] [PubMed] [Google Scholar]
- Ho J, Kong JW, Choong LY, Loh MC, Toy W, Chong PK, et al. (2009). Novel breast cancer metastasis-associated proteins. Journal of Proteome Research, 8(2), 583–594. 10.1021/pr8007368. [DOI] [PubMed] [Google Scholar]
- Huang JJ, Li HR, Huang Y, Jiang WQ, Xu RH, Huang HQ, et al. (2010). Beclin 1 expression: A predictor of prognosis in patients with extranodal natural killer T-cell lymphoma, nasal type. Autophagy, 6(6), 777–783. [DOI] [PubMed] [Google Scholar]
- Huang X, Qi Q, Hua X, Li X, Zhang W, Sun H, et al. (2014). Beclin 1, an autophagy-related gene, augments apoptosis in U87 glioblastoma cells. Oncology Reports, 31(4), 1761–1767. 10.3892/or.2014.3015. [DOI] [PubMed] [Google Scholar]
- Huettenbrenner S, Maier S, Leisser C, Polgar D, Strasser S, Grusch M, et al. (2003). The evolution of cell death programs as prerequisites of multicellularity. Mutation Research, 543(3), 235–249. [DOI] [PubMed] [Google Scholar]
- Jiang H, & Fisher PB (1993). Use of a sensitive and efficient subtraction hybridization protocol for the identification of genes differentially regulated during the induction of differentiation in human melanoma cells. Molecular and Cellular Differentiation, 1(3), 285–299. [Google Scholar]
- Jiang H, Lin JJ, Su ZZ, Goldstein NI, & Fisher PB (1995). Subtraction hybridization identifies a novel melanoma differentiation associated gene, mda-7, modulated during human melanoma differentiation, growth and progression. Oncogene, 11(12), 2477–2486. [PubMed] [Google Scholar]
- Kim YM, Jung CH, Seo M, Kim EK, Park JM,Bae SS, et al. (2015). mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Molecular Cell, 57(2), 207–218. 10.1016/j.molcel.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, & Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology, 13(2), 132–141. 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Yoo NJ, & Lee SH (2008). Somatic mutation of pro-cell death Bif-1 gene is rare in common human cancers. APMIS, 116(10), 939–940. 10.1111/j.1600-0463.2008.01091.x. [DOI] [PubMed] [Google Scholar]
- Klein SR, Piya S, Lu Z, Xia Y, Alonso MM, White EJ, et al. (2015). C-Jun N-terminal kinases are required for oncolytic adenovirus-mediated autophagy. Oncogene, 34, 5295–5301. 10.1038/onc.2014.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ (2007). Autophagy: From phenomenology to molecular understanding in less than a decade. Nature Reviews. Molecular Cell Biology, 8(11), 931–937. 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- Knudson AG Jr. (1971). Mutation and cancer: Statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America, 68(4), 820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko YH, Cho YS, Won HS, Jeon EK, An HJ, Hong SU, et al. (2013). Prognostic significance of autophagy-related protein expression in resected pancreatic ductal adenocarcinoma. Pancreas, 42(5), 829–835. 10.1097/MPA.0b013e318279d0dc. [DOI] [PubMed] [Google Scholar]
- Koukourakis MI, Giatromanolaki A, Sivridis E, Pitiakoudis M, Gatter KC, & Harris AL (2010). Beclin 1 over- and underexpression in colorectal cancer: Distinct patterns relate to prognosis and tumour hypoxia. British Journal of Cancer, 103(8), 1209–1214. 10.1038/sj.bjc.6605904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. (2009). Classification of cell death: Recommendations of the nomenclature committee on cell death 2009. Cell Death and Differentiation, 16(1), 3–11. 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung CP, Budina A, Balaburski G, Bergenstock MK, & Murphy M (2011). Autophagy in tumor suppression and cancer therapy. Critical Reviews in Eukaryotic Gene Expression, 21(1), 71–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, et al. (2007). Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. The EMBO Journal, 26(15), 3699–3708. 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva IV, Sauane M, Gopalkrishnan RV, Sarkar D, Su ZZ, Gupta P, et al. (2005). mda-7/IL-24: Exploiting cancer’s Achilles’ heel. Molecular Therapy, 11(1), 4–18. 10.1016/j.ymthe.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Lee J, Giordano S, & Zhang J (2012). Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. The Biochemical Journal, 441(2), 523–540. 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Kim HP, Jin Y, Choi AM, & Ryter SW (2011). Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy, 7(8), 829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, & Klionsky DJ (2004). Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Developmental Cell, 6(4), 463–477. [DOI] [PubMed] [Google Scholar]
- Li Z, Chen B, Wu Y,Jin F, Xia Y, & Liu X (2010). Genetic and epigenetic silencing of the beclin 1 gene in sporadic breast tumors. BMC Cancer, 10, 98 10.1186/1471-2407-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BX, Li CY, Peng RQ, Wu XJ, Wang HY, Wan DS, et al. (2009). The expression of beclin 1 is associated with favorable prognosis in stage IIIB colon cancers. Autophagy, 5(3), 303–306. [DOI] [PubMed] [Google Scholar]
- Li X, Lu Y, Pan T, & Fan Z (2010). Roles of autophagy in cetuximab-mediated cancer therapy against EGFR. Autophagy, 6(8), 1066–1077. 10.4161/auto.6.8.13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yan J, Wang L, Xiao F, Yang Y, Guo X, et al. (2013). Beclin1 inhibition promotes autophagy and decreases gemcitabine-induced apoptosis in Miapaca2 pancreatic cancer cells. Cancer Cell International, 13(1), 26 10.1186/1475-2867-13-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang J, Chen X, Liu T, He W, Chen Y, et al. (2012). Molecular machinery of autophagy and its implication in cancer. The American Journal of the Medical Sciences, 343(2), 155–161. 10.1097/MAJ.0b013e31821f978d. [DOI] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. (1999). Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature, 402(6762), 672–676. 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, et al. (1998). Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. Journal of Virology, 72(11), 8586–8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Yu J, Brown K, & Levine B (2001).Beclin 1 contains aleucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Research, 61(8), 3443–3449. [PubMed] [Google Scholar]
- Lin HX, Qiu HJ, Zeng F, Rao HL, Yang GF, Kung HF, et al. (2013). Decreased expression of Beclin 1 correlates closely with Bcl-xL expression and poor prognosis of ovarian carcinoma. PLoS One, 8(4), e60516 10.1371/journal.pone.0060516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, & Debnath J (2016). The evolving, multifaceted roles of autophagy in cancer. Advances in Cancer Research, 130, 1–53. 10.1016/bs.acr.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Ma K, Zhang C, Huang MY, Guo YX, & Hu GQ (2016). Crosstalk between Beclin-1-dependent autophagy and caspasedependent apoptosis induced by tanshinone IIA in human osteosarcoma MG-63 cells. Oncology Reports, 36(4), 1807–1818. 10.3892/or.2016.5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison WP, & Maddison DR (2017). Mesquite: A modular system for evolutionary analysis. Version 3.2. http://mesquiteproject.org.
- Martin M, Hinojar A, Cerezo L, Garcia J, Lopez M, Prada J, et al. (2016). Aldehyde dehydrogenase isoform 1 (ALDH1) expression as a predictor of radiosensitivity in laryngeal cancer. Clinical & Translational Oncology, 18(8), 825–830. 10.1007/s12094-015-1445-1. [DOI] [PubMed] [Google Scholar]
- Masuda M, Takeda S, Sone M, Ohki T, Mori H, Kamioka Y, et al. (2006). Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. The EMBO Journal, 25(12), 2889–2897. 10.1038/sj.emboj.7601176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. (2009). Autophagy suppresses tumorigenesis through elimination of p62. Cell, 137(6), 1062–1075. 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, & White E (2011). Autophagy in tumorigenesis and energy metabolism: Friend by day, foe by night. Current Opinion in Genetics & Development, 21(1), 113–119. 10.1016/j.gde.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miracco C, Cevenini G, Franchi A, Luzi P, Cosci E, Mourmouras V, et al. (2010). Beclin 1 and LC3 autophagic gene expression in cutaneous melanocytic lesions. Human Pathology, 41(4), 503–512. 10.1016/j.humpath.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Miracco C, Cosci E, Oliveri G, Luzi P, Pacenti L, Monciatti I, et al. (2007). Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. International Journal of Oncology, 30(2), 429–436. [PubMed] [Google Scholar]
- Mizushima N (2007). Autophagy: Process and function. Genes & Development, 21(22), 2861–2873. 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, & Klionsky DJ (2008). Autophagy fights disease through cellular self-digestion. Nature, 451(7182), 1069–1075. 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan CD, Srinivasa V, Rangappa S, Mervin L, Mohan S, Paricharak S, et al. (2016). Trisubstituted-imidazoles induce apoptosis in human breast cancer cells by targeting the oncogenic PI3K/Akt/mTOR signaling pathway. PLoS One, 11(4), e0153155 10.1371/journal.pone.0153155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau K, Luo S, & Rubinsztein DC (2010). Cytoprotective roles for autophagy. Current Opinion in Cell Biology, 22(2), 206–211. 10.1016/j.ceb.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morran DC, Wu J, Jamieson NB, Mrowinska A, Kalna G, Karim SA, et al. (2014). Targeting mTOR dependency in pancreatic cancer. Gut, 63(9), 1481–1489. 10.1136/gutjnl-2013-306202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowers EE, Sharifi MN, & Macleod KF (2017). Autophagy in cancer metastasis. Oncogene, 36(12), 1619–1630. 10.1038/onc.2016.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, et al. (2013). mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nature Cell Biology, 15(4), 406–416. 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Ramesh R, Munshi A, Chada S, & Meyn RE (2004). Adenovirus-mediated mda-7 (IL24) gene therapy suppresses angiogenesis and sensitizes NSCLC xenograft tumors to radiation. Molecular Therapy, 9(6), 818–828. 10.1016/j.ymthe.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Noble CG, Dong JM, Manser E, & Song H (2008). Bcl-xL and UVRAG cause a monomer-dimer switch in Beclin1. The Journal of Biological Chemistry, 283(38), 26274–26282. 10.1074/jbc.M804723200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Tougeron D, Huang S, Okamoto K, & Sinicrope FA (2014). Beclin 1 and UVRAG confer protection from radiation-induced DNA damage and maintain centrosome stability in colorectal cancer cells. PLoS One, 9(6), e100819 10.1371/journal.pone.0100819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, et al. (2004). BAR domains as sensors of membrane curvature: The amphiphysin BAR structure. Science, 303(5657), 495–499. 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, & Codogno P (2000). Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. The Journal of Biological Chemistry, 275(2), 992–998. [DOI] [PubMed] [Google Scholar]
- Pierrat B, Simonen M, Cueto M, Mestan J, Ferrigno P, & Heim J (2001). SH3GLB, a new endophilin-related protein family featuring an SH3 domain. Genomics, 71(2), 222–234. 10.1006/geno.2000.6378. [DOI] [PubMed] [Google Scholar]
- Pirtoli L, Cevenini G, Tini P, Vannini M, Oliveri G, Marsili S, et al. (2009). The prognostic role of Beclin 1 protein expression in high-grade gliomas. Autophagy, 5(7), 930–936. [DOI] [PubMed] [Google Scholar]
- Pradhan AK, Talukdar S, Bhoopathi P, Shen XN, Emdad L, Das SK, et al. (2017). mda-7/IL-24 mediates cancer cell-specific death via regulation of miR-221 and the Beclin-1 Axis. Cancer Research, 77(4), 949–959. 10.1158/0008-5472.CAN-16-1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. (2003). Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. The Journal of Clinical Investigation, 112(12), 1809–1820. 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radwan SM, Hamdy NM, Hegab HM, & El-Mesallamy HO. (2016). Beclin-1 and hypoxia-inducible factor-1alpha genes expression: Potential biomarkers in acute leukemia patients. Cancer Biomarkers, 16(4), 619–626. 10.3233/CBM-160603. [DOI] [PubMed] [Google Scholar]
- Ramesh R, Mhashilkar AM, Tanaka F, Saito Y, Branch CD, Sieger K, et al. (2003). Melanoma differentiation-associated gene 7/interleukin (IL)-24 is a novel ligand that regulates angiogenesis via the IL-22 receptor. Cancer Research, 63(16), 5105–5113. [PubMed] [Google Scholar]
- Ren J, & Taegtmeyer H (2015). Too much or not enough of a good thing—The Janus faces of autophagy in cardiac fuel and protein homeostasis. Journal of Molecular and Cellular Cardiology, 84, 223–226. 10.1016/j.yjmcc.2015.03.001. [DOI] [PubMed] [Google Scholar]
- Riley BE, Kaiser SE, Shaler TA, Ng AC, Hara T, Hipp MS, et al. (2010). Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: A potential role for protein aggregation in autophagic substrate selection. The Journal of Cell Biology, 191(3), 537–552. 10.1083/jcb.201005012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, & DeSalle R (2012). E value cutoff and eukaryotic genome content phylogenetics. Molecular Phylogenetics and Evolution, 63(2), 342–350. [DOI] [PubMed] [Google Scholar]
- Runkle KB, Meyerkord CL, Desai NV, Takahashi Y, &Wang HG (2012). Bif-1 suppresses breast cancer cell migration by promoting EGFR endocytic degradation. Cancer Biology & Therapy, 13(10), 956–966. 10.4161/cbt.20951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SE, Hickey GI, Lowry WS, White P, & Atkinson RJ (1990). Allele loss from chromosome 17 in ovarian cancer. Oncogene, 5(10), 1581–1583. [PubMed] [Google Scholar]
- Saito H, Inazawa J, Saito S, Kasumi F, Koi S, Sagae S, et al. (1993). Detailed deletion mapping of chromosome 17q in ovarian and breast cancers: 2-cM region on 17q21.3 often and commonly deleted in tumors. Cancer Research, 53(14), 3382–3385. [PubMed] [Google Scholar]
- Sarkar D, Su ZZ, Lebedeva IV, Sauane M, Gopalkrishnan RV, Valerie K, et al. (2002). mda-7 (IL-24) mediates selective apoptosis in human melanoma cells by inducing the coordinated overexpression of the GADD family of genes by means of p38 MAPK. Proceedings of the National Academy of Sciences of the United States of America, 99(15), 10054–10059. 10.1073/pnas.152327199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauane M, Su ZZ, Gupta P, Lebedeva IV, Dent P, Sarkar D, et al. (2008). Auto-crine regulation of mda-7/IL-24 mediates cancer-specific apoptosis. Proceedings of the National Academy of Sciences of the United States of America, 105(28), 9763–9768. 10.1073/pnas.0804089105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Lim HT, Minai-Tehrani A, Noh MS, Kim JE, Kim JH, et al. (2012). Aerosol delivery of beclin1 enhanced the anti-tumor effect of radiation in the lungs of K-rasLA1 mice. Journal of Radiation Research, 53(4), 506–515. 10.1093/jrr/rrs005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SP, Pandey T, Srivastava R, Verma PC, Singh PK, Tuli R, et al. (2010). BECLIN1 from Arabidopsis thaliana under the generic control of regulated expression systems, a strategy for developing male sterile plants. Plant Biotechnology Journal, 8(9), 1005–1022. 10.1111/j.1467-7652.2010.00527.x. [DOI] [PubMed] [Google Scholar]
- Su Z, Emdad L, Sauane M, Lebedeva IV, Sarkar D, Gupta P, et al. (2005). Unique aspects of mda-7/IL-24 antitumor by stander activity: Establishing a role for secretion of MDA-7/IL-24 protein by normal cells. Oncogene, 24(51), 7552–7566. 10.1038/sj.onc.1208911. [DOI] [PubMed] [Google Scholar]
- Su ZZ, Lebedeva IV, Sarkar D, Gopalkrishnan RV, Sauane M, Sigmon C,et al. (2003). Melanoma differentiation associated gene-7, mda-7/IL-24, selectively induces growth suppression, apoptosis and radiosensitization in malignant gliomas in a p53-independent manner. Oncogene, 22(8), 1164–1180. 10.1038/sj.onc.1206062. [DOI] [PubMed] [Google Scholar]
- Su ZZ, Madireddi MT, Lin JJ, Young CS, Kitada S, Reed JC, et al. (1998). The cancer growth suppressor gene mda-7 selectively induces apoptosis in human breast cancer cells and inhibits tumor growth in nude mice. Proceedings of the National Academy of Sciences of the United States of America, 95(24), 14400–14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WL (2016). Ambra1 in autophagy and apoptosis: Implications for cell survival and chemotherapy resistance. Oncology Letters, 12(1), 367–374. 10.3892/ol.2016.4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Liu JH, Jin L, Lin SM, Yang Y, Sui YX, et al. (2010). Over-expression of the Beclin1 gene upregulates chemosensitivity to anti-cancer drugs by enhancing therapy-induced apoptosis in cervix squamous carcinoma CaSki cells. Cancer Letters, 294(2), 204–210. 10.1016/j.canlet.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Sun Y, Liu JH, Sui YX, Jin L, Yang Y, Lin SM, et al. (2011). Beclin1 over-expression inhibitis proliferation, invasion and migration of CaSki cervical cancer cells. Asian Pacific Journal of Cancer Prevention, 12(5), 1269–1273. [PubMed] [Google Scholar]
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, et al. (2007). Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nature Cell Biology, 9(10), 1142–1151. 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Thapa N, Sun Y, &Anderson RA. (2015). A kinase-independent role for EGF receptor in autophagy initiation. Cell, 160(1–2), 145–160. 10.1016/j.cell.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Wang M, Yu M, Zhang J, Bristow RG, Hill RP, et al. (2016). Role of autophagy as a survival mechanism for hypoxic cells in tumors. Neoplasia, 18(6), 347–355. 10.1016/j.neo.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Tangir J, Muto MG, Berkowitz RS, Welch WR, Bell DA, & Mok SC (1996). A 400 kb novel deletion unit centromeric to the BRCA1 gene in sporadic epithelial ovarian cancer. Oncogene, 12(4), 735–740. [PubMed] [Google Scholar]
- Tong AW, Nemunaitis J, Su D, Zhang Y, Cunningham C, Senzer N, et al. (2005). Intratumoral injection of INGN 241, a nonreplicating adenovector expressing the melanoma-differentiation associated gene-7 (mda-7/IL24): Biologic outcome in advanced cancer patients. Molecular Therapy, 11(1), 160–172. 10.1016/j.ymthe.2004.09.021. [DOI] [PubMed] [Google Scholar]
- Valente G, Morani F, Nicotra G, Fusco N, Peracchio C, Titone R, et al. (2014). Expression and clinical significance of the autophagy proteins BECLIN 1 and LC3 in ovarian cancer. BioMed Research International, 2014, 462658 10.1155/2014/462658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Ling S, & Lin WC (2010). 14-3-3Tau regulates Beclin 1 and is required for autophagy. PLoS One, 5(4), e10409 10.1371/journal.pone.0010409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, et al. (2012). Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science, 338(6109), 956–959. 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Pattingre S, Sinha S, Bassik M, & Levine B (2008).JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Molecular Cell, 30(6), 678–688. 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, et al. (2013). EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell, 154(6), 1269–1284. 10.1016/j.cell.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E, & DiPaola RS (2009). The double-edged sword of autophagy modulation in cancer. Clinical Cancer Research, 15(17), 5308–5316. 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EN, Bristol ML, Di X, Maltese WA, Koterba K, Beckman MJ, et al. (2011). A switch between cytoprotective and cytotoxic autophagy in the radiosensitization of breast tumor cells by chloroquine and vitamin D. Hormones and Cancer, 2(5), 272–285. 10.1007/s12672-011-0081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirawan E, Lippens S, Vanden Berghe T, Romagnoli A, Fimia GM, Piacentini M, et al. (2012). Beclin1: A role in membrane dynamics and beyond. Autophagy, 8(1), 6–17. 10.4161/auto.8.1.16645. [DOI] [PubMed] [Google Scholar]
- Xu F, Fang Y, Yan L, Xu L, Zhang S, Cao Y, et al. (2017). Nuclear localization of Beclin 1 promotes radiation-induced DNA damage repair independent of autophagy. Scientific Reports, 7, 45385 10.1038/srep45385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoub A, Mitchell C, Lister A, Lebedeva IV, Sarkar D, Su ZZ, et al. (2003). Melanoma differentiation-associated 7 (interleukin 24) inhibits growth and enhances radiosensitivity of glioma cells in vitro and in vivo. Clinical Cancer Research, 9(9), 3272–3281. [PubMed] [Google Scholar]
- Yamaguchi H, Woods NT, Dorsey JF, Takahashi Y, Gjertsen NR, Yeatman T, et al. (2008). SRC directly phosphorylates Bif-1 and prevents its interaction with Bax and the initiation of anoikis. The Journal of Biological Chemistry, 283(27), 19112–19118. 10.1074/jbc.M709882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Ju JH, Lee KM, Nam K, Oh S, & Shin I (2013). Protein kinase B/Akt1 inhibits autophagy by down-regulating UVRAG expression. Experimental Cell Research, 319(3), 122–133. 10.1016/j.yexcr.2012.11.014. [DOI] [PubMed] [Google Scholar]
- Yin X, Cao L, Peng Y, Tan Y, Xie M, Kang R, et al. (2011). A critical role for UVRAG in apoptosis. Autophagy, 7(10), 1242–1244. 10.4161/auto.7.10.16507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Qu D, Liu C, Ying T, Lv J, Jin S, et al. (2015). Chemoresistance is associated with Beclin-1 and PTEN expression in epithelial ovarian cancers. Oncology Letters, 9(4), 1759–1763. 10.3892/ol.2015.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Shao Z, Xiong L, Che B, Deng C, & Xu W (2009). Expression of Beclin1 in osteosarcoma and the effects of down-regulation of autophagy on the chemotherapeutic sensitivity. Journal of Huazhong University of Science and Technology Medical Sciences, 29(6), 737–740. 10.1007/s11596-009-0613-3. [DOI] [PubMed] [Google Scholar]
- Zhang D, Wang W, Sun X, Xu D, Wang C, Zhang Q, et al. (2016). AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy, 12(9), 1447–1459. 10.1080/15548627.2016.1185576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhang J, Zhang A, Wang Y, Han L, You Y, et al. (2010). PUMA is a novel target of miR-221/222 in human epithelial cancers. International Journal of Oncology, 37(6), 1621–1626. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Oh S, Li D, Ni D, Pirooz SD, Lee JH, et al. (2012). A dual role for UVRAG in maintaining chromosomal stability independent of autophagy. Developmental Cell, 22(5), 1001–1016. 10.1016/j.devcel.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YY, Li Y, Jiang WQ, & Zhou LF. (2015). MAPK/JNK signalling: A potential autophagy regulation pathway. Bioscience Reports, 35(3), e00199 10.1042/BSR20140141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Yue C, Deng J, Hu R, Xu J, Feng L, et al. (2013). Autophagic protein Beclin 1 serves as an independent positive prognostic biomarker for non-small cell lung cancer. PLoS One, 8(11), e80338 10.1371/journal.pone.0080338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, et al. (2009). Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy, 5(6), 816–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.