

Abstract
The receptor‐binding domain (RBD) of the SARS‐CoV‐2 spike protein is a candidate vaccine antigen that binds angiotensin‐converting enzyme 2 (ACE2), leading to virus entry. Here, it is shown that rapid conversion of recombinant RBD into particulate form via admixing with liposomes containing cobalt‐porphyrin‐phospholipid (CoPoP) potently enhances the functional antibody response. Antigen binding via His‐tag insertion into the CoPoP bilayer results in a serum‐stable and conformationally intact display of the RBD on the liposome surface. Compared to other vaccine formulations, immunization using CoPoP liposomes admixed with recombinant RBD induces multiple orders of magnitude higher levels of antibody titers in mice that neutralize pseudovirus cell entry, block RBD interaction with ACE2, and inhibit live virus replication. Enhanced immunogenicity can be accounted for by greater RBD uptake into antigen‐presenting cells in particulate form and improved immune cell infiltration in draining lymph nodes. QS‐21 inclusion in the liposomes results in an enhanced antigen‐specific polyfunctional T cell response. In mice, high dose immunization results in minimal local reactogenicity, is well‐tolerated, and does not elevate serum cobalt levels. Taken together, these results confirm that particulate presentation strategies for the RBD immunogen should be considered for inducing strongly neutralizing antibody responses against SARS‐CoV‐2.
Keywords: antigens, COVID‐19, liposomes, nanoparticles, SARS‐CoV‐2, vaccines
The receptor‐binding domain (RBD) of SARS‐CoV‐2 is a target for COVID‐19 vaccine development. Using a next‐generation vaccine adjuvant system that rapidly converts soluble antigens into stable particles, the neutralizing antibody response induced by the RBD is greatly enhanced, demonstrating merit for particle‐based approaches toward RBD‐based vaccines.

SARS‐CoV‐2 (severe acute respiratory syndrome coronavirus 2) has caused a disruptive worldwide viral pandemic.[ 1 ] The quest for effective vaccine countermeasures is an active pursuit in the biomedical research community.[ 2 ] The spike (S) protein on the virus surface is instrumental for binding, fusing, and entry into host cells, and is also the lead immunogen for several advanced vaccine candidates.[ 3 ] The S protein contains the receptor‐binding domain (RBD) that binds to the host receptor, angiotensin‐converting enzyme 2 (ACE2).[ 4 ] The RBD is an appealing antigen for vaccine development, as most neutralizing antibodies generated during a SARS‐CoV‐2 infection are directed against it.[ 5 ]
SARS‐CoV‐2 RBD has been shown to be a viable immunogen in preclinical studies, conferring protection in non‐human primates from viral challenge.[ 6 ] However, as a relatively small and compact immunogen with 4 internal disulfide bonds, the RBD is expected to exhibit hapten‐like properties that limit its immunogenicity, which could necessitate the use of higher antigen doses that would complicate the large scale roll‐out of a RBD vaccine. Indeed, it has been shown that immunogenicity is enhanced by engineering the protein construct into dimeric[ 7 ] and oligomeric structures,[ 8 ] and another approach necessitated conjugation of the RBD onto a carrier protein.[ 9 ] While effective, such approaches may be time‐consuming and can confound downstream characterization of the RBD during the development process. The polyhistidine tag (His‐tag) has been transformative in its simplicity and efficacy in binding to immobilized metals for protein purification. We have shown that lipid bilayers containing porphyrin–phospholipid conjugates that are chelated with cobalt, but not with other metals, can effectively capture soluble His‐tagged proteins and peptides. Simple mixing of liposomes containing cobalt‐porphyrin‐phospholipid (CoPoP) with His‐tagged soluble proteins results in rapid and stable particle‐formation.[ 10 ] This approach enhanced the functional immunogenicity of Pfs25, a small compact malaria immunogen.[ 11 ] In the present study, we assess whether particulate presentation of SARS‐CoV‐2 RBD leads to enhanced immunogenicity and induces virus‐neutralizing antibody responses.
Recombinant RBD proteins bearing a C‐terminus His‐tag were obtained from mammalian (HEK293; spike residues 319–541) and insect (Sf9; spike residues 330–530) expression systems. Liposomes containing CoPoP, along with the clinical‐stage lipid adjuvants monophosphoryl lipid A (MPLA) and, optionally, QS‐21 were mixed with the RBD for 3 h at room temperature (RT) at a 4:1 mass ratio of CoPoP:protein and RBD binding to liposomes was then assessed. Control liposomes that lacked cobalt within the PoP molecule, but were otherwise identical, were also tested.
Figure 1A shows particle formation of the RBD based on a competition assay with Ni‐NTA beads. The free protein is captured by the beads (“B”), whereas liposome‐bound RBD is not and remains in the supernatant (“S”). A schematic representation of this assay is depicted in Figure S1, Supporting Information. The HEK293‐ and Sf9‐ produced RBD exhibited nearly identical binding patterns, showing full binding to liposomes containing CoPoP, but virtually no binding to identical liposomes lacking cobalt (but still containing the PoP moiety). The presence of QS‐21 in the bilayer did not impact RBD binding. Cobalt‐specific binding of the RBD to CoPoP liposomes was also shown using an independent high‐speed centrifugation assay (Figure 1B). A fluorescence resonance energy transfer assay was developed using a fluorescent‐labeled RBD, which is quenched upon binding to liposomes due to energy transfer to the CoPoP chromophore. Figure 1C shows the RBD particlization kinetics, with approximately 80% of the antigen forming particles within just 15 min of incubation. Rapid particle‐formation was also verified using the Ni‐NTA bead competition assay (Figure 1D).
Figure 1.
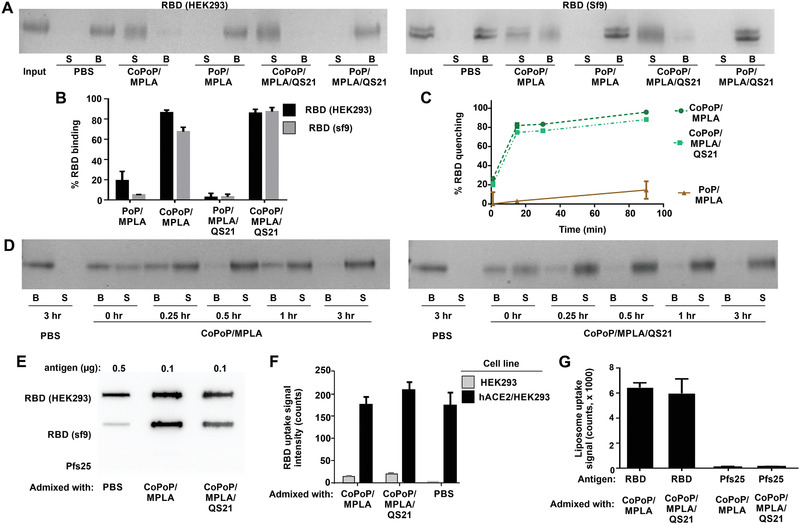
Recombinant receptor‐binding domain (RBD) binds to CoPoP liposomes with intact conformation. A) Nickel‐nitrilotriacetic acid (Ni‐NTA) bead competition assay. RBD antigen produced in the indicated expression system was incubated with liposomes for 3 h, and then Ni‐NTA beads were added and then isolated. Protein that was stably bound to liposomes is in the supernatant (“S”) lanes, whereas unbound protein is in the bead (“B”) fraction. B) Binding of RBD to CoPoP liposomes after 3 h incubation as assessed by a high‐speed centrifugation assay. C) Binding kinetics of a fluorophore‐labeled RBD to CoPoP liposomes. When the fluorophore‐labeled RBD binds CoPoP liposomes, energy transfer results in fluorophore quenching. D) Binding kinetics of the RBD with CoPoP/MPLA or CoPoP/MPLA/QS‐21 liposomes using RBD produced in HEK293 cells, based on Ni‐NTA bead competition. E) Slot blot detection of ACE2 binding to adsorbed RBD or Pfs25 (an unrelated control antigen) in soluble or particulate form. F) Binding of fluorophore‐labeled RBD, in soluble or particulate form, to hACE2‐expressing cells. G) Binding of liposomes themselves (based on PoP signal) decorated with the RBD or the unrelated Pfs25 control antigen to hACE2‐coated plate.
The conformational integrity of the RBD in particle form was next assessed. A slot blot was developed using ACE2, the binding target of the RBD, which was incubated with the RBD in either soluble or particle form, adsorbed on nitrocellulose. A secondary antibody was then used to detect ACE2. As expected, ACE2 did not recognize a Pfs25 control antigen included in the assay. ACE2 recognized the RBD more strongly in particulate form relative to the soluble form, so that a fivefold reduced amount of particlized RBD was used in the assay (Figure 1E). The reason for this behavior is not immediately apparent, but the soluble RBD potentially adsorbs to the membrane in such a way that ACE2 became less accessible. Regardless, this result shows that the RBD maintains the capacity for binding its target receptor in particle form. Figure S2A, Supporting Information, compares the slot blot at varying doses of RBD in soluble or particle form. Figure S2B, Supporting Information, shows ACE2 reactivity at a fixed RBD amount. Particlized RBD could also be recognized by the CR3022 neutralizing monoclonal antibody (Figure S3, Supporting Information), which is known to interact with the SARS‐CoV‐2 RBD.[ 12 ]
When HEK293 cells that overexpress human ACE2 (HEK293/hACE2) were incubated with a fluorescently labeled RBD in either soluble or particle form, strong uptake was observed, as assessed by a whole cell lysate assay (Figure 1F). The same cell line that lacked hACE2 expression exhibited minimal RBD uptake. A similar trend was observed using flow cytometry (Figure S4, Supporting Information). Not only was the RBD taken up preferentially by hACE2 expressing cells, but the CoPoP liposomes themselves showed strong uptake when decorated with the RBD (Figure 1G). In contrast, liposomes decorated with Pfs25 showed minimal uptake. Overall, these biochemical data show that His‐tagged RBD rapidly forms particles when incubated with CoPoP liposomes while maintaining RBD structural integrity.
The nature of the particles themselves was next further assessed. There was a marginal increase in the size of CoPoP/MPLA liposomes following binding to the RBD, whereas CoPoP/MPLA/QS‐21 liposomes remained the same size with or without RBD (Figure 2A, Figure S5, Supporting Information). Neither liposome type exhibited aggregation upon antigen binding. Cryo‐electron microscopy revealed that the RBD particles were spherical, with the QS‐21 liposomes showing a slightly smaller size (Figure 2B), consistent with the dynamic light scattering (DLS) results. As shown in Figure 2C, following particle formation, the labeled RBD formed serum‐stable antigen particles, based on fluorescence quenching, indicating that the antigen was still maintained in the form of intact particles after 1‐week incubation with 20% human serum at 37 °C. To investigate the uptake of antigen particles by antigen‐presenting cells (APCs), in vitro studies were performed with RAW264.7 murine macrophages and bone marrow derived dendritic cells (BMDC) obtained from outbred mice. APCs were incubated with fluorochrome‐labeled RBD, and uptake was assessed (Figure 2D). When the RBD was admixed with CoPoP liposomes, but not identical PoP liposomes (that did not induce particle formation), a higher RBD uptake by both macrophages and BMDCs was observed. However, when those cells were treated with cytochalasin B (a phagocytosis inhibitor) or chlorpromazine (an endocytosis inhibitor), particlized RBD uptake was inhibited. These data are consistent with previous studies showing that a primary mechanism for the adjuvant efficacy of CoPoP liposomes is related to improved antigen delivery APCs.[ 11 , 13 ] To investigate whether enhanced uptake occurred in vivo, mice were immunized intramuscularly with a fluorescent‐labeled RBD admixed with various adjuvant formulations. Two days later, draining lymph nodes were collected and resident APCs were examined for RBD uptake by flow cytometry using the surface markers B220 (for B‐cells), F4/80 (for macrophages), CD11c (for dendritic cells), and I‐A/I‐E (for MHCII‐expressing cells). As shown in Figure S6, Supporting Information, although the antigen fluorescence signal was low, the RBD appeared to be better taken up by all the major types of APCs when presented in particulate form by admixing with CoPoP liposome. On the other hand, when adjuvanted with alum or non‐particlizing AS01‐like liposomes, minimal antigen uptake by the APCs was observed. Thus, the RBD is preferentially taken up in vitro and in vivo by APCs when in particle form.
Figure 2.
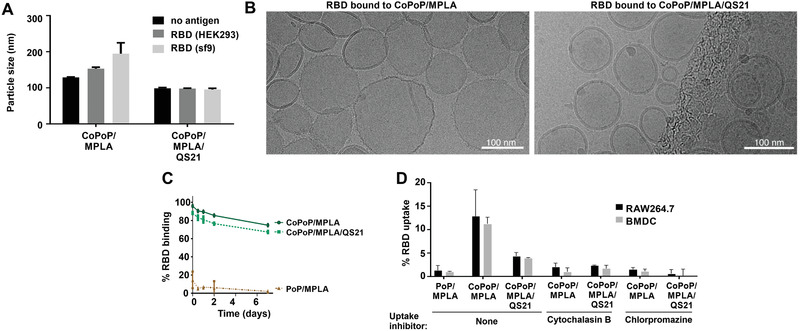
CoPoP/RBD particles are small, stable, and are preferentially taken up by immune cells. A) Size of liposomes following RBD binding, measured by DLS. B) Cryo‐electron microscopy images of the RBD bound to indicated liposomes. A 100 nm scale bar is shown. C) Particle stability with a week‐long incubation in 20% human serum incubated at 37 °C, as measured by the association of a fluorophore labeled RBD to the liposomes. D) RBD uptake in vitro following incubation with murine RAW264.7 cells or BMDC. Cytochalasin B was used as a phagocytosis inhibitor, and chlorpromazine was used as an endocytosis inhibitor. Graphs show mean +/− std. dev. for n = 3 measurements.
The CoPoP adjuvant system has previously been shown to work well with as little as 5–100 ng antigen doses in mice using recombinant antigens related to malaria[ 11 , 14 ] and Lyme disease.[ 13 ] Mice were therefore immunized intramuscularly with 100 ng of RBD (prepared from either insect or mammalian expression systems), admixed prior to immunization with the commercially‐obtained vaccine adjuvants alum, Montanide ISA720, or Addavax, or the lab‐made CoPoP/MPLA, CoPoP/MPLA/QS‐21, PoP/MPLA, or AS01‐like liposomes. No additional purification was carried out after mixing antigen and adjuvants. The MPLA dose in the CoPoP vaccine was 160 ng. A significant increase in RBD‐specific IgG was observed with the CoPoP adjuvants prior to boosting on day 14 (Figure S7, Supporting Information). Figure 3A shows the day 28 endpoint anti‐RBD titer, demonstrating that admixing with CoPoP increased the anti‐RBD titer levels compared to other adjuvants, as well as PoP/MPLA liposomes (which lack cobalt but are otherwise identical to CoPoP/MPLA liposomes) by 2–3 orders of magnitude. Responses induced by mammalian‐ and insect‐produced RBD were similar. The adjuvant itself, without the inclusion of the antigen, did not induce any RBD antibodies. The magnitude of the antibody response for the CoPoP shows the advantage of delivering RBD in a particle format, and is likely due in part due to enhanced delivery to APCs (Figure 2D, Figure S6, Supporting Information).
Figure 3.
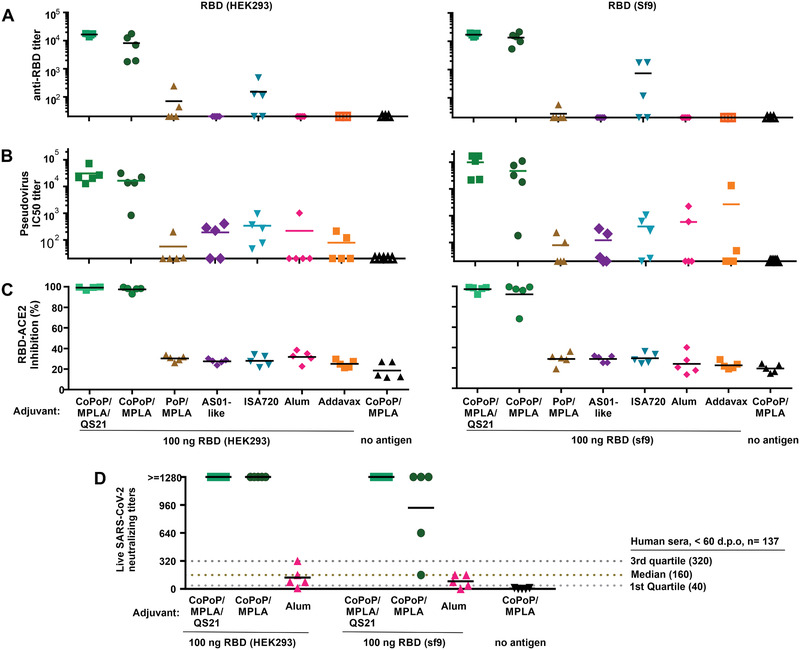
Functional assessment of mouse antibodies induced by the RBD admixed with various vaccine adjuvants. Outbred mice were immunized with 100 ng RBD admixed with indicated adjuvant on day 0 and day 14 prior to serum collection on day 28. A) Anti‐RBD IgG titer. B) PsV IC50 inhibition titer. C) Inhibition in a sVNT that measures interaction between the RBD and hACE2. D) Live SARS‐CoV‐2 virus neutralizing titers in post immune mouse sera. Solid lines show arithmetic mean. Dashed lines in (D) show virus neutralization quartiles and median in the exact same assay in the serum from n = 137 SARS‐CoV‐2‐infected humans, taken <60 days post onset of symptoms.[ 16 ] For (A) and (B), log10 transformed titer was analyzed by one‐way analysis of variance (ANOVA) test followed by Tukey's comparisons, there was no statistical difference between CoPoP liposomes with or without QS‐21; other adjuvants all show significant difference with p < 0.005 when compared to CoPoP liposomes. For (C) and (D), data were analyzed by one‐way ANOVA followed by Tukey's comparisons; there is no statistical difference between CoPoP liposomes with or without QS‐21; other adjuvants all show significant difference with p < 0.001 when compared to CoPoP liposomes.
The short His‐tag on the C‐terminal of the RBD antigen is potentially immunogenic. Prior studies using CoPoP showed that various his‐tagged antigens could induce specific responses with minimal cross‐reactivity, implying a very limited anti‐His‐tag response.[ 11 ] The anti‐His‐tag IgG titer was assessed in the post‐immune sera, by coating enzyme‐linked immunosorbent assay (ELISA) plates with a commercial His‐tag peptide. Overall, mice elicited minimal levels of anti‐His‐tag IgG titer in all the adjuvanted groups, with detected anti‐His IgG levels being similar to sera from untreated mice (Figure S8A–C, Supporting Information). Therefore, RBD particles formed by CoPoP liposomes induced a strong anti‐RBD IgG titer, with little if any detectable anti‐His‐tag antibodies.
Antibody function was initially assessed with a pseudovirus (PsV) assay. A murine leukemia virus‐based PsV expressing luciferase and gag/pol proteins pseudotyped with the S protein of SARS‐CoV‐2 was produced in HEK293T cells and was found to selectively enter cells expressing hACE2 (Figure S9, Supporting Information). As shown in Figure 3B, compared with other vaccine adjuvant groups, the sera from the RBD/CoPoP group potently inhibited viral entry. The NT50 (50% of neutralizing antibody titers) of the mice immunized with the mammalian produced RBD admixed with CoPoP/MPLA liposomes was 16430, whereas the NT50 with CoPoP/MPLA/QS‐21 was 30827. These NT50 values were orders of magnitude higher than most other the adjuvants including ISA720 (NT50: 339); Addavax (NT50: 78); PoP/MPLA (NT50: 56.6); alum (NT50: 219); AS01 (NT50: 191). The PsV neutralizing titers were similar for the insect‐produced RBD. Figure S10A,B show the full PsV entry inhibition curves for mammalian‐ and insect‐ produced RBD.
A SARS‐CoV‐2 surrogate virus neutralization test (sVNT) was used to further assess the nature of the functional antibodies. The sVNT assay is an in vitro, cell‐free method that detects antibodies that block the interaction of hACE2 and the RBD and has been used to predict neutralizing antibody titers in clinical specimens. As shown in Figure 3C, at 100‐fold dilution, post‐immune sera for CoPoP‐immunized mice inhibited 99% of the interaction between RBD and ACE2. In contrast, all the other vaccine adjuvants produced an inhibition at the baseline level of approximately 30%, the same level of serum of mice that did not receive any RBD‐immunogen at all. Again, sVNT results were similar between mammalian‐ and insect‐ produced RBD.
Next, a live virus neutralization test (VNT) was carried out with the SARS‐CoV‐2 strain, USA‐WA1/2020. Sera from mice immunized with the mammalian or insect produced RBD admixed with CoPoP liposomes prevented pathogenic cellular infection at 1:1280 diluted sera, the highest dilution assessed (Figure 3D). Convalescent sera therapy recommends the use of sera with a 1:160 VNT titer.[ 15 ] Thus, nanogram particlized RBD dosing in mice induced strongly functional antibodies. As indicated in the figure, these neutralization levels were substantially higher than the convalescent sera from 137 SARS‐CoV‐2‐infected humans that were within 2 months of the onset of symptoms, tested with the same protocol.[ 16 ] When admixed with alum, the RBD induced antibodies with limited capacity for virus neutralization, with neutralizing levels below recommendations for convalescent sera. Taken together, multiple antibody tests show that the RBD benefits by immunization in particle format. Tables S3 and S4, Supporting Information, tabulate all antibody characterization for mice and rabbits, respectively. Prior work in mice has shown that three immunizations with 5 µg RBD adjuvanted with alum induced neutralizing antibodies.[ 6 ] However, in the present study a dose of 0.1 µg with two immunizations did not. While this shows the CoPoP system holds dose‐sparing potential for RBD‐based vaccines, additional work is required to better understand the relationship between the antigen dose, the adjuvant used, and the resulting antibody quality.
Next, to assess RBD immunization in a second animal species, rabbits were immunized with a 20 µg dose of RBD admixed with either CoPoP liposomes or alum, intramuscularly, on day 0 and day 21. The post‐immune sera showed anti‐RBD IgG presence on day 21 followed by a boosting effect that led to the final day 42 antibody levels to be approximately tenfold higher for CoPoP compared to alum (Figure 4A). This is less than the 2–3 full orders of magnitude enhancement over alum observed in mice. As shown in Figure 4B and Figure S12, Supporting Information, high PsV neutralization activity was evident in the sera of rabbits immunized with CoPoP liposomes. Interestingly, the inclusion of QS‐21 in the liposomes significantly enhanced neutralization titer after boosting. Again, this result differs from the mouse data, where QS‐21 benefits appeared more modest. Similar results were observed in the sVNT assay where QS‐21 inclusion enhanced the blocking of the interaction between ACE2 and the RBD (Figure 4C). When live SARS‐CoV‐2 neutralization was assessed with VNT, post‐immune sera from rabbits immunized with CoPoP/MPLA/QS‐21 liposomes prevented infection at the highest dilution tested (1:1280). In contrast, only 1 of the 4 rabbits immunized with CoPoP/MPLA liposomes had neutralizing antibodies with comparable efficacy (Figure 4D). The reason behind the antibody enhancement with QS‐21 in rabbits warrants further investigation but could relate to the higher antigen dose used in rabbits, or more likely, immunological differences between species. Examination of the mouse antibody data reveals that the QS‐21 CoPoP post‐immune sera also generally had improved viral inhibition function, although the enhancement was subtle.
Figure 4.
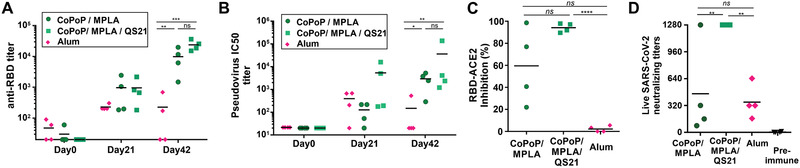
Rabbit RBD immunization. Rabbits were immunized with 20 µg RBD admixed with the indicated adjuvants on day 0 and 21, and day 42 serum was collected. A) Anti‐RBD IgG titer and B) PsV neutralization at indicated time points. C) Inhibition in a sVNT that measures interaction between the RBD hACE2. D) Live SARS‐CoV‐2 virus neutralization using post‐immune sera. For (A) and (B), log10 transformed titer were analyzed by one‐way ANOVA followed by Tukey's comparisons. For (C) and (D), data were analyzed by one‐way ANOVA followed by Tukey's comparisons. p*< 0.05, p**< 0.01, p***< 0.005, p****< 0.001.
Immune cell recruitment was assessed two days after mouse intramuscular immunization with 100 ng of RBD admixed with CoPoP/MPLA liposomes, CoPoP/MPLA/QS‐21 liposomes, alum or PBS. Flow cytometry was used to discriminate various cells in the draining lymph nodes.[ 11 , 17 ] As shown in Figure 5A and Figure S13, Supporting Information, CoPoP/MPLA liposomes and CoPoP/MPLA/QS‐21 liposomes induced enhanced recruitment of macrophages and monocytes, compared to alum. An increased level of CD11b− DCs was shown for all the adjuvant groups compared to control mice. Cd11b− DC cells play a role in cellular adaptive immune responses.[ 18 ] An increased level of CD11blow DCs was observed in mice treated with CoPoP/MPLA/QS‐21, but not CoPoP/MPLA liposomes nor alum. Therefore, a second factor by which CoPoP appears to lead to potent immunization, besides improving antigen delivery to APCs, is by enhanced recruitment of APCs.
Figure 5.
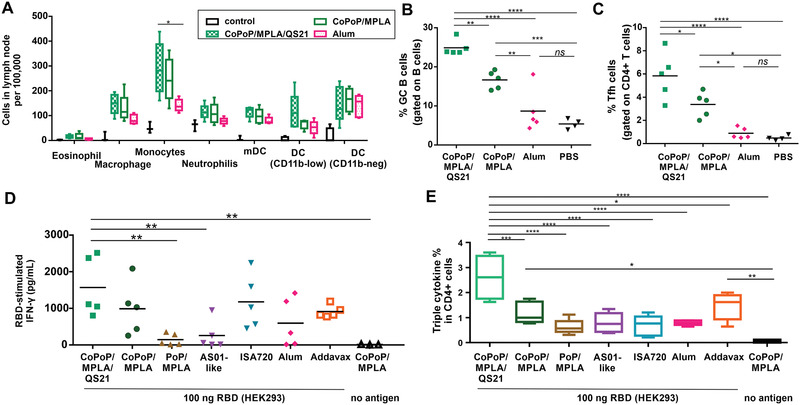
Antibody and cellular immune activation. A) Recruitment of immune cells in draining lymph nodes of mice, 48 h after intramuscular administration. B) GC B cell and C) Tfh cell populations were measured from collected lymph nodes 1 week after immunization with 100 ng of RBD admixed with CoPoP liposomes or alum. Splenocytes were collected from immunized mice and stimulated with RBD antigen prior to flow cytometry. D) IFN‐γ secretion from splenocytes of immunized mice after stimulation with the RBD. E) Intracellular staining triple cytokines (IFNγ, IL‐2 and TNFα) in CD4+ T cells. Data are from n = 4‐5 mice/group and are analyzed by one‐way ANOVA test followed by Tukey's comparisons. p*< 0.05, p**< 0.01, p***< 0.005, p****< 0.001). For (A) and (E), the line in the box represents the median, and the whiskers issuing from the box extend to the group minimum and maximum value. The length of the box represents the interquartile range.
Germinal center (GC) B cell formation was assessed following immunization. CoPoP liposomes enhanced the population of GC B cells, as well as the population of T follicular helper cells (Tfh cells). QS‐21 induced a higher degree of GC B cell formulation compared to similar liposomes lacking QS‐21 and to alum (Figure 5B,C, Figure S14, Supporting Information). Tfh cells play a significant role in protective immunity by helping B cells generate neutralizing antibodies.[ 19 ] This result may account for the enhanced immunity of the QS‐21‐containing liposomes observed in rabbits. Figure S15A,B, Supporting Information, show that mice immunized with CoPoP/MPLA, as well as CoPoP/MPLA/QS‐21 liposomes elicited higher levels of IgG2a antibodies than IgG1 antibodies, suggesting the immune response was biased toward a Th1 response.
Following RBD immunization on day 0 and day 14, splenocytes were isolated and assessed for induction of interferon gamma (IFNγ) secretion following exposure to the antigen (Figure 5D). Splenocytes from the mice immunized with CoPoP secreted a higher level of IFNγ relative to other adjuvants. This reflects the higher antigen‐specific T cell populations that were produced with the CoPoP adjuvant. Again, QS‐21 addition appeared to be beneficial in enhancing T cell responses. Polyfunctional T cells which express multiple cytokines have been shown as a protective immunity in viral infection.[ 20 ] Antigen‐specific CD4+ T cells that secrete IFNγ, IL‐2, and TNFα are desirable to protect against infection. Antigen‐specific CD8+ and CD4+ T cells that secrete IFNγ and TNFα are indicators of memory phenotype that could lead to long‐term protection for SARS‐CoV.[ 21 ] To address the induction of polyfunctional T cells, splenocytes were collected from immunized mice, followed by RBD stimulation in vitro. The cells were assessed with flow cytometry, first gating live/dead cells, followed by gating TCRβ+ CD4+ CD44hiFoxp3− for memory CD4+ T cells and TCRβ+CD8+CD44hi for memory CD8+ T cells (Figure S16, Supporting Information). As shown in Figure S17A,B, Supporting Information, single cytokine‐producing populations in CD4+ T cells and CD8+ T cells were observed for IFNγ, IL‐2, and TNFα. Later, cells were gated to assess all three cytokines, showing that splenocytes from mice immunized with the RBD and CoPoP/MPLA/QS‐21 generated stronger triple cytokine‐producing populations in CD4+ T cells (Figure 5E), as well as CD8+ T cells (Figure S17C, Supporting Information).
Local reactogenicity of the RBD admixed with various adjuvants was assessed in mice using a footpad swelling assay following a single intradermal vaccine injection. This approach has been used previously to gauge the reactogenicity of vaccines.[ 22 ] CoPoP/MPLA liposomes produced the least amount of local reactogenicity of all the adjuvants assessed, which included AS01‐like liposomes, alum, Addavax, and ISA720 (Figure 6A). The MPLA and QS‐21 content of CoPoP liposomes used throughout all the experiments in this work is 60% less than the AS01‐like formulation, which may contribute to the relatively decreased reactogenicity.
Figure 6.
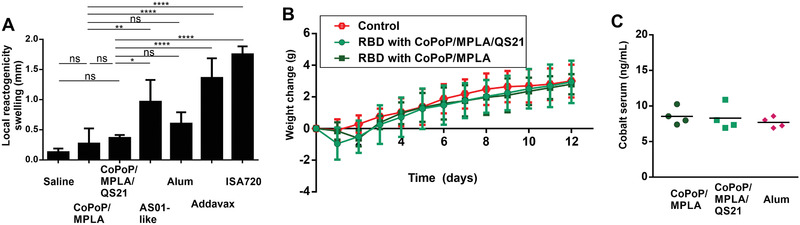
RBD immunization with CoPoP is well‐tolerated. A) Local reactogenicity of CoPoP liposomes compared to other adjuvants (all mixed prior to injection) based on footpad swelling. B) Weight change of mice after a single immunization with 1 µg RBD (tenfold higher than the functional dose used) with n = 5 mice per group. C) Cobalt level in serum a week following 1 µg RBD immunization with the indicated adjuvants. Bar graphs in (A) show mean +/− std. dev. for n = 4 mice/group. For (A), data were analyzed by one‐way ANOVA test followed by Tukey's comparisons. p*< 0.05, p**< 0.01, p****< 0.001).
Safety studies were carried out in mice using 1 µg RBD with CoPoP/MPLA or CoPoP/MPLA/QS‐21 liposomes (along with an MPLA and QS‐21 dose of 1.6 µg). This is a dose tenfold higher than that used for the immunogenicity studies. Mice immunized with CoPoP exhibited normal weight gain compared to untreated mice (Figure 6B). A complete blood cell count (Table S2, Figure S18A, Supporting Information) and serum chemistry panel analysis (Table S3, Figure S18B, Supporting Information) two weeks following treatment revealed test values within the normal ranges for virtually all of the 28 parameters assessed. While most parameters assessed were not different between control and immunized mice, there were differences in white blood cells, neutrophils, lymphocytes, glucose, cholesterol (CHOL), and alkaline phosphatase levels when comparing CoPoP/MPLA/QS‐21 to untreated mice, and differences of the eosinophils and CHOL levels for CoPoP/MPLA‐treated compared to untreated mice. However, these measurements were within the normal range for the tests and thus we do not consider the measurements to reflect any sign of toxicity, even at the elevated dosing use. The use of cobalt in CoPoP is a potential concern for a vaccine, although it is worth noting that vitamin B12, a cobalt tetrapyrrole has been shown to be safe in humans with 5 gram intravenous doses,[ 23 ] a level approximately 50 000‐fold higher than anticipated for CoPoP human dosing. Following mouse immunization, serum cobalt levels were not elevated relative to mice that received the RBD with alum (thus lacking any exogenous cobalt) (Figure 6C).
When recombinant RBD was admixed with CoPoP liposomes, antigen‐particles formed rapidly and spontaneously. Particles were stable in serum and the RBD maintained conformational integrity. RBD immunization with CoPoP drastically enhanced neutralizing antibody generation in mice and rabbits compared to identical liposomes that lacked cobalt, as well as a range of commercial vaccine adjuvants that also lack particle‐formation capacity. The inclusion of QS‐21 appeared to enhance the immune response based on providing T cell help. Although immunization with CoPoP was well‐tolerated in mice and rabbits, and a mouse safety study did not reveal any evidence of toxicity, further safety studies are needed. Overall, these data show that particle‐based presentation of the RBD using CoPoP liposomes is a potent vaccination strategy for SARS CoV‐2 and warrants further investigation.
Experimental Section
Materials
His‐tagged RBD expressed in the human embryonic kidney 293 cells (HEK293) cell line was purchased from RayBiotech (Cat # 230‐30162) and His‐tagged RBD expressed in sf9 cells was purchased from Genscript (Cat # Z03479). CoPoP and PoP were produced as previously described.[ 10 ] The following adjuvants were obtained: Montanide ISA720 (SEPPIC) and Alhydrogel 2% aluminum gel (Accurate Chemical and Scientific Corporation; Cat # A1090BS), Addavax (InVivoGen Cat # vac‐adx‐10). The following lipids were used: 1,2‐dipalmitoyl‐sn‐glycero‐3‐phosphocholine (DPPC, Corden Cat # LP‐R4‐057), 1,2‐Dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC, Avanti Cat # 850 375), cholesterol (PhytoChol, Wilshire Technologies), synthetic monophosphoryl Hexa‐acyl Lipid A, 3‐Deacyl (PHAD‐3D6A, Avanti Cat # 699 855). QS‐21 was obtained from Desert King. Granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) was obtained from Shenandoah Biotechnology (GM‐CSF; Cat # 200‐15‐AF). Cytochalasin B was obtained from ThermoFisher Scientific (cat # 14930‐96‐2). Antibodies for flow cytometry were obtained from Biolegend unless otherwise noted: CD11c‐APC Cy7 (Clone: N418; Cat # 117323; Lot B237078), CD3 PerCP/Cy5.5 (Clone: 17A2; Cat # 100217; Lot B233419), I‐A/I‐E Alex Fluor 700 (Clone: M5/114.15.2; Cat # 107621; Lot B24168), F4/80 Pacific Blue (Clone: BM8; Cat # 123123; Lot B217177), Ly‐6G PE (Clone: 1A8; Cat # 127607; Lot B235376), Ly‐6C (Clone: HK1.4; Cat # 128021: Lot B221000), CD11b PE/Cy7 (Clone: M1/70; Cat # 101215; Lot B249267). For antigen uptake into draining lymph node immune cells, the following antibodies were obtained from Biolegend: I‐A/I‐E Pacific Blue (Clone: M5/114.15.2; Cat # 107619; Lot: B252426), CD11c APC (Clone: N418; Cat # 117310; Lot: B253461), F4/80 PE (Clone: BM8; Cat # 123109; Lot: B251636) were used. For GC B cells staining, the following antibodies against GL7 Pacific Blue (Clone: GL7; Cat # 144613; Lot: B244647), CD95 PE (Clone: SA367H8; Cat # 152607; Lot: B239352), B220 APC (Clone: RA3‐6B2; Cat # 103211; Lot: B205878) were used. For assessing Tfh cells, the following antibodies were obtained from Biolegend: CXCR5 APC (Clone: L138D7 Cat # 145505; Lot B243491), PD‐1 PE (Clone: 29F.1A12; Cat # 135205; Lot: B251877), Alexa Fluor 488 CD4 (Clone: GK1.5; Cat # 100425; Lot: B238433). For intracellular cytokine staining: Surface markers to identified CD4+ and CD8+ T cells including, TCRβ APC/Cy7 (Clone: H57‐597; Cat # 109219), CD4 PE/Cy7 (Clone: RM4‐4; Cat # 116015), CD8 PreCP/Cy5.5 (Clone: 53–5.8; Cat # 140417), CD44 BV605 (Clone: IM7; Cat # 563058), Live/Dead marker (Cat # L34957); Intracellular markers included: IFNγ Pacific Blue (Clone: XMG1.2; Cat # 505817), TNFα PE (Clone: MP6‐XT22; Cat # 506305), Foxp3 Alex Fluor 488 (Clone: MF‐14, Cat # 126405), IL2 PE/TexasRed.(Clone: JES6‐5H4; Cat # 503839).
Cell Culture
For all experiments, cells were cultured and maintained at 37 °C in a humidified atmosphere containing 5% CO2. RAW264.7 murine macrophage cells were obtained from ATCC and cultured in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Pen/Strep). HEK293T cells were cultured in DMEM with 10% FBS, 1% Pen/Strep and 10 × 10−3 m sodium pyruvate. HEK293T‐hACE2 cells were kindly provided by Dr. Michael Farzan, and were cultured in DMEM with 10% FBS and 1% Pen/Strep, 10 × 10−3 m sodium pyruvate and 2 µg mL−1 of puromycin. BMDCs were derived from naive CD‐1 mice and cultured in RPMI 1640 medium with 10% FBS, 1% Pen/Strep and 20 ng mL−1 of GM‐CSF. Bone marrow was collected from the femurs and tibia of mice. The concentration of cells was seeded at 107 cells mL−1 and cultured in a 10 cm Petri dish in RPMI 1640 culture medium with 10% FBS and 20 ng mL−1 of recombinant GM‐CSF on day 0. On day 3, an additional 10 mL RPMI 1640 medium containing GM‐CSF was added, so the final volume of the medium was 20 mL. On day 6, non‐adherent cells were collected and cultured in a 24‐well plate at 5 × 105 cell mL−1 in RPMI 1640 culture medium containing 10% FBS and 1% Pen/Strep.
Liposome Preparation
Liposomes were prepared by an ethanol injection method, followed by nitrogen‐pressurized lipid extrusion in phosphate buffered saline (PBS) carried out at 60 °C.[ 11 ] The remaining ethanol was removed by dialysis against PBS twice at 4 °C. For liposomes containing QS‐21, QS‐21 (1 mg mL−1) was added to the liposomes after formation at an equal mass ratio as MPLA. Final liposome concentration was adjusted to 320 µg mL−1 CoPoP and liposomes were passed through a 0.2 µm sterile filter and stored at 4 °C. Liposome sizes and polydispersity index were determined by DLS with a NanoBrook 90 plus PALS instrument after 200‐fold dilution in PBS. The CoPoP/MPLA liposome formulation had a mass ratio of [DPPC: CHOL: MPLA: CoPoP] [4:2:0.4:1], CoPoP/MPLA/QS‐21 liposome formulation had a mass ratio of [DOPC: CHOL: MPLA: CoPoP: QS‐21] [20:5:0.4:1:0.4], PoP/MPLA liposomes served as the control liposomes which have a similar formulation as CoPoP/MPLA liposomes but lack of cobalt in the porphyrin‐phospholipid, this formulation had a mass ratio of [DPPC: CHOL: MPLA: PoP] [4:2:0.4:1] and AS01‐like liposome formation had a mass ratio of [DOPC: CHOL: MPLA: QS‐21] [20:5:0.4:0.4].
Slot Blot for Antigen Conformation
Liposomal samples (320 μg mL−1 of CoPoP) were mixed with RBD (80 μg mL−1) at antigen: CoPoP = 1:4 mass ratio. Pfs25 (a malaria antigen) with CoPoP liposomes served as a negative control in this experiment. A 48‐well slot blot apparatus (Cat # M1706545 from Bio‐Rad) was set up as described in the manufacturer instructions. The gasket support plate was placed onto the vacuum manifold and the sealing gasket was put on top of the support plate. A nitrocellulose membrane was pre‐wetted in PBS for 10 min at RT, and then placed on top of the sealing gasket. The 24‐well sample template was put on top of the membrane and secured by tightening the screws. Fifty microliter of mixed samples were slowly applied into each well, and the entire sample was allowed to flow through the membrane by gravity. The membrane was removed and blocked using 5% BSA in PBS for 30 min at RT, followed by incubating with 1000× diluted hACE2, Fc Tag (cat # AC2‐H5257 from ACRObiosystems) for 1 h at RT. The membrane was washed with PBS for 5 min twice, followed by incubation with HRP anti‐human IgG (cat # 109‐035‐098 from Jackson ImmunoResearch) for 30 min at RT. After incubation, the membrane was washed for 5 min with PBS 2 times. The membrane was imaged using a Bio‐Rad ChemiDoc Imager.
High‐Speed Centrifugation Binding Assay
CoPoP liposomes (320 μg mL−1 of CoPoP) mixed with RBD antigens (80 μg mL−1) at antigen: CoPoP = 1:4 mass ratio and the mixture were incubated for 3 h at RT. Liposomal samples were pelleted by high‐speed centrifugation (27 000 rcf) for 1.5 h at 4 °C, and unbound antigens were remained in the supernatant. After centrifugation, supernatant was collected and measured with the absorbance 562 nm by micro‐BCA assay. The percentage of antigen binding to the liposome was calculated based on the absorbance signal of soluble RBD in PBS: % antigen binding = [1 − OD562RBD+liposomes/OD562RBD] × 100% (1)where OD = optical density.
Ni‐NTA Competition Binding Test
To check RBD antigen‐binding stability in particle form, Ni‐NTA magnetic beads (ThermoFisher cat # 88831) were used to compete with pre‐bound proteins to the liposomes (1:4 mass ratio of total protein: CoPoP). Sufficient beads were added to ensure full binding of the free proteins in the sample. The samples were incubated with the beads for 30 min at RT before the supernatant and magnetic beads were separated and collected using a magnetic separator (ThermoFisher Cat # 12321D). The beads were then resuspended in PBS. Denaturing reducing loading dye was then added to all samples (supernatant and beads) and heated at 95 °C for 10 min. The samples were then loaded into a Novex 4–12% Bis‐Tris acrylamide gel (Invitrogen Cat # NP0321BOX) and subjected to PAGE and bands were visualized with Coomassie staining.
Fluorophore‐Labeled RBD
RBD was labeled with DY‐490‐NHS‐Ester (Dyomics cat # DY‐490) at RT. Labeling was carried out with DY‐490 to RBD at a molar ratio of 10:1. 100 µg of RBD was first dialysed against 100 × 10−3 m sodium bicarbonate buffer (pH 9) for 4–6 h at 4 °C twice, and then labeled with DY‐490 for 1 h at RT with continuous stirring. Free dye was removed by dialysis against PBS three times at 4 °C.
Fluorescent Quenching Assay
DY‐490‐labeled RBD was carried out by incubating antigens and liposomes with a 1:4 mass ratio of RBD: CoPoP or PoP at the final antigen concentration at 40 µg mL−1 at RT and the quenching of each sample was assessed at 0.5, 1, 2, and 3 h. To check the fluorescence signal, each of the incubation samples were diluted 1:200 in PBS in a 96‐well plate, and the fluorescence signal was measured at excitation/emission of 491/515 nm using a TECAN microplate reader. The percentage of binding was calculated based on the following formula: % antigen binding = [1 – FL liposomes+antigen/FL antigen] × 100% (2)where FL = fluorescent intensity.
Serum Stability
The mixture of DY‐490 labeled RBD (80 µg mL−1) with CoPoP liposomes (320 µg mL−1 CoPoP) were incubated for 3 h at RT followed by adding the same amount of 40% human serum in PBS into the sample to achieve a final concentration at 20% human serum. Samples were incubated at 37 °C for the indicated durations.
RBD Binding Assay to HEK293T‐ACE2 Cells
5 × 105 cells of HEK293T cells or HEK293T/hACE2 cells were incubated with labeled RBD (0.5 µg mL−1) with CoPoP/MPLA liposomes or CoPoP/MPLA/QS‐21 liposomes or PBS alone for 20 min on ice. After incubation, the cells were washed with ice‐cold PBS twice. The cells were lysed with lysis buffer (0.1% triton with 20 µg mL−1 proteinase K) at 60 °C for 10 min. The samples were placed in a 96‐well plate, and fluorescence signal were check at excitation/emission at 491/515 for DY‐490 labeled RBD.
Liposome Binding to hACE2‐Coated Plates
1 µg mL−1 of hACE2 in coating buffer (3.03g Na2CO3; 6 g NaHCO3 in 1 L distilled water, pH 9.6) was coated on the plate for 2 h at 37 °C. Wells were washed and blocked with 2% BSA in PBS for 2 h at 37 °C. At the meantime, RBD with CoPoP/MPLA* liposomes and CoPoP/MPLA/QS‐21* liposomes were incubated for 3 h. RBD (0.4 µg mL−1 of RBD) with CoPoP liposomes were added into each well and incubated for 1 h at RT. The wells were wash with PBS for 4 time, and 200 µL of PBS containing 0.5% Triton X were added into each well to break the liposomes. The CoPoP liposomes in this assay contained a small amount of PoP, which is highly fluorescent and the liposomal formulation for CoPoP/MPLA* liposomes was (DPPC: Chol: MPLA: CoPoP: PoP = 4:2::0.4:0.8:0.2) and CoPoP/MPLA/QS‐21* liposomes was (DOPC:Chol:MPLA:QS‐21: CoPoP: PoP = 20:5:0.4:0.4:0.8:0.2)
PsV Production
HEK293T cells were seeded at 5 × 105 cells mL−1 in a T75 flask overnight with DMEM medium with 10% FBS, and cultured at 37 °C in a humidified atmosphere containing 5% CO2. When the cells were approximately 60% confluent they were transfected with the retroviral vector pQCXIX encoding firefly luciferase, a plasmid expressing MLV gag and pol proteins, and a plasmid expressing the S protein of SARS‐CoV‐2 at a ratio of 5:5:1 by mass. 11 µg of total DNA was mixed with 44 µg of polyethylenimine at RT for 20 min, and then the mixture was slowly added to the cells. After 6 h of incubation at 37 °C, the medium was replaced with 10 mL of complete DMEM medium and the culture was incubated at 32 °C. After 48 h post transfection, the cultured medium containing PsV was harvested, and passed through a 0.45 µm pore size filter and the virus supernatant was supplemented with 10 × 10–3 m HEPES, aliquoted, and stored at −80 °C.
Cryo‐Electron Microscopy
20 μL of RBD‐HEK293 (80 µg mL−1) was mixed with 2 μL of CoPoP/MPLA liposomes or CoPoP/MPLA/QS‐21 liposomes in PBS. A volume of 3.6 μL of each sample was applied to holey carbon grids (c‐flat CF‐2/2‐3C‐T) previously glow discharged at 5mA for 15 seconds immediately before the application of the sample. Vitrification was performed in a Vitrobot Mark IV (Thermo Fisher Scientific Inc.) by blotting the grids once for three seconds and blot force +1 before they were plunged into liquid ethane. Temperature and relative humidity during the vitrification process were maintained at 25 °C and 100%, respectively. Data acquisition was performed using SerialEM software on the Titan Krios electron microscope at FEMR‐McGill, operated at 300 kV. Images were collected with a Gatan K3 direct electron detector equipped with a Bioquantum imaging filter. All images were collected using 3 s exposures and are the sum of 15 frames. Defocus ranged from −1.75 to −3 μm. Images were collected in counting mode using a total exposure of 50 e− A−2 at a nominal magnification of 42,000× corresponding to a calibrated pixel size of 2.12 Å. Images were cropped and prepared using the Adobe Photoshop.
Murine Immunization and Serum Analysis
Five‐week‐old female CD‐1 mice (ordered from Envigo RMS LLC) received intramuscular injections on days 0 and 14 containing 100 ng RBD combined with the following liposomal adjuvants: CoPoP/MPLA liposomes with the following formulation, [DPPC:CHOL:MPLA:CoPoP] of [4:2:0.4:1], CoPoP/MPLA/QS‐21 liposomes with the following formulation, [DOPC:CHOL:MPLA:CoPoP:QS‐21] of [4:2:0.4:1:0.4], PoP/MPLA liposomes with the following formulation, [DPPC:CHOL:MPLA:PoP] of [4:2:0.4:1], AS01‐like liposomes with the following formulation, [DOPC:CHOL:MPLA:CoPoP:QS‐21] of [4:2:1:1:1]. The following commercial adjuvants were used for comparison: ISA720, alum, and Addavax. CoPoP vaccines were prepared by incubating the RBD at a concentration of 80 µg mL−1 with liposomes (CoPoP or equivalent concentration of 320 µg mL−1) for 3 h at room temperature prior to dilution for immunization. ISA720 was prepared by incubating the diluted antigen with ISA720 at a 3:7 volume ratio and vortexing for 40 min at RT. Alum was diluted with PBS to 3 mg mL−1 concentration and then mixed with an equal volume of diluted antigen. Diluted antigens were mixed with an equal volume of Addavax.
Splenocyte Assay
Splenocytes were harvested from the immunized mice on day 28. Spleens were collected and passed through a 70 µm cell strainer in a 50 mL tube to collect single cells. Cells were centrifuged at 500 rcf, and red blood lysis buffer was added for 5 min on ice to lyse red blood cells. After incubation, 20 mL of PBS were added to dilute the lysis buffer, and samples were centrifuged at 500 rcf for 5 min. In a 96‐well culture plate, 2.5 × 105 cells/well were stimulated with 1 μg mL−1 of RBD and cultured in RPMI 1640 medium, with 10% FBS, 1% Pen/Strep, 1 × 10−3 m pyruvate, non‐essential amino acids, and 50 × 10−6 m 2‐mercaptoethanol, at 37 °C in a humidified atmosphere containing 5% CO2. In order to check IFNγ secretion, cultured medium was collected after 48 h, and IFNγ secretion level were measured based on IFNγ mouse ELISA kit (Fisher Scientific, Cat # 50‐183‐06). In order to check cytokines in antigen‐specific T cells, splenocytes were stimulated with 1 µg mL−1 of RBD for 18 h, followed by incubation with brefeldin A (BD Biosciences, Cat. # 555029) for another 6 h to block the cytokine secretion from the cells. Cells were stained for the surface markers using TCRβ APC/Cy7, CD4 PE/Cy7, CD8 PreCP/Cy5.5, CD44 BV605, Live/Dead marker (Cat. L34957) diluted in FASC buffer (cold‐PBS containing 0.5% BSA and 0.05% sodium azide) for 25 min on ice. The cells were washed with FASC buffer twice, and then fixed with the fixation/permeabilization buffer (BD cytofix/perm kit; Biosciences Cat. # 555028) for 10 min on ice. The cells were washed twice with FASC buffer, and permeabilization buffer (BD cytofix/perm kit; BD Biosciences Cat. # 555028) were added into each well for 20 min on ice. Intracellular markers including IFNγ Pacific Blue, TNFα PE, Foxp3 Alex Fluor 488, and IL2 PE/TexasRed were diluted in permeabilization buffer, and cells were stained for 25 min on ice. Stained cells were washed twice with permeabilization buffer, and then resuspended in FASC buffer prior to BD LSRFortessa TM X‐20 flow cytometry.
New Zealand White Rabbit Immunization
10–12‐week‐old female rabbits received intramuscular injections on days 0 and 21 of 20 µg of RBD‐HEK293 with CoPoP/MPLA liposomes with a [DPPC: CHOL: MPLA: CoPoP = 4: 2: 0.4: 1] mass ratio or with CoPoP/MPLA/QS‐21 liposomes with a [DPPC: CHOL: MPLA: CoPoP: QS‐21 = 20: 5: 0.4: 1: 0.4] mass ratio. Serum was collected on day 0, 21, and 42.
ELISA Assay
Anti‐RBD IgG titer was assessed by ELISA in 96‐well plates. 2.5 µg mL−1 of RBD in coating buffer (3.03g Na2CO3; 6 g NaHCO3 in 1 L distilled water, pH 9.6) were coated on the plate for 2 h at 37 °C. Wells were washed and blocked with 2% BSA in PBS containing 0.1% Tween‐20 (PBS‐T) for 2 h at 37 °C. Mouse sera (diluted in PBS‐T containing 1% BSA) were incubated in the wells for 1 h at 37 °C, followed by washing with PBS‐T. Goat anti‐mouse IgG‐HRP was added. Wells were washed again with PBS‐T before addition of tetramethylbenzidine solution. Titers were defined as the reciprocal serum dilution at which the absorbance at 450 nm exceeded background by greater than 0.5 absorbance units.
Anti‐His tag IgG titer was assessed by ELISA in 96‐well plates. 25 µg mL−1 of Hexa His peptide (Genscript Cat # RP11737) in coating buffer (3.03g Na2CO3; 6 g NaHCO3 in 1L distilled water, pH 9.6) were coated on the plate for 2 h at 37 °C. Wells were washed and blocked with 2% BSA in PBS containing 0.1% Tween‐20 (PBS‐T) for 2 h at 37 °C. Mouse sera (diluted in PBS‐T containing 1% BSA at 20 times dilution, following 100 time serial dilution) were incubated in the wells for 1 h at 37 °C, followed by washing with PBS‐T. Goat anti‐mouse IgG‐HRP was added. Wells were washed again with PBS‐T before addition of tetramethylbenzidine solution. Titers were defined as the reciprocal serum dilution at which the absorbance at 450 nm exceeded background by greater than 0.5 absorbance units.
PsV‐Based Neutralization Assay
HEK293T‐hACE2 cells were seeded into 96‐well plate at a density of 2 × 105 cells/well for overnight. Immunized sera from mice and rabbit with serial dilution were incubated with PsV at RT for 30 min, then 50 µL of pesudovirus with sera at different dilutions were added to each well after removing 50 µL of cultured medium, and the cells were cultured for 48 h. The medium was removed from each well and the cells were washed with 200 µL PBS, followed by adding 30 µL of lysis buffer (Promega E1500) for 10 min. The lysate was transferred into a white plate, and 100 µL of substrate were added. CentroPRO (Cat. # LB 962) was used to measure luciferase activity.
RBD‐hACE2 Inhibition Assay
SARS‐CoV‐2 cPass sVNT Kit (GenScript, Cat. L00847) was used to check if post immune sera could block the interaction between hACE2 and HRP‐RBD antigen. Mice sera were diluted 100× and rabbit sera were diluted 20× with sample dilution buffer. Positive and negative controls were included in the kit, and the control vials were diluted 10×. The diluted positive and negative controls, as well as the diluted samples were mixed with HRP‐RBD solution at a 1:1 volume, then incubated at 37 °C for 30 min. 100 µL of these mixtures were loaded into the wells of an ELISA plate pre‐coated with hACE2 and incubated at 37 °C for 15 min. The plate was washed 4 times to remove unbound HRP‐RBD. The percentage of inhibition was calculated as % = (1 − OD450 post immune sera/OD450 negative control) × 100% (3)
VNT Assay
The live VNT assay protocol was carried out at the same site in the way that recently tested for protective levels of neutralizing antibodies in convalescent sera from SARS‐CoV‐2‐infected human individuals.[ 16 ] The ability of plasma samples to neutralize SARS‐CoV‐2 host‐cell infection was determined with a traditional VN assay using a SARS‐CoV‐2 isolate deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS‐Related Coronavirus 2, Isolate USA‐WA1/2020, NR‐52281.[ 24 ] The assay was performed in triplicate, and a series of eight twofold serial dilutions of the serum was assessed. One‐hundred tissue culture infective dose 50 (TCID50) units of SARS‐CoV‐2 were added to twofold dilutions of serum and incubated for 1 h at 37 °C/5% CO2. The virus and serum mixture were added to Vero E6 cells grown in a 96‐well microtiter plates and incubated at 37 °C/5% CO2 for 3 d, after which the host cells were treated for 1 h with crystal violet‐formaldehyde stain (0.013% crystal violet, 2.5% ethanol, and 10% formaldehyde in 0.01 m PBS). The endpoint of the microneutralization assay was designated as the highest plasma dilution at which all three or two of three wells were not protected from virus infection, as assessed by visual examination.[ 25 ]
Lymph Node Studies for RBD Uptake
Mice were immunized with 1 µg of RBD‐DY490 with CoPoP/MPLA, CoPoP/MPLA/QS‐21, alum or AS01‐like liposome. After 48 h, mice were sacrificed and inguinal lymph nodes were collected. Lymph nodes were pass through a 70 µm cell strainer and 5 × 105 cells per tube were stained with the following murine antibodies against I‐A/I‐E, B220, CD11c or F4/80 (all from BioLegend) for 30 min at RT. The samples were washed with FASC buffer twice prior to BD LSRFortessa TM X‐20 flow cytometry. Flowjo (version 10) software was used for data analysis.
GC Cells and Tfh Cell Populations
Mice received 100 ng of RBD adjuvanted with CoPoP/MPLA, CoPoP/MPLA/QS‐21 or alum. Seven days after immunization, mice were sacrificed and the inguinal LN were collected. Lymph nodes were pass through a 70 µm cell strainer and 5 × 105 cells per tube were then stained with antibodies against B220, CD95, GL7. CD4, CXCR5, or PD‐1 for 30 min on ice. The samples were washed with FASC buffer twice prior to BD LSRFortessa TM X‐20 flow cytometry. Flowjo (version 10) software was used for data analysis.
Lymph Node Cell Recruitment
Mice were injected intramuscularly with CoPoP/PHAD liposomes or alum with 100 ng of Pfs25. 48 h after injection, mice were sacrificed and lymph nodes were collected for cell extraction. Cells were stained with combination antibodies against Ly6C, CD11b, Ly6G, CD11c, CD3, I‐A/I‐E, and F4/80, for 30 min on ice. The samples were washed with FASC buffer twice prior to BD LSRFortessa TM X‐20 flow cytometry. Flowjo (version 10) software was used for data analysis. Cells were first gated with CD11c and CD11b, then immune cells were identified based on surface marker in CD11chigh and CD11blow, NEU (Ly6Ghigh), eosinophils (Ly6Gint, F4/80int, SSC), monocytes (Ly6C high) and macrophage (F4/80high). Three types of DC cells were gated; for myeloid DC, we first gate Cd11chigh and CD11bhigh, then gated MHC‐II positive cells.
Acute Toxicity
8‐week‐old female CD‐1 mice were treated with intramuscular injection of CoPoP/MPLA/RBD or CoPoP/MPLA/QS‐21/RBD with 1 µg RBD (along with 4 µg CoPoP, 1.6 µg MPLA and optionally 1.6 µg QS‐21) and weight was monitored daily. Two weeks later, blood and serum were collected and subjected to standard complete blood cell count and a serum panel (IDEXX Cat # 98‐20590‐00). Cobalt levels were assessed in a separate study with 1 µg RBD combined with adjuvants as indicated (including 4 µg CoPoP for CoPoP‐based adjuvants) and sera was collected one week after intramuscular immunization and analyzed using ICP‐MS with a primary trace nutrient panel (50701) at Michigan State University Veterinary Diagnostic lab.
Local Reactogenicity
Mice received 1 µg of RBD admixed with different types of adjuvants, prepared in the same manner as for functional immunization. The mice received 50 µL of RBD and adjuvant injected in their left footpad and 50 µL of PBS into their right footpad as a control. Thickness of the footpads was measured by caliper 48 h after footpad injection and swelling was calculated by the following formula: Swelling = Thicknessleft‐footpad – Thicknessright‐footpad (4)
Conflict of Interest
J. F. L. and W.‐C. H. declare interest in POP Biotechnologies. The other authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
The authors acknowledge assistance from Dr. Michael Farzan and Dr. Huihui Mu for providing the reagents and protocols to generate pseudovirus, as well as providing HEK293 cells expressing hACE2. This study was supported by the National Institutes of Health (R01AI148557 and R01CA247771). The authors thank Kelly Sears and other staff members of the Facility for Electron Microscopy Research (FEMR) at McGill University. FEMR is supported by the Canadian Foundation for Innovation, Quebec Government and McGill University. All animal experiments were carried out in accordance to protocols approved by the University at Buffalo or the Pocono Rabbit Farm Institutional Animal Care and Use Committee.
Huang W.‐C., Zhou S., He X., Chiem K., Mabrouk M. T., Nissly R. H., Bird I. M., Strauss M., Sambhara S., Ortega J., Wohlfert E. A., Martinez‐Sobrido L., Kuchipudi S. V., Davidson B. A., Lovell J. F., SARS‐CoV‐2 RBD Neutralizing Antibody Induction is Enhanced by Particulate Vaccination. Adv. Mater. 2020, 32, 2005637. 10.1002/adma.202005637
References
- 1. Petersen E., Koopmans M., Go U., Hamer D. H., Petrosillo N., Castelli F., Storgaard M., Al Khalili S., Simonsen L., Lancet Infect. Dis. 2020, 20, e238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poland G. A., Ovsyannikova I. G., Crooke S. N., Kennedy R. B., Mayo Clinic Proceedings 2020, 95, 2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Funk C. D., Laferrière C., Ardakani A., Front. Pharmacol. 2020, 11, 937; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F., Proc. Natl. Acad. Sci. USA 2020, 117, 11727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tai W., He L., Zhang X., Pu J., Voronin D., Jiang S., Zhou Y., Du L., Cell Mol. Immunol. 2020, 17, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Wang C., Li W., Drabek D., Okba N. M. A., van Haperen R., Osterhaus A., van Kuppeveld F. J. M., Haagmans B. L., Grosveld F., Bosch B. J., Nat. Commun. 2020, 11, 2251; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) ter Meulen J., van den Brink E. N., Poon L. L., Marissen W. E., Leung C. S., Cox F., Cheung C. Y., Bakker A. Q., Bogaards J. A., van Deventer E., Preiser W., Doerr H. W., Chow V. T., de Kruif J., Peiris J. S., Goudsmit J., PLoS Med. 2006, 3, e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang J., Wang W., Chen Z., Lu S., Yang F., Bi Z., Bao L., Mo F., Li X., Huang Y., Hong W., Yang Y., Zhao Y., Ye F., Lin S., Deng W., Chen H., Lei H., Zhang Z., Luo M., Gao H., Zheng Y., Gong Y., Jiang X., Xu Y., Lv Q., Li D., Wang M., Li F., Wang S., Wang G., Yu P., Qu Y., Yang L., Deng H., Tong A., Li J., Wang Z., Yang J., Shen G., Zhao Z., Li Y., Luo J., Liu H., Yu W., Yang M., Xu J., Wang J., Li H., Wang H., Kuang D., Lin P., Hu Z., Guo W., Cheng W., He Y., Song X., Chen C., Xue Z., Yao S., Chen L., Ma X., Chen S., Gou M., Huang W., Wang Y., Fan C., Tian Z., Shi M., Wang F.‐S., Dai L., Wu M., Li G., Wang G., Peng Y., Qian Z., Huang C., Lau J. Y.‐N., Yang Z., Wei Y., Cen X., Peng X., Qin C., Zhang K., Lu G., Wei X., Nature 2020, 586, 572. [DOI] [PubMed] [Google Scholar]
- 7. Dai L., Zheng T., Xu K., Han Y., Xu L., Huang E., An Y., Cheng Y., Li S., Liu M., Yang M., Li Y., Cheng H., Yuan Y., Zhang W., Ke C., Wong G., Qi J., Qin C., Yan J., Gao G. F., Cell 2020, 182, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walls A. C., Fiala B., Schaefer A., Wrenn S., Pham M. N., Murphy M., Tse L. V., Shehata L., O'Connor M. A., Chen C., Navarro M. J., Miranda M. C., Pettie D., Ravichandran R., Kraft J. C., Ogohara C., Palser A., Chalk S., Lee E.‐C., Kepl E., Chow C. M., Sydeman C., Hodge E. A., Brown B., Fuller J. T., Dinnon K. H. III, Gralinski L. E., Leist S. R., Gully K. L., Lewis T. B., Guttman M., Chu H. Y., Lee K. K., Fuller D. H., Baric R. S., Kellam P., Carter L., Pepper M., Sheahan T. P., Veesler D., King N. P., biorXiv 2020, 10.1101/2020.08.11.247395. [DOI] [Google Scholar]
- 9. Quinlan B. D., Mou H., Zhang L., Guo Y., He W., Ojha A., Parcells M., Luo G., Li W., Zhong G., Choe H., Farzan M., biorXiv 2020, 10.1101/2020.04.10.036418. [DOI] [Google Scholar]
- 10. Shao S., Geng J., Yi H. A., Gogia S., Neelamegham S., Jacobs A., Lovell J. F., Nat. Chem. 2015, 7, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang W.‐C., Deng B., Lin C., Carter K. A., Geng J., Razi A., He X., Chitgupi U., Federizon J., Sun B., Long C. A., Ortega J., Dutta S., King C. R., Miura K., Lee S.‐M., Lovell J. F., Nat. Nanotechnol. 2018, 13, 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yuan M., Wu N. C., Zhu X., Lee C. D., So R. T. Y., Lv H., Mok C. K. P., Wilson I. A., Science 2020, 368, 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Federizon J., Frye A., Huang W. C., Hart T. M., He X., Beltran C., Marcinkiewicz A. L., Mainprize I. L., Wills M. K. B., Lin Y. P., Lovell J. F., Vaccine 2020, 38, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Mabrouk M. T., Huang W.‐C., Deng B., Li‐Purcell N., Seffouh A., Ortega J., Atilla‐Gokcumen G. E., Long C. A., Miura K., Lovell J. F., Int. J. Pharm. 2020, 589, 119843; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang W.‐C., Deng B., Seffouh A., Ortega J., Long C. A., Suresh R. V., He X., Miura K., Lee S.‐M., Wu Y., Lovell J. F., npj Vaccines 2020, 5, 23; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Huang W.‐C., Deng B., Mabrouk M. T., Seffouh A., Ortega J., Long C., Miura K., Wu Y., Lovell J. F., Malar. J. 2020, 19, 119843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. U. S. FDA, Investigational COVID‐19 Convalescent Plasma Guidance, retrieved from , https://www.fda.gov/media/136798/download 2020.
- 16. Salazar E., Kuchipudi S. V., Christensen P. A., Eagar T., Yi X., Zhao P., Jin Z., Long S. W., Olsen R. J., Chen J., Castillo B., Leveque C., Towers D., Lavinder J. J., Gollihar J., Cardona J. A., Ippolito G. C., Nissly R. H., Bird I., Greenawalt D., Rossi R. M., Gontu A., Srinivasan S., Poojary I., Cattadori I. M., Hudson P., Josleyn N. M., Prugar L., Huie K. E., Herbert A. S., Bernard D. W., Dye J. M., Kapur V., Musser J. M., J. Clin. Invest. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Calabro S., Tortoli M., Baudner B. C., Pacitto A., Cortese M., O'Hagan D. T., De Gregorio E., Seubert A., Wack A., Vaccine 2011, 29, 1812; [DOI] [PubMed] [Google Scholar]; b) Liang F., Lindgren G., Sandgren K. J., Thompson E. A., Francica J. R., Seubert A., De Gregorio E., Barnett S., O'Hagan D. T., Sullivan N. J., Koup R. A., Seder R. A., Lore K., Sci. Transl. Med. 2017, 9, eaal2094. [DOI] [PubMed] [Google Scholar]
- 18.a) Manh T. P., Alexandre Y., Baranek T., Crozat K., Dalod M., Eur. J. Immunol. 2013, 43, 1706; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ginhoux F., Liu K., Helft J., Bogunovic M., Greter M., Hashimoto D., Price J., Yin N., Bromberg J., Lira S. A., Stanley E. R., Nussenzweig M., Merad M., J. Exp. Med. 2009, 206, 3115; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Edelson B. T., Kc W., Juang R., Kohyama M., Benoit L. A., Klekotka P. A., Moon C., Albring J. C., Ise W., Michael D. G., Bhattacharya D., Stappenbeck T. S., Holtzman M. J., Sung S. S., Murphy T. L., Hildner K., Murphy K. M., J. Exp. Med. 2010, 207, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang J., Liu W., Wen B., Xie T., Tang P., Hu Y., Huang L., Jin K., Zhang P., Liu Z., Niu L., Qu X., Sci. Rep. 2019, 9, 10090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duvall M. G., Precopio M. L., Ambrozak D. A., Jaye A., McMichael A. J., Whittle H. C., Roederer M., Rowland‐Jones S. L., Koup R. A., Eur. J. Immunol. 2008, 38, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wherry E. J., Ahmed R., J. Virol. 2004, 78, 5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Honda‐Okubo Y., Saade F., Petrovsky N., Vaccine 2012, 30, 5373; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kataoka M., Yamamoto A., Ochiai M., Harashima A., Nagata N., Hasegawa H., Kurata T., Horiuchi Y., Vaccine 2009, 27, 1881. [DOI] [PubMed] [Google Scholar]
- 23. Sauer S. W., Keim M. E., Ann. Emerg. Med. 2001, 37, 635. [DOI] [PubMed] [Google Scholar]
- 24.a) Harcourt J., Tamin A., Lu X., Kamili S., Sakthivel S. K., Murray J., Queen K., Tao Y., Paden C. R., Zhang J., Li Y., Uehara A., Wang H., Goldsmith C., Bullock H. A., Wang L., Whitaker B., Lynch B., Gautam R., Schindewolf C., Lokugamage K. G., Scharton D., Plante J. A., Mirchandani D., Widen S. G., Narayanan K., Makino S., Ksiazek T. G., Plante K. S., Weaver S. C., Lindstrom S., Tong S., Menachery V. D., Thornburg N. J., Emerging Infect. Dis. 2020, 26, 1266; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gralinski L. E., Menachery V. D., Viruses 2020, 12, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sui J., Li W., Murakami A., Tamin A., Matthews L. J., Wong S. K., Moore M. J., Tallarico A. S., Olurinde M., Choe H., Anderson L. J., Bellini W. J., Farzan M., Marasco W. A., Proc. Natl. Acad. Sci. USA 2004, 101, 2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information